Chapter 19. Metabolic disease, dysplasias, osteochondritis and neurological disorders
Abnormalities of bone structure
Many disorders of bone occur as a result of the way it is made rather than attack by a disease. These conditions are complex, but learning can be simplified by looking at the different components of bone and the factors that influence their growth.
Bone growth is influenced by the following hormones and vitamins:
Factors affecting bone growth
1. Growth hormone.
2. Sex hormones.
3. Thyroid hormones.
4. Parathyroid hormone.
5. Vitamin C.
6. Vitamin D.
7. Calcitonin.
Growth hormone is responsible for growth until the epiphyses close. Excess secretion after growth is complete causes the thickening of the bones seen in acromegaly; excess secretion before growth is complete causes gigantism.
Sex hormones are involved in the growth spurt of puberty. Release of testosterone causes a rapid increase in growth followed by epiphyseal closure. If testosterone is not released, the epiphyses grow for longer than normal and the patients are tall; hence the legendary giant eunuchs.
Thyroid hormone permits normal growth and deficiency retards it, which is the reason that hypothyroid cretins are small. Thyrotoxicosis does not cause an increase in size but can lead to osteoporosis.
Parathyroid hormone is a polypeptide released when the serum calcium falls. Parathormone acts to increase the serum calcium in two ways:
1. Mobilization of calcium from bone.
2. Increased tubular resorption of calcium.
Vitamin C is necessary for collagen synthesis. Without it, osteoid cannot be properly formed at the epiphyses and children with scurvy have transradiant bands at the epiphyses.
Vitamin D has three actions:
1. It is essential for the absorption of calcium from the gut.
3. It affects muscle tone; patients with vitamin D deficiency have muscle weakness.
Calcitonin is secreted in the thyroid. Its exact function is unclear but it is secreted if the serum calcium is high and it affects release of calcium from bone.
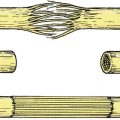
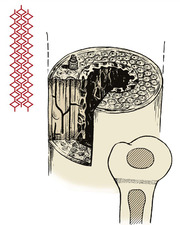
Bones have four main constituents which can affect growth. Disorders of growth can be considered according to the structure affected (Fig. 19.1).
 |
| Fig. 19.1
Constituents of bone. Bone consists of crystals arranged along collagen fibres, cartilage, and osteons or haversian systems.
|
Constituents of bone
1. Collagen.
2. Crystals of calcium hydroxyapatite arranged in an orderly manner along the collagen fibres. Osteoid is the uncalcified precursor of normal bone.
3. Osteons. Long bones are made up of long tubular units – the haversian systems or osteons. The osteocytes lie in cavities inside dense cortical bone and communicate with other osteocytes by prolongations of the cell body which run through canaliculi. This contact enables the osteocytes to react to abnormal stresses.
4. Cartilage. Growing bones develop from cartilage, which contains proteoglycans.
Abnormalities of collagen
Scurvy
The manifestations of scurvy include abnormal calcification at the epiphyses, and capillary fragility which leads to subperiosteal haemorrhage.
Treatment. Ascorbic acid (vitamin C).
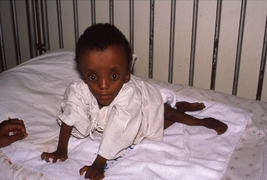
Osteogenesis imperfecta
Osteogenesis imperfecta, or fragilitas ossium, is often sporadic, but can be inherited through either autosomal recessive or autosomal dominant genes (Fig. 19.2). Although the brittleness of bone is the most obvious feature, it is essentially a disorder of type 1 collagen synthesis.
 |
| Fig. 19.2
Gross osteogenesis imperfecta.
|
There are four common types:
Type 1. Mildest form with fractures in childhood but less frequent with age. Patients have a slight blue discoloration to the sclera of the eyes. Hearing loss is common in adults.
Type 2. Most severe form with patients having fractures in utero or being stillborn.
Type 3. Severe. Frequent fractures in childhood and adults. Short stature, hearing loss and blue sclera common. Patients will often also have problems with their teeth development.
Type 4. Moderate form with fractures in child and adulthood. Sclera are often white but there is often hearing loss, short stature and possible teeth problems.
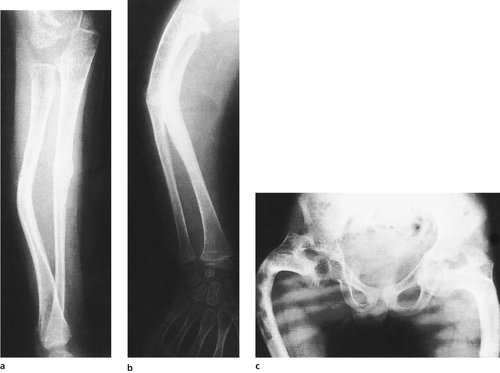
The brittleness of the bones is only part of the syndrome (Fig. 19.3). The teeth may also be affected and are often discoloured and abnormally thick – dentogenesis imperfecta. The ground substance in the sclerae of the eye may be abnormal, so that the retinal pigmentation is seen through the sclerae, producing a characteristic bluish discoloration. This is characteristic of the condition but not present in every patient. Mutations in the COL1A1, COL1A2, CRTAP and LEPRE1 genes appear to be the cause.
 |
| Fig. 19.3
(a), (b) Fractures in osteogenesis imperfecta (note the bowing). (c) Gross bowing of the femora in osteogenesis imperfecta.
|
Bones affected by osteogenesis imperfecta are more plastic than normal and bowing is a recurring problem needing correction by multiple osteotomy and internal fixation.
There are three grades of severity:
1. Some patients are so badly affected that they have multiple fractures in utero and do not survive.
2. Others have multiple fractures during childhood and develop a pigeon chest and scoliosis.
Although fragilitas ossium does occur, many parents believe that their accident-prone children must have a bony abnormality to account for their fractures and require firm reassurance. In other patients the diagnosis of a non-accidental injury must be considered.
Abnormalities of mineralization
Bone loss
Bone can be lost in three ways:
1. Osteomalacia – decreased mineralization.
2. Osteolysis – increased removal by osteoclasts.
3. Osteopenia – decrease in osteoid tissue. In practice, ‘osteopenia’ is more often used in the description of radiologically thin bones without implying a specific cause.
These three processes usually occur together in varying degree. The resulting loss of bone is called osteoporosis, of which there are three common types:
1. Idiopathic osteoporosis.
2. Disuse osteoporosis.
3. Steroid osteoporosis.
Idiopathic osteoporosis
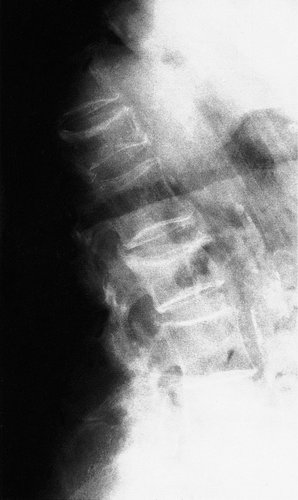
Lack of oestrogen causes a reduction in the amount of collagen in the bones of postmenopausal women, which become thin and ‘porotic’. Affected bone, particularly cancellous bone, is weaker than normal and susceptible to fractures (Fig. 19.4). Fractures of the femoral neck and crush fractures of the vertebrae are common after trivial injuries in elderly women with senile osteoporosis.
 |
| Fig. 19.4
Osteoporosis with crush fractures of the lumbar vertebrae.
|
Clinical and radiological features. The patients experience pain in the bones, especially the back. A kyphosis gradually develops. Radiologically, the bones look ‘thinner’ than normal and pathological fractures may be present.
Treatment. The treatment of osteoporosis is generally unsuccessful because the condition takes so long to appear that, by the time it is recognized, it is too late to treat it. If recognized early, hormonal treatment can be instituted, which can, at best, reverse the osteoporosis and restore normal bone texture.
The first line of treatment has been hormone replacement therapy (HRT) with special emphasis on oestrogen. The second line is low dose bisphosphonates (etidronate) given intermittently.
The ideal treatment has yet to be found but there has been a drive to make people and clinicians aware of the problems. Screening programmes have been started and these will hopefully allow earlier detection; this, together with the newer treatments, may help reduce the morbidity suffered by patients.
Disuse osteoporosis
Disuse osteoporosis is seen in bones that are not stressed normally. Patients confined to bed are particularly affected, as are paralysed limbs and fractures that are treated as non-weight-bearing. Astronauts undergo gross disuse osteoporosis.
Treatment is by mobilization and load-bearing.
Steroid osteoporosis
Steroid osteoporosis is seen in patients receiving large doses of steroids for rheumatoid arthritis or following transplantation, and in Cushing’s disease. The consequences include pathological fractures and vertebral collapse.
Treatment is by reducing the dose of steroids or treating the underlying disorder.
Rickets
Rickets is due to deficiency of calcium and phosphate in childhood (Fig. 19.5). Although much less common than in former times, rickets still occurs widely in developing countries. There are four causes:
1. The commonest is vitamin D deficiency due to inadequate diet or lack of natural vitamin D from exposure to sunlight.
2. Malabsorption of calcium due to steatorrhoea.
3. Renal osteodystrophy due to renal abnormality, which affects vitamin D metabolism and causes renal failure.
4. Hypophosphataemia due to a renal tubular abnormality. This condition causes vitamin D-resistant rickets.
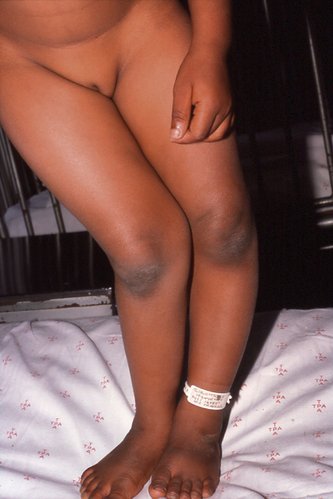 |
| Fig. 19.5
Rickets.
|
Clinical and radiological features. Rickets can be recognized clinically by the curved bones and the prominent epiphyses, which are due to an excess of unmineralized osteoid tissue. Radiologically, the osteoid seams at the epiphyses are widened and there is cupping of the epiphyses (Fig. 19.6).
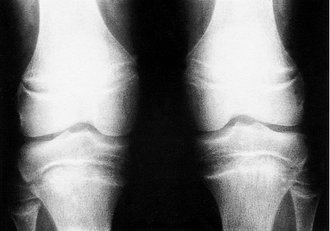 |
| Fig. 19.6
Rickets. Note the wide osteoid seams.
|
Investigations. Calcium levels are usually normal, but phosphate levels are low and alkaline phosphatase high.
Treatment. Vitamin D will produce a marked improvement but residual deformities may need correction by osteotomy.
Osteomalacia
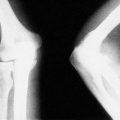
Osteomalacia, or softening of the bone, is caused in the adult by vitamin D deficiency. Osteomalacic bone is less rigid than normal bone. The long bones become bowed and small fractures occur on the tension surfaces of these bones.
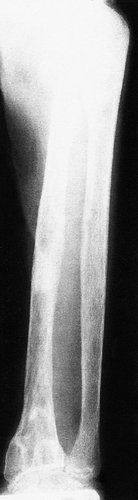
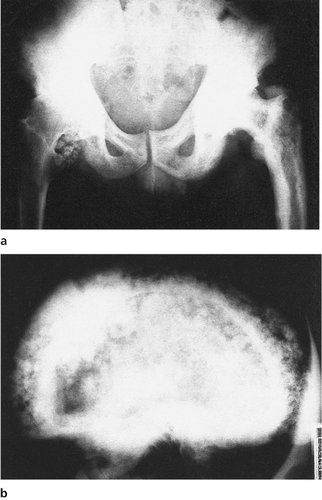
Clinical and radiological features. The patients are usually malnourished, unwell and complain of bone pain. Crush fractures of the vertebrae may be present. The fractures on the tension surfaces of the bones are visible radiologically as Looser’s zones (Fig. 19.7). The pelvis becomes trefoil in outline as the acetabula and the walls of the pelvis move inwards (Fig. 19.8).
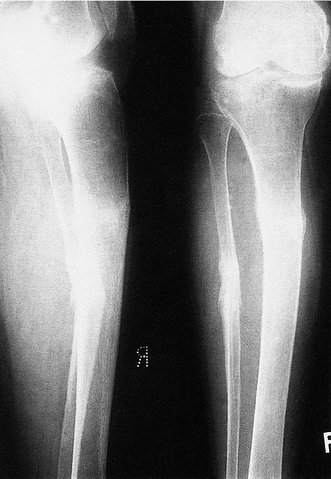 |
| Fig. 19.7
Pathological fracture of the tibia and fibula in osteomalacia. Note the Looser’s zone on the front of the tibia.
|
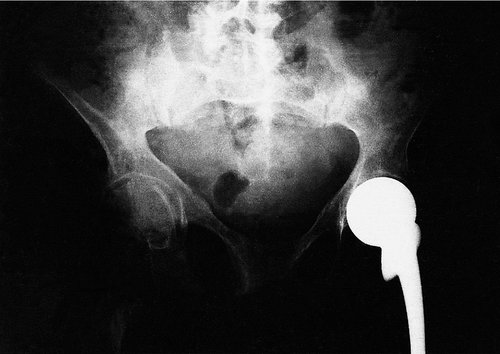 |
| Fig. 19.8
Osteomalacia. Note the trefoil pelvis, fractures of the inferior public rami and right femur and a Thompson prosthesis inserted for a pathological fracture of the neck of the femur.
|
Investigations. Serum calcium and phosphate may be lowered but alkaline phosphatase is raised. Bone biopsy shows wide osteoid seams.
Treatment is by correcting the dietary insufficiency and administering vitamin D.
Hyperparathyroidism
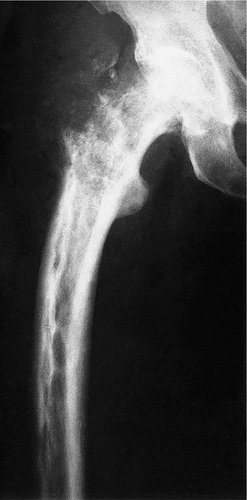
Hyperparathyroidism, also known as von Recklinghausen’s disease of bone and osteitis fibrosa cystica, is due to excess production of parathormone (Fig. 19.9). There is excessive resorption of calcium from the skeleton, and cysts full of brownish tissue (‘brown tumours’) form within the bone in severe disease.
 |
| Fig. 19.9
Hyperparathyroidism with cyst formation.
|
Hyperparathyroidism can be primary, secondary or tertiary:
1. Primary hyperparathyroidism is due to excess parathyroid production by a hormone-secreting adenoma.
2. Secondary hyperparathyroidism is due to excessive secretion of parathormone in order to mobilize calcium from the bones in response to low calcium levels from renal disease or malabsorption.
3. Tertiary hyperparathyroidism occurs when the parathyroid still secretes too much hormone, even when the reason for the excess secretion has been cured. The usual reason is an autonomous nodule of parathyroid tissue which develops while the patient has secondary hyperparathyroidism.
Clinical and radiological features. Classically, the patients have ‘moans, sore bones and abdominal groans’. The bone pain is due to softening and resorption of the bones and the moans to personality changes. The cause of abdominal pain is unknown.
Investigations. Serum calcium is high, phosphate low and alkaline phosphatase high in primary hyperparathyroidism. The levels vary in other types according to renal and other pathology.
Treatment. If the excess production of parathormone can be corrected, the bones return to normal. Surgical removal of the parathyroids is usually required.
Disorders of osteon architecture
Paget’s disease
Paget’s disease, or osteitis deformans, was described by Sir James Paget in 1879 and the cause is still unknown. It is the commonest bone dysplasia.
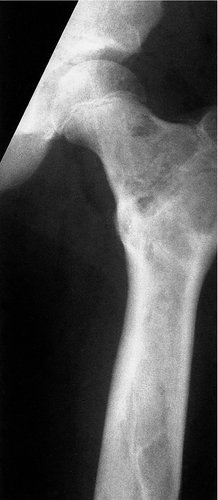
Bones affected by Paget’s disease increase in width, lose their normal architecture and develop an increased blood supply (Fig. 19.10). The increase in size distinguishes the condition from metastases and other disorders which look similar. The increase in blood supply makes the bone warm to touch and in some circumstances can lead to high output cardiac failure. Paget’s bone can also become bowed; hence the name, osteitis ‘deformans’. The bones can be abnormally soft or excessively hard and break easily.
 |
| Fig. 19.10
Paget’s disease of: (a) the pelvis and left femur; (b) the skull.
|
Paget’s disease does not cross joint spaces and is usually confined to individual bones, although many bones may be involved in the same patient.
Clinical and radiological features. The bones are often painful but many patients remain symptom-free, despite gross radiological changes, until they develop osteoarthritis in joints or suffer a pathological fracture. The bones lack the normal arrangement of trabeculae and may be larger than normal. Pseudofractures, like Looser’s zones (p. 322), are seen on the tension side of bones.
Investigations. Alkaline phosphatase is high and isotope scans show up affected areas as ‘hot’.
Complications. Paget’s bone is not as strong as normal bone and pathological fractures occur, but the fractures usually unite as quickly, if not more quickly, than fractures through ordinary bone.
A sinister complication is the development of a particularly malignant tumour, known as Paget’s sarcoma, in a small percentage of cases (Fig. 19.11). It is not known why some patients develop this tumour.
 |
| Fig. 19.11
Paget’s disease with pseudofractures along the convex side of the bone and sarcoma at the greater trochanter.
|
Treatment. Simple analgesics are not always effective for the pain. Parenteral calcitonin and oral diphosphonates are effective in 50–70% of patients but this figure is rising as newer biphosphonates are introduced.
Fibrous dysplasia
Fibrous dysplasia causes the formation of fibrous areas within bone (Fig. 19.12). Affected areas are fragile and pathological fractures occur. The cause is not known.
 |
| Fig. 19.12
Fibrous dysplasia of the femur.
|
Marble bone disease (Albers–Schönberg disease, osteopetrosis)
The normal tubulation of a long bone may be lost and the bone appears as a solid stick on the radiographs, while the vertebrae have a characteristic striped appearance (Fig. 19.13). The bones look very solid but are in fact very brittle. When they break it is almost impossible to fix the fractures internally because the bone is so hard that it cannot be drilled with ordinary equipment. The condition is usually inherited through an autosomal dominant gene but there is a more severe form inherited through an autosomal recessive gene.
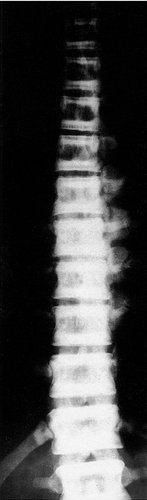 |
| Fig. 19.13
The rugger jersey spine of osteopetrosis (Albers–Schönberg disease).
|
Other dysplasias
There are countless other dysplasias, all interesting and obscure, and many with interesting radiological abnormalities. Striped bones, spotted bones (Fig. 19.14) and candlestick bones (melorheostosis) are all seen.
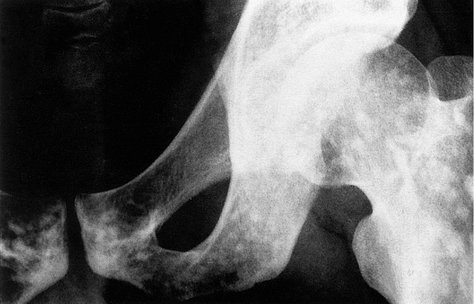 |
| Fig. 19.14
Osteopoikilosis, or ‘spotted bones’.
|
Abnormalities of cartilage
Mucopolysaccharidoses
Mucopolysaccharidoses are due to congenitally determined abnormalities in the composition of cartilage.
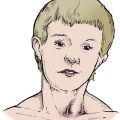
Hurler’s disease, or ‘gargoylism’, includes facial deformity, corneal opacities, epiphyseal and vertebral deformity and mental retardation. The disease is inherited through an autosomal recessive gene.
Morquio’s disease is similar to Hurler’s disease but does not include facial deformity, corneal opacities or mental retardation. The disease is inherited through an autosomal recessive gene.
Achondroplasia
Achondroplasia, or dyschondroplasia, in which the long bones do not grow as much as normal and the patients are very short, is inherited as an autosomal dominant (Fig. 19.15). The families of dwarves seen in circuses have achondroplasia. Achondroplastic bones are of normal strength and the patients are of normal intelligence. A similar genetic disorder in dogs is responsible for short-legged breeds such as dachshunds, basset hounds and corgis.
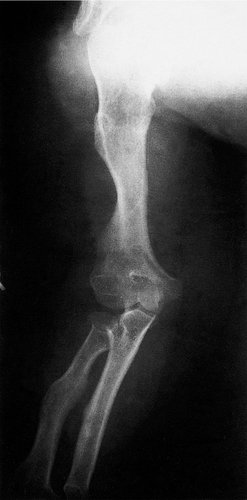 |
| Fig. 19.15
Achondroplasia.
|
Apart from the short stature and the social problems that this can bring, there are other complications of achondroplasia. The pedicles of the vertebrae are shorter than normal and the spinal canal is therefore abnormally narrow, which leads to spinal stenosis (p. 456) and neurological impairment.
Craniocleidodysostosis
This melodiously named condition is the ‘opposite’ of achondroplasia. Cartilage bones are normal but membrane bones, particularly the skull and clavicle, are imperfectly developed. No treatment is effective. The disease is inherited through an autosomal dominant gene.
Diaphyseal aclasis
Diaphyseal aclasis is a generalized failure of bone remodelling inherited as an autosomal dominant. It is easy to understand how a long bone grows from an epiphyseal plate but it is not so easy to understand why the shaft is narrower than the epiphysis. What force makes the bone become narrow? Whatever the mechanism, it sometimes fails and produces the appearance in Figure 19.16.
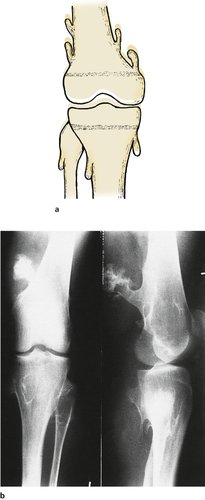 |
| Fig. 19.16
(a) Multiple exostoses pointing away from the growth plates in diaphyseal aclasis. (b) Radiograph of diaphyseal aclasis.
|
Dysplasia epiphysealis multiplex
Dysplasia epiphysealis multiplex is an inherited disorder which affects the epiphyseal plates. Growth is irregular and deformities involve many bones (Fig. 19.17).
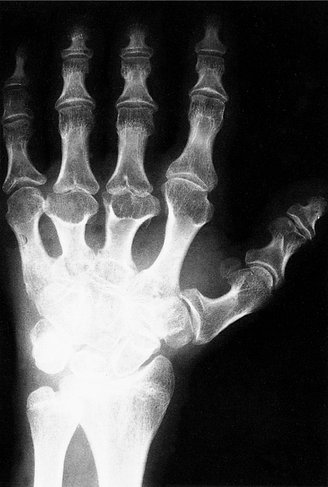 |
| Fig. 19.17
The hand in a patient with multiple epiphyseal dysplasia.
|
There are many other rare types of epiphyseal dysplasias affecting the behaviour of the growth plate in different ways.
Osteochondritis
Osteochondritis is a bad term, used to describe conditions which look a little similar but are caused by several different pathological processes:
1. Vascular abnormalities.
2. Damage to apophyses.
3. Conditions of unknown origin.
Vascular abnormalities
Many osteochondritides are caused by a transient disturbance of vascularity (Fig. 19.18). The cause is almost certainly partial venous occlusion, and is quite different from aseptic necrosis.
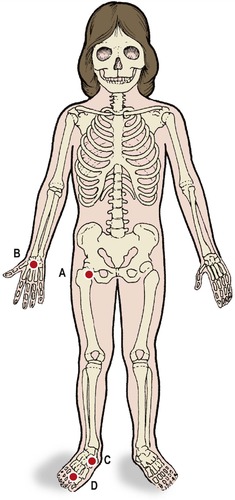 |
| Fig. 19.18
Sites of vascular osteochondritis: ( A) Perthes’ disease at the hip; ( B) Kienböck’s disease of the lunate; ( C) Köhler’s disease of the navicular; ( D) Freiberg’s disease of the metatarsal heads.
|
Perthes’ disease
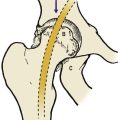
Perthes’ disease is an osteochondritis of the upper femoral epiphysis in which the femoral head becomes soft and then gradually reforms over a period of several years (Fig. 19.19). The condition is seen in children between the ages of 5 and 10 years and is commoner in boys. The primary pathology is an interference with the venous drainage of the femoral head. The reformed head is larger and flatter than the original.
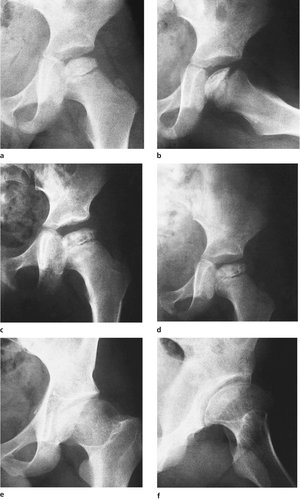 |
| Fig. 19.19
(a), (b) Perthes’ disease with increased density of the femoral head and a sequestrum within it; (c) 3 months later the changes have progressed; (d) 12 months later, the same femoral head is beginning to reform. (e), (f) The end result of Perthes’ disease, sometimes known as ‘coxa plana’ or ‘coxa magna’.
|
Treatment is directed to containing the femoral head within the acetabulum until it reforms. This can usually be achieved by conservative means, with splints if necessary, but osteotomy is sometimes required.
Kienböck’s disease
Kienböck’s disease is similar to Perthes’ disease but affects the lunate, which collapses, becomes dense and gradually reforms (Fig. 19.20). The disease presents with pain on the dorsum of the wrist over the lunate. The pain usually resolves after 1 year, but can recur when the wrist is twisted awkwardly or stressed.
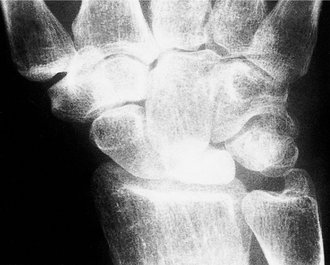 |
| Fig. 19.20
Kienböck’s disease of the lunate.
|
Köhler’s disease
Köhler’s disease affects the navicular bone in the foot and presents with pain on the dorsum (p. 432).
Other sites
Other epiphyses can be affected by the same cycle of rarefaction, collapse and reformation, including Freiberg’s disease of the second and third metatarsal heads.
Damage to apophyses
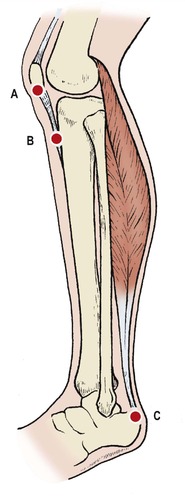
Muscles attached to an apophysis can lift all or part of the apophysis away from the bone during the adolescent growth spurt (Fig. 19.21). The bone is painful at first but the pain subsides gradually as the apophysis becomes reattached with growth. In some patients a sliver or spicule of bone remains detached and causes pain, rather like a splinter or foreign body. A better name for these disorders would be ‘traction apophysitis’.
 |
| Fig. 19.21
Sites of osteochondritis involving apophyses: ( A) Sinding Larsen’s disease; ( B) Osgood–Schlatter disease; ( C) Sever’s disease.
|
Osgood–Schlatter disease
The commonest traction apophysitis is Osgood–Schlatter disease, in which the apophysis of the tibial tubercle is lifted off the tibia. This happens most often in vigorous teenagers at the stage of growth when the quadriceps has enlarged, but before the apophysis has fused to the tibia – about 12–13 years in boys and a year less in girls.
There was no such person as Osgood–Schlatter. Osgood described the condition in America in 1903, the same year that Schlatter described it in Germany. Confusion about the name of the condition was settled by calling it ‘Osgood–Schlatter’s disease’. Although this shares the honours evenly, it sounds so serious that parents need reassurance that their child will survive.
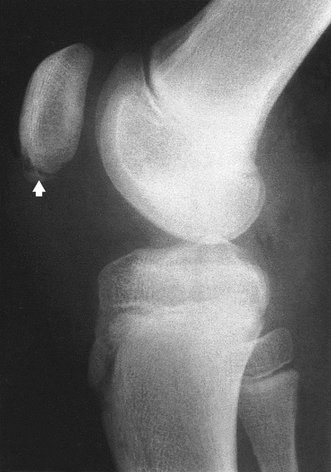
Treatment is conservative, by restriction of activities that cause pain. Plaster immobilization is not required. Most lesions heal as growth proceeds, but about 5% have persistent pain from an ununited spicule of bone within the tubercle (Fig. 19.22). Some of these respond to steroid injection, but a few need the spicule excised.
 |
| Fig. 19.22
Osgood–Schlatter disease of the tibial tubercle. A fragment of the apophysis has become detached from the tibial tubercle.
|
The bump at the tibial tubercle never diminishes in size and persists into adult life. Apart from the appearance, it causes few problems.
Sever’s disease
At the heel, pain and tenderness can arise at the upper edge of the calcaneal apophysis from traction apophysitis, which is an injury caused by overuse of the Achilles tendon (p. 433).
Sinding Larsen’s disease
Sinding Larsen (one person) described pain at the lower pole of the patella caused by traction of the patellar tendon. The disease is similar to Osgood–Schlatter disease but occurs 1 or 2 years earlier (Fig. 19.23).
 |
| Fig. 19.23
Sinding Larsen’s disease. The lower part of the patella shows a separate fragment of apophysis.
|
Scheuermann’s disease
The ring apophyses of the thoracic vertebrae can be damaged at their anterior border, producing pain during the growing period and growth arrest. The cause is unknown but traction is not involved and this separates the condition from the other apophyseal osteochondritides. The condition affects many levels and causes a rounded kyphosis of the thoracic spine (p. 459).
Treatment. No treatment is effective, and there is no disability apart from the shape of the back. Pain ceases when growth is complete. Parents should be told that the child cannot help standing with rounded shoulders and that nagging to ‘stand up straight’ is unhelpful.
Others
Calvé’s disease
A collapse of the vertebral body in children and young adults was formerly described as Calvé’s disease but many cases have since been shown to be the result of an eosinophilic granuloma of bone. Calvé’s disease does not exist as a separate entity.
Osteochondritis dissecans
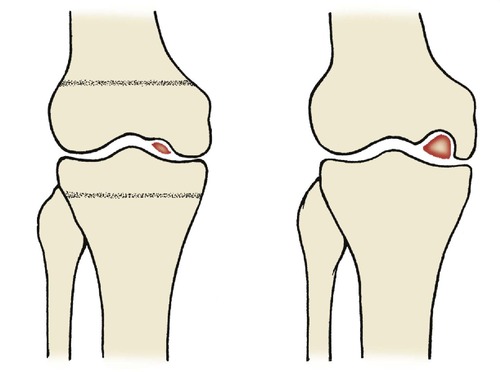
Osteochondritis dissecans consists of a gradual dissection (not desiccation) or separation of a block of bone from its bed (Fig. 19.24 and Fig. 19.25). The medial femoral condyle is by far the commonest site. Osteochondritis dissecans of other joints is rare. Progress should be assessed radiologically. The lesions may require reattachment drilling to encourage union, or removal of the affected area if it separates.
 |
| Fig. 19.24
Osteochondritis dissecans. The lesion begins as a small defect on the medial femoral condyle in the growing skeleton and increases in size, sometimes producing a loose body.
|
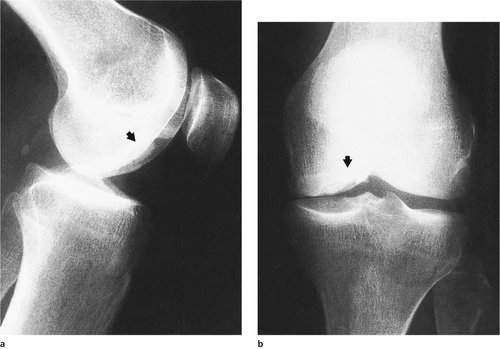 |
| Fig. 19.25
(a), (b) Osteochondritis dissecans. The end result in an adult, showing a potential loose body lying in a crater beneath intact articular cartilage.
|
Neuromuscular disorders
Although neuromuscular disorders fall within the province of neurologists, many present first to the orthopaedic clinic with abnormal gait. They must not be forgotten just because they are not ‘surgical’ diseases.
Duchenne muscular dystrophy
Duchenne muscular dystrophy is inherited through an X-linked recessive gene and therefore affects boys almost exclusively. The boy will have passed the early developmental milestones normally but then develops muscular weakness during childhood and presents with a broad-based waddle that can be mistaken for instability of the hips. The diagnosis can be confirmed by high creatinine phosphokinase levels and muscle biopsy.
Apart from splintage to prevent contractures and aids to minimize the impact of the disease, such as an electric wheelchair, there is no treatment to offer for this distressing disease. Progressive and severe scoliosis is common and spinal fusion may be needed to stabilize the spine.
Death from intercurrent infection in early adult life is usual.
Friedreich’s ataxia
Friedreich’s ataxia often presents with weakness of the ankles and is easily mistaken for a recurrent ankle sprain. The disease is familial and of variable severity.
Peroneal muscular atrophy
This familial condition involves both hands and feet, but principally the feet. There is a high arched foot, absent ankle jerk and an extensor plantar response.
A combination of soft tissue release and arthrodesis is required according to the severity of the disease. Some patients need no treatment.
Acute poliomyelitis
Because poliomyelitis is so rare in the West it is likely to be missed if the patient first presents to an orthopaedic clinic. Think of poliomyelitis if the patient has developed localized muscular weakness after a febrile illness.
Late poliomyelitis
The imbalance of muscle power produces deformity. The treatment of late poliomyelitis consists of balancing the remaining active muscles and splinting joints. Arthrodesis may be required to prevent deformity.
Weakness of the ankle dorsiflexors, for example, can be treated with a toe-raising splint or transfer of a plantarflexor from the calf. The variety of possible problems in poliomyelitis is infinite, and each case must be considered as if it were unique.