[level-membership-for-surgery-category]
1 Metabolic response to injury, fluid and electrolyte balance and shock
The metabolic response to injury
Factors mediating the metabolic response to injury
The metabolic response is a complex interaction between many body systems.
The acute inflammatory response
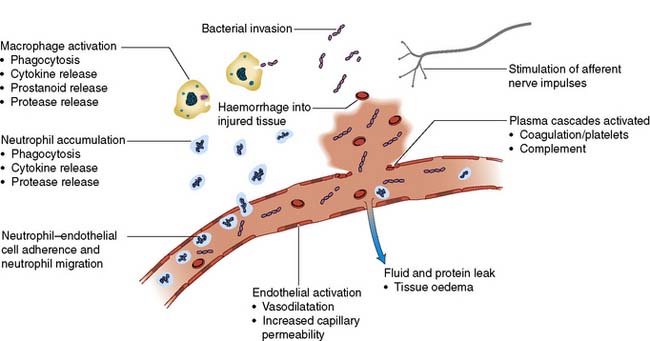
Inflammatory cells and cytokines are the principal mediators of the acute inflammatory response. Physical damage to tissues results in local activation of cells such as tissue macrophages which release a variety of cytokines (Table 1.1). Some of these, such as interleukin-8 (IL-8), attract large numbers of circulating macrophages and neutrophils to the site of injury. Others, such as tumour necrosis factor alpha (TNF-α), IL-1 and IL-6, activate these inflammatory cells, enabling them to clear dead tissue and kill bacteria. Although these cytokines are produced and act locally (paracrine action), their release into the circulation initiates some of the systemic features of the metabolic response, such as fever (IL-1) and the acute-phase protein response (IL-6, see below) (endocrine action). Other pro-inflammatory (prostaglandins, kinins, complement, proteases and free radicals) and anti-inflammatory substances such as antioxidants (e.g. glutathione, vitamins A and C), protease inhibitors (e.g. α2-macroglobulin) and IL-10 are also released (Fig. 1.1). The clinical condition of the patient depends on the extent to which the inflammation remains localized and the balance between these pro- and anti-inflammatory processes.
Table 1.1 Cytokines involved in the acute inflammatory response
| Cytokine | Relevant actions |
|---|---|
| TNF-α | Proinflammatory; release of leucocytes by bone marrow; activation of leucocytes and endothelial cells |
| IL-1 | Fever; T-cell and macrophage activation |
| IL-6 | Growth and differentiation of lymphocytes; activation of the acute-phase protein response |
| IL-8 | Chemotactic for neutrophils and T cells |
| IL-10 | Inhibits immune function |
(TNF = tumour necrosis factor; IL = interleukin)
The endothelium and blood vessels
The expression of adhesion molecules upon the endo-thelium leads to leucocyte adhesion and transmigration (Fig. 1.1). Increased local blood flow due to vasodilatation, secondary to the release of kinins, prostaglandins and nitric oxide, as well as increased capillary permeability increases the delivery of inflammatory cells, oxygen and nutrient substrates important for healing. Colloid particles (principally albumin) leak into injured tissues, resulting in oedema.
Afferent nerve impulses and sympathetic activation
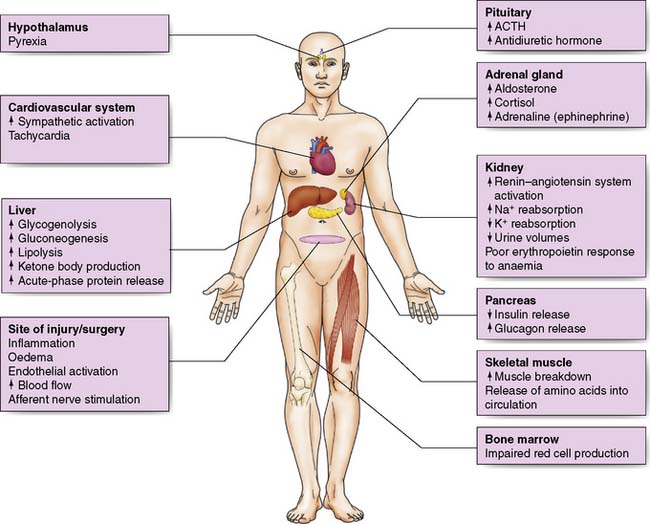
1. Activation of the sympathetic nervous system leads to the release of noradrenaline from sympathetic nerve fibre endings and adrenaline from the adrenal medulla resulting in tachycardia, increased cardiac output, and changes in carbohydrate, fat and protein metabolism (see below). Interventions that reduce sympathetic stimulation, such as epidural or spinal anaesthesia, may attenuate these changes.
The endocrine response to surgery
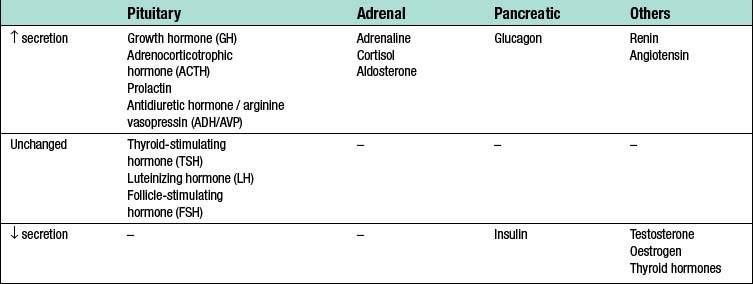
Surgery leads to complex changes in the endocrine mechanisms that maintain the body’s fluid balance and substrate metabolism, with changes occurring to the circulating concentrations of many hormones following injury (Table 1.2). This occurs either as a result of direct gland stimulation or because of changes in feedback mechanisms.
Consequences of the metabolic response to injury
Hypovolaemia
Reduced circulating volume often characterizes moderate to severe injury, and can occur for a number of reasons (Table 1.3):
• Loss of blood, electrolyte-containing fluid or water.
• Sequestration of protein-rich fluid into the interstitial space, traditionally termed “third space loss”, due to increased vascular permeability. This typically lasts 24–48 hours, with the extent (many litres) and duration (weeks or even months) of this loss greater following burns, infection, or ischaemia–reperfusion injury.
| Nature of fluid | Mechanism | Contributing factors |
|---|---|---|
| Blood | Haemorrhage | Site and magnitude of tissue injury Poor surgical haemostasis Abnormal coagulation |
| Electrolyte-containing fluids | Vomiting | Anaesthesia/analgesia (e.g. opiates) Ileus |
| Nasogastric drainage | Ileus Gastric surgery |
|
| Diarrhoea | Antibiotic-related infection Enteral feeding |
|
| Sweating | Pyrexia | |
| Water | Evaporation | Prolonged exposure of viscera during surgery |
| Plasma-like fluid | Capillary leak/sequestration in tissues | Acute inflammatory response Infection Ischaemia–reperfusionsyndrome |
Summary Box 1.1 Factors mediating the metabolic response to injury
Decreased circulating volume will reduce oxygen and nutrient delivery and so increase healing and recovery times. The neuroendocrine responses to hypovolaemia attempt to restore normovolaemia and maintain perfusion to vital organs.
Fluid-conserving measures
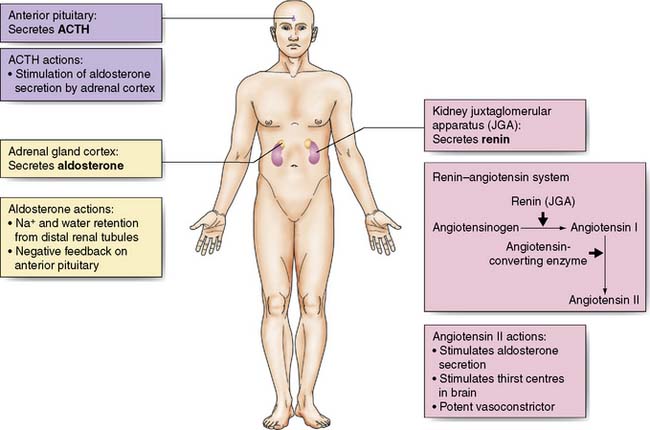
Oliguria, together with sodium and water retention – primarily due to the release of antidiuretic hormone (ADH) and aldosterone – is common after major surgery or injury and may persist even after normal circulating volume has been restored (Fig. 1.2).
Secretion of ADH from the posterior pituitary is increased in response to:
• Afferent nerve impulses from the site of injury
• Atrial stretch receptors (responding to reduced volume) and the aortic and carotid baroreceptors (responding to reduced pressure)
• Increased plasma osmolality (principally the result of an increase in sodium ions) detected by hypothalamic osmoreceptors
• Input from higher centres in the brain (responding to pain, emotion and anxiety).
Aldosterone secretion from the adrenal cortex is increased by:
• Activation of the renin–angiotensin system. Renin is released from afferent arteriolar cells in the kidney in response to reduced blood pressure, tubuloglomerular feedback (signalling via the macula densa of the distal renal tubules in response to changes in electrolyte concentration) and activation of the renal sympathetic nerves. Renin converts circulating angiotensinogen to angiotensin (AT)-I. AT-I is converted by angiotensin-converting enzyme (ACE) in plasma and tissues (particularly the lung) to AT-II which causes arteriolar vasoconstriction and aldosterone secretion.
• Increased adrenocorticotropic hormone (ACTH) secretion by the anterior pituitary in response to hypovolaemia and hypotension via afferent nerve impulses from stretch receptors in the atria, aorta and carotid arteries. It is also raised by ADH.
• Direct stimulation of the adrenal cortex by hyponatraemia or hyperkalaemia.
Blood flow-conserving measures
Hypovolaemia reduces cardiac preload which leads to a fall in cardiac output and a decrease in blood flow to the tissues and organs. Increased sympathetic activity results in a compensatory increase in cardiac output, peripheral vasoconstriction and a rise in blood pressure. Together with intrinsic organ autoregulation, these mechanisms act to try to ensure adequate tissue perfusion (Fig. 1.3).
Increased energy metabolism and substrate cycling
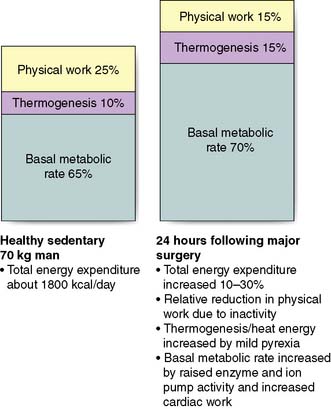
Although physical work usually decreases following surgery due to inactivity, overall energy expenditure may rise by 50% due to increased thermogenesis, fever and BMR (Fig. 1.4).
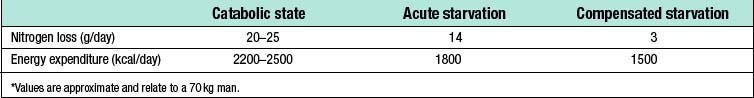
Catabolism and starvation
Catabolism
Protein metabolism
Skeletal muscle is broken down, releasing amino acids into the circulation. Amino acid metabolism is complex, but glucogenic amino acids (e.g. alanine, glycine and cysteine) can be utilized by the liver as a substrate for gluconeogenesis, producing glucose for re-export, while others are metabolized to pyruvate, acetyl CoA or intermediates in the Krebs cycle. Amino acids are also used in the liver as substrate for the ‘acute-phase protein response’. This response involves increased production of one group of proteins (positive acute-phase proteins) and decreased production of another (negative acute-phase proteins) (Table 1.4). The acute-phase response is mediated by pro-inflammatory cytokines (notably IL-1, IL-6 and TNF-α) and although its function is not fully understood, it is thought to play a central role in host defence and the promotion of healing.
| Positive acute-phase proteins (↑ after injury) |
Changes in red blood cell synthesis and coagulation
• endothelial cell injury and activation with subsequent activation of coagulation cascades
• platelet activation in response to circulating mediators (e.g. adrenaline and cytokines)
• venous stasis secondary to dehydration and/or immobility
• increased concentrations of circulating procoagulant factors (e.g. fibrinogen)
• decreased concentrations of circulating anticoagulants (e.g. protein C).
Summary Box 1.3 Physiological changes in catabolism
Factors modifying the metabolic response to injury
The magnitude of the metabolic response to injury depends on a number of different factors (Table 1.6) and can be reduced through the use of minimally invasive techniques, prevention of bleeding and hypothermia, prevention and treatment of infection and the use of locoregional anaesthesia. Factors that may influence the magnitude of the metabolic response to surgery and injury are summarised in table 1.6.
Table 1.6 Factors associated with the magnitude of the metabolic response to injury
| Factor | Comment |
|---|---|
| Patient-related factors | |
| Genetic predisposition | Gene subtype for inflammatory mediators determines individual response to injury and/or infection |
| Coexisting disease | Cancer and/or pre-existing inflammatory disease may influence the metabolic response |
| Drug treatments | Anti-inflammatory or immunosuppressive therapy (e.g. steroids) may alter response |
| Nutritional status | Malnourished patients have impaired immune function and/or important substrate deficiencies. Malnutrition prior to surgery is associated with poor outcomes |
| Acute surgical/trauma-related factors | |
| Severity of injury | Greater tissue damage is associated with a greater metabolic response |
| Nature of injury | Some types of tissue injury cause a disproportionate metabolic response (e.g. major burns), |
| Ischaemia–reperfusion injury | Reperfusion of ischaemic tissues can trigger an injurious inflammatory cascade that further injures organs. |
| Temperature | Extreme hypo- and hyperthermia modulate the metabolic response |
| Infection | Infection is associated with an exaggerated response to injury. It can result in systemic inflammatory response syndrome (SIRS), sepsis or septic shock. |
| Anaesthetic techniques | The use of certain drugs, such as opioids, can reduce the release of stress hormones. Regional anaesthetic techniques (epidural or spinal anaesthesia) can reduce the release of cortisol, adrenaline and other hormones, but has little effect on cytokine responses |
Fluid and Electrolyte Balance
• ADH and aldosterone secretion as described above
• Loss from the gastrointestinal tract (e.g. bowel preparation, ileus, stomas, fistulas)
• Insensible losses (e.g. sweating secondary to fever)
• Third space losses as described above
• Medications (e.g. diuretics)
• Underlying chronic illness (e.g. cardiac failure, portal hypertension).
Normal water and electrolyte balance
Water forms about 60% of total body weight in men and 55% in women. Approximately two-thirds is intracellular, one-third extracellular. Extracellular water is distributed between the plasma and the interstitial space (Fig. 1.5A).
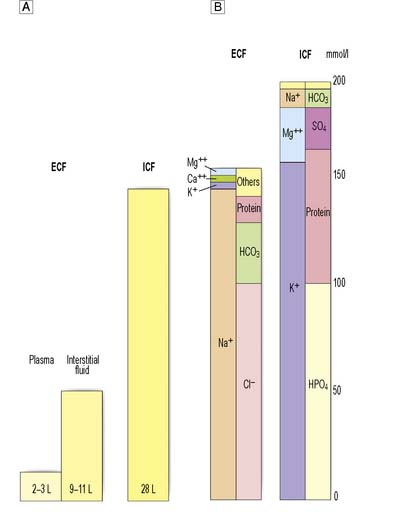
The differential distribution of ions across cell membranes is essential for normal cellular function. The principal extracellular ions are sodium, chloride and bicarbonate, with the osmolality of extracellular fluid (normally 275–295 mOsmol/kg) determined primarily by sodium and chloride ion concentrations. The major intracellular ions are potassium, magnesium, phosphate and sulphate (Fig. 1.5B).
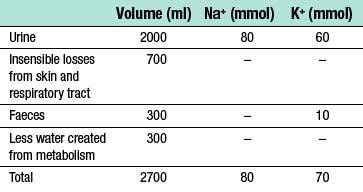
• 2500 to 3000 ml of fluid is lost via the kidneys, gastrointestinal tract and through evaporation from the skin and respiratory tract
• fluid losses are largely replaced through eating and drinking
• a further 200–300 ml of water is provided endogenously every 24 hours by the oxidation of carbohydrate and fat.
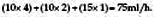
The daily requirement for both sodium and potassium in children is about 2–3 mmol/kg.
Assessing losses in the surgical patient
Only by accurately estimating (Table 1.8) and, where possible, directly measuring fluid and electrolyte losses can appropriate therapy be administered.
| Typical losses per 24 hrs | Factors modifying volume | |
|---|---|---|
| Insensible losses | 700–2000 ml | ↑ Losses associated with pyrexia, sweating and use of non-humidified oxygen |
| Urine | 1000–2500 ml | ↓ With aldosterone and ADH secretion; ↑ With diuretic therapy |
| Gut | 300–1000 ml | ↑ Losses with obstruction, ileus, fistulae and diarrhoea (may increase substantially) |
| Third-space losses | 0–4000 ml | ↑ Losses with greater extent of surgery and tissue trauma |
The effect of surgery
Loss from the gastrointestinal tract
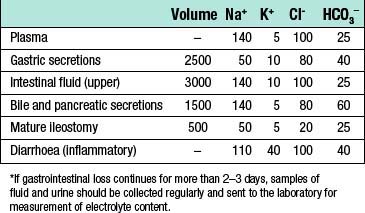
The magnitude and content of gastrointestinal fluid losses depends on the site of loss (Table 1.9):
• Intestinal obstruction. In general, the higher an obstruction occurs in the intestine, the greater the fluid loss because fluids secreted by the upper gastrointestinal tract fail to reach the absorptive areas of the distal jejunum and ileum.
• Paralytic ileus. This condition, in which propulsion in the small intestine ceases, has numerous causes. The commonest is probably handling of the bowel during surgery, which usually resolves within 1–2 days of the operation. Occasionally, paralytic ileus persists for longer, and in this case other causes should be sought and corrected if possible. During paralytic ileus the stomach should be decompressed using nasogastric tube drainage, and fluid losses monitored by measuring nasogastric aspirates.
• Intestinal fistula. As with obstruction, fistulae occurring high in the gut are associated with the greatest fluid and electrolyte losses. As well as volume, it may be useful to measure the electrolyte content of the fluid lost in order to determine the fluid replacement required.
• Diarrhoea. Patients may present with diarrhoea or develop it during the perioperative period. Fluid and electrolyte losses may be considerable.
Intravenous fluid administration
When choosing and administering intravenous fluids (Table 1.10) it is important to consider:
• what fluid deficiencies are present
• the fluid compartments requiring replacement
Types of intravenous fluid
Crystalloids
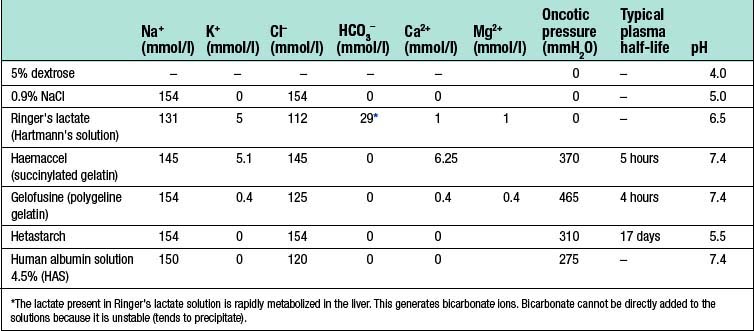
Dextrose 5% contains 5 g of dextrose (d-glucose) per 100 ml of water. This glucose is rapidly metabolized and the remaining free water distributes rapidly and evenly throughout the body’s fluid compartments. So, shortly after the intravenous administration of 1000 ml 5% dextrose solution, about 670 ml of water will be added to the intracellular fluid compartment (IFC) and about 330 ml of water to the extracellular fluid compartment (EFC), of which about 70 ml will be intravascular (Fig. 1.6). Dextrose solutions are therefore of little value as resuscitation fluids to expand intravascular volume. More concentrated dextrose solutions (10%, 20% and 50%) are available, but these solutions are irritant to veins and their use is largely limited to the management of diabetic patients or patients with hypoglycaemia.
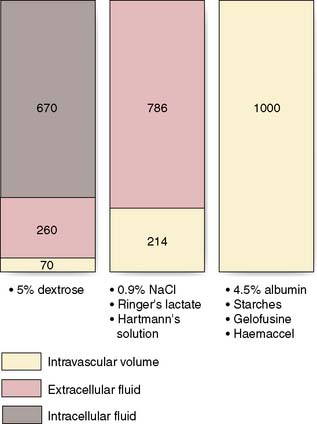
Sodium chloride 0.9% and Hartmann’s solution are isotonic solutions of electrolytes in water. Sodium chloride 0.9% (also known as normal saline) contains 9 g of sodium chloride dissolved in 1000 ml of water; Hartmann’s solution (also known as Ringer’s lactate) has a more physiological composition, containing lactate, potassium and calcium in addition to sodium and chloride ions. Both normal saline and Hartmann’s solution have an osmolality similar to that of extracellular fluid (about 300 mOsm/l) and after intravenous administration they distribute rapidly throughout the ECF compartment (Fig. 1.6). Isotonic crystalloids are appropriate for correcting EFC losses (e.g. gastrointestinal tract or sweating) and for the initial resuscitation of intravascular volume, although only about 25% remains in the intravascular space after redistribution (often less than 30–60 minutes).
Colloids
The theoretical advantage of colloids over crystalloids is that, as they remain in the intravascular space for several hours, smaller volumes are required. However, overall, current evidence suggests that crystalloid and colloid are equally effective for the correction of hypovolaemia (EBM 1.1).
Maintenance fluid requirements
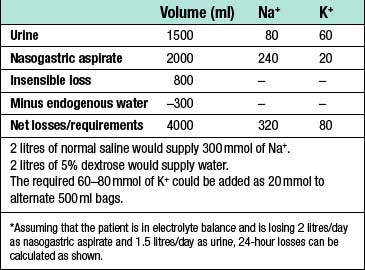
An example of a suitable 24-hour fluid prescription for an uncomplicated patient is shown in Table 1.11; the process of adjusting this for a hypothetical patient with an ileus is shown in Table 1.12.
Table 1.11 Provision of normal 24-hour fluid and electrolyte requirements by intravenous infusion
| Intravenous fluid | Additive | Duration (hrs) |
|---|---|---|
| 500 ml 0.9% NaCl | 20 mmol KCI | 4 |
| 500 ml 5% dextrose | – | 4 |
| 500 ml 5% dextrose | 20 mmol KCI | 4 |
| 500 ml 0.9% NaCl | – | 4 |
| 500 ml 5% dextrose | 20 mmol KCI | 4 |
| 500 ml 5% dextrose | – | 4 |
Specific water and electrolyte abnormalities
Sodium and water
Hyponatraemia
Treatment depends on correct identification of the cause:
• If ECF volume is normal or increased, the most likely cause is excessive intravenous water administration and this will correct spontaneously if water intake is reduced. Although less common in surgical patients, inappropriate ADH secretion promotes the renal tubular reabsorption of water independently of sodium concentration, resulting in inappropriately concentrated urine (osmolality > 100 mOsm/l) in the face of hypotonic plasma (osmolality < 290 mOsm/l). The urine osmolality helps to distinguish inappropriate ADH secretion from excessive water administration.
• In patients with decreased ECF volume, hyponatraemia usually indicates combined water and sodium deficiency. This is most frequently the result of diuresis, diarrhoea or adrenal insufficiency and will correct if adequate 0.9% sodium chloride is administered.
Potassium
Hyperkalaemia
Summary Box 1.4 Aetiology of hyper- and hyponatraemia
Hypernatraemia
result of tissue damage or changes in the Na+/K+-ATPase function, or impaired renal excretion.
Mild hyperkalaemia (K+ < 6 mmol/l) is often asymptomatic, but as serum levels rise there is progressive slowing of electrical conduction in the heart and the development of significant cardiac arrhythmias. All patients suspected of having hyperkalaemia should have an ECG for this reason. Tall ‘tented’ T-waves in the precordial leads are the earliest ECG changes observed, but as hyperkalaemia progresses more significant ECG changes occur, with flattening (or loss) of the P waves, a prolonged PR interval, widening of the QRS complex and eventually, asystole. Severe hyper-kalaemia (K+ > 7 mmol/l) requires immediate treatment to prevent this (Table 1.13).
Table 1.13 Management of severe hyperkalaemia (K+ >7 mmol/l)
Hypokalaemia
Transcellular shift – efflux of potassium from cells
• Rhabdomyolysis (e.g. crush and/or compartment syndromes)*
Impaired excretion
.
Gastrointestinal tract losses
Urinary losses
Transcellular shift–influx of potassium into cells
* Common causes in the surgical patient are denoted by an asterisk.
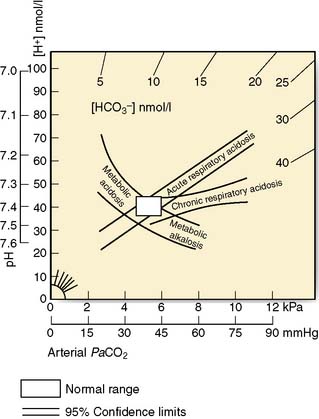
Acid–base balance
There are two broad types of acid–base disturbance: acidosis (‘acidaemia’ if plasma pH < 7.35 or H+ > 45) or alkalosis (‘alkalaemia’ if plasma pH > 7.45 or H+ < 35). Both acidosis and alkalosis may be respiratory or metabolic in origin. While some meaningful data pertaining to acid–base balance can be derived from the analysis of venous blood, accurate assessment of acid–base disturbance relies on the measurement of arterial blood gases. This is frequently coupled with measurement of blood lactate concentration. Arterial blood gas analysis is a straightforward technique, with samples typically taken from the radial artery (Fig. 1.7) and rapidly analysed by near-patient or laboratory-based machines.
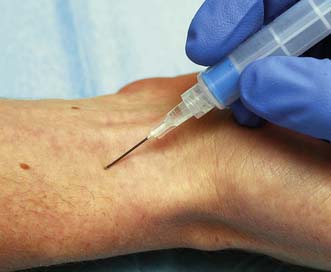
Common disturbances of acid–base balance encountered in the surgical patient are discussed below.
Metabolic acidosis
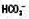 ↓
↓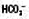 ↓
↓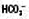 ↓
↓Respiratory acidosis
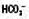 ↑
↑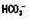 ↑
↑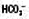 ↑
↑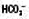 ↑
↑Mixed patterns of acid–base imbalance
Mixed patterns of acid–base disturbance are common, particularly in very sick patients. In this situation acid–base nomograms can be very useful in clarifying the contributing factors (Fig. 1.8).
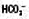 ↔ or ↑
↔ or ↑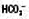 ↔
↔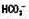 ↑
↑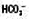 ↔ or ↓
↔ or ↓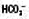 ↔
↔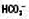 ↓
↓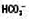 ↓
↓Shock
Types of shock
Hypovolaemic shock
This is probably the commonest and most readily corrected cause of shock encountered in surgical practice and results from a reduction in intravascular volume secondary to the loss of blood (e.g. trauma, gastrointestinal haemorrhage), plasma (e.g. burns) or water and electrolytes (e.g. vomiting, diarrhoea, diabetic ketoacidosis) (Table 1.14).
| Gastrointestinal haemorrhage |
Septic shock
Septic shock results from complex disturbances in oxygen delivery and oxygen consumption and can be defined as sepsis-induced hypotension (systolic BP < 90 mmHg, mean arterial blood pressure [MAP] < 70 mmHg) and/or tissue hypoperfusion (elevated lactate or oliguria) that persist despite adequate fluid resuscitation (~30 ml/kg) (Fig. 1.9).
Anaphylactic shock
Summary Box 1.11 Sepsis – definitions
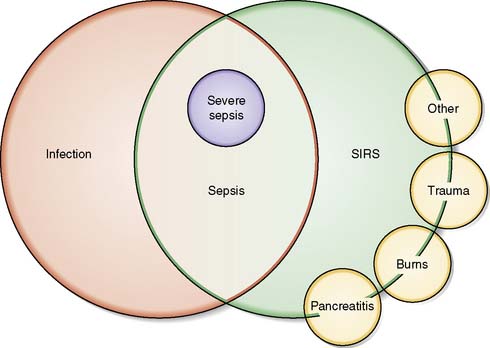
Systemic inflammatory response (SIRS)
SIRS is defined as 2 or more of the following criteria:
• Temperature > 38C° or < 36°C
• Heart rate > 90 beats per minute
• Respiratory rate > 20 per minute or PaCO2 < 4.5 kPa
• White cell count > 12 or < 4 × 109/l or > 10% immature neutrophils
and non-allergic anaphylaxis may be identical, with shock a frequent manifestation of both. Anaphylactic shock results from vasodilation, intravascular volume redistribution, capillary leak and a reduction in cardiac output. Common causes of anaphylaxis include drugs (e.g. neuromuscular blocking drugs, β-lactam antibiotics), colloid solutions (e.g. gelatin containing solutions, dextrans), radiological contrast media, foodstuffs (peanuts, tree nuts, shellfish, dairy products), hymenoptera stings and latex.
Pathophysiology
Macrocirculation
When assessing a patient with shock, it is useful to remember that mean arterial blood pressure (MAP) is equal to the product of cardiac output (CO) and systemic vascular resistance (SVR) (Table 1.15).
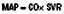 |
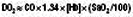 |
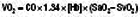 |
MAP = mean arterial pressure; CO = cardiac output; SVR = systemic vascular resistance; DO2 = oxygen delivery; [Hb] = haemoglobin concentration in g/dl; SaO2 = arterial oxygen saturations; VO2 = oxygen consumption; SvO2 mixed venous oxygen saturations (sampled from pulmonary artery)
Microcirculation
In sepsis, there is up-regulation of inducible NO synthetase and smooth muscle cells lose their adrenergic sensitivity resulting in pathological arterio–venous shunting. Endothelial and inflammatory cell activation results in the generation of reactant oxidant species, disruption of barrier function in the microcirculation and widespread activation of coagulation. Microthombi occlude capillary blood flow and the consumption of platelets and coagulation factors leads to thrombocytopenia, coagulopathy and DIC (Fig. 1.10).
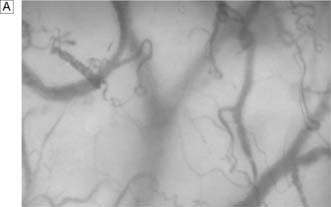
Cellular function
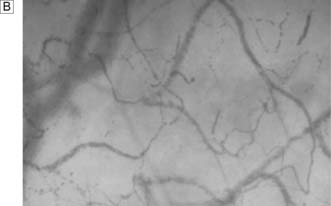
The oxidative metabolism of glucose is energy efficient, yielding up to 38 moles of ATP for each mole of glucose, but requires a continuous supply of oxygen to the cell. Hypoxaemia blocks mitochondrial oxidative phosphorylation, inhibiting ATP synthesis. This leads to a decrease in the intracellular ATP/ADP ratio, an increase in the NADH/NAD+ ratio and an accumulation of pyruvate that is unable to enter the TCA cycle. The cytosolic conversion of pyruvate to lactate allows the regeneration of some NAD+, enabling the limited production of ATP by anaerobic glycolysis. However, anaerobic glycolysis is significantly less efficient, generating only 2 moles of ATP per mole of glucose and predisposing cells to ATP depletion (Fig. 1.11).
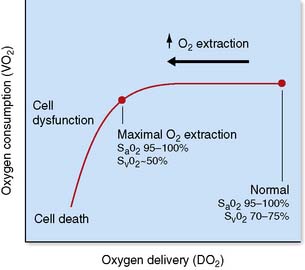
Under normal conditions, the tissues globally extract about 25% of the oxygen delivered to them, with the normal oxygen saturation of mixed venous blood being 70–75%. As oxygen delivery falls, cells are able to increase the proportion of oxygen extracted from the blood, but this compensatory mechanism is limited, with a maximal oxygen extraction ratio of about 50%. At this point, further reductions in oxygen delivery lead to a critical reduction in oxygen consumption and anaerobic metabolism, a state described as dysoxia (Fig. 1.12).
The effect of shock on individual organ systems
Hepatobiliary
Summary Box 1.12 Clinical effects of shock
• Renal hypoperfusion → activation of rennin–angiotensin system
• Oliguria (< 0.5 ml/kg/h urine) → anuria
• Acute renal failure → ↑ urea, ↑ creatinine, ↑ K+ & metabolic acidosis
• ↑ Ventilation/perfusion (V/Q) mismatch & ↑ shunt → hypoxia
• Pulmonary oedema (common in cardiogenic shock) → hypoxia
• Acute lung injury and acute respiratory distress syndrome → hypoxia
occurs 1–3 days following the ischaemic insult. Increases in prothrombin time and/or hypoglycaemia are markers of more severe injury. Significant ischaemic hepatitis is more frequent in patients with underlying cardiac disease and a degree of hepatic venous congestion.
Management
General principles
The management of shock is based upon the following principles:
The early recognition and treatment of potentially reversible causes (e.g. bleeding, intra-abdominal sepsis, myocardial ischaemia, pulmonary embolus, cardiac tamponade) is essential and may be facilitated by a detailed history, a thorough clinical examination (Table 1.16) and focused investigations.
| Conscious level | Restlessness, anxiety, stupor and coma are common features and suggest cerebral hypoperfusion |
| Pulse | Low volume, thready pulse consistent with low-output state; high volume, bounding pulse with high-output state |
| Blood pressure | Changes in diastolic may precede a fall in systolic blood pressure, with ↓ diastolic in sepsis and ↑ in hypovolaemic and cardiogenic shock |
| Peripheral perfusion | Cold peripheries suggest vasoconstriction (↑ SVR); warm peripheries suggest vasodilation (↓ SVR) |
| Pulse oximetry | Hypoxemia common association of all forms of shock and ↓tissue O2 delivery |
| ECG monitoring | Myocardial ischaemia commonest cause of cardiogenic shock but common in all forms of shock |
| Urine output | < 0.5 ml/kg/h suggestive of renal hypoperfusion |
| CVP measurement | Low CVP with collapsing central veins consistent with hypovolaemia |
| Arterial blood gas | Metabolic acidosis and ↑ lactate consistent with tissue hypoperfusion |
In isolation, single measurements are not helpful. Measurements are far more useful when used in combination with the findings of a detailed clinical examination. Observation of trends over time, together with the response to therapeutic interventions (e.g. a fluid challenge) is key to the successful management of shock.
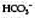 and lactate). In patients with severe hypoxaemia, cardiovascular instability, depressed conscious level or exhaustion, intubation and ventilatory support may be required.
and lactate). In patients with severe hypoxaemia, cardiovascular instability, depressed conscious level or exhaustion, intubation and ventilatory support may be required.Circulation
Initial resuscitation should be targeted at arresting haemorrhage and providing fluid (crystalloid or colloid) to restore intravascular volume and optimize cardiac preload. It is common practice to use blood to maintain a haemoglobin concentration > 10 g/dl (haematocrit around 0.3) during the initial resuscitation of shock if there is evidence of inadequate oxygen delivery, such as a raised lactate concentration or low central venous saturations (measured from a central venous catheter). A reduction in tachycardia, increasing blood pressure, and improving peripheral perfusion and urine output in response to a series of 250–500 ml fluid challenges indicate ‘fluid responsiveness’ and suggest that further fluid and optimization of preload may be required. Once parameters stop improving it is unlikely that further fluid will be beneficial, particularly if there is an associated fall in oxygen saturation and the development of pulmonary oedema. As resuscitation continues, more invasive monitoring allows the acid–base status, central venous pressure (CVP), pulmonary artery wedge pressure (PAWP), CO and mixed (SvO2) or central (ScvO2) venous oxygen saturations to be used to further assess the response to fluid (Fig. 1.13).
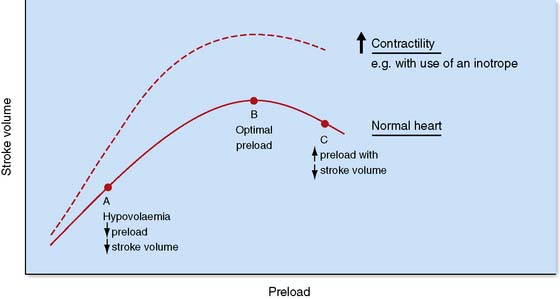
Fig. 1.13 Frank–Starling curve.
Demonstrating the relationship between ventricular preload and stroke volume.
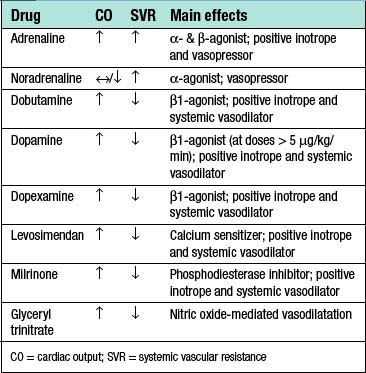
If blood pressure remains low and/or signs of inadequate tissue oxygen delivery persist despite fluid resuscitation and the optimization of preload, then inotropes and/or vasopressors may be required. Although there is a degree of crossover in their mechanism of action, vasopressors (e.g. noradrenaline) cause peripheral vasoconstriction and an increase SVR while inotropes (e.g. dobutamine) increase myocardial contractility, stroke volume and cardiac output. The initial choice of inotrope or vasopressor therefore depends upon the underlying aetiology of shock and an understanding of the main physiological derangements (Table 1.17). Adrenaline, which has both vasopressor and inotropic effects, is a useful first line drug in the emergency treatment of shock. Vasoactive drug administration should be continuously titrated against specific physiological end-points (e.g. blood pressure or cardiac output).
Hypovolaemic shock
The commonest cause of acute hypovolaemic shock in surgical practice is bleeding due to trauma, ruptured aortic aneurysm, gastrointestinal and obstetric haemorrhage (Table 1.14).
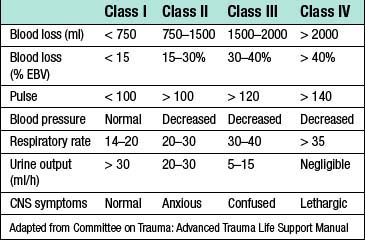
Normal adult blood volume is about 7% of body weight, with a 70 kg man having an estimated blood volume (EBV) of around 5000 ml. The severity of haemorrhagic shock is frequently classified according to percentage of EBV lost where class I (< 15%) represents a compensated state (as may occur following the donation of a unit of blood) and class IV (> 40%) is immediately life threatening (Table 1.18). The term ‘massive haemorrhage’ has a number of definitions including: loss of EBV in 24 hours; loss of 50% EBV in 3 hours; blood loss at a rate ≥ 150 ml/min.
Septic shock
The principles guiding the management of septic shock are:
• the identification and treatment of underlying infection
• early goal-directed therapy to optimize tissue oxygen delivery.
The Surviving Sepsis Campaign has published evidence-based guidelines on the management of severe sepsis and septic shock: http://www.survivingsepsis.org .
As with all forms of shock, the initial assessment and management of septic shock should follow an A, B, C approach. However, in patients with septic shock there is evidence that protocolized early goal-directed therapy (EGDT) improves survival (EBM 1.2) and this should be started as soon as signs of sepsis-induced tissue hypoperfusion are recognized (hypotension, elevated lactate, low central venous saturations or oliguria). The widely accepted resuscitation goals for the first 6 hours of this strategy are:
• Central venous pressure (CVP) of 8–12 mmHg
• Mean arterial blood pressure ≥ 65 mmHg
• Central venous (superior vena cava) O2 saturation (ScvO2) ≥ 70% or mixed venous (pulmonary artery) O2 saturation (SvO2) ≥ 65%.
Intravenous antibiotics must be administered as soon as possible (EBM 1.3), preferably in discussion with a microbiologist. The choice depends on the history, the likely source of infection, whether the infection is community- or hospital-acquired and local patterns of pathogen susceptibility. Covering all likely pathogens (bacterial and/or fungal) usually involves the use of empirical broad-spectrum antibiotics in the first instance, with these rationalized or changed to reduce the spectrum of cover once the results of microbiological investigations become available.
Anaphylactic shock
The management of anaphylactic shock is illustrated in Table 1.19.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
1 Metabolic response to injury, fluid and electrolyte balance and shock
The metabolic response to injury
Factors mediating the metabolic response to injury
The metabolic response is a complex interaction between many body systems.
The acute inflammatory response
Inflammatory cells and cytokines are the principal mediators of the acute inflammatory response. Physical damage to tissues results in local activation of cells such as tissue macrophages which release a variety of cytokines (Table 1.1). Some of these, such as interleukin-8 (IL-8), attract large numbers of circulating macrophages and neutrophils to the site of injury. Others, such as tumour necrosis factor alpha (TNF-α), IL-1 and IL-6, activate these inflammatory cells, enabling them to clear dead tissue and kill bacteria. Although these cytokines are produced and act locally (paracrine action), their release into the circulation initiates some of the systemic features of the metabolic response, such as fever (IL-1) and the acute-phase protein response (IL-6, see below) (endocrine action). Other pro-inflammatory (prostaglandins, kinins, complement, proteases and free radicals) and anti-inflammatory substances such as antioxidants (e.g. glutathione, vitamins A and C), protease inhibitors (e.g. α2-macroglobulin) and IL-10 are also released (Fig. 1.1). The clinical condition of the patient depends on the extent to which the inflammation remains localized and the balance between these pro- and anti-inflammatory processes.
Table 1.1 Cytokines involved in the acute inflammatory response
| Cytokine | Relevant actions |
|---|---|
| TNF-α | Proinflammatory; release of leucocytes by bone marrow; activation of leucocytes and endothelial cells |
| IL-1 | Fever; T-cell and macrophage activation |
| IL-6 | Growth and differentiation of lymphocytes; activation of the acute-phase protein response |
| IL-8 | Chemotactic for neutrophils and T cells |
| IL-10 | Inhibits immune function |
(TNF = tumour necrosis factor; IL = interleukin)
The endothelium and blood vessels
The expression of adhesion molecules upon the endo-thelium leads to leucocyte adhesion and transmigration (Fig. 1.1). Increased local blood flow due to vasodilatation, secondary to the release of kinins, prostaglandins and nitric oxide, as well as increased capillary permeability increases the delivery of inflammatory cells, oxygen and nutrient substrates important for healing. Colloid particles (principally albumin) leak into injured tissues, resulting in oedema.
Afferent nerve impulses and sympathetic activation
1. Activation of the sympathetic nervous system leads to the release of noradrenaline from sympathetic nerve fibre endings and adrenaline from the adrenal medulla resulting in tachycardia, increased cardiac output, and changes in carbohydrate, fat and protein metabolism (see below). Interventions that reduce sympathetic stimulation, such as epidural or spinal anaesthesia, may attenuate these changes.
The endocrine response to surgery
Surgery leads to complex changes in the endocrine mechanisms that maintain the body’s fluid balance and substrate metabolism, with changes occurring to the circulating concentrations of many hormones following injury (Table 1.2). This occurs either as a result of direct gland stimulation or because of changes in feedback mechanisms.
Consequences of the metabolic response to injury
Hypovolaemia
Reduced circulating volume often characterizes moderate to severe injury, and can occur for a number of reasons (Table 1.3):
• Loss of blood, electrolyte-containing fluid or water.
• Sequestration of protein-rich fluid into the interstitial space, traditionally termed “third space loss”, due to increased vascular permeability. This typically lasts 24–48 hours, with the extent (many litres) and duration (weeks or even months) of this loss greater following burns, infection, or ischaemia–reperfusion injury.
| Nature of fluid | Mechanism | Contributing factors |
|---|---|---|
| Blood | Haemorrhage | Site and magnitude of tissue injury Poor surgical haemostasis Abnormal coagulation |
| Electrolyte-containing fluids | Vomiting | Anaesthesia/analgesia (e.g. opiates) Ileus |
| Nasogastric drainage | Ileus Gastric surgery |
|
| Diarrhoea | Antibiotic-related infection Enteral feeding |
|
| Sweating | Pyrexia | |
| Water | Evaporation | Prolonged exposure of viscera during surgery |
| Plasma-like fluid | Capillary leak/sequestration in tissues | Acute inflammatory response Infection Ischaemia–reperfusionsyndrome |
Summary Box 1.1 Factors mediating the metabolic response to injury
Decreased circulating volume will reduce oxygen and nutrient delivery and so increase healing and recovery times. The neuroendocrine responses to hypovolaemia attempt to restore normovolaemia and maintain perfusion to vital organs.
Fluid-conserving measures
Oliguria, together with sodium and water retention – primarily due to the release of antidiuretic hormone (ADH) and aldosterone – is common after major surgery or injury and may persist even after normal circulating volume has been restored (Fig. 1.2).
Secretion of ADH from the posterior pituitary is increased in response to:
• Afferent nerve impulses from the site of injury
• Atrial stretch receptors (responding to reduced volume) and the aortic and carotid baroreceptors (responding to reduced pressure)
• Increased plasma osmolality (principally the result of an increase in sodium ions) detected by hypothalamic osmoreceptors
• Input from higher centres in the brain (responding to pain, emotion and anxiety).
Aldosterone secretion from the adrenal cortex is increased by:
• Activation of the renin–angiotensin system. Renin is released from afferent arteriolar cells in the kidney in response to reduced blood pressure, tubuloglomerular feedback (signalling via the macula densa of the distal renal tubules in response to changes in electrolyte concentration) and activation of the renal sympathetic nerves. Renin converts circulating angiotensinogen to angiotensin (AT)-I. AT-I is converted by angiotensin-converting enzyme (ACE) in plasma and tissues (particularly the lung) to AT-II which causes arteriolar vasoconstriction and aldosterone secretion.
• Increased adrenocorticotropic hormone (ACTH) secretion by the anterior pituitary in response to hypovolaemia and hypotension via afferent nerve impulses from stretch receptors in the atria, aorta and carotid arteries. It is also raised by ADH.
• Direct stimulation of the adrenal cortex by hyponatraemia or hyperkalaemia.
Blood flow-conserving measures
Hypovolaemia reduces cardiac preload which leads to a fall in cardiac output and a decrease in blood flow to the tissues and organs. Increased sympathetic activity results in a compensatory increase in cardiac output, peripheral vasoconstriction and a rise in blood pressure. Together with intrinsic organ autoregulation, these mechanisms act to try to ensure adequate tissue perfusion (Fig. 1.3).
Increased energy metabolism and substrate cycling
Although physical work usually decreases following surgery due to inactivity, overall energy expenditure may rise by 50% due to increased thermogenesis, fever and BMR (Fig. 1.4).
Catabolism and starvation
Catabolism
Protein metabolism
Skeletal muscle is broken down, releasing amino acids into the circulation. Amino acid metabolism is complex, but glucogenic amino acids (e.g. alanine, glycine and cysteine) can be utilized by the liver as a substrate for gluconeogenesis, producing glucose for re-export, while others are metabolized to pyruvate, acetyl CoA or intermediates in the Krebs cycle. Amino acids are also used in the liver as substrate for the ‘acute-phase protein response’. This response involves increased production of one group of proteins (positive acute-phase proteins) and decreased production of another (negative acute-phase proteins) (Table 1.4). The acute-phase response is mediated by pro-inflammatory cytokines (notably IL-1, IL-6 and TNF-α) and although its function is not fully understood, it is thought to play a central role in host defence and the promotion of healing.
| Positive acute-phase proteins (↑ after injury) |
Changes in red blood cell synthesis and coagulation
• endothelial cell injury and activation with subsequent activation of coagulation cascades
• platelet activation in response to circulating mediators (e.g. adrenaline and cytokines)
• venous stasis secondary to dehydration and/or immobility
• increased concentrations of circulating procoagulant factors (e.g. fibrinogen)
• decreased concentrations of circulating anticoagulants (e.g. protein C).
Summary Box 1.3 Physiological changes in catabolism
Factors modifying the metabolic response to injury
The magnitude of the metabolic response to injury depends on a number of different factors (Table 1.6) and can be reduced through the use of minimally invasive techniques, prevention of bleeding and hypothermia, prevention and treatment of infection and the use of locoregional anaesthesia. Factors that may influence the magnitude of the metabolic response to surgery and injury are summarised in table 1.6.
Table 1.6 Factors associated with the magnitude of the metabolic response to injury
| Factor | Comment |
|---|---|
| Patient-related factors | |
| Genetic predisposition | Gene subtype for inflammatory mediators determines individual response to injury and/or infection |
| Coexisting disease | Cancer and/or pre-existing inflammatory disease may influence the metabolic response |
| Drug treatments | Anti-inflammatory or immunosuppressive therapy (e.g. steroids) may alter response |
| Nutritional status | Malnourished patients have impaired immune function and/or important substrate deficiencies. Malnutrition prior to surgery is associated with poor outcomes |
| Acute surgical/trauma-related factors | |
| Severity of injury | Greater tissue damage is associated with a greater metabolic response |
| Nature of injury | Some types of tissue injury cause a disproportionate metabolic response (e.g. major burns), |
| Ischaemia–reperfusion injury | Reperfusion of ischaemic tissues can trigger an injurious inflammatory cascade that further injures organs. |
| Temperature | Extreme hypo- and hyperthermia modulate the metabolic response |
| Infection | Infection is associated with an exaggerated response to injury. It can result in systemic inflammatory response syndrome (SIRS), sepsis or septic shock. |
| Anaesthetic techniques | The use of certain drugs, such as opioids, can reduce the release of stress hormones. Regional anaesthetic techniques (epidural or spinal anaesthesia) can reduce the release of cortisol, adrenaline and other hormones, but has little effect on cytokine responses |
Fluid and Electrolyte Balance
• ADH and aldosterone secretion as described above
• Loss from the gastrointestinal tract (e.g. bowel preparation, ileus, stomas, fistulas)
• Insensible losses (e.g. sweating secondary to fever)
• Third space losses as described above
• Medications (e.g. diuretics)
• Underlying chronic illness (e.g. cardiac failure, portal hypertension).
Normal water and electrolyte balance
Water forms about 60% of total body weight in men and 55% in women. Approximately two-thirds is intracellular, one-third extracellular. Extracellular water is distributed between the plasma and the interstitial space (Fig. 1.5A).
The differential distribution of ions across cell membranes is essential for normal cellular function. The principal extracellular ions are sodium, chloride and bicarbonate, with the osmolality of extracellular fluid (normally 275–295 mOsmol/kg) determined primarily by sodium and chloride ion concentrations. The major intracellular ions are potassium, magnesium, phosphate and sulphate (Fig. 1.5B).
• 2500 to 3000 ml of fluid is lost via the kidneys, gastrointestinal tract and through evaporation from the skin and respiratory tract
• fluid losses are largely replaced through eating and drinking
• a further 200–300 ml of water is provided endogenously every 24 hours by the oxidation of carbohydrate and fat.
The daily requirement for both sodium and potassium in children is about 2–3 mmol/kg.
Assessing losses in the surgical patient
Only by accurately estimating (Table 1.8) and, where possible, directly measuring fluid and electrolyte losses can appropriate therapy be administered.
| Typical losses per 24 hrs | Factors modifying volume | |
|---|---|---|
| Insensible losses | 700–2000 ml | ↑ Losses associated with pyrexia, sweating and use of non-humidified oxygen |
| Urine | 1000–2500 ml | ↓ With aldosterone and ADH secretion; ↑ With diuretic therapy |
| Gut | 300–1000 ml | ↑ Losses with obstruction, ileus, fistulae and diarrhoea (may increase substantially) |
| Third-space losses | 0–4000 ml | ↑ Losses with greater extent of surgery and tissue trauma |
The effect of surgery
Loss from the gastrointestinal tract
The magnitude and content of gastrointestinal fluid losses depends on the site of loss (Table 1.9):
• Intestinal obstruction. In general, the higher an obstruction occurs in the intestine, the greater the fluid loss because fluids secreted by the upper gastrointestinal tract fail to reach the absorptive areas of the distal jejunum and ileum.
• Paralytic ileus. This condition, in which propulsion in the small intestine ceases, has numerous causes. The commonest is probably handling of the bowel during surgery, which usually resolves within 1–2 days of the operation. Occasionally, paralytic ileus persists for longer, and in this case other causes should be sought and corrected if possible. During paralytic ileus the stomach should be decompressed using nasogastric tube drainage, and fluid losses monitored by measuring nasogastric aspirates.
• Intestinal fistula. As with obstruction, fistulae occurring high in the gut are associated with the greatest fluid and electrolyte losses. As well as volume, it may be useful to measure the electrolyte content of the fluid lost in order to determine the fluid replacement required.
• Diarrhoea. Patients may present with diarrhoea or develop it during the perioperative period. Fluid and electrolyte losses may be considerable.
Intravenous fluid administration
When choosing and administering intravenous fluids (Table 1.10) it is important to consider:
• what fluid deficiencies are present
• the fluid compartments requiring replacement
Types of intravenous fluid
Crystalloids
Dextrose 5% contains 5 g of dextrose (d-glucose) per 100 ml of water. This glucose is rapidly metabolized and the remaining free water distributes rapidly and evenly throughout the body’s fluid compartments. So, shortly after the intravenous administration of 1000 ml 5% dextrose solution, about 670 ml of water will be added to the intracellular fluid compartment (IFC) and about 330 ml of water to the extracellular fluid compartment (EFC), of which about 70 ml will be intravascular (Fig. 1.6). Dextrose solutions are therefore of little value as resuscitation fluids to expand intravascular volume. More concentrated dextrose solutions (10%, 20% and 50%) are available, but these solutions are irritant to veins and their use is largely limited to the management of diabetic patients or patients with hypoglycaemia.
Sodium chloride 0.9% and Hartmann’s solution are isotonic solutions of electrolytes in water. Sodium chloride 0.9% (also known as normal saline) contains 9 g of sodium chloride dissolved in 1000 ml of water; Hartmann’s solution (also known as Ringer’s lactate) has a more physiological composition, containing lactate, potassium and calcium in addition to sodium and chloride ions. Both normal saline and Hartmann’s solution have an osmolality similar to that of extracellular fluid (about 300 mOsm/l) and after intravenous administration they distribute rapidly throughout the ECF compartment (Fig. 1.6). Isotonic crystalloids are appropriate for correcting EFC losses (e.g. gastrointestinal tract or sweating) and for the initial resuscitation of intravascular volume, although only about 25% remains in the intravascular space after redistribution (often less than 30–60 minutes).
