[level-membership-for-neurosurgery-category]
Chapter 65 Methods of Cranial Vault Reconstruction for Craniosynostosis
Cranial sutures are essential components in the development of the skull. Nonfunctional sutures during the evolution of the cranial vault and the skull base lead to evolving deformities that may end in neurologic sequel. Although the term craniosynostosis was first used by Bertolotti in 1914, referring to the premature closure of a cranial suture, it was Sommerring who described in 1791 the anatomy of the suture and postulated not only its role in normal skull growth but also the effects of early closure.50 During the 19th century Otto, and later Virchow, asserted that premature closure of sutures (craneostenosis) prevented growth perpendicular to the suture and was accompanied by compensatory growth at others. Premature closure may affect a single suture, but several sutures may be involved and severe deformities can develop, including the orbits and anterior fossa in the process. This is even more evident in craniofacial syndromes, where the main difference between single-suture and nonsyndromic craniosynostosis is the alterations not only in neurocranium but also in viscerocranium, resulting in anomalies in the midface skeleton.
The true incidence of craniosynostosis is not known, but it is estimated to occur in 1 of 2500 newborns. Sagittal synostosis is the most frequent (40%-60% of all cases), followed by metopic, coronal, and lambdoid, which is unusual.1,2 More than 150 associated syndromes have been described, but in most cases craniosynostosis is an isolated phenomenon. Several theories have been postulated to explain the appearance of premature synostosis and posterior deformities. Virchow was the first to suggest in 1852 that the premature closure of the suture was the primary event, while the vault deformity was a consequence of this closure.71 In 1959, Moss suggested that the deformity in the cranial base was the primary event. For this author, dura mater is an important regulator in the activity of the sutures and vault development and the main factor for the alteration in the cranial growth and suture dysfunction. The abnormal mechanical strengths driven through dural structures from certain maldeveloped points at the skull base (crista galli, petrous pyramids, or sphenoidal wings) would be responsible for cranial deformities and suture dysfunction.3 Park and Powers referred to mesenchymal abnormalities in bone structures, related to genetic anomalies, that would explain more thoroughly hypoplasia affecting the craniofacial region in syndromic cases.67,68,70
In the end, all of these mechanisms and changes produce a cosmetic deformity, as well as incompetence of the cranial and facial structures—unable to properly contain the organs inside the vault (brain and cerebellum) and the orbits (optic nerves and eyeballs)—and hypoplasia of the midface and oropharyngeal region. All of these factors are related, and depending on the affected region, there is a predominance of one abnormality. However, as explained later, in craniofacial syndromes the situation is more complicated. The presence of raised intracranial pressure (ICP) (multiple suture closure), hydrocephalus and cerebrospinal fluid (CSF) circulation anomalies, venous hypertension (skull base sutures closure affecting jugular foramina drainage, as well as genetic factors including endothelial proliferation in dural venous sinuses), chronic hypoxia and hypercapnia (obstructive airway in relation to midface retrusion and amygdalar hyperplasia), and chronic tonsillar herniation leads to a complex situation in which staged treatment has to be the rule, particularly after careful and proper understanding of the pathophysiology underlying these cases and always in a multidisciplinary team.
Scaphocephaly
Sagittal synostosis is the most common form of craniosynostosis. The premature closure of the sagittal synostosis has an estimated prevalence of 2 in 1000 births. Up to 80% of the cases are sporadic, and there is a male-to-female ratio of 3:1 to 4:1.1 Sporadic cases are mainly related to intrauterine constraint, but some may have a heterogeneous cause. In particular cases, a dominant autosomal inheritance has been described, with 38% penetrance.1 The frequency of twinning in a series was 4.8%, with only one monozygotic twin pair being concordant for sagittal synostosis.4
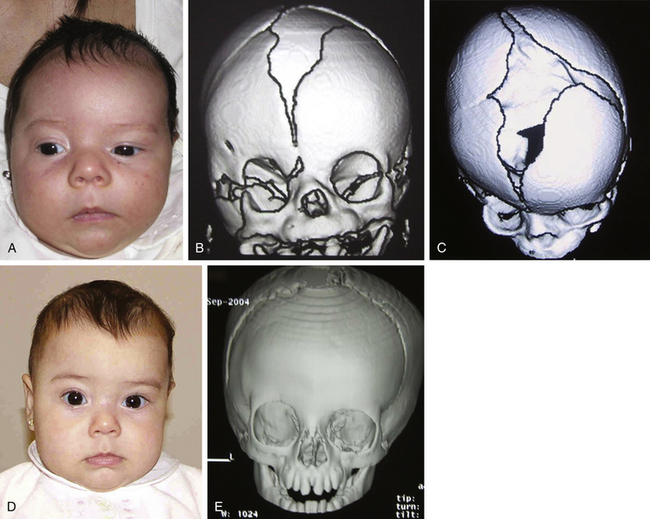
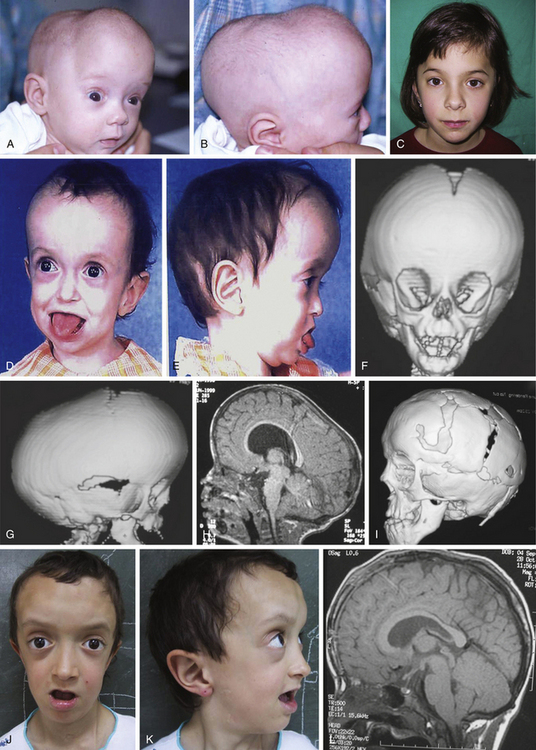
Histology usually shows synostotic ridging on the posterior part of the fused suture, which is consistent with the sagittal suture being thicker on the ectocranial cortex than on the endocranial side, and the bony edge of the suture becomes progressively thicker from anterior to posterior under normal conditions.5 When the sagittal suture is the only involved suture, the head becomes long from front to back (Figs. 65-1 to 65-3). It is narrow from side to side, and frontal compensational bossing may occur when the anterior fontanel is patent. When both fontanels are closed and the posterior part of the suture is predominantly affected, there is a posterior deformity typically referred to as an occipital knob. It is usually accompanied by a thin forehead with severe pterional indentation (leptoscaphocephaly). In the severest forms of scaphocephaly, the head adopts the shape of a saddle, and it is named bathrocephaly. Accurate diagnosis of isolated sagittal synostosis can be made clinically, and operative correction can proceed without a need for radiologic investigations, unless the clinical features are not completely typical.6 This could be the case in which Crouzon syndrome in some patients can resemble, early in evolution, an isolated scaphocephaly.
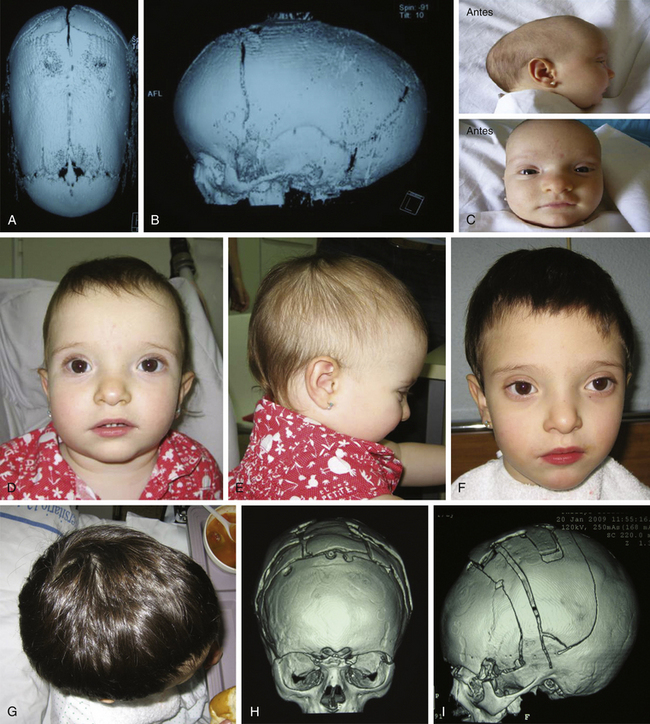
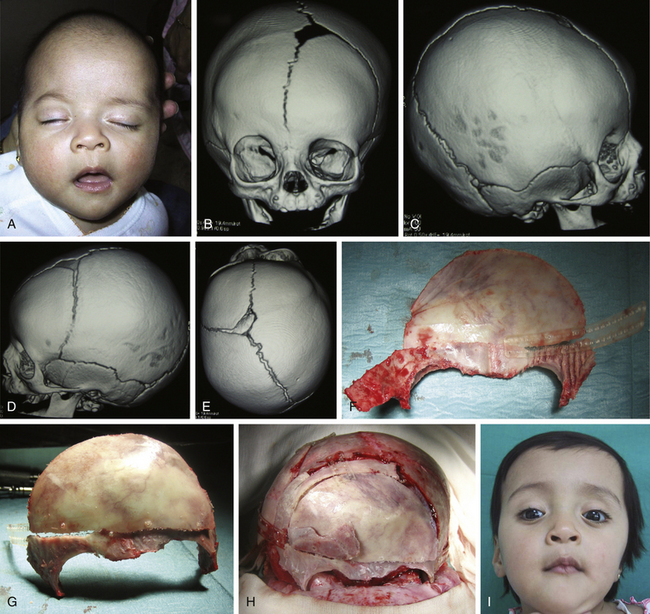
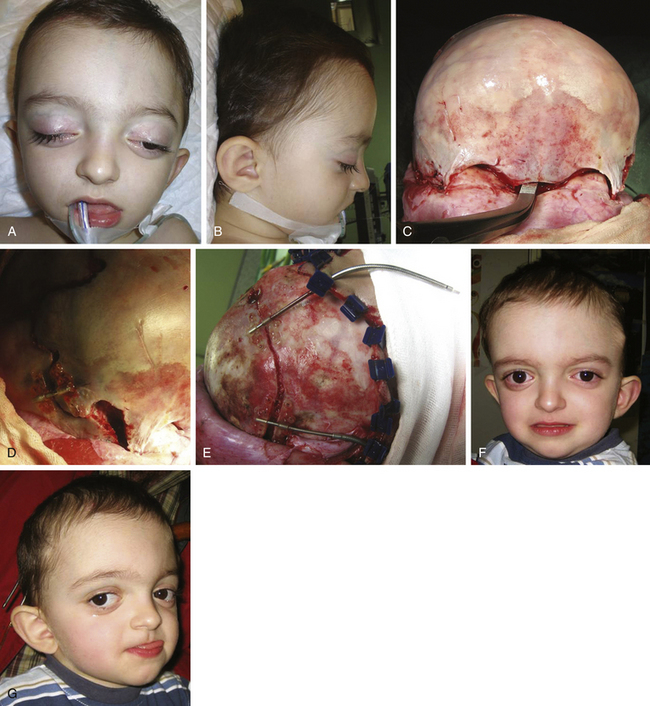
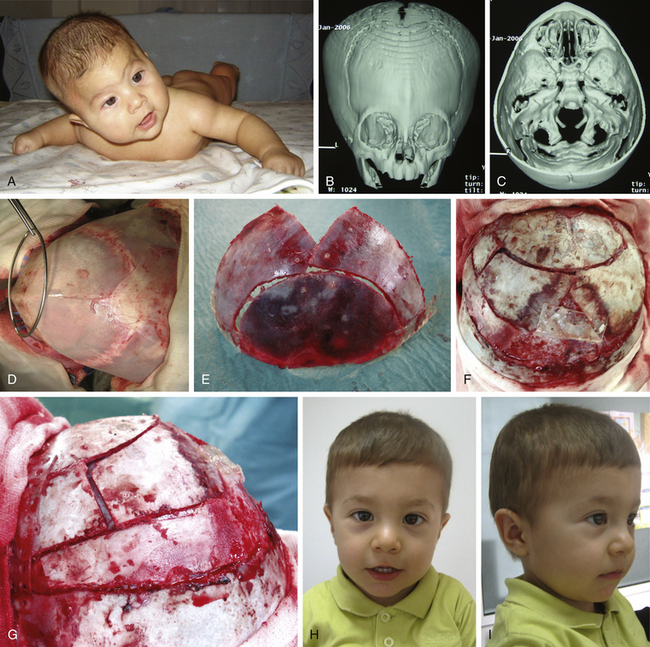
There is a plethora of techniques for the treatment of scaphocephaly7–11 but most of them include the excision of the sagittal suture with different kinds of parietal and occipital osteotomies with or without frontal osteotomies and remodeling. Surgical correction, whatever the technique, when performed early in infancy, results in satisfactory and persisting reshaping of the skull (Figs. 65-1 to 65-3). Delays in diagnosis, as well as treatment, result in a small subset of patients with uncorrected craniosynostosis that require the development of more extensive calvarial reconstruction techniques (Fig. 65-4). Due to calvarial maturity, these more advanced cases would not be amenable to treatment using less aggressive techniques. After 1 year of age the child’s cranial vault has developed abnormally, resulting in a severe skull base deformity, as shown in the figure. Subsequently, the skull is significantly thicker and less pliable to reshaping. At this age, cranial defects due to aggressive surgical remodeling procedures may not reossify, and there is a lack of the driving mechanism that leads to closure of the sutures. For these patients, an approach with a more comprehensive calvarial reconstruction includes multiple interleaving osteotomies crossing the midline, leading to improvements in biparietal narrowing combined with a bifrontal reconstruction.
Trigonocephaly
Trigonocephaly is considered a fairly common type of craniosynostosis. The term was first coined by Welcker in 1862.12 Formerly denoted as unusual,12,13 it is now considered to represent about 10% of all patients with deformities related to craniofacial centers12,14,15 and its frequency is increasing.2 Its incidence is estimated to be 1 in 2500 births,16 and there is a male predominance of 65% to 85%.14
Closure of the metopic suture starts under normal circumstances at the end of the first year and may last until the end of the second year.12 When premature suture closure occurs, usually before birth, a typical craniofacial malformation develops. The premature arrest of growth of the metopic suture may present as a spectrum of manifestations, depending on the timing and extent of the suture closure. Its mildest form is a familial and ethnically inherited facial morphology.17 Milder forms of metopic synostosis consist of only a somewhat prominent metopic ridge that does not need surgical intervention. In severe forms, a characteristic keel-shaped forehead can be observed, with absence of frontal eminences, retruded orbital rims, hypotelorism, and epicanthus giving a peculiar craniofacial appearance to these children. Different degrees of involvement of the anterior chondrocranial structures seem possible, such as presphenoid, mesoethmoid, and ectoethmoid structures.18 Another explanation for the broad phenotype of affected children could be a nonspecific process acting at different evolutive ages. Trigonocephaly may appear either as an isolated anomaly or as part of syndromes involving prosencephalic or rhinencephalic structures (holoprosencephaly), such as Opitz syndrome,19,20 Say-Meyer syndrome, or Frydman syndrome.12 Recently a new fronto-ocular syndrome has been described with trigonocephaly.21
Several genetic defects also have been described associated with trigonocephaly. Abnormalities including deletions and duplications of chromosomes 3p, 9p, 1, lq, and 13q have been published.22–25 Isolated trigonocephaly has an unknown etiology. Autosomal dominant inheritance with very low penetrance has been proposed in 2% to 5% of the cases for familial cases.15,26 The possible involvement of mutated FGFRs in newborns affected by trigonocephaly has recently been investigated by molecular screening in a search for mutations in FGFR2, which has been implicated in complex craniofacial syndromes. However, none of the cases studied carried mutations, except one that evolved later toward a Crouzon syndrome–like profile.25
Mental delay may occur in isolated trigonocephaly and has been observed in as many as 10% of these children.12 Development delays may consist of subtle changes in the time of acquisition of head control or sitting position or include more severe forms of mental retardation, with or without intracranial hypertension.
Di Rocco et al. proposed two subgroups on the basis of clinical and radiologic findings.18 Group I presents with bilateral frontal bone hypoplasia associated with extreme retrusion of the supraorbital margins. In this group, hypotelorism is associated with abnormally deep position of the cribriform plate, giving the ethmoidal region a hollow appearance. In these patients, the nasion–pterional angle is severely restricted and the nasion–clinoidal distance significantly increased. Group II also shows bilateral frontal hypoplasia with hypotelorism, supraorbital retrusion, and a reduced nasopterional angle. However, the nasion–clinoidal distance is almost normal, and pterional evidence is scarcely evident.18 Moreover, patients in group II showed a lesser degree of temporal compensatory expansion. The authors postulate that in some patients a compensatory elongation of the nasion–clinoidal distance and an incomplete synostosis of the frontoethmoidal sutures allows partial lateral expansion of the anterior cranial fossa, which could diminish the need for posterior calvarial expansion. On the other hand, children with more severe involvement of the nasoethmoidal sutures, resulting in diminished lateral expansion of the anterior fossa, need compensatory changes in the temporal and parietal regions. Patients in group II accomplished good correction of associated hypotelorism, whereas patients in group I did not reach normal interorbital values when treated with the same surgical procedure, which did not include specific treatment of hypotelorism. Other authors have addressed the importance of the moment and degree of involvement of pathologic changes in the anterior chondrocranial structures.14,27–31 Milder forms of trigonocephaly would affect only the upper metopic suture, whereas more severe forms include involvement of presphenoid, mesoethmoid, and ectoethmoid structures.
Surgical Technique
Before starting osteotomies, the obliquity of frontal bones, retrusion of the lateral portions of the orbital bandeau, and pterional indentation are assessed. A bifrontal craniotomy is performed after a supraorbital bandeau is created. The marks are extended from the sphenosquamosal suture to its contralateral equivalent prepared for a tongue-in-groove advancement.18 The bifrontal flap includes the anterior part of the bregmatic fontanelle and both coronal sutures. The anterior fossa is exposed extradurally with visualization of the anterior two thirds of the orbital roof.
There is not yet a consensus on hypotelorism correction. Several papers have addressed the importance of associated hypotelorism17,28 and the need for direct surgical approach. Different authors17,27,28,36,37 propose adding a nasofrontal osteotomy and an interpositional bone graft to the supraorbital bar and nasoethmoidal area to correct hypotelorism simultaneously with lateral orbitary wall expansion17 or three quarter orbital wall osteotomies.27 On the other hand, there are others13,34 who do not advocate direct approach to the hypotelorism in the belief that internasalis grafting widens only nasal bones without increasing interorbital width, as far as osteotomies usually remain anterior and superior to nasoethmoidal complex.36 I recommend a standard approach without grafting and thus consider the latter ineffective and unnecessary. Both techniques have achieved statistically significant improvement in hypotelorism, greater than that expected from normal growth curves. However, undercorrection of hypotelorism and persistence of abnormally low interorbitary distances occur frequently when orbital widening is not addressed.18,27,35
Anterior Plagiocephaly
Premature closure of a single coronal suture produces the most complex set of craniofacial deformities due to the peculiarities of the fronto-orbital region and the close relationship between the sutures of the anterior cranial base and the orbits and maxillary complex. The incidence of coronal craniosynostosis has been reported to be variable but can be approximated to be 0.4 per 1000.40 Although most cases are sporadic, a familiar incidence of up to 8% in uncomplicated coronal synostosis has been reported.40
Frontal or anterior synostotic plagiocephaly has been described in association with unilateral coronal synostosis,41–44 but it is known to occur with synostosis of other parts of the coronal ring, such as the frontosphenoidal or frontoethmoidal sutures.41,45–47 In some rare cases, it has been reported to be the result of fusion of the frontosphenoidal48 or the frontozygomatic sutures alone.49 The deformity is manifest as a progressive cranial, orbital, and facial asymmetry of varying severity, with secondary deformities, considered to be compensatory, existing on the contralateral side (Figs. 65-5 and 65-6). Associated cranial anomalies affect the development of all facial bones and have been referred to as “the most complete presentation of craniofacial asymmetry.”50 There is a flat and retruded frontal bone in the affected side with a contralateral bossing. The orbital rim on the synostotic side is also elevated and retruded, while the ipsilateral orbit is elevated and twisted. There is a variable degree of vertical orbital dystopia. The nasal root is deviated toward the closed sutured with an oblique nasal axis. The tip of the nose points to the nonsynostotic side. The zygoma and maxilla are hypoplastic on the affected side, and the ipsilateral temporal bone is malpositioned in an anterior and descended configuration.
Different reconstructive techniques have been described,42,43,46,47,50–53 but most of them can be divided between unilateral and bilateral approaches. There is a long-standing controversy on the timing and type of surgical approach. Some authors have advocated early surgery (younger than 6 months),40,54 whereas others support treating these patients later (between 9 and 10 months).41,45,51 The former group uses the concept of rapid brain growth leading to advancement of the frontal bones and supraorbital rims.
Proponents of late surgery argue that delayed surgery allows for further maturation and growth of the craniofacial skeleton. There is less reliance on brain growth, and blood loss is better tolerated. Also, there is immediate correction of the deformity. Surgical procedures have expanded from simple suturectomies (Fig. 65-5) to frontal–calvarial vault remodeling consisting of bifrontal craniotomes and orbital frontal bandeau advancement (Fig. 65-6). Aggressive surgical approaches have been supported by the theory of “coronal ring” synostosis, which states that there is an extension of the coronal synostosis into the cranial base.55,56 However, in most cases of isolated, nonsyndromic, unilateral coronal synostosis, the sphenofrontal, sphenoethmoidal, ethmoidofrontal, and pterional sutures are patent. Furthermore, review of the literature of these patients who have undergone more extensive procedures indicate that the results are inconsistent and mixed. Often, there is restriction of forward anterior skull base growth and advancement, and late correction (older than 9 months) fails to improve vertical dystopia, which persists into adulthood.40 Most craniofacial centers use the classic fronto-orbital advancement with unilateral or bilateral correction. Undoubtedly, in cases with severe contralateral compensation (frontal and/or temporal bossing), a bilateral correction is preferred. The correction of orbital dystopia warrants careful preoperative evaluation. Orbital hypoplasia is a consequence of the elevated orbital roofs, frontal development of the greater sphenoidal wings, and descent of the ethmoidal position, secondary to early closure of the coronal sutures. Orbital axes are also altered and point downward and outward. In the severest cases, failure to achieve early treatment or undercorrection may lead to strabismus, amblyopia, and anomalies in the binocular vision.
Multiple Suture Craniosynostosis
Brachycephaly
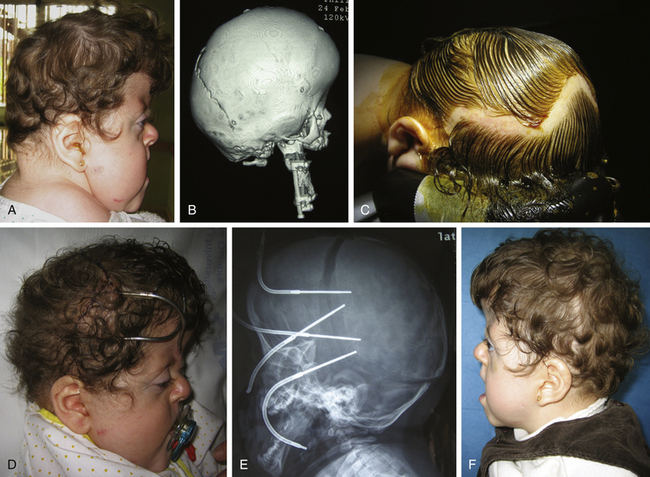
Brachycephaly is most commonly the result of premature synostosis of both coronal sutures. The anterior fossa adopts a characteristic deformation, and there are retruded frontal bones and orbital rims with a vertical, broad, flat forehead and a high bregmatic point. Brachycephaly may appear as an isolated synostosis and has then a favorable prognosis. Often, it is the common final appearance of different syndromic craniosynostoses that share a premature closure of bicoronal sutures and similar phenotype (e.g., some cases of Saehtre-Chotzen or Pfeiffer type I). For this reason, every patient with brachycephaly should be assessed by a clinical geneticist (Figs. 65-7 and 65-8).
Crouzon Syndrome
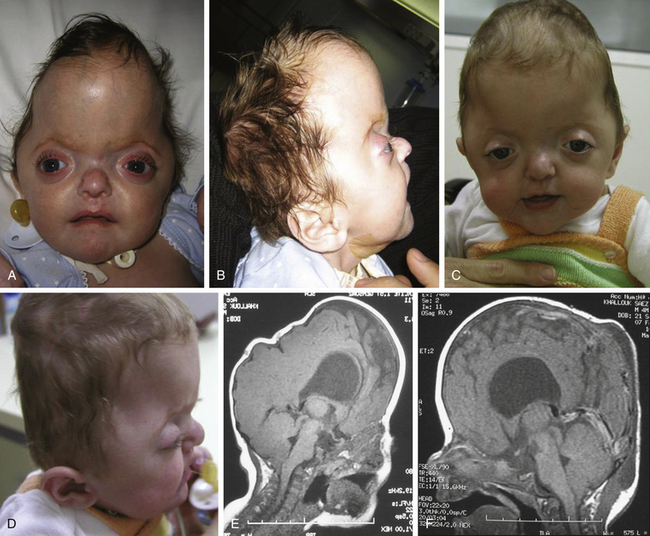
Crouzon syndrome is an autosomal dominant disorder characterized by craniosynostosis that causes secondary alterations of the facial bones and facial structure. Common features include hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism. First described by Crouzon in 1912,59 it was not until 1959 that Shiller observed an autosomal dominant transmission.60 Crouzon syndrome represents approximately 4.8% of cases of craniosynostosis at birth, and the birth prevalence is estimated to be 16.5 per million births. Crouzon craniofacial dysostosis is linked to a high number of different mutations, but most of them are located on IgIII of FGFR2 (exons 7 and 9) on chromosome 10q. An association between Crouzon syndrome and acanthosis nigricans has been described and is related to an A391E mutation in the FGFR3 gene on chromosome 4p.
Crouzon syndrome is characterized by a premature synostosis of both coronal sutures, with a resultant brachycephalic shape of the skull. Sagittal, metopic, or lambdoid sutures may also be prematurely affected, isolated, or combined (Figs. 65-9 and 65-10). The cranial base and upper facial sutures are involved with a variable degree of midface hypoplasia and malocclusion. The orbits are hypoplastic, and the orbital floor is hollow, resulting in proptosis and additional orbital dystopia that may produce mild to moderate orbital hypertelorism and divergent strabismus. Maxillary hypoplasia results in pseudoprognatism. Nasal septum deviation, together with maxillary hypoplasia, may produce a chronic obstruction to respiratory flow and may be associated with choanal atresia, velopharyngeal incompetence, and relative macroglossia. All of these malformations may lead to severe respiratory obstructions and apneas, which may cause a chronic raised ICP by an increase in venous pressure after hypercapnia and even contribute as an etiopathogenic mechanism in the development of hydrocephalus.
The initial treatment for Crouzon syndrome generally requires cranio-orbital decompression, including bicoronal suture release and osteotomies of the anterior cranial vault and upper orbits with reshaping and advancement. They are usually performed by the age of 8 to 11 months unless signs of increased ICP are found earlier. Sometimes, it is necessary to perform a combined approach, including midface advancement.61 When Chiari malformation (CM) is identified precociously, an occipital–parietal calvarial decompression may be preferable to achieve a bigger expansion of the intracranial volume, with or without suboccipital decompression.62 After proper release of the ICP, fronto-orbital remodeling can be achieved via an anterior approach.
Apert Syndrome
Apert syndrome, also known as acrocephalosyndactyly type I, is a congenital disorder characterized by multiple craniosynostoses, facial hypoplasia, and osseous syndactyly of the hands and feet. Approximately 1 in 65,000 to 165,000 of live births is affected. It is usually classified among a group of craniofacial syndromes with Crouzon, Pfeiffer, and Saehtre-Chotzen syndromes, all of which are allelic disorders with similar clinic manifestations and common genetic background. Wheaton first noted the coincidence of craniosynostosis and syndactyly (1894), but it was Apert who fully characterized the syndrome in 1906.63,64 Apert syndrome consists of a craniosynostosis in the shape of an acrocephaly or a brachycephaly due to the premature closure of several cranial sutures, typically coronal sutures and later those of the anterior cranial base and posterior fossa. The sagittal suture is typically widened and opened. It is always accompanied by osseous syndactyly of hands and feet and by fusion of the distal phalanxes. There is usually severe hypoplasia of the midface with an ogival-fissured hard palate. The orbital rims are retruded and elevated, and the skin possesses a characteristic acneiform appearance, mostly over the nasal bridge, shoulders, and back. Midface anomalies and anterior fossa craniosynostoses produce a decrease in the orbital volume that may lead to proptosis, strabismus (V syndrome), hypermetropia, or astigmatism. Most of these patients present with central nervous system anomalies, including hypoplasia of the corpus callosum and limbic and mesial temporal structure malformations. Approximately 10% of these children develop hydrocephalus, but only 2% of them suffer from a CM, in contrast to Crouzon syndrome, where 75% of the patients develop a hindbrain herniation.61 If left untreated, the incidence of raised ICP has previously been reported as 45%.64,65 Untreated intracranial hypertension may result in insidious optic atrophy, visual loss, and possible developmental delay.
Early cranial decompression with occipital expansion or fronto-orbital advancement is the treatment of choice, although some authors have advocated avoiding routine vault expansion in the first year of life. Instead, careful clinical, ophthalmologic, and respiratory monitoring would allow raised ICP to be treated in the most appropriate manner only when it occurs.65 Midface hypoplasia accounts for the exorbitism, strabismus, and respiratory difficulties seen in this condition. In the absence of any clinical sequelae from this hypoplasia, midface advancement is usually postponed until an age of 4 to 6 years. In cases of functional or clinical compromise, it may be necessary to intervene earlier with different techniques, such as monobloc advancement, maxillotomies, and/or mandible distraction. Facial hypoplasia and ogival palate abnormalities are responsible for phonetic disorders, abnormal dental eruption, and malocclusion.
Apert syndrome patients present with a variable degree of mental retardation. Early treatment of the craniosynostosis has been related to better outcomes. Some authors have shown that up to 50% of patients with Apert syndrome have a normal or near-normal intelligence quotient (IQ), with the rest being moderately to severely retarded. Renier et al.72 found an IQ greater than 70 in 50% of Apert syndrome patients when they were treated by cranial expansion in the first year of life but only in 7.1% of those who were treated later.
Pfeiffer Syndrome
Initially described by Pfeiffer in 1964,67,75 Pfeiffer syndrome is characterized by turribrachycephaly, maxillary hypoplasia, and antimongoloid slant of the orbits. There is hypertelorism and a marked degree of proptosis due to the bicoronal synostosis and subsequent recession of supraorbital rim and short anterior fossa. Extremities are notable for broad thumbs and large toes. There may be a variable degree and number of soft tissue syndactyly (most commonly between the second and the third digits), in comparison to Apert syndrome, where bony syndactyly is the hallmark. There may also be symphalangism (phalangeal fusion), ankylosis of the elbow joints, and cervical vertebral fusions (all of them also possible in Apert syndrome).
There are three clinical subtypes of Pfeiffer syndrome,73 which have clinical and prognostic significance. Type I (classic) is a lesser craniosynostosis that has a typical appearance of brachycephaly but usually is associated with normal or near-normal intelligence. Children with this type of Pfeiffer syndrome can develop normally, but there is often increased ICP if the synostoses are left uncorrected. Type II is associated with cloverleaf deformity, and although compatible with life, prognosis is poor. Type III is characterized by striking proptosis, but kleeblattschädel is absent. Long-term prognosis is poor.
Cloverleaf Skull (Kleeblattschädel)
Kleeblattschädel syndrome is characterized by a trilobar skull caused by frontal and bitemporal bossing. The first description of a trilobar cranial malformation in a newborn was made by Vrolik in 1849,77 but the term kleeblattschädel syndrome was not introduced until 1960 by Holtermuller and Wiederman.77,78 These authors reported a series of children with a trilobar or “cloverleaf” cranium associated with multiple craniosynostoses. In 1965, Commings et al. published the first case in the United States and translated the term kleeblattschädel syndrome into cloverleaf skull.78 This malformation has been described in children with Crouzon, Apert, Carpenter, Beare-Stevenson, or type II Pfeiffer syndrome, the latter being most frequently associated (up to 20% in some series).75,77 The etiology of this syndrome is still unknown and has been attributed to abnormalities of both the calvarium and the skull base, probably due to the presence of a combination of prematurely fused cranial sutures and hydrocephalus.
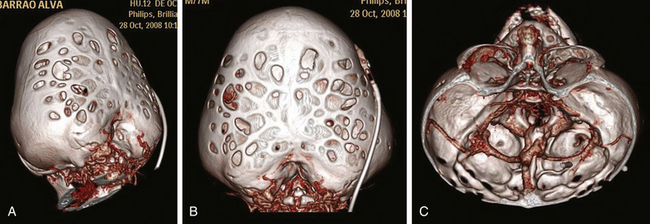
The surgical management of cloverleaf skull remains one of the most formidable challenges for the craniofacial surgeon (Fig. 65-8). The natural history of this deformity without surgery was well described in the 1960s and 1970s. Just a few of these children survived beyond infancy, and those who did developed severe neurologic deficits. The first attempt at surgical correction was reported by Angle in 1967.77 Since then, a number of surgical series have been reported, without a clear conclusion on prognosis and outcome. Review of the literature reveals a lack of consistent surgical strategies.74–76 Postoperative data are sparse, and reoperation is the rule in these patients, with a consequent increased morbidity. In addition to the significant cosmetic deformity, the constant coexistence of intracranial hypertension, hydrocephalus, hindbrain herniation, severe skull base dysplasia, and anomaly in venous drainage (Fig. 65-11) imposes important neurosurgical considerations throughout treatment.
Hydrocephalus in Craniosynostosis
Ventriculomegaly and ventricular asymmetry are frequent even in children with single-suture craniosynostosis, but they require treatment only in selected cases. Abnormal dilatations of the subarachnoid spaces are a common finding and may imply some disturbance in CSF absorption.77 In sagittal synostosis, for example, the frequent encasing of the superior sagittal sinus in the bone groove formed by the fused suture may account for some impairment of CSF absorption. This may lead to mild ventricular dilatation and enlarged CSF subarachnoid spaces over the cerebral hemispheres. In comparison, hydrocephalus is a common feature in patients with complex craniosynostosis. Ventricular dilatation has been reported in 30% to 70% of patients with Crouzon and Pfeiffer syndromes and in 40% to 90% of Apert syndrome patients.66,77,78 With Apert syndrome, most cases of enlarged ventricles remain stable without a shunt, but with Crouzon syndrome and the severest Pfeiffer syndrome cases, shunting is frequently necessary at some stage of treatment. True hydrocephalus has seldom been reported in single-suture pathologies and can almost always be attributed to coincidental disorders independent of craniosynostosis, such as ventricular hemorrhage, meningitis, aqueductal stenosis, or neural tube defects.
Some patients develop rapidly evolving hydrocephalus before any surgical intervention, and this is expected mainly in kleeblattschädel or a severe Pfeiffer condition. In most of the remaining patients, ventricular dilatation only develops following decompressive surgery for craniosynostosis. In these patients, the indication for shunting is mainly based on progressive ventricular dilatation or evidence of persistent intracranial hypertension, which may need to be ascertained by direct pressure monitoring. After cranial surgery, the artificially created spaces are quite often accommodated by some enlargement of the intra- and extracerebral CSF spaces, and the possibility of a compensated or even slowly progressive hydrocephalic state should be taken into account.77,79
Two combined pathogenic factors have been offered as an explanation for the frequent association of hydrocephalus and complex craniosynostosis: a mechanical increase of the CSF outflow resistance due to constricted growth of the posterior fossa62,80–82 and an impaired CSF absorption resulting from venous outflow obstruction.82–84 Crowding of the posterior fossa appears to be an acquired disorder secondary to deficient occipital cranial expansion.79,85 It has been related to the timing of fusion of the lambdoid suture, which in Crouzon and Pfeiffer syndromes is completed at an earlier age than in Apert syndrome. This would explain the delayed appearance of hydrocephalus in patients with Apert syndrome. The mechanical restriction to CSF outflow would worsen after the development of tonsillar herniation in a small posterior fossa. This theory alone fails to explain the incidence of hydrocephalus, because hindbrain herniation is missing in a number of cases of progressive hydrocephalus.83,86 And while it is present in many other cases not affected by hydrocephalus, posterior fossa decompression has often failed to sufficiently restore normal CSF circulation.80 Therefore, constriction of the posterior fossa may not be a single causative mechanism for hydrocephalus in craniosynostosis.
Venous hypertension has been accepted as another major pathogenic mechanism by several authors, with descriptions of venous sinus hypertension caused by a stenosis of the jugular foramen in hydrocephalic Crouzon syndrome patients.84 The venous obstructions could be secondary to the abnormalities of bone growth that affect the base of the skull in children who have craniosynostosis, with early closure of lambdoid and petro-occipital sutures leading to decreased venous outflow through the jugular foramina.79 Another possible explanation is that the disordered venous anatomy is primary rather than secondary and results from the same dysplastic processes that affect the cranial vault sutures, the basicranium, and the facial skeleton. The expression of FGFR gene products in the infant craniosynostotic sutures has been described; these products also have been localized by immunohistochemistry to the cranial vascular endothelia of patients with syndromic craniosynostosis.79,86,87 Therefore, premature endothelial proliferation and subsequent differentiation in the sigmoid and jugular sinuses may result in the narrowed lumen.86 Most patients with progressive hydrocephalus simultaneously exhibit signs of venous outflow obstruction and crowded posterior fossa, favoring a combined action of both mechanisms77 by assuming that venous hypertension causes a CSF absorption deficit, as well as brain swelling resulting in tonsillar herniation,82 or that it aggravates the preexistent cephalocranial disproportion by venous engorgement.79,81–83
Finally, the contribution of upper airway obstruction to raised ICP has long been recognized; in particular, an increase in ICP during episodes of respiratory obstruction has been documented, as has an improvement in the ophthalmologic signs of increased ICP after nasal airway dilation, nocturnal positive airway pressure, or maxillofacial advancement procedures. Possible underlying mechanisms include carbon dioxide retention during obstructive episodes in nocturnal apneas and cerebral flow changes during active sleep, particularly if they occur in the presence of reduced cerebral compliance.82,86,88 All of these factors would lead in the end to a venous hypertension that would ultimately interfere with the CSF absorption.
A percentage of craniosynostosis patients goes on to require hydrocephalus treatment.79 Improvement of ventricular dilatation has been anecdotally reported following removal of constricting bony ridges or decompressive craniectomies alone.81,86 Some authors (Genitori, personal communication) have discussed the use of endoscopic third ventriculostomy for selected cases with the rationale that an acquired stenosis of the aqueduct may develop due to periaqueductal compression during progression of the multiple synostoses. In the presence of hindbrain herniation, there is certainly no place for lumbar peritoneal shunts. Therefore, despite other proposed treatment options, ventriculoperitoneal shunting is widely accepted as the most effective treatment for these patients. Overdrainage must be avoided when selecting the shunting system, because it may induce a pseudo–tumor-like state of venous origin, worsening the preexisting venous problem.86
When shunting is needed soon after cranial reconstruction, the stability of synostosis surgery may be endangered if the dural envelope does not rapidly expand because of artificial depletion of CSF spaces86 and failure of cranial contents to support the bone plates. If shunting is addressed before cranial reconstruction, there is a real possibility of developing new suture closure due to sustained low ICP.
There is little evidence that dilated ventricles per se have an adverse effect on intelligence,72,77 except that severe congenital hydrocephalus, as observed in the most complex craniofacial syndromes, carries an increased risk of a lower performance level. As in other hydrocephalic states, the prognosis mainly depends on coincidental cerebral abnormalities and on the detrimental effect of long-standing elevated CSF pressure.77
Chiari Malformation in Craniosynostosis
The association between CM and premature craniofacial synostosis was first noted by Saldino et al.89 in 1972 in one patient with Pfeiffer syndrome. Since then, numerous authors have reported the incidence of chronic tonsillar herniation in multiple suture craniosynostosis,62,81,90–92 and it is a frequent finding in syndromic and multisuture craniosynostosis, characterized by early fusion of lambdoid sutures and cranial base synchondroses. The incidence of CM has been reported as high as 70% in Crouzon syndrome,90,92,94 75% in nonsyndromic oxycephaly,95 50% in Pfeiffer syndrome, and 100% in kleeblattschädel deformity.62 CM was found in other types of syndromic craniosynostosis, in some cases of nonsyndromic complex craniosynostosis involving the lambdoid suture, and in some rare cases of scaphocephaly.85,92
Downward herniation of neural tissue through the foramen magnum is usually an acquired malformation in craniosynostosis and may be secondary to a disproportion between the posterior fossa and the growing hindbrain structures.96,97 In most cases of craniosynostosis, hindbrain herniation is not present at birth; rather, it develops in response to the changes in the skull base and posterior fossa secondary to premature closure of the lambdoid and cranial base sutures (usually between 3 and 6 months of age), supporting the pathogenetic hypothesis of overcrowding of the posterior fossa secondary to premature suture fusion.90,97 The excellent review by Cinalli et al. gives a comprehensive overview on the pathogenic mechanisms involved in the development of hindbrain herniation in craniosynostosis.97 The progressive fusion of the lambdoid suture produces alteration in the skull base and stenosis of the jugular foramina (if the petro-occipital synchondroses are primarily involved). The first result would be a small posterior fossa, with consequent herniation of the cerebellum into the cervical canal during the phase of rapid neural growth in the first months of life. The second result would be venous hypertension, induced both by jugular foramen stenosis (if the petro-occipital synchondroses are involved) and by crowding of the posterior fossa, with consequent compression of the sigmoid sinus. These factors can obstruct and alter CSF circulation at the level of the posterior fossa and impair CSF reabsorption at the level of the arachnoid granulations, with the overall final result of increased CSF outflow resistance. Severe crowding of the foramen magnum may result not only in hindbrain herniation but also in brain stem compression and deformation of the fourth ventricle. Thus, hindbrain herniation can be considered as a condition creating or aggravating a hydrocephalic state, not a consequence of hydrocephalus. This explains the cases of CM without hydrocephalus that are frequently observed in craniosynostosis without primary involvement of skull base synchondrosis (e.g., oxycephaly) or in the first stages of Crouzon syndrome, where CM is a frequent finding without hydrocephalus.
Under normal conditions, the posterior cranial fossa grows in length in early childhood along the intraoccipital, petro-occipital, and spheno-occipital synchondroses. Growth in the intraoccipital synchondrosis ceases in early childhood, whereas growth in the spheno-occipital synchondrosis continues after puberty.98 In syndromic and complex multisutural craniosynostosis, unlike monosutural craniosynostosis, the facial skeleton and the cartilaginous cranial base are primarily involved.98 In Crouzon and Apert syndromes, the degree of involvement of the skull base synchondroses and the timing of fusion are different. In Apert syndrome, the spheno-occipital, petro-occipital, and occipital synchondrosis are fused later, beginning after 12 to 48 months of life and ending at the age of 4 years, than in Crouzon syndrome, where they can be completely fused in the first year of life. All this could be conditioned by a different genetic pattern. In syndromic craniosynostosis, the genetic mutations responsible for the disease are located mainly in FGFR1 (Pfeiffer syndrome), FGFR2 (Crouzon, Apert, and Pfeiffer syndromes), and FGFR3 (Crouzon syndrome and acanthosis nigricans). Some authors have suggested a correlation between the mutation observed and the presence or absence of CM.83,85 In Crouzon syndrome, the patients affected by CM and syringomyelia would present with a variety of mutations that spread over exons IIIa and IIIc of the FGFR2 gene.92,98 This could explain the significant differences found in the final anatomy of the skull base and the posterior cranial fossa. The Apert basiocciput is larger than normal, whereas in Crouzon syndrome it is smaller.99 According to Cinalli et al., Crouzon syndrome patients preferentially expand along a superoinferior axis, whereas little or no growth is allowed along an anteroposterior axis97; the foramen and the basion–opisthion area incur only small changes, whereas the more significant alterations are in the cranial base posterior to the foramen.99 Previous conditions would result in altered dimensions of the posterior cranial fossa: normal or larger than normal in the Apert syndrome99 and smaller, shortened in the anteroposterior axis, and elongated in the superoinferior and lateral axis in Crouzon syndrome.
If the petro-occipital synchondroses are involved, their premature closure may lead to stenosis or atresia of the jugular foramen. Venous hypertension, induced by jugular foramen stenosis,100 increases the sagittal sinus pressure and results in a higher CSF pressure request to maintain CSF balance. In patients with closed sutures, ICP may rise to very high levels, overcoming the high sagittal sinus pressure and permitting absorption of CSF, with normal-sized or small ventricles, as seen in some cases of pseudotumor cerebri.83,84 In contrast, in infants and children with open sutures (or following surgical cranial suture release), increased CSF pressure induces progressive head enlargement and dilatation of the ventricles and subarachnoid spaces.83,84 This is usually followed by the development of collateral venous drainage, through the foramen magnum and/or through emissary veins and scalp veins. This venous pattern highlights a crucial point: In some cases, the main venous drainage occurs through emissary veins rather than through the usual jugular pathway, and the distortion of the previous could lead to a fatal outcome after surgery.101 Enography should form part of the routine magnetic resonance imaging (MRI) protocol for diagnostic assessment of children with complex and syndromic craniosynostosis.100 Quantification of venous hemodynamics may also have a role, allowing more accurate, noninvasive monitoring of change at follow-up studies.102
The rest of the sutures (lambdoid, sagittal, coronal, and metopic) could also play a role in the development of CM. In the setting of in utero closure of sagittal and coronal sutures, a cephalocranial disproportion in the supratentorial compartment occurs early, forcing the neural growth to be directed posteriorly and inferiorly and pushing down the tentorium. This can induce a lower attachment of the tentorium (near the foramen magnum), reducing the size of the posterior fossa and increasing the risk of CM, especially if premature lambdoid synostosis is also involved. The precocity of coronal and sagittal suture synostosis does not seem to play a role in the pathophysiology of CM in Apert (where the sagittal suture is wide open) and Crouzon syndromes. The premature closure of the lambdoid suture reflects the primary closure of the spheno-occipital synchondrosis and could be a reliable radiologic indicator of synostosis of the posterior cranial base sutures. In Crouzon syndrome, the sagittal and lambdoid sutures close early (median 6 and 21 months, respectively), and they close significantly earlier in the cases of Crouzon syndrome associated with CM compared to in Crouzon syndrome patients without CM.90 On the contrary, in Apert syndrome, where CM only occurs in approximately 2% to 5% of the cases, the cranial vault synostosis occurs early for the coronal suture (median 5 months) and significantly later for the sagittal and lambdoid sutures (median 51 and 60 months, respectively).
More than one third of patients with hindbrain herniation become symptomatic for CM or develop syringomyelic cavities. Usually, the onset of symptoms occurs late in life, but they may be dramatic—especially in very young children—with appearance of respiratory problems such as central apnea, bilateral vocal cord paralysis, bulbar palsy, ventilatory control abnormalities, persistent cyanosis, and breath-holding spells.97 Careful radiologic and clinical follow-up is needed in patients with syndromic or complex craniosynostosis to assess the presence and evolution of CM. MRI with venous angiography is the gold standard for the evaluation of these patients.100,103
Management of CM in Craniosynostosis
In cases of multiple suture synostosis—mainly in patients with a Crouzon syndrome or kleeblattschädel deformity, where cranial vault (bicoronal and both lambdoid) and skull base sutures are affected—the correction of a CM may need to be treated by a complete calvarial reconstruction. This may be performed as a one-step procedure, as described by Pollack et al.104 The child is set in the “modified prone position,” allowing exposure from the orbital ridge to the foramen magnum. This approach, however, requires marked hyperextension of the neck, and it is contraindicated for patients who present with anomalies of the craniovertebral junction, because hyperextension of the neck for several hours104 may lead to prolonged severe compression on the spinal cord and medulla. I have used the same approach (described later in the section on type X holocranial dismantling) but in two steps: first fronto-orbital advancement with the child supine, and then, after closure of the skin incisions, occipital expansion with the patient prone in the same surgical session.
For patients with severe syndromes and marked flattening of the posterior vault and overcrowding of the posterior fossa, occipital expansion and suboccipital craniectomy may be indicated.62 It is possible to perform occipital craniectomy and remodeling through a bicoronal incision. Dissection proceeds in the subperiosteal plane, elevating the occipital musculature over the external protuberance and exposing all suboccipital bone that can be removed and then reaching the posterior and lateral margins of the foramen magnum. The infratentorial compartment is left uncovered, and the occipitoparietal region is conformed, repositioning the bone. In cases such as cloverleaf malformation, a parietal craniectomy can only be performed piecemeal, and extreme care must be exercised when dissecting free the thin external layer of the superior sagittal sinus, which is often imbibed or pinched by bone spikes that enter the sinus.75,76,94 In these cases, it is often not possible to perform an intradural approach, but even if some degree of tonsillar herniation remains after suboccipital craniectomy, crowding of the foramen magnum is reduced and more CSF can be observed around the medulla on postoperative MRI. Dural opening and cervical laminectomy may be indicated in cases of severe compression of the medulla, but this carries the risk of severe bleeding from the dural edges because of the abnormal anatomy of dural venous sinuses, which usually includes a prominent occipital longitudinal sinus over the suboccipital bone and a dominant marginal occipital sinus around the foramen magnum and frequently includes significant collateral circulation in the muscular plane. Computed tomography (CT) and/or magnetic resonance angiograms/venograms are mandatory studies in these patients to avoid catastrophic introperative bleeding.
Surgical Techniques
Type I. Endoscopically Assisted Suturectomies and Osteotomies
Different authors have proposed minimally invasive techniques106 and endoscopic-assisted osteotomies9,11,29,40,106 applied to younger patients using the concept of surgically created open sutures that take advantage of the rapid brain growth during the first months of life. Craniectomies and osteotomies (Figs. 65-1, 65-3, 65-5, and 65-12) are designed to mimic new cranial sutures and are followed by active head molding using custom-made helmets. These techniques are used for the treatment of single-suture premature synostosis, although good results have been reported when they are applied to multiple suture synostosis and syndromic involvement.136 Small skin incisions (between 1.5 and 2 cm for anterior plagiocephaly and 3.5 and 4 cm for scaphocephaly) are placed over the suture to be resected.
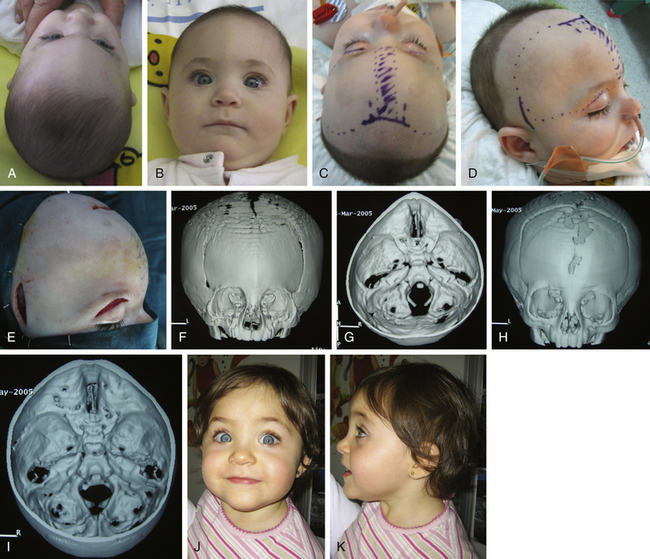
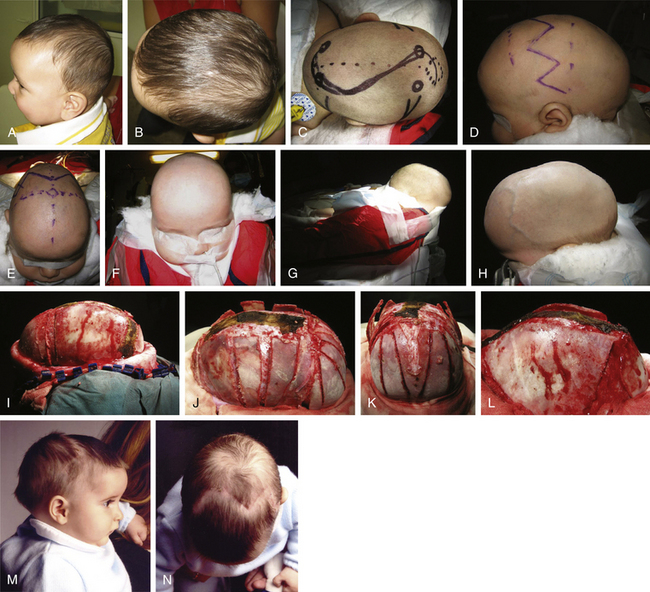
In scaphocephaly (Fig. 65-3), the anterior incision is placed 2 cm behind the anterior fontanel, while a posterior incision is over the posterior fontanel. A solution with 0.25% bupivacaine and 1:100,000 epinephrine is injected over the area of dissection. The two small transverse incisions (approximately 2-3 cm length) are lengthened, and a subgaleal plane is developed over the pericranium with the assistance of a rigid endoscope (lens 0 degrees, 2.7 mm) and with electrocautery at a low setting. Bur holes at both sides of the sagittal suture are fashioned at each incision site. The dura is dissected, and bur holes are connected with epidural rongeuring. The sagittal sinus is then separated from the skull using the endoscope, suction, and a Penfield dissector. This moment is particularly helpful in the direct vision of the emissary veins, which can be coagulated with bipolar cautery. Bur holes are longitudinally connected with bone scissors to complete a sagittal suturectomy measuring approximately 3 to 4 cm in width and between 6 and 11 cm in length. The sagittal bone plate is then freed and can be removed. Hemostasis is performed over the bone cuts with tip-insulated electrocautery and with oxidized cellulose and bipolar cautery over the superior sagittal sinus and dura mater. Further dissection is carried out under endoscopic assistance laterally over the parietal areas to achieve wedge osteotomies on both sides, immediately posterior to the coronal suture and immediately anterior to the lambdoid suture, reaching the squamosal sutures. Hemostasis is performed, and resorbable sutures are used for closure. No bandages are needed, and the patient is positioned in a strict supine position during the postoperative stay in the hospital.
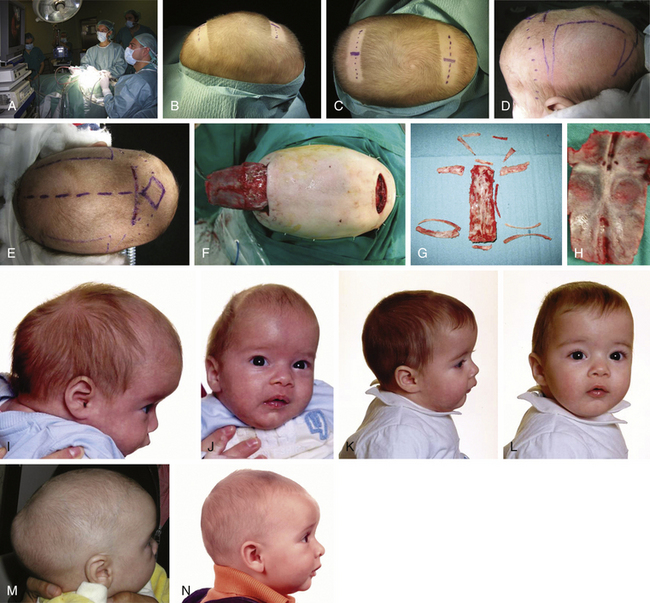
For trigonocephaly (Fig. 65-4), the patient is placed supine on a horseshoe headrest. A 2- to 3-cm transverse incision is made just behind the hair line, approximately 3 cm in front of the anterior fontanelle. A subgaleal dissection over the metopic ridge is carried down to the nasion and frontonasal suture with endoscopic assistance and electrocautery. Two bur holes are placed along both sides of the metopic suture at the level of the skin incision and connected crossing the midline. Metopic suturectomy starts posterior to the bregmatic fontanelle. Then, with the aid of the rigid endoscope, epidural dissection under the fused metopic suture proceeds down to the nasofrontal suture. Suturectomy is completed with rongeurs or heavy bone scissors. At this point, some help with a high-speed drill may be needed to cross the thick glabella and may be used for smoothing the frontal bone edges. In some cases, I added frontal oblique-releasing osteotomies. For the severest cases, I advocated a variation of the technique previously published by Cohen et al.107 The ultimate point of this approach is to achieve a complete fronto-orbitary osteotomy, including both orbital roofs and resection of the entire pterional region, together with metopic suturectomy.
For anterior plagiocephaly (Fig. 65-5), the patient is positioned supine in a neutral position. A 2.5-cm incision is placed halfway along the coronal suture, closer to the bregmatic fontanelle than to the ipsilateral pterion. A dissection plane is carried between the galea and the pericranium with electrocautery, exposing the whole affected coronal suture under endoscopic vision with a rigid lens (0 or 30 degrees). A bur hole is created under the skin incision, and a 1- to 2-cm broad suturectomy is done with rongeurs or bone scissors up to the anterior fontanelle. Then, epidural dissection proceeds downward under endoscopic magnification, exposing the pterion and lesser wing of the sphenoid. Suturectomy is carried out behind the pterional region until the squamosal suture is reached. The osteotomy must be extended down to the pterion and allow the orbit to move forward.9,40,108 Most patients undergoing endoscopic techniques are later sent for helmet molding therapy.
Type II. Scaphocephaly: Standard Technique
The standard scaphocephaly technique is applied in cases of scaphocephaly in children between 5 and 9 months of age, regardless of the presence of severe compensating frontal bossing (Fig. 65-2). It consists of a sagittal suturectomy with expanding green-stick parietotemporal and occipital osteotomies. The patient is placed in a beanbag horseshoe with the head extended in prone position. A modified retroauricular bicoronal incision (zigzag) is usually preferred for all craniofacial procedures for cosmetic purposes. This approach promotes healing without resulting in wide scars. A subperiosteal dissection separates the calvaria from the coronal sutures to the occipital knob. After sagittal suturectomy, I proceed with biparietal and temporo-occipital wedge osteotomies, expanding the biparietal diameter. In cases of severe occipital prominence, an “island” occipital craniectomy helps correct the posterior deformity (Fig. 65-2L).
Type III. Scaphocephaly: Standard Technique Plus Frontal Remodeling
In children older than 10 months with scaphocephaly and severe anterior bulging, a two thirds anterior cranial vault reconstruction is achieved by obtaining a new frontal bone from a parietal donor site and transposition or remodeling of the former frontal bone by midline split and repositioning. At the same time, bitemporal and parietal osteotomies and subsequent fixation are conducted to expand laterally the pterional indentation.
Type IV. Scaphocephaly: Total Cranial Vault Remodeling (Holocranial Dismantling)
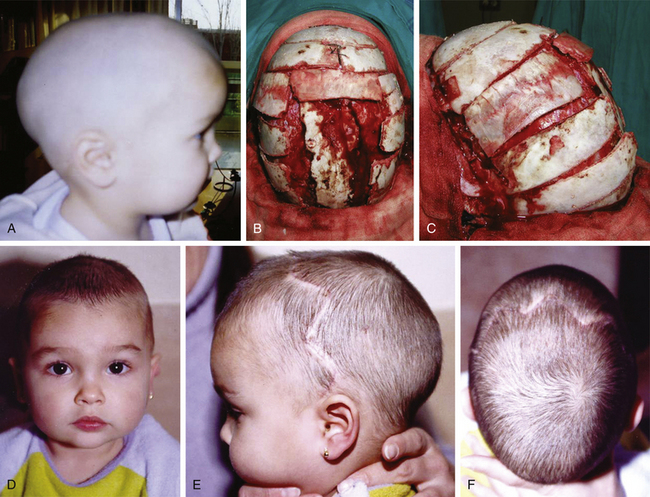
In children older than 12 to 14 months with scaphocephaly, a complete calvarial reconstruction is necessary to achieve an adequate cosmetic result (Fig. 65-4). Severe cranial base distortion and the necessity to change the three diameters (vertical, posterior–anterior, and biparietal) precludes the use of more aggressive techniques that include the dismantling and transposition of multiple bone fragments in the shape of arches (clamshell technique), crosses (holocranial dismantling), or plates and posterior fixation with resorbable material.
Type V. Trigonocephaly: Frontal Remodeling without Fronto-Orbital Bandeau
For patients with trigonocephaly (Fig. 65-13), I routinely use a technique that has been previously reported from my department.30,31 With the patient in a supine position and through the standard modified retroauricular bicoronal incision, the anterior two thirds of the calvaria; temporal region; both supraorbital margins, including the anterior roof of the orbita; and nasion are exposed subgaleally. A very low bilateral fronto-orbital craniotomy is performed, reaching the frontonasal junction, and pterional regions are rongeured away. Both supraorbital eminences and glabella are then drilled and contoured. Finally, a donor parietal bone is selected with the Marchac template, transposed and fixed with resorbable material.
Type VI. Anterior Plagiocephaly/Coronal Suture Synostosis: Frontal Remodeling without Fronto-Orbital Bandeau
Type VII. Anterior Plagiocephaly/Coronal Suture Synostosis: Frontal Bilateral Remodeling with Fronto-Orbital Bandeau
Frontal bilateral remodeling with fronto-orbital bandeau is the technique that I select for most cases of anterior plagiocephaly, where vertical orbital dystopia, short and oblique anterior fossa, and compensating bossing in the contralateral side are consistent findings (Fig. 65-6).
Type VIII. Occipital Remodeling: Occipital Advancement
Single synostosis of the lambdoid suture seldom happens, and surgical indication must be done only in cases of severe cosmetic deformation. In these cases, I performed a bilateral occipital craniotomy with complete posterior remodeling. In younger patients, an endoscopically assisted resection followed by helmet therapy may render good results.11
Posterior occipital advancement, however, is frequently used as the starting procedure for severe craniofacial syndromes, such as Pfeiffer syndrome, Crouzon syndrome, or kleeblattschädel with early elevated ICP when a vault expansion is needed (Fig. 65-7), or in cases of CM associated with multiple craniosynostoses when a single stage procedure for complete calvarial remodeling is not advisable due to elevated venous pressure.62 Occipital advancement has been shown to be superior to fronto-orbital advancement in terms of total volume expansion62,109 and has shown efficacy in correcting the fronto-orbital appearance, even prior to anterior advancement (Fig. 65-8).
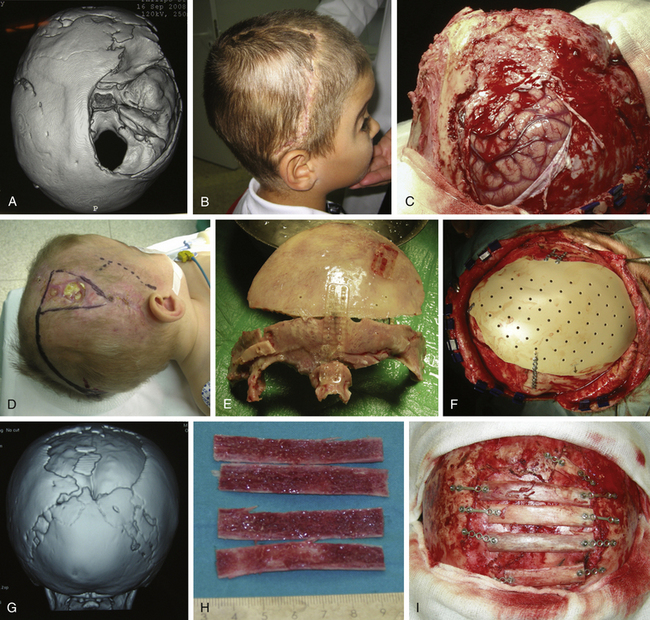
Type X. Holocranial Dismantling (Total Vault Remodeling) in Multiple Craniosynostosis
Holocranial dismantling in multiple craniosynostosis consists of a complete cranial vault remodeling for correction of multiple synostosis to achieve adjustment of the three skull diameters (vertical, interparietal, and posterior–anterior distances) (Fig. 65-10). It can be done in a single surgical step with the patient in a prone position (Persing’s technique)110 or in two steps in the same surgical session (first supine and then prone after closing the bicoronal incision). The two-step variation, commonly used in my department, is preferred over Persing’s technique when venous hypertension, induced both by jugular foramen stenosis and crowding of the posterior fossa with subsequent compression of the sigmoid sinus, makes the hyperextended prone position unadvisable.
With the patient in a supine position, standard bilateral fronto-orbital advancement and frontoparietal remodeling are performed (type IX). A crossbar is designed over the bregmatic region, which allows decrease of the vertical diameter (Fig. 65-10C). After positioning the patient prone, occipital remodeling and advancement are completed (type VIII) (Fig. 65-8). These constitute an extensive procedure with higher morbidity rates that must be reserved for the complex cases in which multiple sutures are involved and early expansion of the whole cranial vault is needed.
Type XI. Fronto-Orbital Advancement by Distraction
The standard fronto-orbital advancement procedure requires elevation of both frontal and orbital bones (Fig. 65-9). This can result in partially resorbed advanced bone and convoluted dura mater that is herniated through the inner table of the skull. If a second operation is performed via the intracranial approach, the dura may be easily torn during calvarectomy and the bone that is advanced can be further resorbed and deformed. Distraction osteogenesis has been used in fronto-orbital advancement for patients with coronal synostosis (nonsyndromic brachycephaly)111 or for those patients with a craniofacial syndrome in whom fronto-orbital advancement is needed at any moment. The main advantage of this technique is the possibility to avoid cranial vault defects, because separation between the dura mater and the bone flap is not necessary and therefore deterioration of the dura mater and resorption of the advanced fronto-orbital bone are minimized.111,112 The concomitant expansion of the scalp prevents postoperative relapse and provides the advancement of soft tissues, thus helping in stability and the final postoperative result. In syndromic patients, “harmonic” Crouzon or Pfeiffer cases (those who present without severe frontal bossing or orbital dystopia) are best candidates. A small but nondisfigured fronto-orbital region may be advanced to avoid intracranial hypertension caused by closure of the anterior cranial fossa and to diminish typical proptosis. The whole fronto-orbital console is advanced to the new position without a change in the shape of the fronto-orbital bandeau. Apert patients usually present with severe retrusion of the supraorbital bar and a large lacunae over the metopic region, which makes them worse candidates for this technique. However, my colleagues and I use it in these patients when preliminary fronto-orbital advancement is needed and a second surgery is planned over the anterior region of the skull. This technique helps us prevent iatrogenic lacunae (Fig. 65-14) and pseudomeningoceles that occur through the standard osteotomies.
Among its disadvantages are the necessity for a second surgery to remove devices and the inability to reshape deformed fronto-orbital and temporosquamousal bones. For example, the surgeon could not simultaneously reduce an increased bitemporal distance or lower a turricephaly as with the standard techniques. In addition, fronto-orbital advancement by distraction has the disadvantage of technical difficulty in the osteotomy at the anterior cranial base (orbital roof). When the surgical technique is not exquisite, orbital meningoceles resulting from intracranial hypertension have been described.94,103
Recurrences
As a rule, any craniosynostosis may recur, even after proper treatment, during development. This is particularly true in cases of multiple synostosis and craniofacial syndromes, where the dysplastic processes that affect the cranial vault sutures, the basicranium, and the facial skeleton by the expression of FGFR gene products remain as an underlying condition. This is also a consequence of the cranial base, which remains unchanged despite treatment, because most surgical procedures address the cranial vault but have little effect on the skull base. Recurrences consist mainly of a progressive vault deformation, but the presence of an evolving fracture or the appearance of a new hydrocephalic condition should alert the surgical team to the possibility of unresolved craniosynostosis. Secondary deformations and the appearance or progression of a new suture closure may also indicate overdrainage and a pseudotumor state after shunting in a craniosynostosis patient with hydrocephalus.
Prognosis
Prognosis in craniofacial surgery depends mostly on the underlying condition. The majority of simple craniosynostosis patients with a single affected suture do not develop mental retardation, and surgical treatment on a proper schedule and with the right technique by an experienced team renders good results in terms of cosmetic and functional outcome. Although it has been stressed that the occurrence of raised ICP in single-suture synostosis is low,109,110,113 up to 15% to 20% of patients with single-suture craniosynostosis have a documented increase in ICP.86 This is particularly true in patients with severe trigonocephaly,117 and special attention must be paid to these children. The possibility of elevated ICP is higher as the number of affected sutures increases. The risk is very high in untreated patients with a craniofacial syndrome where craniosynostosis is an evolving condition that worsens during the first years of life and frequently leads to intracranial hypertension. Restricted skull volume contributes to this elevation in ICP, but it is not the only factor responsible. Venous congestion (sometimes with anomalous venous drainage), hydrocephalus, and/or upper airway obstruction are sometimes associated in these patients and are capable of increasing ICP.
Early detection of intracranial hypertension is important to reduce the risks for brain development and visual function. However, in children with craniosynostosis, the clinical manifestations of abnormally increased ICP are difficult to detect, as the majority of patients may have neither warning signs nor symptoms for a long period.86 As a result, these children warrant extensive clinical surveillance.
Complications
Mortality rates in craniofacial surgery literature are fortunately very low.118,119 The most frequent complication in surgical series is postoperative hyperthermia (13.17%), possibly related to more extensive tissue dissection and a higher amount of blood component transfusion, followed by cranial infection (8.2%).99 In the series of my department, infection was below 10%.119–122 We also encountered subgaleal hematomas (6.0%), dural tears (5.1%), and CSF leaks (2.7%) with significantly lower incidence than that given in prior reports.120
Several issues have been related to infectious complications: type of craniosynostosis,119 age and surgical timing,120 number of surgeons and intensive care unit time,123 and proportion of surgical reoperations.121 My department found two important factors related not to just infections but to all complications: (1) surgical reoperations and (2) type of surgical procedure.
Surgical Reoperations
The rate of relapse in the series of my department (12.8%) is similar to that of other authors.124,125 In a pattern reminiscent of previous reports,121 my department found that of the 8.2% rate of total infection for craniofacial surgery patients, 62.5% belonged to the group that underwent reoperations. There is an even stronger apparent relationship with dural tears (5.1%), of which 93% took place at the time of reintervention. Adhesions within the cleavage plane, mostly in the extradural space, make surgery more difficult and may predispose to a patient complications, including dural tears. The use of resorbable material may promote adherences in the epidural space, making reintervention more difficult and increasing the possibility of dural tears.
Type of Surgical Procedure
Fronto-orbital distraction by internal devices and combined fronto-orbital and midface advancement126–129 are techniques most often associated with complications. Recently, Pelo et al.130 performed an exhaustive literature review of 130 cases and added 8 personal patients. Complications included local infection, device breakage, and CSF leakage, though the patient’s clinical condition is not mentioned in this paper. In the series of my department, complications were frequent in patients under distraction.94,103 In the 26 patients treated, there were eight CSF leaks and three dural tears, as well as eight local infections around the distracting devices. There was only one case of device breakage. This relatively high morbidity may be related to the role of distraction as a “rescue technique” in cases of relapses after the first standard fronto-orbital advancement. Dissection of the pterional and anterior fossa region is not easy without elevation of the frontal bones, and the possibility for dural tears is higher among reoperated patients, where multiple previous adherences and resorbing miniplates make dissection more hazardous. Leptomeningeal cysts,131 encephalocele,94 and growing fractures132 have also been described.
Cinalli G., Chumas P., Arnaud E., et al. Occipital remodeling and suboccipital decompression in severe craniosynostosis associated with tonsillar herniation. Neurosurg. 1998;42:66-73.
Cinalli G., Sainte-Rose C.H., Kollar E.M., et al. Hydrocephalus and craniosynostosis. J Neurosurg. 1998;88:209-214.
Cinalli G., Spennato P., Sainte-Rose C., et al. Chiari malformation in craniosynostosis. Childs Nerv Syst. 2005;21:889-901.
Cohen M. Pfeiffer syndrome: update, clinical subtype and guidelines for differential diagnosis. Am J Gen. 1993;45:300-307.
Collman H., Sörensen N., Krauß J. Hydrocephalus in craniosynostosis: a review. Childs Nerv Syst. 2005;21:902-912.
Di Rocco C., Velardi F., Ferrario A., Marchese E. Metopic synostosis: in favour of a “simplified” surgical treatment. Child’s Nerv Syst. 1996;12:654-663.
Esparza J., Hinojosa J. Complications in the surgical treatment of craniosynostosis and craniofacial syndromes: a propos of 306 transcranial procedures. Child’s Nerv Syst. 2008;24:1421-1430.
Goodrich J.T. Skull base growth in craniosynostosis. Child’s Nervs Syst. 2005;21:871-879.
Hayward R. Venous hypertension and craniosynostosis. Child’s Nervs Syst. 2005;21:880-888.
Hinojosa J., Esparza E., Muñoz M.J. Endoscopic-assisted osteotomies for the treatment of craniosynostosis. Child’s Nerv Syst. 2007;23:1421-1430.
Hirabayashi S., Sugawara Y., Sakurai A., et al. Fronto-orbital advancement by distraction: the latest modification. Ann Plast Surg. 2002;49:447-451.
Jiménez D.F., Barone C.M., Cartwright C.C., Baker L. Early management of craniosynostosis using endoscopic-assisted strip craniectomies and cranial othrotic molding therapy. Pediatrics. 2002;110:97-104.
Marucci D.D., Dunaway D.J., Jones B.M., Hayward R.D. Raised intracranial pressure in Apert syndrome. Plast and Reconstruct Surg. 2008;122:1162-1168.
McCarthy J.G., Glasberg S.B., Cutting C.B., et al. Twenty-year experience with early surgery for craniosynostosis: II. The craniofacial synostosis syndromes and pansynostosis—results and unsolved problems. Plast Reconstr Surg. 1995;96(2):284-295.
Pollack I.F., Losken H.W., Hurwitz D.J. A combined frontoorbital and occipital advancement technique for use in total calvarial reconstruction. J Neurosurg. 1996;84:424-429.
Posnick J.C., Ruiz R.L. The craniofacial dysostosis syndromes: current surgical thinking and future directions. Cleft Palate Craniof J 37. 2000;434(5):1-24.
Renier D., Arnaud E., Cinalli G., et al. Prognosis for mental function in Apert’s syndrome. J Neurosurg. 1996;85:66-72.
Renier D., Cinalli G., Lajeunie E., et al. L’oxycéphalie, une craniosténose sévère. A propos d’une série de 129 cas. Arch Pediatr. 1997;4:722-729.
Sgouros S., Goldin J.H., Hockley A.D., Wake M.J.C. Posterior skull surgery in craniosynostosis. Child’s Nerv Syst. 1996;12:727-733.
Sun P.P., Persing J.A. Craniosynostosis. In: Albright A.L., Pollack I.F., Adelson P.D. Operative Techniques in Pediatric Neurosurgery. New York: Thieme; 2001:51-64.
Tamburrini G., Caldarelli M., Massimi L., et al. Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Childs Nerv Syst. 2005;21:913-921.
Taylor W.J., Hayward R.D., Lasjaunias P., et al. Enigma of raised intracranial pressure in patients with complex craniosynostosis: the role of abnormal intracranial venous drainage. J Neurosurg. 2001;94:377-385.
Thompson D.N., Harkness W., Jones B.M., Hayward R.D. Aetiology of herniation of the hindbrain in craniosynostosis. Pediatr Neurosurg. 1997;26:288-295.
1. Cohen M.M. Epidemiology of craniosynostosis. In: Michael Cohen M.Jr., MacLean Ruth E. Craniosynostosis: Diagnosis, Evaluation and Management. Oxford University Press, 2000.
2. Van der Meulen J., Arnaud E., Hinojosa J., et al. The increase of metopic synostosis, a pan-European observation. J Craniofac Surg. 2009;20(2):283-286.
3. Moss M.L. The pathogenesis of premature cranial synostosis in man. Acta Anat. 1959;37:351-370.
4. Lajeunie E., Le Merrer M., Bonäiti-Pellie C., et al. Renier: Genetic study of scaphocephaly. Am J Med Genet. 1988;62:282-285.
5. Albright A.L., Byrd R.P. Suture pathology in craniosynostosis. J Neurosurg. 1981;54:384-387.
6. Agrawal D., Steinbok P., Cochrane D.D. Diagnosis of isolated sagittal synostosis: are radiographic studies necessary? Childs Nerv Syst. 2006;22:375-378.
7. Albright A.L., Towbin R.B., Shultz B.L. Long-term outcome after sagittal synostosis operations. Pediatr Neurosurg. 1996;25:78-82.
8. Hudgins R.J., Burstein F.D., Boydston W.R. Total calvarial reconstruction for sagittal synostosis in older infants and children. J Neurosurg. 1993;78:199-204.
9. Jiménez D.F., Barone C.M., Cartwright C.C., Baker L. Early management of craniosynostosis using endoscopic-assisted strip craniectomies and cranial othrotic molding therapy. Pediatrics. 2002;110:97-104.
10. Sun P.P., Persing J.A. Craniosynostosis. In: Albright A.L., Pollack I.F., Adelson P.D. Operative Techniques in Pediatric Neurosurgery. New York: Thieme; 2001:51-64.
11. Hinojosa J., Esparza E., Muñoz M.J. Endoscopic-assisted osteotomies for the treatment of craniosynostosis. Child’s Nerv Syst. 2007;23:1421-1430.
12. Collman H., Sörensen N., Krauß J. Consensus: trigonocephaly. Child’s Nerv Syst. 1996;12:664-668.
13. Fearon J.A., Kolar J.C., Munro J.R. Trigonocephaly-associated hypotelorism: is treatment necessary? Plast Reconstr Surg. 1996;97:503-509.
14. Genitori L., Caveiheiro, Lena G., et al. Skull base in trigonocephaly. Pediatr Neurosurg. 1992;17:175-181.
15. Lajeunie E., Le Merrer M., Renier D. Genetics of craniosynostosis: update 1994. Rome: Consensus Conference on Craniosynostosis; 1995.
16. Lorenzini M., Mazza C., Barisoni D. Frontal Vault and Orbital Remodelling in Trigonocephaly. Rome: Consensus Conference on Craniosynostosis; 1995.
17. Goodrich J.T., Craig D.H. Surgical management of trigonocephaly. Techniques in Neurosurgery. 1997;Vol. 3(no 3):190-197.
18. Di Rocco C., Velardi F., Ferrario A., Marchese E. Metopic synostosis: in favour of a “simplified” surgical treatment. Child’s Nerv Syst. 1996;12:654-663.
19. Opitz J.M., Johnson R.C., McCreadie S.R., Smith D.W. The C syndrome of multiple congenital anomalies. Birth Defects. 1969;5:161-166.
20. Weber P., Kuwertz-Broking E., Majewski F., et al. Retinitis pigmentosa, terminal renal insufficiency and Caroli syndrome: new associations with Opitz trigonocephaly syndrome. Klin Padiatr. 2000;212(3):1-4.
21. Schneider E.N., Bogdanow A., Goodrich J.T., et al. Frontoocular syndrome: newly recognized trigonocephaly syndrome. Am J Genet. 2000;17:89-93.
22. Obregon M.G., Mingarelli R., Diglio M.C., et al. Deletion 1 1q2397qter (Jacobsen syndrome). Report of three new patients. Ann Genet. 1992;35:208-212.
23. Preus M., Vekemans M., Kaplan P. Diagnosis of chromosome 3 duplication q23→quter, deletion p25→pter in a patient with the C (trigonocephaly) syndrome. Am J Med Genet. 1986;23:935-943.
24. Teebi A.S., Gibson L., McGrath J., et al. Molecular and cytogenetic characterization of 9p abnormalities. Am J Med Genet. 1993;46:288-292.
25. Tartaglia M., Bordoni V., Velardi F., et al. Fibroblast growth factor receptor mutational screening in newborns affected by metopic synostosis. Child’s Nerv Syst. 1999;15(3):89-394.
26. Hennekam R.C., Van den Boogard M.J. Autosomal dominant craniosynostosis of the sutura metopica. Clin Genet. 1990;38:374-377.
27. Posnick J.C., Lin K.Y., Chen P., Armstrong D. Metopic synostosis: quantitative assessment of presenting deformity and surgical results based on CT scans. Plast Reconstr Surg. 1994;93:16-24.
28. Sadove A., Kalsbeck J.E., Eppley B.L., Javed T. Modifications in surgical correction of trigonocephaly. Plast Reconstr Surg. 1990;85:853.
29. Hinojosa J., Esparza J., García-Recuero I., Romance A. Remodelación fronto-orbitaria endoscópicamente asistida en la trigonocefalia. Cir Pediatr. 2007;20:33-38.
30. Muñoz M.J., Esparza J., Hinojosa J., et al. Fronto-orbital remodeling without orbito-naso-frontal bandeau. Child’s Nerv Syst. 2003;19:353-358.
31. Hinojosa J., Esparza J., Muñoz M.J., et al. Surgical treatment of trigonocephalies and associated hypoteleorbitism. Neurocirugía. 13, 2003. 447–45
32. Czorny A., Forlodou P., Stricker M., Ricbourg B. Les cranes triangulaires. A propos de 87 cas de trigonocéphalies. Neurochirurgie. 1994;40:209-221.
33. David D.J., Posswillo D., Simpson D. Trigonocephaly. In: In: The Craniosynostosis. Causes, Natural History and Management. Springer-Verlag; 1992:133-140.
34. Hansmann C.F. Growth of interorbital distance and skull thickness as observed in roentgenographic measurements. Radiology. 1968;86:87.
35. Havlik R.J., Azurin D.J., Bartlett S.P., Whitaker L.A. Analysis and treatment of severe trigonocephaly. Plast Reconstr Surg. 1999;103(2):381-390.
36. Longaker M.T., Posnick I.C., Rekate H.L. Symposium: craniosynostosis and skull molding. J Craniofac Surg. 1998;9(6):572-600.
37. Friede H., Alberius P., Lilja J., Lauritzen C. Trigonocephaly: clinical and cephalometric assessment of craniofacial morphology in operated and nontreated patients. Cleft Palate J. 1990;27:362-367.
38. McCarthy J.G., Bradley J.P., Longaker M.T. Step expansion of the frontal bar: correction of trigonocephaly. J Craniofac Surg. 1996;7:333-335.
39. Waitzman A.A., Posnick J.C., Armstrong D.C., Pron G.E. Craniofacial skeletal measurements based on computed tomography: normal values and growth trends. Cleft palate Craniofac J. 1992;29:118.
40. Barone C.M., Jiménez D.F. Endoscopic approach to coronal craniosynostosis. Clin Plastic Surg. 2004;31:415-422.
41. Bartlett S.P., Whitaker L.A., Marchac D. The operative treatment of isolated craniofacial dysostosis (plagiocephaly): a comparison of the unilateral and bilateral techniques. Plast Reconstr Surg. 1990;85:677-683.
42. Cohen S.R., Kawamoto K.H., Burstein F., Peacock W.J. Advancement onlay: an improved technique of fronto-orbital remodeling in craniosynostosis. Child’s Nerv Syst. 1991;7:264-271.
43. Posnik J.C. Unilateral coronal synostosis (anterior plagiocephaly): current clinical perspectives. Ann Plast Surg. 1996;12:430-447.
44. Genitori L., Zanon N., Denis D., et al. The skull base in plagiocephaly. Child’s Nerv Syst. 1994;10:217-223.
45. Hoffman H.J., Mohr G. Lateral canthal advancement of the supraorbital margin. A new corrective technique in the treatment of coronal synostosis. J Neurosurg. 1976;45:376-381.
46. Jane J.A., Park T.S., Zide B.M., et al. Alternative techniques in the treatment of unilateral coronal synostosis. J Neurosurg. 1984;61:550-556.
47. Mühlbauer W., Anderl H., Schmidt A., et al. Asymmetrical cranioorbital facial stenosis. Ann Plast Surg. 1991;26:45-51.
48. Francel P.C., Park T.S., Marsch J.L., Kaufman B.A. Frontal plagiocephaly secondary to synostosis of the frontosphenoidal suture. J Neurosurg. 1995;83:733-736.
49. Currarino G. Premature closure of the frontozygomatic suture: unusual frontoorbital dysplasia mimicking unilateral coronal synostosis. AJNR. 1985;6:643-646.
50. Esparza J., Muñoz M.J., Hinojosa J., et al. Operative treatment of the anterior synostotic plagiocephaly. Analysis of 45 cases. Child’s Nervous System. 1998;14:448-454.
51. Hoffman H.J. Procedure of lateral canthal advancement for the treatment of coronal synostosis. Child’s Nerv Syst. 1996;12:678-682.
52. Persing J.A., Jane J.A., Park T.S. Floating C-shaped orbital osteotomy for orbital rim advancement in craniosynostosis: preliminary report. J Neurosurg. 1990;72:22-26.
53. Whitaker L.A., Bartlett S.P., Shut L., Bruce D. Craniosynostosis: an analysis of the timing, treatment and complications in 164 consecutive patients. Plast Reconstr Surg. 1987;80:195-206.
54. Marchac D. Craniosynostosis: an analysis of the timing treatment and complications in 164 consecutive patients [discussion]. Plast Recontr Surg. 1987;20:207-212.
55. Bertelsen T.I. The premature synostosis of the cranial sutures. Acta Opthalmologica (Suppl 51). 1958:1-176.
56. Seeger J.F., Gabrielson T.O. Premature closure of the frontosphenoidal suture. Radiology. 1971;101:631-635.
57. Di Rocco C., Velardi F. Nosographic identification and classification of plagiocephaly. Child’s Nerv Syst. 1988;4:9-15.
58. Marchac D., Renier D., Broumand S. Timing of treatment for craniosynostosis and faciocraniosynostosis: a 20 years experience. Br J Plast Surg. 1994;47:211-222.
59. Crouzon O. Dysostose cranio-faciale hereditaire. Paris: Bull Mem Soc Med Hop; 1912;33:. 545-555
60. Shiller J.G. Craniofacial dysostosis of Crouzon: a case report and pedigree with emphasis on heredity. Pediatrics. 1959;23:107-112.
61. Meling A.C., Due-Tonnessen B.J., Hogevold H.E., et al. Monobloc distraction osteogenesis in pediatric patients with severe syndromal craniosynostosis. J Craniofac Surg. 2004;15(6):990-1000.
62. Cinalli G., Chumas P., Arnaud E., et al. Occipital remodeling and suboccipital decompression in severe craniosynostosis associated with tonsillar herniation. Neurosurg. 1998;42:66-73.
63. Wheaton S.W. Two specimens of congenital cranial deformity in infants associated with fusion of the fingers and toes. Trans. Path. Soc. Lon. 1894;45:238-241.
64. Apert M.E. De l’acrocephalosyndactylie. Paris: Bull Mem Soc Med. Hop; 1906;23:. 1310-1330
65. Marucci D.D., Dunaway D.J., Jones B.M., Hayward R.D. Raised intracranial pressure in Apert syndrome. Plast and Reconstruct Surg. 2008;122:1162-1168.
66. Cohen S.R., Dauser R.C., Gorski J.L. Insidious onset of familial craniosynostosis. Cleft-Palate Craniofac J. 1993;30:401-405.
67. Glaser R.L., Jiang W., Boyadjiev S.A., et al. Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome. Am J Hum Genet. 2000;66:768-777.
68. Reardon W., Winter R.M., Rutland P., et al. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nature Genet. 1994;8:98-103.
69. Reddy K., Hoffman H., Armstrong D. Delayed and progressive multiple suture craniosynostosis. Neurosurgery. 1990;26:442-448.
70. Posnick J.C., Ruiz R.L. The craniofacial dysostosis syndromes: current surgical thinking and future directions. Cleft Palate Craniof J. 2000;37, 434(5):1-24.
71. Sgouros S. Skull vault growth in craniosynostosis. Childs Nerv Syst. 2005;21:861-870.
72. Renier D., Arnaud E., Cinalli G., et al. Prognosis for mental function in Apert’s syndrome. J Neurosurg. 1996;85:66-72.
73. Cohen M. Pfeiffer syndrome: update, clinical subtype and guidelines for differential diagnosis. Am J Gen. 1993;45:300-307.
74. Blount J.P., Louis R.G.Jr., Tubbs R.S., Grant J.H. Pansynostosis: a review. Childs Nerv Syst. 2007;23:1103-1109.
75. Gosain A.K., Moore F.O., Hemmy D.C. The kleeblattschädel anomaly in Apert syndrome: intracranial anatomy, surgical correction, and subsequent cranial vault development. J Plast Reconstr Surg. 1997;100:1796-1802.
76. Thompson D.N., Hayward R.D., Harkness W.J., et al. Lessons from a case of kleeblattschädel. Case report. J Neurosurg. 1995;82:1071-1074.
77. Collman H., Sörensen N., Krauß J. Hydrocephalus in craniosynostosis: a review. Childs Nerv Syst. 2005;21:902-912.
78. Noetzel M.J., Marsh J.L., Palkes H., Gado M. Hydrocephalus and mental retardation in craniosynostosis. J Pediatr. 1985;107:885-892.
79. Hayward R. Venous hypertension and craniosynostosis. Child’s Nervs Syst. 2005;21:880-888.
80. Cinalli G., Renier D., Sebag G., et al. Chronic tonsillar herniation in Crouzon’s and Apert’s syndromes: the role of premature synostosis of the lambdoid suture. J Neurosurg. 1995;83:575-582.
81. Francis P.M., Beals S., Rekate H.L., et al. Chronic tonsillar herniation and Crouzon’s syndrome. Pediatr Neurosurg. 1992;18:202-206.
82. Thompson D.N., Harkness W., Jones B.M., Hayward R.D. Aetiology of herniation of the hindbrain in craniosynostosis. Pediatr Neurosurg. 1997;26:288-295.
83. Cinalli G., Sainte-Rose C.H., Kollar E.M., et al. Hydrocephalus and craniosynostosis. J Neurosurg. 1998;88:209-214.
84. Sainte-Rose C., Lacombe J., Pierre-Kahn A., et al. Intracranial venous sinus hypertension: cause or consequence of hydrocephalus in infants? J Neurosurg. 1984;60:727-736.
85. Goodrich J.T. Skull base growth in craniosynostosis. Child’s Nervs Syst. 2005;21:871-879.
86. Tamburrini G., Caldarelli M., Massimi L., et al. Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Childs Nerv Syst. 2005;21:913-921.
87. Gray J.L., Kang S.S., Zenni G.C., et al. FGF-1 affixation stimulates ePTFE endothelialization without intimal hyperplasia. J Surg Res. 1994;57:596-612.
88. Taylor W.J., Hayward R.D., Lasjaunias P., et al. Enigma of raised intracranial pressure in patients with complex craniosynostosis: the role of abnormal intracranial venous drainage. J Neurosurg. 2001;94:377-385.
89. Saldino R.M., Steinbach H.L., Epstein C.J. Familial acrocephalosyndactyly (Pfeiffer syndrome). AJR. 1972;116:609-622.
90. Sainte-Rose C., Arnaud E., Pierre-Kahn A. Chronic tonsillar herniation in Crouzon and Apert syndrome: the role of the premature synostosis of the lambdoid suture. J Neurosurg. 1995;83:575-582.
91. Frim D.M., Jones D., Goumnerova L. Development of symptomatic Chiari malformation in a child with craniofacial dysmorphism. Pediatr Neurosurg. 1990;16:228-231.
92. Mulliken J.B., Steinberger D., Kunze S., et al. Molecular diagnosis of bilateral bicoronal synostosis. Plast Reconstr Surg. 1999;104:1603-1615.
93. Venes J.L. Arnold-Chiari malformation in an infant with kleeblattschädel: an acquired deformity? Neurosurgery. 1998;23:360-362.
94. Esparza J., Hinojosa J. Complications in the surgical treatment of craniosynostosis and craniofacial syndromes: a propos of 306 transcranial procedures. Child’s Nerv Syst. 2008;24:1421-1430.
95. Renier D., Cinalli G., Lajeunie E., et al. L’oxycéphalie, une craniosténose sévère. A propos d’une série de 129 cas. Arch Pediatr. 1997;4:722-729.
96. Nishikawa M., Sakamoto H., Hakuba A., et al. Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg. 1997;86:40-47.
97. Cinalli G., Spennato P., Sainte-Rose C., et al. Chiari malformation in craniosynostosis. Childs Nerv Syst. 2005;21:889-901.
98. Fujisawa H., Hasegawa M., Kida S., Yamashita J. A novel fibroblast growth factor receptor 2 mutation in Crouzon syndrome associated with Chiari type I malformation and syringomyelia. J Neurosurg. 2002;97:396-400.
99. Kreiborg S. Postnatal growth and development of the craniofacial complex in premature craniosynostosis. In: Cohen M.M.Jr., editor. Craniosynostosis: diagnosis, evaluation, and management. New York: Raven; 1986:157-189.
100. Rollins N., Booth T., Shapiro K. MR venography in children with complex craniosynostosis. Pediatr Neurosurg. 2000;32:308-315.
101. Richtsmeier J.T. Comparative study of normal, Crouzon and Apert craniofacial morphology using finite element scaling analysis. Am J Phys Anthropol. 1987;74:473-493.
102. Rich P.M., Cox T.C., Hayward R.D. The jugular foramen in complex and syndromic craniosynostosis and its relationship to raised intracranial pressure. AJNR Am J Neuroradiol. 2003;24:45-51.
103. Esparza J., Hinojosa J., García-Recuero I., et al. Surgical treatment of isolated and syndromic craniosynostosis. Results and complications in 283 consecutive cases. Neurocirugía. 2008;19:509-529.
104. Pollack I.F., Losken H.W., Hurwitz D.J. A combined frontoorbital and occipital advancement technique for use in total calvarial reconstruction. J Neurosurg. 1996;84:424-429.
105. Di Rocco C. How to decrease the impact of surgical scar in the correction of sagittal synostosis. Childs Nerv Syst. 2003;19:42-45.
106. Stelnicki E.J. Endoscopic treatment of craniosynostosis. Atlas Oral Maxillofacial Surg Clin N Am. 2002;10:57-72.
107. Cohen S.R., Holmes R.E., Ozgur B.M., et al. Fronto-orbital and cranial osteotomies with resorbable fixation using an endoscopic approach. Clin Plastic Surg. 2004;31:429-442.
108. Jiménez D.F., Barone C.M. Early treatment of anterior calvarial craniosynostosis using endoscopic-assisted minimally invasive techniques. Child’s Nerv Syst. 2007;23(12):1411-1419.
109. Sgouros S., Goldin J.H., Hockley A.D., Wake M.J.C. Posterior skull surgery in craniosynostosis. Child’s Nerv Syst. 1996;12:727-733.
110. Persing J.A., Jane J.A., Delashaw J.B. Treatment of bilateral coronal synostosis in infancy: a holistic approach. J Neurosurg. 1990;72:171-175.
111. Hirabayashi S., Sugawara Y., Sakurai A., et al. Fronto-orbital advancement by gradual distraction. J Neurosurg. 1998;89:1058-1061.
112. Hirabayashi S., Sugawara Y., Sakurai A., et al. Fronto-orbital advancement by distraction: the latest modification. Ann Plast Surg. 2002;49:447-451.
113. Roddi R., Jansen M.A., Vaandrager J.M., Van der Meulen J.C.H. Plagiocephaly—new classification and clinical study of a series of 100 patients. J Craniomaxillofac Surg. 1995;23:347-354.
114. Renier D. Intracranial pressure in craniosynostosis: pre- and postoperative recordings correlation with functional results. In: Persing J.A., Edgerton M.T., Jane J.A. Scientific Foundations and Surgical Treatment of Craniosynostosis. Baltimore: Williams & Wilkins; 1989:263-269.
115. Renier D., Sainte-Rose C., Marchac D., Hirsch J.F. Intracranial pressure in craniosynostosis. J Neurosurg. 1982;57:370-377.
116. Thompson D.N.P., Harkness W., Jones B., et al. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst. 1995;11:269-275.
117. Takeyoshi Shimoji T., Naoki N. Mild trigonocephaly and intracranial pressure: report of 56 patients. Childs Nerv Syst. 2004;20:749-756.
118. Breugem C.C., van R Zeeman B.J. Retrospective study of nonsyndromic craniosynostosis treated over a 10-year period. J Craniofac Surg. 1999;10(2):140-143.
119. Sloan G.M., Wells K.C., Raffel C., McComb J.G. Surgical treatment of craniosynostosis: outcome analysis of 250 consecutive patients. Pediatrics. 1997;100(1):E2.
120. David J.D., Cooter R.D. Craniofacial Infection in 10 years of transcranial surgery. Plast Reconstr Surg. 1987;80:213-225.
121. Fearon J.A., Bartlett S.P., Munro I.R., et al. Infections in craniofacial surgery: a combined report of 567 procedures from two centers. Plast Recosntr Surg. 1997;100(4):862-868.
122. Whitaker L.A., Munro I.R., Salyer K.E., et al. Combined report of problems and complications in 793 craniofacial operations. Plast Reconstr Sur. 1979;64:198-203.
123. Yeung L.C., Cunningham M.L., Allpress A.L., et al. Surgical site infections after pediatric intracranial surgery for craniofacial malformations: frequency and risk factors. Neurosurgery. 2005;56(4):733-739.
124. Foster K.A., Frim D.M. McKinnon. Recurrence of synostosis following surgical repair of craniosynostosis. Plast Reconstr Surg. 2008;121(3):70-76.
125. McCarthy J.G., Glasberg S.B., Cutting C.B., et al. Twenty-year experience with early surgery for craniosynostosis: II. The craniofacial synostosis syndromes and pansynostosis—results and unsolved problems. Plast Reconstr Surg. 1995;96(2):284-295.
126. Bradley J.P., Gabbay J.S., Taub P.J., et al. Monobloc advancement by distraction osteogenesis decreases morbidity and relapse. Plast Reconsr Surg. 2006;118(7):1585-1597.
127. Cohen S.R., Boydston W., Hudgins R., Burstein F.D. Monobloc and facial bipartition distraction with internal devices. J Craniofac Surg. 1999;10(3):244-251.
128. Kubler A.C., Speder B., Zoller J.E. Fronto-orbital advancement with simultaneous LeFort III-distraction. J Craniomaxillofac Surg. 2004;32(5):291-295.
129. Nadal E., Doghliotti P.L., Rodríguez J.C., Zuccaro G. Craniofacial distraction osteogenesis en bloc. J Craniofac Surg. 2000;11(3):246-251.
130. Pelo S., Gasparini G., Di Petrillo A., et al. Distraction osteogenesis in the surgical treatment of craniostenosis: a comparison of internal and external craniofacial distractor devices. Child’s Nerv Syst. 2007;23:1447-1453.
131. Aryan H.E., Meltzer H.S., Gerras G.G., et al. Leptomeningeal cyst development after endoscopic craniosynostosis repair: case report. Neurosurgery. 2004;55(1):235-237.
132. Yamamoto M., Moore M.H., Hanieh A. Growing skull fracture after cranial vault reshaping in infancy. J Craniofac Surg. 1998;9(1):73-75.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 65 Methods of Cranial Vault Reconstruction for Craniosynostosis
Cranial sutures are essential components in the development of the skull. Nonfunctional sutures during the evolution of the cranial vault and the skull base lead to evolving deformities that may end in neurologic sequel. Although the term craniosynostosis was first used by Bertolotti in 1914, referring to the premature closure of a cranial suture, it was Sommerring who described in 1791 the anatomy of the suture and postulated not only its role in normal skull growth but also the effects of early closure.50 During the 19th century Otto, and later Virchow, asserted that premature closure of sutures (craneostenosis) prevented growth perpendicular to the suture and was accompanied by compensatory growth at others. Premature closure may affect a single suture, but several sutures may be involved and severe deformities can develop, including the orbits and anterior fossa in the process. This is even more evident in craniofacial syndromes, where the main difference between single-suture and nonsyndromic craniosynostosis is the alterations not only in neurocranium but also in viscerocranium, resulting in anomalies in the midface skeleton.
The true incidence of craniosynostosis is not known, but it is estimated to occur in 1 of 2500 newborns. Sagittal synostosis is the most frequent (40%-60% of all cases), followed by metopic, coronal, and lambdoid, which is unusual.1,2 More than 150 associated syndromes have been described, but in most cases craniosynostosis is an isolated phenomenon. Several theories have been postulated to explain the appearance of premature synostosis and posterior deformities. Virchow was the first to suggest in 1852 that the premature closure of the suture was the primary event, while the vault deformity was a consequence of this closure.71 In 1959, Moss suggested that the deformity in the cranial base was the primary event. For this author, dura mater is an important regulator in the activity of the sutures and vault development and the main factor for the alteration in the cranial growth and suture dysfunction. The abnormal mechanical strengths driven through dural structures from certain maldeveloped points at the skull base (crista galli, petrous pyramids, or sphenoidal wings) would be responsible for cranial deformities and suture dysfunction.3 Park and Powers referred to mesenchymal abnormalities in bone structures, related to genetic anomalies, that would explain more thoroughly hypoplasia affecting the craniofacial region in syndromic cases.67,68,70
In the end, all of these mechanisms and changes produce a cosmetic deformity, as well as incompetence of the cranial and facial structures—unable to properly contain the organs inside the vault (brain and cerebellum) and the orbits (optic nerves and eyeballs)—and hypoplasia of the midface and oropharyngeal region. All of these factors are related, and depending on the affected region, there is a predominance of one abnormality. However, as explained later, in craniofacial syndromes the situation is more complicated. The presence of raised intracranial pressure (ICP) (multiple suture closure), hydrocephalus and cerebrospinal fluid (CSF) circulation anomalies, venous hypertension (skull base sutures closure affecting jugular foramina drainage, as well as genetic factors including endothelial proliferation in dural venous sinuses), chronic hypoxia and hypercapnia (obstructive airway in relation to midface retrusion and amygdalar hyperplasia), and chronic tonsillar herniation leads to a complex situation in which staged treatment has to be the rule, particularly after careful and proper understanding of the pathophysiology underlying these cases and always in a multidisciplinary team.
Scaphocephaly
Sagittal synostosis is the most common form of craniosynostosis. The premature closure of the sagittal synostosis has an estimated prevalence of 2 in 1000 births. Up to 80% of the cases are sporadic, and there is a male-to-female ratio of 3:1 to 4:1.1 Sporadic cases are mainly related to intrauterine constraint, but some may have a heterogeneous cause. In particular cases, a dominant autosomal inheritance has been described, with 38% penetrance.1 The frequency of twinning in a series was 4.8%, with only one monozygotic twin pair being concordant for sagittal synostosis.4
Histology usually shows synostotic ridging on the posterior part of the fused suture, which is consistent with the sagittal suture being thicker on the ectocranial cortex than on the endocranial side, and the bony edge of the suture becomes progressively thicker from anterior to posterior under normal conditions.5 When the sagittal suture is the only involved suture, the head becomes long from front to back (Figs. 65-1 to 65-3). It is narrow from side to side, and frontal compensational bossing may occur when the anterior fontanel is patent. When both fontanels are closed and the posterior part of the suture is predominantly affected, there is a posterior deformity typically referred to as an occipital knob. It is usually accompanied by a thin forehead with severe pterional indentation (leptoscaphocephaly). In the severest forms of scaphocephaly, the head adopts the shape of a saddle, and it is named bathrocephaly. Accurate diagnosis of isolated sagittal synostosis can be made clinically, and operative correction can proceed without a need for radiologic investigations, unless the clinical features are not completely typical.6 This could be the case in which Crouzon syndrome in some patients can resemble, early in evolution, an isolated scaphocephaly.
There is a plethora of techniques for the treatment of scaphocephaly7–11 but most of them include the excision of the sagittal suture with different kinds of parietal and occipital osteotomies with or without frontal osteotomies and remodeling. Surgical correction, whatever the technique, when performed early in infancy, results in satisfactory and persisting reshaping of the skull (Figs. 65-1 to 65-3). Delays in diagnosis, as well as treatment, result in a small subset of patients with uncorrected craniosynostosis that require the development of more extensive calvarial reconstruction techniques (Fig. 65-4). Due to calvarial maturity, these more advanced cases would not be amenable to treatment using less aggressive techniques. After 1 year of age the child’s cranial vault has developed abnormally, resulting in a severe skull base deformity, as shown in the figure. Subsequently, the skull is significantly thicker and less pliable to reshaping. At this age, cranial defects due to aggressive surgical remodeling procedures may not reossify, and there is a lack of the driving mechanism that leads to closure of the sutures. For these patients, an approach with a more comprehensive calvarial reconstruction includes multiple interleaving osteotomies crossing the midline, leading to improvements in biparietal narrowing combined with a bifrontal reconstruction.
Trigonocephaly
Trigonocephaly is considered a fairly common type of craniosynostosis. The term was first coined by Welcker in 1862.12 Formerly denoted as unusual,12,13 it is now considered to represent about 10% of all patients with deformities related to craniofacial centers12,14,15 and its frequency is increasing.2 Its incidence is estimated to be 1 in 2500 births,16 and there is a male predominance of 65% to 85%.14
Closure of the metopic suture starts under normal circumstances at the end of the first year and may last until the end of the second year.12 When premature suture closure occurs, usually before birth, a typical craniofacial malformation develops. The premature arrest of growth of the metopic suture may present as a spectrum of manifestations, depending on the timing and extent of the suture closure. Its mildest form is a familial and ethnically inherited facial morphology.17 Milder forms of metopic synostosis consist of only a somewhat prominent metopic ridge that does not need surgical intervention. In severe forms, a characteristic keel-shaped forehead can be observed, with absence of frontal eminences, retruded orbital rims, hypotelorism, and epicanthus giving a peculiar craniofacial appearance to these children. Different degrees of involvement of the anterior chondrocranial structures seem possible, such as presphenoid, mesoethmoid, and ectoethmoid structures.18 Another explanation for the broad phenotype of affected children could be a nonspecific process acting at different evolutive ages. Trigonocephaly may appear either as an isolated anomaly or as part of syndromes involving prosencephalic or rhinencephalic structures (holoprosencephaly), such as Opitz syndrome,19,20 Say-Meyer syndrome, or Frydman syndrome.12 Recently a new fronto-ocular syndrome has been described with trigonocephaly.21
Several genetic defects also have been described associated with trigonocephaly. Abnormalities including deletions and duplications of chromosomes 3p, 9p, 1, lq, and 13q have been published.22–25 Isolated trigonocephaly has an unknown etiology. Autosomal dominant inheritance with very low penetrance has been proposed in 2% to 5% of the cases for familial cases.15,26 The possible involvement of mutated FGFRs in newborns affected by trigonocephaly has recently been investigated by molecular screening in a search for mutations in FGFR2, which has been implicated in complex craniofacial syndromes. However, none of the cases studied carried mutations, except one that evolved later toward a Crouzon syndrome–like profile.25
Mental delay may occur in isolated trigonocephaly and has been observed in as many as 10% of these children.12 Development delays may consist of subtle changes in the time of acquisition of head control or sitting position or include more severe forms of mental retardation, with or without intracranial hypertension.
Di Rocco et al. proposed two subgroups on the basis of clinical and radiologic findings.18 Group I presents with bilateral frontal bone hypoplasia associated with extreme retrusion of the supraorbital margins. In this group, hypotelorism is associated with abnormally deep position of the cribriform plate, giving the ethmoidal region a hollow appearance. In these patients, the nasion–pterional angle is severely restricted and the nasion–clinoidal distance significantly increased. Group II also shows bilateral frontal hypoplasia with hypotelorism, supraorbital retrusion, and a reduced nasopterional angle. However, the nasion–clinoidal distance is almost normal, and pterional evidence is scarcely evident.18 Moreover, patients in group II showed a lesser degree of temporal compensatory expansion. The authors postulate that in some patients a compensatory elongation of the nasion–clinoidal distance and an incomplete synostosis of the frontoethmoidal sutures allows partial lateral expansion of the anterior cranial fossa, which could diminish the need for posterior calvarial expansion. On the other hand, children with more severe involvement of the nasoethmoidal sutures, resulting in diminished lateral expansion of the anterior fossa, need compensatory changes in the temporal and parietal regions. Patients in group II accomplished good correction of associated hypotelorism, whereas patients in group I did not reach normal interorbital values when treated with the same surgical procedure, which did not include specific treatment of hypotelorism. Other authors have addressed the importance of the moment and degree of involvement of pathologic changes in the anterior chondrocranial structures.14,27–31 Milder forms of trigonocephaly would affect only the upper metopic suture, whereas more severe forms include involvement of presphenoid, mesoethmoid, and ectoethmoid structures.
Surgical Technique
Before starting osteotomies, the obliquity of frontal bones, retrusion of the lateral portions of the orbital bandeau, and pterional indentation are assessed. A bifrontal craniotomy is performed after a supraorbital bandeau is created. The marks are extended from the sphenosquamosal suture to its contralateral equivalent prepared for a tongue-in-groove advancement.18 The bifrontal flap includes the anterior part of the bregmatic fontanelle and both coronal sutures. The anterior fossa is exposed extradurally with visualization of the anterior two thirds of the orbital roof.
There is not yet a consensus on hypotelorism correction. Several papers have addressed the importance of associated hypotelorism17,28 and the need for direct surgical approach. Different authors17,27,28,36,37 propose adding a nasofrontal osteotomy and an interpositional bone graft to the supraorbital bar and nasoethmoidal area to correct hypotelorism simultaneously with lateral orbitary wall expansion17 or three quarter orbital wall osteotomies.27 On the other hand, there are others13,34 who do not advocate direct approach to the hypotelorism in the belief that internasalis grafting widens only nasal bones without increasing interorbital width, as far as osteotomies usually remain anterior and superior to nasoethmoidal complex.36 I recommend a standard approach without grafting and thus consider the latter ineffective and unnecessary. Both techniques have achieved statistically significant improvement in hypotelorism, greater than that expected from normal growth curves. However, undercorrection of hypotelorism and persistence of abnormally low interorbitary distances occur frequently when orbital widening is not addressed.18,27,35
Anterior Plagiocephaly
Premature closure of a single coronal suture produces the most complex set of craniofacial deformities due to the peculiarities of the fronto-orbital region and the close relationship between the sutures of the anterior cranial base and the orbits and maxillary complex. The incidence of coronal craniosynostosis has been reported to be variable but can be approximated to be 0.4 per 1000.40 Although most cases are sporadic, a familiar incidence of up to 8% in uncomplicated coronal synostosis has been reported.40
Frontal or anterior synostotic plagiocephaly has been described in association with unilateral coronal synostosis,41–44 but it is known to occur with synostosis of other parts of the coronal ring, such as the frontosphenoidal or frontoethmoidal sutures.41,45–47 In some rare cases, it has been reported to be the result of fusion of the frontosphenoidal48 or the frontozygomatic sutures alone.49 The deformity is manifest as a progressive cranial, orbital, and facial asymmetry of varying severity, with secondary deformities, considered to be compensatory, existing on the contralateral side (Figs. 65-5 and 65-6). Associated cranial anomalies affect the development of all facial bones and have been referred to as “the most complete presentation of craniofacial asymmetry.”50 There is a flat and retruded frontal bone in the affected side with a contralateral bossing. The orbital rim on the synostotic side is also elevated and retruded, while the ipsilateral orbit is elevated and twisted. There is a variable degree of vertical orbital dystopia. The nasal root is deviated toward the closed sutured with an oblique nasal axis. The tip of the nose points to the nonsynostotic side. The zygoma and maxilla are hypoplastic on the affected side, and the ipsilateral temporal bone is malpositioned in an anterior and descended configuration.
Different reconstructive techniques have been described,42,43,46,47,50–53 but most of them can be divided between unilateral and bilateral approaches. There is a long-standing controversy on the timing and type of surgical approach. Some authors have advocated early surgery (younger than 6 months),40,54 whereas others support treating these patients later (between 9 and 10 months).41,45,51 The former group uses the concept of rapid brain growth leading to advancement of the frontal bones and supraorbital rims.
Proponents of late surgery argue that delayed surgery allows for further maturation and growth of the craniofacial skeleton. There is less reliance on brain growth, and blood loss is better tolerated. Also, there is immediate correction of the deformity. Surgical procedures have expanded from simple suturectomies (Fig. 65-5) to frontal–calvarial vault remodeling consisting of bifrontal craniotomes and orbital frontal bandeau advancement (Fig. 65-6). Aggressive surgical approaches have been supported by the theory of “coronal ring” synostosis, which states that there is an extension of the coronal synostosis into the cranial base.55,56 However, in most cases of isolated, nonsyndromic, unilateral coronal synostosis, the sphenofrontal, sphenoethmoidal, ethmoidofrontal, and pterional sutures are patent. Furthermore, review of the literature of these patients who have undergone more extensive procedures indicate that the results are inconsistent and mixed. Often, there is restriction of forward anterior skull base growth and advancement, and late correction (older than 9 months) fails to improve vertical dystopia, which persists into adulthood.40 Most craniofacial centers use the classic fronto-orbital advancement with unilateral or bilateral correction. Undoubtedly, in cases with severe contralateral compensation (frontal and/or temporal bossing), a bilateral correction is preferred. The correction of orbital dystopia warrants careful preoperative evaluation. Orbital hypoplasia is a consequence of the elevated orbital roofs, frontal development of the greater sphenoidal wings, and descent of the ethmoidal position, secondary to early closure of the coronal sutures. Orbital axes are also altered and point downward and outward. In the severest cases, failure to achieve early treatment or undercorrection may lead to strabismus, amblyopia, and anomalies in the binocular vision.
Multiple Suture Craniosynostosis
Brachycephaly
Brachycephaly is most commonly the result of premature synostosis of both coronal sutures. The anterior fossa adopts a characteristic deformation, and there are retruded frontal bones and orbital rims with a vertical, broad, flat forehead and a high bregmatic point. Brachycephaly may appear as an isolated synostosis and has then a favorable prognosis. Often, it is the common final appearance of different syndromic craniosynostoses that share a premature closure of bicoronal sutures and similar phenotype (e.g., some cases of Saehtre-Chotzen or Pfeiffer type I). For this reason, every patient with brachycephaly should be assessed by a clinical geneticist (Figs. 65-7 and 65-8).
Crouzon Syndrome
Crouzon syndrome is an autosomal dominant disorder characterized by craniosynostosis that causes secondary alterations of the facial bones and facial structure. Common features include hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism. First described by Crouzon in 1912,59 it was not until 1959 that Shiller observed an autosomal dominant transmission.60 Crouzon syndrome represents approximately 4.8% of cases of craniosynostosis at birth, and the birth prevalence is estimated to be 16.5 per million births. Crouzon craniofacial dysostosis is linked to a high number of different mutations, but most of them are located on IgIII of FGFR2 (exons 7 and 9) on chromosome 10q. An association between Crouzon syndrome and acanthosis nigricans has been described and is related to an A391E mutation in the FGFR3 gene on chromosome 4p.
Crouzon syndrome is characterized by a premature synostosis of both coronal sutures, with a resultant brachycephalic shape of the skull. Sagittal, metopic, or lambdoid sutures may also be prematurely affected, isolated, or combined (Figs. 65-9 and 65-10). The cranial base and upper facial sutures are involved with a variable degree of midface hypoplasia and malocclusion. The orbits are hypoplastic, and the orbital floor is hollow, resulting in proptosis and additional orbital dystopia that may produce mild to moderate orbital hypertelorism and divergent strabismus. Maxillary hypoplasia results in pseudoprognatism. Nasal septum deviation, together with maxillary hypoplasia, may produce a chronic obstruction to respiratory flow and may be associated with choanal atresia, velopharyngeal incompetence, and relative macroglossia. All of these malformations may lead to severe respiratory obstructions and apneas, which may cause a chronic raised ICP by an increase in venous pressure after hypercapnia and even contribute as an etiopathogenic mechanism in the development of hydrocephalus.
The initial treatment for Crouzon syndrome generally requires cranio-orbital decompression, including bicoronal suture release and osteotomies of the anterior cranial vault and upper orbits with reshaping and advancement. They are usually performed by the age of 8 to 11 months unless signs of increased ICP are found earlier. Sometimes, it is necessary to perform a combined approach, including midface advancement.61 When Chiari malformation (CM) is identified precociously, an occipital–parietal calvarial decompression may be preferable to achieve a bigger expansion of the intracranial volume, with or without suboccipital decompression.62 After proper release of the ICP, fronto-orbital remodeling can be achieved via an anterior approach.
Apert Syndrome
Apert syndrome, also known as acrocephalosyndactyly type I, is a congenital disorder characterized by multiple craniosynostoses, facial hypoplasia, and osseous syndactyly of the hands and feet. Approximately 1 in 65,000 to 165,000 of live births is affected. It is usually classified among a group of craniofacial syndromes with Crouzon, Pfeiffer, and Saehtre-Chotzen syndromes, all of which are allelic disorders with similar clinic manifestations and common genetic background. Wheaton first noted the coincidence of craniosynostosis and syndactyly (1894), but it was Apert who fully characterized the syndrome in 1906.63,64 Apert syndrome consists of a craniosynostosis in the shape of an acrocephaly or a brachycephaly due to the premature closure of several cranial sutures, typically coronal sutures and later those of the anterior cranial base and posterior fossa. The sagittal suture is typically widened and opened. It is always accompanied by osseous syndactyly of hands and feet and by fusion of the distal phalanxes. There is usually severe hypoplasia of the midface with an ogival-fissured hard palate. The orbital rims are retruded and elevated, and the skin possesses a characteristic acneiform appearance, mostly over the nasal bridge, shoulders, and back. Midface anomalies and anterior fossa craniosynostoses produce a decrease in the orbital volume that may lead to proptosis, strabismus (V syndrome), hypermetropia, or astigmatism. Most of these patients present with central nervous system anomalies, including hypoplasia of the corpus callosum and limbic and mesial temporal structure malformations. Approximately 10% of these children develop hydrocephalus, but only 2% of them suffer from a CM, in contrast to Crouzon syndrome, where 75% of the patients develop a hindbrain herniation.61 If left untreated, the incidence of raised ICP has previously been reported as 45%.64,65 Untreated intracranial hypertension may result in insidious optic atrophy, visual loss, and possible developmental delay.
Early cranial decompression with occipital expansion or fronto-orbital advancement is the treatment of choice, although some authors have advocated avoiding routine vault expansion in the first year of life. Instead, careful clinical, ophthalmologic, and respiratory monitoring would allow raised ICP to be treated in the most appropriate manner only when it occurs.65 Midface hypoplasia accounts for the exorbitism, strabismus, and respiratory difficulties seen in this condition. In the absence of any clinical sequelae from this hypoplasia, midface advancement is usually postponed until an age of 4 to 6 years. In cases of functional or clinical compromise, it may be necessary to intervene earlier with different techniques, such as monobloc advancement, maxillotomies, and/or mandible distraction. Facial hypoplasia and ogival palate abnormalities are responsible for phonetic disorders, abnormal dental eruption, and malocclusion.
Apert syndrome patients present with a variable degree of mental retardation. Early treatment of the craniosynostosis has been related to better outcomes. Some authors have shown that up to 50% of patients with Apert syndrome have a normal or near-normal intelligence quotient (IQ), with the rest being moderately to severely retarded. Renier et al.72 found an IQ greater than 70 in 50% of Apert syndrome patients when they were treated by cranial expansion in the first year of life but only in 7.1% of those who were treated later.
Pfeiffer Syndrome
Initially described by Pfeiffer in 1964,67,75 Pfeiffer syndrome is characterized by turribrachycephaly, maxillary hypoplasia, and antimongoloid slant of the orbits. There is hypertelorism and a marked degree of proptosis due to the bicoronal synostosis and subsequent recession of supraorbital rim and short anterior fossa. Extremities are notable for broad thumbs and large toes. There may be a variable degree and number of soft tissue syndactyly (most commonly between the second and the third digits), in comparison to Apert syndrome, where bony syndactyly is the hallmark. There may also be symphalangism (phalangeal fusion), ankylosis of the elbow joints, and cervical vertebral fusions (all of them also possible in Apert syndrome).
There are three clinical subtypes of Pfeiffer syndrome,73 which have clinical and prognostic significance. Type I (classic) is a lesser craniosynostosis that has a typical appearance of brachycephaly but usually is associated with normal or near-normal intelligence. Children with this type of Pfeiffer syndrome can develop normally, but there is often increased ICP if the synostoses are left uncorrected. Type II is associated with cloverleaf deformity, and although compatible with life, prognosis is poor. Type III is characterized by striking proptosis, but kleeblattschädel is absent. Long-term prognosis is poor.
Cloverleaf Skull (Kleeblattschädel)
Kleeblattschädel syndrome is characterized by a trilobar skull caused by frontal and bitemporal bossing. The first description of a trilobar cranial malformation in a newborn was made by Vrolik in 1849,77 but the term kleeblattschädel syndrome was not introduced until 1960 by Holtermuller and Wiederman.77,78 These authors reported a series of children with a trilobar or “cloverleaf” cranium associated with multiple craniosynostoses. In 1965, Commings et al. published the first case in the United States and translated the term kleeblattschädel syndrome into cloverleaf skull.78 This malformation has been described in children with Crouzon, Apert, Carpenter, Beare-Stevenson, or type II Pfeiffer syndrome, the latter being most frequently associated (up to 20% in some series).75,
[/not-level-membership-for-neurosurgery-category]
