[level-membership-for-internal-medicine-category]
Chapter 2 Molecular cell biology and human genetics
Cell biology
Cells consist of cytoplasm enclosed within a lipid sheath (the plasma membrane). The cytoplasm contains a variety of organelles (sub-cellular compartments enclosed within their own membranes) in a mixture of salts and organic compounds (the cytosol). These are held within an adaptive internal scaffold (the cytoskeleton) that radiates from the nucleus outwards to the cell surface (Fig. 2.1). Many cells have special functions and their size, shape and behaviour adapt to meet their physiological roles. Cells can be organized into tissues and organs in which the individual component cells are in contact and able to send and receive messages, both directly and indirectly. Coordinated cellular responses can be achieved through systemic signalling, e.g. via hormones.
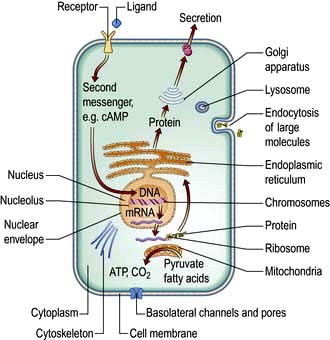
Cell structure
Cellular membranes
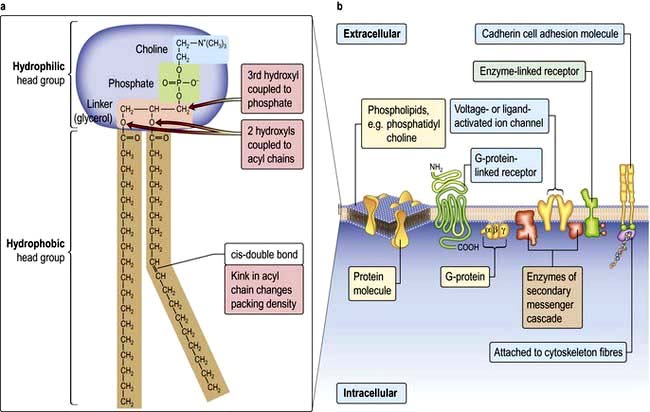
 Lipid bilayers separate the cell contents from the external environment and compartmentalize distinct cellular activities into organelles. These consist of a large variety of glycerophospholipids and sphingolipids. Membrane lipids usually have two hydrophobic acyl chains linked via glycerol or serine, to polar hydrophilic head groups (Fig. 2.2). This amphiphilic nature, with a ‘water-loving’ head and a ‘water-hating’ tail, means that in aqueous solution membrane lipids self-associate into a tail-to-tail bilayer with their hydrophobic chains separated from the aqueous phase by their polar head groups.
Lipid bilayers separate the cell contents from the external environment and compartmentalize distinct cellular activities into organelles. These consist of a large variety of glycerophospholipids and sphingolipids. Membrane lipids usually have two hydrophobic acyl chains linked via glycerol or serine, to polar hydrophilic head groups (Fig. 2.2). This amphiphilic nature, with a ‘water-loving’ head and a ‘water-hating’ tail, means that in aqueous solution membrane lipids self-associate into a tail-to-tail bilayer with their hydrophobic chains separated from the aqueous phase by their polar head groups.

 Plasma membranes are more complicated than liposomes. Their lipids are organized asymmetrically in the bilayer. For example, the outer leaflet of the plasma membrane is enriched in phosphatidyl-choline (PC) and the sphingolipids, whereas the inner leaflet is enriched in phosphatidyl-serine (PS) and phosphatidyl-ethanolamine (PE). This arrangement is necessary in normal physiology and in disease, not just for barrier function. For example, PC is extracted from the outer-leaflet of the canalicular membrane of hepatocytes to form the lipid/bile-salt micelles of bile. One of the sphingolipids, GM1-ganglioside, is the receptor for cholera toxin. The appearance of PS in the outer leaflet of the membrane is an early step in the apoptotic pathway and signals to macrophages to clear the dying cell, while PE, once cleaved by phospholipase, produces two signalling molecules as second messengers (see p. 25). Cholesterol is also an essential component of the plasma membrane and cannot be substituted by plant sterols, which have a subtly different shape. For this reason, the liver secretes plant sterols back into the gut.
Plasma membranes are more complicated than liposomes. Their lipids are organized asymmetrically in the bilayer. For example, the outer leaflet of the plasma membrane is enriched in phosphatidyl-choline (PC) and the sphingolipids, whereas the inner leaflet is enriched in phosphatidyl-serine (PS) and phosphatidyl-ethanolamine (PE). This arrangement is necessary in normal physiology and in disease, not just for barrier function. For example, PC is extracted from the outer-leaflet of the canalicular membrane of hepatocytes to form the lipid/bile-salt micelles of bile. One of the sphingolipids, GM1-ganglioside, is the receptor for cholera toxin. The appearance of PS in the outer leaflet of the membrane is an early step in the apoptotic pathway and signals to macrophages to clear the dying cell, while PE, once cleaved by phospholipase, produces two signalling molecules as second messengers (see p. 25). Cholesterol is also an essential component of the plasma membrane and cannot be substituted by plant sterols, which have a subtly different shape. For this reason, the liver secretes plant sterols back into the gut.
Membrane proteins
Cells can absorb gases or small hydrophobic compounds directly across the plasma membrane by passive diffusion, but membrane proteins are required to take-up hydrophilic nutrients or secrete hydrophilic products, to mediate cell–cell communication and to respond to endocrine signals. Membrane proteins can be integral to the membrane (i.e. their protein chain traverses the membrane one or multiple times) or they can be anchored to the membrane by an acyl chain (Fig. 2.2).
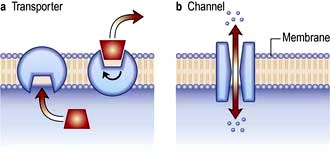
 Membrane channel proteins (Fig. 2.3): membrane proteins that form solute channels through the membrane can only work downhill and only to equilibrium. Solute actually moves down its electrochemical gradient, which is the combined force of the electric potential and the solute concentration gradient across the membrane. The bulk flow can be very high, the opening and closing of the channel can be regulated, and they can be selective for specific solutes. For example, the cystic fibrosis transmembrane regulator (CFTR; Fig. 2.22), the protein whose malfunction causes cystic fibrosis, is a chloride channel found on the apical surface of epithelial cells. CFTR functions to regulate the fluidity of the extra-epithelial mucous layer. When the channel opens, millions of negatively-charged chloride ions flow out of the cell down their electrochemical gradient. This induces positively-charged sodium ions to flow between the cells of the epithelium (via a paracellular pathway) to balance the electrical charge. Water follows the efflux of sodium chloride by osmosis, thus maintaining the fluidity of the mucus.
Membrane channel proteins (Fig. 2.3): membrane proteins that form solute channels through the membrane can only work downhill and only to equilibrium. Solute actually moves down its electrochemical gradient, which is the combined force of the electric potential and the solute concentration gradient across the membrane. The bulk flow can be very high, the opening and closing of the channel can be regulated, and they can be selective for specific solutes. For example, the cystic fibrosis transmembrane regulator (CFTR; Fig. 2.22), the protein whose malfunction causes cystic fibrosis, is a chloride channel found on the apical surface of epithelial cells. CFTR functions to regulate the fluidity of the extra-epithelial mucous layer. When the channel opens, millions of negatively-charged chloride ions flow out of the cell down their electrochemical gradient. This induces positively-charged sodium ions to flow between the cells of the epithelium (via a paracellular pathway) to balance the electrical charge. Water follows the efflux of sodium chloride by osmosis, thus maintaining the fluidity of the mucus.
 Transporters (Fig. 2.3): in contrast to channels, transporters have a low capacity and work by binding solute on one side of the membrane which induces a conformational change that exposes the solute binding site on the other side of the membrane for release.
Transporters (Fig. 2.3): in contrast to channels, transporters have a low capacity and work by binding solute on one side of the membrane which induces a conformational change that exposes the solute binding site on the other side of the membrane for release.
 Receptors: there are three major receptor categories: receptors that mediate endocytosis, anchorage receptors (e.g. integrins, see p. 23) and signalling receptors (see cell signalling p. 24). There are two forms of receptor-mediated endocytosis:
Receptors: there are three major receptor categories: receptors that mediate endocytosis, anchorage receptors (e.g. integrins, see p. 23) and signalling receptors (see cell signalling p. 24). There are two forms of receptor-mediated endocytosis:
Organelles
Cytoplasmic organelles
 Endoplasmic reticulum (ER) is an array of interconnecting tubules or flattened sacs (cisternae) that is contiguous with the outer nuclear membrane (Fig. 2.1). There are three types of ER:
Endoplasmic reticulum (ER) is an array of interconnecting tubules or flattened sacs (cisternae) that is contiguous with the outer nuclear membrane (Fig. 2.1). There are three types of ER:
 Golgi apparatus has flattened cisternae similar to those of the ER but arranged in a stack (Fig. 2.1). Vesicles that bud from the ER with cargo destined for secretion, for the plasma membrane or for other organelles, fuse with the Golgi stack. The proteins, lipids and sterols synthesized in the ER are exported to the Golgi apparatus to complete maturation (e.g. the final stages of membrane protein glycosylation occurs here). The mature products are then sorted into vesicles that bud from the Golgi for transport to their final destination (Fig. 2.4b,c). Mutation in the Golgin protein GMAP-210, with a probable role in tethering of the Golgi cisternae, causes achondrogenesis type 1A, where Golgi architecture is disrupted, particularly in bone cells.
Golgi apparatus has flattened cisternae similar to those of the ER but arranged in a stack (Fig. 2.1). Vesicles that bud from the ER with cargo destined for secretion, for the plasma membrane or for other organelles, fuse with the Golgi stack. The proteins, lipids and sterols synthesized in the ER are exported to the Golgi apparatus to complete maturation (e.g. the final stages of membrane protein glycosylation occurs here). The mature products are then sorted into vesicles that bud from the Golgi for transport to their final destination (Fig. 2.4b,c). Mutation in the Golgin protein GMAP-210, with a probable role in tethering of the Golgi cisternae, causes achondrogenesis type 1A, where Golgi architecture is disrupted, particularly in bone cells.
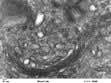
 Lysosomes mature from vesicles (endosomes) that bud from the Golgi. They contain digestive enzymes such as lipases, proteases, nucleases and amylases that work in an acidic environment. The membrane of the lysosome therefore includes a proton ATPase pump to acidify the lumen of the organelle. Lysosomes fuse with phagocytotic vesicles to digest their contents. This is crucial to the function of macrophages and polymorphs (neutrophils and eosinophils) in killing and digesting infective agents, in tissue remodelling during development, and osteoclast remodelling of bone. Not surprisingly, many metabolic disorders result from impaired lysosomal function (p. 1040).
Lysosomes mature from vesicles (endosomes) that bud from the Golgi. They contain digestive enzymes such as lipases, proteases, nucleases and amylases that work in an acidic environment. The membrane of the lysosome therefore includes a proton ATPase pump to acidify the lumen of the organelle. Lysosomes fuse with phagocytotic vesicles to digest their contents. This is crucial to the function of macrophages and polymorphs (neutrophils and eosinophils) in killing and digesting infective agents, in tissue remodelling during development, and osteoclast remodelling of bone. Not surprisingly, many metabolic disorders result from impaired lysosomal function (p. 1040).

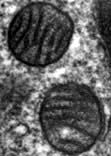
 Mitochondria are the engines of the cell, providing energy in the form of ATP. Mitochondria can be small, discrete and few in number in cells with low energy demand, or large and abundant in cells with a high energy demand like hepatocytes or muscle cells. The mitochondrion has its own genome encoding 13 proteins. The other proteins (~1000) required for mitochondrial function are encoded by the nuclear genome and imported into the mitochondrion. The mitochondrion has a double membrane surrounding a central matrix. The central matrix contains the enzymes for the Krebs cycle, which accepts the products of sugar and fatty acid catabolism and uses it to produce cofactors that donate their electrons into the electron transport chain of the inner membrane (see pp. 20, 31). The inner membrane is highly folded into cristae to increase its effective surface area. The protein complexes of the electron transport chain accept and donate electrons in redox reactions, releasing energy to efflux protons (H+) into the inter-membrane space. ATP synthase, another integral membrane protein, uses this H+ electrochemical gradient to drive formation of ATP. Mitochondria have many additional functions, including roles in apoptosis (see p. 32) and supply of substrates for biosynthesis. Mitochondria are also necessary for the synthesis of porphyrin, deficiency of which causes a range of diseases collectively called porphyrias (p. 1043).
Mitochondria are the engines of the cell, providing energy in the form of ATP. Mitochondria can be small, discrete and few in number in cells with low energy demand, or large and abundant in cells with a high energy demand like hepatocytes or muscle cells. The mitochondrion has its own genome encoding 13 proteins. The other proteins (~1000) required for mitochondrial function are encoded by the nuclear genome and imported into the mitochondrion. The mitochondrion has a double membrane surrounding a central matrix. The central matrix contains the enzymes for the Krebs cycle, which accepts the products of sugar and fatty acid catabolism and uses it to produce cofactors that donate their electrons into the electron transport chain of the inner membrane (see pp. 20, 31). The inner membrane is highly folded into cristae to increase its effective surface area. The protein complexes of the electron transport chain accept and donate electrons in redox reactions, releasing energy to efflux protons (H+) into the inter-membrane space. ATP synthase, another integral membrane protein, uses this H+ electrochemical gradient to drive formation of ATP. Mitochondria have many additional functions, including roles in apoptosis (see p. 32) and supply of substrates for biosynthesis. Mitochondria are also necessary for the synthesis of porphyrin, deficiency of which causes a range of diseases collectively called porphyrias (p. 1043).
The cytoskeleton
 Microtubules (20–25 nm diameter) are polymers of α and β tubulin. These tubular structures resist bending and stretching, and are polar with plus and minus ends. They emanate from the microtubule organizing centre (MTOC), a complex of centrioles, γ-tubulin and other proteins, with their plus ends extending into the cell. At their plus ends repeated cycles of assembly and disassembly permit rapid changes in length. Microtubules form a ‘highway’, transporting organelles and vesicles through the cytoplasm. The two major microtubule-associated motor proteins (kinesin and dynein) allow movement of cargo to the plus and minus ends, respectively. During cell division the MTOC forms the mitotic spindle (see p. 28). Drugs that disrupt microtubule assembly (e.g. colchicine and vinca alkaloids) or stabilize microtubules (taxanes) preferentially kill dividing cells by preventing mitosis.
Microtubules (20–25 nm diameter) are polymers of α and β tubulin. These tubular structures resist bending and stretching, and are polar with plus and minus ends. They emanate from the microtubule organizing centre (MTOC), a complex of centrioles, γ-tubulin and other proteins, with their plus ends extending into the cell. At their plus ends repeated cycles of assembly and disassembly permit rapid changes in length. Microtubules form a ‘highway’, transporting organelles and vesicles through the cytoplasm. The two major microtubule-associated motor proteins (kinesin and dynein) allow movement of cargo to the plus and minus ends, respectively. During cell division the MTOC forms the mitotic spindle (see p. 28). Drugs that disrupt microtubule assembly (e.g. colchicine and vinca alkaloids) or stabilize microtubules (taxanes) preferentially kill dividing cells by preventing mitosis.
 Intermediate filaments (~10 nm) form a network around the nucleus extending to the periphery of the cell. They make cell-to-cell contacts with adjacent cells via desmosomes, and with basement matrix via hemidesmosomes (Fig. 2.5; see also Fig. 24.27). Their function appears to be structural integrity; they are prominent in cellular tissues under stress and their disruption in genetic disease can cause structural defects or cell collapse. More than 40 different types of proteins polymerize to form intermediate filaments specific to particular cell types. For example keratin intermediate fibres are only found in epithelial cells whilst vimentin is in mesothelial (fibroblastic) cells. However, lamin intermediate filaments form the nuclear membrane skeleton in most cells.
Intermediate filaments (~10 nm) form a network around the nucleus extending to the periphery of the cell. They make cell-to-cell contacts with adjacent cells via desmosomes, and with basement matrix via hemidesmosomes (Fig. 2.5; see also Fig. 24.27). Their function appears to be structural integrity; they are prominent in cellular tissues under stress and their disruption in genetic disease can cause structural defects or cell collapse. More than 40 different types of proteins polymerize to form intermediate filaments specific to particular cell types. For example keratin intermediate fibres are only found in epithelial cells whilst vimentin is in mesothelial (fibroblastic) cells. However, lamin intermediate filaments form the nuclear membrane skeleton in most cells.
 Microfilaments (3–6 nm) are polymers of actin, one of the most abundant proteins in all cells. The actin microfilament network controls cell shape, prevents cellular deformation, is involved in cell–cell and cell–matrix adhesion, in cell movements such as crawling and cytokinesis (cell division), and in intracellular vesicle transport. Bundles of actin filaments form the structural core of cellular protrusions such as microvilli, lamellipodia and filopodia (see below). Actin microfilament bundles within the cell can associate with myosin II to form contractile stress fibres, similar to muscle sarcomeres. Stress fibres are often found as circumferential belts around the apical surfaces of epithelial cells where cells associate with adjacent cells via adherens junctions, permitting reaction to external stresses as a cellular sheet. Stress fibres also form where actin interacts via accessory proteins with the extracellular matrix at sites of focal adhesion (see Fig. 2.8c). This occurs during cell movements during inflammation, wound healing and metastasis. During cytokinesis actin-myosin II bundles form the contractile ring separating dividing cells. Like microtubules, microfilaments are polar, so can be used to transport secretory vesicles, endosomes and mitochondria, powered by motor proteins, including myosin I and V.
Microfilaments (3–6 nm) are polymers of actin, one of the most abundant proteins in all cells. The actin microfilament network controls cell shape, prevents cellular deformation, is involved in cell–cell and cell–matrix adhesion, in cell movements such as crawling and cytokinesis (cell division), and in intracellular vesicle transport. Bundles of actin filaments form the structural core of cellular protrusions such as microvilli, lamellipodia and filopodia (see below). Actin microfilament bundles within the cell can associate with myosin II to form contractile stress fibres, similar to muscle sarcomeres. Stress fibres are often found as circumferential belts around the apical surfaces of epithelial cells where cells associate with adjacent cells via adherens junctions, permitting reaction to external stresses as a cellular sheet. Stress fibres also form where actin interacts via accessory proteins with the extracellular matrix at sites of focal adhesion (see Fig. 2.8c). This occurs during cell movements during inflammation, wound healing and metastasis. During cytokinesis actin-myosin II bundles form the contractile ring separating dividing cells. Like microtubules, microfilaments are polar, so can be used to transport secretory vesicles, endosomes and mitochondria, powered by motor proteins, including myosin I and V.
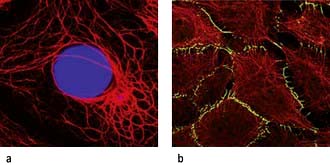
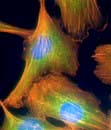
Cell shape and motility
The cytoskeleton determines cell shape and surface structures.
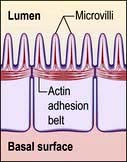
Microvilli. The apical surface of some epithelial cells is covered in tiny microvilli (~1 µm long) forming a brush border of thousands of small finger-like projections of the plasma membrane that increase the surface area for uptake or efflux (Fig. 2.6). At their core are 20–30 cross-linked actin microfilaments.

Motile cilia are also fine, finger-like protrusions but these are longer (~10–20 µm long) (Fig. 2.6). At their core is an axoneme, a bundle of nine cross-linked tubulin microtubule doublets surrounding a central pair. The action of the motor domain dynein serves to bend the cilium. Neighbouring cilia tend to beat in unison generating waves of motion that move fluid over the cell surface in the gut and airways (see Fig. 15.9), and also in the fallopian tubes.
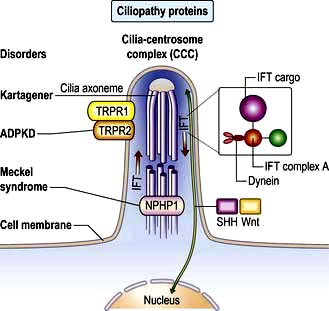
Non-motile or primary cilia. Most cells also have a single primary cilium. These cilia have a variant axoneme with no central pair of microtubules and while they have dynein they are non-motile (the dynein is used to traffic cargo along the axoneme). Primary cilia are used for signalling during development and in the adult. Other related non-motile cilia are found in specialized cells, e.g. in the photoreceptors of the retina, the sensory neurones of the olfactory system, and in the sensory hair cells of the cochlea. A range of human ciliopathies (Fig. 2.7) have been described with pleiotropic symptoms depending on which cilia are affected. These include polycystic kidney disease, Bardet–Biedl syndrome (p. 1007), Joubert’s syndrome and Ellis–van Creveld syndrome.
 Filopodia: if remodelled essentially in one dimension into a long actin filament, the leading edge of the plasma membrane is pushed forward as spikes, similar to long thin villi.
Filopodia: if remodelled essentially in one dimension into a long actin filament, the leading edge of the plasma membrane is pushed forward as spikes, similar to long thin villi.


Movement. A similar mechanism involving the coordinated remodelling of the cytoskeleton and the formation and release of cell adhesions underlies all three modes of migration. Essentially, actin is polymerized at the leading edge extending the plasma membrane forward. New adhesions are formed with the substratum (cells and/or extracellular matrix) at the leading edge to provide purchase. Release of attachments and depolymerization of the actin filaments at the trailing edge then allows the cell to move forward. Myosin and myosin motor proteins may also be involved at the trailing edge providing the tractive force to pull the cell body forward. The complex coordination of these processes is controlled via signalling pathways involving members of the Rho protein family of GTPases (see p. 21). Key signalling targets are the WASp family of proteins which stimulate actin polymerization. The significance of cell motility in humans is illustrated by mutation of the WASp expressed in blood cell lineages, which causes Wiskott–Aldrich syndrome (p. 66), and is characterized by severe immunodeficiency and thrombocytopenia (platelet deficiency).
The cell and its environment
 Epithelial tissues comprise layers of cells held tightly together by intercellular junctions and are usually separated from underlying tissue by specialized ECM called basal lamina. Epithelia cover surfaces (e.g. epidermis, tongue surface) and line passageways (airways, digestive tract, blood vessels), providing protection and regulating absorption and secretion.
Epithelial tissues comprise layers of cells held tightly together by intercellular junctions and are usually separated from underlying tissue by specialized ECM called basal lamina. Epithelia cover surfaces (e.g. epidermis, tongue surface) and line passageways (airways, digestive tract, blood vessels), providing protection and regulating absorption and secretion.

Extracellular matrix
The gel or ground substance of the ECM is made from polysaccharides (glycosaminoglycans or GAGs), usually bound to proteins to form proteoglycans (p. 494). These are a diverse group of molecules conferring different matrix properties in different tissues. They form hydrated gels which can resist compression yet permit diffusion of metabolites and signalling molecules.
 Hyaluronan, a very large hydrated GAG, is secreted into the joint space in synovial joints (p. 493), where it aids lubrication and helps reduce compressive forces.
Hyaluronan, a very large hydrated GAG, is secreted into the joint space in synovial joints (p. 493), where it aids lubrication and helps reduce compressive forces.
 Aggrecan, a very large proteoglycan, forms part of the articular cartilage of joints (p. 494) also contributing to compression resistance.
Aggrecan, a very large proteoglycan, forms part of the articular cartilage of joints (p. 494) also contributing to compression resistance.

Fibrous proteins of ECM (p. 495) include collagens and tropoelastin, which polymerize into collagen and elastin fibres, and fibronectin which is insoluble in many tissues but soluble in plasma. Collagen provides tensile strength, elastin confers elasticity, while the widely distributed fibronectin adheres to both cells and ECM, and thus positions cells within the ECM. Collagens, the most abundant proteins in the body, are widely distributed and play a structural role in skin and bone, where collagen defects and disorders often manifest. Elastin fibres are abundant in arteries, lung and skin. Elastic fibres have a fibrillin sheath and fibrillin mutations underlie Marfan’s syndrome (p. 760). The ECM can be degraded and remodelled by proteins of the matrix metalloproteinase (MMP) family. These are needed for angiogenesis and morphogenesis and are also involved in the pathophysiology of cancer, cirrhosis and arthritis.
Basal lamina or basement membrane (lamina propria) is a specialized form of ECM, which separates cells from underlying tissue and provides a supportive, anchoring and protective role. Basal lamina can also act as molecular filters (e.g. glomerular filtration barrier, p. 636) and mediate signalling between adjacent tissues (e.g. epidermal-dermal signalling in skin). Type IV collagen, heparan sulphate proteoglycan, laminin and nidogen are key basal lamina proteins. Inherited abnormalities in these proteins cause skin blistering diseases (see Fig. 24.27). Breach of the basal lamina by invading cancer cells is a key stage in progression of epithelial carcinoma in situ to a malignant carcinoma.
Cell–cell adhesion
Cell–cell adhesion proteins (Fig. 2.8a)
 Immunoglobulin-like cell adhesion molecules (iCAMs or CAMs) (Fig. 2.8a) are structurally related to antibodies. The neural cell adhesion molecule (N-CAM) is found predominantly in the nervous system. It mediates a homophilic (like-like) adhesion. When bound to an identical molecule on another cell, N-CAM can also associate laterally with a fibroblast growth factor receptor and stimulate its tyrosine kinase activity to induce neurite growth thus triggering cellular responses by indirect activation of the recipient.
Immunoglobulin-like cell adhesion molecules (iCAMs or CAMs) (Fig. 2.8a) are structurally related to antibodies. The neural cell adhesion molecule (N-CAM) is found predominantly in the nervous system. It mediates a homophilic (like-like) adhesion. When bound to an identical molecule on another cell, N-CAM can also associate laterally with a fibroblast growth factor receptor and stimulate its tyrosine kinase activity to induce neurite growth thus triggering cellular responses by indirect activation of the recipient.
 Selectins. Unlike most adhesion molecules which bind to other proteins, the selectins interact with carbohydrate ligands or mucin complexes on leucocytes and endothelial cells (vascular and haematological systems). Leucocyte-selectin (CD62L) mediates the homing of lymphocytes to lymph nodes. Endothelial-selectin (CD62E) is expressed after activation by inflammatory cytokines; the small basal amount of E-selectin in many vascular beds appears to be necessary for the migration of leucocytes. Platelet-selectin (CD62P) is stored in the alpha granules of platelets and the Weibel–Palade bodies of endothelial cells, but it moves rapidly to the plasma membrane upon stimulation of these cells. All three selectins play a part in leucocyte rolling (p. 63).
Selectins. Unlike most adhesion molecules which bind to other proteins, the selectins interact with carbohydrate ligands or mucin complexes on leucocytes and endothelial cells (vascular and haematological systems). Leucocyte-selectin (CD62L) mediates the homing of lymphocytes to lymph nodes. Endothelial-selectin (CD62E) is expressed after activation by inflammatory cytokines; the small basal amount of E-selectin in many vascular beds appears to be necessary for the migration of leucocytes. Platelet-selectin (CD62P) is stored in the alpha granules of platelets and the Weibel–Palade bodies of endothelial cells, but it moves rapidly to the plasma membrane upon stimulation of these cells. All three selectins play a part in leucocyte rolling (p. 63).

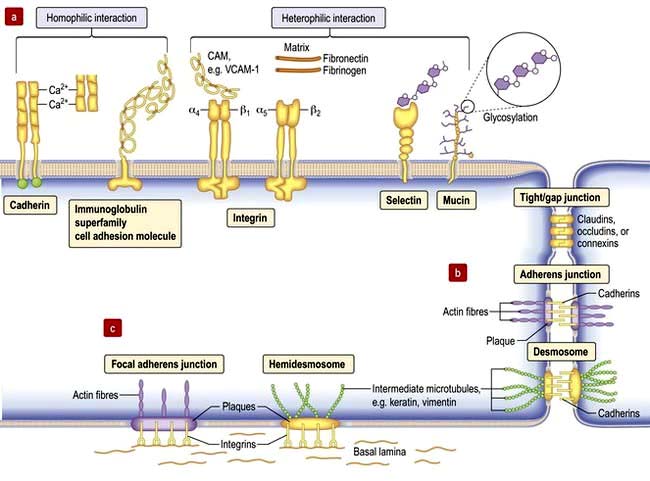
Tight junctions (zonula occludens)
These are mediated by the integral membrane proteins, claudins and occludens; they hold cells together. They form at the top (apical) side of epithelial cells including intestinal, skin and kidney cells, and endothelial cells of blood vessels (Fig. 2.8) to provide a regulated barrier to the movement of ions and solutes through the epithelia or endothelia but also between cells (paracellular transport). Tight junctions also confer polarity to cells by acting as a gate between the apical and the baso-lateral membranes, preventing diffusion of membrane lipids and proteins. Twenty-four claudins (the protein in the junction) are differentially expressed in different cell types to regulate paracellular transport. For example, changes in claudin expression in the kidney nephron correlate with permeability changes. Mutations in claudins 16 (previously named parcellin-1) and 19, expressed in the thick ascending limb in the loop of Henle in the kidney, cause an inherited renal disorder, familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC; p. 657).
Gap junctions
Gap junctions (Fig. 2.8) allow low molecular weight substances to pass directly between cells, permitting metabolic and electric coupling (e.g. in cardiomyocytes). Protein channels made of six connexin proteins (as well as claudins and occludens) are aligned between adjacent cells and allow the passage of solutes up to 1000 kDa (e.g. amino acids, sugars, ions, chemical messengers). The channels are regulated by many factors such as intracellular Ca2+, pH, voltage. Gap junctions form in almost all interacting cells, but connexin family members are differentially expressed. Mutant connexins cause many inherited disorders, such as the X-linked form of Charcot–Marie–Tooth disease (GJB1; p. 1147) and are also a major cause of genetic hearing loss (GJB2).
Adherens junctions
Adherens junctions are multiprotein intercellular adhesive structures, prominent in epithelial tissues (Fig. 2.8b). They attach principally to actin microfilaments inside the cell with the aid of multiple additional proteins, and also attach and stabilize microtubules. At the apical sides of epithelial cells a prominent type of adherens junction, the zonula adherens, attaches to the circumferential actin stress fibres. The fascia adherens in cardiac muscle is also an adherens junction. Transmembrane proteins of the cadherin family provide the adhesion through interaction of their extracellular domains. Downregulation of cadherins is a feature of cancer progression in many cells.
Desmosomes (macula adherens)
Desmosomes provide strong attachment between cells and are prominent in tissues subject to stress such as skin and cardiac muscle (see Fig. 2.5, Fig. 2.8b and Fig. 24.1). Like adherens junctions, they are multiprotein complexes, where adhesion is provided by transmembrane cadherin proteins, desmogleins and desmocollins. However, within the cell desmosomes interact principally with intermediate filaments rather than microfilaments and microtubules. Germline mutations in genes encoding desmosomes are a cause of cardiomyopathy with/without cutaneous features and in pemphigus vulgaris and pemphigus foliaceus (p. 1222).
Basement membrane adhesion
Cells adhere (Fig. 2.8c) to non-basal lamina ECM via secreted proteins such as fibronectin and collagen, and to basal lamina proteins via focal adhesion and hemidesmosome multiprotein complexes (e.g. keratin or vimentin). Here, integrins replace cadherins as surface adhesion molecules as the key adhesive proteins. Integrins are transmembrane sensors or receptors, which change shape upon binding to ECM, a process called ‘outside-in’ signalling. Inside the cell, integrins interact with the cytoskeleton and a complex array of over 150 proteins that influence intracellular signalling pathways affecting proliferation, survival, shape, mobility and gene expression.
 Outside-in signalling: forms the basis for anoikis or apoptotic death, such as occurs in cancer cells that inappropriately lose cell-substratum adhesion.
Outside-in signalling: forms the basis for anoikis or apoptotic death, such as occurs in cancer cells that inappropriately lose cell-substratum adhesion.
 Inside-out signalling: intracellular changes can also be communicated extracellularly via integrins whereby intracellular changes cause integrins to change from an inactive to an actively adhesive conformation. This ‘inside-out’ signalling occurs when platelet integrins glycoprotein IIb-IIIa (GPIIb-IIa) are activated to bind fibrinogen at sites of vessel injury, resulting in platelet aggregation (p. 415 and Fig. 8.41).
Inside-out signalling: intracellular changes can also be communicated extracellularly via integrins whereby intracellular changes cause integrins to change from an inactive to an actively adhesive conformation. This ‘inside-out’ signalling occurs when platelet integrins glycoprotein IIb-IIIa (GPIIb-IIa) are activated to bind fibrinogen at sites of vessel injury, resulting in platelet aggregation (p. 415 and Fig. 8.41).
Defective integrins are associated with many immunological and clotting disorders such as Bernard–Soulier syndrome and Glanzmann’s thrombasthenia (p. 420).
FURTHER READING
De Matteis MA, Luini A. Mendelian disorders of membrane trafficking. N Engl J Med 2011; 365:927–928.
Jean C, Gravelle P, Fournie JJ et al. Influence of stress on extracellular matrix and integrin biology. Oncogene 2011; 30:2697–2706.
Thomason HA, Scothern A, McHarg S et al. Desmosomes: adhesive strength and signalling in health and disease. Biochem J 2010; 429:419–433.
Cellular mechanisms
Cell signalling
Signalling or communication between cells is often via extracellular molecules or ligands which can be proteins (e.g. hormones, growth factors), small molecules (e.g. lipid-soluble steroid hormones such as oestrogen and testosterone) or dissolved gases such as nitric oxide. The signal is usually received by membrane protein receptors, although some signals such as steroid hormones, enter the target cell where they interact with intracellular receptors (Fig. 2.9). Some signalling, especially in the immune system, relies on cell–cell contact, where the signalling molecule (ligand) and receptor are on adjacent cells.
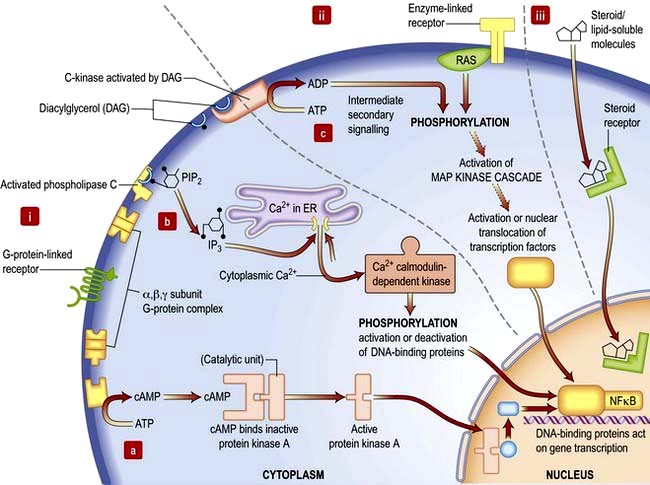
Receptors transduce signals across the membrane to an intracellular pathway or second messengers to change cell behaviour, often ultimately affecting gene expression (Figs 2.9, 2.10). The membrane-bound receptors fall into three main groups based on downstream signalling pathways:
 Ion channel linked receptors (voltage or ligand activated ion channels; see Fig. 2.3). At synaptic junctions between neurones (Fig. 22.1), these receptors open in response to neurotransmitters such as glutamate, epinephrine (adrenaline) or acetylcholine to cause a rapid depolarization of the membrane.
Ion channel linked receptors (voltage or ligand activated ion channels; see Fig. 2.3). At synaptic junctions between neurones (Fig. 22.1), these receptors open in response to neurotransmitters such as glutamate, epinephrine (adrenaline) or acetylcholine to cause a rapid depolarization of the membrane.
 G-protein-linked receptors such as the odorant and light (opsin) family of receptors belong to a large family of seven-pass transmembrane proteins (see Figs 2.2 and 2.9). On activation by ligand G-protein-linked receptors bind a GTP-binding protein (G-protein), which activates adjacent enzyme complexes or ion channels (Figs 2.9 and 22.1). The adjacent enzymes can be adenylcyclase (see below).
G-protein-linked receptors such as the odorant and light (opsin) family of receptors belong to a large family of seven-pass transmembrane proteins (see Figs 2.2 and 2.9). On activation by ligand G-protein-linked receptors bind a GTP-binding protein (G-protein), which activates adjacent enzyme complexes or ion channels (Figs 2.9 and 22.1). The adjacent enzymes can be adenylcyclase (see below).
 Enzyme-linked receptors (Figs 2.2 and 2.9) typically have an extracellular ligand-binding domain, a single transmembrane-spanning region, and a cytoplasmic domain that has intrinsic enzyme activity or which will bind and activate other membrane-bound or cytoplasmic enzyme complexes. This group of receptors is highly variable but many have kinase activity or associate with kinases, which act by phosphorylating substrate proteins usually on a tyrosine (e.g. the platelet-derived growth factor (PDGF) receptor) or a serine/threonine (e.g. the transforming growth factor-beta (TGF-β) receptor).
Enzyme-linked receptors (Figs 2.2 and 2.9) typically have an extracellular ligand-binding domain, a single transmembrane-spanning region, and a cytoplasmic domain that has intrinsic enzyme activity or which will bind and activate other membrane-bound or cytoplasmic enzyme complexes. This group of receptors is highly variable but many have kinase activity or associate with kinases, which act by phosphorylating substrate proteins usually on a tyrosine (e.g. the platelet-derived growth factor (PDGF) receptor) or a serine/threonine (e.g. the transforming growth factor-beta (TGF-β) receptor).
Signal transduction
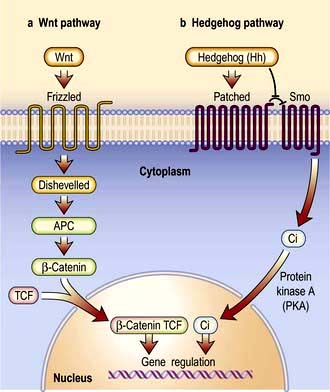
Signal transduction from the receptor to the site of action in the cell is mediated by small signalling molecules called second messengers, or by signalling proteins (Fig. 2.9). Changes to activity of signalling proteins by acquired mutation occur in cancer, and many anti-cancer drugs target signalling pathways. For example, the Hedgehog pathway is involved in human development, tissue repair and cancer (Fig. 2.10). Inhibitors of this pathway are being developed for therapeutic interventions. The Wnt pathway is also involved in bone formation (p. 550).
 Second messengers include cAMP and lipid-derived inositol triphosphate (IP3) and diacylglycerol (Fig. 2.9). These molecules diffuse from the receptor to bind and change the activity of downstream proteins propagating the signal. cAMP triggers a protein signalling cascade by activating a cAMP-dependent protein kinase. Diacylglycerol activates protein kinase C while IP3 mobilizes calcium from intracellular stores (e.g. from the ER; Fig. 14.9).
Second messengers include cAMP and lipid-derived inositol triphosphate (IP3) and diacylglycerol (Fig. 2.9). These molecules diffuse from the receptor to bind and change the activity of downstream proteins propagating the signal. cAMP triggers a protein signalling cascade by activating a cAMP-dependent protein kinase. Diacylglycerol activates protein kinase C while IP3 mobilizes calcium from intracellular stores (e.g. from the ER; Fig. 14.9).
 G-proteins or GTP-binding proteins are signalling proteins which switch between an active state when GTP is bound and an inactive state when bound to GDP. The most well-known members are the Ras superfamily, comprising Ras, Rho, Rab, Arf and Ran families. Activation of Ras members by somatic mutation is found in ~33% of human cancers. Ras members are often bound downstream of tyrosine kinase receptors, where they transmit signals by activating a cascade of downstream protein kinase activity (Fig. 2.9). Ras signalling molecules have roles in many cellular activities, including regulation of cell cycle, intracellular transport, and apoptosis.
G-proteins or GTP-binding proteins are signalling proteins which switch between an active state when GTP is bound and an inactive state when bound to GDP. The most well-known members are the Ras superfamily, comprising Ras, Rho, Rab, Arf and Ran families. Activation of Ras members by somatic mutation is found in ~33% of human cancers. Ras members are often bound downstream of tyrosine kinase receptors, where they transmit signals by activating a cascade of downstream protein kinase activity (Fig. 2.9). Ras signalling molecules have roles in many cellular activities, including regulation of cell cycle, intracellular transport, and apoptosis.
 Kinase and phosphatase signalling proteins are enzymes that phosphorylate or dephosphorylate residues on downstream proteins to alter their activity. Chains of kinase activity (phosphorylation cascades) consisting of sequential phosphorylation of proteins can transduce signals from the membrane receptor to the site of action in the cell. The tyrosine kinase receptors phosphorylate each other when ligand binding brings the intracellular receptor components into close proximity (see Fig. 2.9). The inner membrane and cytoplasmic targets of these activated receptor complexes are ras, protein kinase C and ultimately the MAP (mitogen-activated protein) kinase, Janus-Stat pathways or phosphorylation of IκB causing it to release its DNA-binding protein, nuclear factor kappa B (NFκB). For example, activated Ras binds and activates the kinase Raf, the first of a set of three mitogen-activated protein (MAP) kinases, which transmit signals by successive phosphorylation of target proteins which can ultimately effect transcription (Fig. 2.9). Kinases and phosphatases are frequently mutated in cancers. Somatic mutations in one Raf member, B-Raf, occur in ~60% of malignant melanomas (usually the mutation V600E) and are common in other cancers (p. 1225).
Kinase and phosphatase signalling proteins are enzymes that phosphorylate or dephosphorylate residues on downstream proteins to alter their activity. Chains of kinase activity (phosphorylation cascades) consisting of sequential phosphorylation of proteins can transduce signals from the membrane receptor to the site of action in the cell. The tyrosine kinase receptors phosphorylate each other when ligand binding brings the intracellular receptor components into close proximity (see Fig. 2.9). The inner membrane and cytoplasmic targets of these activated receptor complexes are ras, protein kinase C and ultimately the MAP (mitogen-activated protein) kinase, Janus-Stat pathways or phosphorylation of IκB causing it to release its DNA-binding protein, nuclear factor kappa B (NFκB). For example, activated Ras binds and activates the kinase Raf, the first of a set of three mitogen-activated protein (MAP) kinases, which transmit signals by successive phosphorylation of target proteins which can ultimately effect transcription (Fig. 2.9). Kinases and phosphatases are frequently mutated in cancers. Somatic mutations in one Raf member, B-Raf, occur in ~60% of malignant melanomas (usually the mutation V600E) and are common in other cancers (p. 1225).
Nuclear control
DNA and RNA structure
Hereditary information is contained in the sequence of the building blocks of double-stranded deoxyribonucleic acid (DNA) (Fig. 2.11). Each strand of DNA is made up of a deoxyribose-phosphate backbone and a series of purine (adenine (A) and guanine (G)) and pyrimidine (thymine (T) and cytosine (C)) bases, and because of the way the sugar phosphate backbone is chemically coupled, each strand has a polarity with a phosphate at one end (the 5′ end) and a hydroxyl at the other (the 3′ end). The two strands of DNA are held together by hydrogen bonds between the bases. A can only pair with T, and G can only pair with C, therefore each strand is the antiparallel complement of the other (Fig. 2.11b). This is key to DNA replication because each strand can be used as a template to synthesize the other.
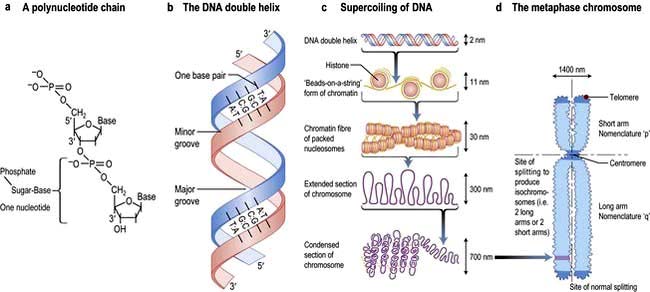
The two strands twist to form a double helix with a major and a minor groove, and the large stretches of helical DNA are coiled around histone proteins to form nucleosomes (Fig. 2.11c). They can be condensed further into the chromosomes that can be visualized by light microscopy at metaphase (see below; Fig. 2.11, Fig. 2.19).
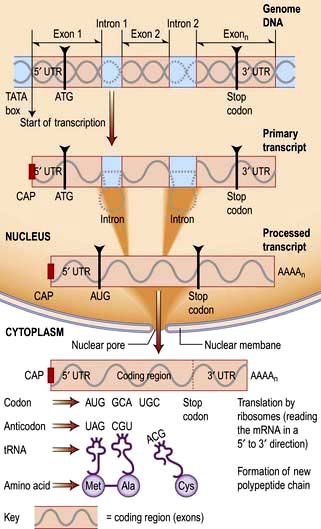
To express the information in the genome, cells first transcribe the code into the single strand ribonucleic acid (RNA). RNA is similar to DNA in that it comprises four bases A, G and C but with uracil (U) instead of T, and a sugar phosphate backbone with ribose instead of deoxyribose. Several types of RNA are made by the cell. Messenger RNA (mRNA) codes for proteins that are translated on ribosomes. Ribosomal RNA (rRNA) is a key catalytic component of the ribosome and amino acids are delivered to the nascent peptide chain on transfer RNA (tRNA) molecules. There are also a variety of RNAs that regulate gene expression or RNA processing. These include microRNA (miRNA) and small interfering RNA (siRNA) (see p. 27) that typically bind to a subset of mRNAs and inhibit their translation, or initiate their degradation, respectively. Other non-coding RNAs are involved in X-inactivation and telomere maintenance or RNA splicing and maturation.
DNA transcription
RNA is transcribed from the DNA template by an enzyme complex of more than one hundred proteins including RNA polymerase, transcription factors and enhancers. Promoter regions upstream of the gene dictate the start point and direction of transcription. The complex binds to the promoter region, the nucleosomes are remodelled to allow access, and a DNA helicase unwinds the double helix. RNA, like DNA, is synthesized in the 5′ to 3′ direction as ribonucleotides are added to the growing 3′ end of a nascent transcript. RNA polymerase does this by base-pairing the ribonucleotides to the DNA template strand running in the 3′ to 5′ direction. Messenger RNA is modified as it is synthesized (Fig. 2.12). It is capped at the 5′ end with a modified guanine that is required for efficient processing of the mRNA and efficient translation, and introns are spliced from the nascent chain. Finally, the 3′ of the mRNA is modified with up to 200 A nucleotides by the enzyme poly-A polymerase. This 3′ poly-A tail is essential for nuclear export (through the nuclear pores), stability and efficient translation into protein by the ribosome.
Human protein coding sequences (exons) are interrupted by intervening sequences that are non-coding (introns) at multiple positions (Fig. 2.12). These have to be spliced from the nascent message in the nucleus by an RNA/protein complex called a spliceosome. Differential splicing describes the process by which two or more introns and their intervening exons are spliced from the mRNA. This contributes significantly to the complexity of the human transcriptome as proteins translated from these messages lack particular domains. This exon skipping can produce different protein activities.
Control of gene expression
The genome of all cells in the body encodes the same genetic information, yet different cell types express a very different subset of proteins and respond to external signals to switch on a new set of genes or to switch off a pathway. Gene expression can be controlled at many steps from transcription to protein degradation. However, for many genes transcription is the key point of regulation. This is controlled primarily by proteins which bind to short sequences within the promoter regions that either repress or activate transcription, or to more distant sequences where proteins bind to enhance expression. These transcription factors and enhancers are often the end points of signalling pathways that transduce extracellular signals to changes in gene expression (Fig. 2.9).
Often this involves the translocation of an activated factor from the cytoplasm to the nucleus. In the nucleus the DNA binding proteins recognize the shape and position of hydrogen bond acceptor and donor groups within the major and minor grooves of the double helix (i.e. the double helix does not need to be unwound). There are several classes of DNA binding protein that differ in the protein structural motif that allows them to interact with the double helix. These primarily include helix-turn-helix, zinc finger and leucine zipper motifs, although protein loops and β-sheets are used by some proteins. More permanent control of gene expression patterns can be achieved epigenetically. These are modifications (typically methylation and/or acetylation) of the DNA, or the histones of the nucleosome, that silence genes. Epigenetic modification is also heritable meaning that a dividing liver cell, for example, can give rise to two daughter cells with the same epigenetic signals such that they express the appropriate transcriptome for a liver cell. Epigenetic change forms the basis of genetic imprinting (see p. 42).
Most of the genome is transcribed but only a minority of transcripts encode proteins (see Human Genetics, p. 34). The non-coding RNAs (ncRNAs) include a group that regulate gene expression (see DNA and RNA structure). miRNAs and siRNAs are short ncRNAs (19–29 bp) that are known to regulate expression of approximately 30% of genes by degradation of transcripts or repression of protein synthesis. With further annotation of the genome a growing range of additional regulatory ncRNA classes are being identified, many of which control gene expression by epigenetic mechanisms.
The cell cycle and mitosis
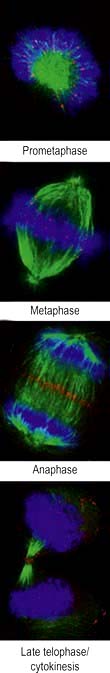
The cell duplication cycle has four phases, G1, S, G2 and mitosis (Fig. 2.13), and takes about 20–24 hours to complete for a rapidly dividing adult cell. G1, S and G2 are collectively known as interphase during which the cells double in mass (the two gap phases are used for growth) and duplicate their 46 chromosomes (S phase). Mitosis describes, in four sub-phases (prophase, metaphase, anaphase and telophase), the process of chromosome separation and nuclear division before cytokinesis (division of the cytoplasm into two daughter cells).
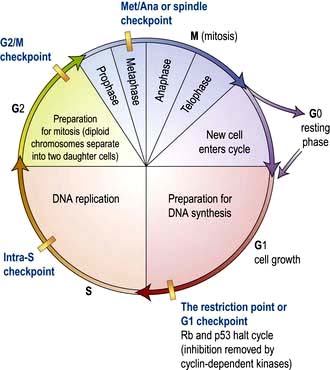
Figure 2.13 The cell cycle. Cells are stimulated to leave non-cycle G0 to enter G1 phase by growth factors. During G1, transcription of the DNA synthesis molecules occurs. Rb is a ‘checkpoint’ (inhibition molecule) between G1 and S phases and must be removed for the cycle to continue. This is achieved by the action of the cyclin-dependent kinase produced during G1. During the S phase, any DNA defects will be detected and p53 will halt the cycle (see p. 46). Following DNA synthesis (S phase), cells enter G2, a preparation phase for cell division. Mitosis takes place in the M phase. The new daughter cells can now either enter G0 and differentiate into specialized cells, or re-enter the cell cycle.
Synthesis phase; DNA replication
 DNA helicase which hydrolyses ATP to unwind the double helix and expose each strand as a template for replication. The two strands are antiparallel, and because DNA can only be extended by addition of nucleotide triphosphates to the 3′-hydroxyl end of the growing chain, replication of each strand must be treated differently. For one strand, called the leading template strand, the replication fork is moving in a 3′ to 5′ direction along the template, meaning that the newly synthesized strand is being synthesized in a 5′ to 3′ direction.
DNA helicase which hydrolyses ATP to unwind the double helix and expose each strand as a template for replication. The two strands are antiparallel, and because DNA can only be extended by addition of nucleotide triphosphates to the 3′-hydroxyl end of the growing chain, replication of each strand must be treated differently. For one strand, called the leading template strand, the replication fork is moving in a 3′ to 5′ direction along the template, meaning that the newly synthesized strand is being synthesized in a 5′ to 3′ direction.



Control of the cell cycle and checkpoints
Cyclin-dependent kinases (Cdks), Retinoblastoma (Rb) and p53
Progression through the cell cycle is tightly controlled and punctuated by three key checkpoints when the cell interprets environmental and cellular signals to determine whether it is appropriate or safe to proceed (Fig. 2.13). The switches that allow progression beyond these checkpoints are a family of small protein complexes called cyclin-dependent kinases (Cdks) that phosphorylate serines or threonines in key target proteins at each stage. It is the regulatory cyclin subunit of the Cdks that oscillates during the cell cycle (the actual kinase domain may be present throughout but only activated by the transient expression of its cognate cyclin).
Checkpoints
The restriction point (G1 checkpoint)
 to phosphorylate their target proteins to initiate helix unwinding of the DNA at origins of replication allowing the replication complex to begin DNA synthesis
to phosphorylate their target proteins to initiate helix unwinding of the DNA at origins of replication allowing the replication complex to begin DNA synthesis

 G1-Cdk responds positively to mitogenic (progrowth) environmental signals like platelet-derived growth factor (PDGF) or epidermal growth factor (EGF). Activated G1-Cdk phosphorylates and inactivates the retinoblastoma (Rb) protein which releases the transcription factor E2F to stimulate G1/S-Cdk and S-Cdk synthesis that are necessary for progression.
G1-Cdk responds positively to mitogenic (progrowth) environmental signals like platelet-derived growth factor (PDGF) or epidermal growth factor (EGF). Activated G1-Cdk phosphorylates and inactivates the retinoblastoma (Rb) protein which releases the transcription factor E2F to stimulate G1/S-Cdk and S-Cdk synthesis that are necessary for progression.
 G1/S-Cdk and S-Cdk are also responsive to DNA damage via the p53 pathway. On DNA damage, the transcription factor p53 is phosphorylated and stimulates transcription of the p21 gene. p21 protein is an inhibitor of both G1/S-Cdk and S-Cdk. Both the Rb and p53 are regulators of the restriction-point. Loss of function of either disables aspects of the negative control pathways. Rb and p53 are commonly mutated in cancer and both are therefore considered ‘tumour suppressor genes’ (see p. 46).
G1/S-Cdk and S-Cdk are also responsive to DNA damage via the p53 pathway. On DNA damage, the transcription factor p53 is phosphorylated and stimulates transcription of the p21 gene. p21 protein is an inhibitor of both G1/S-Cdk and S-Cdk. Both the Rb and p53 are regulators of the restriction-point. Loss of function of either disables aspects of the negative control pathways. Rb and p53 are commonly mutated in cancer and both are therefore considered ‘tumour suppressor genes’ (see p. 46).



Synthesis and secretion
Protein translation
The mature mRNA is transported through the nuclear pore into the cytoplasm for translation into protein by ribosomes (Fig. 2.12).
 The two subunits of ribosomes (the 40S and 60S) are formed in the nucleolus from multiple proteins and several rRNAs, before transport to the cytoplasm.
The two subunits of ribosomes (the 40S and 60S) are formed in the nucleolus from multiple proteins and several rRNAs, before transport to the cytoplasm.




Lipid synthesis
Fatty acids, molecules with a hydrocarbon chain with 4–28 carbons, are central to cellular life and human metabolism. They form the hydrophobic moiety of membrane lipids (see p. 17), they are precursors for short-lived, near acting lipid paracrines such as leukotrienes and prostaglandins, and they are energy stores particularly in the form of triglycerides.
Fatty acids as an energy store
Long chain fatty acids can be incorporated into triglycerides, which are relatively inert and lipophilic compounds that can be stored as fat droplets in cells (particularly adipocytes). When blood glucose is low, these triglycerides are hydrolysed, secreted into the bloodstream as free fatty acids, and distributed as an energy source for the cells of the body. In the recipient cell, fatty acids are metabolized in the mitochondrion to produce acetyl-CoA for the Krebs cycle (see p. 31). This is a particularly efficient storage system as gram for gram, triglyceride produces six times the amount of energy than glycogen and occupies less volume in the cell.
Essential fatty acids
Unsaturated fatty acids (UFAs) have carbon–carbon double bonds that are introduced by desaturase enzymes by removal of the hydrogens. The remaining hydrogens on either side of the double bond can be on the same side of the chain (cis) or on opposite sides (trans). The acyl chain of cis UFAs is kinked, which influences the packing of membrane lipids and the function of the membrane barrier. Humans have desaturases that can introduce some double bonds but lack a desaturase required to make linoleic acid or alpha-linolenic acid. These fatty acids have double bonds 6 and 3 carbons from their respective omega ends (the methyl end of the chain). Omega-6 and omega-3 UFAs are essential fatty acids that must be obtained from the diet (see Ch. 5). They are precursors of arachidonic acid and eicosapentaenoic acid, respectively, from which cyclo-oxygenase 1 and 2 (cox-1 and 2) (see p. 826) produce the paracrines that play a role in inflammation, pain, fever, and airway constriction.
Intracellular trafficking, exocytosis (secretion) and endocytosis
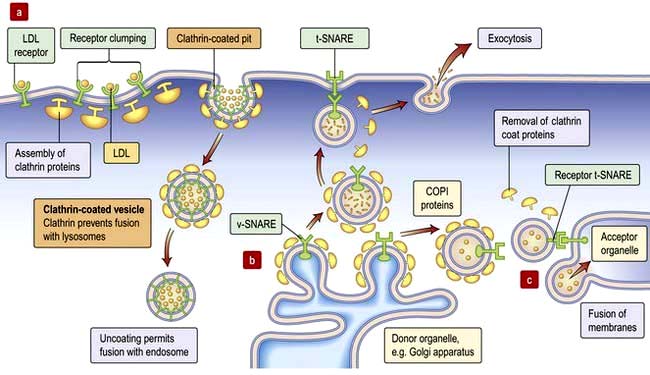
 Budding of vesicles involves recruitment of coat proteins and adaptors to the membrane. Thus, a receptor on binding to its ligand may stimulate a kinase to phosphorylate neighbouring phosphatidyl-inositol, or activate an associated small GTPase (Arf or SarI), increasing their affinities for a coat protein or adaptor. The coat protein (clathrin at the plasma membrane, COPI at the Golgi, COPII in the ER) forms a mesh around the developing vesicle (Fig. 2.4). Fully-formed vesicles normally shed their coat (often triggered by GTP hydrolysis by the GTPase), leaving the adaptor/receptor/lipid combination to identify the vesicle.
Budding of vesicles involves recruitment of coat proteins and adaptors to the membrane. Thus, a receptor on binding to its ligand may stimulate a kinase to phosphorylate neighbouring phosphatidyl-inositol, or activate an associated small GTPase (Arf or SarI), increasing their affinities for a coat protein or adaptor. The coat protein (clathrin at the plasma membrane, COPI at the Golgi, COPII in the ER) forms a mesh around the developing vesicle (Fig. 2.4). Fully-formed vesicles normally shed their coat (often triggered by GTP hydrolysis by the GTPase), leaving the adaptor/receptor/lipid combination to identify the vesicle.

 Fusion is accomplished by membrane-fusion SNARES (Fig. 2.4). The v-SNARE protein on the vesicle (often associated with the Rab effector) interacts with the t-SNARE on the target membrane to facilitate fusion of the two compartments (distinct combinations of v-SNARE and t-SNARE specify particular pathways).
Fusion is accomplished by membrane-fusion SNARES (Fig. 2.4). The v-SNARE protein on the vesicle (often associated with the Rab effector) interacts with the t-SNARE on the target membrane to facilitate fusion of the two compartments (distinct combinations of v-SNARE and t-SNARE specify particular pathways).
Vesicles that fuse with the plasma membrane replenish membrane lipids and proteins and also release cargo extracellularly (exocytosis; Fig. 2.4). Clathrin-coated vesicles are also used to recycle protein from the plasma membrane, and import extracellular cargo to internal compartments called endosomes. From endosomes cargo such as receptors is recycled back to the membrane, or cargo is sent for degradation in the lysosome in the process called endocytosis.
Pinocytosis and phagocytosis (see p. 19) are forms of endocytosis. Endocytosis can also occur via plasma membrane microdomains or lipid rafts called caveolae which pinch in to form uncoated vesicles that fuse with endosomes. Endocytosed vesicles can also be transported across the cell in a process called transcytosis. For example, cargo can be endocytosed at the apical surface of an epithelial cell and exocytosed across the basolateral membrane.
Energy production
The lipids and polysaccharides provide the most energy in a human diet, although protein can also be used. Enzymes secreted into the gut break down these polymers to their respective building blocks of fatty acids and sugars that are absorbed by the apical membrane of the gut epithelium (the transporters involved in the transcellular transport of glucose across the enterocyte are described in Figure 6.24). Fatty acids and sugars are further catabolized by enzyme pathways inside the cell to produce an array of activated carrier molecules.
Glycolysis
The six-carbon glucose is primarily catabolized in 10 steps by enzymes of the glycolytic pathway (see Fig. 8.25) to produce two three-carbon molecules of the carboxylic acid pyruvate. Glycolysis occurs in the cytosol and the first three steps actually consume energy (2×ATP), but the remaining six steps generate 4×ATP and 2×NADH, giving a net return of 2×ATP and 2×NADH.
Oxidative phosphorylation
Cellular degradation and death
Cell dynamics
Cell components are continually being formed and degraded, and most of the degradation steps involve ATP-dependent multienzyme complexes. Old cellular proteins are mopped up by a small cofactor molecule called ‘ubiquitin’, which interacts with these worn proteins via their exposed hydrophobic residues. Ubiquitin is a small 8.5 kDa regulating protein present universally in all living cells. Cells mark the destruction of a protein by attaching molecules to the protein. This ‘ubiquitination’ signals the protein to move to lysosomes or proteosomes for destruction. A complex containing more than five ubiquitin molecules is rapidly degraded by a large proteolytic multienzyme array termed ‘26S proteosome’. Ubiquitin also plays a role in regulation of the receptor tyrosine kinase in the cell cycle and in repair of DNA damage. The failure to remove worn proteins can result in the development of chronic debilitating disorders. For example, Alzheimer’s and frontotemporal dementias are associated with the accumulation of ubiquinated proteins (prion-like proteins), which are resistant to ubiquitin-mediated proteolysis. Similar proteolytic-resistant ubiquinated proteins give rise to inclusion bodies found in myositis and myopathies. This resistance can be due to point mutation in the target protein itself (e.g. mutant p53 in cancer; see p. 46) or as a result of an external factor altering the conformation of the normal protein to create a proteolytic-resistant shape, as in the prion protein of variant Creutzfeldt–Jakob disease (vCJD). Other conditions include von Hippel–Lindau syndrome (p. 634) and Liddle’s syndrome (p. 653).
Free radicals
Free radical scavengers bind reactive oxygen species. Alpha-tocopherol, urate, ascorbate and glutathione remove free radicals by reacting directly and non-catalytically. Severe deficiency of α-tocopherol (vitamin E deficiency) causes neurodegeneration. There is evidence that cardiovascular disease and cancer can be prevented by a diet rich in substances that diminish oxidative damage (p. 211). The principal dietary antioxidants are vitamin E, vitamin C, β-carotene and flavonoids.
Autophagy
Cells continually recycle material. For example, cellular proteins targeted for degradation can be ubiquitinated and degraded by the proteasome (p. 31), and mRNA can be de-tailed and degraded by the exosome or decapping complex. Cells respond to stresses like starvation by degrading much of their cytoplasmic contents in order to recycle components and survive.
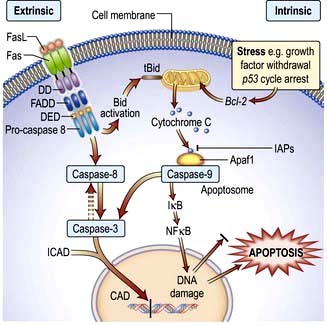
Apoptotic cell death
Apoptosis has characteristic features:
 Shrinkage of the cell and its nucleus
Shrinkage of the cell and its nucleus
 Chromatin aggregation into membrane-bound vesicles called apoptotic bodies
Chromatin aggregation into membrane-bound vesicles called apoptotic bodies


 signals from outside the cell (the extrinsic apoptotic pathway or the death receptor pathway) and
signals from outside the cell (the extrinsic apoptotic pathway or the death receptor pathway) and
 internal signals, such as DNA damage (the intrinsic apoptotic pathway or the mitochondrial pathway) (Fig. 2.14).
internal signals, such as DNA damage (the intrinsic apoptotic pathway or the mitochondrial pathway) (Fig. 2.14).


FURTHER READING
Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer 2011; 11(4):268–277.
Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med 2011; 17(3):313–319.
Forraz N, McGuckin CP. The umbilical cord: a rich and ethical stem cell source to advance regenerative medicine. Cell Prolif 2011; 44(Suppl 1):60–69.
Robbins RD, Prasain N, Maier BF et al. Inducible pluripotent stem cells: not quite ready for prime time? Curr Opin Organ Transplant 2010; 15:61–67.
Wu SM, Hochedlinger K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat Cell Biol 2011; 13:497–505.
Human genetics
FURTHER READING
ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007; 447(7146):799–816.
Lander ES. Initial impact of the sequencing of the human genome. Nature 2011; 470:187–197.
Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell 2011; 145:178–181.
Zhou H, Hu H, Lai M. Non-coding RNAs and their epigenetic regulatory mechanisms. Biol Cell 2010; 102:645–655.
Tools for human genetic analysis
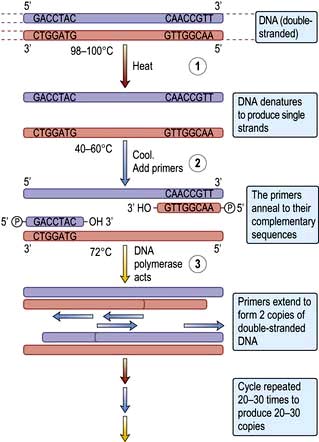
The polymerase chain reaction (PCR)
This technique revolutionized genetic research because minute amounts of DNA, e.g. from buccal cell scrapings, blood spots or single embryonic cells, can be amplified over a million times within a few hours. The exact DNA sequence to be amplified needs to be known because the DNA is amplified between two short (generally 17–25 bases) single-stranded DNA fragments (‘oligonucleotide primers’) that are complementary to the sequences on different strands at each end of the DNA of interest (Fig. 2.15).
Hybridization arrays
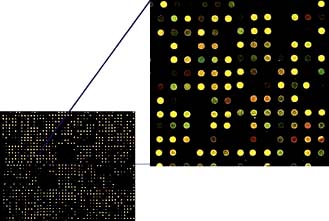
A fundamental property of DNA is that when two strands are separated, e.g. by heating, they will always re-associate and stick together again because of their complementary base sequences. Therefore, the presence or position of a particular gene can be identified using a gene ‘probe’ consisting of DNA or RNA, with a base sequence that is complementary to that of the sequence of interest. A DNA probe is thus a piece of single-stranded DNA that can locate and bind to its complementary sequence. Hybridization is utilized in array-based platforms, where thousands and thousands of probes can be analysed in one experiment to investigate global gene expression, large-scale genotyping, gene methylation status and/or for chromosomal aberrations, including for small chromosomal deletion/insertion events or copy number changes (Fig. 2.16).
DNA sequencing
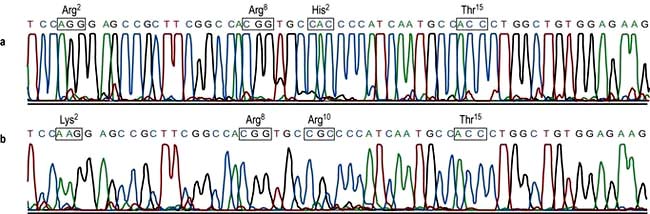
A chemical process known as dideoxy-sequencing or Sanger sequencing (after its inventor) allows the identification of the exact nucleotide sequence of a piece of DNA. As in PCR, an oligonucleotide primer is annealed adjacent to the region of interest. This primer acts as the starting point for a DNA polymerase to build a new DNA chain that is complementary to the sequence under investigation. Chain extension can be prematurely interrupted when a dideoxynucleotide becomes incorporated (because they lack the necessary 3′-hydroxyl group). As the dideoxynucleotides are present at a low concentration, not all the chains in a reaction tube will incorporate a dideoxynucleotide in the same place; so the tubes contain sequences of different lengths but which all terminate with a particular dideoxynucleotide. Each base dideoxynucleotide (G, C, T, A) has a different fluorochrome attached, and thus each termination base can be identified by its fluorescent colour. As each strand can be separated efficiently by capillary electrophoresis according to its size/length, simply monitoring the fluorescence as the reaction products elute from the capillary will give the gene sequence (Fig. 2.17).
Identification of gene function
RNAi
RNAi takes advantage of the cellular machinery that allows microRNAs encoded by the genome to regulate the expression of many genes at the level of messenger RNA stability and translation (see Control of gene expression, above). This phenomenon has been exploited in the laboratory to study the function of a gene of interest or, on a much larger scale, the function of each gene in the genome. In such an RNAi screen, a small interfering (si) RNA specific for each gene in the genome is introduced into cells grown in vitro, in effect knocking down expression of each gene in ~20 000 separate experiments. The phenotype of the cells in each experiment is then monitored to test the effect of loss of gene expression.
Animal models
FURTHER READING
Kim IY, Shin JH, Seong JK. Mouse phenogenomics, toolbox for functional annotation of human genome. BMB Rep 2010; 43:79–90.
Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet 2007; 8(5):353–367.
Markaki M, Tavernarakis N. Modeling human diseases in Caenorhabditis elegans. Biotechnology Journal 2010; 5:1261–1276
Perrimon N, Ni JQ, Perkins L. In vivo RNAi: today and tomorrow. Cold Spring Harb Perspect Biol 2010; 2(8):a003640.
Genetic polymorphisms and linkage studies
Techniques have been developed to identify and quantitate genetic polymorphisms such as single nucleotide polymorphisms (SNPs; p. 34), microsatellites and copy number variants (CNVs). For example, SNPs consist usually of two nucleotides at a particular site and vary between populations and ethnic groups. They must occur in at least 1% of the population to be a SNP. SNPs can be in coding or non-coding regions of the genes or be between genes and thus may not change the amino acid sequence of the protein.


SIGNIFICANT WEBSITES
National Center for Biotechnology Information: http://www.ncbi.nlm.nih.gov
UCSC Genome Bioinformatics: http://genome.ucsc.edu
Ensemble: http://www.ensembl.org/index.html
The Online Mendelian Inheritance in Man website, for information on gene products and their disease association: http://www.ncbi.nlm.nih.gov/omim
The biology of chromosomes
Human chromosomes
The nucleus of each diploid cell contains 6 × 109 bp of DNA in long molecules called chromosomes (Fig. 2.11). Chromosomes are massive structures containing one linear molecule of DNA that is wound around histone proteins into small units called nucleosomes, and these are further wound to make up the structure of the chromosome itself.
The chromosomes are classified according to their size and shape, the largest being chromosome 1. The constriction in the chromosome is the centromere, which can be in the middle of the chromosome (metacentric) or at one extreme end (acrocentric). The centromere divides the chromosome into a short arm and a long arm, referred to as the p arm and the q arm, respectively (Fig. 2.11d).
Chromosomes can only be seen easily in actively dividing cells. Typically, lymphocytes from the peripheral blood are stimulated to divide and are processed to allow the chromosomes to be examined. Cells from other tissues can also be used for chromosomal analysis, e.g. amniotic fluid, placental cells from chorionic villus sampling, bone marrow and skin (Box 2.1).
 Box 2.1
Box 2.1
Indications for chromosomal analysis
Chromosome studies may be indicated in the following circumstances:














Telomeres and immortality
The ends of chromosomes, telomeres (Fig. 2.11d), do not contain genes but many repeats of a hexameric sequence TTAGGG. Replication of linear chromosomes starts at coding sites (origins of replication) within the main body of chromosomes and not at the two extreme ends. The extreme ends are therefore susceptible to single-stranded DNA degradation back to double-stranded DNA. Thus, cellular ageing can be measured as a genetic consequence of multiple rounds of replication, with consequential telomere shortening. This leads to chromosome instability and cell death.
The mitochondrial chromosome
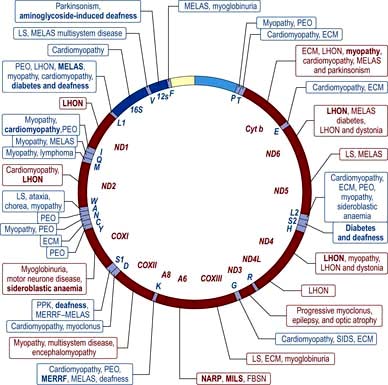
In addition to the 23 pairs of chromosomes in the nucleus of every diploid cell, the mitochondria in the cytoplasm of the cell also have their own genome. The mitochondrial chromosome is a circular DNA (mtDNA) molecule of approximately 16 500 bp, and every base-pair makes up part of the coding sequence. These genes principally encode proteins or RNA molecules involved in mitochondrial function. These proteins are components of the mitochondrial respiratory chain involved in oxidative phosphorylation producing ATP. They also have a critical role in apoptotic cell death. Every cell contains several hundred mitochondria, and therefore several hundred mitochondrial chromosomes. Virtually all mitochondria are inherited from the mother as the sperm head contains no (or very few) mitochondria. Disorders mapped to the mitochondrial chromosome are shown in Figure 2.18 and discussed on page 40.
Genetic disorders
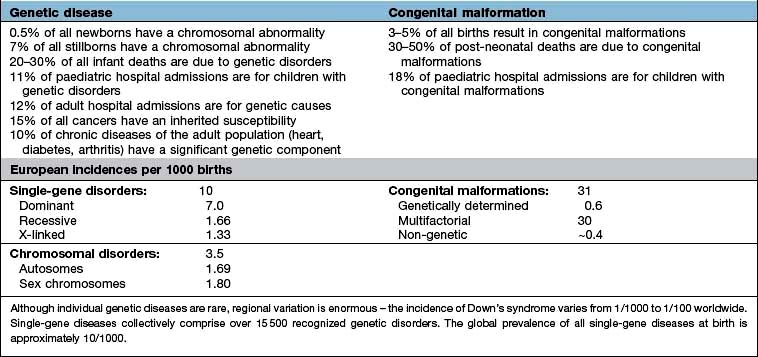
The spectrum of inherited or congenital genetic disorders can be classified as the chromosomal disorders, including mitochondrial chromosome disorders, the Mendelian and sex-linked single-gene disorders, a variety of non-Mendelian disorders, and the multifactorial and polygenic disorders (Table 2.1 and Box 2.2). All are a result of a mutation in the genetic code. This may be a change of a single base-pair of a gene, resulting in functional change in the product protein (e.g. thalassaemia) or gross rearrangement of the gene within a genome (e.g. chronic myeloid leukaemia). These mutations can be congenital (inherited at birth) or somatic (arising during a person’s life).
 Box 2.2
Box 2.2















Chromosomal disorders
Abnormal chromosome numbers
 either an extra chromosome, so resulting in a fetus that is ‘trisomic’ and has three instead of two copies of the chromosome;
either an extra chromosome, so resulting in a fetus that is ‘trisomic’ and has three instead of two copies of the chromosome;
 or no chromosome, so the fetus is ‘monosomic’ and has one instead of two copies of the chromosome.
or no chromosome, so the fetus is ‘monosomic’ and has one instead of two copies of the chromosome.
Non-disjunction can occur with autosomes or sex chromosomes. However, only individuals with trisomy 13, 18 and 21 survive to birth, and most children with trisomy 13 and trisomy 18 die in early childhood. Trisomy 21 (Down’s syndrome) is observed with a frequency of 1 in 650 live births, regardless of geography or ethnic background. This should be reduced with widespread screening (p. 43). Full autosomal monosomies are extremely rare and very deleterious. Sex-chromosome trisomies (e.g. Klinefelter’s syndrome, XXY) are relatively common. The sex-chromosome monosomy in which the individual has an X chromosome only and no second X or Y chromosome is known as Turner’s syndrome and is estimated to occur in 1 in 2500 live-born girls.
Abnormal chromosome structures
 Deletions of a portion of a chromosome may give rise to a disease syndrome if two copies of the genes in the deleted region are necessary, and the individual will not be normal with just the one normal copy remaining on the non-deleted homologous chromosome. Many deletion syndromes have been well described. For example, Prader–Willi syndrome (p. 198) is the result of cytogenetic events resulting in deletion of part of the long arm of chromosome 15; Wilms’ tumour is characterized by deletion of part of the short arm of chromosome 11; and microdeletions in the long arm of chromosome 22 give rise to the DiGeorge’s syndrome.
Deletions of a portion of a chromosome may give rise to a disease syndrome if two copies of the genes in the deleted region are necessary, and the individual will not be normal with just the one normal copy remaining on the non-deleted homologous chromosome. Many deletion syndromes have been well described. For example, Prader–Willi syndrome (p. 198) is the result of cytogenetic events resulting in deletion of part of the long arm of chromosome 15; Wilms’ tumour is characterized by deletion of part of the short arm of chromosome 11; and microdeletions in the long arm of chromosome 22 give rise to the DiGeorge’s syndrome.
 Duplications occur when a portion of the chromosome is present on the chromosome in two copies, so the genes in that chromosome portion are present in an extra dose. A form of neuropathy, Charcot–Marie–Tooth disease (p. 1105), is due to a small duplication of a region of chromosome 17.
Duplications occur when a portion of the chromosome is present on the chromosome in two copies, so the genes in that chromosome portion are present in an extra dose. A form of neuropathy, Charcot–Marie–Tooth disease (p. 1105), is due to a small duplication of a region of chromosome 17.
 Inversions involve an end-to-end reversal of a segment within a chromosome, e.g. ‘abcdefgh’ becomes ‘abcfedgh’, e.g. haemophilia (p. 421).
Inversions involve an end-to-end reversal of a segment within a chromosome, e.g. ‘abcdefgh’ becomes ‘abcfedgh’, e.g. haemophilia (p. 421).
 Translocations occur when two chromosome regions join together, when they would not normally. Chromosome translocations in somatic cells may be associated with tumorigenesis (see p. 451 and Fig. 9.16).
Translocations occur when two chromosome regions join together, when they would not normally. Chromosome translocations in somatic cells may be associated with tumorigenesis (see p. 451 and Fig. 9.16).
 Reciprocal translocations occur when any two non-homologous chromosomes break simultaneously and rejoin, swapping ends. In this case, the cell still has 46 chromosomes but two of them are rearranged. Someone with a balanced translocation is likely to be normal (unless a translocation breakpoint interrupts a gene); but at meiosis, when the chromosomes separate into different daughter cells, the translocated chromosomes will enter the gametes and any resulting fetus may inherit one abnormal chromosome and have an unbalanced translocation, with physical manifestations.
Reciprocal translocations occur when any two non-homologous chromosomes break simultaneously and rejoin, swapping ends. In this case, the cell still has 46 chromosomes but two of them are rearranged. Someone with a balanced translocation is likely to be normal (unless a translocation breakpoint interrupts a gene); but at meiosis, when the chromosomes separate into different daughter cells, the translocated chromosomes will enter the gametes and any resulting fetus may inherit one abnormal chromosome and have an unbalanced translocation, with physical manifestations.

Table 2.2 shows some of the syndromes resulting from chromosomal abnormalities.
Mitochondrial chromosome disorders
The mitochondrial chromosome (see Fig. 2.18, p. 37) carries its genetic information in a very compact form; e.g. there are no introns in the genes. Therefore, any mutation has a high chance of having an effect. However, as every cell contains hundreds of mitochondria, a single altered mitochondrial genome will not be noticed. As mitochondria divide, there is a statistical likelihood that there will be more mutated mitochondria, and at some point, this will give rise to a mitochondrial disease.
Myopathies include chronic progressive external ophthalmoplegia (CPEO); encephalomyopathies include myoclonic epilepsy with ragged red fibres (MERRF) and mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) (see p. 1153).
Analysis of chromosome disorders
The cell cycle can be arrested at mitosis with colchicine and, following staining, the chromosomes with their characteristic banding can be seen and any abnormalities identified (Fig. 2.19). This is an automated process with computer scanning software searching for metaphase spreads and then automatic binning of each chromosome to allow easy scoring of chromosome number and banding patterns. Another approach utilizes genome-wide array based platforms (comparative genomic hybridization (CGH) or chromosomal microarray analysis (CMA)) to identify changes in chromosome copy number and can identify very small interstitial deletions and insertions (<1 Mb in size).
Large region specific probes are labelled with fluorescently tagged nucleotides and used to allow rapid identification of metaphase chromosomes. This approach allows easy identification of chromosomal translocations (Fig. 2.20). High-throughput sequencing is another method to identify deletions, insertions and translocation breakpoints.
Gene defects
Single-gene disease
Some monogenetic disorders show a racial or geographical prevalence, e.g. thalassaemia (see p. 390) is seen mainly in Greeks, South-east Asians and Italians; porphyria variegata in the South African white population; and Tay–Sachs disease (p. 1042) in Ashkenazi Jewish people. Thus, although the prevalence of some single-gene diseases is very low worldwide, it is much higher in specific populations.
Autosomal dominant disorders
Each diploid cell contains two copies of all the autosomes. An autosomal dominant disorder (Fig. 2.21a) occurs when one of the two copies has a mutation and the protein produced by the normal form of the gene cannot compensate. In this case, a heterozygous individual who has two different forms (or alleles) of the same gene will manifest the disease. The offspring of heterozygotes have a 50% chance of inheriting the chromosome carrying the disease allele, and therefore also of having the disease. However, estimation of risk to offspring for counselling families can be difficult because of three factors:



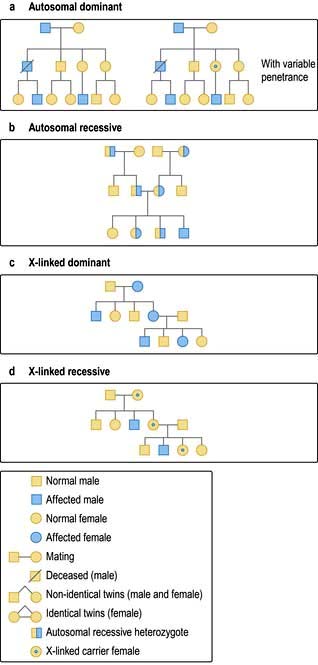
Autosomal recessive disorders
These disorders (Fig. 2.21b) manifest themselves only when an individual is homozygous or a compound heterozygote for the disease allele, i.e. both chromosomes carry the same gene mutation (homozygous) or different mutations in the same gene (compound heterozygote). The parents are unaffected carriers (heterozygous for the disease allele). If carriers marry, the offspring have a one in four chance of carrying both mutant copies of the gene and being affected, a one in two chance of being a carrier, and a one in four chance of being genetically normal. Consanguinity increases the risk.
Sex-linked disorders
Genes carried on the X chromosome are said to be ‘X-linked’, and can be dominant or recessive in the same way as autosomal genes (Fig. 2.21c,d).
X-linked recessive disorders
These disorders present in males and present only in homozygous females (usually rare). X-linked recessive diseases are transmitted by healthy female carriers or affected males if they survive to reproduce. An example of an X-linked recessive disorder is haemophilia A (see p. 421), which is caused by a mutation in the X-linked gene for factor VIII. It has been shown that in 50% of cases there is an intrachromosomal rearrangement (inversion) of the tip of the long arm of the X chromosome (one break point being within intron 22 of the factor VIII gene).


Other single-gene disorders
Triplet repeat mutations
In the gene responsible for myotonic dystrophy (p. 1153), the mutated allele was found to have an expanded 3′UTR region in which three nucleotides, CTG, were repeated up to about 200 times. In families with myotonic dystrophy, people with the late-onset form of the disease had 20–40 copies of the repeat, but their children and grandchildren who presented with the disease from birth had vast increases in the number of repeats, up to 2000 copies. It is thought that some mechanism during meiosis causes this ‘triplet repeat expansion’ so that the offspring inherit an increased number of triplets. The number of triplets affects mRNA and protein function (Table 2.3). See also page 43 for the phenomenon of ‘anticipation’.
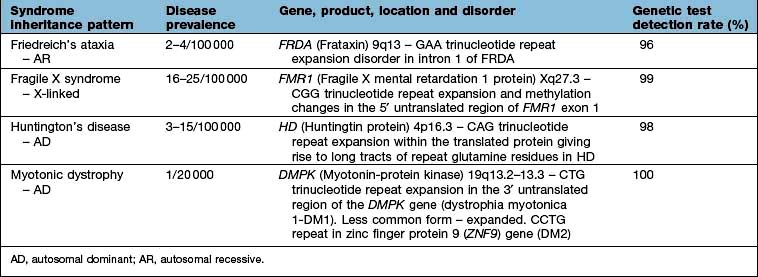
Mitochondrial disease
As discussed on pages 37 and 40, various mitochondrial gene mutations can give rise to complex disease syndromes with incomplete penetrance maternal inheritance (Fig. 2.18).
Complex traits: multifactorial and polygenic inheritance
Most human diseases, such as heart disease, diabetes and common mental disorders, are multifactorial traits (Table 2.4).
Table 2.4 Examples of disorders that may have a polygenic inheritance
| Disorder | Frequency (%) | Heritability (%)a |
|---|---|---|
|
Hypertension |
5 |
62 |
|
Asthma |
4 |
80 |
|
Schizophrenia |
1 |
85 |
|
Congenital heart disease |
0.5 |
35 |
|
Neural tube defects |
0.5 |
60 |
|
Congenital pyloric stenosis |
0.3 |
75 |
|
Ankylosing spondylitis |
0.2 |
70 |
|
Cleft palate |
0.1 |
76 |
a Percentage of the total variation of a trait which can be attributed to genetic factors.
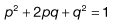
Clinical genetics and genetic counselling
Genetic counselling should have the following aims:
 Obtaining a full history. The pregnancy history, drug and alcohol ingestion during pregnancy and maternal illnesses (e.g. diabetes) should be detailed.
Obtaining a full history. The pregnancy history, drug and alcohol ingestion during pregnancy and maternal illnesses (e.g. diabetes) should be detailed.







Genetic anticipation
It has been noted that successive generations of people with, e.g. dystrophia myotonica and Huntington’s chorea, present earlier and with progressively worse symptoms. This ‘anticipation’ is due to unstable mutations occurring within the disease gene. Trinucleotide repeats such as CTG (dystrophia myotonica) and CAG (Huntington’s chorea) expand within the disease gene with each generation, and somatic expansion with cellular replication is also observed. This type of genetic mutation can occur within the translated region or untranslated (and presumably regulatory) regions of the target genes. This genetic distinction has been used to subclassify a number of genetic diseases which have now been shown to be caused by trinucleotide repeat expansion and display phenotypic ‘anticipation’ (Table 2.3).
Prenatal diagnosis for chromosomal disorders
Investigations
The choice of investigation depends on gestational age:
7–11 Weeks (vaginal ultrasound)
Ultrasound is used to confirm viability, fetal number and gestation by crown-rump measurement.
11–13 Weeks and 6 days (combined test)
 Ultrasound for nuchal translucency measurement (normal fold <6 mm) to attempt to detect major chromosomal abnormalities (e.g. trisomies and Turner’s syndrome)
Ultrasound for nuchal translucency measurement (normal fold <6 mm) to attempt to detect major chromosomal abnormalities (e.g. trisomies and Turner’s syndrome)

The combined test is more accurate than the triple test alone at 16 weeks (see below).
14–20 Weeks (serum triple or quadruple test)
The triple test for chromosomal abnormalities consists of testing maternal serum for:



The α-fetoprotein is high for neural tube defects.
The quadruple test also measures inhibin A – high in Down’s syndrome.
Genomic medicine
Gene therapy
Two major factors are involved in gene therapy:
 The introduction of the functional gene sequence into target cells
The introduction of the functional gene sequence into target cells
 The expression and permanent integration of the transfected gene into the host cell genome.
The expression and permanent integration of the transfected gene into the host cell genome.
Cystic fibrosis (see also p. 821)
CFTR, the cystic fibrosis transmembrane regulator, is an unusual ABC transporter in that it does not function as a primary active transporter but as a ligand-gated chloride channel (Fig. 2.22). The common CF mutation is a 3 bp deletion in exon 10 resulting in the removal of a codon specifying phenylalanine (F508del). In this mutation the CFTR protein is misfolded, thereby causing ineffective biosynthesis and consequently disrupting the delivery of the protein to the cell surface. In the mutation G551 D-CFTR, glycine in position 551 is replaced by aspartate; the CFTR channel reaches the cell surface but fails to open. This has introduced a new era of treatment. VX-770, a potentiating agent which can be given orally, has been developed. It increases the fraction of time that the phosphorylated G551 D-CFTR channel is open allowing bicarbonate and chloride flow across the membrane. Early clinical results are encouraging.
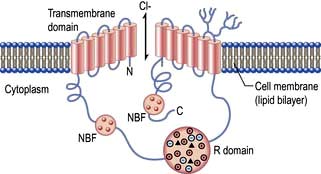
Two other routes of gene therapy have been tried, either with placing the wild-type CFTR cDNA into an adenovirus vector (see Fig. 15.28) to allow infection of human cells or into a plasmid (an engineered circle of DNA) that is then encapsulated into a liposome to allow transfection of human cells. The latter can be conveyed via an aerosol spray to the lung where the liposome fuses with the cell membrane to deliver the CFTR cDNA into the cell. However, neither is yet a treatment option. An alternative method is to suppress premature termination codons and thus permit translation to continue; topical nasal gentamicin (an aminoglycoside antibiotic) has been shown to result in the expression of functional CFTR channels.
Stem cell therapy
Stem cell therapy has the potential to radically change the treatment of human disease (see p. 33). A number of adult stem cell therapies already exist, particularly bone marrow transplants. It is currently anticipated that technologies derived from stem cell research can be used to treat a wider variety of diseases in which replacement of destroyed specialist tissues is required, such as in Parkinson’s disease, spinal cord injuries and muscle damage.
The genetic basis of cancer
Autosomal dominant inheritance
The following are examples of cancer syndromes (see Table 9.3, p. 433) that exhibit dominant inheritance:
 Retinoblastoma is an eye tumour found in young children. It occurs in both hereditary (40%) and non-hereditary (60%) forms. The 40% of people with the hereditary form have a germline mutation in the Retinoblastoma gene (RB1) and are also at risk for developing other tumours, particularly osteosarcoma.
Retinoblastoma is an eye tumour found in young children. It occurs in both hereditary (40%) and non-hereditary (60%) forms. The 40% of people with the hereditary form have a germline mutation in the Retinoblastoma gene (RB1) and are also at risk for developing other tumours, particularly osteosarcoma.


 Multiple-endocrine-adenomatosis syndromes (see p. 997). Multiple endocrine neoplasia type 1 is associated with the MEN1 gene and type 2 (MEN2) is associated with mutations in the RET proto-oncogene on chromosome 10 and as such are the exception to all the other syndromes which involve tumour suppressor genes.
Multiple-endocrine-adenomatosis syndromes (see p. 997). Multiple endocrine neoplasia type 1 is associated with the MEN1 gene and type 2 (MEN2) is associated with mutations in the RET proto-oncogene on chromosome 10 and as such are the exception to all the other syndromes which involve tumour suppressor genes.



Oncogenes
The genes coding for growth factors, growth factor receptors, secondary messengers or even DNA-binding proteins would act as promoters of abnormal cell growth if mutated. This concept was verified when viruses were found to carry genes which, when integrated into the host cell, promoted oncogenesis. These were originally termed viral or ‘v-oncogenes’, and later their normal cellular counterparts, c-oncogenes, were found. Thus, oncogenes encode proteins that are known to participate in the regulation of normal cellular proliferation e.g. erb-A on chromosome 17q11-q12 encodes for the thyroid hormone receptor. See Table 2.5.
Table 2.5 Examples of acquired/somatic mutations and proto-oncogenes
|
Point mutation |
|
|
K-RAS |
Colorectal and pancreatic cancer |
|
B-RAF |
Melanoma, thyroid |
|
ALK |
Lung cancer |
|
DNA amplification |
|
|
MYC |
Neuroblastoma |
|
HER2-neu |
Breast cancer |
|
Chromosome translocation |
|
|
BCR/ABL |
CML, ALL |
|
PML/RARA |
APL |
|
BCL2/IGH |
Follicular lymphoma |
|
IGH/CCND1 |
Mantle cell lymphoma |
|
MYC/IgH |
Burkitt’s lymphoma |
CML, chronic myeloid leukaemia; ALL, acute lymphoblastic leukaemia; APL, acute promyelocytic leukaemia.
Activation of oncogenes
Chromosomal translocation
An example of such a fusion gene (the Philadelphia chromosome) occurs in chronic myeloid leukaemia (CML, see p. 451). Similarly in Burkitt’s lymphoma, a translocation causes the regulatory segment of the myc oncogene to be replaced by a regulatory segment of an unrelated immunoglobulin.
FURTHER READING
Croce CM. Molecular origins of cancer: oncogenes and cancer. N Engl J Med 2008; 358:502–511.
Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148–1159.
Gearhart J, Pashos EE, Prasad MK. Pluripotency redux – advances in stem-cell research. N Engl J Med 2007; 357:1469–1472.
Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009; 361:1475–1485.
Tumour suppressor genes
These genes restrict undue cell proliferation (in contrast to oncogenes), and induce the repair or self-destruction (apoptosis) of cells containing damaged DNA. Therefore, mutations in these genes which disable their function, lead to uncontrolled cell growth in cells with active oncogenes. An example is the germline mutations in genes found in non-polyposis colorectal cancer, which are responsible for repairing DNA mismatches (p. 288).
The RB gene was the first tumour suppressor gene to be described (p. 433). In the familial variety, the first mutation is inherited and by chance, a second somatic mutation occurs with the formation of a tumour. In the sporadic variety, by chance both mutations occur in both the RB genes in a single cell.
Since the finding of RB, other tumour suppressor genes have been described, including the gene p53. Mutations in p53 have been found in almost all human tumours, including sporadic colorectal carcinomas, carcinomas of breast and lung, brain tumours, osteosarcomas and leukaemias. The protein encoded by p53 is a cellular 53 kDa nuclear phosphoprotein that plays a role in DNA repair and synthesis, in the control of the cell cycle and cell differentiation and programmed cell death – apoptosis. p53 is a DNA-binding protein which activates many gene expression pathways but it is normally only short-lived. In many tumours, mutations that disable p53 function also prevent its cellular catabolism. Although in some cancers there is a loss of p53 from both chromosomes, in most cancers (particularly colorectal carcinomas; see Fig. 9.1) such long-lived mutant p53 alleles can disrupt the normal alleles’ protein. As a DNA-binding protein, p53 is likely to act as a tetramer.
Thus, a mutation in a single copy of the gene can promote tumour formation because a hetero-tetramer of mutated and normal p53 subunits would still be dysfunctional. p53 and RB are involved in normal regulation of the cell cycle. Other cancer–associated genes are also intimately involved in control of the cell cycle (Fig. 2.13).


[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 2 Molecular cell biology and human genetics
Cell biology
Cells consist of cytoplasm enclosed within a lipid sheath (the plasma membrane). The cytoplasm contains a variety of organelles (sub-cellular compartments enclosed within their own membranes) in a mixture of salts and organic compounds (the cytosol). These are held within an adaptive internal scaffold (the cytoskeleton) that radiates from the nucleus outwards to the cell surface (Fig. 2.1). Many cells have special functions and their size, shape and behaviour adapt to meet their physiological roles. Cells can be organized into tissues and organs in which the individual component cells are in contact and able to send and receive messages, both directly and indirectly. Coordinated cellular responses can be achieved through systemic signalling, e.g. via hormones.
Cell structure
Cellular membranes
Lipid bilayers separate the cell contents from the external environment and compartmentalize distinct cellular activities into organelles. These consist of a large variety of glycerophospholipids and sphingolipids. Membrane lipids usually have two hydrophobic acyl chains linked via glycerol or serine, to polar hydrophilic head groups (Fig. 2.2). This amphiphilic nature, with a ‘water-loving’ head and a ‘water-hating’ tail, means that in aqueous solution membrane lipids self-associate into a tail-to-tail bilayer with their hydrophobic chains separated from the aqueous phase by their polar head groups.
Plasma membranes are more complicated than liposomes. Their lipids are organized asymmetrically in the bilayer. For example, the outer leaflet of the plasma membrane is enriched in phosphatidyl-choline (PC) and the sphingolipids, whereas the inner leaflet is enriched in phosphatidyl-serine (PS) and phosphatidyl-ethanolamine (PE). This arrangement is necessary in normal physiology and in disease, not just for barrier function. For example, PC is extracted from the outer-leaflet of the canalicular membrane of hepatocytes to form the lipid/bile-salt micelles of bile. One of the sphingolipids, GM1-ganglioside, is the receptor for cholera toxin. The appearance of PS in the outer leaflet of the membrane is an early step in the apoptotic pathway and signals to macrophages to clear the dying cell, while PE, once cleaved by phospholipase, produces two signalling molecules as second messengers (see p. 25). Cholesterol is also an essential component of the plasma membrane and cannot be substituted by plant sterols, which have a subtly different shape. For this reason, the liver secretes plant sterols back into the gut.
Membrane proteins
Cells can absorb gases or small hydrophobic compounds directly across the plasma membrane by passive diffusion, but membrane proteins are required to take-up hydrophilic nutrients or secrete hydrophilic products, to mediate cell–cell communication and to respond to endocrine signals. Membrane proteins can be integral to the membrane (i.e. their protein chain traverses the membrane one or multiple times) or they can be anchored to the membrane by an acyl chain (Fig. 2.2).
Membrane channel proteins (Fig. 2.3): membrane proteins that form solute channels through the membrane can only work downhill and only to equilibrium. Solute actually moves down its electrochemical gradient, which is the combined force of the electric potential and the solute concentration gradient across the membrane. The bulk flow can be very high, the opening and closing of the channel can be regulated, and they can be selective for specific solutes. For example, the cystic fibrosis transmembrane regulator (CFTR; Fig. 2.22), the protein whose malfunction causes cystic fibrosis, is a chloride channel found on the apical surface of epithelial cells. CFTR functions to regulate the fluidity of the extra-epithelial mucous layer. When the channel opens, millions of negatively-charged chloride ions flow out of the cell down their electrochemical gradient. This induces positively-charged sodium ions to flow between the cells of the epithelium (via a paracellular pathway) to balance the electrical charge. Water follows the efflux of sodium chloride by osmosis, thus maintaining the fluidity of the mucus.
Transporters (Fig. 2.3): in contrast to channels, transporters have a low capacity and work by binding solute on one side of the membrane which induces a conformational change that exposes the solute binding site on the other side of the membrane for release.
Receptors: there are three major receptor categories: receptors that mediate endocytosis, anchorage receptors (e.g. integrins, see p. 23) and signalling receptors (see cell signalling p. 24). There are two forms of receptor-mediated endocytosis:
Organelles
Cytoplasmic organelles
Endoplasmic reticulum (ER) is an array of interconnecting tubules or flattened sacs (cisternae) that is contiguous with the outer nuclear membrane (Fig. 2.1). There are three types of ER:
Golgi apparatus has flattened cisternae similar to those of the ER but arranged in a stack (Fig. 2.1). Vesicles that bud from the ER with cargo destined for secretion, for the plasma membrane or for other organelles, fuse with the Golgi stack. The proteins, lipids and sterols synthesized in the ER are exported to the Golgi apparatus to complete maturation (e.g. the final stages of membrane protein glycosylation occurs here). The mature products are then sorted into vesicles that bud from the Golgi for transport to their final destination (Fig. 2.4b,c). Mutation in the Golgin protein GMAP-210, with a probable role in tethering of the Golgi cisternae, causes achondrogenesis type 1A, where Golgi architecture is disrupted, particularly in bone cells.
Lysosomes mature from vesicles (endosomes) that bud from the Golgi. They contain digestive enzymes such as lipases, proteases, nucleases and amylases that work in an acidic environment. The membrane of the lysosome therefore includes a proton ATPase pump to acidify the lumen of the organelle. Lysosomes fuse with phagocytotic vesicles to digest their contents. This is crucial to the function of macrophages and polymorphs (neutrophils and eosinophils) in killing and digesting infective agents, in tissue remodelling during development, and osteoclast remodelling of bone. Not surprisingly, many metabolic disorders result from impaired lysosomal function (p. 1040).
Mitochondria are the engines of the cell, providing energy in the form of ATP. Mitochondria can be small, discrete and few in number in cells with low energy demand, or large and abundant in cells with a high energy demand like hepatocytes or muscle cells. The mitochondrion has its own genome encoding 13 proteins. The other proteins (~1000) required for mitochondrial function are encoded by the nuclear genome and imported into the mitochondrion. The mitochondrion has a double membrane surrounding a central matrix. The central matrix contains the enzymes for the Krebs cycle, which accepts the products of sugar and fatty acid catabolism and uses it to produce cofactors that donate their electrons into the electron transport chain of the inner membrane (see pp. 20, 31). The inner membrane is highly folded into cristae to increase its effective surface area. The protein complexes of the electron transport chain accept and donate electrons in redox reactions, releasing energy to efflux protons (H+) into the inter-membrane space. ATP synthase, another integral membrane protein, uses this H+ electrochemical gradient to drive formation of ATP. Mitochondria have many additional functions, including roles in apoptosis (see p. 32) and supply of substrates for biosynthesis. Mitochondria are also necessary for the synthesis of porphyrin, deficiency of which causes a range of diseases collectively called porphyrias (p. 1043).
The cytoskeleton
Microtubules (20–25 nm diameter) are polymers of α and β tubulin. These tubular structures resist bending and stretching, and are polar with plus and minus ends. They emanate from the microtubule organizing centre (MTOC), a complex of centrioles, γ-tubulin and other proteins, with their plus ends extending into the cell. At their plus ends repeated cycles of assembly and disassembly permit rapid changes in length. Microtubules form a ‘highway’, transporting organelles and vesicles through the cytoplasm. The two major microtubule-associated motor proteins (kinesin and dynein) allow movement of cargo to the plus and minus ends, respectively. During cell division the MTOC forms the mitotic spindle (see p. 28). Drugs that disrupt microtubule assembly (e.g. colchicine and vinca alkaloids) or stabilize microtubules (taxanes) preferentially kill dividing cells by preventing mitosis.
Intermediate filaments (~10 nm) form a network around the nucleus extending to the periphery of the cell. They make cell-to-cell contacts with adjacent cells via desmosomes, and with basement matrix via hemidesmosomes (Fig. 2.5; see also Fig. 24.27). Their function appears to be structural integrity; they are prominent in cellular tissues under stress and their disruption in genetic disease can cause structural defects or cell collapse. More than 40 different types of proteins polymerize to form intermediate filaments specific to particular cell types. For example keratin intermediate fibres are only found in epithelial cells whilst vimentin is in mesothelial (fibroblastic) cells. However, lamin intermediate filaments form the nuclear membrane skeleton in most cells.
Microfilaments (3–6 nm) are polymers of actin, one of the most abundant proteins in all cells. The actin microfilament network controls cell shape, prevents cellular deformation, is involved in cell–cell and cell–matrix adhesion, in cell movements such as crawling and cytokinesis (cell division), and in intracellular vesicle transport. Bundles of actin filaments form the structural core of cellular protrusions such as microvilli, lamellipodia and filopodia (see below). Actin microfilament bundles within the cell can associate with myosin II to form contractile stress fibres, similar to muscle sarcomeres. Stress fibres are often found as circumferential belts around the apical surfaces of epithelial cells where cells associate with adjacent cells via adherens junctions, permitting reaction to external stresses as a cellular sheet. Stress fibres also form where actin interacts via accessory proteins with the extracellular matrix at sites of focal adhesion (see Fig. 2.8c). This occurs during cell movements during inflammation, wound healing and metastasis. During cytokinesis actin-myosin II bundles form the contractile ring separating dividing cells. Like microtubules, microfilaments are polar, so can be used to transport secretory vesicles, endosomes and mitochondria, powered by motor proteins, including myosin I and V.
Cell shape and motility
The cytoskeleton determines cell shape and surface structures.
Microvilli. The apical surface of some epithelial cells is covered in tiny microvilli (~1 µm long) forming a brush border of thousands of small finger-like projections of the plasma membrane that increase the surface area for uptake or efflux (Fig. 2.6). At their core are 20–30 cross-linked actin microfilaments.
Motile cilia are also fine, finger-like protrusions but these are longer (~10–20 µm long) (Fig. 2.6). At their core is an axoneme, a bundle of nine cross-linked tubulin microtubule doublets surrounding a central pair. The action of the motor domain dynein serves to bend the cilium. Neighbouring cilia tend to beat in unison generating waves of motion that move fluid over the cell surface in the gut and airways (see Fig. 15.9), and also in the fallopian tubes.
Non-motile or primary cilia. Most cells also have a single primary cilium. These cilia have a variant axoneme with no central pair of microtubules and while they have dynein they are non-motile (the dynein is used to traffic cargo along the axoneme). Primary cilia are used for signalling during development and in the adult. Other related non-motile cilia are found in specialized cells, e.g. in the photoreceptors of the retina, the sensory neurones of the olfactory system, and in the sensory hair cells of the cochlea. A range of human ciliopathies (Fig. 2.7) have been described with pleiotropic symptoms depending on which cilia are affected. These include polycystic kidney disease, Bardet–Biedl syndrome (p. 1007), Joubert’s syndrome and Ellis–van Creveld syndrome.
Filopodia: if remodelled essentially in one dimension into a long actin filament, the leading edge of the plasma membrane is pushed forward as spikes, similar to long thin villi.
Movement. A similar mechanism involving the coordinated remodelling of the cytoskeleton and the formation and release of cell adhesions underlies all three modes of migration. Essentially, actin is polymerized at the leading edge extending the plasma membrane forward. New adhesions are formed with the substratum (cells and/or extracellular matrix) at the leading edge to provide purchase. Release of attachments and depolymerization of the actin filaments at the trailing edge then allows the cell to move forward. Myosin and myosin motor proteins may also be involved at the trailing edge providing the tractive force to pull the cell body forward. The complex coordination of these processes is controlled via signalling pathways involving members of the Rho protein family of GTPases (see p. 21). Key signalling targets are the WASp family of proteins which stimulate actin polymerization. The significance of cell motility in humans is illustrated by mutation of the WASp expressed in blood cell lineages, which causes Wiskott–Aldrich syndrome (p. 66), and is characterized by severe immunodeficiency and thrombocytopenia (platelet deficiency).
The cell and its environment
Epithelial tissues comprise layers of cells held tightly together by intercellular junctions and are usually separated from underlying tissue by specialized ECM called basal lamina. Epithelia cover surfaces (e.g. epidermis, tongue surface) and line passageways (airways, digestive tract, blood vessels), providing protection and regulating absorption and secretion.
Extracellular matrix
The gel or ground substance of the ECM is made from polysaccharides (glycosaminoglycans or GAGs), usually bound to proteins to form proteoglycans (p. 494). These are a diverse group of molecules conferring different matrix properties in different tissues. They form hydrated gels which can resist compression yet permit diffusion of metabolites and signalling molecules.
Hyaluronan, a very large hydrated GAG, is secreted into the joint space in synovial joints (p. 493), where it aids lubrication and helps reduce compressive forces.
Aggrecan, a very large proteoglycan, forms part of the articular cartilage of joints (p. 494) also contributing to compression resistance.
Fibrous proteins of ECM (p. 495) include collagens and tropoelastin, which polymerize into collagen and elastin fibres, and fibronectin which is insoluble in many tissues but soluble in plasma. Collagen provides tensile strength, elastin confers elasticity, while the widely distributed fibronectin adheres to both cells and ECM, and thus positions cells within the ECM. Collagens, the most abundant proteins in the body, are widely distributed and play a structural role in skin and bone, where collagen defects and disorders often manifest. Elastin fibres are abundant in arteries, lung and skin. Elastic fibres have a fibrillin sheath and fibrillin mutations underlie Marfan’s syndrome (p. 760). The ECM can be degraded and remodelled by proteins of the matrix metalloproteinase (MMP) family. These are needed for angiogenesis and morphogenesis and are also involved in the pathophysiology of cancer, cirrhosis and arthritis.
Basal lamina or basement membrane (lamina propria) is a specialized form of ECM, which separates cells from underlying tissue and provides a supportive, anchoring and protective role. Basal lamina can also act as molecular filters (e.g. glomerular filtration barrier, p. 636) and mediate signalling between adjacent tissues (e.g. epidermal-dermal signalling in skin). Type IV collagen, heparan sulphate proteoglycan, laminin and nidogen are key basal lamina proteins. Inherited abnormalities in these proteins cause skin blistering diseases (see Fig. 24.27). Breach of the basal lamina by invading cancer cells is a key stage in progression of epithelial carcinoma in situ to a malignant carcinoma.
Cell–cell adhesion
Cell–cell adhesion proteins (Fig. 2.8a)
Immunoglobulin-like cell adhesion molecules (iCAMs or CAMs) (Fig. 2.8a) are structurally related to antibodies. The neural cell adhesion molecule (N-CAM) is found predominantly in the nervous system. It mediates a homophilic (like-like) adhesion. When bound to an identical molecule on another cell, N-CAM can also associate laterally with a fibroblast growth factor receptor and stimulate its tyrosine kinase activity to induce neurite growth thus triggering cellular responses by indirect activation of the recipient.
Selectins. Unlike most adhesion molecules which bind to other proteins, the selectins interact with carbohydrate ligands or mucin complexes on leucocytes and endothelial cells (vascular and haematological systems). Leucocyte-selectin (CD62L) mediates the homing of lymphocytes to lymph nodes. Endothelial-selectin (CD62E) is expressed after activation by inflammatory cytokines; the small basal amount of E-selectin in many vascular beds appears to be necessary for the migration of leucocytes. Platelet-selectin (CD62P) is stored in the alpha granules of platelets and the Weibel–Palade bodies of endothelial cells, but it moves rapidly to the plasma membrane upon stimulation of these cells. All three selectins play a part in leucocyte rolling (p. 63).
Tight junctions (zonula occludens)
These are mediated by the integral membrane proteins, claudins and occludens; they hold cells together. They form at the top (apical) side of epithelial cells including intestinal, skin and kidney cells, and endothelial cells of blood vessels (Fig. 2.8) to provide a regulated barrier to the movement of ions and solutes through the epithelia or endothelia but also between cells (paracellular transport). Tight junctions also confer polarity to cells by acting as a gate between the apical and the baso-lateral membranes, preventing diffusion of membrane lipids and proteins. Twenty-four claudins (the protein in the junction) are differentially expressed in different cell types to regulate paracellular transport. For example, changes in claudin expression in the kidney nephron correlate with permeability changes. Mutations in claudins 16 (previously named parcellin-1) and 19, expressed in the thick ascending limb in the loop of Henle in the kidney, cause an inherited renal disorder, familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC; p. 657).
Gap junctions
Gap junctions (Fig. 2.8) allow low molecular weight substances to pass directly between cells, permitting metabolic and electric coupling (e.g. in cardiomyocytes). Protein channels made of six connexin proteins (as well as claudins and occludens) are aligned between adjacent cells and allow the passage of solutes up to 1000 kDa (e.g. amino acids, sugars, ions, chemical messengers). The channels are regulated by many factors such as intracellular Ca2+, pH, voltage. Gap junctions form in almost all interacting cells, but connexin family members are differentially expressed. Mutant connexins cause many inherited disorders, such as the X-linked form of Charcot–Marie–Tooth disease (GJB1; p. 1147) and are also a major cause of genetic hearing loss (GJB2).
Adherens junctions
Adherens junctions are multiprotein intercellular adhesive structures, prominent in epithelial tissues (Fig. 2.8b). They attach principally to actin microfilaments inside the cell with the aid of multiple additional proteins, and also attach and stabilize microtubules. At the apical sides of epithelial cells a prominent type of adherens junction, the zonula adherens, attaches to the circumferential actin stress fibres. The fascia adherens in cardiac muscle is also an adherens junction. Transmembrane proteins of the cadherin family provide the adhesion through interaction of their extracellular domains. Downregulation of cadherins is a feature of cancer progression in many cells.
Desmosomes (macula adherens)
Desmosomes provide strong attachment between cells and are prominent in tissues subject to stress such as skin and cardiac muscle (see Fig. 2.5, Fig. 2.8b and Fig. 24.1). Like adherens junctions, they are multiprotein complexes, where adhesion is provided by transmembrane cadherin proteins, desmogleins and desmocollins. However, within the cell desmosomes interact principally with intermediate filaments rather than microfilaments and microtubules. Germline mutations in genes encoding desmosomes are a cause of cardiomyopathy with/without cutaneous features and in pemphigus vulgaris and pemphigus foliaceus (p. 1222).
Basement membrane adhesion
Cells adhere (Fig. 2.8c) to non-basal lamina ECM via secreted proteins such as fibronectin and collagen, and to basal lamina proteins via focal adhesion and hemidesmosome multiprotein complexes (e.g. keratin or vimentin). Here, integrins replace cadherins as surface adhesion molecules as the key adhesive proteins. Integrins are transmembrane sensors or receptors, which change shape upon binding to ECM, a process called ‘outside-in’ signalling. Inside the cell, integrins interact with the cytoskeleton and a complex array of over 150 proteins that influence intracellular signalling pathways affecting proliferation, survival, shape, mobility and gene expression.
Outside-in signalling: forms the basis for anoikis or apoptotic death, such as occurs in cancer cells that inappropriately lose cell-substratum adhesion.
Inside-out signalling: intracellular changes can also be communicated extracellularly via integrins whereby intracellular changes cause integrins to change from an inactive to an actively adhesive conformation. This ‘inside-out’ signalling occurs when platelet integrins glycoprotein IIb-IIIa (GPIIb-IIa) are activated to bind fibrinogen at sites of vessel injury, resulting in platelet aggregation (p. 415 and Fig. 8.41).
Defective integrins are associated with many immunological and clotting disorders such as Bernard–Soulier syndrome and Glanzmann’s thrombasthenia (p. 420).
FURTHER READING
De Matteis MA, Luini A. Mendelian disorders of membrane trafficking. N Engl J Med 2011; 365:927–928.
Jean C, Gravelle P, Fournie JJ et al. Influence of stress on extracellular matrix and integrin biology. Oncogene 2011; 30:2697–2706.
Thomason HA, Scothern A, McHarg S et al. Desmosomes: adhesive strength and signalling in health and disease. Biochem J 2010; 429:419–433.
Cellular mechanisms
Cell signalling
Signalling or communication between cells is often via extracellular molecules or ligands which can be proteins (e.g. hormones, growth factors), small molecules (e.g. lipid-soluble steroid hormones such as oestrogen and testosterone) or dissolved gases such as nitric oxide. The signal is usually received by membrane protein receptors, although some signals such as steroid hormones, enter the target cell where they interact with intracellular receptors (Fig. 2.9). Some signalling, especially in the immune system, relies on cell–cell contact, where the signalling molecule (ligand) and receptor are on adjacent cells.
Receptors transduce signals across the membrane to an intracellular pathway or second messengers to change cell behaviour, often ultimately affecting gene expression (Figs 2.9, 2.10). The membrane-bound receptors fall into three main groups based on downstream signalling pathways:
Ion channel linked receptors (voltage or ligand activated ion channels; see Fig. 2.3). At synaptic junctions between neurones (Fig. 22.1), these receptors open in response to neurotransmitters such as glutamate, epinephrine (adrenaline) or acetylcholine to cause a rapid depolarization of the membrane.
G-protein-linked receptors such as the odorant and light (opsin) family of receptors belong to a large family of seven-pass transmembrane proteins (see Figs 2.2 and 2.9). On activation by ligand G-protein-linked receptors bind a GTP-binding protein (G-protein), which activates adjacent enzyme complexes or ion channels (Figs 2.9 and 22.1). The adjacent enzymes can be adenylcyclase (see below).
Enzyme-linked receptors (Figs 2.2 and 2.9) typically have an extracellular ligand-binding domain, a single transmembrane-spanning region, and a cytoplasmic domain that has intrinsic enzyme activity or which will bind and activate other membrane-bound or cytoplasmic enzyme complexes. This group of receptors is highly variable but many have kinase activity or associate with kinases, which act by phosphorylating substrate proteins usually on a tyrosine (e.g. the platelet-derived growth factor (PDGF) receptor) or a serine/threonine (e.g. the transforming growth factor-beta (TGF-β) receptor).
Signal transduction
Signal transduction from the receptor to the site of action in the cell is mediated by small signalling molecules called second messengers, or by signalling proteins (Fig. 2.9). Changes to activity of signalling proteins by acquired mutation occur in cancer, and many anti-cancer drugs target signalling pathways. For example, the Hedgehog pathway is involved in human development, tissue repair and cancer (Fig. 2.10). Inhibitors of this pathway are being developed for therapeutic interventions. The Wnt pathway is also involved in bone formation (p. 550).
Second messengers include cAMP and lipid-derived inositol triphosphate (IP3) and diacylglycerol (Fig. 2.9). These molecules diffuse from the receptor to bind and change the activity of downstream proteins propagating the signal. cAMP triggers a protein signalling cascade by activating a cAMP-dependent protein kinase. Diacylglycerol activates protein kinase C while IP3 mobilizes calcium from intracellular stores (e.g. from the ER; Fig. 14.9).
G-proteins or GTP-binding proteins are signalling proteins which switch between an active state when GTP is bound and an inactive state when bound to GDP. The most well-known members are the Ras superfamily, comprising Ras, Rho, Rab, Arf and Ran families. Activation of Ras members by somatic mutation is found in ~33% of human cancers. Ras members are often bound downstream of tyrosine kinase receptors, where they transmit signals by activating a cascade of downstream protein kinase activity (Fig. 2.9). Ras signalling molecules have roles in many cellular activities, including regulation of cell cycle, intracellular transport, and apoptosis.
Kinase and phosphatase signalling proteins are enzymes that phosphorylate or dephosphorylate residues on downstream proteins to alter their activity. Chains of kinase activity (phosphorylation cascades) consisting of sequential phosphorylation of proteins can transduce signals from the membrane receptor to the site of action in the cell. The tyrosine kinase receptors phosphorylate each other when ligand binding brings the intracellular receptor components into close proximity (see Fig. 2.9). The inner membrane and cytoplasmic targets of these activated receptor complexes are ras, protein kinase C and ultimately the MAP (mitogen-activated protein) kinase, Janus-Stat pathways or phosphorylation of IκB causing it to release its DNA-binding protein, nuclear factor kappa B (NFκB). For example, activated Ras binds and activates the kinase Raf, the first of a set of three mitogen-activated protein (MAP) kinases, which transmit signals by successive phosphorylation of target proteins which can ultimately effect transcription (Fig. 2.9). Kinases and phosphatases are frequently mutated in cancers. Somatic mutations in one Raf member, B-Raf, occur in ~60% of malignant melanomas (usually the mutation V600E) and are common in other cancers (p. 1225).
Nuclear control
DNA and RNA structure
Hereditary information is contained in the sequence of the building blocks of double-stranded deoxyribonucleic acid (DNA) (Fig. 2.11). Each strand of DNA is made up of a deoxyribose-phosphate backbone and a series of purine (adenine (A) and guanine (G)) and pyrimidine (thymine (T) and cytosine (C)) bases, and because of the way the sugar phosphate backbone is chemically coupled, each strand has a polarity with a phosphate at one end (the 5′ end) and a hydroxyl at the other (the 3′ end). The two strands of DNA are held together by hydrogen bonds between the bases. A can only pair with T, and G can only pair with C, therefore each strand is the antiparallel complement of the other (Fig. 2.11b). This is key to DNA replication because each strand can be used as a template to synthesize the other.
The two strands twist to form a double helix with a major and a minor groove, and the large stretches of helical DNA are coiled around histone proteins to form nucleosomes (Fig. 2.11c). They can be condensed further into the chromosomes that can be visualized by light microscopy at metaphase (see below; Fig. 2.11, Fig. 2.19).
To express the information in the genome, cells first transcribe the code into the single strand ribonucleic acid (RNA). RNA is similar to DNA in that it comprises four bases A, G and C but with uracil (U) instead of T, and a sugar phosphate backbone with ribose instead of deoxyribose. Several types of RNA are made by the cell. Messenger RNA (mRNA) codes for proteins that are translated on ribosomes. Ribosomal RNA (rRNA) is a key catalytic component of the ribosome and amino acids are delivered to the nascent peptide chain on transfer RNA (tRNA) molecules. There are also a variety of RNAs that regulate gene expression or RNA processing. These include microRNA (miRNA) and small interfering RNA (siRNA) (see p. 27) that typically bind to a subset of mRNAs and inhibit their translation, or initiate their degradation, respectively. Other non-coding RNAs are involved in X-inactivation and telomere maintenance or RNA splicing and maturation.
DNA transcription
RNA is transcribed from the DNA template by an enzyme complex of more than one hundred proteins including RNA polymerase, transcription factors and enhancers. Promoter regions upstream of the gene dictate the start point and direction of transcription. The complex binds to the promoter region, the nucleosomes are remodelled to allow access, and a DNA helicase unwinds the double helix. RNA, like DNA, is synthesized in the 5′ to 3′ direction as ribonucleotides are added to the growing 3′ end of a nascent transcript. RNA polymerase does this by base-pairing the ribonucleotides to the DNA template strand running in the 3′ to 5′ direction. Messenger RNA is modified as it is synthesized (Fig. 2.12). It is capped at the 5′ end with a modified guanine that is required for efficient processing of the mRNA and efficient translation, and introns are spliced from the nascent chain. Finally, the 3′ of the mRNA is modified with up to 200 A nucleotides by the enzyme poly-A polymerase. This 3′ poly-A tail is essential for nuclear export (through the nuclear pores), stability and efficient translation into protein by the ribosome.
Human protein coding sequences (exons) are interrupted by intervening sequences that are non-coding (introns) at multiple positions (Fig. 2.12). These have to be spliced from the nascent message in the nucleus by an RNA/protein complex called a spliceosome. Differential splicing describes the process by which two or more introns and their intervening exons are spliced from the mRNA. This contributes significantly to the complexity of the human transcriptome as proteins translated from these messages lack particular domains. This exon skipping can produce different protein activities.
Control of gene expression
The genome of all cells in the body encodes the same genetic information, yet different cell types express a very different subset of proteins and respond to external signals to switch on a new set of genes or to switch off a pathway. Gene expression can be controlled at many steps from transcription to protein degradation. However, for many genes transcription is the key point of regulation. This is controlled primarily by proteins which bind to short sequences within the promoter regions that either repress or activate transcription, or to more distant sequences where proteins bind to enhance expression. These transcription factors and enhancers are often the end points of signalling pathways that transduce extracellular signals to changes in gene expression (Fig. 2.9).
Often this involves the translocation of an activated factor from the cytoplasm to the nucleus. In the nucleus the DNA binding proteins recognize the shape and position of hydrogen bond acceptor and donor groups within the major and minor grooves of the double helix (i.e. the double helix does not need to be unwound). There are several classes of DNA binding protein that differ in the protein structural motif that allows them to interact with the double helix. These primarily include helix-turn-helix, zinc finger and leucine zipper motifs, although protein loops and β-sheets are used by some proteins. More permanent control of gene expression patterns can be achieved epigenetically. These are modifications (typically methylation and/or acetylation) of the DNA, or the histones of the nucleosome, that silence genes. Epigenetic modification is also heritable meaning that a dividing liver cell, for example, can give rise to two daughter cells with the same epigenetic signals such that they express the appropriate transcriptome for a liver cell. Epigenetic change forms the basis of genetic imprinting (see p. 42).
Most of the genome is transcribed but only a minority of transcripts encode proteins (see Human Genetics, p. 34). The non-coding RNAs (ncRNAs) include a group that regulate gene expression (see DNA and RNA structure). miRNAs and siRNAs are short ncRNAs (19–29 bp) that are known to regulate expression of approximately 30% of genes by degradation of transcripts or repression of protein synthesis. With further annotation of the genome a growing range of additional regulatory ncRNA classes are being identified, many of which control gene expression by epigenetic mechanisms.
