CHAPTER 87 Metabolic Bone Disorders of the Spine
Cellular Biology
Bone Cells
Four families of cells combine to create bone tissue that is biomechanically functional and structurally sound. These include the osteoprogenitor cell; its derivative, the osteoblast; the incorporated mature form, the osteocyte; and, finally, the osteoclast. Together, these cells produce new osseous tissue. This tissue is maintained through resorption and remodeling. In short, the cells determine mechanical stability and mineral homeostasis. Healthy bone metabolism results from equilibrating these competing forces. Some forms of metabolic bone disease, therefore, can be viewed as the pathologic imbalance—in number, power, or degree of differentiation—of bone-forming and bone-resorbing cells.1,2
The osteoprogenitor cells are components of the bone marrow stromal system, approximating the surfaces of bone (periosteum and endosteum) and the adjacent bone marrow environment.3 These potentially mitotic cells, under appropriate stimulation and conditions, give rise to either the bone-forming osteoblast or the cartilage-producing chondroblast. The natural history of these osteoprogenitor cells and the precise signals for their differentiation and modulation are not yet fully elucidated.
Osteoblasts sit on the metabolically active surface of bone in an adherent row, where they synthesize and release unmineralized bone matrix called osteoid.4–7 They subsequently participate in the mineralization of the osteoid by releasing packets of ions, as well as by synthesizing regulatory and crystal-nucleating noncollagenous proteins. These cells are characterized by high levels of alkaline phosphatase, the ability to manufacture type I collagen and osteocalcin, and the presence of numerous receptor sites for parathyroid hormone (PTH), estrogen, and other products. Osteoblasts are also thought to govern the actions of osteoclasts (described later), thus regulating and coupling bone formation and bone resorption.
The osteocyte, the terminal cell of the osteogenic cell line, is derived from osteoblasts.4,5 As the osteoblast synthesizes bone matrix, it becomes embedded within its product. The cell is now termed an osteocyte, and the space in which it sits is termed a lacuna. There is one osteocyte per lacuna. Despite being surrounded by bone matrix, the osteocyte communicates with other cells via slender cell processes that it extends for some distance into canaliculi in the surrounding matrix. Thus osteocytes are not completely isolated in their lacunae from other cells but appear to communicate with one another and with surface cells via these cell-to-cell junctions. In this way, osteocytes maintain the matrix that envelops them, regulate local ionic concentrations, and govern the degree of mineralization. In addition, they monitor the local mechanical load and signal changes in the matrix. The surface area covered by the canaliculi is greater than the surface area of osteoblasts and osteocytes combined.
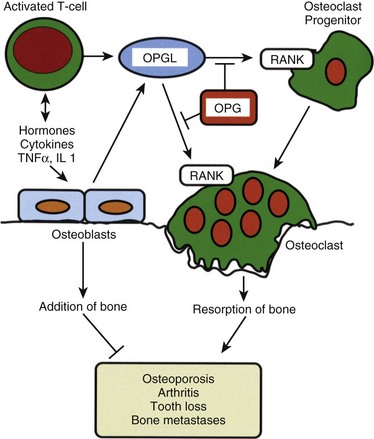
Osteoclasts resorb bone.8,9 Unlike the mesenchymally derived osteoblast lineage, osteoclasts arise from the monocyte line. These large multinucleated cells are found on the resorbing surfaces of bone, where they bind to a bone-specific integrin10 and form an isolated macrocavity. At this attachment cavity site, an acidic microenvironment causes the dissolution of the hydroxyapatite mineral and releases acidic proteases that degrade the organic collagen matrix. The resultant resorption cavity is called a Howship lacuna. Thus by remodeling formed bone and delivering calcium into the circulation, the osteoclasts participate in both the mechanical and biochemical roles of bone tissue. Osteoclasts are devoid of PTH receptors, even though they functionally respond to this hormone. Indeed, the hormonal response is found only in those osteoclasts approximated to functional osteoblasts (which do possess PTH receptors). On stimulation by PTH, osteoblasts produce RANKL (receptor activator of NFκB ligand), which interacts with RANK receptors on osteoclast progenitor cells, leading to the activation of mature osteoclasts. Osteoprotegerin (OPG, also known as NFκB) blocks this pathway. RANKL, RANK, and the decoy receptor OPG are the key regulators for the development and activation of mature osteoclasts (Fig. 87–1). Agents that inhibit RANKL via OPG or anti-RANKL antibodies can be used for the treatment of osteopenic disorders such as osteoporosis as well as bone loss associated with bone metastases.11–14
Bone Matrix
In addition to the cellular component, two matrices—an organic matrix and an inorganic matrix—constitute the remainder of bone tissue (Fig. 87–2). Whereas the inorganic matrix consists of bone mineral, the organic matrix consists of collagenous fibers embedded in a ground substance.
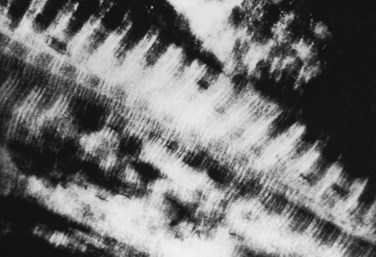
FIGURE 87–2 Collagen-mineral relationship. The hydroxyapatite forms in the hole zone between collagen molecules.
(From Anatomy II. Orthop. Science. Park Ridge, IL, AAOS, 1986.)
Bone Mineral
The inorganic matrix represents two thirds of the dry weight of bone and primarily consists of calcium phosphate,15 which exists in three forms: (1) crystalline hydroxyapatite [Ca10(PO4)6(OH)2], the most abundant; (2) octacalcium phosphate [Ca8H2(PO4)6 · 5H2O)], a rarer form; and (3) brushite [CaHPO4 · 2H2O]. The hydroxyapatite forms platelike crystals 40 nm in length and 3 nm in thickness. Occasional contaminants including carbonate substituted for a phosphate or a fluoride for a hydroxy group (as well as the potential inclusion of lead or arsenic) disrupt the purity of the crystals. These contaminants alter the physical properties of the matrix and may also affect the biologic characteristics. The mineral crystal closely associates with the organic matrix, initially deposited in the hole zones of the collagen fibril. Later, it surrounds itself with ground substance (proteoglycan), as well as with water and other ions.
Collagen
Concentrations of calcium and phosphate alone do not dictate mineral deposition. An organic matrix, consisting of collagenous fibers embedded in ground substance, assists with and regulates mineralization.16 The organic matrix of bone is primarily composed of proteins, approximately 95% of which is collagen in adult humans. Collagen, a rigid macromolecule, employs tropocollagen as its basic structural unit. Tropocollagen, in turn, is composed of three polypeptide chains, each comprising approximately 1000 amino acids.17 In bone collagen (type I), two of these chains share an identical amino acid sequence; a third chain (α2) has a similar, but not identical, sequence. All three chains contain unusually high concentrations of glycine, proline, alanine, hydroxyproline, and hydroxylysine and are wound into right-handed helices.
Type I collagen synthesis occurs in osteoblasts. It is formed as a precursor “procollagen” molecule in the cytoplasm and then extruded from the osteoblast after cleavage of signal sequences. Numerous additional modifications are subsequently made to the secreted procollagen molecule. Notably, extensive nonhelical regions are removed from both its amino and carboxy terminals, and key proline and lysine residues are hydroxylated by an iron-containing enzyme. (The iron in this hydroxylase functions only in the ferrous state; therefore ascorbic acid, functioning as a reducing agent, is necessary.) The hydroxylation of proline and lysine occurs after these amino acids are incorporated into the chain; therefore free hydroxyproline in the serum signifies lysis of collagen or its precursors. Accordingly, measurements of urinary hydroxyapatite excretion provide a qualitative measure of bone turnover. The hydroxylation of proline and lysine assists crosslinking, which, in turn, lowers the solubility of collagen and increases its tensile strength. Collagen is one of the strongest components of bone; the tensile strength of bone collagen exceeds the compressive strength of the bone mineral by 2000 N/cm2. Pyridinoline, deoxypyridinoline, C-telopeptides, and N-telopeptides are crosslink breakdown products that all provide better markers of bone resorption than hydroxyproline.18,19 They are elevated in high-turnover osteoporosis and Paget disease, and they decline rapidly with successful antiresorptive therapy (see later). Adequate nutrition and vitamin C (ascorbic acid) are required for collagen maintenance.
Type I collagen is found not only in the bones of the spine but also in the intervertebral discs, where it coexists with type II collagen. These homologous molecules differ only in the chains that compose their triple helices. Within the disc, each type of collagen maintains a separate anatomic domain: type I collagen is more abundant in the annulus fibrosus, whereas type II predominates in the nucleus pulposus.20
Ground Substance
Proteoglycans form the ground substance of bone. A proteoglycan monomer is composed of two kinds of glycosaminoglycans—keratin sulfate and chondroitin sulfate—joined to a core protein. Keratin and chondroitin sulfate form through the polymerization of disaccharide units, one sugar of which is always a hexosamine possessing an anionic group. This negatively charged group binds water, expanding the tissue volume and imparting resilience to the ground substance. In addition, the presence of water in the ground substance allows for the diffusion of metabolites in the organic matrix. Thus the proteoglycans in bone exert regulatory and structural forces. When the bone mineralizes, the water content is reduced, leading to denser, more compact tissue. The intervertebral discs also contain proteoglycans, which comprise 30% to 60% of the dry weight of the nucleus pulposus. With age, proteoglycan concentration (and, consequently, water concentration) declines. However, even with decreased proteoglycan concentration, osmotic forces allow the proteoglycans to remain somewhat hydrated.21
Osteocalcin is a bone-specific protein that, like type I collagen, is synthesized by osteoblasts.22 Osteocalcin comprises only 2% of the organic matrix by weight. Vitamin D enhances the synthesis of osteocalcin but is not an absolute requirement for synthesis. Osteocalcin prefers to bind to calcium within hydroxyapatite. The precise function of this prevalent osseous protein is unknown, although its levels are correlated with bone mineral content. Possible roles include the attraction of osteoclasts to sites of bone resorption, regulation of the rate of mineral turnover, and determination of the morphology of the mineral crystal. Osteocalcin, derived from new bone synthesis, circulates in plasma at concentrations proportional to osteoblast activity. A correlation between abnormal osteocalcin levels and radiographic evidence of metabolic bone disease has been suggested.
Osteonectin, a 32-kd phosphoprotein secreted by osteoblasts, is the second most prevalent protein in bone. It binds to both collagen and hydroxyapatite,23 as well as to carbohydrate moieties. Putative roles for osteonectin include calcium phosphate nucleation and stabilization of the newly formed crystal.
Bone morphogenetic protein (BMP), as the name implies, induces osteoprogenitor cells to form bone.24 A small protein, BMP accounts for only 0.1% of the total bone protein mass in cortical bone, and even less in the spine. More than 15 BMPs exist, and some are related to the transforming growth factors. Experimentally, BMP implanted in ectopic sites (in the absence of bone collagen) induces perivascular mesenchymal cells to become bone-forming osteoprogenitor cells. The absence of BMPs (or the inhibition of their activity) may contribute to the pathogenesis of diseases of decreased bone mass or impaired bone remodeling or repair. The genes for the expression of BMPs have been cloned,24,25 and the protein has proven to possess a pharmacologic role in repairing bone and inducing spinal arthrodesis.26
Skeletal Homeostasis
Bone growth occurs during all phases of life.4,5 Functionally, this growth can be divided into two processes: modeling and remodeling. The deposition of new bone in regions not first cleared by resorption or the resorption of old bone without deposition of new tissue is termed modeling. Modeling results in changes to the external shape, mass, or volume of existing bone. Alternatively, remodeling retains approximately the external form of bones. During remodeling, bone replaces itself by balancing synthesis in some places with lysis in others. This balanced turnover releases ions into the circulation and prevents accumulation of aged or fatigued bone.
Mineral Homeostasis
Calcium
Bone exchanges calcium, magnesium, and phosphate and participates in acid-base balance. Of all of these, calcium homeostasis is the most important to both the skeletal and metabolic functions of bone.27–30 Calcium fulfills its skeletal role in the hydroxyapatite mineral crystal, where it provides mechanical strength. It serves its metabolic function as a free divalent cation. These functions include transducing hormonal signals within the cytoplasm (“second messenger” function), coupling neural excitation with muscular contraction, and effecting homeostasis by interacting with both vascular smooth muscle and platelets.
Active absorption in the duodenum takes place by means of a protein-dependent transport system31 that is activated by 1,25-dihydroxyvitamin D (1,25[OH]2D). In the jejunum, passive diffusion accounts for the remainder of the calcium absorbed. Clearly, the jejunal phase depends more on concentration gradients and intestinal transit times. In addition, dietary composition affects absorption: Diets high in fiber and oxalate (both found in green vegetables) allow for less calcium absorption. In times of increased calcium demand such as the growth years, or during pregnancy, the duodenal fractional absorption can be augmented by increased carrier protein synthesis, which is in turn stimulated by the active vitamin D metabolite (see later). The efficiency of calcium absorption decreases with aging and is adversely affected with bariatric surgery.29,32
Phosphate
The second important ion in bone metabolism is phosphate.33 Like calcium, phosphate serves multiple functions throughout the body. As a component of adenosine triphosphate, phosphate participates in the interconversion of the energy of metabolism; as a constituent of nucleotides, phosphate partakes in the transmission and expression of genetic information; and in 2,3-diphosphoglycerate, it regulates the oxygen affinity of hemoglobin.
As with calcium, the kidney is responsible for maintaining phosphate balance,34 and it does so by a similar mechanism: proximal sodium bulk-driven flow and distal control. When phosphate intake is high, the kidney excretes increasing amounts by spilling the excess phosphate into the urine. When serum phosphate concentrations are low, the kidney can avidly conserve phosphate by implementing vitamin D–mediated changes in calcium concentration. In addition to calcium balance, dietary load, volume status, and acid-base balance also affect renal phosphate handling.
Regulators of Bone and Mineral Metabolism
Parathyroid Hormone
Parathyroid hormone is an 84-amino acid polypeptide secreted by the chief cells of the parathyroid glands in response to low serum calcium.35 Its physiologic role is to restore a normal calcium concentration by stimulating all three organs of calcium homeostasis. The kidney and the bone are affected directly,29,36 whereas the intestine is affected only indirectly, by means of the synthesis of the active vitamin D metabolite 1,25(OH)2D3.
PTH promotes calcium conservation in the kidney.37 There, increased serum calcium is achieved through a twofold mechanism; both steps depend on stimulation of adenylate cyclase, the enzyme that forms cyclic adenosine monophosphate (cAMP). First, PTH increases calcium resorption in the distal nephron. In addition, it promotes the loss of phosphate. This phosphaturic effect prevents the recently resorbed calcium from being deposited into the bone hydroxyapatite or into ectopic calcium-phosphate deposits. Moreover, the excretion of phosphate lowers the levels of circulating calcium-anion complexes, causing more of the serum calcium to remain in the physiologically useful, free form.
In the bone, PTH affects calcium metabolism by increasing the surface resorption of the mineral by osteoclasts, by promoting ion flux by osteocytes, and by decreasing their bone synthetic activity through the inhibition of calcium “consumption” by osteoblasts. PTH increases the number of resorptive surfaces in bone and, within them, the density of osteoclasts. The precise mechanism of this osteoclastic action is unfolding and appears dependent on the actions of osteoblasts. The osteoblast does possess PTH receptors. It is postulated that PTH induces the osteoblast to activate the RANKL-RANK system, which stimulates osteoclastic bone resorption. The catabolic effects are reversed by low, intermittent doses of PTH that directly stimulate osteoblastic bone formation but do not activate RANKL.21
Vitamin D
Unlike the polypeptide hormone PTH, vitamin D is a steroid.36 As such, it defies rapid proteolytic inactivation and thus functions best as a longer-acting regulator of calcium homeostasis. The principal effect of this hormone is to increase intestinal absorption of calcium from the diet, a process that is usually only 25% efficient. Secondarily, it complements PTH in the promotion of calcium resorption from the bone, again by promoting transport across cell membranes (in this case, bone cells rather than intestinal cells).
Cholecalciferol (inactive vitamin D3) forms in the skin from the ultraviolet light activation of 7-dehydrocholesterol, an intermediate of cholesterol synthesis.38 7-Dehydrocholesterol can degrade and re-form cholecalciferol; therefore once formed, cholecalciferol is removed from the local environment by a transport protein, thus favoring further formation. As little as 15 minutes of sunlight exposure allows the synthesis of a fair-skinned individual’s vitamin D3 requirement. The total need for sunlight increases with melanin concentration. In the absence of sun exposure, cod liver oil and enriched milk and cereals can provide usable precursors of the hormone.
In the liver, vitamin D3 undergoes 25-hydroxylation,39 forming the prehormone 25-hydroxyvitamin D (25[OH]D). Subsequent 1-hydroxylation to the active form, 1,25(OH)2D3, takes place in the kidney and is promoted by PTH. In the presence of low PTH, an alternative hydroxylation occurs at the 24 position, yielding 24,25(OH)2D3, an inactive form of the hormone. Unlike PTH, which is functionally regulated at the level of secretion (calcium levels do not cause minute-to-minute changes in messenger RNA activity but rather control the release of PTH), vitamin D is regulated at the level of biosynthesis. The 1-hydroxylation reaction is the rate-limiting step and is regulated by feedback inhibition, as well as by PTH levels.
Active vitamin D, also known as calcitriol, promotes increased transport across cell borders. As a steroid maturation hormone, vitamin D works by crossing the cell membrane and subsequently entering the nucleus. There it binds to DNA to promote DNA translation, thus increasing the synthesis of the target proteins. Vitamin D mediates increased expression of calcium-transport proteins, although the nucleic receptor has not yet been identified. In the gut, 1,25(OH)2D3 causes increased calcium transport; phosphate absorption increases as well. In the bone, 1,25(OH)2D3 complements the action of PTH by increasing calcium transport across bone cell membranes, thus assisting with the mobilization of calcium from the bone and into the circulation. As such, it functions as an agent of bone resorption. It can indirectly assist with bone formation as well because the tasks of mineral homeostasis and skeletal homeostasis are not always at odds with each other. Calcium needs can usually be met through the diet, not by bone resorption. Vitamin D also functions as a maturation hormone by increasing the villous membrane of the gut and augmenting PTH recruitment of macrophage stem cells, the bone-resorbing osteoclast precursors. Accordingly, vitamin D, which promotes the uptake of dietary calcium, typically also serves as an agent of bone maintenance. In addition, vitamin D has been associated with decreased cancer rates, improved muscle function, and a more normal immune system.40
Calcitonin
Calcitonin is a 32-amino acid polypeptide secreted by the parafollicular cells in the thyroid in response to increased serum calcium concentration. Calcitonin lowers serum calcium levels by its actions in the bone and the kidney. Still, its precise physiologic role awaits determination.41,42
In the bone, calcitonin rapidly inhibits osteoclastic action at pharmacologic doses. Direct calcitonin-binding sites are present on osteoclasts. Calcitonin decreases the number and activity of osteoclasts. Specifically, it decreases the adherence of osteoclasts to bone resorptive surfaces and diminishes their activity once they adhere. Calcitonin also affects osteocytes, causing decreased calcium ion flux across their cell membranes. In the kidney, calcitonin blocks the reuptake from the glomerular fluid of calcium and phosphate, as well as that of other ions, notably sodium (which influences calcium uptake by the passive mechanisms described earlier). Calcitonin may also influence calcium handling in the gut and, at pharmacologic doses, may alter other intestinal processes (such as water balance).43
Osteoporosis
Osteoporosis is a metabolic bone disease characterized by low bone mass and microarchitectural deterioration of bone tissue, leading to increased bone fragility and a consequent increase in fracture risk, especially of the hip, spine, and wrist. Osteoporosis affects an estimated 28 million Americans. Of these, 10 million are estimated to already have the disease and nearly 18 million more are estimated to have low bone mass, placing them at increased risk for osteoporosis.44 It is the most prevalent metabolic bone disease. The estimated national direct expenditures (hospitals and nursing homes) for osteoporotic and associated fractures were $17 billion in 2001 (or $47 million each day) and will likely rise with the expanding elderly population. In postmenopausal women and in the elderly, osteoporosis is a major underlying cause of pathologic fractures. At least 1.5 million fractures each year are attributable to the disease,45 and almost half of these occur in the spine. Moreover, one third of all American women older than 65 years of age suffer vertebral fractures.46
Like all metabolic bone diseases, osteoporosis is not limited to the spine. In addition to the annual 700,000 vertebral fractures caused by osteoporosis, the disease claims 300,000 hip fractures and 250,000 Colles fractures.45 In the elderly, hip fractures, most caused by osteoporosis, result in death, disability, and dependency: The mortality rate of older patients with hip fractures can be as much as 20% higher than in persons of similar age47; up to 50% require long-term nursing home care, and less than 30% return to a lifestyle comparable with their prefracture state.48 Among the vertebral fractures, two thirds are clinically silent; however, these patients have lower quality-of-life function.49 Women with at least one new vertebral fracture had an age-adjusted 32% increased risk of mortality. When these women were stratified by number of incident vertebral fractures, those with two or more incident vertebral fractures had a 1.47-fold increased risk of mortality.50
Generally, osteoporosis is classified as either primary or secondary. Primary osteoporosis is further subdivided on the basis of its pathogenesis. Type I, or postmenopausal osteoporosis, is related to the abrupt decline of estrogen levels that occurs in menopausal women. Type II osteoporosis, known as senile or age-related osteoporosis, is due to the progressive decrease of bone mineral density in both men and women that occurs with aging. Patients may suffer from both subtypes of primary osteoporosis. Primary osteoporosis can occur in both men and women of all ages but often follows menopause in women and occurs later in life in men. Several risk factors for osteoporosis have been identified. A typical patient is a slim, white, postmenopausal woman of northwestern European descent.38 She may have a history of premature menopause, cigarette or excessive alcohol use,27 an eating disorder (e.g., anorexia nervosa), a sedentary lifestyle, use of anticonvulsants,51 or lifelong low calcium intake. Seventy-five percent of women with scoliosis older than 65 years of age have at least one osteopenic wedge fracture.52
Secondary osteoporosis results from any medical condition or medication that contributes to accelerated bone loss. The incidence of secondary osteoporosis is higher in men than in women and is estimated to be 30% to 60%.53 Causes of secondary osteoporosis include endocrine and gastrointestinal disorders, bone marrow dysplasias, and disorders of calcium balance, among others.54,55 Endogenous hyperthyroidism increases cortical bone loss and may accelerate skeletal deterioration.56 Inflammatory bowel disease (i.e., Crohn disease or ulcerative colitis) can cause osteoporosis both from the disease itself and its treatment. Malabsorptive diseases of the gut such as celiac sprue are also associated with low bone density and osteoporosis. Corticosteroid-induced osteoporosis is the most common cause of drug-related osteoporosis and is associated with a high fracture rate. Corticosteroid therapy causes bone loss and fractures because it suppresses bone formation and inhibits intestinal calcium absorption, which leads to secondary hyperparathyroidism and increased osteoclastic bone resorption.57 In addition, osteomalacia (vitamin D deficiency) may mask itself as osteoporosis. The distinction is important because antiresorptive medications for osteoporosis are not appropriate choices to treat osteomalacia. Many secondary causes of osteoporosis are treatable and may necessitate a different management course than primary osteoporosis.
Pathology
Osteoporosis is a disease of decreased bone mass in the absence of a mineralization defect. Bone mass is diminished; however, the bone that remains contains a normal matrix and a normal degree of calcification. Declining bone mass is now thought to be a universal phenomenon of aging.58 Peak bone mass is attained in the mid 30s for both sexes. Gender, nutrition, race, exercise habits, and overall health all influence bone mass. Peak bone mass is higher in men than in women and higher in African Americans than in whites.59 After the fourth decade, both men and women lose bone mass from the skeleton (Fig. 87–3). Two phases of this loss have been identified: slow and accelerated. The slow phase, related to an imbalance between resorption and formation leading to negative calcium balance, is equal in both men and women. It results in an annual basal slow phase rate of bone loss of 0.3% to 0.5%.60 The accelerated phase that occurs with estrogen deficiency—a phenomenon found exclusively in women—is responsible for cortical bone mass loss of 2% to 3% per year. This loss is in addition to the slow phase losses, which continue during the accelerated phase. The accelerated phase begins after surgical or natural menopause and lasts for approximately 5 years.60 Thereafter, bone loss continues at the basal slow phase rate.
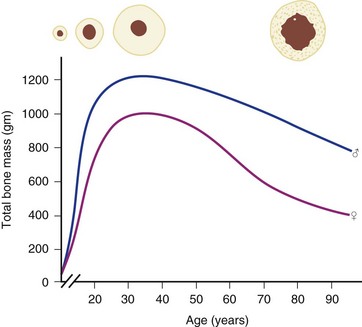
FIGURE 87–3 Bone mass as a function of age in men and women.
(From Disorders of Bone 9. Orthop. Science. Park Ridge, IL, AAOS, 1986.)
Multiple studies have shown the importance of estrogen deficiency in the causation of the accelerated phase.61,62 Estrogen is thought to play a critical role in maintaining bone mass in adult women by suppressing cancellous bone remodeling and maintaining remodeling balance between osteoblastic and osteoclastic activity. With estrogen deficiency, there is increased osteoclastic activity and possibly also impaired osteoblastic activity, resulting in a remodeling imbalance.63 The administration of estrogen to women during this period of rapid bone loss can decrease the loss in all bones, especially those rich in trabecular bone (e.g., the vertebral bodies, the pelvis and other flat bones, in the ends of long bones).64
Riggs and Melton60 have subclassified primary osteoporosis on the basis of patterns of bone loss and fracture (Table 87–1). Type I or “postmenopausal” osteoporosis primarily affects trabecular bone sites and is characterized by fractures of the vertebral bodies and wrist. In patients with type I osteoporosis, the rate of trabecular bone loss is three times above normal but the rate of cortical bone loss is only slightly above normal. In contrast, type II osteoporosis, also known as “senile” osteoporosis, occurs in both men and women aged 75 years and older and involves areas of predominantly cortical bone. Clinically, fractures of the hip, pelvis, proximal humerus, and proximal tibia are seen. The causes of senile osteoporosis are the aging process itself and chronic calcium deficiency. Senile osteoporosis may also involve decreased vitamin D and increased PTH activity or impaired bone formation. Osteoporosis in patients between the ages of 66 and 74 years may represent a combination of these two syndromes.
| Type I | Type II | |
| Female-male ratio | 6 : 1 | 2 : 1 |
| Calcium deficiency | No | Yes |
| Estrogen deficiency | Yes | No |
| Bone loss | Disproportionate loss of trabecular bone | Proportionate loss of both cortical and trabecular bone |
From Riggs BL, Melton LJ 3rd: Evidence for two distinct syndromes of involutional osteoporosis. Am J Med 75:899, 1983.
The spine is composed of primarily trabecular bone. Compared with cortical bone, it has a high surface-to-volume ratio. Because metabolic activity (remodeling) occurs on bone surfaces, trabecular bones in general, and the vertebral bodies in particular, are resorbed preferentially in times of skeletal loss.65 Osteoporosis is thus characterized by trabeculae of decreased size and number. Work by Dempster and colleagues66 has demonstrated that osteoporosis involves a thinning of the cortex, as well as a change in the shape of the trabecular bone from plates to narrow bars. The trabecular bone contains areas in which osteoclasts create a loss of connectivity, leading to a significant and localized weakening of the bone.66,67 Vertebral body density declines before a similar loss is detected in cortical areas.
The body accommodates bone loss by redistribution. As people grow older, the diameter of the long bones gradually increases in both women and in men. Concurrently, the medullary diameter also increases, leading to a net thinning of the cortical bone. A 10% shift of bone mass outward from the epicenter through an enlargement of the bone diameter will compensate for a 30% decrease in the bone mass against applied bending and torque stresses but not against axial loading. This differential resorption explains the timing and patterns of the fracture syndromes seen in osteoporosis.46 The incidence of vertebral compression fractures rises immediately after menopause, whereas the hip (with its higher proportion of cortical bone) fractures later in life, when cortical bone loss accumulates over the next 1 to 2 decades. The distal forearm, like the spine, has high trabecular content, so the incidence of Colles fractures also increases in menopause.
Deficiencies in dietary calcium cause decreased peak bone mass.68 White women on average have less bone than either white men or African Americans of either sex.69 Thus the risk of clinically significant osteoporosis depends on hereditary factors, gender, race, and nutrition, which all contribute to peak bone mass, and aging, which causes progressive bone loss. Although the formation of good bone in sufficient quantity in young adulthood is clearly important, the aging process remains the most important cause of involutional osteoporosis.
Aging leads to bone loss independent of menopause, but the rapid decline of skeletal mass after estrogen deficiency implies that this hormone prevents the dissolution of the skeleton.70 Nonetheless, its precise mechanism is unclear. Some investigators believe that the action is mediated by estrogen receptors on osteoblasts.71 Others contend that estrogen antagonizes PTH activity72 or stimulates endogenous calcitonin release. Decreased calcitonin levels have been found in oophorectomized and postmenopausal patients, and increased calcitonin levels are noted after estrogen administration. Regardless of the mechanism, estrogen deficiency leads to bone loss and plays a major role in the pathogenesis of type I osteoporosis.
The age-related (“slow phase”) bone loss is not affected by estrogen; rather, it likely represents impaired vitamin D metabolism. As an individual ages, the kidney gradually loses its ability to hydroxylate vitamin D into its active form, 1,25(OH)2D.73 This active vitamin D is necessary to transport calcium in the gut; therefore decreased hydroxylation, often coupled with poor dietary intake, leads to lowered serum calcium levels and, in turn, to elevated secretion of PTH. In addition to secondary hyperparathyroidism, elderly people are also more likely to take medications (e.g., diuretics) that further contribute to calcium losses. Furthermore, there is some evidence that the mechanism in remodeling that couples the actions of osteoclasts and osteoblasts functions suboptimally in old age: after the fourth decade, at a given site of remodeling, less bone is laid down than is resorbed.69 Accordingly, normal bone turnover in the elderly leads to calcium depletion as well.
Clinical Course
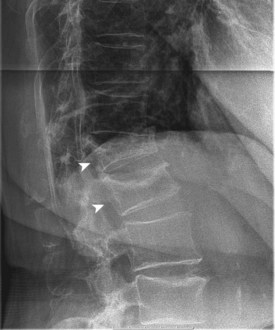
End-stage osteoporosis culminates in fracture.27,29,74 Still, for most of its course osteoporosis is a silently progressive disease. Patients typically present to the physician, usually late in the course, following one of three general scenarios. In the first of these, the patient presents with an acute painful fracture, usually of the spine (Fig. 87–4), but possibly of the rib, wrist, or hip. In the spine, normal activity (even minimal activity) may exceed the depleted bones’ stress tolerance and result in fracture. The acute fracture can be severely painful, with the pain remaining over the affected area or radiating across the thorax. The pain from a vertebral fracture does not radiate down the legs. Symptoms such as leg pain suggest involvement of the spinal cord and obligate the physician to search for another or concomitant process to explain the pain. Osteoporosis, even if established in such a case, is probably not the only disease present. The acute pain usually abates when the fracture heals. Nonetheless, the patient rarely returns to the prefracture status. At a minimum, there may be point tenderness over the fracture site. The patient may note constant abdominal pain, often brought about by the constraining forces on the viscera in the now smaller abdominal cavity. Other patients complain of generalized backache. The backache may be due to changing muscular demands brought on by altered spinal curvature. Furthermore, some patients fear reinjury and strictly limit their activities. Others develop chronic pain syndromes, dysthymic states, or even overt clinical depression. Osteoporosis, therefore, often has a profound effect beyond the acute fracture episode.
Diagnosis
While a good clinical understanding of osteoporosis takes into account the pathophysiology of bone remodeling, mineralization changes, and variable bone quality of the patient, the diagnosis of osteoporosis until recently has relied on a single criterion: the bone mineral density. Bone mineral density testing is currently the clinical standard for diagnosing osteoporosis. Bone mineral density is the best predictor of osteoporotic fractures in postmenopausal women because bone mass accounts for approximately 60% to 80% of bone strength.75–77 Bone mineral density testing offers a painless and noninvasive way to identify osteoporosis, predict a patient’s risk for fractures, and monitor treatment.
The World Health Organization (WHO) definition of osteoporosis is expressed in terms of the T score, defined as the number of standard deviations below the peak adult bone mass of young healthy adults (Table 87–2). Individuals whose T score is within 1 SD of the mean (T score ≥ −1) are considered to have normal bone density. Individuals with osteopenia have T scores that fall between 1 and 2.5 SD below the mean (T score < −1 but > −2.5). An individual with a T score that is at or below –2.5, or 2.5 SD or more below the average peak density of a young adult, is considered to have osteoporosis. Severe osteoporosis defines an individual with bone density that is 2.5 SD or more below the average peak of a young adult in the presence of one or more fragility fractures. In addition, a patient may fracture without “osteoporotic” bone by bone density levels. The WHO set a standard with definitions of “osteopenia” and “osteoporosis,” defining the latter as low bone density with increased fracture risk.
TABLE 87–2 Definition of Osteoporosis Based on Total Hip Bone Mass Density
| Group | T score |
|---|---|
| Normal | ≥−1.0 SD |
| Osteopenia (low bone mass) | Between −1.0 and −2.5 SD |
| Osteoporosis | ≤−2.5 SD |
| Severe osteoporosis | ≤−2.5 SD and fragility fracture |
SD, standard deviation.
Adapted from Kanis JA, Glüer CC: An update on the diagnosis and assessment of osteoporosis with densitometry. Committee of Scientific Advisors, International Osteoporosis Foundation. Osteoporos Int 11:192-202, 2000.
The Z score compares a patient’s bone mineral density to a reference population of the same age, sex, and ethnicity fracture. The Z score indicates how many standard deviations the patient’s bone mass density is below the expected value for his or her own age. The Z score cannot be used to diagnose osteoporosis, but it is useful for screening the patient for secondary causes. A Z score of −1.5 or lower should increase the index of suspicion that underlying medical problems, medications, or other factors may be responsible for the patient’s low bone mass density.78
Although several tests are available to measure bone mineral density, dual-energy x-ray absorptiometry (DEXA) is the technical standard. DEXA is the preferred technique because it measures bone mineral density at the important core sites of osteoporotic fractures including the hip and spine. The WHO has reported that a measurement at either the total hip or femoral neck is preferred, but that posteroanterior (not lateral) vertebral bone mineral density can be used to make the diagnosis.74
The spine is a useful site for bone mineral density measurement because it predicts the risk of any fracture as well as or better than hip bone mineral density in the perimenopausal population in which hip fracture risk is low. With advancing age, however, spine measurements are confounded by osteoarthritis and soft tissue calcification to a greater degree than the hip.79,80 For this reason, the hip is the preferred site, especially in women older than 60. Based on the WHO diagnostic threshold, 15% to 20% of postmenopausal women can be identified as having osteoporosis when measurements using DEXA are made at the spine or hip.74
A number of risk factors for osteoporosis have been identified by the International Society for Clinical Densitometry (ISCD)81 and should be used to guide the screening process in a cost-effective manner. The current indications for bone mineral density testing include any patient who is:
Evaluation for Osteoporosis
Once diagnosed with osteoporosis, a complete medical history should be obtained with particular attention to the risk factors for osteoporosis. These include age of 65 years or more, a history of vertebral fracture or any fracture during childhood, a family history of hip fracture, low body weight (body mass index [BMI] < 21 or weight < 127 pounds), cigarette smoking, and use of corticosteroids for more than 3 months.81 The physical examination should be performed, particularly at the spine region. Height should be measured and compared with the greatest known height to determine height loss, which is an indicator of the presence of vertebral compression fractures. Balance and walking gait should be assessed in each individual.
Laboratory Investigations for Osteoporosis
Generally, laboratory investigations other than bone mass density measurement are not required for the diagnosis of osteoporosis. Some routine tests, however, should be performed to obtain baseline values as part of the initial workup. These include complete blood count with differential, urinalysis, and blood chemistry profiles with serum calcium and phosphate. Although vitamin D deficiency is common among the elderly with a prevalence of approximately 50%, many patients are asymptomatic. In addition, serum calcium and phosphate levels in this group of patients may not necessarily be abnormal. All older individuals, therefore, should be tested for vitamin D deficiency by measuring levels of 25(OH)D. If low, adequate vitamin D supplementation is encouraged. Performance speed and proximal muscle strength were markedly improved when 25(OH)D levels increased from 4 to 16 ng/mL and continued to improve as the levels increased to more than 40 ng/mL.82
Some special laboratory tests are available to measure the balance between bone resorption and bone formation from serum and urine samples. These assays are called “biochemical bone markers.” Biochemical bone markers can be classified into two groups: bone formation and bone resorption markers (Table 87–3). Although bone mineral density is a critical measurement used to evaluate patients for osteoporosis, it represents a static parameter that provides no information regarding the rate of bone turnover in a given patient. Biochemical bone markers are generally noninvasive, widely available, quickly performed, and relatively inexpensive. Unlike DEXAs, they can be used repeatedly over short intervals to assess the dynamic process of bone turnover. In principle, they can be used (along with bone mineral density) to predict future risk of fracture.
| Bone Formation Markers | Bone Resorption Markers |
|---|---|
Adapted from Camacho P: Biochemical markers of bone turnover. In Favus MJ (ed): Primer on the metabolic bone diseases and disorders of mineral metabolism, 6th ed, Washington, DC, American Society for Bone and Mineral Research, 2006, p. 127.
During bone formation, osteoblasts produce type I collagen, which is their major synthetic product. Carboxy-terminal propeptide and amino-terminal propeptide of type I collagen, known as PICP and PINP respectively, are cleaved from the newly formed collagen molecule and therefore can be used as the indices to indicate type I collagen biosynthesis. Osteoblasts also secrete a variety of noncollagenous proteins, two of which are used clinically as markers of osteoblast activity: bone specific alkaline phosphatase and osteocalcin. It is these noncollagenous products that are most useful as markers for bone formation. Although alkaline phosphatase is derived from several tissues, the two most common sources are from liver and bone. The utilization of tissue-specific monoclonal antibodies is able to differentiate between liver and bone isoform; however, the bone isoform has 10% to 20% cross-reactivity with the liver isoform.83
During osteoclast-mediated bone resorption, the collagen structure within bone is degraded. This collagen degradation product is used as an indicator for bone resorption. In general, collagen molecules in bone matrix are staggered to form collagen fibrils by covalent cross-links.84 These cross-links consist of pyridinolines (Pyds) and deoxypyridinolines (Dpds). Pyd and Dpd cross-links occur at two intermolecular sites in collagen molecule: amino-terminal-telopeptide and carboxy-terminal-telopeptide. As part of this degradation process, cross-linked collagen peptides from both the amino-terminal-telopeptide (NTX) and carboxy-terminal-telopeptide (CTX) are released and achieve measurable concentrations in both serum and urine. Therefore when osteoclasts resorb bone, they release a variety of collagen breakdown products into the circulation that are further metabolized by liver and kidney. These include free Pyds, free Dpds, NTX, and CTX.
The use of bone biochemical markers to determine therapy effectiveness is their best established clinical use. Several studies indicate that, within 4 to 6 weeks, there is significant reduction in biochemical markers of bone resorption after initiation of antiresorptive therapy.85–87 Lack of marker reduction may indicate noncompliance or the need to change the dosage or the therapeutic agent. Serial bone mineral density tests should be at least 12 (and possibly 24) months apart; therefore more frequent measurements of biochemical markers offer a significant advantage.
Evaluation for Secondary Osteoporosis
When secondary osteoporosis is suspected on the basis of clinical findings or because the patient is relatively young and presented with fragility fracture, specific tests should be considered to evaluate contributing causes that may require additional medical attention. These include basic laboratory investigation of a complete blood count with differential, erythrocyte sedimentation rate, serum calcium and phosphate levels, liver function tests, thyroid-stimulating hormone level, testosterone level in men, and a serum protein electrophoresis if myeloma is considered (Table 87–4). When abnormalities are detected, the patient should be referred to a specialist for further evaluation and specific treatment.
TABLE 87–4 Laboratory Investigations for Secondary Osteoporosis
| Medical Diseases | Diagnostic Study |
|---|---|
BUN, blood urea nitrogen; CBC, complete blood count; CRP, c-reactive protein; ESR, erythrocyte sedimentation rate; FSH, follicle stimulating hormone; LH, luteinizing hormone; TSH, thyroid stimulating hormone.
Data from Unnanuntana A, Gladnick BP, Lane JM: Osteoporosis and the aging spine: diagnosis and treatment. In Yue JJ, Guyer RD, Hochschuler SH, et al (eds): The Comprehensive Treatment of the Aging Spine: Minimally Invasive and Advanced Techniques. Philadelphia, Elsevier, 2011.
Assess for Risk of Falls and Fracture Risk
To adjust for the disparity, a new vehicle called the Fracture Risk Assessment Tool (FRAX) has been developed that adds risk factors to the calculation and offers a better assessment of fracture risk than DEXA alone. This instrument calculates the patient’s 10-year fracture risk on the basis of (1) age; (2) sex; (3) weight; (4) height; (5) previous fracture; (6) parent with fractured hip; (7) current smoking; (8) use of glucocorticoids; (9) presence of rheumatoid arthritis; (10) secondary osteoporosis; (11) alcohol use (>3 drinks/day); and (12) bone mass density at the femoral neck area. Each risk factor is weighted differently in the calculation depending on its importance. For example, in the absence of bone mass density measurements, a prior history of fracture is considerably more predictive of a patient’s future fracture risk than is a history of smoking and is weighted as such in the FRAX calculation. While the patient accrues more risk factors, the FRAX score increases in a predictable manner.88
Prevention and Treatment
Prevention and therapy of osteoporosis go hand in hand. The end point of both is to build and maintain sufficient bone mass to avoid fracture. Building strong bones during childhood and adolescence can be the best defense against the development of osteoporosis later in life. Other preventive strategies include maintaining a balanced diet with adequate calcium and vitamin D intake; undertaking regular weight-bearing exercise; and limiting the use of certain drugs, tobacco, and excessive alcohol intake that induce bone loss. Smoking one pack per day throughout adult life is associated with a 5% to 10% reduction in bone density.89 In addition, smoking lowers serum estrogen concentrations by accelerating the metabolism of estrogen90 and may negate the beneficial effects of estrogen in postmenopausal women.91,92 For these reasons, smoking cessation is highly recommended to improve skeletal health.
Nutrition, Calcium, and Vitamin D Supplementation
First-line treatment of osteoporosis includes adequate dietary intake of calories, protein, calcium, and vitamin D. Calcium and vitamin D supplementation is the cornerstone of all treatment modalities for osteoporosis. Literature clearly showed that adequate calcium and vitamin D intake reduced the risk of fractures. For optimal treatment, adequate calcium intake of 1000 to 1500 mg/day should be maintained in all patients on any type of treatment.93,94 To maximize the absorption of calcium across the small bowels, no more than 500 to 600 mg of elemental calcium should be taken at any given time.95 Among all calcium formulations, calcium citrate is the preferred form. Calcium citrate binds to oxalate, reducing its intestinal absorption, and citrate in urine inhibits crystal formation, thus reducing the incidence of kidney stones. In addition, calcium citrate does not require low pH for salt dissociation; therefore the absorption of this calcium formulation is reliable in patients taking H2 blockers or proton pump inhibitors.96
The current recommended dosages of vitamin D3 from the Institute of Medicine are 200 to 600 IU/day.93 However, many experts consider these recommendations to be too low and suggest that the minimum adult intake should be 1000 to 2000 IU/day.97 The appropriate amount of vitamin D intake should be evaluated by monitoring 25 (OH) D level and serum PTH. Patients with vitamin D insufficiency will have low levels of 25 (OH) D and elevated serum PTH from secondary hyperparathyroidism. Higher doses of vitamin D are required in these patients to replenish depleted total body stores. Fifty thousand international units of vitamin D2 (ergocalciferol) can be taken orally once a week or every other week for 6 to 8 weeks, followed by a maintenance dose of vitamin D3 of 1000 to 2000 IU/day. Toxicity is rare even if a dosage of 10,000 IU/day is given for up to 5 months.98
Protein is also a significant contributor to bone health. A randomized prospective study that studied hip fractures in 82 elderly women demonstrated that those who took protein supplementation in addition to calcium and vitamin D supplements had an attenuation of the loss of bone at 1 year compared with those who received nonprotein supplements.99 However, high dietary protein and sodium increase urinary calcium excretion and have also been shown to decrease the calcium available to the skeleton.100
Exercise
Regular physical activity reduces the risk for osteoporosis and osteoporotic fractures.101,102 Weight-bearing exercise has beneficial effects on bone mineral density including that of the spine in both premenopausal and postmenopausal women.101,103–106 In one study, young women (aged 20 to 35) were randomized into groups of exercise and calcium supplementation for 2 years and bone density at various sites was evaluated. The group randomized to the aerobics and weight-training program had a 2.5% increase in their spine density compared with the calcium and stretching program.107 Muscle mass is significantly correlated to bone mass,29 suggesting that increased skeletal loading leads to increased bone mass. In a study of postmenopausal women, strength training increased bone density in both the spine and hip, whereas inactivity decreased bone density at both sites after just 1 year.101 As little as 1 hour of exercise two or three times per week can increase vertebral bone mass in postmenopausal women, whereas inactivity results in continued bone loss and increased risk for hip fracture.108
Immobilization results in decreased bone mass through increased activation at remodeling sites and decreased osteoblast stimulation.109 Bone loss with complete immobilization can reach 40% in 1 year but can be prevented with upright positioning and daily postural shifting for 30 minutes.110 Permanent bone loss can be prevented by instituting exercise within the first months of immobilization.109 Therefore although the gains in bone mass with exercise may be small, the decline in bone mass and subsequent increased fracture risk with inactivity can be substantial and irreversible.
Regular physical activity also improves muscle strength and balance, thus reducing the risk of falls.111–114 An exercise program of tai chi, combining traditional Chinese medicine and martial arts and performed while standing, significantly improves balance,115 mitigates the fear of falling, and reduces falls in the elderly by more than 47%.111
A comprehensive general rehabilitation program that stresses spine extension and abdominal strengthening exercises without flexion, as well as impact-loading activities such as walking, should be part of the treatment plan for every osteoporotic patient. Persistent participation in the exercise program is important because the benefits of exercise are quickly lost with cessation of the exercise regimen.105
Antiresorptive Agents
Estrogen
Estrogen is an antiosteoporotic agent that has been shown to increase bone mass and thus decreases the risk of vertebral and hip fracture by approximately 30% to 40% as compared with patients taking placebo.116 Estrogen, however, has been found to increase rates of stroke and deep vein thrombosis, whereas combined estrogen and progesterone therapy is associated with increased risks of cardiovascular disease, breast cancer, dementia, and gallbladder disease. As a consequence, estrogen is mainly used in the early postmenopausal period to treat postmenopausal syndrome and then lowered to the least effective dose to control symptomatology. Because of the risks of estrogen formulations, this precludes their use as primary agents in the treatment of osteoporosis.117,118
Selective Estrogen Receptor Modulators (SERMs)
Selective estrogen receptor modulators (SERMs) are a class of agents that bind to estrogen receptors. They have a significant effect on breast tissue and bone cells; however, they act as antagonists in the other receptor sites. Of the SERMs currently being used for clinical settings, only raloxifene has been approved for the prevention and treatment of osteoporosis. Early data suggest that raloxifene decreases the risk of breast cancer by 70%, which made raloxifene a preferred agent in this particular indication.119 Although raloxifene has been shown to reduce the risk of vertebral fracture, there was no significant reduction in the overall risk of nonvertebral fracture.120,121 In addition, by stimulating estrogen receptors, raloxifene increases the risk of pulmonary emboli and thrombophlebitis and may cause profound postmenopausal symptoms.120–122 Therefore clinicians must weigh the benefits of the reduced risks of vertebral fracture and invasive breast cancer against the lack of protection against hip fracture and the increased risks of venous thromboembolism and fatal stroke when considering this agent for osteoporosis management.
Calcitonin
Calcitonin is available as both a parenteral injection and nasal spray. The intranasal spray is the most commonly used formulation due to its superior compliance and ease of use. Calcitonin reduces the risk of vertebral fracture; however, there is only a modest increase in bone mass density.123,124 Additionally, calcitonin treatment shows no benefit for reducing the risk of hip and other nonvertebral fractures. Some data suggest the analgesic effect of calcitonin. Although there is a hypothesis that calcitonin-induced analgesia may be mediated by increased beta-endorphins and may directly affect pain receptors in the central nervous system, the exact mechanism is still unknown.125 Therefore the current indication for calcitonin treatment is for alleviating painful vertebral compression fractures. It should be discontinued as soon as pain has been controlled because other pharmacologic agents are much more effective in order to prevent future fracture.
Bisphosphonates
Bisphosphonates are a class of antiresorptive agents that have been shown to be extremely efficacious in high-turnover osteoporosis. Currently, four bisphosphonates have been approved by the U.S. Food and Drug Administration for the treatment of postmenopausal osteoporosis: alendronate (Fosamax), risedronate (Actonel), ibandronate (Boniva), and zoledronic acid (Reclast). These drugs differ in their potency, mode of administration, and dosing schedules. Alendronate and risedronate are administered orally, whereas zoledronic acid is administered by intravenous injection. Ibandronate is available in both oral and intravenous formulations (Table 87–5).

The mechanism of action of bisphosphonate involves interposition of a nondegradable drug barrier between osteoclasts and Howship lacunae, thus interfering with resorption. The drug is then ingested by osteoclasts and disrupts the cellular membrane mevalonic synthesis pathway, leading to osteoclasts’ premature cell death.126 Bisphosphonates slow the bone turnover rate, which has been reported to be decreased within 6 weeks with the oral formulations and within 3 days with the intravenous formulations. All oral bisphosphonates reduce the risk of both vertebral and nonvertebral fractures.127–130 The intravenous zoledronic acid appears to be effective in increasing bone mass and decreasing both the vertebral and nonvertebral fracture risk.131,132 In addition, when given within 3 months of an acute hip fracture, zoledronic acid lowers mortality by more than 28% and does not interfere with fracture healing.133
The adverse effects of oral bisphosphonates are due to their inherent toxicity to epithelial cells lining the gastrointestinal tract. The results are irritation of esophagus, acid eructation, nausea, and heartburn. Patients with a significant background of gastrointestinal intolerance, therefore, are not suitable for prescribing oral formulations. However, clinicians may improve patients’ tolerance by giving oral bisphosphonates in small doses and slowly increasing the dosage until the full dose is achieved. This will lead to better compliance even with individuals who have a history of dyspepsia. The other potential complication is osteonecrosis of the jaw. Osteonecrosis of the jaw is defined as exposed bone in the musculofascial region that fails to heal within 8 weeks after identification by a health care provider.134 The epidemiology of osteonecrosis of the jaw in osteoporotic patients is not clear. Postmarketing surveys to mid-2007 indicated an incidence of less than 1.2 per 100,000 patient-years for risedronate and less than 0.5 to 2.5 per 100,000 patient-years for alendronate.135 The group of patients who carry the greatest risk for developing this complication are those with multiple myeloma or metastatic carcinoma of the skeleton who are being treated with relatively high doses of intravenous bisphosphonates concurrently with chemotherapy. Therefore prior to starting bisphosphonate treatment, especially with intravenous formulation, patients should complete their dental work and establish meticulous oral hygiene. In addition, intravenous bisphosphonates can cause a release of cytokines from osteoclasts and result in flulike symptoms. These include fever, muscle pain, headache, and bone pain. Most of the symptoms resolve within 3 days. Coadministration of diphenhydramine hydrochloride (Benadryl) and acetaminophen can minimize these side effects.
The sequelae of long-term bisphosphonates on bone metabolism remain unclear. There is a hypothesis that prolonged treatment with bisphosphonates may lead to adynamic, fragile bone. In this state of oversuppression, microfractures generated through the wear and tear of normal daily life begin to accumulation and coalesce, leading to insufficiency fractures. Accumulation of microdamage is associated with a reduction in bone toughness. Many studies report low-energy subtrochanteric or midshaft fractures after prolonged treatment with alendronate.136–138 This type of fracture is characterized by (1) simple or transverse fracture; (2) beaking of the cortex on one side; (3) hypertrophied diaphyseal cortices; and (4) result from minimal or no trauma.138 Currently, the recommendation for bisphosphonate therapy is a 5-year period of treatment. Once the bone mass density is in its plateau and the urine level of NTX is in therapeutic range (20 to 40 nmol bone collagen equivalents/mmol of creatinine), changes in treatment such as a rest period from bisphosphonates or the use of an anabolic agent should be considered.
Anabolic Agents
Teriparatide, parathyroid hormone (1-34), is the only anabolic agent available for the treatment of postmenopausal osteoporosis in the United States. It is administered subcutaneously once a day. When given intermittently, teriparatide can increase bone mass up to 13% over 2 years of therapy.139 This increase is greater than that achieved with bisphosphonate therapy. The risk of fracture was reduced by 65% and 53% for vertebral and nonvertebral fracture, respectively.140 Other benefits of teriparatide have been proposed. Many studies reported the possible benefits of teriparatide on fracture healing. Callus formation was accelerated by the early stimulation of proliferation and differentiation of osteoprogenitor cells and increases in production of bone matrix proteins. Teriparatide should be considered in these conditions:
Contraindications include active Paget disease of bone, unexplained elevations of alkaline phosphatase, history of skeletal irradiation, and children with open epiphyses. The adverse reactions associated with teriparatide are nausea, headache, dizziness, leg cramps, swelling, pain, weakness, erythema around the injection site, and elevation of serum calcium. There is a concern regarding osteosarcoma due to evidence showing that rodents, exposed to prolonged high doses of teriparatide, developed osteosarcoma.141,142 Therefore teriparatide should be discontinued after 2 years of treatment. After that, bisphosphonate therapy should be initiated to maintain its results.
Pharmacologic Agents and Spinal Fusion
No clinical studies have evaluated pharmacologic agents for osteoporosis in the setting of vertebral fractures or spinal fusion. Bisphosphonates increase fracture callus size during endochondral repair but cause delay in maturation.143 Animal studies in rats and rabbits have clearly demonstrated that bisphosphonates delayed, and at higher doses prevented, spinal fusion.144,145 Therefore bisphosphonates should not be used following spinal fusion or acute vertebral fracture in order to reduce the possible adverse effects to the early biologic processes of fracture healing. Teriparatide in all animal studies assisted fracture healing. In both rat and rabbit models of spinal fusion, the administration of teriparatide significantly enhanced fusion and increased the fusion mass.146,147 On the basis of these animal studies, teriparatide offers superior biology for spinal indications, when compared with bisphosphonates and controls that only took calcium and vitamin D. The application of this agent in patients, however, still awaits evidence-based studies.
Future Directions
The available pharmacologic agents used to treat osteoporosis have their own limitations. Understanding of cellular mechanisms regulating bone formation and remodeling is critically important for developing new agents that will reduce the adverse effects or improve the outcome of treatment. Denosumab, a human monoclonal antibody against RANKL, has been shown in preclinical trials to increase bone mineral density and decrease bone resorption in postmenopausal women with osteoporosis.12–14 It is now approved by the FDA for the treatment of postmenopausal osteoporosis. Cathepsin-K inhibitors are another group of antiresorptive agents, which were designed to reduce the activity of cathepsin K (a powerful osteoclast protease). Theoretically, this agent can limit the enzymatic degradation of bone matrix proteins. It is currently approved in Europe but not in the United States.148
Strontium ranelate is an agent that has both antiresorptive and anabolic action. Treatment of postmenopausal osteoporotic women with strontium ranelate has been shown to decrease fracture risk and increase bone mineral density.149,150 The long-term effects, however, remain unknown. The development of new formulations of teriparatide (noninjectable forms) or development of alternative parathyroid hormone analogs that possess longer half-life, leading to less frequent dosing, are also under investigation.151,152
Paget Disease of Bone (Osteitis Deformans)
Paget disease of the bone is the second most prevalent metabolic bone disease. The overall incidence, however, of Paget disease is decreasing.153,154 The pelvis is the most frequently affected site, but the disease in the pelvis is most often asymptomatic. When Paget disease is clinically apparent, spinal complaints, notably pain, are the most common.155 Paget disease primarily affects older individuals; in the United States it occurs in 2% to 3% of people older than the age of 60 years.156 Paget disease affects men and women equally and has a distinctive geographic distribution. It is found more commonly in the United Kingdom, North America, Australia, New Zealand, France, and Germany and is rarely encountered in Scandinavia, China, Japan, India, Africa, or the Middle East.157,158
Pathology
Paget disease is a focal disorder of accelerated skeletal remodeling that usually involves a single bone (monostotic) or several bones (polyostotic) and rarely affects numerous bones. Initially, there is an increase in the rate of bone resorption at areas of bone remodeling (Fig. 87–5). Pagetic osteoclasts are abnormal in several ways: They are greater in size and number and highly multinucleate, with up to 100 nuclei.159,160 In response to this increased resorptive activity, additional osteoblasts are recruited to remodeling sites, with increased production of qualitatively poor new bone matrix that is deposited in a disorganized woven pattern. The result is increased bone formation and a disruption of the normally organized lamellar collagen architecture.159 Over time this woven bone is incompletely replaced by more organized bone as is the case with a fracture callus.
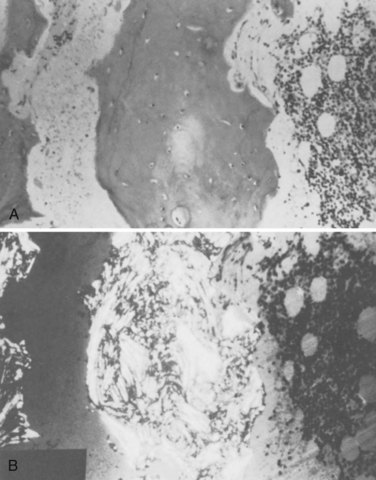
FIGURE 87–5 Histologic appearance of Paget disease. A, Mosaic pattern. B, Polarized view of disorganized collagen.
(From Disorders of Bone 20. Orthop. Science. Park Ridge, IL, AAOS, 1986.)
The etiology of Paget disease remains unknown. Evidence exists for both viral and hereditary causes. Genetic factors seem to play a role as evidence by the epidemiology of British migrants being affected more frequently. Familial clusters of Paget disease have been documented.158,161 A positive family history has been reported in up to 40% of patients.156 A first-degree relative has 7 times greater risk of developing Paget disease.161 Genetic studies have identified several possible loci for Paget disease.162–164 Sequestosome 1 (SQSTM1) is the best studied and encodes for a scaffold protein (p62) in the nuclear factor κB (NFκB) signaling pathway.165 The common area of interruption is the region encoding the binding site for ubiquitin. The loss of the ubiquitin site leads to disregulation of the protein, which is thought to lead to activation of the RANKL pathway and subsequent increased osteoclast activity.165
Environmental factors also may play a role in Paget disease. Initial enthusiasm for a viral etiology was stimulated by the nuclear and cytoplasmic inclusion bodies, similar to paramyxovirus nucleocapsids, detected in affected osteoclasts by numerous methods in several studies.166–169 Several syncytial viruses and canine distemper virus have also been postulated to play a role in the etiology of Paget disease.166,170 A characteristic feature of these viruses is their ability to persist at low levels and invade the host immune system.
Despite decades of research, a pure viral cause for Paget disease has not been proven. No virus has ever been cultured from Pagetic cells. PCR studies have had mixed results attempting to isolate measles virus and canine distemper virus from blood and osteoblasts of patients with the disease.171,172 Also, unlike the distinctive geographic distribution of Paget disease, measles has a similar incidence worldwide and occurs in young patients, whereas Paget disease generally occurs in the elderly.173 Furthermore, similar viral nuclear inclusion bodies have been identified in other skeletal disorders, so the virus may be a nonetiologic cotraveler in an osteoclast altered by some other mechanism.
Clinical Course
Most patients with Paget disease are asymptomatic. The disease is often detected when the patient has a radiograph taken or serum alkaline phosphatase measured for an unrelated reason. In the majority of patients with Paget disease, one or several bones are involved. In decreasing order, the most commonly involved bones include the pelvis, lumbar spine, femur, thoracic spine, skull, tibia, humerus, and cervical spine; however, any bone may be affected. The lumbar, thoracic and cervical vertebrae are affected in 58%, 45%, and 14% of patients, respectively.174,175
Clinical manifestations, when present, cover a wide spectrum. Patients who are symptomatic most often present with back pain,176 although other areas, especially the hip, can be involved. The skull can be implicated in the disease, and when it is the woven bone expansion can encroach on the cranial nerves, most typically the eighth nerve. Both the acoustic and the vestibular branches can be affected; hence the initial presentation of Paget disease might be decreased hearing or difficulty with gait or balance.175
In the spine, the pain is infrequently due to primary Pagetic involvement alone but rather represents related secondary changes (Fig. 87–6).177 These include osteoarthritis of the spinal facets; encroachment on the spinal cord; or in the case of intense pain, pathologic fracture. When the spinal column is involved, most often it is the thoracolumbar regions that are affected but cervical disease may also contribute.178 Involvement of the spinal cord may be secondary to a mechanical or vascular complication. Of the mechanical causes, vertebral collapse, osteophytic overgrowth, or bony volume expansion in the osteosclerotic phase (see later) can all impinge on neural elements.179 The spinal cord’s vascular supply can likewise be disrupted by bone compression of afferent arteries, leading to neural ischemia. In addition, Paget disease causes a reactive vasodilation near diseased areas. This may lead to shunting of blood and ischemia of the deprived areas. In the spine, increased blood flow around pagetic lesions may result in a diversion of blood destined for the cord, causing the so-called arterial steal phenomenon and leading to neurologic signs. In summary, spinal Paget disease can cause not only bone pain and arthritis or pathologic fractures but also, through its effect on nerves, headaches, hearing or vision loss, cerebellar deficits, and even fecal and urinary incontinence.
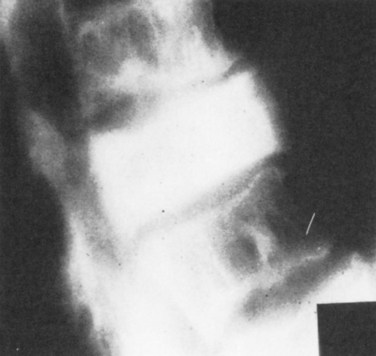
A hallmark of Paget disease is skeletal deformity, which may be manifest as an increase in bone size or an abnormality in their shape. Bone is resorbed and replaced rapidly in Paget disease, and the replacement bone is necessarily less organized and weaker. In weight-bearing bones, this can cause skeletal deformities.180 Although this problem is more common in the femur and tibia, it can occur in the spine, leading to significant kyphotic changes.
The most serious clinical complication of Paget disease heralded by the acute onset of pain or sharp increase in the intensity of chronic pain is osteosarcoma or other types of sarcoma. Although the incidence of osteosarcoma is less than 1% of patients with Paget disease, it is 1000 times higher than in the general population for this age group.181 Moreover, the tumors that do form are particularly aggressive. Fortunately, sarcomatous degeneration is especially infrequent in spinal Paget disease. An unusual form of tumorous degeneration in Paget disease is the giant cell tumor. The lesion is frequently multifocal and has a unique association with individuals from Avellino, Italy.182 The tumor frequently involves the spine and leads to paraplegia unless treated. It responds to dexamethasone therapy, which can be augmented by selective radiation.
Diagnosis
Biochemical Markers
The increase in bone resorption and bone formation is reflected in an increase in biochemical markers of bone turnover. Bone resorption releases fragments of the collagen matrix, as evidenced by an elevated urinary excretion of hydroxyproline and hydroxylysine. Although these provide reasonably accurate measures of bone collagen resorption, more specific tests are used routinely. Serum total alkaline phosphatase is a bone formation marker, and its elevation in Paget disease is the most common index of disease activity. A measure of bone-specific alkaline phosphatase may be preferred in a subset of patients with hepatic disease or monostotic involvement, in which 15% of patients have a normal alkaline phosphatase level174 but appears to offer no advantage in the average patient.183 In all patients, however, changes in the total serum alkaline phosphatase activity are adequate to monitor changes in overall disease activity. Urinary and serum markers of bone resorption including deoxypyridinoline, N-telopeptide, and C-telopeptide provide more immediate measures of response to therapy than bone formation markers.
Treatment
Calcium and Vitamin D
Calcium and vitamin D deficiency should be corrected before the use of any bisphosphonates or treatments for Paget disease. Hypocalcemia is a significant risk, especially with the use of any of the intravenous bisphosphonates.131
Bisphosphonates
The emergence of newer, more potent bisphosphonates has resulted in a major change in the treatment of Paget disease over the past several years. Bisphosphonates, which inhibit osteoclast-mediated bone resorption and induce osteoclast apoptosis,184 are the mainstay of drug treatment for Paget disease. Studies have demonstrated bisphosphonates are effective in reducing bone turnover,185–187 decreasing pain,185,187 and promoting healing of osteolytic lesions.188 On the other hand, studies have yet to demonstrate the long-term efficacy of bisphosphonates to prevent complications of the disease. In order of decreasing potency, the Food and Drug Administration (FDA)-approved bisphosphonates for the treatment of Paget disease are zoledronic acid, risedronate, alendronate, pamidronate, tiludronate, and etidronate. Zoledronic acid was recently shown to be more efficacious than risedronate in controlling pain, decreasing bone turnover markers, and maintenance of suppression after treatment.185
Calcitonin
Calcitonin, the first widely used therapy for Paget disease, also inhibits osteoclastic bone resorption.189 Salmon calcitonin is available in injectable and nasal-spray forms, but only the injectable form is FDA approved for the treatment of Paget disease. Compared with bisphosphonates, calcitonin is less powerful and does not suppress the disease activity for as long after cessation. Calcitonin use today is largely limited to those patients who are unable to tolerate bisphosphonates.190
Summary
Pearls
Pitfalls
Key Points
1 Kanis JA. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int. 2008;19:385-397.
2 Vieth R. Why the optimal requirement for Vitamin D3 is probably much higher than what is officially recommended for adults. Steroid Biochen Mol Biol. 2004;89-90:573-579.
3 Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
4 Huang RC, Khan SN, Sandhu HS, et al. Alendronate inhibits spine fusion in a rat model. Spine. 2005;30:2516-2522.
5 O’Loughlin PF, Cunningham ME, Bukata SV, et al. Parathyroid hormone (1-34) augments spinal fusion, fusion mass volume, and fusion mass quality in a rabbit spinal fusion model. Spine. 2009;34(2):121-130.
1 Bruder SP, Fink DJ, Caplan AI. Mesenchymal stem cells in bone development, bone repair, and skeletal regeneration therapy. J Cell Biochem. 1994;56:283-294.
2 Lazarus HM, Haynesworth SE, Gerson SL, et al. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant. 1995;16:557-564.
3 Owen M. Lineage of osteogenic cells and their relationship to the stromal system. In: Peck WA, editor. Bone and Mineral Research. Amsterdam: Elsevier, 1985.
4 Buckwalter JA, Glimcher MJ, Cooper RR, et al. Bone biology. I: Structure, blood supply, cells, matrix, and mineralization. Instr Course Lect. 1996;45:371-386.
5 Buckwalter JA, Glimcher MJ, Cooper RR, et al. Bone biology. II: Formation, form, modeling, remodeling, and regulation of cell function. Instr Course Lect. 1996;45:387-399.
6 Nijweide PJ, Burger EH, Feyen JH. Cells of bone: proliferation, differentiation, and hormonal regulation. Physiol Rev. 1986;66:855-886.
7 Stein GS, Lian JB. Molecular mechanisms mediating proliferation/differentiation interrelationships during progressive development of the osteoblast phenotype. Endocr Rev. 1993;14:424-442.
8 Chambers TJ. The origin of the osteoclast. In: Peck WA, editor. Bone and Mineral Research, Vol. 6. Amsterdam: Elsevier; 1989:1.
9 Bartkiewicz M, Hernando N, Reddy SV, et al. Characterization of the osteoclast vacuolar H(+)-ATPase B-subunit. Gene. 1995;160:157-164.
10 Teitelbaum SL, Abu-Amer Y, Ross FP. Molecular mechanisms of bone resorption. J Cell Biochem. 1995;59:1-10.
11 Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795-823.
12 Body JJ, Facon T, Coleman RE, et al. A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res. 2006;12:1221-1228.
13 McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821-831.
14 Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res. 2009;24:153-161.
15 Posner AS. Bone mineral and the mineralization process. In: Peck WA, editor. Bone and Mineral Research, Vol. 5. Amsterdam: Elsevier; 1987:65.
16 Raisz LG, Kream BE. Regulation of bone formation. N Engl J Med. 1983;309:29-35.
17 Prockop DJ, Kivirikko KI, Tuderman L, et al. The biosynthesis of collagen and its disorders (first of two parts). N Engl J Med. 1979;301:13-23.
18 Price CP, Thompson PW. The role of biochemical tests in the screening and monitoring of osteoporosis. Ann Clin Biochem. 1995;32(Pt 3):244-260.
19 Sanchez CP, Salusky IB. Biochemical marker in metabolic bone disease. Curr Opin Orthop. 1994;5:43-49.
20 Bullough PG, Boachie-Adjei O. Atlas of Spinal Disease. New York: Gower Medical Publishing; 1988.
21 Kondo H, Guo J, Bringhurst FR. Cyclic adenosine monophosphate/protein kinase A mediates parathyroid hormone/parathyroid hormone-related protein receptor regulation of osteoclastogenesis and expression of RANKL and osteoprotegerin mRNAs by marrow stromal cells. J Bone Miner Res. 2002;17:1667-1679.
22 Price P. Osteocalcin Amsterdam. Peck WA, editor. Bone and Mineral Research. 1983:157.
23 Whyte MP. Alkaline phosphatase: Physiological role explored in hypophosphatasia. In: Peck WA, editor. Bone and Mineral Research, Vol. 6. Amsterdam: Elsevier; 1989:175.
24 Gazdag AR, Lane JM, Glaser D, et al. Alternatives to Autogenous Bone Graft: Efficacy and Indications. J Am Acad Orthop Surg. 1995;3:1-8.
25 Wozney JM, Rosen V, Celeste AJ, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528-1534.
26 Hoffmann A, Weich HA, Gross G, et al. Perspectives in the biological function, the technical and therapeutic application of bone morphogenetic proteins. Appl Microbiol Biotechnol. 2001;57:294-308.
27 Mohler DG, Lane JM, Cole BJ, et al. Skeletal failure in osteoporosis. In: Lane JM, Healey JH, editors. Diagnosis and Management of Pathologic Fractures. New York: Raven, 1993.
28 Boden SD, Kaplan FS. Calcium homeostasis. Orthop Clin North Am. 1990;21:31-42.
29 Lane JM, Riley EH, Wirganowicz PZ. Osteoporosis: diagnosis and treatment. Instr Course Lect. 1997;46:445-458.
30 Minaire P, Neunier P, Edouard C, et al. Quantitative histological data on disuse osteoporosis: comparison with biological data. Calcif Tissue Res. 1974;17:57-73.
31 Wasserman RH, Chandler JS. Molecular mechanisms of intestinal calcium absorption. In: Peck WA, editor. Bone and Mineral Research, Vol. 3. Amsterdam: Elsevier; 1985:181.
32 Johnston CCJr. Slemenda CW: Peak bone mass, bone loss and risk of fracture. Osteoporos Int. 1994;4(Suppl 1):43-45.
33 Marcus R. Endocrine control of bone and mineral metabolism. In: Manolagas SC, Olefsky JM, editors. Metabolic Bone and Mineral Disorders. New York: Churchill Livingstone; 1988:13.
34 Klahr S, Hruska K. Effects of parathyroid hormone on the renal reabsorption of phosphorus and divalent cations. In: Peck WA, editor. Bone and Mineral Research, Vol. 2. Amsterdam: Elsevier; 1983:65.
35 Wong GL. Skeletal effects of parathyroid hormone. In: Peck WA, editor. Bone and Mineral Research, Vol. 4. Amsterdam: Elsevier; 1986:103.
36 Norman AW, Roth J, Orci L. The vitamin D endocrine system: steroid metabolism, hormone receptors, and biological response (calcium binding proteins). Endocr Rev. 1982;3(4):331-366.
37 Potts JTJr. Kronenberg HM, Rosenblatt M: Parathyroid hormone: chemistry, biosynthesis, and mode of action. Adv Protein Chem. 1982;35:323-396.
38 Bell NH. Vitamin D-endocrine system. J Clin Invest. 1985;76:1-6.
39 Compston JE. Hepatic osteodystrophy: vitamin D metabolism in patients with liver disease. Gut. 1986;27(9):1073-1090.
40 Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
41 Hurley DL, Tiegs RD, Wahner HW, et al. Axial and appendicular bone mineral density in patients with long-term deficiency or excess of calcitonin. N Engl J Med. 1987;317:537-541.
42 Tiegs RD, Body JJ, Wahner HW, et al. Calcitonin secretion in postmenopausal osteoporosis. N Engl J Med. 1985;312:1097-1100.
43 Austin LA, Heath H3rd. Calcitonin: physiology and pathophysiology. N Engl J Med. 1981;304:269-278.
44 Osteoporosis Prevention, Diagnosis, and Therapy. NIH Consensus Statement. 2000;17:1-36.
45 National Osteoporosis Foundation. Osteoporosis: Disease Statistics. Vol. 2003. http://nof.org/osteoporosis/stats.htm, 2003.
46 Riggs BL, Melton LJ3rd. Evidence for two distinct syndromes of involutional osteoporosis. Am J Med. 1983;75:899-901.
47 Gallagher JC, Melton LJ, Riggs BL, et al. Epidemiology of fractures of the proximal femur in Rochester, Minnesota. Clin Orthop Relat Res. 1980;150:163-171.
48 Melton LJr, Riggs BL. Epidemiology of age-related fractures. In: Avioli LV, editor. The Osteoporotic Syndrome. New York: Grune & Stratton; 1987:1-30.
49 Silverman SL. Quality-of-life issues in osteoporosis. Curr Rheumatol Rep. 2005;7:39-45.
50 Kado DM, Duong T, Stone KL, et al. Incident vertebral fractures and mortality in older women: a prospective study. Osteoporos Int. 2003;14:589-594.
51 Hunt PA, Wu-Chen ML, Handal NJ, et al. Bone disease induced by anticonvulsant therapy and treatment with calcitriol (1,25-dihydroxyvitamin D3). Am J Dis Child. 1986;140:715-718.
52 Healey JH, Lane JM. Structural scoliosis in osteoporotic women. Clin Orthop Relat Res. 1985;195:216-223.
53 Orwoll ES, Klein RF. Osteoporosis in men. Endocr Rev. 1995;16:87-116.
54 Management of postmenopausal osteoporosis: position statement of the North American Menopause Society. Menopause. 2002;9:84-101.
55 Harper KD, Weber TJ. Secondary osteoporosis. Diagnostic considerations. Endocrinol Metab Clin North Am. 1998;27:325-348.
56 Ben-Shlomo A, Hagag P, Evans S, et al. Early postmenopausal bone loss in hyperthyroidism. Maturitas. 2001;39:19-27.
57 Lukert BP, Raisz LG. Glucocorticoid-induced osteoporosis: pathogenesis and management. Ann Intern Med. 1990;112:352-364.
58 Cummings SR, Kelsey JL, Nevitt MC, et al. Epidemiology of osteoporosis and osteoporotic fractures. Epidemiol Rev. 1985;7:178-208.
59 National Institutes of Health, Osteoporosis and Related Bone Disease, National Resource Center. Information for patients about Paget’s disease of bone. http://www.osteo.org/pdisbone.html, 2003.
60 Riggs BL, Melton LJ3rd. Involutional osteoporosis. N Engl J Med. 1986;314:1676-1686.
61 Richelson LS, Wahner HW, Melton LJ3rd, et al. Relative contributions of aging and estrogen deficiency to postmenopausal bone loss. N Engl J Med. 1984;311:1273-1275.
62 Klibanski A, Neer RM, Beitins IZ, et al. Decreased bone density in hyperprolactinemic women. N Engl J Med. 1980;303:1511-1514.
63 Parfitt AM. Quantum concept of bone remodeling and turnover: implications for the pathogenesis of osteoporosis. Calcif Tissue Int. 1979;28:1-5.
64 Lane JM, Nydick M. Osteoporosis: current modes of prevention and treatment. J Am Acad Orthop Surg. 1999;7:19-31.
65 Riggs BL, Hodgson SF, O’Fallon WM, et al. Effect of fluoride treatment on the fracture rate in postmenopausal women with osteoporosis. N Engl J Med. 1990;322:802-809.
66 Mellish RW, Ferguson-Pell MW, Cochran GV, et al. A new manual method for assessing two-dimensional cancellous bone structure: comparison between iliac crest and lumbar vertebra. J Bone Miner Res. 1991;6:689-696.
67 Compston JE. Connectivity of cancellous bone: assessment and mechanical implications. Bone. 1994;15:463-466.
68 Matkovic V, Kostial K, Simonovic I, et al. Bone status and fracture rates in two regions of Yugoslavia. Am J Clin Nutr. 1979;32:540-549.
69 Lips P, Courpron P, Meunier PJ. Mean wall thickness of trabecular bone packets in the human iliac crest: changes with age. Calcif Tissue Res. 1978;26:13-17.
70 Meema S, Bunker ML, Meema HE. Preventive effect of estrogen on postmenopausal bone loss. Arch Intern Med. 1975;135:1436-1440.
71 Eriksen EF, Colvard DS, Berg NJ, et al. Evidence of estrogen receptors in normal human osteoblast-like cells. Science. 1988;241:84-86.
72 Lane JM, Healey JH, Vigorita VJ, et al. Orthopaedic management of osteoporosis: Effects of nutrition and exercise on the skeleton. In: Uhthoff HK, editor. Current Concepts of Bone Fragility. Berlin: Springer-Verlag; 1986:429-447.
73 Tsai KS, Heath H3rd, Kumar R, et al. Impaired vitamin D metabolism with aging in women. Possible role in pathogenesis of senile osteoporosis. J Clin Invest. 1984;73:1668-1672.
74 Kanis JA, Adami S. Bone loss in the elderly. Osteoporos Int. 1994;4(Suppl 1):59-65.
75 Melton LJr, Chao EY, Lane J. Biomechanical aspects of fractures. In: Riggs BL, Melton LJr, editors. Osteoporosis: Etiology, Diagnosis, and Management. New York: Raven; 1988:111-113.
76 Melton LJ3rd, Riggs BL, Keaveny TM, et al. Structural determinants of vertebral fracture risk. J Bone Miner Res. 2007;22:1885-1892.
77 Lochmuller EM, Eckstein F, Kaiser D, et al. Prediction of vertebral failure loads from spinal and femoral dual-energy X-ray absorptiometry, and calcaneal ultrasound: an in situ analysis with intact soft tissues. Bone. 1998;23:417-424.
78 Stein E, Shane E. Secondary osteoporosis. Endocrinol Metab Clin North Am. 2003;32:115-134. vii
79 Marshall D, Johnell O, Wedel H. Meta-analysis of how well measures of bone mineral density predict occurrence of osteoporotic fractures. BMJ. 1996;312:1254-1259.
80 Masud T, Langley S, Wiltshire P, et al. Effect of spinal osteophytosis on bone mineral density measurements in vertebral osteoporosis. BMJ. 1993;307:172-173.
81 Kaufman JD, Cummings SR. Osteoporosis and prevention of fractures: practical approaches for orthopaedic surgeons. Instr Course Lect. 2002;51:559-565.
82 Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18-28.
83 Camacho P. Biochemical markers of bone turnover. In: Favus MJ, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. 6 ed. Washington, D.C.: American Society for Bone and Mineral Research; 2006:127.
84 Calvo MS, Eyre DR, Gundberg CM. Molecular basis and clinical application of biological markers of bone turnover. Endocr Rev. 1996;17:333-368.
85 Chesnut CH3rd, Bell NH, Clark GS, et al. Hormone replacement therapy in postmenopausal women: urinary N-telopeptide of type I collagen monitors therapeutic effect and predicts response of bone mineral density. Am J Med. 1997;102:29-37.
86 Prestwood KM, Pilbeam CC, Burleson JA, et al. The short-term effects of conjugated estrogen on bone turnover in older women. J Clin Endocrinol Metab. 1994;79:366-371.
87 Garnero P, Shih WJ, Gineyts E, et al. Comparison of new biochemical markers of bone turnover in late postmenopausal osteoporotic women in response to alendronate treatment. J Clin Endocrinol Metab. 1994;79:1693-1700.
88 Kanis JA, Johnell O, Oden A, et al. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int. 2008;19:385-397.
89 Hopper JL, Seeman E. The bone density of female twins discordant for tobacco use. N Engl J Med. 1994;330:387-392.
90 Byrjalsen I, Haarbo J, Christiansen C. Role of cigarette smoking on the postmenopausal endometrium during sequential estrogen and progestogen therapy. Obstet Gynecol. 1993;81:1016-1021.
91 Kiel DP, Baron JA, Anderson JJ, et al. Smoking eliminates the protective effect of oral estrogens on the risk for hip fracture among women. Ann Intern Med. 1992;116:716-721.
92 Komulainen M, Kroger H, Tuppurainen MT, et al. Identification of early postmenopausal women with no bone response to HRT: results of a five-year clinical trial. Osteoporos Int. 2000;11:211-218.
93 Optimal Calcium Intake. NIH Consens Statement. 1994;12:1-31.
94 Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Institute of Medicine. Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, D.C.: National Academy Press; 1997.
95 Levine JP. Pharmacologic and nonpharmacologic management of osteoporosis. Clin Cornerstone. 2006;8:40-53.
96 Lin JT, Lane JM. Osteoporosis: a review. Clin Orthop Relat Res. 2004;425:126-134.
97 Gehrig L, Lane J, O’Connor MI. Osteoporosis: management and treatment strategies for orthopaedic surgeons. J Bone Joint Surg Am. 2008;90(6):1362-1374.
98 Vieth R. Why the optimal requirement for Vitamin D3 is probably much higher than what is officially recommended for adults. J Steroid Biochem Mol Biol. 2004;89-90:575-579.
99 Schurch MA, Rizzoli R, Slosman D, et al. Protein supplements increase serum insulin-like growth factor-I levels and attenuate proximal femur bone loss in patients with recent hip fracture. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1998;128:801-809.
100 Massey LK, Whiting SJ. Dietary salt, urinary calcium, and kidney stone risk. Nutr Rev. 1995;53:131-139.
101 Nelson ME, Fiatarone MA, Morganti CM, et al. Effects of high-intensity strength training on multiple risk factors for osteoporotic fractures. A randomized controlled trial. JAMA. 1994;272:1909-1914.
102 Ernst E. Exercise for female osteoporosis. A systematic review of randomised clinical trials. Sports Med. 1998;25:359-368.
103 Dalsky GP, Stocke KS, Ehsani AA, et al. Weight-bearing exercise training and lumbar bone mineral content in postmenopausal women. Ann Intern Med. 1988;108:824-828.
104 Kerr D, Ackland T, Maslen B, et al. Resistance training over 2 years increases bone mass in calcium-replete postmenopausal women. J Bone Miner Res. 2001;16:175-181.
105 Kohrt WM, Snead DB, Slatopolsky E, et al. Additive effects of weight-bearing exercise and estrogen on bone mineral density in older women. J Bone Miner Res. 1995;10:1303-1311.
106 Layne JE, Nelson ME. The effects of progressive resistance training on bone density: a review. Med Sci Sports Exerc. 1999;31:25-30.
107 Friedlander AL, Genant HK, Sadowsky S, et al. A two-year program of aerobics and weight training enhances bone mineral density of young women. J Bone Miner Res. 1995;10:574-585.
108 Cummings SR, Nevitt MC, Browner WS, et al. Risk factors for hip fracture in white women. Study of Osteoporotic Fractures Research Group. N Engl J Med. 1995;332:767-773.
109 Minaire P. Immobilization osteoporosis: a review. Clin Rheumatol. 1989;8(Suppl 2):95-103.
110 Whalen RT, Carter DR, Steel CR. The relationship between physiological activity and bone density. Trans Orthop Res Soc. 1987;12:464-470.
111 Wolf SL, Barnhart HX, Kutner NG, et al. Reducing frailty and falls in older persons: an investigation of Tai Chi and computerized balance training. Atlanta FICSIT Group. Frailty and Injuries: Cooperative Studies of Intervention Techniques. J Am Geriatr Soc. 1996;44:489-497.
112 Campbell AJ, Robertson MC, Gardner MM, et al. Randomised controlled trial of a general practice programme of home based exercise to prevent falls in elderly women. BMJ. 1997;315:1065-1069.
113 Lord SR, Ward JA, Williams P, et al. The effect of a 12-month exercise trial on balance, strength, and falls in older women: a randomized controlled trial. J Am Geriatr Soc. 1995;43:1198-1206.
114 Lord SR, Ward JA, Williams P. Exercise effect on dynamic stability in older women: a randomized controlled trial. Arch Phys Med Rehabil. 1996;77:232-236.
115 Hain TC, Fuller L, Weil L, et al. Effects of T’ai Chi on balance. Arch Otolaryngol Head Neck Surg. 1999;125:1191-1195.
116 Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701-1712.
117 Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321-333.
118 Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-613.
119 Cauley JA, Norton L, Lippman ME, et al. Continued breast cancer risk reduction in postmenopausal women treated with raloxifene: 4-year results from the MORE trial. Multiple outcomes of raloxifene evaluation. Breast Cancer Res Treat. 2001;65:125-134.
120 Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA. 1999;282:637-645.
121 Siris ES, Harris ST, Eastell R, et al. Skeletal effects of raloxifene after 8 years: results from the continuing outcomes relevant to Evista (CORE) study. J Bone Miner Res. 2005;20:1514-1524.
122 Barrett-Connor E, Mosca L, Collins P, et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med. 2006;355:125-137.
123 Munoz-Torres M, Alonso G, Raya MP. Calcitonin therapy in osteoporosis. Treat Endocrinol. 2004;3:117-132.
124 Chesnut CH3rd, Silverman S, Andriano K, et al. A randomized trial of nasal spray salmon calcitonin in postmenopausal women with established osteoporosis: the prevent recurrence of osteoporotic fractures study. PROOF Study Group. Am J Med. 2000;109:267-276.
125 Gennari C. Analgesic effect of calcitonin in osteoporosis. Bone. 2002;30(5 Suppl):67S-70S.
126 Weinstein RS, Roberson PK, Manolagas SC. Giant osteoclast formation and long-term oral bisphosphonate therapy. N Engl J Med. 2009;360:53-62.
127 Black DM, Cummings SR, Karpf DB, et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet. 1996;348:1535-1541.
128 Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077-2082.
129 Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83-91.
130 Chesnut IC, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241-1249.
131 Reid IR, Brown JP, Burckhardt P, et al. Intravenous zoledronic acid in postmenopausal women with low bone mineral density. N Engl J Med. 2002;346:653-661.
132 Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809-1822.
133 Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
134 Khosla S, Burr D, Cauley J, et al. Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2007;22:1479-1491.
135 Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44:4-10.
136 Stepan JJ, Burr DB, Pavo I, et al. Low bone mineral density is associated with bone microdamage accumulation in postmenopausal women with osteoporosis. Bone. 2007;41:378-385.
137 Odvina CV, Levy S, Rao S, et al. Unusual mid-shaft fractures during long term bisphosphonate therapy. Clin Endocrinol (Oxf). 2009.
138 Neviaser AS, Lane JM, Lenart BA, et al. Low-energy femoral shaft fractures associated with alendronate use. J Orthop Trauma. 2008;22:346-350.
139 Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434-1441.
140 Dempster DW, Cosman F, Kurland ES, et al. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res. 2001;16:1846-1853.
141 Vahle JL, Long GG, Sandusky G, et al. Bone neoplasms in F344 rats given teriparatide [rhPTH(1-34)] are dependent on duration of treatment and dose. Toxicol Pathol. 2004;32:426-438.
142 Vahle JL, Sato M, Long GG, et al. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1-34) for 2 years and relevance to human safety. Toxicol Pathol. 2002;30:312-321.
143 McDonald MM, Dulai S, Godfrey C, et al. Bolus or weekly zoledronic acid administration does not delay endochondral fracture repair but weekly dosing enhances delays in hard callus remodeling. Bone. 2008;43:653-662.
144 Huang RC, Khan SN, Sandhu HS, et al. Alendronate inhibits spine fusion in a rat model. Spine. 2005;30:2516-2522.
145 Lehman RAJr, Kuklo TR, Freedman BA, et al. The effect of alendronate sodium on spinal fusion: a rabbit model. Spine J. 2004;4:36-43.
146 O’Loughlin PF, Cunningham ME, Bukata SV, et al. Parathyroid hormone (1-34) augments spinal fusion, fusion mass volume, and fusion mass quality in a rabbit spinal fusion model. Spine. 2009;34:121-130.
147 Abe Y, Takahata M, Ito M, et al. Enhancement of graft bone healing by intermittent administration of human parathyroid hormone (1-34) in a rat spinal arthrodesis model. Bone. 2007;41:775-785.
148 Yasuda Y, Kaleta J, Bromme D. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005;57:973-993.
149 Reginster JY, Seeman E, De Vernejoul MC, et al. Strontium ranelate reduces the risk of nonvertebral fractures in postmenopausal women with osteoporosis: Treatment of Peripheral Osteoporosis (TROPOS) study. J Clin Endocrinol Metab. 2005;90:2816-2822.
150 Burlet N, Reginster JY. Strontium ranelate: the first dual acting treatment for postmenopausal osteoporosis. Clin Orthop Relat Res. 2006;443:55-60.
151 Morley P. Delivery of parathyroid hormone for the treatment of osteoporosis. Expert Opin Drug Deliv. 2005;2:993-1002.
152 Kostenuik PJ, Ferrari S, Pierroz D, et al. Infrequent delivery of a long-acting PTH-Fc fusion protein has potent anabolic effects on cortical and cancellous bone. J Bone Miner Res. 2007;22:1534-1547.
153 Poor G, Donath J, Fornet B, et al. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res. 2006;21:1545-1549.
154 Cooper C, Schafheutle K, Dennison E, et al. The epidemiology of Paget’s disease in Britain: is the prevalence decreasing? J Bone Miner Res. 1999;14:192-197.
155 Altman RD. Musculoskeletal manifestations of Paget’s disease of bone. Arthritis Rheum. 1980;23:1121-1127.
156 Siris ES. Paget’s disease of bone. J Bone Miner Res. 1998;13:1061-1065.
157 Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull. 1984;40:396-400.
158 Barry HC. Paget’s disease of bone. Edinburgh: Churchill-Livingstone; 1969.
159 Meunier PJ, Coindre JM, Edouard CM, et al. Bone histomorphometry in Paget’s disease. Quantitative and dynamic analysis of pagetic and nonpagetic bone tissue. Arthritis Rheum. 1980;23:1095-1103.
160 Rasmussen H, Bordier P. The cellular basis of metabolic bone disease. N Engl J Med. 1973;289:25-32.
161 Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum. 1994;23:222-225.
162 Cody JD, Singer FR, Roodman GD, et al. Genetic linkage of Paget disease of the bone to chromosome 18q. Am J Hum Genet. 1997;61:1117-1122.
163 Haslam SI, Van Hul W, Morales-Piga A, et al. Paget’s disease of bone: evidence for a susceptibility locus on chromosome 18q and for genetic heterogeneity. J Bone Miner Res. 1998;13:911-917.
164 Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res. 2006;21(Suppl 2):P31-P37.
165 Layfield R, Cavey JR, Najat D, et al. p62 mutations, ubiquitin recognition and Paget’s disease of bone. Biochem Soc Trans. 2006;34(Pt 5):735-737.
166 Gordon MT, Anderson DC, Sharpe PT. Canine distemper virus localised in bone cells of patients with Paget’s disease. Bone. 1991;12:195-201.
167 Mills BG, Singer FR. Critical evaluation of viral antigen data in Paget’s disease of bone. Clin Orthop Relat Res. 1987;217:16-25.
168 Rebel A, Malkani K, Basle M. [Nuclear anomalies in osteoclasts in Paget’s bone disease]. Nouv Presse Med. 1974;3:1299-1301.
169 Reddy SV, Singer FR, Mallette L, et al. Detection of measles virus nucleocapsid transcripts in circulating blood cells from patients with Paget disease. J Bone Miner Res. 1996;11:1602-1607.
170 Mills BG, Singer FR, Weiner LP, et al. Immunohistological demonstration of respiratory syncytial virus antigens in Paget disease of bone. Proc Natl Acad Sci U S A. 1981;78:1209-1213.
171 Ralston SH, Afzal MA, Helfrich MH, et al. Multicenter blinded analysis of RT-PCR detection methods for paramyxoviruses in relation to Paget’s disease of bone. J Bone Miner Res. 2007;22:569-577.
172 Matthews BG, Afzal MA, Minor PD, et al. Failure to detect measles virus ribonucleic acid in bone cells from patients with Paget’s disease. J Clin Endocrinol Metab. 2008;93:1398-1401.
173 Rota JS, Heath JL, Rota PA, et al. Molecular epidemiology of measles virus: identification of pathways of transmission and implications for measles elimination. J Infect Dis. 1996;173:32-37.
174 Schneider D, Hofmann MT, Peterson JA. Diagnosis and treatment of Paget’s disease of bone. Am Fam Physician. 2002;65:2069-2072.
175 Ralston SH, Langston AL, Reid IR. Pathogenesis and management of Paget’s disease of bone. Lancet. 2008;372:155-163.
176 Altman RD, Brown M, Gargano F. Low back pain in Paget’s disease of bone. Clin Orthop Relat Res. 1987;217:152-161.
177 Dinneen SF, Buckley TF. Spinal nerve root compression due to monostotic Paget’s disease of a lumbar vertebra. Spine. 1987;12:948-950.
178 Rosen MA, Wesolowski DP, Herkowitz HN. Osteolytic monostotic Paget’s disease of the axis. A case report. Spine. 1988;13:125-127.
179 Hadjipavlou A, Shaffer N, Lander P, et al. Pagetic spinal stenosis with extradural pagetoid ossification. A case report. Spine. 1988;13:128-130.
180 Barry HC. Orthopedic aspects of Paget’s disease of bone. Arthritis Rheum. 1980;23:1128-1130.
181 Reddy SV, Kurihara N, Menaa C, et al. Paget’s disease of bone: a disease of the osteoclast. Rev Endocr Metab Disord. 2001;2:195-201.
182 Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 1-1986. A 67-year-old man with Paget’s disease and progressive leg weakness. N Engl J Med. 1986;314:105-113.
183 Lyles KW, Siris ES, Singer FR, et al. A clinical approach to diagnosis and management of Paget’s disease of bone. J Bone Miner Res. 2001;16:1379-1387.
184 Hughes DE, Wright KR, Uy HL, et al. Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J Bone Miner Res. 1995;10:1478-1487.
185 Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med. 2005;353:898-908.
186 Altman RD, Johnston CC, Khairi MR, et al. Influence of disodium etidronate on clinical and laboratory manifestations of Paget’s disease of bone (osteitis deformans). N Engl J Med. 1973;289:1379-1384.
187 Fraser WD, Stamp TC, Creek RA, et al. A double-blind, multicentre, placebo-controlled study of tiludronate in Paget’s disease of bone. Postgrad Med J. 1997;73:496-502.
188 Reid IR, Nicholson GC, Weinstein RS, et al. Biochemical and radiologic improvement in Paget’s disease of bone treated with alendronate: a randomized, placebo-controlled trial. Am J Med. 1996;101:341-348.
189 Kallio DM, Garant PR, Minkin C. Ultrastructural effects of calcitonin on osteoclasts in tissue culture. J Ultrastruct Res. 1972;39:205-216.
190 Reginster JY, Lecart MP. Efficacy and safety of drugs for Paget’s disease of bone. Bone. 1995;17(5 Suppl):485S-488S.