Molecular Genetics of Pancreatobiliary Neoplasms
Laura D. Wood
Ralph H. Hruban
Introduction
Decades of research have established that cancer is a genetic disease caused by accumulation of somatic mutations in oncogenes and tumor suppressor genes. Some patients inherit gene mutations that predispose them to cancer, whereas others acquire all alterations somatically. However, in both inherited and sporadic cancers, tumors acquire numerous somatic alterations throughout their development—some are driver alterations that play crucial roles in tumorigenesis, whereas others are passenger alterations with no functional consequences. Knowledge of somatic mutations has greatly advanced understanding of basic tumor biology and has revolutionized the practice of oncology, with mutation-specific diagnostic and treatment strategies now available for multiple tumor types.
Pancreatobiliary neoplasms are among the best genetically characterized tumors. Studies of their molecular features have shown that morphologically defined entities also represent molecularly distinct neoplasms: genetics mirrors morphology. With this newly acquired understanding of the molecular biology of pancreatobiliary neoplasms, the field is primed to move into an era of genomic medicine. In the coming decades, knowledge of the molecular underpinnings of a tumor will be crucial for individual patient care, and pathologists will continue to play a pivotal role in the translation of genetic findings into clinical tests. Therefore, a sound understanding of the molecular genetics of pancreatobiliary neoplasms will be required to care effectively for patients in the era of genomic medicine.
Pancreatic Neoplasms
Ductal Adenocarcinoma
Germline Alterations
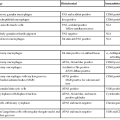
Approximately 10% of pancreatic ductal adenocarcinomas (PDAs) have a familial basis, and several genetic syndromes lead to an increased risk of PDA (Table 34.1). However, the genetic basis for most cases of familial PDA remains unknown.1 Still, the influence of inherited genetics is clear: a family history of PDA significantly increases an individual’s risk for the disease.2
Table 34.1
Genes with Germline Alterations Causing Increased Risk of Pancreatobiliary Neoplasia
| Gene | Chromosome | Syndrome | Neoplasm |
| BRCA2 | 13 | Familial breast cancer | PDA |
| PALB2 (FANCN) | 16 | Familial breast cancer | PDA |
| P16/CDKN2A | 9 | Familial atypical multiple mole melanoma syndrome (FAMMM) | PDA |
| STK11/LKB1 | 19 | Peutz-Jeghers syndrome (PJS) | PDA, IPMN |
| PRSS1, SPINK1 | 7, 5 | Hereditary pancreatitis | PDA |
| hMSH2, hMLH1, hPMS1, hPMS2, hMSH6 (formerly GTBP) | Multiple | Lynch syndrome/hereditary nonpolyposis colorectal cancer (HNPCC) | PDA (medullary variant) |
| ATM | 11 | Ataxia-telangiectasia | PDA |
| FANCC, FANCG | Multiple | Fanconi anemia pathway | PDA (?) |
| APC | 5 | Familial adenomatous polyposis (FAP) | PDA (?) |
| VHL | 3 | von Hippel-Lindau syndrome (VHL) | SCA, PanNET |
| MEN1 | 11 | Multiple endocrine neoplasia type 1 (MEN1) | PanNET |
| TSC1, TSC2 | 9, 16 | Tuberous sclerosis complex (TSC) | PanNET |
| NF1 | 17 | Neurofibromatosis type 1 (NF1) | PanNET |
| Unknown | 11 | Beckwith-Wiedemann syndrome (BWS) | PB |
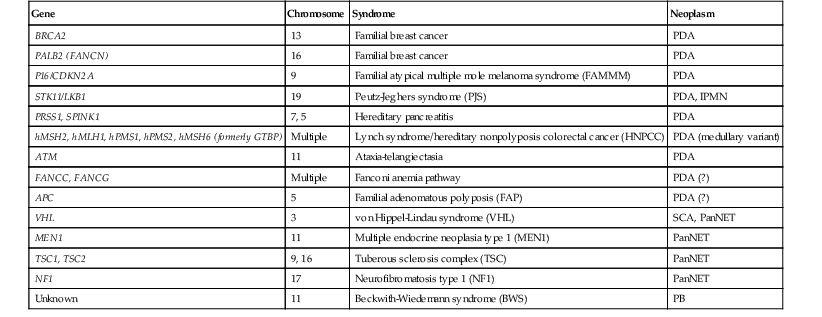
IPMN, Intraductal papillary mucinous neoplasm; PanNET, well-differentiated pancreatic neuroendocrine tumor; PB, pancreatoblastoma; PDA, pancreatic ductal adenocarcinoma; SCA, serous cystadenoma; (?), weak or questionable association
Germline mutations in genes in the Fanconi anemia pathway, which encode proteins involved in repair of DNA cross-linking damage, have been strongly associated with familial PDA.1 Germline mutations in BRCA2, a crucial component of this pathway, result in increased risk of breast, ovarian, pancreatic, and other cancers.3,4 Importantly, some patients with BRCA2-related familial PDA have no personal or family history of breast or ovarian cancer, and the low penetrance of some germline BRCA2 mutations necessitates a high index of suspicion for this genetic alteration in patients with familial PDA.4,5 In addition to germline BRCA2 mutations, germline alterations in other genes in the Fanconi pathway also play important roles in familial pancreatic cancer. The gene PALB2 (also known as FANCN) encodes a protein that interacts with the BRCA2 protein. Germline mutations in PALB2 account for a subset (~3%) of patients with familial PDA.6 Germline mutations in other Fanconi pathway genes, such as FANCC and FANCG, have been reported in young patients with PDA, but their specific role in familial PDA remains to be firmly established.7–10 In addition, although the importance of FANCA in familial PDA has been investigated, germline mutations in this gene do not significantly contribute to familial PDA.11 Finally, germline mutations in BRCA1 may slightly increase the risk of PDA in breast cancer kindreds, but mutations in this gene only minimally contribute to familial PDA in the absence of breast or ovarian cancer.12,13
In addition to syndromes associated with germline mutations in Fanconi pathway genes, increased risk of PDA is also a key feature in several other inherited syndromes (see Table 34.1). Germline mutations in p16/CDKN2A cause familial atypical mole melanoma syndrome (FAMMM); patients with this syndrome have an increased risk of melanoma (with multiple nevi and atypical nevi) and PDA.14–18 Germline mutations in STK11/LKB1 are associated with Peutz-Jeghers syndrome (PJS), a syndrome associated with gastrointestinal hamartomas and pigmented macules on the lips and buccal mucosa, as well as cancer predisposition, including an increased risk of PDA.19 PDAs in these patients show somatic loss of the wild-type STK11/LKB1 allele, indicating the importance of biallelic inactivation of the gene for carcinoma development in these patients.20 Patients with hereditary pancreatitis are also at markedly increased risk for PDA.21,22 Hereditary pancreatitis, caused by germline mutations in PRSS1 and SPINK1, is characterized by young age at onset and continuous or relapsing chronic pancreatic inflammatory disease.23–25 In contrast to patients with germline mutations in tumor suppressor genes, the increased risk of cancer in these patients appears to be the result of repeated bouts of inflammation and repair, and the increased cancer risk is limited to the pancreas.
In other inherited syndromes, the increased risk of PDA is less well established. Hereditary nonpolyposis colorectal cancer (HNPCC or Lynch syndrome) is caused by germline mutations in hMSH2, hMLH1, hPMS1, hPMS2 or hMSH6 (formerly GTBP), which lead to defects in DNA mismatch repair (and thus microsatellite instability), as well as markedly increased risk of carcinomas of the colon and other sites. There appears to be a slight, but real, increased risk of PDA in patients with HNPCC.1,26–28 In addition, microsatellite instability caused by somatic mutations in one of these mismatch repair genes occurs in sporadic PDA and has been associated with a characteristic “medullary” morphology.27
Germline mutations in the ATM gene, which encodes a protein with roles in DNA damage response as well as cell cycle regulation, also rarely occur in familial PDA kindreds.29 Whereas biallelic germline mutations in ATM cause ataxia-telangiectasia, a syndrome characterized by cerebellar ataxia, sensitivity to ionizing radiation, and increased frequency of multiple malignancies, germline heterozygous ATM mutations have been reported in approximately 2% of patients with familial PDA.29 Germline mutations in APC are the cause of familial adenomatous polyposis (FAP), a syndrome with a marked increase in adenomatous colorectal polyps as well as colorectal adenocarcinoma. Although an increased risk of PDA has been reported in patients with FAP, some of this risk probably reflects the increased risk of duodenal carcinomas, which can invade the pancreas and mimic primary PDA.1,30
Somatic Alterations
In sporadic PDA, somatic alterations clearly play a crucial role in tumorigenesis (Table 34.2). Whole exome sequencing of PDA revealed that each carcinoma contains an average of 48 nonsynonymous somatic mutations.31 Aside from known driver genes (discussed later), the individual genes altered in each carcinoma are markedly heterogeneous, but there are 12 core cellular pathways that are altered in the vast majority of carcinomas.31 These pathways, including KRAS signaling, transforming growth factor-β (TGF-β) signaling, DNA damage control, and cell adhesion, represent key cellular processes that are dysregulated in the development of PDA.31
Table 34.2
Somatic Mutation Prevalence of Commonly Altered Genes in Pancreatobiliary Neoplasms
| Neoplasm | Gene | Chromosome | Alteration Prevalence | Mechanisms of Alteration |
| PDA | KRAS | 12 | 95% | Missense mutation |
| p16/CDKN2A | 9 | 95% | Intragenic mutation with LOH, homozygous deletion, promoter methylation | |
| TP53 | 17 | 75% | Intragenic mutation with LOH | |
| SMAD4/DPC4 | 18 | 55% | Intragenic mutation with LOH, homozygous deletion | |
| IPMN | KRAS | 12 | 80% | Missense mutation |
| RNF43 | 17 | 75% | Intragenic mutation with LOH | |
| GNAS | 20 | 60% | Missense mutation | |
| p16/CDKN2A | 9 | Only in HGD/invasive carcinoma | Intragenic mutation with LOH, homozygous deletion, promoter methylation | |
| TP53 | 17 | Only in HGD/invasive carcinoma | Intragenic mutation with LOH | |
| SMAD4/DPC4 | 18 | Only in HGD/invasive carcinoma | Intragenic mutation with LOH, homozygous deletion | |
| PIK3CA | 3 | 10% | Missense mutation | |
| MCN | KRAS | 12 | 80% | Missense mutation |
| RNF43 | 17 | 40% | Intragenic mutation with LOH | |
| p16/CDKN2A | 9 | Only in HGD/invasive carcinoma | Missense mutation with LOH, homozygous deletion, promoter methylation | |
| TP53 | 17 | Only in HGD/invasive carcinoma | Intragenic mutation with LOH | |
| SMAD4/DPC4 | 18 | Only in HGD/invasive carcinoma | Intragenic mutation with LOH, homozygous deletion | |
| SCA | VHL | 3 | 50% | Intragenic mutation with LOH |
| PanNET | MEN1 | 11 | 45% | Intragenic mutation with LOH |
| DAXX/ATRX | 6/X | 45% | Intragenic mutation with LOH | |
| mTOR pathway | Multiple | 15% | Multiple | |
| VHL | 3 | 25% | Promoter methylation, intragenic mutation | |
| SPN | CTNNB1 | 3 | 95% | Missense mutation |
| ACC | CTNNB1 | 3 | 5% | Missense mutation |
| APC | 5 | 15% | Intragenic mutation with LOH | |
| PB | CTNNB1 | 3 | 55% | Missense mutation |
| APC | 5 | 10% | Intragenic mutation with LOH | |
| Unknown | 11 | 85% | Loss of heterozygosity | |
| CCA | KRAS | 12 | Variable | Missense mutation |
| p16/CDKN2A | 9 | 85% | Intragenic mutation with LOH, homozygous deletion, promoter methylation | |
| TP53 | 17 | 50% | Intragenic mutation with LOH | |
| SMAD4/DPC4 | 18 | 50% | Intragenic mutation with LOH, homozygous deletion | |
| PIK3CA | 3 | Variable | Missense mutation | |
| IDH1/IDH2 | 2/15 | 20% | Missense mutation | |
| BAP1 | 3 | 20% | Intragenic mutation with LOH | |
| ARID1A | 1 | 15% | Intragenic mutation with LOH | |
| PBRM1 | 3 | 15% | Intragenic mutation with LOH | |
| GBC | KRAS | 12 | Variable | Missense mutation |
| BRAF | 7 | 30% | Missense mutation | |
| p16/CDKN2A | 9 | 75% | Intragenic mutation with LOH, homozygous deletion, promoter methylation | |
| TP53 | 17 | 60% | Intragenic mutation with LOH | |
| SMAD4/DPC4 | 18 | 20% | Intragenic mutation with LOH, homozygous deletion | |
| PIK3CA | 3 | 10% | Missense mutation | |
| CTNNB1 | 3 | 10% | Missense mutation | |
| KEAP1 | 19 | 30% | Intragenic mutation with LOH |
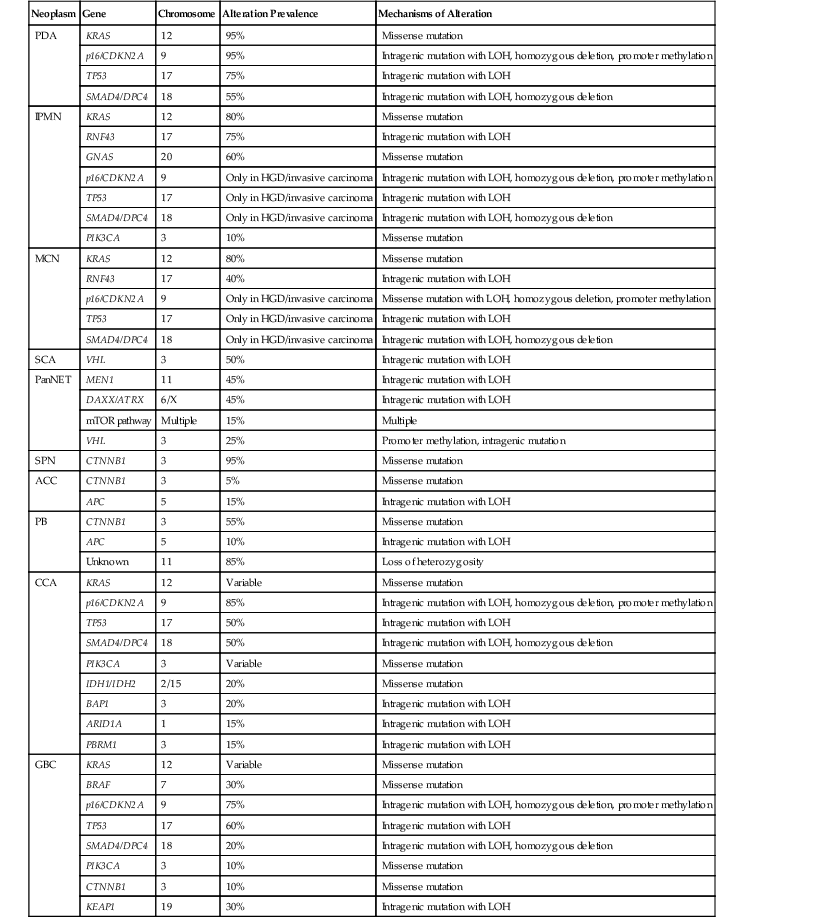
ACC, acinar cell carcinoma; CCA, cholangiocarcinoma; GBC, gallbladder carcinoma; HGD, high-grade dysplasia; IPMN, intraductal papillary mucinous neoplasm; LOH, loss of heterozygosity; MCN, mucinous cystic neoplasm; mTOR, mammalian target of rapamycin; PanNET, well-differentiated pancreatic neuroendocrine tumor; PB: pancreatoblastoma; PDA, pancreatic ductal adenocarcinoma; SCA, serous cystadenoma; SPN, solid-pseudopapillary neoplasm.
Numerous studies have shown that KRAS is the most frequently altered oncogene in PDA (somatic KRAS mutations are present in >90% of cancers), clearly indicating that this gene is a driver of tumorigenesis in the pancreas.31–36 KRAS is a small guanosine triphosphatase (GTPase) that mediates downstream signaling from growth factor receptors. Somatic mutations in KRAS cluster in a few specific hotspots (most commonly in codon 12), confirming the role of KRAS as an oncogene.32,33,37 Mutations have been reported in other oncogenes in the same cell-signaling pathway (including BRAF) in rare KRAS wild-type carcinomas.38,39
Several frequently altered tumor suppressor genes have also been identified in PDA, including p16/CDKN2A, TP53, and SMAD4/DPC4, and these genes also represent drivers in pancreatic tumorigenesis. p16/CDKN2A is the most frequently altered tumor suppressor gene in ductal adenocarcinoma, with loss of p16 protein function identified in more than 90% of carcinomas.31,40,41 Multiple genetic and epigenetic mechanisms underlie the loss of p16 protein expression, including intragenic mutation coupled with loss of the second allele, homozygous deletion of both copies of the gene, and promoter methylation.31,42,43 Loss of p16 results in cell cycle dysregulation, because this protein normally blocks cell cycle progression by preventing inactivation of the retinoblastoma tumor suppressor protein Rb, another important cell cycle regulator.37,44
TP53, which encodes a protein with a pivotal role in the cellular stress response, is another key tumor suppressor gene in PDA. Somatic mutations have been reported in approximately 75% of cases.31,35,37,45,46 These somatic mutations almost always occur through intragenic mutation followed by loss of the wild-type allele.31
Somatic inactivation of SMAD4/DPC4 also occurs frequently in PDA. Homozygous deletion or intragenic mutation followed by loss of the wild-type allele occurs in approximately 55% of carcinomas.31,41,47–49 The Smad4 (Dpc4) protein mediates cellular signaling downstream of the TGF-β receptor. Less frequently, somatic mutations occur in other components of the same signaling pathway, including the TGF-β receptors, TGFBR2 and TGFBR1 (ALK-5).37,50
In addition to these known driver genes, somatic mutations are present at a low prevalence in numerous other genes in PDA.31 However, the role of these genes as drivers or passengers is more difficult to establish when only a few mutations are identified. Infrequent mutations in genes known to be altered in other tumor types are likely to play a functional role in pancreatic neoplasia. For example, ARID1A, which encodes a chromatin remodeling protein, is rarely mutated in PDA (with somatic mutations in approximately 8% of carcinomas) but is frequently mutated in ovarian clear cell carcinomas (somatic mutations in >50% of carcinomas). Although the mutations in the pancreas are infrequent, the gene’s importance in ovarian cancer supports a role for mutations in ARID1A in pancreatic neoplasia.51 Similarly, the histone methyltransferase KMT2C (MLL3) is mutated in only a small percentage of PDA, but somatic mutations in this gene have been reported in several other tumor types, supporting a role for this gene in tumorigenesis in multiple organs.31,52–54
In addition to small somatic mutations, large chromosomal gains and losses and complex karyotypic abnormalities also occur frequently in ductal adenocarcinoma. Some alterations target known driver genes, whereas many others (such as the frequently altered chromosome 6p) identify regions that may contain specific loci with important roles in pancreatic tumorigenesis. Further characterization and validation of specific target genes is necessary.55 Additionally, PDAs also contain alterations in microRNAs, small, noncoding RNAs that regulate gene expression. Differential expression of multiple microRNAs has been reported in PDA compared with nonneoplastic pancreatic tissue.56–58
Although pancreatic intraepithelial neoplasia (PanIN) have been recognized histologically for years, data on the genetic alterations in PanIN lesions firmly established them as noninvasive precursors to PDA in 2001.59 As PanINs acquire increasing cytologic and architectural abnormality with increasing grade, they also sequentially acquire genetic alterations—the same alterations that occur in invasive PDA. Some molecular changes occur early and are present in low-grade PanINs, whereas others are limited to severely dysplastic and invasive lesions. KRAS mutation and loss of p16 expression are early events that are present in low-grade PanINs.60–65 When extremely sensitive techniques are used, KRAS mutations are identified in more than 90% of low-grade PanINs, suggesting that KRAS mutations may represent a key initiating step in pancreatic neoplasia.66 In contrast, loss of Smad4 and TP53 mutation are late events, occurring only in high-grade PanIN and invasive PDA.63,67 For both SMAD4/DPC4 and TP53, allelic loss may occur earlier than somatic mutation, with a subset of intermediate-grade PanINs showing loss of one allele.68
Alterations other than those in known driver genes also occur in PanINs. Telomere shortening is one of the most common early events in pancreatic tumorigenesis, with shortening detected in approximately 90% of low-grade PanINs.69 Early telomere shortening may make the cells susceptible to chromosomal fusion and anaphase bridges, which may produce some of the chromosomal abnormalities observed in invasive PDA.
In addition to expanding knowledge of precursors to PDA, detailed study of somatic mutations in primary tumors and metastases has deepened understanding of the process of clonal evolution within tumors, enabling estimation of evolutionary time in tumors.70 These studies suggest a time window of approximately 15 years between tumor initiation and the acquisition of metastatic ability, providing a broad time window for early detection and subsequent clinical intervention.70
Implications for Pathology
Knowledge of the genetic underpinnings of familial PDA has direct clinical implications for pathologists in several ways. First, the pathologist is the first to have the opportunity to recognize subtypes of pancreatic cancer (e.g., “medullary” carcinoma) that are associated with specific familial syndromes. Second, some familial syndromes have specific treatment implications, which highlights the importance of recognizing them clinically at the time of diagnosis. For example, PDAs in patients with germline alterations in the Fanconi anemia pathway (BRCA2, PALB2) are exquisitely sensitive to drugs that target their specific DNA repair defect, such as mitomycin C and inhibitors of the enzyme poly(ADP-ribose) polymerase (PARP).1,71 Carcinomas in patients with HNPCC also possess specific recommendations for treatment: tumors with microsatellite instability are resistant to fluorouracil-based chemotherapy.72 Finally, clinical recognition of familial PDA syndromes lead to increased screening of at-risk patients. Knowledge of these syndromes is crucial in the interpretation of specimens from these patients.73
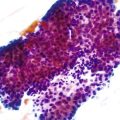
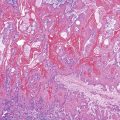
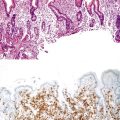
Somatic alterations in sporadic PDA also affect pathology practice. Immunohistochemistry can be used to demonstrate alterations of protein expression that are indicative of characteristic gene mutations. For example, TP53 mutations result in strong diffuse TP53 nuclear labeling by immunohistochemistry, providing a histologic surrogate for the genetic alteration and a potential technique for identifying neoplastic pancreatic cells (Fig. 34.1).41,45,74 In addition, loss of Smad4 protein expression by immunohistochemistry correlates with genetic alterations in the SMAD4/DPC4 gene (Fig. 34.2).75,76 This technique can be used to distinguish PDA (loss of Smad4) from nonneoplastic pancreatic diseases (retention of Smad4) and may suggest that a metastatic adenocarcinoma is of pancreatobiliary origin.75,76 Importantly, this technique can also be used to evaluate prognosis in ductal adenocarcinomas, because mutations in SMAD4/DPC4 have been associated with a worse prognosis.77
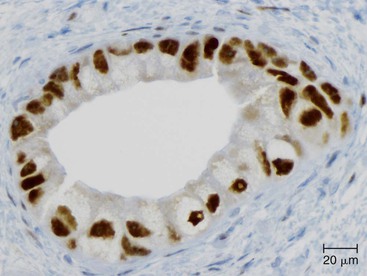
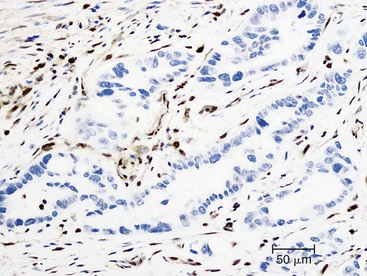
Molecular studies interrogating genetic alterations also demonstrate clinical utility. For example, molecular analyses of KRAS, TP53, and SMAD4/DPC4 can supplement morphologic diagnosis in cytology specimens to increase the sensitivity of fine-needle aspiration.78 In addition, microRNA levels in fine-needle aspiration specimens show diagnostic promise as indicators of ductal adenocarcinoma or mucinous pancreatic cysts. It has been suggested that microRNA profiles can be used to prognosticate in some cases.56,79,80 Finally, with the continuous development of targeted therapies, molecular studies will likely serve a critical role in determining eligibility for therapy.
Variants of Ductal Adenocarcinoma
There are many uncommon morphologic variants of PDA. Although some share molecular features of ductal adenocarcinoma, others contain unique genetic alterations, and these entities have clinical implications.
Compared with PDA, colloid carcinoma (mucinous noncystic carcinoma) has a better prognosis, a lower prevalence of KRAS mutations (approximately 30%) and TP53 mutations (approximately 20%), and a high prevalence of somatic mutations in GNAS, an oncogene that is frequently altered in intraductal papillary mucinous neoplasms (IPMNs) and their associated carcinomas.81 Medullary carcinoma, another variant with a better prognosis than ductal adenocarcinoma, has a high prevalence of microsatellite instability and lacks somatic mutations in KRAS, although oncogenic BRAF mutations have been reported.27,39,82
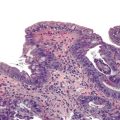
Undifferentiated carcinoma is an aggressive neoplasm with a poor prognosis; in addition to frequent KRAS mutations, these carcinomas exhibit frequent loss of E-cadherin protein expression, which provides a possible explanation for the carcinoma’s discohesive morphology (Fig. 34.3).83–85 Undifferentiated carcinoma with osteoclast-like giant cells, another aggressive carcinoma with poor prognosis, contains two distinct cell populations. Although the neoplastic mononuclear cells contain frequent KRAS mutations and TP53 overexpression, the osteoclast-like giant cells are non-neoplastic and only contain mutant KRAS from phagocytized neoplastic mononuclear cell DNA.86–90 Adenosquamous carcinoma has molecular features similar to ductal adenocarcinoma, showing frequent alterations in KRAS, p16/CDKN2A, SMAD4/DPC4, and TP53, but it is an aggressive neoplasm with poor prognosis.91 Finally, hepatoid carcinoma is an exceedingly rare variant, with too few cases to comprehensively determine clinical outcome or characterize molecular alterations.92
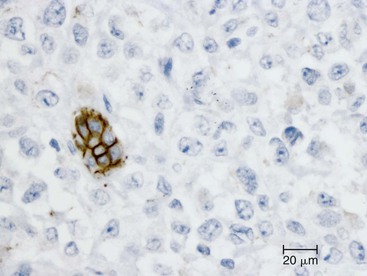
Careful integration of molecular findings with histopathology has helped explain some of the morphologic variants of PDA and is forming the basis for a new classification of pancreatic neoplasia, one that integrates molecular genetics together with tumor histopathology.
Intraductal Papillary Mucinous Neoplasm
Germline Alterations
IPMNs are large, noninvasive, mucin-producing precursor lesions that arise in the larger pancreatic ducts. IPMNs occur rarely in inherited cancer predisposition syndromes (see Table 34.1). For example, they have been reported in patients with PJS, caused by germline mutations in the STK11/LKB1 gene on chromosome 19p.93 IPMNs in patients with PJS frequently undergo loss of heterozygosity (LOH) at the STK11/LKB1 gene locus, suggesting a second somatic hit to the remaining wild-type allele. In addition, IPMNs have been reported in patients with HNPCC (Lynch syndrome) and FAP, suggesting that IPMNs may be uncommon extracolonic manifestations of these syndromes.94,95 Germline alterations in BRCA2 have also been reported in patients with IPMN and a family history of PDA.96
Somatic Alterations
Recent whole exome sequencing of noninvasive sporadic IPMNs identified an average of 26 nonsynonymous mutations per IPMN (approximately half as many as in invasive ductal adenocarcinoma).97 Many of the genes mutated in IPMNs are the same ones commonly mutated in PDAs (see Table 34.2). Somatic point mutations in KRAS occur frequently in IPMNs (30% to 70% of cases), with increasing mutation prevalence in neoplasms with higher grades of dysplasia or invasive adenocarcinoma.98–104 When extremely sensitive techniques are used, KRAS mutations are identified in 80% of IPMNs.81 Somatic mutations in TP53 have also been reported in IPMNs with high-grade dysplasia or invasive carcinoma, and TP53 overexpression similarly occurs in the most dysplastic areas.98,103–105 Smad4 expression is retained in most noninvasive IPMNs but is lost in approximately one third of IPMN-associated invasive carcinomas.106,107 Therefore, the frequency of Smad4 loss is lower in IPMN-associated invasive carcinomas than in invasive ductal adenocarcinomas not arising in association with an IPMN. Loss of p16 protein expression occurs in IPMNs both with and without associated invasive carcinomas, but the frequency of p16 loss is much higher in IPMNs with associated carcinoma (as much as 100% of IPMN-associated invasive carcinomas, compared with 10% of noninvasive IPMNs).107 Promoter hypermethylation of the p16/CDKN2A gene is a frequent mechanism of expression loss; it occurs in more than 50% of all IPMNs, including those with and without associated carcinoma.108
In addition to alterations in genes frequently altered in PDA, IPMNs also contain mutations in genes that are unique among pancreatic neoplasms (see Table 34.2). Somatic mutations in GNAS have been reported in approximately 60% of IPMNs.81,109 The protein encoded by GNAS plays a crucial role in numerous signaling pathways by coupling transmembrane receptors to their downstream signaling proteins.110 The GNAS mutations in IPMNs all occur in a previously described oncogenic hotspot (codon 201), providing strong evidence for their functional importance in IPMNs. Mutations in GNAS are most prevalent in intestinal-type IPMNs but occur in most histologic subtypes, with the possible exception of oncocytic IPMNs.81 In IPMNs with associated invasive carcinoma, GNAS mutations are identified in both noninvasive and invasive components, providing further genetic evidence that IPMNs give rise to invasive carcinoma.81
The gene RNF43 is also frequently altered in IPMNs; approximately 75% of IPMNs contain somatic mutations in RNF43, which encodes an E3 ubiquitin ligase.97 The majority of these alterations are nonsense substitution mutations that lead to the insertion of stop codons and thus to loss of function of the encoded protein. This mutation pattern, along with frequent LOH at the RNF43 locus on chromosome 17q, provides strong evidence that RNF43 is a tumor suppressor gene in IPMNs. Approximately 10% of intestinal-type IPMNs contain somatic mutations in PIK3CA, the catalytic component of a crucial cell signaling pathway known to be an oncogene in several other tumor types, and some somatic mutations in IPMNs occur at previously described oncogenic hotspots in PIK3CA.101,102,104 Approximately 5% of sporadic IPMNs contain mutations in STK11/LKB1 (the locus of PJS), and rare mutations in EGFR (epidermal growth factor receptor) and ERBB2 (also called Her2/Neu) have been reported in IPMNs, although the functional significance of these latter mutations is not yet established.93,103
Genetic mechanisms other than somatic mutation also likely play a role in the development of IPMNs. Large chromosomal alterations have been identified in IPMNs with all grades of dysplasia, but these alterations are more frequent in neoplasms with intermediate- or high-grade dysplasia compared with those with low-grade dysplasia.111,112 Promoter hypermethylation occurs in several genes in IPMNs, and hypermethylation is more prevalent and more extensive in IPMNs with associated invasive adenocarcinoma than in those without adenocarcinoma.108 MicroRNAs may also play a role in IPMN pathogenesis, because significantly higher expression of miR-21, miR-221, and miR-17-3p has been reported in IPMN cyst fluid compared with fluid from nonmucinous cysts.80
Implications for Pathology
These genetic data provide strong evidence for the role of IPMNs as a precursor to PDA, a role strongly supported by clinical data as well. The knowledge of frequent somatic mutations in IPMNs can also be used in the development of diagnostic assays. As in PDAs, neoplastic cells with mutations in TP53 can be identified with immunohistochemical stains. In addition, mutations from neoplastic cells can be detected in aspirated cyst fluid, indicating that molecular analyses of cyst fluid may be useful as a preoperative diagnostic tool in pancreatic cysts.81,113 Importantly, more than 95% of IPMNs contain a somatic mutation in either KRAS or GNAS, suggesting that a molecular assay involving these two genes may be highly sensitive for the identification of IPMNs.81 In addition, the specificity of RNF43 mutations for mucin-producing cystic lesions suggests that assays for these mutations may hold future diagnostic promise.97 Finally, identification of microRNAs shows promise in preoperative diagnosis of pancreatic cysts, because several microRNAs are differentially expressed in mucin-producing versus non–mucin-producing cysts.80
Mucinous Cystic Neoplasm
Germline Alterations
There is no known genetic predisposition or association with particular genetic syndromes for mucinous cystic neoplasm (MCN).
Somatic Alterations
Frequent somatic mutations occur in MCNs. Recent whole exome sequencing of noninvasive MCNs revealed an average of 16 nonsynonymous mutations per MCN, fewer than in IPMN or invasive ductal adenocarcinoma.97 Similar to IPMNs, MCNs contain frequent somatic alterations in genes that are commonly mutated in PDA (see Table 34.2). Somatic mutations in KRAS oncogenic hotspots occur frequently in MCNs. Mutation prevalence correlates with degree of dysplasia (30% KRAS mutation prevalence in MCNs with low-grade dysplasia versus 80% KRAS mutation prevalence in MCNs with high-grade dysplasia or associated invasive carcinoma).100,114–116 Somatic mutation of TP53, as well as TP53 protein overexpression, are limited to MCNs with high-grade dysplasia.97,115,117,118 Loss of Smad4 protein expression, indicating somatic mutation in the SMAD4/DPC4 gene, has been reported primarily in invasive carcinomas associated with MCNs. Loss of Smad4 expression occurs in only 14% of MCN-associated carcinomas, a far lower prevalence rate than in invasive carcinomas not associated with MCNs.117,119 The p16/CKDN2A gene is also infrequently altered in MCNs; Somatic mutation has been reported in a neoplasm with high-grade dysplasia, and promoter hypermethylation occurs in approximately 15% of MCNs.116,118 In addition, aneuploidy has been reported in MCNs and is associated with poor prognosis.120
These findings suggest a stepwise genetic progression in MCNs, from low-grade dysplasia to invasive carcinoma, in which KRAS mutation is an early event and loss of Smad4 is a late event. This concept is supported by the finding in a mouse model of pancreatic neoplasia that oncogenic KRAS mutation and haploinsufficiency of SMAD4/DPC4 led to the development of MCNs, associated with the occurrence of additional somatic alterations in TP53, p16/CDKN2A, and SMAD4/DPC4.121
In addition to mutations in genes frequently altered in PDA, MCNs also share alterations in genes unique to mucin-producing cyst-forming neoplasms of the pancreas (see Table 34.2). Similar to IPMNs, MCNs contain frequent alterations in RNF43; approximately 40% of MCNs harbor somatic mutations in RNF43, and these mutations are enriched for nonsense substitutions.97
Molecular studies have also provided insight into the biphasic nature of MCNs. Gene expression studies suggest different expression profiles in the epithelial and stromal components. Activation of the Notch pathway (JAG1 and HES1) occurs in the epithelial component and activation of estrogen metabolism (STAR and ESR) occurs in the stromal component.122 The genetic or epigenetic bases for these differences remain unclear. In exceedingly rare mixed malignant neoplasms with both epithelial and high-grade “sarcomatous” components, LOH studies suggest a monoclonal origin for the two components with subsequent genetic and morphologic divergence.123
Implications for Pathology
Immunohistochemical staining for TP53 and Smad4 facilitates identification of high-grade neoplastic cells in MCNs, although the prevalence rate of alterations in these genes is far lower in MCNs than in PDAs. As in IPMNs, identification of mutations in DNA from cyst fluid (including mutations KRAS and RNF43) represents a promising preoperative diagnostic test to determine the likelihood of a premalignant mucin-producing cyst.
Serous Cystadenoma
Germline Alterations
Serous cystadenomas (SCAs) occur in approximately 90% of patients with von Hippel-Lindau (VHL) syndrome, an autosomal dominant disorder that is characterized by the development of clear cell neoplasms in multiple organs (see Table 34.1).92,124 VHL is caused by germline mutations in the VHL gene on chromosome 3p, which encodes a regulator of the hypoxia-inducible factor 1 (HIF1α) pathway.92,125,126 SCAs that occur in this syndrome are often combined serous-neuroendocrine neoplasms.92
Somatic Alterations
A recent whole exome sequencing study of sporadic SCAs identified approximately 10 nonsynonymous somatic alterations per tumor, approximately half the number of alterations in IPMN and far fewer than in invasive PDA.97 Importantly, SCAs lack somatic alterations in genes frequently mutated in invasive PDA, such as KRAS and TP53 (see Table 34.2). Sporadic SCAs frequently contain somatic mutations of the VHL gene, which has been reported in as many as 50% of SCAs.97,127,128 LOH at the VHL locus on chromosome 3p also occurs in a large proportion of sporadic SCAs.116,127 In addition to frequent loss of chromosome 3p, allelic loss of chromosome 10q has been reported in 50% of SCAs, and loss of several other chromosomal regions occurs less frequently.128 No target genes for these latter losses have been identified. Aberrant methylation at a few loci has been reported in only a small proportion of SCAs.116
Implications for Pathology
The frequency of SCAs in patients with VHL syndrome requires a high index of suspicion when examining pancreata from these patients. Conversely, the finding of multiple SCAs or a combined serous-neuroendocrine neoplasm should raise the suspicion that the patient has VHL. In addition, VHL mutations have been identified in DNA from pancreatic cyst fluid.97 The specificity of these mutations for SCAs suggest that molecular assays to identify VHL mutations in cyst fluid may provide a useful preoperative diagnostic test, separating cysts with a risk of malignant progression (IPMNs and MCNs) from benign cysts (SCAs).
Pancreatic Neuroendocrine Tumor
Germline Alterations
Pancreatic neuroendocrine tumors (PanNETs) occur as a key feature of many inherited syndromes (see Table 34.1). PanNETs occur in 60% to 70% of patients with multiple endocrine neoplasia syndrome type 1 (MEN1), an autosomal dominant syndrome characterized by neuroendocrine lesions of the parathyroid, pituitary, pancreas, duodenum, and adrenal.129 MEN1 is caused by germline mutations in the MEN1 gene on chromosome 11q. PanNETs also occur in 5% to 10% of patients with VHL syndrome, which is caused by germline mutations in the VHL gene on chromosome 3p.125,130 Patients with VHL are predisposed to neoplasms in various organs, including the adrenal glands, kidneys, and pancreas, many of which are composed of optically clear neoplastic cells.125,126
In addition, PanNETs occur rarely in other genetic syndromes. Tuberous sclerosis complex is caused by germline mutations in TSC1 on chromosome 9q or TSC2 on chromosome 16p. This autosomal dominant clinical syndrome is characterized by the development of hamartomas and rare malignant neoplasms, and PanNETs are a rare feature.131,132 Neurofibromatosis type 1 (NF1) is an autosomal dominant clinical syndrome characterized most prominently by nervous system abnormalities. PanNETs expressing somatostatin (somatostatinomas) are a rare finding in this syndrome, which is caused by germline mutations in the tumor suppressor gene NF1 on chromosome 17q.133,134 Although pancreatic neoplasms have been reported, somatostatinomas occur more frequently in the duodenum of patients with NF1.
Somatic Alterations
In sporadic PanNETs, frequent somatic mutations occur in multiple genes (see Table 34.2). Sequencing of all protein coding genes in 10 PanNETs revealed an average of 16 nonsynonymous somatic mutations per PanNET, far fewer than in invasive PDA.135 Somatic mutations in MEN1 occur in 45% of sporadic PanNETs.135–140 LOH at the MEN1 locus also frequently occurs in PanNETs. Loss occurs in 30% to 70% of sporadic PanNETs, including some without somatic MEN1 mutations.137–140 Somatic mutations in MEN1 cause defects in MEN1 protein function, with loss of expression or aberrant localization.141 Additional mechanisms also likely cause MEN1 protein dysfunction, because some neoplasms without MEN1 mutation show aberrant MEN1 protein localization.141
Somatic mutations in the ATRX and DAXX genes are also common in sporadic PanNETs.135 These somatic mutations, which occur in approximately 45% of sporadic PanNETs, inactivate the encoded proteins that function in a chromatic remodeling complex.135 The proteins encoded by ATRX and DAXX are components of a complex involved in telomere maintenance. Inactivating mutations in PanNETs are associated with the telomerase-independent telomere maintenance mechanism termed “alternative lengthening of telomeres (ALT).”142 Inactivating mutations in ATRX and DAXX are associated with acquisition of the ALT phenotype, indicating that the telomeres of PanNETs are fundamentally different from the telomeres of PDA, which exhibit telomere shortening and reactivation of telomerase.69,142,143 Moreover, mutations in ATRX and DAXX (as well as the ALT phenotype) occur late in the development of PanNETs: they occur only in large tumors (>3 cm) and are absent from microadenomas.144
The cell signaling pathway of mammalian target of rapamycin (mTOR) is also altered in some PanNETs. Mutations in components of this pathway (including PIK3CA, PTEN, and TSC2) occur in approximately 15% of sporadic PanNETs.135 In addition to somatic mutations, LOH at the TSC2 locus on chromosome 16p occurs in approximately 30% of sporadic PanNETs.145
Although rare alterations in some genes have been reported, PanNETs lack frequent alterations in genes commonly mutated in PDA, including KRAS, TP53, SMAD4/DPC4, and p16/CDKN2A.135,146 Somatic mutations in KRAS and BRAF have not been identified in PanNETs.135,146,147 Mutations in TP53 are rare in PanNETs, occurring in approximately 3% of neoplasms, a far lower prevalence rate than in PDA.135 However, dysregulation of the TP53 signaling pathway may still be important in PanNETs, because copy number alterations of TP53 regulators (e.g., MDM2) have also been reported in PanNETs.148 No mutations were identified in SMAD4/DPC4 in a recent whole exome sequencing study of PanNETs.135,149 Neither homozygous deletion nor somatic mutation of p16/CDKN2A has been identified in PanNETs, but promoter hypermethylation of that locus occurs in approximately 50% of gastrinomas.150
Several large chromosomal gains and losses (including some recurrent alterations) have been reported in PanNETs, but the target genes for most alterations remain to be identified.145,151–153 Promoter hypermethylation and deletion of the VHL locus on chromosome 3p occurs in approximately 35% of sporadic PanNETs,154 but no other target genes for aberrant methylation in PanNETs have been identified.
Implications for Pathology
Knowledge of the genetic syndromes predisposing to PanNETs (including MEN1 and VHL) allows recognition of these neoplasms in syndromic patients. In particular, pathologists need to carefully examine the duodenum in patients with MEN1, because small duodenal neuroendocrine tumors (gastrinomas) in these patients can metastasize to a peripancreatic lymph node and mimic a primary pancreatic neoplasm.155 In addition, some somatic alterations have prognostic implications—mutations in MEN1, ATRX, and DAXX have been associated with improved prognosis.135 Immunohistochemistry can be used to show loss of ATRX or DAXX protein expression in mutant cases (Fig. 34.4). In contrast, the presence of multiple chromosomal abnormalities confers a worse prognosis in sporadic PanNETs regardless of the specific loci altered, and patients with these neoplasms are more likely to be seen with metastatic disease.151–153
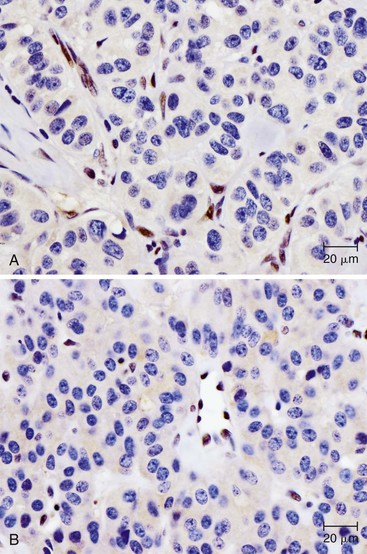
Finally, some somatic mutations in PanNETs may influence therapy. Alterations in the mTOR pathway can have profound clinical significance, because drugs targeting the mTOR pathway have already been developed for clinical use and may be specifically efficacious in patients with somatic alterations in this cellular pathway.156
Solid-Pseudopapillary Neoplasm
Germline Alterations
Solid-pseudopapillary neoplasm of the pancreas (SPN) is not known to occur in association with any particular type of genetic syndrome, and there is no known genetic predilection for this neoplasm.92,124 An SPN occurring in a patient with FAP has been reported, but the significance of this association remains unclear.157
Somatic Alterations
Recent whole exome sequencing of SPNs revealed very few somatic alterations—only the β-catenin gene (CTNNB1) was mutated in more than one SPN (see Table 34.2).135 These studies revealed an average of only three nonsynonymous somatic mutations per SPN, the lowest number for any solid neoplasm sequenced to date. Several of the SPNs studied contained only one or two somatic mutations, but every SPN contained a CTNNB1 mutation. These studies showed that SPNs lack alterations in genes commonly altered in PDA (e.g., KRAS, TP53, SMAD4/DPC4) or in other pancreatic cysts (e.g., GNAS, VHL, RNF43).135
SPNs are characterized by frequent activating somatic mutations of CTNNB1, which occur in approximately 95% of SPNs, leading to the abnormal nuclear accumulation of β-catenin protein in almost 100% of cases.158–161 Importantly, the other common pancreatic neoplasms (including invasive PDA and PanNET) lack somatic mutations in CTNNB1.162 The protein product of this gene has diverse functions in normal cells, regulating cell adhesion through its interaction with E-cadherin and serving as an important target of Wnt signaling.37,163 (β-catenin is normally targeted for degradation by APC and GSK-3β, but when degradation of β-catenin is inhibited by Wnt signaling, the protein translocates to the nucleus, leading to transcription of target genes.) Somatic mutations in CTNNB1 frequently affect key phosphorylation sites, preventing degradation of the β-catenin protein and leading to nuclear translocation of β-catenin and transcriptional activation.158,159
Somatic mutations in CTNNB1 in SPNs lead to overexpression of cyclin D1, a key cell cycle regulator and important downstream target of β-catenin.158,159 In addition, expression of the E-cadherin protein is also dramatically altered in SPNs—expression of the extracellular domain of E-cadherin is lost in these neoplasms, whereas the intracellular component of the E-cadherin protein is abnormally expressed in the nucleus of neoplastic cells.161,164–166 Based on gene expression analyses, SPNs exhibit activation of both the β-catenin pathway (with overexpression of AXIN2, TBX3, SP5, and NOTUM) and the Notch signaling pathway (with the overexpression of HEY1, HEY2, and NOTCH2).167
The role of large chromosomal alterations in SPNs remains controversial, with conflicting reports in the literature. Although there are many case reports of translocations and complex chromosomal abnormalities in single patients, no recurrent abnormalities have been identified.168,169 Similarly, some studies have shown recurrent chromosomal gains and losses in small case series, but others have failed to identify any chromosomal alterations.160,170,171 Therefore, although the importance of activating somatic mutations in CTNNB1 is well established in SPN, the role of other alterations remains unclear.
Implications for Pathology
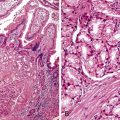
The discovery of a frequent genetic alteration in SPNs (i.e., CTNNB1 mutation) has led to the development of a diagnostic test (β-catenin immunohistochemistry) that, when combined with routine histopathology and other immunostains, helps to distinguish this neoplasm from other solid cellular neoplasms of the pancreas (Fig. 34.5). In the appropriate setting, immunohistochemical labeling for β-catenin to identify aberrant nuclear labeling represents a key step in the diagnostic algorithm for SPN. In addition, abnormalities in E-cadherin expression may explain this lesion’s discohesive morphology.
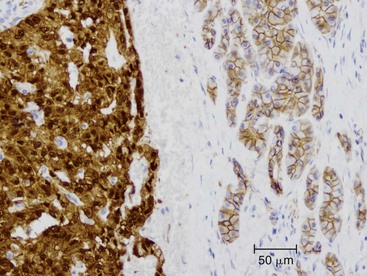
Acinar Cell Carcinoma
Germline Alterations
Acinar cell carcinoma (ACC) is not firmly associated with any known type of genetic syndrome, and there is no known specific genetic predilection.124 However, ACC has been reported in patients with germline mutation in BRCA2, and the ACC exhibited LOH at the BRCA2 locus, suggesting biallelic gene inactivation.172
Somatic Alterations
Recurrent somatic alterations of only a few genes have been identified in ACCs (see Table 34.2). Somatic mutations in the APC/β-catenin pathway occur in 20% to 25% of ACCs; both activating mutations in CTNNB1 and inactivating mutations in APC have been reported.173 Importantly, ACCs lack frequent alterations in genes commonly mutated in PDA. Although rare mutations of KRAS and TP53, as well as rare loss of Smad4 protein expression, have been reported, most studies have identified no alterations in these genes in ACC.146,173–177 A subset of ACCs exhibit microsatellite instability.173
Large chromosomal alterations occur frequently in ACCs. Numerous regions of gain and loss have been reported, including several regions that are altered in multiple ACCs.55,173,177,178 Allelic loss of chromosome 11p is one of the most frequent alterations, occurring in approximately 50% of ACCs.173 Each ACC commonly harbors numerous large chromosomal alterations.55,176,178 Although specific target genes for these alterations have not been identified, the regions altered in ACC differ from those frequently altered in PDA.178 In addition, promoter methylation of several tumor suppressor genes has been identified in a small number of ACCs, although the functional consequences of these epigenetic alterations are unknown.177
Implications for Pathology
Because of frequent alterations in the APC/β-catenin pathway, immunohistochemical labeling for β-catenin shows the characteristic nuclear accumulation in a subset of ACCs.173 However, immunohistochemistry for β-catenin must be interpreted in the context of other immunohistochemical, morphologic, and clinical data to avoid confusion with other neoplasms with aberrant β-catenin staining, such as SPN.
Pancreatoblastoma
Germline Alterations
Several cases of pancreatoblastoma have been reported in patients with Beckwith-Wiedemann syndrome, a disorder of organ overgrowth and predisposition to embryonal tumors.179–180 One case of pancreatoblastoma has been reported in a patient with FAP.181,182
Somatic Alterations
Sporadic pancreatoblastomas show frequent allelic loss at chromosome 11p, occurring in 86% of cases in one study (see Table 34.2).181,182 Loss of 11p has also been reported in other embryonal neoplasms, such as hepatoblastoma183 and Wilms tumor,184 suggesting the possibility of a common genetic pathway in embryonal tumors. In addition, most pancreatoblastomas have somatic alterations in the APC/β-catenin pathway, including activating mutations in CTNNB1 and inactivating mutations in APC.181 These alterations lead to abnormal nuclear accumulation of β-catenin as well as overexpression of cyclin D1, both of which occur most frequently in the squamoid nests of pancreatoblastoma.185 Pancreatoblastomas lack frequent alterations in genes commonly mutated in PDA: although rare loss of Smad4 expression has been reported, KRAS mutations and alterations of TP53 expression have not been identified in pancreatoblastoma.181 In addition, there have been several case reports of complex karyotypes with several large regions of loss and gain, and trisomy 8 has been reported.186–189 However, target genes for these large chromosomal alterations have not been identified.
Implications for Pathology
Because of the associated germline predisposition to pancreatoblastoma, this tumor should be suspected in patients with Beckwith-Wiedemann syndrome. In addition, because of genetic alterations in the APC/β-catenin pathway, many pancreatoblastomas show abnormal nuclear accumulation of β-catenin that can be detected by immunohistochemistry.181 Although nuclear β-catenin staining may be useful diagnostically, if other histopathologic features are not considered, it can also create confusion in the differential diagnosis with SPN and other neoplasms with aberrant β-catenin labeling.
Biliary Neoplasms
Cholangiocarcinoma
Germline Alterations
Cholangiocarcinoma is not associated with any known genetic syndromes.
Somatic Alterations
Whole exome sequencing studies of intrahepatic cholangiocarcinomas have revealed alterations in chromatin remodeling genes.190,191 Three chromatin remodeling genes, BAP1, ARID1A, and PBRM1, contain somatic inactivating mutations in almost half of intrahepatic cholangiocarcinomas (see Table 34.2). Inactivating mutations in these genes, which are the most frequently mutated genes in intrahepatic cholangiocarcinoma, highlight the importance of dysregulated chromatin remodeling in this tumor type. Functional studies have also confirmed the roles of BAP1 and ARID1A as tumor suppressor genes in cholangiocarcinoma cell lines.190
Several studies have also identified a significant number of mutations in isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) in cholangiocarcinoma—mutations in a single codon of these genes occur in approximately 20% of intrahepatic cholangiocarcinomas but not in any other type of gastrointestinal malignancy (see Table 34.2).190–192 These genes encode metabolic enzymes and are frequently altered in tumors of the central nervous system, but intrahepatic cholangiocarcinoma is unique among pancreatobiliary cancers regarding somatic mutations in IDH1 and IDH2.192,193
In addition, sporadic cholangiocarcinomas contain somatic alterations in many of the driver genes altered in pancreatic ductal adenocarcinoma (see Table 34.2). Oncogenic hotspot mutations in KRAS are frequent in cholangiocarcinomas, though the reported prevalence varies from 10% to 70%.192,194–196 Interestingly, multiple studies report differences in KRAS mutation frequency in intrahepatic cholangiocarcinoma based on the tumor location within the liver as well as country of origin, pointing to an explanation for the wide range of KRAS mutation prevalence in pooled data.197,198 A high frequency of KRAS mutations are present in cholangiocarcinomas in patients with pancreatobiliary maljunction—the prevalence of KRAS mutation is approximately 60% in these patients.199 Rare mutations in NRAS have also been identified in cholangiocarcinomas, and BRAF mutations are infrequent, with a prevalence of less than 5% in cholangiocarcinoma.192,196 Alterations of the tumor suppressor gene p16/CDK2NA are also common on cholangiocarcinomas, with p16 alterations reported in 55% to 85% of cholangiocarcinomas by mechanisms including intragenic mutation, homozygous deletion, and promoter methylation.200–203 Many cholangiocarcinomas contain alterations in the TP53 tumor suppressor gene, with aberrant expression reported in 25-50% of carcinomas.200,201,204 Although the prevalence of intragenic mutations in TP53 varies widely among studies, this is likely the result of the limited number of exons assayed, resulting in decreased sensitivity for identification of TP53 alterations compared with assays of aberrant protein expression.192,200,201 Loss of Smad4 (Dpc4) also frequently occurs in cholangiocarcinoma, with a prevalence of 35-50%.204 Intriguingly, the prevalence of Smad4 (Dpc4) loss varies with cholangiocarcinoma location, with higher prevalence in carcinomas of the distal bile duct compared with those of the proximal bile duct.205 Whole exome sequencing studies have revealed infrequent mutations in KRAS, TP53, and SMAD4 in intrahepatic cholangiocarcinomas.190,191
In addition to these genes commonly altered in pancreatic ductal adenocarcinoma, cholangiocarcinoma also contains alterations in unique genes (see Table 34.2). Somatic mutations and rearrangements involving the FGFR2 gene occur in a minority of cholangiocarcinomas.191,206 The PI3K pathway has been reported to be altered in cholangiocarcinoma, but its importance remains controversial. Prevalence of oncogenic mutations in PIK3CA in cholangiocarcinoma varies widely among studies—although some fail to identify any mutations, others identify a prevalence of greater than 30% for PIK3CA mutations in cholangiocarcinomas.191,196,207 However, some authors suggest that the low frequency of somatic alterations in PIK3CA provides a genetic basis from which to distinguish cholangiocarcinoma from gallbladder carcinoma.191 In addition, rare mutations in PTEN and AKT1, the protein products of which act in the same pathway as PIK3CA, have also been reported in cholangiocarcinomas.191 Rare mutations in EGFR kinase as well as CDH1 (E-cadherin gene) have also been reported.208–210 Homozygous deletion of STK11/LKB1 has been reported in a cholangiocarcinoma from the distal common bile duct.20
Microsatellite instability has been reported in more than 60% of cholangiocarcinomas arising in association with pancreatobiliary maljunction, but this percentage seems rather high.199 The significance of microsatellite instability in other sporadic cholangiocarcinomas remains unclear because most studies have identified a low prevalence (less than 5%) of microsatellite instability in sporadic cholangiocarcinomas.198,211 In addition, aberrant promoter methylation of several other genes has been reported in cholangiocarcinomas, but the functional significance of this aberrant methylation remains to be elucidated.212,213
Numerous large chromosomal gains and losses have also been reported in cholangiocarcinomas, but the target genes for many of these alterations are not yet known—still, many studies show clear differences in these alterations between cholangiocarcinoma and hepatocellular carcinoma.203,214–218 However, copy number alterations of a few known driver genes have also been reported. Gene amplification of ERBB2 (Her2Neu) with protein overexpression occurs in a significant proportion of cholangiocarcinomas.219–221 In one study, approximately 20% of cholangiocarcinomas showed ERBB2 gene amplification, whereas protein overexpression occurred in almost 30% of tumors.221 In addition, amplification and overexpression of the cyclin D1 gene (CCND1), a crucial cell cycle regulator, occur in a subset of cholangiocarcinomas, with overexpression in approximately 40% of cases and amplification in approximately 25%.222 Gene amplification of EGFR with associated protein overexpression also occurs rarely in cholangiocarcinomas.220
Although most studies do not separately assess cholangiocarcinomas based on site, some data suggest that intrahepatic cholangiocarcinoma is genetically distinct from extrahepatic cholangiocarcinoma. For example, IDH1 and IDH2 mutations occur exclusively in intrahepatic cholangiocarcinoma, whereas mutations in KRAS and TP53 are more frequent in extrahepatic cholangiocarcinoma. In addition, loss of Smad4 (Dpc4) expression is more common in cholangiocarcinomas of the distal bile ducts compared with proximal and intrahepatic cholangiocarcinomas, further illustrating the anatomic variability of these genetic lesions.205 Further studies that directly compare the two tumor locations will be necessary to better understand their unique genetic features.
One clinically distinct subtype of cholangiocarcinoma is related to infection with the liver fluke (Opisthorchis viverrini), a known risk factor for this carcinoma in areas of Asia where the liver fluke is endemic. Whole exome sequencing of these cholangiocarcinomas related to liver fluke infection reveals unique genetic features.190,223 Similar to other cholangiocarcinomas, liver fluke-associated cancers contain frequent mutations in KRAS, TP53, SMAD4/DPC4, and p16/CDKN2A. However, these genes are altered more frequently in liver fluke-associated cancers compared with cancers not associated with liver fluke infection. In addition, these cancers also contain mutations in GNAS (at the previously identified mutation hotspot) as well as RNF43, genes that are frequently altered in mucin-producing cysts of the pancreas but not previously associated with cholangiocarcinoma. These studies also identified mutations in genes not previously shown to be involved in pancreatobiliary tumorigenesis. Intriguingly, liver fluke-associated cholangiocarcinomas lacked mutations in IDH1 and IDH2. Mutations in BAP1 and ARID1A were much less frequent than in carcinomas not associated with liver fluke infection. In addition, microsatellite instability occurs at a higher frequency in intrahepatic cholangiocarcinomas related to infection by the liver fluke (with a prevalence of almost 25% in liver fluke-associated cancers, compared with less than 5% in other sporadic carcinomas).224 Microsatellite instability in this subset of cholangiocarcinomas is related to poor prognosis and is thought be mediated at least in part by promoter methylation of hMLH1.224,225 Thus, this cholangiocarcinoma subtype initially defined by its unique underlying infection also constitutes a distinct genetic subset of this cancer.
Implications for Pathology
Similar to pancreatic ductal adenocarcinoma, immunohistochemical stains for p53 and Smad4 (Dpc4) have clinical utility in their ability to identify tumors with mutations in these genes. The development of diagnostic assays and therapeutic approaches based on IDH1 mutations will be applicable to cholangiocarcinomas as well as neoplasms of the central nervous system (which also contain frequent IDH1 mutations). Therapies that target FGFR may also be applicable to cholangiocarcinomas that contain mutations or rearrangements in FGFR2. In addition, cholangiocarcinomas also infrequently contain alterations in ERBB2 and EGFR, both of which are targets for mutation-based therapies. Assays that identify tumors with these alterations will help to identify patients most likely to benefit from targeted therapies.
Gallbladder Carcinoma
Germline Alterations
Gallbladder carcinoma is not associated with any known type of genetic syndrome. However, numerous studies have investigated the association between gallbladder carcinoma risk and germline polymorphisms in specific populations. Germline polymorphisms in several gene families, including DNA repair genes and Toll-like receptors, have been associated with a slightly increased risk of gallbladder carcinoma in India, and germline polymorphisms in genes associated with cholesterol metabolism have been associated with an increased risk of gallbladder carcinoma in Chile and China.226–229
Somatic Alterations
Sporadic gallbladder carcinomas contain somatic alterations in many of the driver genes that are altered in PDA (see Table 34.2). The prevalence rate of somatic mutations in KRAS is lower in gallbladder carcinoma than in other pancreatobiliary carcinomas, but the precise rate varies widely among studies (from 0% to 30%).203,230–233 Oncogenic mutations in KRAS also occur in carcinomas from patients with pancreatobiliary maljunction, who are at increased risk for development of biliary cancer. Interestingly, carcinomas from these patients have a higher prevalence rate of KRAS mutation, showing mutations in 50% to 60% of carcinomas.196 Similar oncogenic KRAS mutations also occur in the precursors to gallbladder carcinoma, including 25% of adenomas.228,234 Some experts argue that the difference in KRAS mutation prevalence rate between adenomas and carcinomas provides evidence for the model of multiple tumorigenic pathways in the gallbladder—a more common pathway in which carcinoma arises from flat dysplasia and a less common pathway in which carcinoma arises from an adenoma (which on its own has low malignant potential). In addition to KRAS mutations, somatic mutations at a previously described oncogenic hotspot in BRAF also occur frequently in gallbladder carcinoma (>30% of carcinomas in one study).235
Somatic mutations in tumor suppressor genes known to be drivers in other pancreatobiliary carcinomas also occur in gallbladder carcinoma. p16/CDKN2A alterations are common, occurring in 60% to 75% of gallbladder carcinomas, by mechanisms including point mutation, chromosomal loss, and promoter methylation.199,233 These somatic p16/CKDN2A alterations are associated with a poor prognosis.199 Loss of expression of RB1 (pRb), a protein that functions in the same cell cycle control pathway as p16, occurs infrequently in gallbladder carcinomas, with loss in only 4% of carcinomas.233 Alterations in TP53 are common, with abnormal p53 protein expression present in approximately 60% of carcinomas.201,233 Moreover, whole exome sequencing studies of gallbladder carcinoma identified TP53 as the most frequently altered gene, with somatic mutations in almost half of the tumors sequenced.191 LOH at the TP53 locus also frequently occurs in gallbladder carcinomas.236 Both TP53 loss and mutation have been identified in dysplastic gallbladder mucosa as well as histologically normal mucosa adjacent to carcinomas, and different mutations have been reported in carcinomas and adjacent non-neoplastic mucosa, suggesting the possibility of a field effect in this tumor type.236 TP53 mutations occur both in sporadic carcinomas and in those associated with pancreatobiliary malfunction,196 and serum antibodies to the TP53 protein have been reported in a subset of patients with gallbladder carcinoma.237 Loss of Smad4 expression also occurs in gallbladder carcinomas, although less frequently than in other tumor types; fewer than 20% of gallbladder carcinomas show loss of this protein.201,233
Mutations in genes not frequently altered in other pancreatobiliary neoplasms have also been reported in gallbladder carcinoma. Somatic mutations in CTNNB1 occur in fewer than 10% of gallbladder carcinomas.232,238–240 Gallbladder adenomas exhibit a much higher CTNNB1 mutation prevalence rate (approximately 60%), providing further evidence for the model of separate genetic pathways underlying adenomas and carcinomas of the gallbladder.238–240 Activating somatic mutations in the oncogene PIK3CA occur in approximately 10% of gallbladder carcinomas.192,203 Rare mutations in the EGFR tyrosine kinase domain have been reported.206 Somatic mutations in KEAP1, a regulator of the cellular response to oxidative stress, have been reported in several gallbladder carcinomas, with a mutation prevalence rate of approximately 30%.241
Microsatellite instability has been reported in sporadic gallbladder carcinomas, but reports of its prevalence are conflicting (range, 3% to 35%).242 Microsatellite instability frequently occurs in gallbladder carcinomas in patients with pancreatobiliary maljunction (>70%).196 Some studies point to regional variation in microsatellite instability: In a comparison between carcinomas from Japan and Hungary, microsatellite instability was much more frequent in the Japanese cases (42% versus 7%).243
In addition to somatic point mutations, large chromosomal alterations and epigenetic abnormalities frequently occur in gallbladder carcinoma. Large gains and losses have been reported in several genomic regions, but the target genes for most of these alterations remain to be identified.214,231 A few amplifications with known target genes have been identified: amplifications with overexpression of ERBB2 and of EGFR each occur in approximately 10% of gallbladder carcinomas.217 In addition, a small number of gallbladder carcinomas (<5%) exhibit amplification of the MYC oncogene.244 Aberrant methylation has also been reported in multiple genes, including p16/CDKN2A and APC.245 Aberrant methylation seems to be a progressive process throughout gallbladder tumorigenesis, with increasingly aberrant methylation in dysplasia and carcinoma.246
Small cell carcinoma of the gallbladder represents a distinct clinical, histologic, and molecular entity. These neoplasms, which have histologic features similar to those of small cell carcinomas in other organs, have a dismal prognosis. They frequently occur as one component of a mixed neoplasm, often combined with more conventional adenocarcinoma.247 In contrast to conventional gallbladder adenocarcinoma, all small cell carcinomas of the gallbladder assayed showed inactivation of the p16/pRb pathway, with 67% of tumors showing loss of RB1 and the remaining 33% showing loss of p16.233 These neoplasms also showed very high prevalence rate of TP53 alterations, with abnormal TP53 protein expression in 83%.233 However, KRAS mutation and loss of Smad4 have not been identified in small cell carcinomas of the gallbladder, which indicates significant differences in the molecular features of this tumor type compared with conventional gallbladder adenocarcinoma.
Implications for Pathology
As with other types of pancreatobiliary neoplasms, immunohistochemical assays for TP53, Smad4, and β-catenin have clinical utility in gallbladder carcinoma, reliably identifying tumors with gene mutations. Immunolabeling for β-catenin may help differentiate carcinomas that occur through a separate tumorigenic pathway, because mutations in CTNNB1 are much more common in gallbladder adenomas. However, the precise clinical relevance of this distinction remains to be determined.238–240 As with cholangiocarcinoma, identification of patients with alterations in ERBB2 or EGFR may help identify those who are likely to benefit from specific targeted therapies. In addition, methylation patterns of some genes have prognostic significance. There is an association of methylated DLC1 with poor prognosis and an association of methylated MGMT with improved prognosis.246 Finally, recognition of small cell carcinoma of the gallbladder as a morphologically and genetically distinct entity will facilitate the identification of a subset of patients with gallbladder carcinoma, which carries a far worse prognosis.
Conclusion
Pancreatobiliary neoplasms encompass a broad range of clinical and morphologic entities. These tumors represent some of the best-characterized neoplasms on a genetic level and provide a unique opportunity for integration of clinical, morphologic, and genetic data. Pathologists will be at the forefront of this integration. As the fields of pathology and oncology move into the era of genomic medicine, pathologists must combine morphologic and molecular data in their assessment of pancreatobiliary specimens. This will allow clinicians to provide the best possible care for patients, including diagnosis and treatment based on the specific genetic alterations in an individual patient’s tumor.