Chapter 45 Molecular Imaging of Gene Expression and Cell Therapy
INTRODUCTION
In 1953, Watson and Crick elucidated the structure of deoxyribonucleic acid (DNA) in a 2-page letter published in the journal Nature.1 Over the next 50 years, advances in genomic science and molecular medicine have resulted in a better understanding of the pathophysiology of many diseases. Recently, another revolution has been taking place. A new set of technologies, as part of a new field termed molecular imaging, is now being developed to noninvasively examine the integrative functions of molecules, cells, and organs in intact whole-body systems.2,3 The field remains in its infancy at present and to a large extent is limited to the laboratory environment. Notable exceptions are in the disciplines of oncology, neurology, and cardiology, which have started to make the transition from basic science to clinical application. Cardiac imaging modalities such as computed tomography (CT), magnetic resonance imaging (MRI), single-photon emission computed tomography (SPECT), positron emission tomography (PET), and ultrasound have seen major advances over the past decades. In the clinical setting, these modalities can provide outstanding data regarding organ structure and physiologic function. This chapter serves as an introduction to the field of cardiovascular molecular imaging, with specific emphasis on its utility in evaluating gene and cellular therapy. Because the field may be new to many, we have purposely simplified much of the discussion on technical details to make the context more appealing to a broad range of readers with different backgrounds.
FUNDAMENTALS OF GENE EXPRESSION
The central dogma of molecular biology states that genetic information flows from DNA to ribonucleic acid (RNA) to protein. The human genome has 23 pairs of chromosomes containing approximately 3 billion base pairs of DNA and ~23,000 genes.4,5 Each gene consists of sequences of four different nucleotides: adenine (A), guanine (G), cytosine (C), and thymine (T). The flow of information from DNA to RNA within the cell nucleus is termed transcription. Each messenger RNA (mRNA) is composed of intron and exon sequences. Introns are intervening sequences that are spliced out and removed. Exons are sequences that exit the nucleus to the cytoplasm to serve as templates for protein synthesis in a process called translation. The proteins are formed from a combination of 20 different units called amino acids. Proteins are considered the workhorse of cell machinery, performing activities ranging from gene regulation to cell integrity to receptor signaling. The versatility of molecular biology cloning techniques allows both the DNA and RNA to be manipulated. For example, restriction enzymes can be used to cleave the DNA of interest at specific sites to generate discrete gene fragments.6 They are named after the bacteria from which they were isolated (e.g., Eco RI from Escherichia coli and Hind III from Haemophilus influenza) and serve originally as part of the bacterial immune arsenal to protect itself against invasion by cleaving the DNA of foreign vectors. These techniques have been used to “cut and paste” therapeutic genes and reporter genes, as will be discussed later. For an in-depth discussion on the basics of molecular biology and genomics, please refer to a recent review article.
BACKGROUND OF CARDIOVASCULAR MOLECULAR IMAGING
Traditionally, researchers have monitored cardiac gene transfer by using reporter constructs such as β-galactosidase (β-gal),7 green fluorescent protein (GFP),8 and chloramphenicol-acetyl transferase (CAT).9 Cellular transplant therapies have employed similar techniques as well as newer approaches such as TaqMan reverse transcriptase polymerase chain reaction (RT-PCR),10 Y-chromosome paint probes,11 and antibodies specific to various stem cell types.12 All of these established traditional techniques, however, require invasive biopsies and/or postmortem tissue sampling for analysis. Molecular imaging offers distinct advantages by allowing for noninvasive, quantitative, and repetitive imaging of targeted macromolecules and biological processes in living organisms.2 Although a vast array of molecular imaging techniques are available, they all require two fundamental elements: (1) a molecular probe that can signal confirmation of gene expression by detecting messenger ribonucleic acid (mRNA) transcripts or proteins and (2) a method to monitor these probes or events. Presently, the two most widely used strategies are direct and indirect imaging.
Direct molecular imaging involves direct probe-target interaction. Targets can include receptors, enzymes, or mRNA. For probe-receptor imaging, radiolabeled monoclonal antibodies binding to tumor cell-specific surface antigens have been used for the past 2 decades.13 Recent examples of cardiac application involved imaging αvβ3 integrin receptor14 or vascular endothelial growth factor (VEGF) receptor15 expressed during angiogenesis after myocardial infarction (MI). For probe-enzyme imaging, the most well-known cardiac application is fluorine-18 (18F)-labeled-fluorodeoxyglucose (FDG), used to assess for myocardial tissue viability. After transport across an intact cell membrane, the 18F-labeled glucose analog undergoes phosphorylation by hexokinase and is retained intracellularly in proportion to the rate of cellular glycolysis.16 The radioactive 18F undergoes positron annihilation into two high-energy γ rays (511 keV), which can be detected as coincidence events by PET. For probe-mRNA imaging, radiolabeled antisense oligonucleotide (RASON) probes can be used.17,18 RASON probes are typically 12 to 35 nucleotides long and are complementary to a small segment of the target mRNA. However, the RASON approach is limited at this point due to (1) low number of target mRNA (~1000 copies) per cell compared to proteins (>10,000 copies); (2) limited tracer penetration across the cell membrane; (3) poor intracellular stability; (4) slow washout of unbound oligonucleotide probes; and (5) low target-to-background ratios. On the other hand, direct imaging of DNA (two copies) within the nuclear membrane has proven exceedingly difficult and is not yet feasible. In addition, knowing the activity of gene expression (as reflected in mRNA transcripts or protein levels) rather than the number of DNA copies is more relevant for biological research. The main disadvantage of direct imaging is that it requires synthesizing a customized probe for the product (e.g., receptor, enzyme, or mRNA) of every therapeutic gene of interest, which can be time-consuming and is not generalizable to most applications.
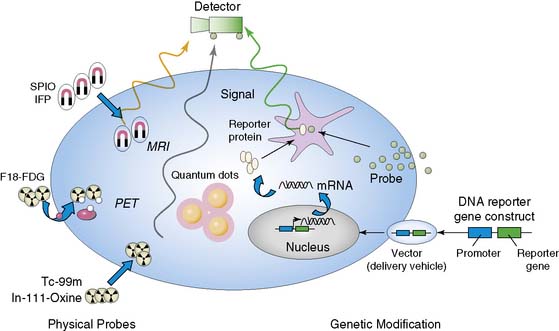
Indirect molecular imaging using reporter genes has only been recently validated. The concept of imaging reporter gene expression is illustrated in Figure 45-1. A reporter gene is first introduced into target tissues by viral or nonviral vectors. Using molecular biology techniques, the promoter or regulatory regions of genes can be cloned into different vectors to drive reporter gene mRNA transcription. Promoter activity can be “constitutive” (always on), “inducible” (turned on or off), or “tissue specific” (expressed only in the heart, liver, or other organs). Translation of mRNA leads to a reporter protein that can interact with the reporter probe. This interaction may be enzyme based or receptor-based. Probe signals can then be detected by various imaging modalities such as an optical charged coupled device (CCD) camera, PET, SPECT, or MRI.
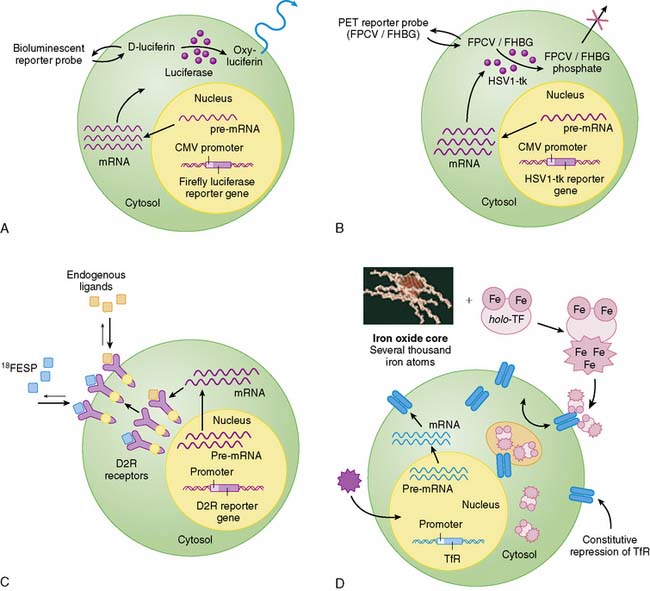
Clearly, the main advantage of the reporter gene system is its flexibility and multiplexing capacity.19 By altering various components, the reporter gene can provide information about the regulation of DNA by upstream promoters, intracellular protein trafficking, and the efficiency of vector transduction on cells. Likewise, the reporter probe itself does not have to be changed in order to study a new biological process, saving time and resources required for synthesis, testing, and validation of new reagents. However, the main disadvantage of indirect imaging is that it remains a surrogate marker for the physiologic/biochemical process of interest, as opposed to directly measuring receptor density, mRNA copies, or intracellular enzymatic activity, which might be more clinically relevant. It should be noted that in the case of monitoring stem cell fate, this is less of a concern, since the focus is on detecting the presence of a population of cells. Finally, the ideal reporter gene and/or reporter probe should have the following characteristics:
IMAGING TECHNIQUES
Optical Imaging
Bioluminescence imaging utilizes the photogenic properties of various luciferase genes cloned from different organisms, such as firefly (Photinus pyralis), jellyfish (Aequorea), coral (Renilla), and dinoflagellates (Gonyaulax). In the case of the firefly, the firefly luciferase enzyme (FL) converts its substrate (D-luciferin) to oxyluciferin via an ATP-dependent pathway. This process emits low levels of photons (2 to 3 eV) that can be detected and counted by a cooled CCD camera (e.g., Xenogen IVIS system).20 Unlike bioluminescence, fluorescence imaging does not require injection of a reporter substrate but relies upon an excitation wavelength that produces an emission wavelength for measurement.21 Recent technologic advances (e.g., eXplore Optix system) has allowed the measurement, quantification, and visualization of fluorescence intensity in small living animals using the time domain approach.22 In general, optical-based imaging is a relatively low-cost endeavor (i.e., typically $100 to $200K versus $500K to $1 million for small-animal PET and MRI systems) with the capacity for high throughput (i.e., several mice can be scanned at once). However, the aforementioned techniques suffer from low spatial resolution, inability to monitor multiple physiologic processes, and photon attenuation/scatter within deep tissues.23 Moreover, optical imaging has yet to be extrapolated into clinical usage. Use of novel intravascular catheter devices capable of detecting bioluminescence and/or fluorescence signals from deeper tissues or organs may be possible, but the general practicality of “invasive imaging” remains to be seen.24
Magnetic Resonance Imaging
Unlike optical imaging, MRI has the advantage of a very high spatial resolution (25 to 100 μm) and the ability to measure more than one physiologic parameter at once by using different radiofrequency pulse sequences.25 This makes MR a very attractive option for imaging reporter gene expression. The imaging signal is generated as a result of spin relaxation effects, which can be altered by atoms with high magnetic moments (e.g., gadolinium and iron). One particularly useful MR imaging signal amplification system is based on the cellular internalization of superparamagnetic probes such as monocrystalline iron oxide nanoparticles (MION) and crosslinked iron oxide (CLIO).26 MIONs or CLIOs can be linked to a variety of biomolecules to produce injectable probes for targets such as hematopoietic and neural progenitor cells,27,28 activated thrombotic factor XIII,29 and endothelial cell surface markers such as E-selectin.30 These studies hold promise for in vivo imaging in humans, given the widespread availability of clinical MR scanners, nontoxic and biodegradable properties of intravenous superparamagnetic particles, and the precedent of similar preparations already in clinical use.31 However, persisting residual signals from superparamagnetic particles may hinder the capacity for quantitative and repetitive imaging. MR is also several log of orders less sensitive (10−3 to 10−5 Molar) for detection of reporter probes compared to optical bioluminescence imaging (10−15 to 10−17) or PET (10−11 to 10−12) imaging.2 Therefore, further strategies for robust signal amplification will be necessary before this modality can be of practical use for imaging cardiac gene expression and detecting small numbers of transplanted cells.
Ultrasound
The scope of ultrasound for cardiac molecular imaging has seen increasing applications over the past decade.32–34 Targeted contrast agents have been constructed by linking ligands of interest to liposomes, perfluorocarbon nanoparticles, and encapsulated microbubbles,35 but these agents are relatively large, precluding their extravasation from the vasculature. While ultrasound-based molecular imaging may play an increasing role in endothelial imaging,36 its utility as a molecular cardiac imaging modality to track gene expression or cell therapy remains to be seen. Newer techniques in photoacoustic molecular imaging where the molecular imaging agents can extravasate from blood vessels may hold a potential solution to limitations of conventional ultrasound.37
Radionuclide Imaging (See Chapter 11)
Radionuclide probes are the first example of molecular probes used in the clinical setting, and this technology represents the evolutionary roots of molecular imaging as it is known today. PET, SPECT, and planar scintigraphy have all been used to detect radionuclide-labeled probes. However, PET exhibits several advantages compared to other modalities. First, PET is more sensitive compared to SPECT and MRI for detection of probe activity, as already discussed. This may allow monitoring of gene delivery by vectors with relatively low transfection efficiencies (e.g., plasmid) or weak promoters (e.g., tissue specific), as well as detection of low numbers of cells (e.g., cardiac stem cell transplants). Second, PET imaging is more quantitative (unlike MRI) and allows for dynamic imaging with tracer kinetic modeling for analysis of rate constants underlying the biochemical processes.38 Third, inasmuch as many PET tracers have a short half-life (e.g., ~110 minutes for 18F), daily repetitive imaging of tracer retention by targeted tissues is possible. Fourth, PET imaging is tomographic (unlike two-dimensional (2D) images from optical imaging), so a relatively precise location of probe signal can be identified within the heart. This is especially apt in the basic research environment, because current generations of small-animal PET scanners have a resolution of 13 to 23 mm3 compared to around 63 mm3 for clinical PET scanners.39 Finally, studies performed in these small-animal PET scanners can potentially be scaled up to human patients using clinical PET scanners with relative ease.3
IMAGING CARDIAC GENE THERAPY
Gene transfer has been heralded as the most promising therapy of molecular medicine in the 21st century. It is usually defined as the transfer and expression of DNA to somatic cells of an individual, with a resulting therapeutic effect (see Fundamentals of Gene Expression section). In cardiovascular diseases, gene therapy offers an exciting new approach to express the therapeutic factors locally in the myocardium.40 In general, the successful application of gene therapy requires three essential elements: (1) a vector for gene delivery, (2) delivery of the vector to the target tissue, and (3) a therapeutic gene to be expressed in a particular patient population.
These encouraging results have led to the initiation of several clinical trials, beginning in the 1990s. Of the 509 ongoing gene therapy trials in the United States, 46 are related to cardiovascular diseases. The majority of these are aimed at testing the safety and efficacy of therapeutic angiogenesis and, to a lesser extent, examining restenosis. In the late 1990s, several phase-1 open-labeled trials involving small numbers of patients with myocardial ischemia and peripheral vascular disease uniformly showed positive results.48–50 However, recent phase-2 randomized, double-blind, placebo-controlled trials have yielded conflicting, if not disappointing, results. The Vascular Endothelial Growth Factor in Ischemia for Vascular Angiogenesis (VIVA),51 FGF Initiating Revascularization Trial (FIRST),52 Adenovirus Fibroblast Growth Factor Angiogenic Gene Therapy (AGENT),53,54 and Kuopio Angiogenesis Trial (KAT)55 tested gene therapy using either VEGF or FGF. Unfortunately, these trials failed to show any consistent improvement in various parameters such as symptomatic improvement, ejection fraction, wall motion scores, myocardial perfusion, and restenosis rate.
Nonetheless, important lessons can be learned from these trials. They showed that angiogenesis is a complex process regulated by the interaction of various growth factors and may be difficult to stimulate using a single protein or gene injection. The ideal injection method, delivery vector, and patient population remain to be determined. The pharmacokinetics and pharmacodynamics of therapeutic gene expression will need to be defined first before gene therapy can proceed further to widespread clinical usage, similar to the research and development of experimental drugs.56 Finally, since there is no available method of assessing gene expression in vivo, the investigators are unable to determine whether the lack of symptomatic improvement is due to poor injection technique, insufficient gene expression, host inflammatory response, or an inappropriate therapeutic gene.
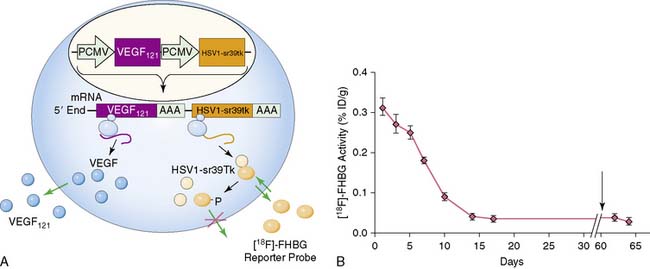
Imaging of cardiac transgene expression has been established in several proof-of-principle studies involving injection of various reporter genes into the myocardium and following the kinetics of transgene expression over time, using optical bioluminescence, microPET, and clinical PET imaging.57–61 The feasibility of linking a PET reporter gene to a therapeutic gene was also demonstrated in two separate studies.62,63 In this case, an adenovirus containing two constitutive cytomegalovirus promoters driving a VEGF121 therapeutic gene and an HSV1-sr39tk PET reporter gene separated by polyadenine sequences was constructed (Ad-CMV-VEGF121-CMV-HSV1-sr39tk). Wu et al. injected the construct into the myocardium of a rat in a myocardial infarction model.63 Reporter gene expression (which indirectly reflects the VEGF121 therapeutic gene expression) persisted for only about 2 weeks, owing to host immune response against the adenovirus. At 2 months, there were no significant improvements in myocardial contractility, perfusion, or metabolism, as measured by echocardiography, nitrogen-13-labeled ammonia (13N-NH3), and FDG imaging, between study and control groups (Fig. 45-2). Thus, this study highlights the importance of monitoring the pharmacokinetics of gene expression. It also demonstrates the proof of principle that any other cardiac therapeutic genes of interest (e.g., HIF-1α, SERCA2a, heat shock protein, or endothelial nitric oxide synthase) can likewise be coupled to a PET reporter gene for subsequent noninvasive monitoring.
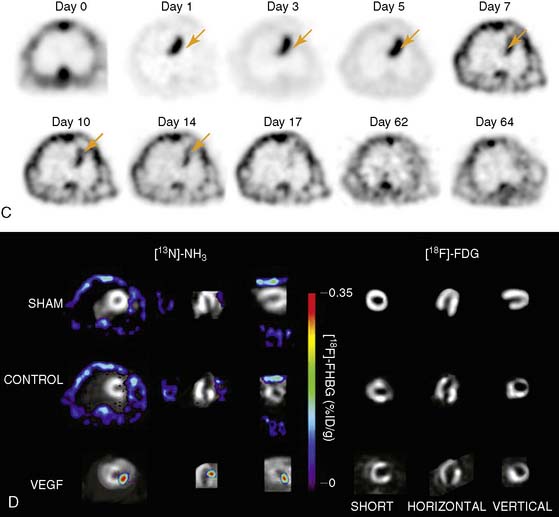
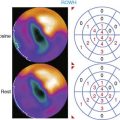
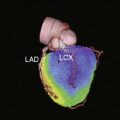
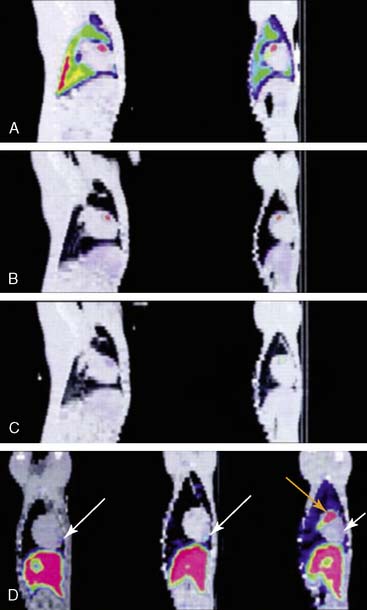
The feasibility of transferring imaging results from a small-animal PET scanner to a clinical PET scanner (ECAT EXACT scanner, Siemens/CTI) has been demonstrated in porcine models as well. Bengel et al. showed that myocardial tissues infected with adenovirus expressing HSV1-tk had significantly higher iodine-124-labeled 2′-fluoro-2′-deoxy-1-beta-d-arabinofuranosyl-5-iodouracil (124I-FIAU) retention during the first 30 minutes following injection.60 The FIAU uptake correlated with ex vivo images, autoradiography, and immunohistochemistry for reporter gene product after euthanasia. However, the signal-to-background ratio at the site of HSV1-tk injection was only around 1.25 during the first 30 minutes following delivery. Afterwards, there was significant washout at 45 to 120 minutes post injection, and the 124I-FIAU retention became similar to control myocardial regions. Recently, Rodriquez-Porcel et al. also demonstrated the feasibility of imaging HSV1-tk reporter gene expression with fluorine-18-labeled fluoro-3-hydroxymethyl butylguanine (18F-FHBG) reporter probe in a porcine model. Following endomyocardial injection using a Biocardia catheter, myocardial gene expression activity was quantified by clinical PET-CT system (Fig. 45-3). The best myocardial signal-to-background (left ventricular) ratio was obtained 3 hours post-injection (180 minutes, 4.63 ± 1.4, versus 90 minutes, 1.78 ± 0.6; P < 0.05). Autoradiography and microPET confirmed the increased 18F-FHBG uptake in the anteroseptum. Overall, these studies suggest that the combination of HSV1-tk/FHBG may be superior to HSV1-tk/FIAU for imaging cardiac transgene expression.64

In the future, it will also be useful to have the following arsenals for cardiac gene therapy: (1) non-immunogenic vectors such as non-viral minicircle plasmids that can prolong transgene expression in the myocardium,65 (2) cardiac tissue-specific promoter (e.g., myosin light chain kinase or troponin) that can diminish unwanted extracardiac activity,66 (3) development of novel gene therapy approaches such as short hairpin RNA interference,67 and (4) multimodality molecular imaging approaches that can monitor the location, magnitude, and duration of transgene expressions, as well as their downstream functional effects.56
IMAGING CARDIAC CELL DELIVERY
In recent years, research in stem cell therapy has rivaled or even surpassed gene therapy as a promising treatment for ischemic heart disease. In fact, stem cell therapy now occupies the lion’s share of scientific presentations, media attention, and the public imagination. By most accounts, this field “started” in 2001 when Orlic et al. published a seminal (albeit still controversial to date) study showing that a particular type of bone marrow cells (lin−/c-kit+) could form de novo myocardium occupying 68% of the infarcted left ventricle just 9 days after transplantation in a mouse model.68 At present, there are many studies reporting the efficacy of fetal and neonatal cardiomyocytes,69 skeletal myoblasts,70,71 bone marrow cells (BMCs),68 and embryonic stem cell derivatives72 for myocardial repair. As with any type of therapy, each cell type also has its own merits and pitfalls.73
Fetal and neonatal cardiomyocytes have been regarded as promising candidates for cell replacement therapy,69 but their restricted availability and ethical dilemma render widespread clinical application impractical. Autologous skeletal myoblasts were the first cell type to be used clinically for cell-based cardiac repair.70 Skeletal myoblasts are attractive candidates because they can be cultured and expanded ex vivo from muscle biopsies, and they survive well after transplantation because of their strong resistance to ischemia. Skeletal myoblast transplantation has been shown to provide functional benefit in animal models of infarction, but a large placebo-controlled, randomized trial in humans did not demonstrate sustained efficacy as defined by the primary end point of left ventricular ejection fraction (LVEF).74 Furthermore, the increased number of early postoperative arrhythmic events after skeletal myoblast transplantation raises serious concerns and warrants further investigation. By contrast, transplantation of BMCs has been shown to improve heart function in animal studies, and no serious complications have been reported in clinical trials to date. However, results from three large clinical trials published in 2006 in the New England Journal of Medicine showed inconsistent benefits that at best led to short-term but no long-term improvement.75–77 In the accompanying editorial, the author urges the scientific community to “guard against both premature declarations of victory and premature abandonment of a promising therapeutic strategy” and that “this strategy is likely to depend on continued and effective coordination of rigorous basic and clinical investigations.”78 More recently, isolation of resident cardiac stem cells from human biopsy samples has been demonstrated.79 Whether these cells can promote cardiac regeneration and improve heart function in humans following transplantation will need to be addressed in future clinical trials.
One of the main limitations of cardiac stem cell therapy is the lack of available methods to assess stem cell survival. In clinical studies, changes in myocardial perfusion, viability, and perfusion are assessed by echocardiography, nuclear SPECT and PET imaging, or MR imaging. These parameters measure the therapeutic effects but do not actually detect the presence or absence of transplanted stem cells. This is an important distinction because most patients also undergo concurrent coronary artery bypass grafting (CABG) or percutaneous coronary intervention (PCI), and it is not clear whether the improvement is due to these procedures or to the transplanted stem cells. Likewise, in animal studies, analysis of stem cell survival is based on postmortem histology, which is invasive and precludes longitudinal monitoring. Thus, the ability to study stem cells in the context of the intact whole-body system rather than by indirect (clinical trials) or invasive (animal studies) means will give better insight into the underlying biology and physiology of stem cells. Several investigators have started addressing this issue via different approaches, including radionuclide labeling, ferromagnetic labeling, and reporter gene labeling (Fig. 45-4).73
Radionuclide Labeling
This technique requires the transplanted cells to be directly labeled with a radioisotope prior to transplantation. The advantages are high sensitivity and immediate translation to clinical practice because indium-111 (111In)-oxine and 18F-FDG are approved by the U.S. Food and Drug Administration (FDA) as clinical radiotracers. The technique has demonstrated utility for tracking cell homing in, on infarcted myocardium and distribution following injection. Aicher et al. demonstrated that 111In-oxine-labeled endothelial and hematopoietic progenitor cells accumulated in infarcted rat myocardium after intraventricular injection.80 However, by 96 hours postinjection, only about 5% of the injected dose of radioactivity was found in the heart. This highlights the sensitivity of the method, because although only a few cells persisted in the heart, they were nevertheless detectable. An inherent limitation of radionuclide labeling is the short half-life of the radioactive probes, which precludes long-term imaging. For radiotracers with a shorter half-life (e.g., 18F-FDG is 110 minutes), one can follow those cells at most, out to 1 day. Even for radiotracers that have longer half-lives (e.g., copper-64 [64Cu], 12.7 hrs; [111In], 67.3 hours; thallium-201 [201Tl], 74 hours; [124I], 96 hours), the longest time period of imaging would still be limited to within the first 1 to 2 weeks. Furthermore, the radiotracer itself has other concerns, specifically potential adverse effects of the tracer on stem cell viability and differentiation and the leakage of the tracer from the labeled cells, which can provide a false-positive signal.81
Nanoparticle Labeling
Semiconductor quantum dots (QDs) are a new class of fluorescent probes that have been used in noninvasive imaging in recent years.82,83 This technique makes use of fluorescent semiconductor nanocrystals to detect membrane molecules of interest. The excitation wavelengths of the QDs can be manipulated, ranging from ultraviolet to near-infrared ranges. Depending on the size and composition of these probes, they can be designed to emit different wavelengths of light. The photostability of QDs, as seen in their resistance to photobleaching and long-lasting fluorescence, makes them appealing for tracking stem cells in vivo.84 However, the effects of QDs on stem cell proliferation and differentiation remain unclear; mixed results have been reported using different origins of stem cells or experimental protocols.82,85 Finally, several other obstacles, including the tendency for aggregation of QDs in the cytosol, difficulty in delivery of QDs into cells, and nonspecific binding to multiple molecules,86 must be overcome before QDs can realize their full potential in clinical imaging. More recently, Raman nanoparticles have also started to be imaged in small living subjects and may provide as much as 100-fold greater sensitivity than QD-based approaches.87,88
Iron Particle Labeling
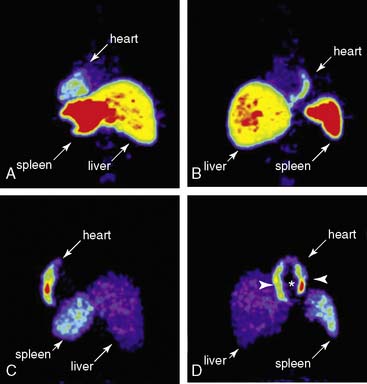
Compared to radionuclide imaging, the main advantage of MRI is its capacity for high anatomic resolution. In general, MRI techniques can be divided into two classes based on the contrast agent being used. Lanthanide gadolinium (Gd3+) is applied for T1-weighted contrast enhanced images, and superparamagnetic iron oxide (SPIO) particles are applied for T2-weighted images. Gadolinium-enhanced images have been used in previous studies to track human neural and mesenchymal stem cells for up to 6 weeks.89 However, SPIO particles are favored and more widely used for stem cell imaging because a lower concentration (5–10 µm/L) is required. The immediate translation of SPIO to clinical studies is feasible because certain superparamagnetic formulations have already been approved by the FDA and found to be biocompatible, safe, and nontoxic. Recent studies have demonstrated the utility of MRI for long-term tracking of bone-marrow-derived mesenchymal stem cells (MSCs). Amado et al. labeled porcine MSCs with iron oxide, induced a myocardial infarct (MI) by balloon occlusion of the left anterior descending (LAD) artery, and then injected MSCs intramyocardially 3 days after MI.90 The iron oxide–labeled cells created hypoenhanced regions. Interestingly, relative to the intensity of hypoenhancement at 2 days posttransplantation, approximately 42% of the signal still remained at 8 weeks. Similarly, Kraitchmen et al. magnetically labeled porcine MSCs with iron oxide, injected the cells into adult swine following MI, and demonstrated that the cells could be tracked for up to 3 weeks using MRI.91 MRI showed that the cell survival trend correlated with proximity to the infarct; specifically, cells injected into the infarct zone had more robust survival than those injected into healthy myocardium. In a related study, Kraitchmen et al. compared the sensitivity of SPECT to MRI to detect MSCs injected intravenously following MI in canines (Fig. 45-5).92 MSCs were co-labeled with [111In]-oxine and iron oxide, thus enabling SPECT and MRI tracking, respectively. SPECT demonstrated MSCs homing and engrafting to the heart for up to 7 days, whereas MRI was insufficiently sensitive to detect the cells in the heart or any other organ. In summary, MRI currently lacks the sensitivity of radionuclide-based labeling (SPECT/PET), which limits its ability to detect small numbers of cells in the heart (>105 cells are needed to be detectable).92 In addition, the ferromagnetic probe may become diluted with cell division, making it difficult to quantify proliferating cells. Last, the detected signal does not necessarily indicate whether the cells are viable. This is because iron particles can remain internalized in nonviable cells, and the iron particle may be engulfed by tissue macrophages, both events creating a false-positive signal.93,94
Reporter Gene Labeling
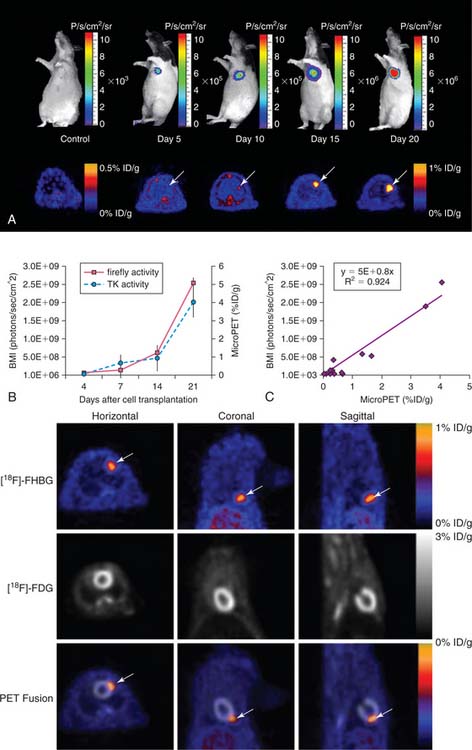
In contrast to physical labeling (radionuclide and iron particles), reporter gene labeling requires genetic modification of the stem cells. In the first proof-of-principle study using reporter genes to track cardiac cell survival, Wu et al. transfected rat H9c2 embryonic cardiomyoblasts with adenovirus carrying either firefly luciferase or HSV1-sr39tk reporter gene before injection into the rat myocardium.95 Cell survival was monitored noninvasively by optical bioluminescence or small-animal PET imaging. Cell signal activity was quantified in units of photons per second per square centimeter per steradian (photons/sec/cm2/sr) or percentage injected dose of 18F-FHBG per gram tissue (%ID/g), respectively. In both cases, drastic reductions in signal activity were seen within the first 1 to 4 days due to acute donor cell death from inflammation, ischemia, and apoptosis. Interestingly, this pattern of cell death was consistent with other reports using traditional ex vivo assays such as TUNEL apoptosis,96 classical histology,97 and TaqMan RT-PCR.10 However, all of these ex vivo techniques required large numbers of animals to be sacrificed at different time points.
Given that several types of reporter genes and reporter probes are now available, it should be possible in the future to perform multimodality imaging of stem cell transplantation. For example, the feasibility of imaging murine embryonic stem cells transplanted into the rodent myocardium has been demonstrated (Fig. 45-6).98 These cells were genetically manipulated to express a triple-fusion reporter that consists of red fluorescence protein (RFP) for fluorescence activated cell sorting (FACS) analysis and single cell fluorescence microscopy, firefly luciferase (Fluc) for high throughput bioluminescence imaging, and herpes simplex virus thymidine kinase (HSVtk) for small-animal PET imaging. Imaging analysis revealed significant intracardiac and extracardiac teratoma formation with transplantation of undifferentiated embryonic stem cells. Thus, future efforts will need to involve differentiated derivatives such as embryonic stem cell–derived endothelial cells99 or cardiomyocytes100 for myocardial repair while avoiding the tumorigenicity issues.101 Follow-up studies have also been demonstrated in a large-animal model using clinical PET/CT imaging of porcine MSCs stably transduced with Renilla luciferase, red fluorescent protein, and herpes simplex virus truncated thymidine kinase (RL-RFP-HSVttk) (Fig. 45-7).102 In addition, several studies have used transgenic mice that constitutively express reporter genes to understand the spatiotemporal kinetics of bone marrow stem cell homing,103 the optimal adult stem cell type for improving cardiac function,104 the efficacy of different biomatrices capable of improving cell survival,105 the fate of resident stem cells isolated from the heart,106 and the effect of acute versus chronic infarction on stem cell viability.107 These multi-reporter gene approaches may be helpful for evaluating other issues relevant to stem cell biology, such as imaging stem cell survival, proliferation, and differentiation using tissue-specific promoters.
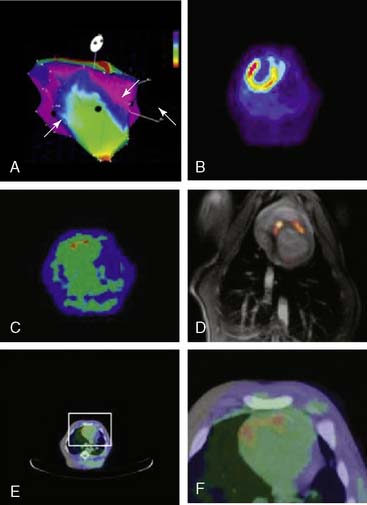
Clinical Imaging
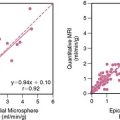
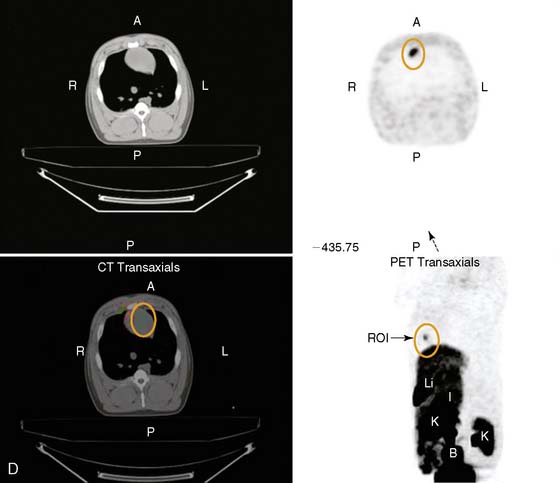
The first steps toward using molecular imaging technology to track cardiac cell therapy in humans have already been undertaken. In a recent study by Hofmann et al., PET imaging was used to track [18F]-FDG-labeled autologous bone marrow cells for treatment of patients following acute MI (Fig. 45-8).108 Unselected bone marrow cells were radiolabeled with 100 MBq [18F]-FDG and infused into the infarct-related coronary artery (3 patients), or injected into the antecubital vein (3 patients). In an additional group of 3 patients, a CD34+-enriched population of bone marrow cells was delivered by intracoronary infusion. In all groups, cells were administered 5 to 10 days following coronary stenting, and PET imaging was carried out 50 to 75 minutes after the procedure. PET successfully detected [18F] signals in all groups, with higher intramyocardial signal in the intracoronary versus intravenous delivery groups. Of the two groups receiving intracoronary infusion, the CD34+-enriched population had a higher myocardial signal than unselected BMSCs (1.3% to 2.6% versus 14% to 39%, P < 0.005), suggesting enhanced homing to the injured myocardial milieu associated with CD34+ stem cells (see Fig. 45-8). Although conducted in a small number of patients, this study demonstrates nicely a potential means to track stem cell therapy on a short-term basis. The fact that differing myocardial signal between groups was observed is encouraging insofar as it helps validate the sensitivity of clinical PET scanning for tracking cellular delivery and homing. As mentioned earlier, the major limitation of this technique is its inability to track long-term survival kinetics and cellular trafficking following therapy, given the half-life of 18F-FDG is only around 110 minutes. Furthermore, radiolabeled cell signals do not provide information regarding cell proliferation, because the radiotracers cannot be “passed on” from mother to daughter cells. It would be most useful, for example, to observe changes in myocardial stem cell populations over weeks and correlate their late-phase survival or proliferation with functional improvement. In the future, genetically engineered constructs integrated into the transplanted stem cells may allow for such measurements, using a variety of molecular imaging techniques discussed here. Very recently, the feasibility of imaging CD8+ T cells genetically engineered to express the HSV1-tk and imaged with 18F-FHBG PET has been performed, and this sets the stage for human imaging of cell populations.109 However, significant challenges remain to be overcome. From the imaging standpoint, the main obstacles are understanding the lowest number of detectable stem cells in the heart and the use of other reporter genes (e.g., human dopamine 2 receptor or human sodium iodide symporter or humanized HSVtk) that may be less immunogenic compared to the viral HSV1-tk.61,110,111
1. Watson J.D., Crick F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737-738.
2. Massoud T.F., Gambhir S.S. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17:545-580.
3. Phelps M.E. Inaugural article: positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci U S A. 2000;97:9226-9233.
4. International Human Genome Sequencing Consortium: Finishing the euchromatic sequence of the human genome. Nature. 2004;431(7011):931-945.
5. International Human Genome Sequencing Consortium: Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921.
6. Tefferi A: Genomic basics: DNA structure, gene expression, cloning, genetic mapping, and molecular tests. Semin Cardiothorac Vasc Anesth. 2006;10(4):282-290.
7. Schröder G., Risch K., Nizze H., et al. Immune response after adenoviral gene transfer in syngeneic heart transplants: effects of anti-CD4 monoclonal antibody therapy. Transplantation. 2000;70:191-198.
8. Hajjar R.J., Schmidt U., Matsui T., et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci U S A. 1998;95:5251-5256.
9. Kass-Eisler A., Falck-Pedersen E., Alvira M., et al. Quantitative determination of adenovirus-mediated gene delivery to rat cardiac myocytes in vitro and in vivo. Proc Natl Acad Sci U S A. 1993;90:11498-11502.
10. Muller-Ehmsen J., Whittaker P., Kloner R.A., et al. Survival and development of neonatal rat cardiomyocytes transplanted into adult myocardium. J Mol Cell Cardiol. 2002;34:107-116.
11. Herzog E.L., Chai L., Krause D.S. Plasticity of marrow-derived stem cells. Blood. 2003;102:3483-3493.
12. Shim W.S., Jiang S., Wong P., et al. Ex vivo differentiation of human adult bone marrow stem cells into cardiomyocyte-like cells. Biochem Biophys Res Commun. 2004;324:481-488.
13. Verel I., Visser G.W., van Dongen G.A. The promise of immuno-PET in radioimmunotherapy. J Nucl Med. 2005;46(Suppl 1):164S-171S.
14. Meoli D.F., Sadeghi M.M., Krassilnikova S., et al. Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction. J Clin Invest. 2004;113:1684-1691.
15. Rodriguez-Porcel M., Cai W., Gheysens O., et al. Imaging of VEGF receptor in a rat myocardial infarction model using PET. J Nucl Med. 2008;49:667-673.
16. Schelbert H.R. 18F-deoxyglucose and the assessment of myocardial viability. Semin Nucl Med. 2002;32:60-69.
17. Shi N., Boado R.J., Pardridge W.M. Antisense imaging of gene expression in the brain in vivo. Proc Natl Acad Sci U S A. 2000;97:14709-14714.
18. Tavitian B., Terrazzino S., Kuhnast B., et al. In vivo imaging of oligonucleotides with positron emission tomography. Nat Med. 1998;4:467-471.
19. Wu J.C., Yla-Herttuala S. Human gene therapy and imaging: cardiology. Eur J Nucl Med Mol Imaging. 2005;32(2):S346-57.
20. Contag P.R., Olomu I.N., Stevenson D.K., et al. Bioluminescent indicators in living mammals. Nat Med. 1998;4:245-247.
21. Lippincott-Schwartz J., Patterson G.H. Development and use of fluorescent protein markers in living cells. Science. 2003;300:87-91.
22. Ramjiawan B., Ariano R.E., Mantsch H.H., et al. Immunofluorescence imaging as a tool for studying the pharmacokinetics of a human monoclonal single chain fragment antibody. IEEE Trans Med Imaging. 2002;21:1317-1323.
23. Wu J.C., Sundaresan G., Iyer M., et al. Noninvasive optical imaging of firefly luciferase reporter gene expression in skeletal muscles of living mice. Mol Ther. 2001;4:297-306.
24. Funovics M.A., Weissleder R., Mahmood U. Catheter-based in vivo imaging of enzyme activity and gene expression: feasibility study in mice. Radiology. 2004;231:659-666.
25. Bogdanov A., Weissleder R. In vivo imaging of gene delivery and expression. Trends Biotechnol. 2002;20:S11-S18.
26. Weissleder R., Moore A., Mahmood U., et al. In vivo magnetic resonance imaging of transgene expression. Nat Med. 2000;6:351-355.
27. Kircher M.F., Allport J.R., Graves E.E., et al. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 2003;63:6838-6846.
28. Lewin M., Carlesso N., Tung C.H., et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18:410-414.
29. Jaffer F.A., Tung C.H., Wykrzykowska J.J., et al. Molecular imaging of factor XIIIa activity in thrombosis using a novel, near-infrared fluorescent contrast agent that covalently links to thrombi. Circulation. 2004;110:170-176.
30. Kang H.W., Josephson L., Petrovsky A., et al. Magnetic resonance imaging of inducible E-selectin expression in human endothelial cell culture. Bioconjug Chem. 2002;13:122-127.
31. Bulte J.W., Kraitchman D.L. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004;17:484-499.
32. Kaufmann B.A., Lewis C., Xie A., et al. Detection of recent myocardial ischaemia by molecular imaging of P-selectin with targeted contrast echocardiography. Eur Heart J. 2007;28:2011-2017.
33. Kaufmann B.A., Sanders J.M., Davis C., et al. Molecular imaging of inflammation in atherosclerosis with targeted ultrasound detection of vascular cell adhesion molecule-1. Circulation. 2007;116:276-284.
34. Pascotto M., Leong-Poi H., Kaufmann B., et al. Assessment of ischemia-induced microvascular remodeling using contrast-enhanced ultrasound vascular anatomic mapping. J Am Soc Echocardiogr. 2007;20:1100-1108.
35. Dayton P.A., Ferrara K.W. Targeted imaging using ultrasound. J Magn Reson Imaging. 2002;16:362-377.
36. Kaufmann B.A., Lindner J.R. Molecular imaging with targeted contrast ultrasound. Curr Opin Biotechnol. 2007;18:11-16.
37. de la Zerda A., Zavaleta C., Keren S., et al. Carbon nanotubes as contrast agents for photoacoustic imaging of living mice. Nat Nanotechnol. 2008;3:557-562.
38. Schelbert H.R., Inubushi M., Ross R.S. PET imaging in small animals. J Nucl Cardiol. 2003;10:513-520.
39. Tai Y.C., Chatziioannou A.F., Yang Y., et al. MicroPET II: design, development and initial performance of an improved microPET scanner for small-animal imaging. Phys Med Biol. 2003;48:1519-1537.
40. Yla-Herttuala S., Alitalo K. Gene transfer as a tool to induce therapeutic vascular growth. Nat Med. 2003;9:694-701.
41. Takeshita S., Pu L.Q., Stein L.A., et al. Intramuscular administration of vascular endothelial growth factor induces dose-dependent collateral artery augmentation in a rabbit model of chronic limb ischemia. Circulation. 1994;90:II228-II234.
42. Brogi E., Wu T., Namiki A., et al. Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation. 1994;90:649-652.
43. Shyu K.G., Wang M.T., Wang B.W., et al. Intramyocardial injection of naked DNA encoding HIF-1alpha/VP16 hybrid to enhance angiogenesis in an acute myocardial infarction model in the rat. Cardiovasc Res. 2002;54:576-583.
44. Steg P.G., Tahlil O., Aubailly N., et al. Reduction of restenosis after angioplasty in an atheromatous rabbit model by suicide gene therapy. Circulation. 1997;96:408-411.
45. Miyamoto M.I., del Monte F., Schmidt U., et al. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A. 2000;97:793-798.
46. Kozarsky K.F., Donahee M.H., Glick J.M., et al. Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler Thromb Vasc Biol. 2000;20:721-727.
47. Matsui T., Li L., del Monte F., et al. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100:2373-2379.
48. Rosengart T.K., Lee L.Y., Patel S.R., et al. Angiogenesis gene therapy: phase I assessment of direct intramyocardial administration of an adenovirus vector expressing VEGF121 cDNA to individuals with clinically significant severe coronary artery disease. Circulation. 1999;100:468-474.
49. Symes J.F., Losordo D.W., Vale P.R., et al. Gene therapy with vascular endothelial growth factor for inoperable coronary artery disease. Ann Thorac Surg. 1999;68:830-836. discussion 836–837
50. Vale P.R., Losordo D.W., Milliken C.E., et al. Left ventricular electromechanical mapping to assess efficacy of phVEGF(165) gene transfer for therapeutic angiogenesis in chronic myocardial ischemia. Circulation. 2000;102:965-974.
51. Henry T.D., Annex B.H., McKendall G.R., et al. The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation. 2003;107:1359-1365.
52. Simons M., Annex B.H., Laham R.J., et al. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation. 2002;105:788-793.
53. Grines C.L., Watkins M.W., Mahmarian J.J., et al. A randomized, double-blind, placebo-controlled trial of Ad5FGF-4 gene therapy and its effect on myocardial perfusion in patients with stable angina. J Am Coll Cardiol. 2003;42:1339-1347.
54. Grines C.L., Watkins M.W., Helmer G., et al. Angiogenic Gene Therapy (AGENT) trial in patients with stable angina pectoris. Circulation. 2002;105:1291-1297.
55. Hedman M., Hartikainen J., Syvanne M., et al. Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: phase II results of the Kuopio Angiogenesis Trial (KAT). Circulation. 2003;107:2677-2683.
56. Pislaru S., Janssens S.P., Gersh B.J., et al. Defining gene transfer before expecting gene therapy: putting the horse before the cart. Circulation. 2002;106:631-636.
57. Wu J.C., Inubushi M., Sundaresan G., et al. Positron emission tomography imaging of cardiac reporter gene expression in living rats. Circulation. 2002;106:180-183.
58. Wu J.C., Inubushi M., Sundaresan G., et al. Optical imaging of cardiac reporter gene expression in living rats. Circulation. 2002;105:1631-1634.
59. Inubushi M., Wu J.C., Gambhir S.S., et al. Positron-emission tomography reporter gene expression imaging in rat myocardium. Circulation. 2003;107:326-332.
60. Bengel F.M., Anton M., Richter T., et al. Noninvasive imaging of transgene expression by use of positron emission tomography in a pig model of myocardial gene transfer. Circulation. 2003;108:2127-2133.
61. Chen I.Y., Wu J.C., Min J.J., et al. Micro-positron emission tomography imaging of cardiac gene expression in rats using bicistronic adenoviral vector-mediated gene delivery. Circulation. 2004;109:1415-1420.
62. Anton M., Wittermann C., Haubner R., et al. Coexpression of herpesviral thymidine kinase reporter gene and VEGF gene for noninvasive monitoring of therapeutic gene transfer: an in vitro evaluation. J Nucl Med. 2004;45:1743-1746.
63. Wu J.C., Chen I.Y., Wang Y., et al. Molecular imaging of the kinetics of vascular endothelial growth factor gene expression in ischemic myocardium. Circulation. 2004;110:685-691.
64. Miyagawa M., Anton M., Haubner R., et al. PET of cardiac transgene expression: comparison of 2 approaches based on herpesviral thymidine kinase reporter gene. J Nucl Med. 2004;45:1917-1923.
65. Huang M., Chen Z.Y., Hu S., Jia F., Li Z., Hoyt G., Robbins R.C., Kay M.A., Wu J.C. Novel minicircle vector for gene therapy in murine myocardial infarction. Circulation. 2009;120:S230-S237.
66. Boecker W., Bernecker O.Y., Wu J.C., et al. Cardiac-specific gene expression facilitated by an enhanced myosin light chain promoter. Mol Imaging. 2004;3:69-75.
67. Huang M., Chan D.A., Jia F., Xie X., Li Z., Hoyt G., Robbins R.C., Chen X., Giaccia A.J., Wu J.C. Short hairpin RNA interference therapy for ischemic heart disease. Circulation. 2008;118:S226-S233.
68. Orlic D., Kajstura J., Chimenti S., et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701-705.
69. Soonpaa M.H., Koh G.Y., Klug M.G., et al. Formation of nascent intercalated disks between grafted fetal cardiomyocytes and host myocardium. Science. 1994;264:98-101.
70. Menasche P., Hagege A.A., Vilquin J.T., et al. Autologous skeletal myoblast transplantation for severe postinfarction left ventricular dysfunction. J Am Coll Cardiol. 2003;41:1078-1083.
71. Taylor D.A., Atkins B.Z., Hungspreugs P., et al. Regenerating functional myocardium: improved performance after skeletal myoblast transplantation. Nat Med. 1998;4:929-933.
72. Laflamme M.A., Chen K.Y., Naumova A.V., et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015-1024.
73. Zhang S.J., Wu J.C. Comparison of imaging techniques for tracking cardiac stem cell therapy. J Nucl Med. 2007;48(12):1916-1919.
74. Menasche P., Alfieri O., Janssens S., et al. The Myoblast Autologous Grafting in Ischemic Cardiomyopathy (MAGIC) trial: first randomized placebo-controlled study of myoblast transplantation. Circulation. 2008;117:1189-1200.
75. Assmus B., Honold J., Schachinger V., et al. Transcoronary transplantation of progenitor cells after myocardial infarction. N Engl J Med. 2006;355:1222-1232.
76. Lunde K., Solheim S., Aakhus S., et al. Intracoronary injection of mononuclear bone marrow cells in acute myocardial infarction. N Engl J Med. 2006;355:1199-1209.
77. Schachinger V., Erbs S., Elsasser A., et al. Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006;355:1210-1221.
78. Rosenzweig A. Cardiac cell therapy: Mixed results from mixed cells. N Engl J Med. 2006;355:1274-1277.
79. Smith R.R., Barile L., Cho H.C., et al. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896-908.
80. Aicher A., Brenner W., Zuhayra M., et al. Assessment of the tissue distribution of transplanted human endothelial progenitor cells by radioactive labeling. Circulation. 2003;107:2134-2139.
81. Adonai N., Nguyen K.N., Walsh J., et al. Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci U S A. 2002;99:3030-3035.
82. Dubertret B., Skourides P., Norris D.J., et al. In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science. 2002;298:1759-1762.
83. Jaiswal J.K., Mattoussi H., Mauro J.M., et al. Long-term multiple color imaging of live cells using quantum dot bioconjugates. Nat Biotechnol. 2003;21:47-51.
84. Lin S., Xie X., Patel M.R., et al. Quantum dot imaging for embryonic stem cells. BMC Biotechnol. 2007:67.
85. Hsieh S.C., Wang F.F., Lin C.S., et al. The inhibition of osteogenesis with human bone marrow mesenchymal stem cells by CdSe/ZnS quantum dot labels. Biomaterials. 2006;27:1656-1664.
86. Jaiswal J.K., Simon S.M. Potentials and pitfalls of fluorescent quantum dots for biological imaging. Trends Cell Biol. 2004;14:497-504.
87. Keren S., Zavaleta C., Cheng Z., et al. Noninvasive molecular imaging of small living subjects using Raman spectroscopy. Proc Natl Acad Sci U S A. 2008;105:5844-5849.
88. Zavaleta C., de la Zerda A., Liu Z., et al. Non-invasive Raman spectroscopy in living mice for evaluation of tumor targeting with carbon nanotubes. Nano Lett. 2008;8:2800-2805.
89. Bulte J.W., Douglas T., Witwer B., et al. Magnetodendrimers allow endosomal magnetic labeling and in vivo tracking of stem cells. Nat Biotechnol. 2001;19:1141-1147.
90. Amado L.C., Saliaris A.P., Schuleri K.H., et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci U S A. 2005;102:11474-11479.
91. Kraitchman D.L., Heldman A.W., Atalar E., et al. In vivo magnetic resonance imaging of mesenchymal stem cells in myocardial infarction. Circulation. 2003;107:2290-2293.
92. Kraitchman D.L., Tatsumi M., Gilson W.D., et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112:1451-1461.
93. Li Z., Suzuki Y., Huang M., et al. Comparison of reporter gene and iron particle labeling for tracking fate of human embryonic stem cells and differentiated endothelial cells in living subjects. Stem Cells. 2008;26:864-873.
94. Chen I.Y., Greve J.M., Gheysens O., Willmann J.K., Rodriguez-Porcel M., Chu P., Sheikh AY, Faranesh A.Z., Paulmurugan R., Yang P.C., Wu J.C., Gambhir S.S. Comparison of optical bioluminescence reporter gene and superparamagnetic iron oxide MR contrast agent as cell markers for noninvasive imaging of cardiac cell transplantation. Mol Imaging Biol. 2009;11(3):178-187.
95. Wu J.C., Chen I.Y., Sundaresan G., et al. Molecular imaging of cardiac cell transplantation in living animals using optical bioluminescence and positron emission tomography. Circulation. 2003;108:1302-1305.
96. Zhang M., Methot D., Poppa V., et al. Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. J Mol Cell Cardiol. 2001;33:907-921.
97. Murry C.E., Wiseman R.W., Schwartz S.M., et al. Skeletal myoblast transplantation for repair of myocardial necrosis. J Clin Invest. 1996;98:2512-2523.
98. Cao F., Lin S., Xie X., et al. In vivo visualization of embryonic stem cell survival, proliferation, and migration after cardiac delivery. Circulation. 2006;113:1005-1014.
99. Li Z., Wu J.C., Sheikh A.Y., et al. Differentiation, survival, and function of embryonic stem cell derived endothelial cells for ischemic heart disease. Circulation. 2007;116:I46-I54.
100. Cao F., Wager R.A., Wilson K.D., et al. Transcriptional and functional profiling of human embryonic stem cell-derived cardiomyocytes. PLoS ONE. 2008:e3474.
101. Cao F., Li Z., Lee A., et al. Noninvasive de novo imaging of human embryonic stem cell-derived teratoma formation. Cancer Res. 2009;69:2709-2713.
102. Gyongyosi M., Blanco J., Marian T., et al. Serial non-invasive in vivo positron emission tomographic (PET) tracking of percutaneously intramyocardially injected autologous porcine mesenchymal stem cells modified for transgene reporter gene expression. Circ Cardiovasc Imaging. 2008;1:94-103.
103. Sheikh A.Y., Lin S.A., Cao F., et al. Molecular imaging of bone marrow mononuclear cell homing and engraftment in ischemic myocardium. Stem Cells. 2007;25:2677-2684.
104. van der Bogt K.E., Sheik A.Y., Schrepfer S., et al. Comparison of different adult stem cell types for treatment of myocardial ischemia. Circulation. 2008;118:S121-S129.
105. Cao F., Sadrzadeh Rafie A.H., Abilez O.J., et al. In vivo imaging and evaluation of different biomatrices for improvement of stem cell survival. J Tissue Eng Regen Med. 2007;1:465-468.
106. Li Z., Chun H., Xie X., et al. Imaging survival and function of transplanted cardiac stem cells. J Am Coll Cardiol. 2009;53:1229-1240.
107. Swijnenburg R.J., Govaert J.A., Stein W., et al. Effect of timing of bone marrow cell delivery on cell viability and cardiac recovery following myocardial infarction [abstract]. Circulation. 2008;118:S_790.
108. Hofmann M., Wollert K.C., Meyer G.P., et al. Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation. 2005;111:2198-2202.
109. Yaghoubi S., Jensen M.C., Satyamurthy N., et al. Non-invasive detection of therapeutic cytolytic T cells with [18F]FHBG positron emission tomography in a glioma patient. Nat Clin Pract Oncol. 2009;6(1):53-58.
110. Ponomarev V., Doubrovin M., Shavrin A., et al. A human-derived reporter gene for noninvasive imaging in humans: mitochondrial thymidine kinase type 2. J Nucl Med. 2007;48:819-826.
111. Terrovitis J., Kwok K.F., Lautamaki R., et al. Ectopic expression of the sodium-iodide symporter enables imaging of transplanted cardiac stem cells in vivo by SPECT or PET. J Am Coll Cardiol. 2008;52(20):1650-1660.