CHAPTER 56 MIGRAINE
Migraine is a primary episodic headache disorder characterized by various combinations of neurological, gastrointestinal, and autonomic changes. The word migraine is derived from the Greek word “hemicrania” (Galen, circa 200 AD).1 Diagnosis is based on the headache’s characteristics and associated symptoms.2 The International Headache Society diagnostic criteria for headache disorders (1988)3 have been revised (2004) and describe a total of seven subtypes of migraine.4 Data in this chapter5 are from the Technical Reports of the Agency for Healthcare Policy and Research,6–9 the U.S. Headache Consortium Guidelines,10,11 and the triptan metaanalysis.12
EPIDEMIOLOGY
Migraine prevalence rates are similar and stable in European countries and in the United States.13 In the United States, according to one study, 17.6% of women and 6% of men had had one migraine attack in the previous year.14 A second study 10 years later had similar prevalence estimates (Fig. 56-1).15 Migraine prevalence varies by age, gender, race, and income. Before puberty, migraine prevalence is approximately 4%.16 After puberty, the prevalence increases more rapidly in girls than in boys. It increases until approximately age 40, then declines. The prevalence is lowest among Asian-Americans, intermediate among African-Americans, and highest among white persons.16 In the United States, migraine prevalence decreases as household income increases.14,16,17
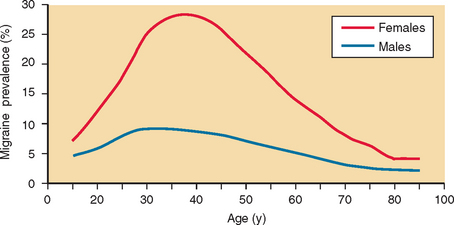
Figure 56-1 Adjusted age-specific prevalence of migraine by sex.
(From Lipton RB, Stewart WF, Diamond S, et al: Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache 2001; 41:646-657.)
Migraine substantially affects quality of life. The World Health Organization ranks migraine among the world’s most disabling medical illnesses.18 Approximately 28 million Americans have severe, disabling migraine headaches.15 Migraine’s annual cost to employers is approximately $13 billion, and related annual medical costs exceed $1 billion.16 Instruments to quantify migraine disability include the Migraine Disability Assessment Scale19 and the Headache Impact Test.20
PATHOPHYSIOLOGY
Genetics
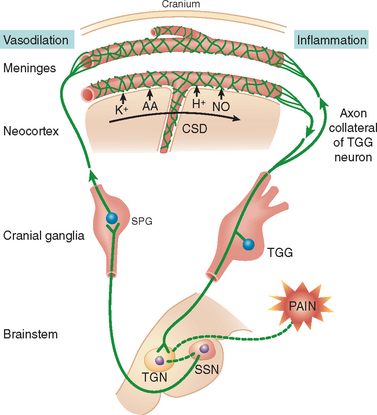
Migraine is a group of familial disorders with a genetic component. Familial hemiplegic migraine (FHM) is an autosomal dominant disorder characterized by attacks of migraine, with and without aura, and hemiparesis. The gene has been mapped to chromosome 19p13 in approximately two thirds of cases.21,22 The defect arises from at least 10 different missense mutations in the CACNA1A gene, which codes for the α1 subunit of a voltage-dependent P/Q Ca2+ channel.23 The same gene is associated with episodic ataxia with cerebellar vermix atrophy.21 P-type neuronal Ca2+ channels mediate 5-hydroxytriptamine (5-HT) and excitatory neurotransmitter release. Dysfunction may impair 5-HT release and predispose patients to migraine attacks or impair their self-aborting mechanism. Voltage-gated P/Q-type calcium channels mediate glutamate release, are involved in cortical spreading depression (CSD), and may be integral in initiating the migraine aura (Fig. 56-2).24 A second gene has been mapped to chromosome 1q21-23. The defect is due to a mutation in the α2 subunit of the Na/K pump.25
Aura
The migraine aura was believed to be caused by cerebral vasoconstriction and the headache by reactive vasodilation.26 This explained the headache’s throbbing quality and its relief by ergots. It is now believed that the migraine aura is caused by neuronal dysfunction, not ischemia; ischemia rarely, if ever, occurs. Headache often begins while cortical blood flow (CBF) is reduced27–29; thus, headache is not caused by simple reflex vasodilation.30,31
The migrainous fortification spectrum corresponds to an event moving across the cortex at 2 to 3 mm/minute.32 Noxious stimulation of the rodent cerebral cortex produced a spreading decrease in electrical activity that moved at 2 to 3 mm/minute (CSD).33 CSD is characterized by shifts in cortical steady-state potential; transient increases in potassium, nitric oxide, and glutamate levels; and transient increases in CBF, followed by sustained decreases.27
The aura is associated with an initial hyperemic phase followed by reduced CBF, which moves across the cortex (spreading oligemia).34 Olesen and colleagues34,35 found 17% to 35% reductions in posterior CBF, which spread anteriorly at 2 to 3 mm/minute. It crossed brain areas supplied by separate vessels and is thus not caused by segmental vasoconstriction.35 Reduced CBF persisted from 30 minutes to 6 hours, and then CBF slowly returned to baseline or even increased. The rates of progression of spreading oligemia are similar to those of migrainous scotoma and CSD, which suggests that they are related.31,33,36
Additional studies28,29,37–40 support the hypothesis that CSD produces the aura.27 During visual auras, CBF decreased 15% to 53%, cerebral blood volume decreased 6% to 33%, and mean transit time increased 10% to 54% in the occipital cortex contralateral to where the aura was experienced. The perfusion defect moved anteriorly.29 The absence of diffusion abnormalities suggests that ischemia does not occur during the aura.41
Blood oxygenation level–dependent (BOLD) functional magnetic resonance imaging (MRI) reflects the relative concentration of deoxyhemoglobin in venous blood. Visual stimulation was used to trigger headache in migraineurs.37 A wave of increased BOLD signal (reflecting hyperoxygenated blood) and then decreased BOLD signal (possibly reflecting neuronal metabolic flow coupling) propagated into the contiguous occipital cortex at 3 to 6 mm/minute. When visual stimulation was used to test the visual cortex response, the BOLD signal and the BOLD response to visual activation diminished after progression of the visual aura.30
Magnetoencephalography demonstrates changes that are consistent with CSD in migraineurs but not in controls.42,43 Using transcranial magnetic stimulation (which applies magnetic fields of increasing intensity) to evaluate occipital cortex excitability, Aurora and associates44 and Young and coworkers,45 but not Afra and colleagues,46 found that phosphenes were generated in migraineurs at lower thresholds than in controls and that it was easier to visually trigger headaches in subjects with lower thresholds. Other evidence of increased central nervous system (CNS) excitability comes from studies of visual and brainstem auditory evoked potentials.47 Migraine with aura may be caused by neuronal hyperexcitability, which perhaps arises from cortical disinhibition.
Headache
Headache probably results from the activation of meningeal and blood vessel nociceptors in combination with a change in central pain modulation. Headache and its associated neurovascular changes are subserved by the trigeminal system. Reflex connections to the cranial parasympathetic nerves form the trigeminoautonomic reflex. Activation results in vasoactive intestinal polypeptide release and vasodilation.31
Trigeminal sensory neurons contain substance P, calcitonin gene–related peptide (CGRP), and neurokinin A.48 Stimulation results in release of substance P and CGRP from sensory C-fiber terminals49 and neurogenic inflammation.50 The neuropeptides interact with the blood vessel wall, producing dilation, plasma protein extravasation, and platelet activation.51 One study suggests that neurogenic inflammation occurs in humans.52 Neurogenic inflammation sensitizes nerve fibers (peripheral sensitization) that then respond to previously innocuous stimuli, such as blood vessel pulsations,53 causing, in part, the pain of migraine.54 Central sensitization can also occur. After meningeal irritation, expression of c-fos (a marker for neuronal activation) occurs in the trigeminal nucleus caudalis55 and in the dorsal horn at the C1 and C2 levels.56,57
Superior sagittal sinus stimulation results in release of CGRP but not substance P.58 This is important: Levels of CGRP, not substance P, is elevated in external jugular venous blood during migraine.59 Sumatriptan reduced elevated CGRP levels in a migraine attack and in experimental animals during trigeminal ganglion stimulation.60,61 CGRP may play a role in migraine headache.62,63 A potent specific CGRP antagonist64 has been reported to be effective in acute migraine treatment.65
Applying an “inflammatory soup” to the dura sensitizes second-order trigeminovascular neurons (increased spontaneous activity and response to mechanical and thermal skin stimulation).53 Triptans administered early prevented central sensitization: Dural and facial receptive fields did not expand; spontaneous activity, mechanical sensitivity, and thermal sensitivity did not increase. Late triptan intervention did not reverse central sensitization but shrank the expanded dural receptive fields and normalized intracranial mechanosensitivity. Central sensitization may play a key role in maintaining the headache.66,67
Affected patients often develop cutaneous allodynia (nonpainful stimuli are experienced as painful) during migraine attacks because of trigeminal sensitization.66 Triptans can prevent but not reverse cutaneous allodynia.67 Cutaneous allodynia can be used to predict triptans’ effectiveness.66 In the absence of allodynia, triptans completely relieved the headache and blocked the development of allodynia. In 90% of attacks with established allodynia, triptans provided little or no headache relief and did not suppress allodynia. However, late triptan therapy eliminated peripheral sensitization (throbbing pain aggravated by movement), even when pain relief was incomplete and allodynia was not suppressed.66 Early intervention may work by preventing cutaneous allodynia and central sensitization.
Brainstem activation occurs in migraine without aura. On positron emission tomography, patients with right-sided migraine headache showed increased regional CBF in the left brainstem.68 Sumatriptan relieved the headache and associated symptoms but did not normalize brainstem regional CBF. This suggests that activation is caused by factors other than, or in addition to, increased activity of the endogenous antinociceptive system. A second report corroborated these findings.69
A link exists between the migraine aura and headache. CSD activates trigeminovascular afferents, causing a long-lasting increase in middle meningeal artery blood flow and plasma protein extravasation within the dura mater.70 CSD results in upregulation of inducible nitric oxide synthetase and inflammatory cytokines. This mechanism couples meningeal blood flow and neurogenic inflammation to CSD but does not explain headache ipsilateral to the aura.31,70
Serotonin (5-HT) Receptors and Migraine Treatment
There are seven classes of 5-HT receptors: 5-HT1, 5-HT2, 5-HT3, 5-HT4, 5-HT5, 5-HT6, and 5-HT7.71 In humans, there are five 5-HT1 receptor subtypes: 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, and 5-HT1F.72 The 5-HT1B receptor is located on intracranial blood vessels and CNS neurons. The 5-HT1D receptor is located on CNS neurons and trigeminal nerve endings. The 5-HT1F receptors are located on trigeminal nerve endings.73 Ergots and triptans act at the 5-HT1B, 5-HT1D, and, in part, 5-HT1F receptors. They constrict extracerebral intracranial vessels, inhibit trigeminal neurons, and block transmission in the trigeminal nucleus. They minimally constrict human coronary arteries. They block plasma protein extravasation50 by activating prejunctional trigeminal 5-HT1D and 5-HT1F heteroreceptors, blocking neuropeptide release. Plasma protein extravasation can be also be blocked by nonsteroidal anti-inflammatory drugs (NSAIDs),74 γ-aminobutyric acid (GABA) agonists,75,76 neurosteroids,77 substance P antagonists,78 and the endothelin antagonist bosentan.79 Dihydroergotamine and the centrally penetrant triptans label the nuclei in the brainstem and spinal cord that are involved in pain transmission and modulation.80 The caudal trigeminal nucleus is activated by stimulation of the sagittal sinus, and this activity is transmitted to the thalamus. Ergots and triptans suppress this activation.
Conclusion
The migraine aura is probably caused by CSD. Headache probably results from activation of meningeal and blood vessel nociceptors in combination with a change in central pain modulation. Headache and its associated neurovascular changes are subserved by the trigeminal system. Stimulation results in the release of substance P and CGRP from sensory C-fiber terminals and neurogenic inflammation.51 Neurogenic inflammation sensitizes nerve fibers (peripheral sensitization), which then respond to previously innocuous stimuli, such as blood vessel pulsations, causing, in part, the pain of migraine. Central sensitization of trigeminal nucleus caudalis neurons can also occur. Central sensitization may play a key role in maintaining the headache. Brainstem activation also occurs in migraine without aura, partly as a result of increased activity of the endogenous antinociceptive system. The migraine aura can trigger headache; CSD activates trigeminovascular afferent vessels. Stress can also activate meningeal plasma cells via a parasympathetic mechanism, leading to nociceptor activation.81
DESCRIPTION OF THE MIGRAINE ATTACK
The migraine attack can consist of premonitory, aura, headache, and resolution phases. Premonitory symptoms occur in 20% to 60% of migraineurs, hours to days before headache onset. They may include psychological, neurological, constitutional, or autonomic features, such as depression, cognitive dysfunction, and bouts of food cravings.82 Migraineurs who reported having premonitory symptoms were able to accurately predict 72% of their full-blown headaches. The most common premonitory symptoms were feeling tired/weary (72%), difficulty concentrating (51%), and stiff neck (50%). Poor functioning was commonly predictive of headache.83
Aura
The migraine aura consists of focal neurological symptoms that precede, accompany, or (rarely) follow an attack. Aura usually develops over 5 to 20 minutes; lasts less than 60 minutes; can be visual, sensory, or motor; and may involve language or brainstem disturbances.3 Headache usually follows within 60 minutes of the end of the aura. Patients can have multiple aura types; most patients with a sensory aura also have a visual aura.84
Auras vary in complexity. Simple auras include scotomata, simple flashes (phosphenes), specks, geometrical forms, and shimmering in the visual field. More complicated visual auras include teichopsia or fortification spectra (the characteristic aura of migraine), metamorphopsia, micropsia, macropsia, zoom vision, and mosaic vision. Paresthesias are often cheiro-aural: Numbness starts in the hand, migrates up the arm, and jumps to involve the face, lips, and tongue.2,85 Weakness is rare, occurs in association with sensory symptoms, and is unilateral.86 Apraxia, aphasia, agnosia, states of altered consciousness associated with déjà vu or jamais vu, and elaborate dreamy, nightmarish, trancelike, or delirious states can occur.82
Headache Phase
The median migraine attack frequency is 1.5 per month.14 The typical headache is unilateral, of gradual onset, throbbing (85%),87 moderate to marked in severity, and aggravated by movement.3 Pain may be bilateral (40%) or may start on one side and become generalized. It lasts 4 to 72 hours in adults and 2 to 48 hours in children.3
During the headache, anorexia is common. Nausea occurs in almost 90% of patients, and vomiting occurs in about one third.88 Sensory hypersensitivity results in patients’ seeking a dark, quiet room.2,88 Blurry vision, nasal stuffiness, hunger, tenesmus, diarrhea, abdominal cramps, polyuria, facial pallor, sensations of heat or cold, and sweating may occur. Depression, fatigue, anxiety, nervousness, irritability, and impairment of concentration are common. Symptom complexes may be generated by linked neuronal modules.89
FORMAL DIAGNOSTIC CRITERIA
The International Headache Society subdivides the disorder into migraine with aura and migraine without aura.4,90 To diagnose migraine without aura (Table 56-1), five attacks must have occurred. No single feature is mandatory, but recurrent episodic attacks must be documented.3 Migraine persisting for more than 3 days defines “status migrainosus.”3,4
Migraine with aura is subdivided into migraine with typical aura, migraine with prolonged aura, hemiplegic migraine, basilar-type migraine, and migraine with acute-onset aura (Table 56-2.) The International Headache Society classification now allows the association of aura with other headache types. Prolonged aura lasts from 1 hour to 1 week; persistent aura lasts for more than 1 week (but resolves). If neuroimaging demonstrates a stroke, a migrainous infarction has occurred.
TABLE 56-2 Migraine with Aura (Classic Migraine): Diagnostic Criteria
Periodic neurological dysfunction (scintillating scotomata and recurrent sensory, motor, and mental phenomena) can occur without headache.91 Visual phenomena, which are usually benign, occurred in 1.33% of women and in 1.08% of men in a general population sample.92 Scintillating scotomata, numbness, aphasia, dysarthria, and motor weakness may occur for the first time after age 45 and be confused with transient ischemic attacks of cerebrovascular origin.93 In general, migrainous symptoms are slower to develop (>5 minutes) and consist of positive and negative phenomena.
MIGRAINE VARIANTS
Basilar-type migraine aura has brainstem symptoms: ataxia, vertigo, tinnitus, diplopia, nausea and vomiting, nystagmus, dysarthria, bilateral paresthesia, and a change in the level of consciousness and cognition.3 The diagnosis should be considered when patients have paroxysmal brainstem disturbances. Some authorities have suggested that hemiplegic migraine should be diagnosed if weakness is present.86
Ophthalmoplegic migraine results from an idiopathic inflammatory neuritis.94 There is enhancement of the cisternal segment of the oculomotor nerve, followed by resolution over several weeks as symptoms resolve.
Hemiplegic migraine can be sporadic or familial.2 Attacks are frequently precipitated by minor head injury.86 FHM is an autosomal dominant, genetically heterogeneous disorder with variable penetration. FHM includes attacks of migraine without aura, migraine with typical aura, and episodes of prolonged aura, fever, meningismus, and impaired consciousness.23 Headache may precede the hemiparesis or be absent. Hemiparesis onset may be abrupt and simulate the effects of a stroke. In 20% of unselected families whose members have FHM, patients have cerebellar symptoms and signs (nystagmus, progressive ataxia). All have mutations within CACNA1A.22
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an inherited arterial disease of the brain caused by a Notch3 gene mutation on chromosome 19.95,96 CADASIL mutations are highly stereotyped missense mutations within epidermal growth factor–like repeats in the extracellular domain of Notch3. Mutations lead to loss or gain of a cysteine, thereby creating an odd number of cysteines within a given epidermal growth factor domain.97 Notch3 expression is restricted to smooth muscle cells, and normal proteolysis of the Notch3 receptor generates a 210-kD extracellular fragment and a 97-kD intracellular fragment. Patients with CADASIL accumulate excess Notch3 ectodomain (i.e., the 210-kD fragment) within the cerebral vasculature, apparently because of impaired clearance of the receptor from the surface of vascular smooth muscle cells and pericytes. Most cases are inherited in an autosomal dominant manner, but a de novo symptomatic mutation, Arg182Cys, has been reported.98 Symptoms include recurrent subcortical lacunar infarctions (84%), progressive or stepwise subcortical dementia with pseudobulbar palsy (31%), migraine with aura (22%), and mood disorders with severe depressive episodes (20%).99 MRI scans of at-risk individuals are often abnormal, with extensive areas of increased white matter T2 signals. The arteriopathy involves the media of small cerebral arteries and, to a lesser extent, extracerebral arteries, including skin arterioles. In skin biopsy, abnormal patches of agranular osmiophilic material within the basal membranes of vascular smooth muscle cells are diagnostic.97 A commercial genetic test is available for the most common CADASIL mutations. It currently has a false-negative rate of at least 20% because it screens only mutational hot spots located in exons 3, 4, 11, and 18. Scanning all 23 exons that encode all 34 epidermal growth factor–like repeat sequences is considered the most accurate test for CADASIL, but it is time-consuming and costly.
TREATMENT
Migraine varies widely in its frequency, severity, and impact on the patient’s quality of life. A treatment plan should account not only for the patient’s diagnosis, symptoms, and any coexistent or comorbid conditions but also for the patient’s expectations, needs, and goals.100,101 Migraine treatment begins with making a diagnosis,2 explaining it to the patient, and developing a treatment plan that considers coincidental or comorbid conditions.101 Headache calendars record headache duration, severity, and treatment response. Comorbidity indicates an association between two disorders that is more than coincidental.
Pharmacotherapy for the Acute Migraine Headache
Acute treatment can be specific (ergots and triptans), or nonspecific (analgesics and opioids) (Table 56-3). Nonspecific medications control the pain of migraine or other pain disorders, whereas specific medications are effective in migraine (and certain other) headache attacks but are not useful for nonheadache pain disorders. Triptans are effective in the range of mild, moderate, and severe migraine attacks.102
TABLE 56-3 Acute Medications: Efficacy, Adverse Events, Relative Contraindications, and Indications
| Comorbid Condition | ||
|---|---|---|
| Drug | Relative Contraindication | Relative Indication |
| Acetaminophen (Paracetamol) | Liver disease | Pregnancy |
| Aspirin | Kidney disease | CAD |
| Ulcer disease | Transient ischemic attack | |
| PUD | ||
| Gastritis (age <15) | ||
| NSAIDs | Kidney disease | Arthritis |
| PUD | ||
| Gastritis | ||
| Butalbital, caffeine, and analgesics | Use of other sedative | — |
| History of medication overuse | ||
| Caffeine adjuvant | Sensitivity to caffeine | — |
| Isometheptene | Uncontrolled HTN, CAD, PVD | — |
| Opioids | Drug or substance abuse | Pregnancy |
| Rescue medication | ||
| Neuroleptics | Parkinson’s disease | Nausea |
| Prolonged QTc | Vomiting | |
| Pregnancy | ||
| Rescue medication | ||
| Dihydroergotamine | ||
| Injections | CAD | Orthostatic hypotension |
| Intranasal instillation | PVD | |
| Uncontrolled HTN | Prominent nausea or vomiting | |
| Ergotamine | ||
| Tablets | Prominent nausea or vomiting | — |
| Suppositories | CAD | |
| PVD | ||
| Uncontrolled HTN | ||
| Triptans | ||
| Almotriptan (tablets) | CAD | — |
| PVD | ||
| Uncontrolled HTN | ||
| Eletriptan (tablets) | CAD | — |
| PVD | ||
| Uncontrolled HTN | ||
| Frovatriptan (tablets) | CAD | — |
| PVD | ||
| Uncontrolled HTN | ||
| Naratriptan (tablets) | CAD | — |
| PVD | ||
| Uncontrolled HTN | ||
| Rizatriptan (tablets) | CAD | — |
| PVD | ||
| Uncontrolled HTN | ||
| Zolmitriptan (tablets, intranasal) | CAD | Prominent nausea or vomiting |
| PVD | ||
| Uncontrolled HTN | ||
| Sumatriptan (subcutaneous injection, intranasal instillation, tablets) | CAD | — |
| PVD | ||
| Uncontrolled HTN | ||
CAD, coronary artery disease; HTN, hypertension; NSAID, nonsteroidal anti-inflammatory drug; PUD, peptic ulcer disease; PVD, peripheral vascular disease; QTc, corrected QT interval.
Treatment choice depends on attack severity, frequency, associated symptoms, coexistent disorders, prior treatment response, and the medication’s efficacy, its potential for overuse, and adverse events. A nonoral route of administration and an antiemetic should be considered for severe nausea or vomiting.11 Injections provide rapid relief. Headaches can be stratified by severity and disability (on the Migraine Disability Assessment Scale or Headache Impact Test). Analgesics are used for mild to moderate headaches.11 Triptans and dihydroergotamine are first-line drugs for severe attacks and for less severe attacks that do not adequately respond to analgesics.11 Patients with moderate or severe headaches with moderate or severe disability (according to the Migraine Disability Assessment Scale score) who were given a triptan did better than patients given aspirin and metoclopramide.103
Early intervention prevents escalation and may increase efficacy.104 Triptans can prevent the development of cutaneous allodynia, and cutaneous allodynia is predictive of triptans’ effectiveness.66 Before the clinician decides that a drug is ineffective, at least two attacks should be treated. It may be necessary to change the dose, formulation, or route of administration or add an adjuvant. When the response is inadequate, the headache recurs, or adverse events are bothersome, a medication change may be needed. Limiting acute treatment to 2 to 3 days a week can prevent medication-overuse headache. When headaches are very frequent, early intervention may not be appropriate.
Nonspecific Medication
Analgesics and Nonsteroidal Anti-inflammatory Drugs
Aspirin; ibuprofen; tolfenamic acid; naproxen sodium; acetaminophen; and acetaminophen, aspirin, and caffeine (Excedrin) combination are effective in acute migraine treatment.11,105,106
Barbiturate Hypnotics
No randomized, placebo-controlled studies have established the efficacy of butalbital-containing agents.107 They are used in the United States but have been withdrawn from the market in many European countries. Because of the potential for medication-overuse headache and withdrawal, their use should be limited and carefully monitored.
Opioids
Opioids are effective.2,11 However, because of the risk of medication overuse, they should be limited (<2 days a week) to patients who have severe, relatively infrequent headaches.108 They are used in the United States for patients who do not respond to simple analgesics (or cannot take ergots or a triptan) and as a rescue medication. They are often prescribed for pregnant women in the absence of controlled data.109
Neuroleptics and Antiemetics
Prochlorperazine is relatively safe and effective for the treatment of migraine and associated nausea and vomiting.7,9,11,110,111 Droperidol, a parenteral neuroleptic, was effective in one placebo-controlled, double-blind trial, at a dose of 2.75 mg intramuscularly. Sedation, akathisia, and other extrapyramidal reactions can be treated with diphenhydramine or benzotropine.112
Specific Medication
Ergots and triptans are potent 5-HT1B, 5-HT1D, and, in some cases, 5-HT1F receptor agonists. Ergots have much greater receptor affinity at 5-HT1A, 5-HT2, adrenergic, and dopaminergic receptors than do triptans, and thus lead to more adverse events. All are indicated for acute migraine treatment. If the initial (appropriate) dose is not effective, it is unlikely that subsequent doses will be effective during the same attack, and rescue medications should be used. Contraindications include documented or suspected ischemic heart disease, Prinzmetal’s angina, uncontrolled hypertension, basilar or hemiplegic migraine, and pregnancy. Patients with sepsis, renal or hepatic failure, and cerebral or peripheral vascular disease should avoid ergots. There is little consensus as to how many risk factors preclude triptan use and what constitutes an appropriate evaluation.113
Selective 5-HT1 Agonists (Triptans)
Sumatriptan was the first triptan to be introduced. It was followed by zolmitriptan, naratriptan, rizatriptan, almotriptan, frovatriptan, and eletriptan, all of which are more centrally penetrant than is sumatriptan. Eletriptan’s central penetrance is limited, inasmuch as it is a substrate for the P-glycoprotein pump114: P-glycoprotein pump inhibitors allow higher central penetrance. Data for all formulations of triptans are given in Table 56-4.11,12 All are effective, even if given after the onset of migraine, and may be more effective when pain is mild.115 They relieve head pain and nausea and vomiting. Efficacy is measured by 2-hour response rates and therapeutic gain (the difference between active drug and placebo; see Table 56-4). The drug is used to compensate for differences in placebo rates in different trials. Other measures include 2-hour pain-free rates and recurrence rates. Common adverse events include pain, tingling, flushing, and burning sensation at the subcutaneous injection site; warm or hot sensations; dizziness; paresthesias; somnolence; fatigue; sensation of heaviness; neck pain; and dysphoria.116,117 In the text, mainly nonoral formulations are discussed.

Sumatriptan is available in 6-mg subcutaneous injection; 20-mg nasal spray; 25, 50, or 100-mg tablets; and 25-mg suppository.12,118 Subcutaneous sumatriptan has a rapid onset of action, reaching peak plasma concentrations within 12 minutes. The time of maximal concentration (Tmax) for oral sumatriptan is 2 hours. Bioavailability is 97% for subcutaneous injection, 19.2% for suppository, 15.8% for intranasal instillation, and 14.3% for tablet.116 The terminal elimination half-life is 2 hours. Sumatriptan is metabolized principally by the monoamine oxidase (MAO)-A; therefore, the oral, nasal, and suppository formulations are contraindicated in patients using MAO inhibitors.
Subcutaneous sumatriptan (6 mg) produces a 1-hour response rate of 69%, with a therapeutic gain in 50%.119 The 1-hour pain-free rate was 48% to 49%.120 Intranasal sumatriptan (20 mg) produced a 2-hour response rate of 61%, with a therapeutic gain in 31%.119,121 The 2-hour efficacy for the 25-mg sumatriptan suppository ranges from 64 to 72 (five studies).9 The pain-free rate ranged from 34% to 50%. In a comparative study, a combination (ergotamine, 2 mg, and caffeine, 100 mg) suppository was more effective than sumatriptan (73% versus 63%) but produced more adverse events.117 In Ferrari and associates’ metaanalysis of oral triptans, 100-mg sumatriptan was used as the standard.12
Zolmitriptan is available in 2.5- and 5-mg regular and orally disintegrating tablets and a 5-mg nasal spray. Zolmitriptan has a 40% oral bioavailability, has a Tmax of about 2.5 hours, and is metabolized by the cytochrome P-450 system to an active metabolite that is degraded by MAO-A. Patients taking MAO inhibitors are limited to a total dose of 5 mg/day. The nasal spray is absorbed in the nasopharynx; detectable blood levels appear within 5 minutes, and 40% of maximal concentration is achieved within 10 to 15 minutes.122,123 First-attack 2-hour headache response rates for 5 mg nasal spray are 69%, with a therapeutic gain in 38%.122–124
Naratriptan is available in 1- and 2.5-mg oral tablets. The recommended dose is 2.5 mg. Bioavailability is 60% to 70%, Tmax is 2 hours (outside an attack), and the terminal elimination half-life is 5 hours. Naratriptan is excreted largely as unchanged drug in the urine.125
Frovatriptan is available in 2.5-mg tablets. The daily limit is three tablets. Frovatriptan has a bioavailability of 22% in men and 30% in women. The Tmax is 2 to 3 hours. It is metabolized by P4540 (CYP-1A2) and excreted in the urine.126 The mean half-life is 26 hours.
Eletriptan is available in 40 and 80-mg oral tablets. It has a 50% bioavailability and a half-life of 5 hours and is rapidly absorbed.127 Eletriptan interacts with drugs that are metabolized by cytochrome P-450 (including CYP-3A4). Adverse events are more common with 80-mg eletriptan than with other triptans.
These seven triptans are safe (for patients without cardiovascular risk factors), effective, and appropriate first-line therapy for patients who have a moderate to severe migraine headache or for whom analgesics have failed to provide adequate relief. Although they are safe, no evidence supports their effectiveness during the aura phase of a migraine attack.128
The oral formulations can be divided into two classes. Almotriptan, eletriptan, rizatriptan, sumatriptan, and zolmitriptan have the highest 2-hour efficacy, can provide headache relief within 30 to 60 minutes, and would be the first choice when patients require efficacy and speed of onset and do not have multiple recurrences. The metaanalysis suggests that almotriptan, eletriptan, and rizatriptan are most effective.12
Frovatriptan and naratriptan have lower 2-hour efficacy but produce fewer adverse events (as does almotriptan). Whether headache recurs less often with these drugs is controversial. Almotriptan, frovatriptan, and naratriptan are choices for patients who are prone to adverse events.129
Ergotamine and Dihydroergotamine
The evidence supporting ergotamine’s efficacy is inconsistent.130 Some patients respond preferentially to rectal ergotamine.131 Dihydroergotamine can be administered intramuscularly, subcutaneously, or intravenously. The headache recurrence rate may be low (<20%), there are fewer adverse events, and it may be less likely than ergotamine to produce rebound headache.130 Limited efficacy evidence exists for dihydroergotamine nasal spray. No placebo-controlled trials have demonstrated the efficacy and safety of dihydroergotamine subcutaneously, intramuscularly, or intravenously as monotherapy. Repetitive intravenous dihydroergotamine is commonly used in North America to treat intractable headache.130,132
PREVENTIVE TREATMENT
Preventive therapy is given in an attempt to reduce the frequency, duration, or severity of attacks. Additional benefits include improving responsiveness to acute attack treatment, improving function, and reducing disability. Preventive treatment may prevent episodic migraine’s progression to chronic migraine and result in health care cost reductions. Silberstein and colleagues retrospectively analyzed resource utilization information in a large claims database. The addition of migraine preventive drug therapy to therapy that consisted of only an acute medication was effective in reducing resource consumption. During the second 6 months after the initial preventive medication, as compared with the 6 months preceding preventive therapy, office and other outpatient visits with a migraine diagnosis decreased by 51.1%, emergency department visits with a migraine diagnosis decreased 81.8%, computed tomographic scans with a migraine diagnosis decreased 75.0%, MRI scans with a migraine diagnosis decreased 88.2%, and other migraine medication dispensations decreased 14.1%.133
Preventive medications reduce attack frequency, duration, or severity.2,134 According to the U.S. Headache Consortium Guidelines,10 indications for preventive treatment include the following:






Preventive measures are not used to nearly the extent they should be. In the American Migraine Study II, 25% of all people with migraine, or more than 7 million people, experienced more than three attacks per month, and 53% of those surveyed reported either having severe impairment because of their attacks or needing bed rest.17 However, only 5% of all migraineurs currently use preventive therapy to control their attacks.135
Preventive medication groups include β-adrenergic blockers, antidepressants, calcium channel antagonists, serotonin antagonists, anticonvulsants, and NSAIDs. Choice is based on efficacy, adverse events, and coexistent and comorbid conditions (Table 56-5). It is started at a low dose and increased slowly until therapeutic effects develop or the ceiling dose is reached. A full therapeutic trial may take 2 to 6 months. Acute headache medications should not be overused. If headaches are well controlled, medication can be tapered and discontinued. Dose reduction may provide a better risk/benefit ratio. Women of childbearing potential should practice adequate contraception.
TABLE 56-5 Choices of Preventive Treatment in Migraine: Influence of Comorbid Conditions
| Comorbid Condition | ||
|---|---|---|
| Drug | Relative Contraindication | Relative Indication |
| β Blockers | Asthma, depression | HTN |
| CHF | Angina | |
| Raynaud’s disease | ||
| Diabetes | ||
| Antiserotonin Agents | ||
| Pizotifen | Obesity | Orthostatic HTN |
| Methysergide | Angina | |
| PVD | ||
| Calcium-Channel Blockers | ||
| Verapamil | Constipation | Migraine with aura |
| Hypotension | HTN | |
| Angina | ||
| Asthma | ||
| Flunarizine | Parkinson’s disease | Hypertension |
| FHM | ||
| Antidepressants | ||
| TCAs | Mania | Other pain disorders |
| Urinary retention | Depression | |
| Heart block | Anxiety disorders | |
| Insomnia | ||
| SSRIs | Mania | Depression |
| OCD | ||
| MAOIs | Unreliability of patient | Refractory depression |
| Anticonvulsants | ||
| Divalproex/valproate | Liver disease | Mania |
| Bleeding disorders | Epilepsy | |
| Anxiety disorders | ||
| Gabapentin | Liver disease | Mania |
| Bleeding disorders | Epilepsy | |
| Anxiety disorders | ||
| Topiramate | Kidney stones | Mania |
| Epilepsy | ||
| Anxiety disorders | ||
| NSAIDs | ||
| Naproxen | Ulcer disease | Arthritis |
| Gastritis | Other pain disorders | |
CHF, congestive heart failure; FHM, familial hemiplegic migraine; HTN, hypertension; MAOI, monoamine oxidase inhibitor; NSAID, nonsteroidal anti-inflammatory drug; OCD, obsessive-compulsive disorder; PVD, peripheral vascular disease; SSRI, serotonin-specific reuptake inhibitor; TCA, tricyclic antidepressant.
Behavioral and psychological interventions used for prevention include relaxation training, thermal biofeedback combined with relaxation training, electromyographic biofeedback, and cognitive-behavioral therapy.136
Mechanism of Action of Preventive Medications
Most migraine-preventive drugs were designed to treat other disorders. Serotonin antagonists were developed on the basis of the concept that migraine is caused by excess 5-HT. Antidepressants downregulate 5-HT2 and β-adrenergic receptors. Anticonvulsant medications decrease glutamate and enhance GABAA. Potential mechanisms of migraine-preventive medications include raising the threshold to migraine activation by stabilizing a more reactive nervous system; enhancing antinociception; inhibiting CSD; inhibiting peripheral and central sensitization; blocking neurogenic inflammation; and modulating sympathetic, parasympathetic or serotonergic tone. Oshinsky showed that descending control from the upper brainstem, through serotonergic and noradrenergic systems, modulates the trigeminal nucleus caudalis and prevents central sensitization (personal communication). Moskowitz has recently shown that preventive medications given chronically, but not acutely, block CSD.137
Medication
β Blockers
β-Adrenergic blockers (also called β blockers), the most widely used class of drugs in prophylactic migraine treatment, are approximately 50% effective in producing a greater than 50% reduction in attack frequency. Rabkin and coworkers138 serendipitously discovered propranolol’s effectiveness in headache treatment in patients who were being treated for angina.139 Propranolol, nadolol, atenolol, metoprolol, and timolol are effective.8 Their relative efficacy has not been established; choice is based on β-adrenergic selectivity, convenience, adverse events, and patients’ reactions.2 β Blockers can produce behavioral adverse events, such as drowsiness, fatigue, lethargy, sleep disorders, nightmares, depression, memory disturbance, and hallucinations; they should be avoided by patients who are depressed. Decreased exercise tolerance limits their use by athletes. Less common adverse events include impotence, orthostatic hypotension, and bradycardia. β Blockers are useful for patients with angina or hypertension. They are relatively contraindicated in patients with congestive heart failure, asthma, Raynaud’s disease, and insulin-dependent diabetes.
Antidepressants
Antidepressants consist of a number of different classes of drugs with different mechanisms of action. The tricyclic antidepressants (TCAs) most commonly used for headache prophylaxis include amitriptyline, nortriptyline, doxepin, and protriptyline. Imipramine and desipramine have been used at times. With the exception of amitriptyline, the TCAs have not been vigorously evaluated; their use is based on anecdotal or uncontrolled reports. None have been approved for migraine. Antidepressants are especially useful for patients with comorbid depression and anxiety disorders.
Amitriptyline (a TCA) is the only antidepressant whose efficacy has limited support.8 Adverse events include increased appetite, weight gain, dry mouth, and sedation; cardiac toxicity and orthostatic hypotension occasionally occur.140 There has been one trial with positive results for fluoxetine. Sexual dysfunction is a common adverse event.141
Calcium-Channel Blockers
The Agency for Healthcare Policy and Research analyzed 45 controlled trials.8 Flunarizine was effective, nimodipine produced mixed results, and the results of nifedipine were difficult to interpret. Verapamil was more effective than placebo in two of three trials, but both positive trials had high dropout rates, rendering the findings uncertain.2 Its most common adverse event is constipation. Flunarizine is the most effective drug of this class. It also has dopamine-blocking properties.
Anticonvulsant Medications
Anticonvulsant medication is increasingly recommended for migraine prevention because of placebo-controlled, double-blind trials that have proved it to be effective. With the exception of valproic acid, topiramate, and zonisamide, anticonvulsants may interfere substantially with the efficacy of oral contraceptives.142,143 Nine controlled trials of five different anticonvulsants were included in the Agency for Healthcare Policy and Research Technical Report.145–151,153,154
Divalproex sodium (500 to 1000 mg) and sodium valproate are effective, as is the extended-release formulation.8 Five studies provided strong and consistent support for the efficacy of divalproex sodium (approved by the U.S. Food and Drug Administration)146,147 and sodium valproate.150,151 Nausea, vomiting, and gastrointestinal distress are the most common adverse events of valproate therapy. These are generally self-limited and are slightly less common with divalproex sodium than with sodium valproate. On rare occasions, valproate administration is associated with severe adverse events, such as hepatitis or pancreatitis. The frequency varies with the number of concomitant medications used, the patient’s age and general state of health, and the presence of genetic and metabolic disorders. Valproate is teratogenic and should not be used by pregnant women or women considering pregnancy.152 In clinical trials, the most frequent adverse events were nausea (42%), alopecia (31%), tremor (28%), asthenia (25%), dyspepsia (25%), somnolence (25%), and weight gain (19%).153 Baseline liver function studies should be obtained, but follow-up studies are probably not needed for adults receiving monotherapy.152
Valproic acid is available as 250-mg capsules and as a syrup (250 mg/5 mL). Divalproex sodium is a stable coordination complex composed of sodium valproate and valproic acid in a 1:1 molar ratio. An enteric-coated form of divalproex sodium is available as 125, 250, and 500-mg capsules and a sprinkle formulation. It should be started at a dosage of 250 to 500 mg/day in divided doses, and then the dosage should be slowly increased. Serum levels should be monitored if there is a question of toxicity or compliance. (The usual therapeutic level is from 50 to 100 mg/mL.) The maximum recommended dosage is 60 mg/kg/day. An extended-release form of divalproex sodium demonstrated efficacy comparable with that of the tablet formulation. The adverse event profile in the clinical trial, however, showed almost identical adverse event rates for placebo and active treatment.154
Gabapentin (1800 to 2400 mg) showed efficacy in a placebo-controlled, double-blind trial only in a modified intent-to-treat analysis. Migraine attack frequency was reduced by 50% in about one third of patients.155 The most common adverse events were dizziness or giddiness and drowsiness.
Topiramate was originally synthesized as part of a research project to discover structural analogs of fructose-1,6-diphosphate capable of inhibiting the enzyme fructose-1,6- bisphosphatase, thereby blocking gluconeogenesis, but it has no hypoglycemic activity. Topiramate is a derivative of the naturally occurring monosaccharide D-fructose and contains a sulfamate functionality. Topiramate was originally marketed for the treatment of epilepsy156; it is now approved by the U.S. Food and Drug Administration for treatment of migraine.
It is associated with weight loss, not weight gain. In two large, double-blind, placebo-controlled, multicenter trials, topiramate, both 100 and 200 mg, was effective in reducing migraine attack frequency by 50% in half of the patients.157,158 Withdrawals from the study because of adverse events were common among the topiramate recipients but did not affect statistical significance. The most common adverse event was paresthesia, which was rated as mild to moderate in the majority of patients and limited treatment in only 8% of these subjects. The other most common adverse events were fatigue, decreased appetite, nausea, diarrhea, weight decrease, taste perversion, hypoesthesia and abdominal pain. The most common CNS adverse events were somnolence, insomnia, difficulty with memory, language problems, difficulty with concentration, mood problems, and anxiety. Renal calculi occur with a reported incidence of about 1.5%. A rare syndrome consisting of acute myopia in association with secondary angle-closure glaucoma has been reported.
Serotonin Antagonists
The antiserotonin migraine-preventive drugs are potent 5-HT2B and 5-HT2C receptor antagonists. Methysergide is a semisynthetic ergot alkaloid that is structurally related to methylergonovine. It is a 5-HT2 receptor antagonist and 5-HT1B/5-HTD agonist. It was probably the first drug developed for migraine prevention,159 but its usefulness is limited by reports of retroperitoneal and retropleural fibrosis associated with long-term, mostly uninterrupted, administration.160
Methysergide is effective.8,161 Adverse events include transient muscle aching, claudication, abdominal distress, nausea, weight gain, and hallucinations. The major complication is rare (1 per 2500) retroperitoneal, pulmonary, or endocardial fibrosis.161 To prevent this, a 4-week medication-free interval is recommended after 6 months of continuous treatment.2,161
Methysergide is indicated for the treatment of migraine and cluster headache. The dosage ranges from 2 to 8 mg/day, the higher doses being given two or three times a day. Clinicians find that some patients can take higher doses, up to 14 mg a day, without adverse events and with higher efficacy.162 To minimize early adverse events, patients can start with a dose of 1 mg/day and increase the dose gradually by 1 mg every 2 to 3 days. Methysergide, in general, should not be taken continuously for long periods, because this may produce retroperitoneal fibrosis.160,163,164 Instead, the drug should be given for 6 months, stopped for 1 month, and then restarted. To avoid an increase in headache when methysergide is stopped, the patient should be weaned off the drug over a 1-week period. Some authorities use methysergide on a continuous basis with careful monitoring,162 which includes auscultation of the heart and yearly echocardiography, chest radiography, and abdominal MRI. The drug should be discontinued immediately on suspicion of pulmonary or cardiac retroperitoneal fibrosis.162
Cyproheptadine, an antagonist at the 5-HT2, histamine H1, and muscarinic cholinergic receptors, is widely used in the prophylactic treatment of migraine in children.162,165,166 Cyproheptadine is available as 4-mg tablets. The total dosage ranges from 12 to 36 mg/day (given two to three times a day or at bedtime). Common adverse events are sedation and weight gain; dry mouth, nausea, lightheadedness, ankle edema, aching legs, and diarrhea are less common. Cyproheptadine may inhibit growth in children167 and reverse the effects of selective serotonin reuptake inhibitors.
Pizotifen, a benzocycloheptathiophene derivative, is a 5-HT2 receptor antagonist structurally similar to cyproheptadine.10 It is not available everywhere. According to the United States Headache Consortium guidelines,118 evidence was inconsistent for its efficacy. Analysis of the placebo-controlled trials suggested a large clinical effect that was statistically significant. Pizotifen was generally poorly tolerated.8 Substantial weight gain, tiredness, drowsiness, or a combination of these was frequently reported. Pizotifen was associated with a high rate of withdrawals from the study because of adverse events. Controlled and uncontrolled studies in Europe168 have shown this drug to be of benefit in 40% to 79% of patients. The dosage recommendation is 0.5 to 1 mg, one three times daily by titration. Adverse events include drowsiness, increased appetite, and weight gain.169
Natural Products
Feverfew (Tanacetum parthenium) is a medicinal herb whose effectiveness has not been totally established.170 Riboflavin (400 mg) was effective in one placebo-controlled, double-blind trial; more than half the patients responded.171 A standardized extract (75 mg bid) of Petasites hybridus root (butterbur), a perennial shrub,16 was effective in a double-blind, placebo-controlled study.172 The most common adverse event was belching. Coenzyme Q10 was effective in one double-blind, placebo-controlled trial.173
Newer Treatments
Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Antagonists
Schrader and associates174 conducted a double-blind, placebo-controlled, crossover study of lisinopril, an angiotensin-converting enzyme inhibitor. The treatment period was 12 weeks, with one 10-mg lisinopril tablet taken once daily for 1 week and then two 10-mg lisinopril tablets taken once daily for 11 weeks, followed by a 2-week washout period. Hours with headache, days with headache, days with migraine, and headache severity index were significantly reduced by 20% (95% confidence interval, 5% to 36%), 17% (5% to 30%), 21% (9% to 34%), and 20% (3% to 37%), respectively, with lisinopril, in comparison with placebo. Tronvik and colleagues175 performed a randomized, double-blind, placebo-controlled crossover study of candesartan (16-mg), an angiotensin II receptor blocker. The number of candesartan responders (reduction of 50% or more in comparison with placebo) was 18 (31.6%) of 57 for days with headache and 23 (40.4%) of 57 for days with migraine. Adverse events were similar in the two periods. In this study, candesartan was effective, with a tolerability profile comparable with that of placebo.
Botulinum toxin type A (Botox) in dosages of 0, 25, or 75 U showed promising results in one placebo-controlled, double-blind trial. It was injected into glabellar, frontalis, and temporalis muscles. The 25-U treatment was significantly better than placebo in reducing mean frequency of moderate to severe migraines during days 31 to 60, 50% reduction of incidence of all migraine at days 61 to 90, and reduction in all migraine at days 61 to 90.176
Setting Treatment Priorities
The goals of preventive treatment are to reduce the frequency, duration, or severity of attacks, improve responsiveness to acute attack treatment, improve function, and reduce disability. Preventive treatment may also prevent episodic migraine’s progression to chronic migraine and result in health care cost reductions. The preventive medications with the best documented efficacy are the β blockers, divalproex, and topiramate. The choice of drug is based on a drug’s proven efficacy, the physician’s informed belief about medications not yet evaluated in controlled trials, the drug’s adverse events, the patient’s preferences and headache profile, and the presence or absence of coexisting disorders.2 The drug chosen should be the one that has the best risk/benefit ratio for the individual patient and takes advantage of the drug’s side effect profile.177,178
Comorbid and coexistent diseases have important implications for treatment. The presence of a second illness provides therapeutic opportunities but also imposes certain therapeutic limitations. An underweight patient would be a candidate for one of the medications that commonly produce weight gain, such as a TCA; in contrast, these drugs should be avoided by an overweight patient and the use of topiramate considered instead. Tertiary TCAs that have a sedating effect would be useful at bedtime for patients with insomnia. Older patients with cardiac disease or patients with significant hypotension may not be able to use TCAs or calcium channel or β blockers, but they could use divalproex or topiramate. In the athletic patient, β blockers should be used with caution. Medication that can impair cognitive functioning should be avoided when patients are dependent on their wits.177,178
In some instances, two or more conditions may be treated with a single drug. When migraine occurs with hypertension and/or angina, β blockers or calcium channel blockers may be effective for all conditions.179 For the patient with migraine and depression, TCAs or selective serotonin reuptake inhibitors may be especially useful.180 For the patient with migraine and epilepsy181 or migraine and bipolar illness,152,182 divalproex and topiramate are the drugs of choice. The pregnant migraineur who has a comorbid condition that necessitates treatment should be given a medication that is effective for both conditions and has the lowest potential for adverse events on the fetus. When individuals have more than one disease, certain categories of treatment may be relatively contraindicated. For example, β blockers should be used with caution in depressed migraineurs, whereas TCAs, neuroleptics, or sumatriptan may lower the seizure threshold and should be used with caution in the epileptic migraineur.
Although monotherapy is preferred, it is sometimes necessary to combine preventive medications. Antidepressants are often used with β blockers or calciums-channel blockers, and topiramate or divalproex sodium may be used in combination with any of these medications. Pascual and colleagues183 found that combining a β blocker and sodium valproate could lead to an increased benefit for patients with migraine that had previously been resistant to either drug alone. Fifty-two patients (43 women) with a history of episodic migraine with or without aura and previously unresponsive to β blockers or sodium valproate monotherapy were treated with a combination of propranolol (or nadolol) and sodium valproate in an open-label manner. Fifty-six percent had a greater than 50% reduction in days with migraine. This open-label trial supports the practice of combination therapy. Controlled trials are needed to determine the true advantage of this combination treatment in episodic and chronic migraine.
1 Critchley M. Migraine: from Cappadocia to Queen Square. London: Heinemann, 1967.
2 Silberstein SD, Saper JR, Freitag F. Migraine: diagnosis and treatment. In: Silberstein SD, Lipton RB, Dalessio DJ, editors. Wolff’s Headache and Other Head Pain. 7th ed. New York: Oxford University Press; 2001:121-237.
3 Headache Classification Committee of the International Headache Society. Classification and diagnostic criteria for headache disorders, cranial neuralgia, and facial pain. Cephalalgia. 1988;8:1-96.
4 Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 2nd edition. Cephalalgia. 2004;24(Suppl 1):9-160.
5 Ferrari MD. Migraine. Lancet. 1998;351:1043-1051.
6 Goslin RE, Gray RN, McCrory DC, et al: Behavioral and physical treatments for migraine headache. Prepared for the Agency for Healthcare Policy and Research, Contract No. 290–94–2025. Available from the National Technical Information Service, Accession No. 127946, February 1999.
7 Gray RN, McCrory DC, Eberlein K, et al: Parenteral drug treatments for acute migraine headache. Prepared for the Agency for Healthcare Policy and Research, Contract No. 290–94–2025. Available from the National Technical Information Service, Accession No. 127862 (Technical Review 2.5), February 1999.
8 Gray RN, Goslin RE, McCrory DC, et al: Drug treatments for the prevention of migraine headache. Prepared for the Agency for Healthcare Policy and Research, Contract No. 290–94–2025. Available from the National Technical Information Service, Accession No. 127953 (Technical Review 2.3), February 1999.
9 Gray RN, McCrory DC, Eberlein K, et al: Self-administered drug treatments for acute migraine headache. Prepared for the Agency for Healthcare Policy and Research, Contract No. 290–94–2025. Available from the National Technical Information Service, Accession No. 127854 (Technical Review 2.4), February 1999.
10 Ramadan NM, Silberstein SD, Freitag FG, et al. Evidence-based guidelines of the pharmacological management for prevention of migraine for the primary care provider. Neurology. 1999.
11 Matchar DB, Young WB, Rosenberg JA, et al. Evidence-based guidelines for migraine headache in the primary care setting: pharmacological management of acute attacks. Neurology. 2000.
12 Ferrari MD, Roon KI, Lipton RB, et al. Oral triptans (serotonin 5-HT1B/1D agonists) in acute migraine treatment: a metaanalysis of 53 trials. Lancet. 2001;358:1668-1675.
13 Stewart WF, Shechter A, Rasmussen RK. Migraine prevalence. A review of population-based studies. Neurology. 1994;44:S17-S23.
14 Stewart WF, Lipton RB, Celentano DD, et al. Prevalence of migraine in the United States. JAMA. 1992;267:64-69.
15 Lipton RB, Diamond S, Reed M, et al. Migraine diagnosis and treatment: results from the American Migraine Study II. Headache. 2001;41:638-645.
16 Lipton RB, Hamelsky SW, Stewart WF. Epidemiology and impact of headache. In: Silberstein SD, Lipton RB, Dalessio DJ, editors. Wolff’s Headache and Other Head Pain. 7th ed. New York: Oxford University Press; 2001:85-107.
17 Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41:646-657.
18 World Health Organization. World Health Report. Available at www.who.int/whr/en/, 2001.
19 Stewart WF, Lipton RB, Kolodner KB, et al. Validity of the Migraine Disability Assessment (MIDAS) score in comparison to a diary-based measure in a population sample of migraine sufferers. Pain. 2000;88:41-52.
20 Ware JE, Bjorner JB, Kosinski M. Practical implication of item response theory and computerized adaptive testing. A brief summary of ongoing studies of widely used headache impact scales. Med Care. 2000;38:73-82.
21 Ophoff RA, Terwindt GM, Vergouwe MN. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNLA4.. Cell Tiss Res. 1996;87:543-552.
22 Joutel A, Bousser MG, Biousee V. A gene for familial hemiplegic migraine maps to chromosome 19. Nat Genet. 1993;5:40-45.
23 Ducros A, Deiner C, Joutel A, et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17-24.
24 Ferrari MD, Haan J. Genetics of headache. In: Silberstein SD, Lipton RB, Dalessio DJ, editors. Wolff’s Headache and Other Head Pain. 7th ed. New York: Oxford University Press; 2001:73-84.
25 De Fusco M, Marconi R, Silvestri L, et al. Haploinsufficiency of ATP1A2 encoding the Na(+)/K(+) pump α2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192-196.
26 Wolff HG. Headache and other head pain. New York: Oxford University, 1963.
27 Olesen J, Friberg L, Skyhoj-Olsen T. Timing and topography of cerebral blood flow, aura and headache during migraine attacks. Ann Neurol. 1990;28:791-798.
28 Sanchez-del Rio M, Bakker D, Wu O, et al. Perfusion weighted imaging during migraine: spontaneous visual aura and headache. Cephalalgia. 1999;19:701-707.
29 Cutrer FM, Sorenson AG, Weisskoff RM, et al. Perfusion-weighted imaging defects during spontaneous migrainous aura. Ann Neurol. 1998;43:25-31.
30 Hadjikhani N, Sanchez delRio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687-4692.
31 Pietrobon D, Striessnic J. Neurobiology of migraine. Nat Rev Neurosci. 2003;4:386-398.
32 Lashley KS. Patterns of cerebral integration indicated by the scotomas of migraine. Arch Neurol. 1941;46:331-339.
33 Leão AAP. Spreading depression of activity in cerebral cortex. J Neurophysiol. 1944;7:359-390.
34 Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of RCBF in classic migraine. Ann Neurol. 1981;9:344-352.
35 Olesen J. Cerebral and extracranial circulatory disturbances in migraine: pathophysiological implications. Cerebrovasc Brain Metab Rev. 1991;3:1-28.
36 Milner PM. Note on a possible correspondence between the scotomas of migraine and spreading depression of Leão. Electroencephalogr Clin Neurophysiol. 1958;10:705.
37 Cao Y, Welch KM, Aurora S, et al. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch Neurol. 1999;56:548-554.
38 Andersen AR, Friberg L, Skyloj-Olsen T, et al. Delayed hyperemia following hypoperfusion in classic migraine. Single photon emission tomographic demonstration. Arch Neurol. 1988;45:154-159.
39 Seto H, Shimizu M, Futatsuya R, et al. Basilar artery migraine. Reversible ischemia demonstrated by Tc-99m HMPAO brain SPECT. Clin Nucl Med. 1994;19:215-218.
40 Woods RP, Iacoboni M, Mazziotta JC. Bilateral spreading cerebral hypoperfusion during spontaneous migraine headaches. N Engl J Med. 1994;331:1689-1692.
41 Cutrer FM, O’Donnell A. Recent advances in functional neuroimaging. Curr Opin Neurol. 1999;12:255-259.
42 Barkley GL, Tepley N, Nagel L, et al. Magnetoencephalographic studies of migraine. Headache. 1990;30:428-434.
43 Bowyer SM, Aurora KS, Moran JE, et al. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol. 2001;50:582-587.
44 Aurora SK, Cao Y, Bowyer SM, et al. The occipital cortex is hyperexcitable in migraine: experimental evidence. Headache. 1999;39:469-476.
45 Young WB, Oshinsky ML, Shechter AL, et al. Consecutive transcranial magnetic stimulation induced phosphene thresholds in migraineurs and controls [Abstract]. Neurology. 2001;56:A142.
46 Afra J, Mascia A, Gerard P, et al. Interictal cortical excitability in migraine: a study using transcranial magnetic stimulation of motor and visual cortices. Ann Neurol. 1998;44:209-215.
47 Schoenen J, Thomsen LL. Neurophysiology and autonomic dysfunction in migraine. In: Olesen J, Tfelt-Hansen P, Welch KMA, editors. The Headaches. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2000:301-312.
48 Uddman R, Edvinsson L, Ekman R, et al. Innervation of the feline cerebral vasculature by nerve fibers containing calcitonin gene–related peptide: trigeminal origin an co-existence with substance P. Neurosci Lett. 1985;62:131-136.
49 Buzzi MG, Moskowitz MA, Shimizu T, et al. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology. 1991;30:1193-1200.
50 Markowitz S, Saito K, Moskowitz MA. Neurogenically mediated plasma extravasation in dura mater: effect of ergot alkaloids. A possible mechanism of action in vascular headache. Cephalalgia. 1988;8:83-91.
51 Dimitriadou V, Buzzi MG, Theoharides TC, et al. Ultrastructural evidence for neurogenically mediated changes in blood vessels of the rat dura mater and tongue following antidromic trigeminal stimulation. Neuroscience. 1992;48:187-203.
52 Pappagalo M, Szabo Z, Esposito G, et al. Imaging neurogenic inflammation inpatients with migraine headaches. Neurology. 2002;52:274-275.
53 Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560-564.
54 Moskowitz MA, Cutrer FM. Sumatriptan: a receptor-targeted treatment for migraine. Annu Rev Med. 1993;44:145-154.
55 Nozaki K, Boccalini P, Moskowitz MA. Expression of c-fos–like immunoreactivity in brainstem after meningeal irritation by blood in the subarachnoid space. Neuroscience. 1992;49:669-680.
56 Kaube H, Keay K, Hoskin KL, et al. Expression of c-fos like immunoreactivity in the trigeminal nucleus caudalis and high cervical cord following stimulation of the sagittal sinus in the cat. Brain Res. 1993;629:95-102.
57 Goadsby PJ, Hoskin KL. The distribution of trigeminovascular afferents in the non-human primate brain. J Anat. 1997;190:367-375.
58 Zagami AS, Goadsby PJ, Edvinsson L. Stimulation of the superior sagittal sinus in the cat causes release of vasoactive peptides. Neuropeptides. 1990;16:69-75.
59 Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28:183-187.
60 Goadsby PJ, Edvinsson L. The trigeminovascular system in migraine: Studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48-56.
61 Edvinsson L, Goadsby PJ. Neuropeptides in headache. Eur J Neurol. 1998;5:329-341.
62 O’Connor TP, Van der Kooy D. Enrichment of vasoactive neuropeptide (calcitonin gene related peptide) in trigeminal sensory projection to the intracranial arteries. J Neurosci. 1988;8:2468-2476.
63 O’Connor TP, vanderKooy D. Pattern of intracranial and extracranial projections of trigeminal ganglion cells. J Neurosci. 1986;6:2200-2207.
64 Doods H, Hallermayer G, Wu D, et al. Pharmacological profile of BIBN-4096BS, the first selective small molecule CGRP antagonist. Br J Pharmacol. 2000;129:420-423.
65 Olesen J, Diener HC, Husstedt IW, et al. Calcitonin gene–related peptide (CGRP) receptor antagonist BIBN4096BS is effective in the treatment of migraine attacks [Abstract]. Cephalalgia. 2003;23:579.
66 Burstein R, Collins B, Bajwa Z, et al. Triptan therapy can abort migraine attacks if given before the establishment or in the absence of cutaneous allodynia and central sensitization: clinical and preclinical evidence [Abstract]. Headache. 2002;42:390-391.
67 Burstein R, Cutrer MF, Yarnitsky D. The development of cutaneous allodynia during a migraine attack: clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain. 2001;123:1703-1709.
68 Weiller C, May A, Limmroth V, et al. Brainstem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658-660.
69 Bahra A, Matharu MS, Buchel C, et al. Brainstem activation specific to migraine headache. Lancet. 2001;357:1016-1017.
70 Bolay H, Reuter U, Dunn AK, et al. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136-142.
71 Hoyer D, Clarke DE, Fozard JR, et al. VII International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacol Rev. 1994;46:157-203.
72 Hartig PR, Hoyer D, Humphrey PPA, et al. Alignment of receptor nomenclature with the human genome: classification of 5HT-1B and 5HT1D receptor subtypes. Trends Pharmacol Sci. 1996;17:103-105.
73 Longmore J, Shaw D, Smith D, et al. Differential distribution of 5HT(1D)- and 5HT(1B)-immunoreactivity within the human trigeminocerebrovascular system: implications for the discovery of new antimigraine drugs. Cephalalgia. 1997;17:833-842.
74 Buzzi MG, Sakas DE, Moskowitz MA. Indomethacin and acetyl salicylic acid block neurogenic plasma protein extravasation in rat dura mater. Eur J Pharmacol. 1989;165:251-258.
75 Cutrer FM, Limmroth V, Ayata G, et al. Attenuation by valproate of c-fos immunoreactivity in trigeminal nucleus caudalis induced by intracisternal capsaicin. Br J Pharmacol. 1995;116:3199-3204.
76 Lee WS, Limmroth V, Ayata C, et al. Peripheral GABA-A receptor mediated effects of sodium valproate on dural plasma protein extravasation to substance P and trigeminal stimulation. Br J Pharmacol. 1995;116:1661-1667.
77 Limmroth V, Lee WS, Cutrer FM, et al. GABAA-receptor–mediated effects of progesterone, its ring-A–reduced metabolites and synthetic neuroactive steroids on neurogenic edema in the rat meninges. Br J Pharmacol. 1996;117:99-104.
78 Lee WS, Moussaoui SM, Moskowitz MA. Blockade by oral or parenteral RPR100893 (a nonpeptide NK1 receptor antagonist) of neurogenic plasma protein extravasation in guinea-pig dura mater and conjunctiva. Br J Pharmacol. 1994;112:920-924.
79 May A, Gijsman HJ, Wallnoefer A, et al. Endothelin antagonist bosentan blocks neurogenic inflammation, but is not effective in aborting migraine attacks. Pain. 1996;67:375-378.
80 Goadsby PJ, Gundlach AL. Localization of [3H]-dihydroergotamine binding sites in the cat central nervous system: relevance to migraine. Ann Neurol. 1991;29:91-94.
81 Kandere-Grzybowska K, Gheorghe D, Priller J, et al. Stress-induced dura vascular permeability does not develop in mast cell–deficient and neurokinin-1 receptor knockout mice. Brain Res. 2003;980:213-220.
82 Silberstein SD, Young WB. Migraine aura and prodrome. Semin Neurol. 1995;45:175-182.
83 Giffin NJ, Ruggiero L, Lipton RB, et al. A novel approach to the study of premonitory symptoms in migraine using an electronic diary. Neurology. 2003;60:935-940.
84 Russell MB, Olesen J. A nosographic analysis of the migraine aura in a general population. Brain. 1996;119:355-361.
85 Silberstein SD, Lipton RB. Overview of diagnosis and treatment of migraine. Neurology. 1994;44:6-16.
86 Thomsen LL, Eriksen MK, Roemer SF, et al. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain. 2002;125:1379-1399.
87 Stewart WF, Schechter A, Lipton RB. Migraine heterogeneity: disability, pain intensity, attack frequency, and duration. Neurology. 1994;44:S24-S39.
88 Silberstein SD. Migraine symptoms: results of a survey of self-reported migraineurs. Headache. 1995;35:387-396.
89 Young WB, Peres MF, Rozen TD. Modular headache theory. Cephalalgia. 2001;21:842-849.
90 Draft International Headache Society Classification. Available at: www.I-H-S.org (accessed February 9, 2006).
91 Whitty CWM. Migraine without headache. Lancet. 1967;2:283-285.
92 Wijman C, Wolf PA, Kase CS, et al. Migrainous visual accompaniments are not rare in late life: the Framingham Study. Stroke. 1998;29:1539-1543.
93 Fisher CM. Late-life migraine accompaniments—further experience. Stroke. 1986;17:1033-1042.
94 Mark AS, Casselman J, Brown D, et al. Ophthalmoplegic migraine: reversible enhancement and thickening of the cisternal segment of the oculomotor nerve on contrast-enhanced MR images. AJNR Am J Neuroradiol. 1998;19:1887-1891.
95 Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707-710.
96 Tournier-Lasserve E, Joutel A, Melki J. Cerebral autosomal arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosomes. Nat Genet. 1993;3:256-259.
97 Joutel A, Vahedi K, Corpechot C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. 1997;350:1511-1515.
98 Meschia JF, Brott TG, Brown RDJr. Genetics of cerebrovascular disorders. Mayo Clin Proc. 2005;80:122-132.
99 Chabriat H, Vahedi K, Iba-Zizen MT, et al. Clinical spectrum of CADASIL: a study of seven families. Lancet. 1995;346:934-939.
100 Silberstein SD. Migraine. Lancet. 2004;363:381-391.
101 Lipton RB, Silberstein SD. Why study the comorbidity of migraine? Neurology. 1994;44:4-5.
102 Lipton RB, Cady RK, O’Quinn S, et al. Sumatriptan treats the full spectrum of headache in individuals with disabling IHS migraine. Headache. 1999;40:783-791.
103 Lipton RB, Stewart WF, Stone AM, et al. Stratified care vs step care strategies for migraine. The disability in strategies of care (DISC) study: a randomized trial. JAMA. 2000;284:2599-2605.
104 Cady RK, Sheftell F, Lipton RB, et al. Effect of early intervention with sumatriptan on migraine pain: retrospective analyses of data from three clinical trials. Clin Therap. 2000;22:1035-1048.
105 Lipton RB, Stewart WF, Ryan RE, et al. Efficacy and safety of the nonprescription combination of acetaminophen, aspirin, and caffeine in alleviating headache pain of an acute migraine attack: three double-blind, randomized, placebo-controlled trials. Arch Neurol. 1998;55:210-217.
106 Lipton RB, Baggish JS, Stewart WF, et al. Efficacy and safety of acetaminophen in the treatment of migraine: results of a randomized, double-blind, placebo-controlled, population-based study. Arch Intern Med. 2000;160:3486-3492.
107 Silberstein SD, McCrory DC. Butalbital in the treatment of headache: history, pharmacology, and efficacy. Headache. 2001;41:953-967.
108 Silberstein SD, McCrory DC. Opioids. Cephalalgia. 2000;20:854-864.
109 Silberstein SD. Migraine and pregnancy. Neurol Clin. 1997;15:209-231.
110 Callaham M, Raskin N. A controlled study of dihydroergotamine in the treatment of acute migraine headache. Headache. 1986;26:168-171.
111 Jones J, Sklar D, Dougherty J, et al. Randomized double-blind trial of intravenous prochlorperazine for the treatment of acute headache. JAMA. 1989;261:1174-1176.
112 Silberstein SD, Young WB, Mendizabal JE, et al. Acute migraine treatment with the dopamine receptor antagonist, droperidol: results of a randomized, double-blind, placebo-controlled, multicenter trial. Neurology. 2003;60:315-321.
113 Young WB, Mannix L, Adelman JU, et al. Cardiac risk factors and the use of triptans: a survey study. Headache. 2000;40:587-591.
114 Tatsuta T, Naito M, Oh-Hara T, et al. Functional involvement of P-glycoprotein in blood-brain barrier. J Biol Chem. 1992;267:20383-20391.
115 Lipton RB, Stewart WF, Cady R, et al. 2000 Wolfe Award. Sumatriptan for the range of headaches in migraine sufferers: results of the Spectrum Study. Headache. 2000;40:783-791.
116 Duquesnoy C, Mamet JP, Sumner D, et al. Comparative clinical pharmacokinetics of single doses of sumatriptan following subcutaneous, oral, rectal, and intranasal administration. Eur J Pharm Sci. 1998;6:99-104.
117 Dahlöf C. Clinical efficacy and tolerability of sumatriptan tablet and suppository in the acute treatment of migraine: a review of data from clinical trials. Cephalalgia. 2001;21:9-12.
118 Silberstein SD. Practice Parameter—Evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology for the United States Headache Consortium. Neurology. 2000;55:754-762.
119 Tfelt-Hansen P. Efficacy and adverse events of subcutaneous, oral, and intranasal sumatriptan used for migraine treatment: a systematic review based on number needed to treat. Cephalalgia. 1998;18:532-538.
120 Thomson Healthcare. Physicians’ Desk Reference, 57th ed. Montvale, NJ: Thomson PDR, 2003.
121 Dahlöf C. Sumatriptan nasal spray in the acute treatment of migraine: a review of clinical studies. Cephalalgia. 1999;19:769-778.
122 Shakra S, Yates Ra, Sorensen J, et al. Distribution and pharmacokinetics of zolmitriptan following administration by nasal spray [Abstract]. Neurology. 2002;58:A91.
123 Shakra S, Becker WJ, Lee D. Zolmitriptan nasal spray is effective, fast-acting, and well tolerated during both short and long-term treatment [Abstract]. Neurology. 2002;58:A414.
124 Charlesworth BR, Dowson AJ, Purdy A, et al. Speed of onset and efficacy of zolmitriptan nasal spray in the acute treatment of migraine: a randumized, double blind, placebo-controlled, dose-ranging study versus zolmitriptan tablet. CNS Drugs. 2003;17:653-667.
125 Gunasekara NS, Wiseman LR. Naratriptan. CNS Drugs. 1997;8:402-408.
126 Silberstein SD. Pharmacological profile and clinical characteristics of frovatriptan in the acute treatment of migraine: introduction. Headache. 2002;42:S45-S99.
127 Färkkilä M. A dose-finding study of eletriptan (UK-116,044) (5–30 mg) for the acute treatment of migraine. Cephalalgia. 1996;16:387-388.
128 Bates D, Ashford E, Dawson R, et al. Subcutaneous sumatriptan during the migraine aura. Neurology. 1994;44:1587-1592.
129 Silberstein SD. Pharmacological profile and clinical characteristics of frovatriptan in the acute treatment of migraine. Headache. 2002;42:S45-S93.
130 Silberstein SD, McCrory DC. Ergotamine and dihydroergotamine: history, pharmacology, and efficacy. Headache. 2003;43:144-166.
131 Tfelt-Hansen P, Saxena PR, Dahlöf C, et al. Ergotamine in the acute treatment of migraine: a review and European consensus. Brain. 2000;123:9-18.
132 Silberstein SD, Schulman EA, Hopkins MM. Repetitive intravenous DHE in the treatment of refractory headache. Headache. 1990;30:334-339.
133 Silberstein SD, Winner PK, Chmiel JJ. Migraine preventive medication reduces resource utilization. Headache J Head Face Pain. 2003;43:171-178.
134 Silberstein SD, Goadsby PJ. Migraine: preventive treatment. Cephalalgia. 2002;22:491-512.
135 Lipton RB, Scher AI, Kolodner K, et al. Migraine in the United States: epidemiology and patterns of health care use. Neurology. 2002;58:885-894.
136 Campbell JK, Penzien D, Wall EM. Evidenced-based guidelines for migraine headache: behavioral and physical treatments. Available at: http://www.aan.org/professionals/practice/pdfs/g10089.pdf.
137 Ayata C, Jin H, Kudo C, et al. Suppression of cortical spreading depression in migraine prophylasis. Ann Neurol. 2006;59:652-661.
138 Rabkin R, Stables DP, Levin NW, et al. The prophylactic value of propranolol in angina pectoris. Am J Cardiol. 1966;18:370-383.
139 Weber RB, Reinmuth OM. The treatment of migraine with propranolol. Neurology. 1972;22:366-369.
140 Saper JR, Silberstein SD, Lake AE, et al. Double-blind trial of fluoxetine: chronic daily headache and migraine. Headache. 1994;34:497-502.
141 Stein DJ, Hollander E. Sexual dysfunction associated with the drug treatment of psychiatric disorders: incidence and treatment. CNS Drugs. 1994;2:78-86.
142 Coulam CB, Annagers JR. New anticonvulsants reduce the efficacy of oral contraception. Epilepsia. 1979;20:519-525.
143 Hansten PP, Horn JR. Drug interaction. Newsletter. 1985;5:7-10.
144 Kuritzky A, Hering R. Prophylactic treatment of migraine with long acting propranolol: a comparison with placebo. Cephalalgia. 1987;7:457-458.
145 Jensen R, Brinck T, Olesen J. Sodium valproate has prophylactic effect in migraine without aura: a triple-blind, placebo-controlled crossover study. Neurology. 1994;44:241-244.
146 Klapper JA. An open label crossover comparison of divalproex sodium and propranolol HCl in the prevention of migraine headaches. Headache Q. 1995;5:50-53.
147 Mathew NT, Saper JR, Silberstein SD, et al. Migraine prophylaxis with divalproex. Arch Neurol. 1995;52:281-286.
148 Rompel H, Bauermeister PW. Aetiology of migraine and prevention with carbamazepine (Tegretol). S Afr Med J. 1970;44:75-80.
149 Klapper JA. Divalproex sodium in migraine prophylaxis: a dose-controlled study. Cephalalgia. 1997;17:103-108.
150 Hering R, Kuritzky A. Sodium valproate in the prophylactic treatment of migraine: a double-blind study versus placebo. Cephalalgia. 1992;12:81-84.
151 Jensen R, Brinck T, Olesen J. Sodium valproate has a prophylactic effect in migraine without aura. Neurology. 1994;44:647-651.
152 Silberstein SD. Divalproex sodium in headache—literature review and clinical guidelines. Headache. 1996;36:547-555.
153 Silberstein SD, Collins SD. Safety of divalproex sodium in migraine prophylaxis: an open-label, long-term study. Long-Term Safety of Depakote in Headache Prophylaxis Study Group. Headache. 1999;39:633-643.
154 Freitag FG, Collins SD, Carlson HA, et al. A randomized trial of divalproex sodium extended-release tablets in migraine prophylaxis. Neurology. 2002;58:1652-1659.
155 Mathew NT, Rapoport A, Saper J, et al. Efficacy of gabapentin in migraine prophylaxis. Headache. 2001;41:119-128.
156 Shank RP, Gardocki JF, Vaught JL, et al. Topiramate: preclinical evaluation of structurally novel anticonvulsant. Epilepsia. 1994;35:450-460.
157 Brandes JL, Jacobs DJ, Neto W, et al. Topiramate in the prevention of migraine headache: a randomized, double-blind, placebo-controlled, parallel study (MIGR-002) [Abstract]. Neurology. 2003;60:A238.
158 Mathew NT, Schmit TJ, Neto W, et al. Topiramate in migraine prevention: MIGR 001 [Abstract]. Neurology. 2003;60:A336.
159 Sicuteri R. Prophylactic and therapeutic properties of 1-methyl-lysergic acid butanolamide in migraine. Int Arch Allergy Appl Immunol. 1959;15:300-307.
160 Graham JR, Suby HI, LeCompte PR, et al. Fibrotic disorders associated with methysergide therapy for headache. N Engl J Med. 1966;274:360-368.
161 Silberstein SD. Methysergide. Cephalalgia. 1998;18:421-435.
162 Raskin NH. Headache, 2nd ed. New York: Churchill-Living-stone, 1988.
163 Graham J. Cardiac and pulmonary fibrosis during methysergide therapy for headache. Am J Med Sci. 1967;254:1-12.
164 Bana DS, MacNeal PS, LeCompte PM, et al. Cardiac murmurs and endocardial fibrosis associated with methysergide therapy. Am Heart J. 1974;88:640-655.
165 Barlow CF. Headaches and Migraine in Children. Philadelphia: Lippincott, 1984.
166 Forsythe I, Hockaday JM. Management of childhood migraine. In: Hockaday JM, editor. Migraine in Childhood. London: Butterworths; 1988:63-74.
167 Smyth GA, Lazarus L. Suppression of growth hormone secretion by melatonin and cyproheptadine. J Clin Invest. 1974;54:116-121.
168 Peatfield R. Drugs acting by modification of serotonin function. Headache. 1986;26:129-131.
169 Capildeo R, Rose FC. Single-dose pizotifen, 1.5 mg nocte: a new approach in the prophylaxis of migraine. Headache. 1982;22:272-275.
170 Vogler BK, Pittler MH, Ernst E. Feverfew as a preventive treatment for migraine: a systematic review. Cephalalgia. 1998;18:704-708.
171 Schoenen J, Jacquy J, Lenaerts M. Effectiveness of high-dose riboflavin in migraine prophylaxis. A randomized controlled trial. Neurology. 1998;50:466-470.
172 Lipton RB, Gobel H, Wilks K, et al. Efficacy of petasites (an extract from petasites rhizome) 50 and 75 mg for prophylaxis of migraine: results of a randomized, double-blind, placebo-controlled study [Abstract]. Neurology. 2002;58:A472.
173 Sandor PS, diClemente L, Coppola G, et al. Coenzyme Q10 for migraine prophylaxis: a randomized controlled trial [Abstract]. Cephalalgia. 2003;23:577.
174 Schrader H, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with angiotensin converting enzyme inhibitor (lisinopril): randomized, placebo-controlled, crossover study. BMJ. 2001;322:19-22.
175 Tronvik E, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with an angiotensin II receptor blocker: a randomized controlled trial. JAMA. 2003;289:65-69.
176 Silberstein SD, Mathew N, Saper J, et al. Botulinum toxin type A as a migraine preventive treatment. Headache. 2000;40:445-450.
177 Silberstein SD, Lipton RB, Goadsby PJ. Migraine: diagnosis and treatment. In: Silberstein SD, Lipton RB, Goadsby PJ, editors. Headache in Clinical Practice. Oxford, UK: Isis Medical Media; 1998:61-90.
178 Silberstein SD. Preventive treatment of migraine: an overview. Cephalalgia. 1997;17:67-72.
179 Solomon GD. Management of the headache patient with medical illness. Clin J Pain. 1989;5:95-99.
180 Silberstein SD, Lipton RB, Breslau N. Migraine: association with personality characteristics and psychopathology. Cephalalgia. 1995;15:337-369.
181 Mathew NT, Saper JR, Silberstein SD, et al. Prophylaxis of migraine headaches with divalproex sodium. Arch Neurol. 1995;52:281-286.
182 Bowden CL, Brugger AM, Swann AC. Efficacy of divalproex vs lithium and placebo in the treatment of mania. JAMA. 1994;271:918-924.
183 Pascual J, Leira R, Lainez JM. Combined therapy for migraine prevention? Clinical experience with a beta-blocker plus sodium valproate in 52 resistant migraine patients. Cephalalgia. 2003;23:961-962.






