[level-membership-for-anesthesiology-category]
18 Medications for Hemostasis
This chapter will examine specific treatments aimed at correcting coagulopathy and at reducing the requirement for transfusion of blood products by encouraging hemostasis. Although similar, these objectives are not the same. During many types of surgery, in vitro tests may only demonstrate mild coagulation defects, and yet the use of hemostatic medications may reduce blood loss and obviate the need to transfuse red cells.1,2 In the absence of these medications, bleeding itself is often unlikely to be life-threatening. In comparison, severe coagulopathic bleeding is, by its nature, life-threatening. Transfusion of blood products cannot be avoided and the hemostatic medications used are often the blood products themselves. Fundamentally, the equation of risk and benefit is very different in these two situations. In the former case, benefit will occur if the risk reduction associated with the avoidance of blood products is greater than any risk from the medication itself. In the latter case, the use of medications that carry a high risk of causing adverse effects is more acceptable if the aim is to prevent death from uncontrolled bleeding. As an example of this, aprotinin has been largely rejected for use during adult cardiac surgery as a strategy to reduce transfusion,3 whereas use of recombinant factor VIIa (almost certainly a more “hazardous” drug) is still considered appropriate for the treatment of life-threatening bleeding.
Avoidance of transfusion of human blood products seems to be a fundamentally good notion. However, many common ideas about the potential harm of transfusion may be mistaken. A national audit of the serious hazards of transfusion (SHOT) in the United Kingdom reported 3239 adverse events over a 10-year period; this is only 0.013% of all blood transfusions.4 These adverse events were more common in infants, but were only reported in 0.037% of infants given transfusions. The true incidence of events is likely to be more common because of underreporting, though many of these “events” were procedural errors not associated with actual harm. It is clear that the risk of immediate harm directly attributable to transfusion is exceptionally low. The risk of direct transmission of infection (the most feared risk in the public perception) is particularly small; the risk of transmission of HIV from transfusion of blood in the UK is 1 in 5 million, and no child has contracted a viral infection from transfusion in the last 4 years.4 The greatest hazard of immediate harm is of a mistake leading to transfusion of incompatible blood.5 However, subtle negative effects of transfusion on children’s outcomes may be considerably more important.6 Transfusion of blood may lead to deterioration in pulmonary function and immunologic effects that predispose children to infection. In addition, if the objective of transfused red cells is to boost tissue oxygenation by improving oxygen carrying capacity, then transfused blood is less effective than the child’s own blood for this function.7 Additional considerations include the increasing costs of transfused blood products, the logistical difficulties of maintaining a secure blood supply (in both well-resourced and less well developed health systems), substantially greater risks of transfusion (in poorly developed health systems), and the possibility of new, unrecognized infective agents entering the blood supply.
There are 2.3 million transfusions each year (37 per 1000 population) in the UK; 4.2% are given to children under 18 years of age (7.1 per 1000) and 1.7% to infants (52 per 1000). An audit of pediatric blood transfusion from Australia revealed 41% of blood transfused was given perioperatively,8 and blood was transfused during 6.3% of instances of anesthesia. The majority of units were used during heart surgery (58% of perioperative use) while a very small minority, 4% were used during major trauma surgery. Heart surgery (on and off bypass), craniosynostosis surgery, and liver transplantation were all commonly associated with blood transfusion. The epidemiology of major bleeding is less certain. Reports of blood use may be misleading (e.g., neonates undergoing cardiac surgery to correct congenital defects may receive few blood units despite significant bleeding), and blood is also used for purposes such as priming the bypass circuit. Observational studies have reported very heavy blood loss associated with heart surgery in children, with increased (per kg) blood loss in smaller children and more complex surgery.9,10 Cardiac surgery can probably be considered the main cause of major blood loss in children and will account for most severe bleeding events in children less than 1 year of age.
Physiology of Coagulation
The understanding of coagulation has shifted greatly in the last few years. The “traditional” model, which emphasizes the importance of a cascade of proteolytic enzymes has given way to the “cell based” model of coagulation that emphasizes the importance of cellular elements in coagulation and presents coagulation as a complex web of interactions rather than a linear process.11,12
To achieve effective hemostasis, a platelet plug must form at the site of vessel injury. In addition, the procoagulant factors need to remain localized to the injured site to avoid widespread clotting activation. This is achieved through changes at the cell surface and localization of the procoagulant reactions on the surfaces of specific cells. Different cells possess different procoagulant and anticoagulant properties; these are incompletely understood, but platelets and cells bearing tissue factor (TF) are central to the process. Intact endothelium is also vital to normal control of coagulation, as it keeps these different procoagulant components apart and modifies coagulation through expression of inhibitory proteins such as thrombomodulin. The described phases of coagulation are overlapping and involve initiation, amplification, and propagation.12
Initiation
Coagulation is initiated by a membrane-bound lipoprotein called tissue factor. This is usually expressed on subendothelial TF-bearing cells, such as stromal fibroblasts; in the absence of vessel injury it is separated from the vessel lumen. After injury, a complex is formed between TF and factor VIIa (TF/VIIa), which activates factors IX and X. Factor Xa, in association with cofactor Va, forms “prothrombinase” complexes on the surface of the TF-bearing cell, which activates a small amount of thrombin (factor IIa)12 (Fig. 18-1). The purpose of this small generation of thrombin and Xa is to activate platelets and factors V and VIII.
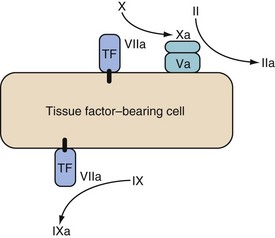
Low levels of IX and X activation occur in the absence of tissue injury without causing clot formation. The process only leads to amplification when damage to the vasculature allows intravascular platelets and a complex formed by factor VIII and von Willebrand factor (VIII/vWF) to adhere to the extravascular TF-bearing cells.11
Amplification
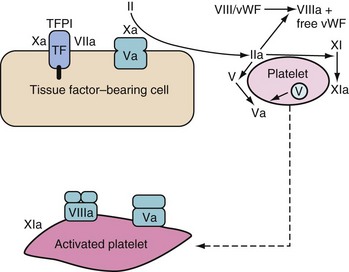
Small quantities of thrombin are generated on the TF-bearing cells. This sets up the subsequent propagation phase, during which thrombin is generated in large quantities (Fig. 18-2). This thrombin has many major functions:
 Activation of platelets, exposing receptors and binding sites for clotting factors
Activation of platelets, exposing receptors and binding sites for clotting factors

 Activation of factor XI to XIa13
Activation of factor XI to XIa13
 Activation of factor XIII (fibrin stabilizing factor) and promotion of fibrin crosslinking
Activation of factor XIII (fibrin stabilizing factor) and promotion of fibrin crosslinking
 Cleaving fibrinopeptides A and B from fibrinogen (forming fibrin)
Cleaving fibrinopeptides A and B from fibrinogen (forming fibrin)
Propagation
Propagation occurs on the surface of activated platelets that are recruited to the site in large numbers. Activated factor IX (from both initiation phase and provided by factor XI on the platelet) binds to VIIIa. The resultant IXa/VIIIa complex activates factor X on the platelet surface. This factor Xa associates with factor Va and forms the prothrombinase complex. The prothrombinase complex causes a “burst” of thrombin generation to cause clotting via fibrinogen (Fig. 18-3).11
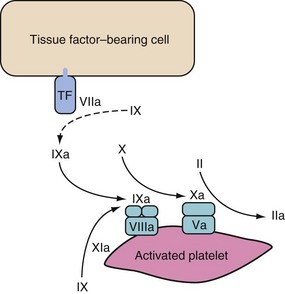
This sequence explains why children with hemophilia bleed despite having a normal TF/VIIa complex; the factor Xa during the initiation phase is broken down by ATIII and TFPI if it dissociates from the TF-bearing cell. It is therefore unable to activate the “burst” of thrombin generation that normally occurs in this propagation phase. The factor Xa needs to be generated on the platelet itself via the IXa/VIIIa complex.11
Of note, an alternative pathway is initiated by contact factors (XII, XI, prekallikrein, and high-molecular-weight kininogen [HMWK]). It is of no physiologic importance in terms of coagulation activation; however, it provides important acceleration loops through feedback activation of factors VIII, IX, and XI14 and is important in fibrinolytic and inflammatory pathways.15
Clot Inhibition
Several indirect systems inhibit thrombin, such as the protein C–protein S–thrombomodulin (TM) and the TFPI systems. Thrombin binds to TM on the surface of intact endothelial cells and can no longer cleave fibrinogen to form fibrin. The TM/thrombin complex is neither able to activate platelets nor activate factors V and VIII. Instead, this complex activates protein C, which binds to the cofactor protein S and inactivates factors Va (on the surface of endothelial cells and platelets) and VIIIa.11,14 TFPI bound to endothelial surfaces can form complexes with factor Xa that inhibit factor VIIa. Thrombin generation is therefore inhibited.14
Fibrinolysis
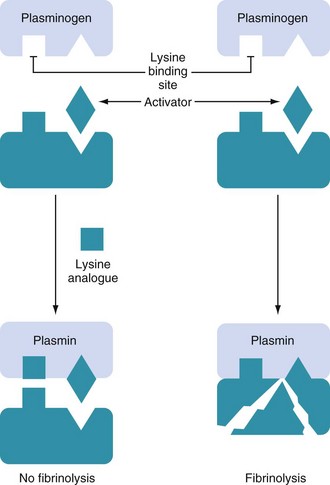
Fibrinolysis (the breakdown of fibrin into soluble degradation products) is mediated by the proteolytic enzyme, plasmin. Plasmin is formed from an inactive zymogen, plasminogen, which is produced in the liver. This process is controlled by activators and inhibitors. The principal plasminogen activators are tissue plasminogen activator (tPA) and urokinase (uPA). Although overlap exists between the functions of these activators, they have distinct physiologic roles. uPA is primarily involved in a wide variety of extracellular processes, including remodeling of tissues. tPA is the major intravascular activator of fibrinolysis and is discussed in more detail later.16 The main inhibitory proteins are plasminogen activator inhibitor-1 (PAI-1), antiplasmins (α2-PI and α2-macroglobulin), and thrombin activated fibrinolysis inhibitor (TAFI)17 (Fig. 18-4).
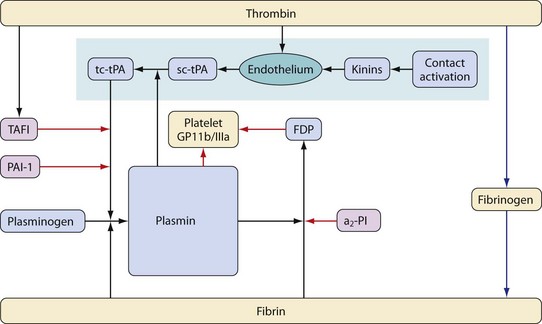
As well as cleaving fibrin, plasmin metabolizes a number of other proteins, including the platelet receptor for fibrinogen (glycoprotein IIb/IIIa) and fibrinogen.18 In addition, plasmin accelerates its own production by metabolizing the conversion of single chain plasminogen activators to more active two-chain versions. The action of plasmin on fibrin produces a series of degradation products some of which convey anticoagulant properties; this effect is achieved by preventing polymerization of fibrinogen and by inhibition of platelet function.
tPA is released by vascular endothelium of small blood vessels. Release is increased in the presence of stimuli such as trauma, endotoxins, ischemia, or normal exercise. This effect is mediated via contact activation (through the kallikrein system) and also by a series of other substances, including thrombin. Once released, tPA is rapidly metabolized by the liver with a half-life of approximately 5 minutes.17 Fibrin binds both plasminogen and tPA and greatly accelerates the conversion of plasminogen to plasmin (facilitating its own degradation, but also localizing the process to areas of clot). An alternative mechanism for plasminogen activation by tPA exists through binding to receptors expressed by certain cells (endothelium, white cells and some tumor cells); the importance of this in health or disease is unclear.
Excessive fibrinolysis can directly result from excess production of fibrin, as in disseminated intravascular coagulation. This is termed secondary hyperfibrinolysis and in this context the fibrinolysis is considered beneficial because it prevents widespread vascular occlusion. Therapy is directed at replacement of consumed clotting factors, inhibition of excessive coagulation, and treatment of the underlying cause. Primary hyperfibrinolysis can occur during cardiac bypass, massive blood loss, trauma, and liver transplantation.19 During the anhepatic stage of liver transplant surgery, there is hyperfibrinolysis because of failure to metabolize tPA. On reperfusion of the liver, a further surge of tPA occurs that can take several hours to return to normal. In coagulopathic patients, reduced thrombin formation may lead to reduced production of TAFI (important in inhibition of membrane bound plasmin), whereas conversion of single- to two-strand tPA by plasmin may further sustain the process. Individual susceptibility is likely to be important and may have a genetic component.20,21
Two groups of drugs are used clinically to inhibit fibrinolysis:
Synthetic lysine analogues are fairly specific inhibitors of plasminogen activation, working by competitively binding to lysine-binding sites on the plasminogen molecule (Fig. 18-5). This blocks the binding of plasminogen to fibrin; a required step for the conversion of plasminogen to plasmin by plasminogen activators.22 At larger doses, they may have additional effects through direct inhibition of plasmin; this includes inhibition of the plasmin mediated effects on platelets.
Aprotinin is a less specific inhibitor of proteolytic enzymes; it has actions on the kallikrein–kinin (contact) system, as well as enzymes involved in coagulation and fibrinolysis. In addition, aprotinin may be associated with greater preservation of platelet function, as well as an antiinflammatory effect. This wider spectrum of effects from aprotinin may have additional benefits over the antifibrinolytic lysine analogues. Some additional effects may not necessarily be of benefit; for example, the contact system may have protective effects against ischemic reperfusion injury that are inhibited by aprotinin.15
Developmental Coagulation
The hemostatic system in the neonate rapidly matures towards that of the adult (see also Chapter 9).23–25 One must consider how the coagulation system matures in the child to interpret coagulation tests and use appropriate modalities to manipulate hemostasis in vivo.
All fetal coagulation factors are produced independently of the mother; fibrinogen starts to be formed as early as 5.5 weeks gestation, and blood can clot at 11 weeks. The introduction of microassays in the 1980s allowed for the determination of reference ranges for the coagulation factors beginning at 19 weeks gestational age.14,23,24 In general, there are four fundamental differences between the coagulation systems in the infant and the adult26:
 Concentrations of components of the hemostatic system
Concentrations of components of the hemostatic system
 The turnover rate of various components of the coagulation cascade
The turnover rate of various components of the coagulation cascade

 Differences in the overall ability to generate and regulate the key enzymes: thrombin and plasmin
Differences in the overall ability to generate and regulate the key enzymes: thrombin and plasmin
Despite the upregulation of coagulation factors at birth, the vitamin K–dependent factors II, VII, IX, and X in the neonate are only 50% of adult values; this leads to a slightly prolonged prothrombin time (PT) or international normalized ratio (INR).14 The contact factors HMWK, prekallikrein, and factors XI and XII are also approximately 50% of adult values.14,26 The reduced contact factors account for a disproportionally prolonged activated partial thromboplastin time (aPTT). The reduced concentration of factors at birth is probably explained by the reduced synthesis of factors by the liver; however, concentrations increase rapidly, reaching approximately 80% of adult values by 6 months of age.14,27
In contrast, the plasma concentrations of fibrinogen and factors V and VIII at birth are similar to those in adults, although the fetal form of fibrinogen differs in structure from that of the adult. The physiologic significance of this is not clear.28 Concentrations of vWF in the first 2 months of life are greater than those in adults.29
The inhibitor systems of coagulation also differ from adults. At birth, plasma protein C and S are 35% of adult values, although the fetal forms of the proteins differ from the adult forms. The concentration of protein C does not reach adult values until adolescence. Neonatal concentrations of ATIII and HCII are 50% of adult values; they reach adult concentrations by 6 months of age. The α2-macroglobulin value, however, is increased at birth and remains increased throughout childhood; it is postulated that this may be one of the mechanisms that protects young children from thromboembolic complications.30
Finally, thrombin generation in vitro is reduced in children to approximately 75% of adult values,30 but there is difficulty in interpreting the apparent discrepancy between the sufficient hemostasis seen in vivo and these in vitro laboratory thrombin generation assays. It still remains to be clarified whether neonates and children are more susceptible to bleeding14; however, the risk of thromboembolic complications appears to increase with age.30
Despite reduced plasma concentrations of many procoagulant and anticoagulant proteins in infants, there still appears to be an effective hemostatic balance; healthy fetuses, neonates, and children do not suffer excessive hemorrhage in the presence of minor challenges. This is consistent with the thromboelastogram (TEG) studies of healthy children younger than 2 years of age; no defects in coagulation were noted using this test compared with adults, indicating an intact hemostatic system.31 Another TEG study reported that infants younger than 1 year of age with complex congenital heart disease have an intact and balanced coagulation–fibrinolytic system but at a “lower level” than healthy children. This has been interpreted as a reduction in hemostatic potential with less reserve.32
Genetics of Bleeding
The hemophilias are a group of genetic diseases that cause excessive bleeding, often in response to minor trauma. von Willebrand disease and hemophilia A are the most common variants (see also Chapter 9), associated with low levels of von Willebrand factor and factor VIII respectively. A wide range of single gene defects resulting in deficiencies of single clotting proteins or regulatory proteins have now been described. A further group of single gene disorders may result in thrombophilic disorders, associated with abnormalities of inhibitory proteins.
Unexplained variation has been observed in bleeding between apparently similar patients in the absence of specific factor deficiency. The causes of this variation are likely to be multifold according to nuances of surgical technique and subtle difference in disease process and therapy. It might appear counterintuitive that genetic factors have a significant role in acquired bleeding resulting from surgery; however, recently there has been increased interest in the interplay of genetic and environmental factors in the progress of acquired diseases. The genetics of most clotting proteins has been described, and common variations within populations have been revealed for some. Of these, a common polymorphism of PAI-1 has been well described. PAI-1 is an important endogenous inhibitor of fibrinolysis, and deficiency is associated with increased bleeding.33 The G5/G5 polymorphism is common (about 20% in European populations) and is associated with lower levels of PAI-1. It has been linked (though not consistently) to bleeding after heart surgery and to increased benefit from use of antifibrinolytics.20,21 Should these findings be substantiated, then PAI-1 may still be an exceptional example. In general, the influence and consequences of genetic determinants of bleeding are likely to be more subtle. In infancy an additional factor in the mix may be the relationship between developmental and genetic factors. Many clotting proteins are present in infants as isoforms distinct from those in adults. This implies a different gene expression in the young. It is possible that understanding genetically determined variations in bleeding will increase our understanding of bleeding and, perhaps, allow us to guide therapy for the individual patient. The practical applications of such techniques, however, remain speculative.
Coagulopathy and Major Surgery
Coagulation changes during major surgery and bleeding are complex34 and depend on the clinical context in which bleeding occurs. Coagulation changes that occur in surgical patients have some similarities to those who present after severe trauma; however, the balance of pathophysiologic factors is likely to be very different. The factors underlying these coagulation changes include:
 Dilution. Components of the coagulation system are lost in shed blood. The volume of blood lost is then replaced by crystalloid, colloid, or blood products lacking these components, leading to progressively smaller concentrations of these coagulation components. To some degree such changes are balanced, as the concentration of coagulation inhibitors also falls. In addition, coagulation components may be produced or released in response to trauma, which limits the reduction in concentration. To complicate this issue, a reduction in the concentration of one component of the coagulation system may not have the same clinical effect as the same reduction of another component. For example, substantial decreases in the concentration of many clotting proteins will not result in severe bleeding, while even modest decreases in platelet numbers or in the fibrinogen concentration may be significant (see also Chapter 10).
Dilution. Components of the coagulation system are lost in shed blood. The volume of blood lost is then replaced by crystalloid, colloid, or blood products lacking these components, leading to progressively smaller concentrations of these coagulation components. To some degree such changes are balanced, as the concentration of coagulation inhibitors also falls. In addition, coagulation components may be produced or released in response to trauma, which limits the reduction in concentration. To complicate this issue, a reduction in the concentration of one component of the coagulation system may not have the same clinical effect as the same reduction of another component. For example, substantial decreases in the concentration of many clotting proteins will not result in severe bleeding, while even modest decreases in platelet numbers or in the fibrinogen concentration may be significant (see also Chapter 10).

 Physiologic derangement associated with blood loss. Acidosis, hypothermia, and hypocalcemia are associated with excess bleeding.35 Hypothermia will result in a general slowing of proteolytic enzyme activity, reduced fibrin synthesis, and reduced platelet function. These effects are largely reversible on rewarming. Acidosis is associated with a marked reduction in activity of coagulation proteins.36 These effects are not fully reversed by correcting acidosis. The degree to which coagulation changes reflect acidosis itself or reflect other underlying factors is unclear. During major bleeding, calcium ion concentration should be monitored and replaced as needed.
Physiologic derangement associated with blood loss. Acidosis, hypothermia, and hypocalcemia are associated with excess bleeding.35 Hypothermia will result in a general slowing of proteolytic enzyme activity, reduced fibrin synthesis, and reduced platelet function. These effects are largely reversible on rewarming. Acidosis is associated with a marked reduction in activity of coagulation proteins.36 These effects are not fully reversed by correcting acidosis. The degree to which coagulation changes reflect acidosis itself or reflect other underlying factors is unclear. During major bleeding, calcium ion concentration should be monitored and replaced as needed.
 Effects of treatment. The use of some synthetic colloids may worsen bleeding to an extent greater than might be expected by dilution. The use of hydroxyethyl starch leads to increased risk of coagulation abnormalities and of acute kidney injury when compared to albumin, gelatins, or crystalloids. Whether differences exist in coagulation effects between different starches is controversial.37–39 Caution should be exercised in the use of starches in children at risk of coagulation problems, and in larger volumes.
Effects of treatment. The use of some synthetic colloids may worsen bleeding to an extent greater than might be expected by dilution. The use of hydroxyethyl starch leads to increased risk of coagulation abnormalities and of acute kidney injury when compared to albumin, gelatins, or crystalloids. Whether differences exist in coagulation effects between different starches is controversial.37–39 Caution should be exercised in the use of starches in children at risk of coagulation problems, and in larger volumes.
 Use of specific techniques during surgery. The important effects of cardiac bypass and anticoagulation are discussed later. Liver transplant surgery (see also Chapter 29) and major trauma (see also Chapter 38) are discussed in detail elsewhere in this book.
Use of specific techniques during surgery. The important effects of cardiac bypass and anticoagulation are discussed later. Liver transplant surgery (see also Chapter 29) and major trauma (see also Chapter 38) are discussed in detail elsewhere in this book.
Alterations in Hemostasis during Pediatric Cardiac Surgery
Routine Anticoagulation
Heparin
Heparin remains the most effective anticoagulant used to facilitate cardiopulmonary bypass (CPB).40 The binding of heparin to lysine sites on ATIII causes a conformational change in ATIII. This results in an increase in ATIII potency; the inhibition of thrombin and factors IXa, Xa, XIa, and XIIa are increased by a factor of 1000.41,42 In infants ATIII levels are low until 3 to 6 months of age and other heparin cofactors, in particular, α2-macroglobulin may have greater importance. However neonates requiring surgery for congenital heart disease have unusually reduced concentrations of all major heparin cofactors, and this may explain the greater concentrations of thrombin production in these patients.43
There are limitations to the use of the ACT; first, the ACT is altered by hypothermia, hemodilution, platelet activation, activation of the hemostatic system, and aprotinin therapy.44,45 Accordingly, it does not accurately reflect the heparin concentrations. One study found that as soon as children go on CPB, the heparin concentrations decreased by 50% as a result of hemodilution, even though the ACT doubled.46 Second, in the bleeding child, the ACT is unable to differentiate between bleeding resulting from excess heparin or from other acquired hemostatic defects.46 The gold standard for measurement of heparin concentration is considered to be the antifactor Xa assay; however, this test remains too cumbersome for routine clinical use. A further method is protamine titration, which has been available as a point-of-care test for some years (Medtronic Hepcon HMS Plus, Medtronic, Minneapolis, USA). In adult patients, use of this system has been shown to lead to reduced thrombin formation47; however, the accuracy of the device has been questioned.48 Reduced bleeding and reduced thrombin generation in children has been demonstrated with the use of this system, whereas other studies have demonstrated reduced thrombin formation22,49 in infants. A trial in small infants was terminated early when increased bleeding and increased length of stay was demonstrated in children in whom the Hepcon device was used.49 The device underestimated heparin concentration in this group, leading to excess dosing of heparin and inadequate reversal with protamine. After modification of their protocol, use of the device demonstrated reduced bleeding, length of stay, and reduced thrombin formation compared to standard treatment.49 Agreement between protamine titration, measures of heparin concentration, and laboratory measures has been demonstrated, but the Hepcon devices tended to underestimate heparin concentrations in infants.50
A common feature of the pediatric studies using the Hepcon device is increased heparin use compared to regimes based on units/kg dosing or ACT. This is consistent with other studies of traditional dosing regimes. Using common pediatric heparin regimens (300 units/kg before CPB, then 100 units/kg to keep the ACT above 450 sec), 50% of children on CPB had low levels of heparin (less than 2 units/mL).46 It is suggested that reduced heparin concentrations during CPB is a major factor responsible for activation of coagulation and fibrinolysis. It is likely that widely used regimens for dosing of heparin in children lead to inadequate dosing, and that units/kg dosing fails to allow for important pharmacokinetic (PK) and pharmacodynamic (PD) differences in children.
Even effective dosing of heparin does not completely abolish the production of thrombin. Low-grade ongoing thrombin production leads to ongoing activation of the coagulation cascade, platelets, fibrinolysis, and the endothelium. The continued thrombin generation and activity during CPB reflects the inability of the heparin–ATIII complex to inactivate fibrin-bound thrombin or to inhibit thrombin-induced platelet activation.51 Theoretically, direct thrombin inhibition may be free of these limitations. Practically, the use of the current generation of thrombin inhibitors (such as hirudin and bivalirudin) is limited because of a lack of effective monitoring and reversal. Currently few reports exist of the use of hirudin in children, although it would be indicated in children in whom heparin use is not possible.52
Reversal of Anticoagulation with Protamine
Protamine is a positively charged polypeptide derived from salmon sperm. It neutralizes heparin by forming an ionic bond with heparin. The resultant complex is removed by the reticuloendothelial system. The most appropriate dosing regimen has yet to be determined. Current dosing used in pediatric practice fails to take into account the range of concentrations of heparin that occurs in infants and children.40,49 The administered heparin dose is often used to guide the dose of protamine; however, it is unclear how this should be modified by various factors, such as additional doses of heparin administered (to prime or during bypass), duration of bypass, techniques (e.g., ultrafiltration), or developmental coagulation differences in children.53
Excessive protamine has been associated with catastrophic pulmonary hypertension and hemorrhagic pulmonary edema. It is also known that protamine can be associated with coagulation abnormalities; an increasing ACT occurs at a protamine-heparin ratio of 2.6 : 1, and platelet aggregation occurs with a minimal excess in protamine.54 Although some studies on titrating protamine regimens in adults demonstrated encouraging results in terms of reduced bleeding,55 others failed to demonstrate differences in transfusion requirements.56
The clearance of protamine is greater than that of heparin, and “heparin rebound” is described as tissue-bound heparin redistributes.40 The diagnosis of residual heparin effect or heparin rebound is challenging. The ACT is not a specific measure of excessive heparin and is also poor at detecting heparin at low concentrations (less than 0.5 unit/mL).57 The aPTT and PT are similarly nonspecific and may be increased after CPB in the absence of heparin.58 An unmodified TEG does not reliably detect heparin rebound if the heparin concentration is small.59 The sensitivity of these tests may be improved by performing similar tests in parallel and comparing the results, such as a reptilase time (unaffected by heparin), or by eliminating residual heparin in vitro with heparinase or protamine. Protamine titration can be used to guide protamine dose in children; however, in infants protocols will require modification (50% higher than calculated dose).49 In practice most anesthesiologists continue to give protamine empirically at a protamine-heparin ratio of between 1 and 1.3 to 1.
Failure of Hemostasis Associated with Cardiac Surgery
Complex abnormalities occur in the coagulation system in cardiac surgery, owing to the profound surgical insult, hypothermia, acid-base disturbance, blood transfusion, anticoagulants, CPB (contact activation, platelet dysfunction, and hemodilution), and in some cases, deep hypothermic cardiorespiratory arrest. In addition children and infants with congenital heart disease may have preexisting coagulation defects or be taking medications that affect coagulation before surgery (Table 18-1).
TABLE 18-1 Causes of Excessive Bleeding after Pediatric Cardiac Surgery
Antiplatelet drugs such as aspirin or clopidogrel may exacerbate bleeding. The clinician must balance the risk of perioperative bleeding against the risk of drug discontinuation when deciding if and when to withhold the drug before surgery. In most cases aspirin can be safely discontinued 5 days before surgery. Prostaglandin E1 can inhibit platelet aggregation, acting in synergy with endothelial cell–derived factors (nitric oxide and prostacyclin) at clinically relevant concentrations,60 although this effect was too subtle to detect in vitro using the TEG.32 It is usually not possible to stop the prostaglandin E1 infusions, because neonates are dependent on its use to maintain ductal patency. For elective surgery in children on oral anticoagulant therapy, it is usually possible to transfer to heparin before surgery. An INR of less than 1.5 is usually considered acceptable for surgery. If urgent correction of anticoagulants is required, prothrombin complex concentrates, together with vitamin K, are more effective than FFP.61 FFP should only be used if prothrombin complex concentrates are not available.
In the child with cyanotic congenital heart disease, hemostasis is further impaired because of polycythemia, low platelet count and altered function, reduced factors V, VII, and VIII, and increased fibrinolysis.32,62 The degree of derangement is related to the degree of cyanosis.63 Preexisting coagulopathy may also occur in children with severe underlying illness or poor nutritional status.
Despite improvements in design and materials of the CPB circuit, abnormal activation of the coagulation and fibrinolytic systems persist. The normal balance of coagulation and fibrinolysis is particularly delicate in children, and more susceptible to exogenous perturbations.64 Shortly after CPB is established in children, all hemostatic protein concentrations are decreased (to a variable degree) as a result of dilution.65 Because of the relative volume of the circuit in relation to the size of the child, it is not surprising that the magnitude of the effect of hemodilution is greater than in adults.46
Reductions in coagulation factor concentrations after cardiac surgery contribute to bleeding in adult cardiac patients,66 and a reduction in platelet count is well described on initiation of bypass. In adults, the platelet count is reduced on CPB by approximately 50%.67 This is also attributed primarily to dilution, although other factors include organ sequestration, mechanical disruption, and adhesion to the circuit.68 A relatively greater reduction will occur in smaller children, although newer combined filter and oxygenators should considerably reduce the circuit volume and, hence, the degree of dilution. Significant platelet dysfunction also occurs.46,69 Low platelet counts have been strongly associated with increased bleeding.10,31,70
Further activation of platelets and denaturing of plasma proteins will occur at interfaces between blood and air, or on contact with extravascular tissue. CPB pump suction and spilling of blood into the pericardium will further contribute to coagulopathy. Greater coagulopathy is also seen during prolonged bypass and when deep hypothermic circulatory arrest is employed.71
The major mechanism for activation of the coagulation cascade during CPB is thought to be the “extrinsic” TF pathway, which is activated as a result of surgical trauma and inflammation.42,72,73 Inflammatory mediators such as tumor necrosis factor (TNF) and interleukin-1 induce expression of TF on endothelial cells and monocytes. The “intrinsic” coagulation system is also activated when factor XII is adsorbed onto the surface of the CPB circuit, causing activation of complement, neutrophils, and the fibrinolytic system via kallikrein.42 Although the intrinsic system has little role in initiating coagulation, activations of kinins will lead to increased fibrinolysis and inflammation.15
The blood loss in the first 24 hours after cardiac surgery can vary between 15 and 110 mL/kg, although the risk of excessive blood loss is greatest among those weighing less than 8 kg, younger than 1 year of age, and children undergoing complex surgery.10 The common preoperative and intraoperative risk factors for excessive bleeding are summarized in Table 18-2.
| Preoperative | Intraoperative |
|---|---|
| Age <1 year or weight <8 kg | Individual surgeon |
| High hematocrit | Complex surgery |
| Congestive heart failure | Low platelet count during CPB |
| Repeat sternotomy | Prolonged CPB |
| Congenital and preoperative acquired coagulopathy | Duration of hypothermia on CPB |
| Cyanotic congenital heart disease | Deep hypothermic cardiac arrest |
CPB, Cardiopulmonary bypass.
Williams GD, Bratton SL, Ramamoorthy C. Factors associated with blood loss and blood product transfusions: a multivariate analysis in children after open-heart surgery. Anesth Analg 1999;89:57-64.
Blood loss and transfusion requirements in pediatric cardiac surgery vary inversely with age; neonates bleed more and receive more products per kilogram than any other age group.74 There are some correlations between coagulation tests before and during CPB and postoperative blood component transfusion requirements, although the sensitivity and specificity of the tests are not sufficient to justify routine coagulation tests while on CPB. Of all the tests, a platelet count of less than 108,000/mm3 while on CPB yielded the greatest sensitivity (83%) and specificity (58%) for predicting excessive blood loss.70
Medications Used for Hemostasis
The pathophysiology of coagulopathies and bleeding in children during surgery are complex, and the etiology is multifactorial (see Table 18-1). No single blood component or drug treatment can reverse the abnormal clotting profile. Initially an attempt should be made to identify causes of bleeding that can be remedied by surgical interventions. The anesthesiologist should attempt to ensure adequate reversal of heparin and restore normal physiologic variables, such as body temperature, serum [Ca2+], and acid-base balance. In the postoperative period, platelets, and other blood products, such as fresh frozen plasma (FFP) and cryoprecipitate, remain the mainstay of treatment for excessive bleeding.75 Blood products (see also Chapter 10) are briefly discussed here, primarily within the context of pediatric heart surgery.
Blood Products
Frequently it is necessary to administer blood products on a largely empirical basis; laboratory tests describe only parts of the coagulation process32 and are practically too slow to direct the anesthesiologist in real time as to the requirement for blood components. The TEG is a dynamic whole-blood test that is frequently used as a near-patient test to assess clot elasticity properties and, hence, more precisely delineate the bleeding and homeostasis profile. The role of TEG during pediatric heart surgery has been recently reviewed.76 The use of treatment algorithms based on TEG can limit transfusion of blood products in adults77–81 and children.82 In children, it may not be appropriate to use protocols originally designed for adults. An alternative approach has recently been proposed.76
Platelets
Platelet dysfunction and thrombocytopenia are common after cardiac bypass in infants. Hence, in the presence of bleeding, transfusion of platelets is logical. Coagulation variables that relate to platelet number and function (such as TEG maximum amplitude) are corrected by infusion of platelets, and clinical experience indicates that platelet transfusion reduces bleeding. This has been confirmed in a small study of children after cardiac surgery.31 A platelet count of less than 108,000/mm3 has been identified as a predictor of bleeding, whereas clot strength measured by TEG reduces steeply at platelet counts below 120,000/mm3. The current recommendation of targeting a platelet count of approximately 100,000/mm3 appears to be logical. It would also appear reasonable to use platelets for the initial treatment of presumed coagulopathic bleeding (in the absence of specific clotting tests).
Fresh Frozen Plasma
The case for use of fresh frozen plasma (FFP), either as empirical treatment of bleeding, or guided by coagulation tests, is considerably weaker than for platelets. The use of FFP in cardiac surgery is based on the observation that the concentration of clotting factors are often low in bleeding patients, especially in the period following bypass. The PT (with its derived measure, INR) is the most common test used to detect the presence and gauge the severity of clotting factor–deficient coagulopathy. Unfortunately, observation studies have shown that the PT correlates poorly with clinical bleeding, and that transfusion of plasma often achieves no measurable change in the INR, nor is of any known clinical benefit (particularly if the INR was only marginally raised to less than 1.7).83 Despite this, FFP is frequently transfused in the absence of either bleeding or significantly raised PT.84 Administration to patients with higher PT values will be more effective in correcting clotting tests, however large volumes may be required,83 even in the absence of ongoing loss of clotting factors. The situation is further complicated, in that coagulation tests that are initially normal will deteriorate during severe bleeding.
A systemic review of the use of FFP to treat or prevent bleeding resulting from acquired coagulopathy failed to demonstrate benefit, although the trials examined were small, used a wide range of outcomes, and were conducted in heterogeneous populations.85 In a small observational study of children undergoing heart surgery, a number of patients had coagulopathic bleeding after transfusion of platelets; if these patients were then given FFP, the bleeding increased, whereas if cryoprecipitate was given bleeding decreased.31
It is possible that the lack of efficacy may be related to the dose of FFP used. The dose of FFP most frequently recommended (10 to 15 mL/kg)84,86 may be inadequate to restore factor concentration.83 Of note, dosage of plasma is inexact. Hence, interinstitution practices differ considerably in defining both a threshold INR for prophylactic plasma administration and the actual dosage of plasma prescribed.87
Modeling of FFP administration in major trauma suggests that larger doses (30 to 40 mL/kg) may be required to increase or maintain factor concentrations88; however, administration of such a large volume is often undesirable in the absence of severe ongoing bleeding. Addition of FFP to the bypass circuit or during modified ultrafiltration may allow administration of much larger doses; benefit appears to be confined to younger children.89,90 It is always important to document the specific reasons for FFP administration.
Cryoprecipitate
Not all coagulation factors are of equal importance during bleeding. Fibrinogen is present in much greater concentrations than other clotting factors, and while other factors are mainly involved in initiating or amplifying thrombin formation, fibrinogen is a substrate for the production of fibrin. Deficiencies of fibrinogen are reflected in reduced strength of clot and are associated with increased bleeding.91 Cryoprecipitate is the most concentrated fibrinogen replacement widely available, with a fibrinogen concentration 4 to 8 times that of FFP; however, a unit of cryoprecipitate contains less fibrinogen than a unit of FFP, and multiple cryoprecipitate units will be required in larger patients. One cryoprecipitate unit for each 10 kg should raise fibrinogen concentration by 0.5 to 1.0 g/L. As well as fibrinogen, cryoprecipitate is a source of von Willebrand factor and factors VIII and XIII, although the clinical significance of this to bleeding is unclear. Cryoprecipitate has been effective in treating bleeding resistant to platelet concentrates alone during pediatric heart surgery.31
Fractionated Human Blood Products
Products such as FFP or cryoprecipitate can be considered as only crudely purified blood products. It is possible to produce more refined products containing high concentrations of only a single clotting protein or relatively standardized concentrates of selected proteins. Examples of these are prothrombin complex concentrates and fibrinogen concentrate (e.g., Riastap, CSL Behring, King of Prussia, Pa., USA; Haemocomplettan P, CSL Behring, Marburg, Germany). These products are human blood products produced from pooled plasma; they are presented as a powder requiring reconstitution for use. They do not require freezing or cross matching, which simplifies their supply, storage, and administration. Risk of viral transmission should be low92 because of sourcing of plasma from low-risk populations, pooling of plasma from many individual donors (reducing viral load resulting from a single infected donor), and pasteurization. Although the effect of pasteurization on prion infection is less certain, the risk of transmission would be expected to be similarly low. Recombinant factor concentrates (as opposed to factor concentrates of human origin) are increasingly available and avoid cross infection. One such agent, recombinant activated factor VII (rVIIa), is discussed later in this chapter (see also Chapter 10). Although the agents discussed in this section are used in a similar way to traditional factor supplementation to reestablish near physiologic concentrations, rVIIa is used very differently to produce factor levels greatly in excess of “normal.”
Prothrombin complex concentrates (PCCs) are mixtures of factors II (prothrombin), VII, IX, and X, plus the coagulation inhibitors Protein S and C. They are indicated when urgent reversal of oral anticoagulation (with warfarin or related drugs) is required. Dose is specific for each make of PCC, and is dependent on the child’s INR and the target for correction (generally an INR of 1.5). Older reports of lack of efficacy or risk of thrombosis probably relate to older brands of PCCs with low levels of factor VII or of inhibitors.93 PCCs are more efficacious than FFP for warfarin reversal.61 They have also been used in other causes of acquired coagulopathy and bleeding, including after adult heart surgery.94,95 Potentially these products are superior to FFP in such situations; however, routine use for treatment of acquired bleeding in children (other than reversal of warfarin) cannot be recommended until further evaluation is conducted. Dosing in this situation is also uncertain.
The importance of fibrinogen depletion in coagulopathy and bleeding has been increasingly appreciated.96,97 It has also been recognized that traditional cut-off values for supplementation of fibrinogen during bleeding (1.0 g/L) are likely to be too low and that further advantage can be gained from higher targets; possibly as high as 2.0 g/L. This may be difficult to achieve with traditional blood products. FFP contains only physiologic concentrations of fibrinogen and only modest increases may be achieved, while cryoprecipitate will require administration of product from several donors to achieve significant increases in fibrinogen concentration (especially in larger patients). Human fibrinogen concentrate has been available in continental Europe for some time and has recently been licensed within the United Kingdom and the United States for treatment of congenital fibrinogen deficiency. Unlike cryoprecipitate, they contain only fibrinogen and a direct comparison in other forms of coagulopathy has not been made. Recent reports have demonstrated advantages to its use in adult cardiac surgery and other causes of bleeding.98,99 Reports of its use in children with acquired coagulopathy are limited. Combining use of fibrinogen concentrates with rapid measurement of fibrinogen concentration (using modified TEG) is an attractive approach.100
The most obvious use for fibrinogen concentrates is as an alternative to cryoprecipitate. Their apparent safety and ease of administration may, however, make other applications possible. A small randomized study comparing prophylactic administration of fibrinogen concentrate to placebo in adult cardiac patients demonstrated a reduction in bleeding.99 Alternatively, it has been proposed that targeting larger fibrinogen concentrations with these agents may reduce the requirement for platelet transfusion. Animal work has demonstrated the effectiveness of this approach in models of bleeding and thrombocytopenia.101 The product of fibrinogen concentration and platelet count may be a better predictor of bleeding in children than either measure alone.91 If the concentration of fibrinogen is high, the amplitude of TEG recordings can be normal, even in the absence of platelets. In vivo, it would be expected that there are limits to this phenomenon; at some point, platelets are required for formation of clot. Such an approach requires proper clinical evaluation before being more widely adopted. Recombinant fibrinogen in fibrin sealants is currently undergoing phase II trials, although widespread availability of systemic recombinant fibrinogen is still some years away.102 With proper assessment, recombinant fibrinogen has the potential to greatly alter the management of severe bleeding and coagulopathy.
Deficiency of factor XIII during and after cardiopulmonary bypass has been described, although the importance of this finding is uncertain.103–105 Supplementation with human or recombinant factor XIII has been demonstrated to reduce bleeding after adult heart surgery, although possibly only in the presence of factor deficiency.104,106,107 The action of factor XIII is to catalyze the formation of cross bridges between fibrin molecules. It also has a wider function in promoting fibroblast proliferation, and it has been used in the treatment of chylothorax after pediatric heart surgery (with possibly only minor benefit).108 Recombinant factor XIII is currently being considered for approval by the FDA, and wider availability may lead to greater use of this agent. Evidence to support widespread use is currently not available.
Pharmaceutical Agents to Reduce Bleeding
The Effectiveness of Synthetic Antifibrinolytics during Pediatric Heart Surgery
A meta-analysis comparing TXA and placebo in children undergoing heart surgery2 concluded that antifibrinolytics reduced bleeding and that TXA was as effective as aprotinin. Figure 18-6 shows a summary of the studies included in this meta-analysis109–116; the figure has been updated to include a more recent trial.115 There were 1061 children entered into nine studies, and results demonstrated a mean reduction in bleeding of approximately 14 mL/kg (95% confidence interval [CI] of 12.7 to 15.6 mL). This is a clinically useful reduction in blood loss. The meta-analysis also demonstrated a small reduction in transfusion of red blood cells with use of TXA.
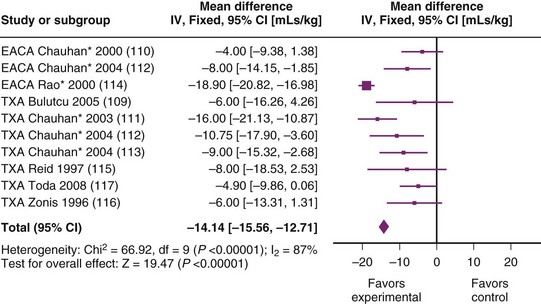
Data concerning more clinically important outcomes (e.g., reexploration or “life-threatening” hemorrhage) are lacking. Analyses have a high degree of statistical heterogeneity (I2 = 78%), indicating differences in the populations, interventions, or study methodology.115–117 This makes pooling of results less valid and makes it impossible to exclude factors that could introduce bias to conclusions. The patients and protocols are clinically variable with differing drugs, dosing regimens and patient populations. Only 163 of the patients did not have cyanotic heart disease; data on 6-hour blood loss was available for 144 of these children and did not demonstrate a significant reduction in bleeding (mean reduction 1. 8 mL/kg, 95% CI of −0.8 to 4.5 mL/kg).115–117 The majority of studies come from a single institution (The All Indian Institute of Medical Science),111–114 and these patients represented a specific population undergoing relatively late correction of cyanotic disease. Polycythemia, and presumably severe cyanosis, was common, with a mean hemoglobin concentration of 19 g/dL.
Another feature of the studies115–117 is that only one study recruited infants less than 2 months of age.116 The mean age of patients was 40 months. This may be important, because the fibrinolytic system in neonates differs from adults; plasminogen levels are about 50% of adult values, plasminogen activation occurs more slowly, and levels of inhibitor (PAI-1) are normal.25 This may make neonates relatively resistant to fibrinolysis. In turn, this may mean that antifibrinolytic medication is less effective, although in vitro experiments using neonatal plasma do not demonstrate a difference in the ability of synthetic antifibrinolytics to inhibit fibrinolysis.118,119
In conclusion, current pediatric data support the use of synthetic antifibrinolytics in cyanotic children older than 2 months of age, but data supporting more widespread use are unclear. While there is substantial evidence to support the use of these drugs in adults,120 extrapolation of conclusions to children is uncertain and has implications with regard to drug development.121 If it can be demonstrated that there are similarities for both the disease process (e.g., hyperfibrinolysis as a contributor to hemorrhage) and drug effect at different age groups (e.g., hyperfibrinolysis reduction), then adult data for efficacy could more reliably be used in children. Hyperfibrinolysis is most likely part of an inflammatory response to cardiopulmonary bypass and would be expected to occur in children (other than neonates) in a similar way to adults. It would also be expected that antifibrinolytic drugs function in a similar way in children and adults. It seems reasonable to extrapolate adult data of efficacy to children. A more difficult question is whether this reduction in bleeding is worthwhile when balanced against adverse effects (discussed later). Small children have more bleeding (relative to body weight) compared to adults. This does not in itself imply that an intervention to reduce bleeding will have greater effect in children. It has been suggested that factors other than fibrinolysis make a greater contribution to bleeding in children.91
Synthetic Antifibrinolytic Medications
There is considerable uncertainty about the dose of these drugs. A recent survey of pediatric cardiac anesthetists in the UK demonstrated a 50-fold variation in TXA dose used during surgery (from a single dose of 5 mg/kg to a cumulative dose of 250 mg/kg).122 In the studies summarized in Figure 18-6 there was also wide variation.
Rational dosing of a drug is determined by its PK and how the resulting plasma (or tissue) concentration translates to a clinical effect (PD). TXA has a volume of distribution (Vd) of 9 to 12 L in adults. The drug is water soluble and poorly protein bound. Most of the drug is excreted unchanged by the kidneys, so the dose should be reduced in renal failure. The terminal elimination half-life is about 2 hours,123 although CPB will affect this. In adults a loading dose of 12.5 mg/kg followed by 6.5 mg/kg/hour with 1 mg/kg added to the pump prime maintains the concentration of TXA greater than 52.5 µg/mL.124 However, neonates and infants will be expected to have a relatively larger Vd and a greater change in Vd with initiation of CPB. In addition, clearance will be reduced in neonates because of renal immaturity. The effects of the surgical insult, CPB, hypothermia, ultrafiltration, and fluid resuscitation on the PK and PD of TXA remain to be established.
The target concentration of TXA is not clear. A plasma concentration of greater than 100 µg/mL will produce complete inhibition of fibrinolysis in vitro, however a concentration of 30 µg/mL accounts for most of the pharmacologic effect of the drug.125 It is difficult to translate these in vitro effects into clinical dosing recommendations. Dosing regimens that involved multiple doses were more effective than a larger single bolus at the start of cardiac surgery, in terms of sternal closure time, blood loss, and blood product requirement, despite similar total doses.113 The administration of a loading dose (50 mg/kg repeated on bypass) in addition to an infusion of 15 mg/kg/hour achieved mean tranexamic concentrations above 100 µg/mL, although concentrations may be below this concentration after bypass.126 This dosing regimen reduced bleeding compared with placebo, and greater tranexamic concentrations were associated with less bleeding (although this does not demonstrate causality). Dosing regimens using a smaller total dose (30 to 50 mg/kg in divided doses) have also been effective. A detailed PK study of TXA in children undergoing heart surgery is currently under way and may help to improve decision making about dosing.
The use of EACA during surgery to correct a congenital heart defect was first described in 1969.127 The dose of EACA is based on a small PK study in children undergoing heart surgery.128 Most of the drug is excreted unchanged by the kidneys, while about 35% of EACA undergoes hepatic metabolism. The terminal elimination half-life is 1 to 2 hours.129 Children have a greater weight-adjusted clearance and Vd compared with adults before, during, and after CPB. Consequently, larger doses are used in children: 75 mg/kg over 10 minutes, 75 mg/kg in the CPB circuit, and 75 mg/kg/hour as an infusion. This regimen maintains concentrations of EACA above the presumed “therapeutic” concentration of 260 µg/mL.128 While the target concentration is not clear, this triple-dosing regimen appears to be clinically effective in children. One study (using three doses of 100 mg/kg EACA before, during, and after CPB) demonstrated a reduction in postoperative blood loss, reduced blood and platelet transfusion requirements, and reduced reexploration rate compared with control subjects.114 The PK have not been studied in infants and neonates.
Adverse Effects of Synthetic Lysine Analogues
Patients following any major surgery or trauma can be hypercoagulable and at risk of thrombosis. Unlike aprotinin, the lysine analogues (TXA or EACA) have not shown an increased risk of renal, cardiac, or cerebral events in adult cardiac surgery patients.130 Several meta-analyses in adults concluded that prophylactic lysine analogues in cardiac surgery did not increase the incidence of thromboembolic complications.120,131,132 However, thrombosis has been reported in nonsurgical hypercoagulable states.133 Despite occasional anecdotes, there is no clear-cut evidence that the incidence of thrombosis is increased with either of the lysine analogues. Both TXA and EACA appear to have a very low incidence of other serious adverse effects, such as anaphylaxis.
The most common acute adverse effect of EACA is hypotension, usually associated with rapid intravenous (IV) administration. Rash, nausea, vomiting, weakness, retrograde ejaculation, myopathy, and rhabdomyolysis have been less frequently reported and are associated with longer-term use.123 EACA is teratogenic and therefore contraindicated in pregnancy.
Rapid IV administration of TXA can cause hypotension. Oral administration can be associated with gastrointestinal adverse effects. In an adult study, prolonged infusion was associated with renal dysfunction.134 There is a possible association between TXA and seizures (without any neurologic injury). TXA is found in the cerebrospinal fluid following IV administration (though at concentrations much lower than in plasma),135 and when applied directly to an animal’s brain can initiate seizures. Two observational studies (one in children) have reported an association with seizures136,137; however, the incidence of seizures (in both those treated and not treated) was much greater than that which occurred at the authors’ institutions. The significance of this association is unclear.
Serine Protease Inhibitor: Aprotinin
The use of aprotinin has become a source of considerable controversy since publication of data that appeared to demonstrate increased morbidity and mortality with its use in adults undergoing heart surgery.3,130 This is discussed further, later, but has lead to a reduced availability of the drug in most countries. If aprotinin remains available, the decision to continue to use it must rest on a balance of the benefits (principally reduction in bleeding) and uncertain risks. More precisely, one must consider how this risk–benefit balance compares with that of the synthetic antifibrinolytics, which have more certain risk profiles.
Effectiveness of Aprotinin for Reducing Bleeding
Two meta-analyses demonstrated that aprotinin reduces bleeding and volume of red cell transfusion in children undergoing heart surgery.2,138 However, similar difficulties arise for studies of synthetic antifibrinolytic drugs: statistical heterogeneity, suboptimal methodology, varied dosing regimes, different study populations, and small, underpowered studies. In particular, most studies are extremely small: 12 of these studies (67%) had less than 50 children in total and only one study had more than 100 children. A meta-analysis of studies in adult patients detailed 107 studies and a total of 11,000 patients.120 There were 94 trials (just less than 10,000 patients) that examined aprotinin use in cardiac surgery. Risk of blood transfusion, volume of blood transfused, volume of blood loss, and requirement for reexploration were all reduced. As with lysine analogues, similar caveats exist with extrapolation of adult data to pediatrics. It appears a reasonable conclusion that aprotinin is effective at reducing blood loss during cardiac surgery and that this conclusion also applies to children. Importantly, there is evidence for efficacy in neonates, where a controlled trial demonstrated reduced blood loss, exposure to allogeneic blood products, and reexploration after arterial switch procedures.139 A further randomized trial of aprotinin in neonates was terminated early (because of the concerns about toxicity in adults), but it failed to show any advantage in indices of bleeding or postoperative recovery.140
Secondary Benefits of Aprotinin
While synthetic antifibrinolytics are selective inhibitors of fibrinolysis, aprotinin is a less specific inhibitor of proteolytic enzymes.15 Proteolytic enzymes are important mediators of inflammation via contact activation and the compliment system; it is postulated that aprotinin may exert a beneficial antiinflammatory effect. A number of studies have demonstrated a reduction in markers of inflammation; however, other studies have failed to demonstrate this effect.141 A recent trial demonstrated reduced inflammatory markers postoperatively in infants treated with aprotinin in comparison to infants treated with TXA.142 A further small study in older children undergoing mainly low complexity surgery failed to demonstrate any effect of aprotinin on a series of inflammatory markers compared to control.143 There are no convincing clinical data to show any benefit from an antiinflammatory action of aprotinin. A trial designed to detect a reduction in ventilation days (as a marker of organ dysfunction) in neonates was terminated before an adequate number of patients had been recruited to identify a significant effect.140
Adverse Effects of Aprotinin
There is great concern about the adverse effects of aprotinin in adult patients. This followed the publication of an observational study in adults with acute coronary syndromes, which concluded that rates of renal failure, serious intravascular thrombosis (myocardial infarction and stroke), and of death (at 5 years) were increased if aprotinin was used.3,130 The rate of these complications was not increased with the use of synthetic antifibrinolytics, whereas all agents appeared effective for reducing bleeding. This was followed by the early termination of a large randomized trial (BART trial), comparing synthetic antifibrinolytics and aprotinin (in “high risk” adults undergoing heart surgery), when interim analysis showed an increased mortality in the group treated with aprotinin.144 Excess deaths appeared to result from heart failure and myocardial infarction. Rates of renal failure were not increased in those treated with aprotinin, although an increase in plasma creatinine was common. The relative risk of death in adults treated with aprotinin compared to those treated with either TXA or EACA was 1.53 with a 95% CI of 1.06 to 2.22. Data on bleeding in this study were inconclusive, although they suggested a small benefit for aprotinin for preventing severe bleeding. The conclusion of these papers was that aprotinin should not be used in the patient populations studied. This has lead to differing responses from drug regulators and pharmaceutical companies in different countries. The drug is effectively unavailable in a number of countries; it has remained available in the UK on a restricted, named patient basis.
The implication of these studies for children and young adults with congenital heart disease is controversial. The use of aprotinin has reduced dramatically within the UK.122 An important large observation series (over 30,000 patients) demonstrated no increased risk of death or dialysis in children,145 and there was a significant reduction in length of stay for those undergoing reoperation. The main cause of death reported in adult patients was from stroke and myocardial infarction. Although thrombotic complications do occur in children, the pathogenesis and underlying risk factors are very different from those in adults (especially in adults with ischemic heart disease). Renal failure occurs in children after high-risk operations, and may share pathogenic factors with postoperative renal failure in adults; aprotinin accumulates within renal tissue and affects local autoregulation of blood flow in response to ischemia. The evidence is unclear as to whether aprotinin causes any more than temporary derangement of renal function tests in adults or children. A further retrospective series demonstrated no increase in the incidence of renal failure in neonates when corrected for confounding variables.146
As with many polypeptides, anaphylactic reactions can occur with aprotinin. The reported incidence of adverse reactions in children varies, but the largest reported experience with aprotinin found 1 reaction from 2202 primary exposures (0.05%) and 6 reactions from 453 reexposures (1.3%). There have been no severe reactions to aprotinin since 1998 and this is attributed to the elimination of its use in recently exposed patients.147,148 Studies report the incidence and severity of reaction to be less than that of adults. This may be due to the immature neonatal immune system.149 It has been recommended that:


Dosing of Aprotinin
The lack of clarity regarding the efficacy of aprotinin in children may reflect differing doses used in clinical studies. Aprotinin rapidly redistributes into the extracellular space after IV administration. It is metabolized in the proximal renal tubules and eliminated in a biphasic pattern: a distribution half-life of 40 minutes and an elimination half-life of 7 hours.129,150 A PK study of aprotinin in children undergoing CPB examined aprotinin concentrations after administration of weight-based dosing (25,000 KIU/kg bolus pre-CPB, 35,000 KIU/kg in CPB prime, and 12,500 KIU/kg/hour infusion).151 Although there was considerable variation in plasma concentration of aprotinin, there was a correlation between concentration and weight at 5 minutes after administration and 5 minutes after cardiopulmonary bypass. This study can provide one possible explanation as to why there are such inconsistent results from aprotinin trials in children. Using a dose-per-weight regimen, the smaller children may fail to achieve therapeutic concentrations of aprotinin, in terms of plasmin inhibition and the possible antiinflammatory effects (a low concentration of aprotinin, e.g., less than 200 KIU/mL, would be insufficient to inhibit contact activation on the CPB circuit). This is consistent with greater clearance (expressed as per kilogram) in children (see also Chapter 6, Fig. 6-4). To achieve target concentrations, aprotinin should be given according to nonlinear functions (e.g., body surface area or an allometric  -power model; see Chapter 6), as opposed to the linear dose per kilogram. This would result in a 2.5 times greater dose in neonates than would otherwise be given.48 A recent qualitative literature review suggested that an initial loading dose of at least 30,000 KIU/kg is required, followed by an infusion. The pump prime dose should be based on the volume of the pump rather than the weight of the child.149
-power model; see Chapter 6), as opposed to the linear dose per kilogram. This would result in a 2.5 times greater dose in neonates than would otherwise be given.48 A recent qualitative literature review suggested that an initial loading dose of at least 30,000 KIU/kg is required, followed by an infusion. The pump prime dose should be based on the volume of the pump rather than the weight of the child.149
Which Antifibrinolytic Drug for Pediatric Cardiac Surgery?
A meta-analysis of trials in adult patients demonstrated no difference between EACA and TXA. Aprotinin was more effective than either TXA or EACA, and although that effect appeared marginal, only aprotinin was demonstrated to reduce the risk of reoperation.120 Direct comparison between TXA and EACA has demonstrated no difference in children.112,152 Low-dose aprotinin (10,000 KIU/kg followed by 10,000 KIU/kg/hour) has been compared with EACA (100 mg/kg on induction, 100 mg/kg in the pump prime and 100 mg/kg on weaning CPB).110 There was a reduction in postoperative blood loss and in transfusion requirements in both groups compared with control but no significant difference between the two drugs. These data are difficult to interpret because substantially larger doses of aprotinin are frequently used in current practice. More recently, TXA (100 mg/kg before, during, and after CPB) was compared with aprotinin (30,000 KIU after induction, 30,000 KIU in the pump, and 30,000 KIU after weaning off bypass). There was a reduction in the time to sternal closure, transfusion requirements, and blood loss compared with control, but no significant difference between the two drugs. When the two drugs were combined at the same doses, they showed no additional benefit.109 A recent retrospective study confirmed these findings.153
Use of Antifibrinolytic Drugs in Noncardiac Surgery
A recent trend has been towards increased use of antifibrinolytic drugs (especially TXA) during noncardiac surgery; principally major orthopedic surgery (e.g., spinal instrumentation, see also Chapter 30) and craniofacial surgery (see also Chapter 33). Aprotinin is less commonly used because of reduced availability and the safety concerns discussed previously.
There are 27 trials that have examined the use of TXA during orthopedic surgery in adults120; they report a 50% reduction in the relative risk of requiring a blood transfusion. There was a reduction in the incidence of total blood loss of over 400 mL in 20 of these trials. Almost all these studies were conducted in adults undergoing major joint replacement surgery. In one study of adults (n = 147) undergoing posterior spinal fusion, TXA reduced total blood loss by 25%; however, the reduction in blood products transfused was not statistically significant.154 The popularity of antifibrinolytic use during scoliosis surgery in children and adolescents is reflected in two surveys, which show use in 70% to 80% of hospitals.155,156 It is unclear whether this use is mostly confined to higher-risk cases; however, in the authors’ institution TXA is currently used during all scoliosis surgery as part of a program aimed at reducing the use of blood products. Two meta-analyses have examined this question (looking at 6 and 7 studies, respectively); no individual study had more than 45 patients.2,157 These meta-analyses conclude that antifibrinolytic drugs reduce bleeding and volume of red cells transfused. To date there has been no single prospective trial that compares different antifibrinolytics. A retrospective comparison of TXA and EACA appeared to show superiority of TXA; however, doses used were not comparable.158 The two meta-analyses failed to establish any difference between the drugs.2,157 It is also not possible to determine whether reported benefits hold for all children undergoing scoliosis repair. Bleeding is greater in children with secondary scoliosis (including patients with Duchenne muscular dystrophy),159 those with hemostatic disorders, and in those undergoing more extensive surgery; these might, therefore, be expected to see a greater advantage in terms of bleeding reduction. At least one study examined children with idiopathic scoliosis and demonstrated a reduction in blood loss with use of aprotinin (see also Chapter 30).160 As with cardiac studies there is a considerable range in the dose of drug used; in a survey of practice in the UK, TXA dose ranged from three- to fivefold, whereas in published trials, the dose varied 10-fold.155
Antifibrinolytics are also used widely in craniosynostosis surgery. In a survey of North American practice, they were used in 20% of hospitals for strip craniotomy, rising to 30% for more complex repairs.161 Four small trials (total 141 children) have addressed the use of these agents for this indication; three using TXA162–164 and one using aprotinin.165 All of these trials have demonstrated reductions in blood transfused and in bleeding. A recent publication demonstrated a greater than 50% reduction in the volume of blood transfused and in the proportion of children transfused (70% vs. 37%).162 Although all these studies were relatively small, it would appear likely that antifibrinolytics produce a useful reduction in blood loss for craniosynostosis surgery.
Antifibrinolytic Medication and Trauma
There is no evidence in children concerning the use of antifibrinolytic medications to treat massive hemorrhage following trauma. The CRASH 2 study (a blinded randomized control trial of over 20,000 patients) demonstrated a reduction in mortality with TXA in adult trauma patients with, or at risk of, significant hemorrhage; mortality was reduced from 16% to 14.5%.166 Mortality due to bleeding was reduced from 5.7% to 4.9%. No difference was seen in rates of vascular occlusive events. Despite the size and complexity of the study, it appears to have been properly randomized and blinded. It is difficult to see an explanation for the reduction in mortality, other than a reduction in bleeding. It is therefore likely that TXA can be efficacious in the treatment of established bleeding (at least in the context of trauma). Ideally, further studies would be required to establish the effectiveness of TXA in the treatment of established bleeding of other causes in children; however, it is unlikely a controlled trial of pediatric trauma would be large enough to be adequately powered. In the absence of such studies, and in the absence of definite evidence of toxicity, it would be reasonable to consider TXA as part of the treatment of established severe bleeding resulting from trauma or surgery. Some caution is required, and given the uncertainty concerning dosing, it would be premature to recommend the widespread use of the drug in young children. When used, it should be reserved for severe bleeding; for example, bleeding requiring treatment with non–red cell blood products. An important caveat to this is that administration greater than 3 hours after injury appeared to increase mortality.167 The significance of this is unclear; however, such late administration should be avoided.
Authors’ Recommendations for Use of Antifibrinolytics
 Antifibrinolytics are used routinely during heart surgery in children with cyanotic heart disease; especially in the presence of polycythemia, reduced saturations for a prolonged period, or iron deficiency.
Antifibrinolytics are used routinely during heart surgery in children with cyanotic heart disease; especially in the presence of polycythemia, reduced saturations for a prolonged period, or iron deficiency.
 Antifibrinolytics are used during other cardiac procedures in children to help reduce the use of blood products. Greater benefit is likely (but not proven) in children with greater tendency to bleeding, including smaller children (when prolonged bypass times are anticipated) and repeat surgery.115,168
Antifibrinolytics are used during other cardiac procedures in children to help reduce the use of blood products. Greater benefit is likely (but not proven) in children with greater tendency to bleeding, including smaller children (when prolonged bypass times are anticipated) and repeat surgery.115,168

 Synthetic antifibrinolytics are used in preference to aprotinin, outside of well-conducted trials.
Synthetic antifibrinolytics are used in preference to aprotinin, outside of well-conducted trials.



Desmopressin
Desmopressin acetate (1-desamino-8-d-arginine; DDAVP) is a synthetic analogue of vasopressin. The production of desmopressin involves alteration in the chemical structure of naturally occurring vasopressin. In the process, the antidiuretic effect is enhanced and the vasopressor effect is virtually eliminated. Desmopressin is more resistant to enzymatic cleavage, and hence the duration of action is prolonged to 6 to 24 hours.123 Desmopressin potently causes endothelial release of factor complexes VIII/protein C and VIII/vWf. It has been used in mild hemophilia, von Willebrand disease, coagulopathy of uremia, liver failure, and in adults undergoing cardiac and spinal fusion surgery (see also Chapter 10).
During the early phase of coagulation, platelets bind via the glycoprotein receptor Ib to vWf on the damaged endothelium. IV desmopressin releases vWf, thus enhancing the binding of platelets to damaged endothelium. The maximum effect of desmopressin is observed at a dose of 0.3 µg/kg. It must be given after cessation of the extracorporeal circuit to prevent unwanted platelet activation.48
Several meta-analyses of its use in adults have been published, the most recent showing that desmopressin was associated with a small reduction in blood loss, but no benefit in terms of repeat sternotomy, proportion of patients requiring transfusion, or mortality.131 It was also associated with a 2.4-fold increase in risk of myocardial infarction.
In children, desmopressin at 0.3 µg/kg failed to demonstrate any benefit for both non–high-risk169 and high-risk cardiac surgery.170 Younger children are not as capable of releasing vWf from endothelial storage sites as are older children, and the maximal release of vWf caused by the operative stimulus cannot be enhanced by desmopressin.171 Potential adverse sequelae from desmopressin include fluid retention, hyponatremia, tachyphylaxis, tachycardia, and mild hypotension.172 Desmopressin is not currently recommended in pediatric surgery,64 although one could postulate that it could be reserved for those with ongoing bleeding with evidence of platelet function abnormalities, such as prolonged bleeding time or decreased maximum amplitude values on a TEG.173
Factor VIIa
Recombinant activated factor VII (rVIIa) is known to be safe and effective for treatment and prevention of hemorrhage in children with hemophilia who have circulating inhibitors to replacement factors (see also Chapters 9 and 10). It is also used in patients with Glanzmann thrombasthenia refractory to platelet transfusion, and in patients with Factor VII deficiency. Regulatory approval has been granted in Europe and the United States for these conditions.
Off-label use of this drug has grown steadily over the last few years and now accounts for 97% of in-hospital use.125 This has encompassed a number of indications involving treatment or prevention of acquired bleeding or coagulopathy174–186 of varying severity. The most common off-label indications are treatment of bleeding related to cardiovascular surgery, trauma, and intracranial bleeds. Off-label use in children is also widespread. One retrospective study recorded 3655 administrations (in 39 U.S. pediatric hospitals over a 7-year period); 46% were in children admitted to a surgical specialty or to a pediatric intensive care unit, over 20% to cardiac surgery or cardiology. Administration was most common in children under 1 year of age.
For rVIIa to exert a beneficial action, it must generate a burst of thrombin at the sight of injury. Two mechanisms are likely to work in synergy to produce this. First, the TF pathway described previously is stimulated to augment generation of factor Xa. Second, high concentrations of rVIIa will bind directly to the surface of activated platelets, again activating factor Xa, leading to thrombin production (Figs. 18-7 and 18-8).187
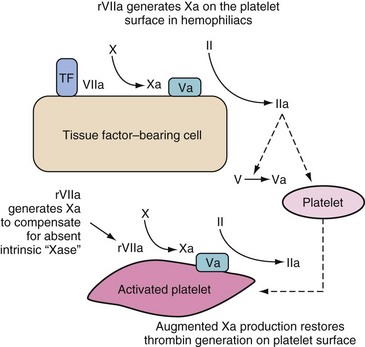
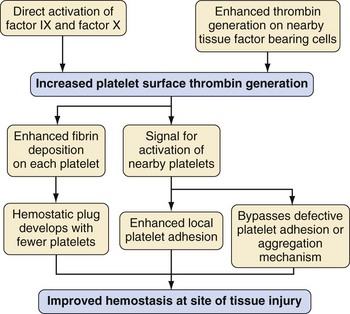
Usually such processes will be confined to the site of injury; however, in markedly proinflammatory states, expression of tissue factor on activated monocytes and platelets increases the risk of thrombosis distant from the injury. For this reason rVIIa is contraindicated in disseminated intravascular coagulation and should be used with caution in children on extracorporeal life support.188 The importance of this to more routine bypass is unknown. The therapeutic effects of factor rVIIa begin at doses up to 10 times greater than physiologic concentrations of the endogenous factor. It is therefore not simply a “replacement” therapy of a deficient factor.147
The action of rVIIa is to produce a surge of thrombin; however, because clots comprise fibrin, platelets, and red cells, it is unlikely that rVIIa will be effective in the absence of these substrates. Measures should be taken to ensure adequate fibrinogen concentration and platelet numbers before giving rVIIa. In addition, factors such as acidosis and hypothermia will reduce the efficiency of rVIIa (although mild to moderate hypothermia has only minor effects in vitro).148 Given the expense of rVIIa, in addition to doubts over safety, it would appear to be wise to employ more established measures before its use, along with (as dictated by local agreements) consultation with a hematologist.
Most controlled trials of rVIIa have focused on prophylaxis use or use in less severe bleeding. There are 26 randomized controlled trials that have examined off-label use of rVIIa, and 5 have focused on surgical use.150,189–192 A meta-analysis of these trials demonstrated no reduction in mortality and only relatively modest reductions in bleeding and transfusion.193 A study190 that examined patients with moderately severe bleeding (more than 200 mL/hr or greater than 2 mL/kg for 2 consecutive hours) following heart surgery in adults reported that although 2619 patients had consented to the trial, only 179 subsequently had sufficient bleeding to be randomized. The results demonstrated a reduction in reexploration, bleeding, and in transfusion requirements. The objective of the trial was to examine the safety of using rVIIa. Unfortunately, the trial was terminated early; although a trend to increased adverse effects was seen, the result was inconclusive. The authors of this study cautioned against wider use before further trials. A single randomized trial (n = 76) that examined prophylactic rVIIa administration (40 µg/kg) in infants undergoing heart surgery,191 demonstrated neither efficacy nor toxicity in this group. The study was small but larger than several of the adult studies that demonstrated an effect. Whether this demonstrates a genuine difference between adults and infants or is related to the relatively low dose rVIIa used is unclear. The use of rVIIa in children has been recently reviewed.188
Concerns about adverse effects of this drug have grown. Two studies have attempted to systematically review the data on toxicity.194,195 The rate of thrombosis was greater (10.2% vs. 8.7%) in those treated with rVIIa, but this increase was largely confined to the elderly. The second study examined data from observational trials with comparator groups; mortality was not affected by treatment with rVIIa, although the risk of thrombosis was increased in some groups (including adult cardiac patients). Both these reviews combined data from studies on very different patient populations, and some caution is required in their interpretation. The risk of thrombosis in children is uncertain and data inconclusive.188 Neonates treated with either FFP or rVIIa for similar indications had a similar incidence of thrombosis (7%).196 Thrombotic complications occurred in 10.8% of children who received off-label rVIIa; the overall mortality in those receiving rVIIa was 34%.197 It is not possible to know how this was affected by use of rVIIa. It is clear that both thrombotic complications and mortality are common in patients with bleeding serious enough to warrant use of this drug. Use of rVIIa will very likely increase the risk of thrombosis, although the impact of this (and the benefit of the drug) will vary in different clinical situations.
The dose regimen for off-label use of rVIIa is yet to be firmly established. One of the difficulties is that there is no satisfactory laboratory test to monitor its effectiveness,198,199 and factor VII activity does not always predict efficacy.200 Although it is known that the PT, aPTT, and TEG improve after rVIIa is given in liver surgery,201 they cannot reliably be used to determine a dosing regimen. If the main effect of rVIIa is at the site of injury, clinical observation still remains the best indication of effect.198 The dose used often closely relates to the dose indicated in hemophilia patients (90 µg/kg), although in practice doses are often rounded to the nearest vial.201 Although Warren et al. recommended a lower dose of 40 to 60 µg/kg, a dose of 40 µg/kg has been shown to have no effect.191 Larger doses of 60 to 90 µg/kg may prove more effective.
The justification to use larger doses is supported by differences in PK. In older children with hemophilia, the terminal elimination half-life of rVIIa is less than in adults (1.3 vs. 2.7 hours), and the clearance is greater (67 vs. 37 mL/kg/hour).202,203 When prophylactic rVIIa at 100 µg/kg was given to 10 preterm infants between 23 and 28 weeks gestational age every 4 hours for 72 hours, two umbilical artery catheter thromboses were reported and no embolic events or increase in intraventricular hemorrhage rate compared with those not given rVIIa.204 There were no other treatment-related side effects, despite this large repeated dose, which suggests that a large study in neonates is warranted.
Summary
We began this chapter by reviewing the mechanism of coagulation and how this alters during normal development and disease. This understanding of coagulation has lead to the development of new treatments, most notably rVIIa.12 Complex systems such as the coagulation system behave in unanticipated ways and while mechanisms can be found to justify many interventions, not all interventions will bring a clinically useful improvement for the patient. The coagulation model becomes more complicated when we consider the interactions between coagulation and other systems (noticeably the kinin–kallikrein system15 and other proinflammatory cascades), as well as the effects of transfusion of allogeneic blood products. Mechanistic justifications for use of any treatment are therefore limited. Direct evidence that a specific treatment brings a benefit to the child is required. Although the evidence available is still lacking, and at times appear contradictory, we have aimed to give clear advice to the practicing anesthesiologist when possible.
The problems of research into this subject are demonstrated by aprotinin studies. Despite relatively large numbers of patients (including children) recruited into trials, many important questions remain incompletely resolved. Concerns over safety in an entirely different population have led to the effective withdrawal of what may (or may not) have been a useful medication in children. Beyond moderate reductions in blood loss, few advantages have been convincingly demonstrated for this drug.2 For therapies aimed at treatment rather than prevention of bleeding, the challenges of conducting well-designed trials are even greater. Currently, several therapeutic approaches are proposed (rVIIa, fibrinogen concentrates, transfusion protocols based on TEG) that differ from traditional treatment. The requirement for well-conducted trials, with meaningful outcome measures, is greater than ever.
Eaton MP. Antifibrinolytic therapy in surgery for congenital heart disease. Anesth Analg. 2008;106:1087–1100.
Guzzetta NA, Miller BE. Principles of hemostasis in children: models and maturation. Paediatr Anaesth [Review]. 2011;21:3–9.
Hoffman M. Remodeling the blood coagulation cascade. J Thromb Thrombolysis. 2003;16:17–20.
Mangano DT, Tudor IC, Dietzel C. The risk associated with aprotinin in cardiac surgery. N Engl J Med. 2006;354:353–365.
Brown JR, Birkmeyer NJO, O’Connor GT. Aprotinin in cardiac surgery. N Engl J Med. 2006;354:1953–1957. (Correspondence)
Ferraris VA, Bridges CR, Anderson RP. Aprotinin in cardiac surgery. N Engl J Med. 2006;354:1953–1957. (Correspondence)
Levy JH, Ramsay JG, Guyton RA. Aprotinin in cardiac surgery. N Engl J Med. 2006;354:1953–1957. (Correspondence)
Mangano DT. Aprotinin in cardiac surgery. N Engl J Med. 2006;354:1953–1957. (Correspondence)
Schouten ES, van de Pol AC, Schouten AN, et al. The effect of aprotinin, tranexamic acid, and aminocaproic acid on blood loss and use of blood products in major pediatric surgery: a meta-analysis. Pediatr Crit Care Med. 2009;10:182–190.
A systemic review and meta-analysis of the role of antifibrinolytic drugs during pediatric surgery
Warren OJ, Rogers PL, Watret AL, et al. Defining the role of recombinant activated factor VII in pediatric cardiac surgery: where should we go from here? Pediatr Crit Care Med. 2009;10:572–582.
A thorough review of the use of rVIIa in pediatric heart surgery.
Webber TP, Grosse Hartlange MA, Van Aken H, Brooke M. Anaesthetic strategies to reduce perioperative blood loss in paediatric surgery. Eur J Anaesthesiol. 2003;20:175–181.
Welsby IJ, Monroe DM, Lawson JH, Hoffmann M. Recombinant activated factor VIIa and the anaesthetist. Anaesthesia. 2005;60:1203–1212.
This is a very good overview of the mechanisms and use of rVIIa.
1 Brenn BR, Theroux MC, Dabney KW, Miller F. Clotting parameters and thromboelastography in children with neuromuscular and idiopathic scoliosis undergoing posterior spinal fusion. Spine (Phila Pa 1976). 2004;29:E310–E314.
2 Schouten ES, van de Pol AC, Schouten AN, et al. The effect of aprotinin, tranexamic acid, and aminocaproic acid on blood loss and use of blood products in major pediatric surgery: a meta-analysis. Pediatr Crit Care Med. 2009;10:182–190.
3 Mangano DT, Miao Y, Vuylsteke A, et al. Mortality associated with aprotinin during 5 years following coronary artery bypass graft surgery. JAMA. 2007;297:471–479.
4 Harrison E, Bolton P. Serious hazards of transfusion in children (SHOT). Paediatr Anaesth. 2011;21:10–13.
5 Hendrickson JE, Hillyer CD. Noninfectious serious hazards of transfusion. Anesth Analg. 2009;108:759–769.
6 Kneyber MC, Hersi MI, Twisk JW, Markhorst DG, Plotz FB. Red blood cell transfusion in critically ill children is independently associated with increased mortality. Intensive Care Med. 2007;33:1414–1422.
7 Guzzetta NA. Benefits and risks of red blood cell transfusion in pediatric patients undergoing cardiac surgery. Paediatr Anaesth. 2011;21:504–511.
8 Keung CY, Smith KR, Savoia HF, Davidson AJ. An audit of transfusion of red blood cell units in pediatric anesthesia. Paediatr Anaesth. 2009;19:320–328.
9 Szekely A, Cserep Z, Sapi E, et al. Risks and predictors of blood transfusion in pediatric patients undergoing open heart operations. Ann Thorac Surg. 2009;87:187–197.
10 Williams GD, Bratton SL, Ramamoorthy C. Factors associated with blood loss and blood product transfusions: a multivariate analysis in children after open-heart surgery. Anesth Analg. 1999;89:57–64.
11 Hoffman M. Remodeling the blood coagulation cascade. J Thromb Thrombolysis. 2003;16:17–20.
12 Hoffman M, Monroe DM. A cell-based model of hemostasis. Thromb Haemost. 2001;85:958–965.
13 Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol. 1999;19:170–177.
14 Kuhle S, Male C, Mitchell L. Developmental hemostasis: pro- and anticoagulant systems during childhood. Semin Thromb Hemost. 2003;29:329–338.
15 Saxena P, Thompson P, d’Udekem Y, Konstantinov IE. Kallikrein-kinin system: a surgical perspective in post-aprotinin era. J Surg Res. 2011;167:70–77.
16 Lijnen RH, Collen D. Fibrinolytic system and its disorders. In: Handin RI, Lux SE, Stossel TP, eds. Blood: principles and practice of hematology. Philadelphia: Lippincott Williams & Wilkins; 2003:1249–1274.
17 Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307–321.
18 Hunt BJ, Segal H. Hyperfibrinolysis. J Clin Pathol. 1996;49:958.
19 Carey, Cressey. Hyperfibrinolysis—is it common? Measurement and treatment including the role of thromboelastography. Clinical Risk. 2009;15:188–191.
20 Sirgo G, Morales P, Rello J. PAI-1 gene: pharmacogenetic association of 4G/4G genotype with bleeding after cardiac surgery—pilot study. Eur J Anaesthesiol. 2009;26:404–411.
21 Iribarren JL, Jimenez JJ, Hernandez D, et al. Postoperative bleeding in cardiac surgery: the role of tranexamic acid in patients homozygous for the 5G polymorphism of the plasminogen activator inhibitor-1 gene. Anesthesiology. 2008;108:596–602.
22 Guzzetta NA, Bajaj T, Fazlollah T, et al. A comparison of heparin management strategies in infants undergoing cardiopulmonary bypass. Anesth Analg. 2008;106:419–425.
23 Andrew M, Paes B, Milner R, et al. Development of the human coagulation system in the full-term infant. Blood. 1987;70:165–172.
24 Andrew M, Vegh P, Johnston M, et al. Maturation of the hemostatic system during childhood. Blood. 1992;80:1998–2005.
25 Guzzetta NA, Miller BE. Principles of hemostasis in children: models and maturation. Paediatr Anaesth. 2011;21:3–9.
26 Andrew M, Paes B, Johnston M. Development of the hemostatic system in the neonate and young infant. Am J Pediatr Hematol Oncol. 1990;12:95–104.
27 Chalmers EA. Neonatal coagulation problems. Arch Dis Child Fetal Neonatal Ed. 2004;89:F475–F478.
28 Miller BE, Tosone SR, Guzzetta NA, Miller JL, Brosius KK. Fibrinogen in children undergoing cardiac surgery: is it effective? Anesth Analg. 2004;99:1341–1346.
29 Weinstein MJ, Blanchard R, Moake JL, Vosburgh E, Moise K. Fetal and neonatal von Willebrand factor (vWF) is unusually large and similar to the vWF in patients with thrombotic thrombocytopenic purpura. Br J Haematol. 1989;72:68–72.
30 Andrew M, Mitchell L, Vegh P, Ofosu F. Thrombin regulation in children differs from adults in the absence and presence of heparin. Thromb Haemost. 1994;72:836–842.
31 Miller BE, Mochizuki T, Levy JH, et al. Predicting and treating coagulopathies after cardiopulmonary bypass in children. Anesth Analg. 1997;85:1196–1202.
32 Haizinger B, Gombotz H, Rehak P, Geiselseder G, Mair R. Activated thrombelastogram in neonates and infants with complex congenital heart disease in comparison with healthy children. Br J Anaesth. 2006;97:545–552.
33 Schleef RR, Higgins DL, Pillemer E, Levitt LJ. Bleeding diathesis due to decreased functional activity of type 1 plasminogen activator inhibitor. J Clin Invest. 1989;83:1747–1752.
34 Bolliger D, Gorlinger K, Tanaka KA. Pathophysiology and treatment of coagulopathy in massive hemorrhage and hemodilution. Anesthesiology. 2010;113:1205–1219.
35 Thorsen K, Ringdal KG, Strand K, et al. Clinical and cellular effects of hypothermia, acidosis and coagulopathy in major injury. Br J Surg. 2011;98:894–907.
36 Engstrom M, Schott U, Romner B, Reinstrup P. Acidosis impairs the coagulation: a thromboelastographic study. J Trauma. 2006;61:624–628.
37 Groeneveld AB, Navickis RJ, Wilkes MM. Update on the comparative safety of colloids: a systematic review of clinical studies. Ann Surg. 2011;253:470–483.
38 Sumpelmann R, Kretz FJ, Luntzer R, et al. Hydroxyethyl starch 130/0.42/6:1 for perioperative plasma volume replacement in 1130 children: results of an European prospective multicenter observational postauthorization safety study (PASS). Paediatric Anaesthesia. 2012;22:371–378.
39 Chong Sung K, Kum Suk P, Mi Ja Y, Kyoung Ok K. Effects of intravascular volume therapy using hydroxyethyl starch (130/0.4) on post-operative bleeding and transfusion requirements in children undergoing cardiac surgery: a randomized clinical trial. Acta Anaesthesiologica Scandinavica. 2006;50:108–111.
40 Oliiver WC. Overview of heparin and protamine management and dosing regimens in pediatric cardiac surgical patients. Semin Cardiothorac Vasc Anesth. 2003;7:387–410.
41 Hirsh J. Heparin. N Engl J Med. 1991;324:1565–1574.
42 Maslow A, Schwartz C. Cardiopulmonary bypass-associated coagulopathies and prophylactic therapy. Int Anesthesiol Clin. 2004;42:103–133.
43 Guzzetta NA, Miller BE, Todd K, et al. Clinical measures of heparin’s effect and thrombin inhibitor levels in pediatric patients with congenital heart disease. Anesth Analg. 2006;103:1131–1138.
44 Gu YJ, Huyzen RJ, van Oeveren W. Intrinsic pathway-dependent activated clotting time is not reliable for monitoring anticoagulation during cardiopulmonary bypass in neonates. J Thorac Cardiovasc Surg. 1996;111:677–678.
45 Despotis GJ, Filos KS, Levine V, Alsoufiev A, Spitznagel E. Aprotinin prolongs activated and nonactivated whole blood clotting time and potentiates the effect of heparin in vitro. Anesth Analg. 1996;82:1126–1131.
46 Chan AK, Leaker M, Burrows FA, et al. Coagulation and fibrinolytic profile of paediatric patients undergoing cardiopulmonary bypass. Thromb Haemost. 1997;77:270–277.
47 Koster A, Fischer T, Praus M, et al. Hemostatic activation and inflammatory response during cardiopulmonary bypass: impact of heparin management. Anesthesiology. 2002;97:837–841.
48 Garvin S, FitzGerald DC, Despotis G, Shekar P, Body SC. Heparin concentration-based anticoagulation for cardiac surgery fails to reliably predict heparin bolus dose requirements. Anesth Analg. 2010;111:849–855.
49 Codispoti M, Ludlam CA, Simpson D, Mankad PS. Individualized heparin and protamine management in infants and children undergoing cardiac operations. Ann Thorac Surg. 2001;71:922–927. discussion 27-8
50 Guzzetta NA, Monitz HG, Fernandez JD, et al. Correlations between activated clotting time values and heparin concentration measurements in young infants undergoing cardiopulmonary bypass. Anesth Analg. 2010;111:173–179.
51 Dietrich W. Cardiac surgery and the coagulation system. Curr Opin Anaesthesiol. 2000;13:27–34.
52 Iannoli ED, Eaton MP, Shapiro JR. Bidirectional Glenn shunt surgery using lepirudin anticoagulation in an infant with heparin-induced thrombocytopenia with thrombosis. Anesth Analg. 2005;101:74–76.
53 Williams GD, Ramamoorthy C, Totzek FR, Oakes RL. Comparison of the effects of red cell separation and ultrafiltration on heparin concentration during pediatric cardiac surgery. J Cardiothorac Vasc Anesth. 1997;11:840–844.
54 Mochizuki T, Olson PJ, Szlam F, Ramsay JG, Levy JH. Protamine reversal of heparin affects platelet aggregation and activated clotting time after cardiopulmonary bypass. Anesth Analg. 1998;87:781–785.
55 Despotis GJ, Joist JH, Hogue CW, et al. The impact of heparin concentration and activated clotting time monitoring on blood conservation. A prospective, randomized evaluation in patients undergoing cardiac operation. J Thorac Cardiovasc Surg. 1995;110:46–54.
56 Shore-Lesserson L, Reich DL, DePerio M. Heparin and protamine titration do not improve haemostasis in cardiac surgical patients. Can J Anaesth. 1998;45:10–18.
57 Murray DJ, Brosnahan WJ, Pennell B, et al. Heparin detection by the activated coagulation time: a comparison of the sensitivity of coagulation tests and heparin assays. J Cardiothorac Vasc Anesth. 1997;11:24–28.
58 Nuttall GA, Oliver WC, Beynen FM, et al. Determination of normal versus abnormal activated partial thromboplastin time and prothrombin time after cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 1995;9:355–361.
59 Martin P, Horkay F, Gupta NK, Gebitekin C, Walker DR. Heparin rebound phenomenon—much ado about nothing? Blood Coagul Fibrinolysis. 1992;3:187–191.
60 Koga T, Az-ma T, Yuge O. Prostaglandin E1 at clinically relevant concentrations inhibits aggregation of platelets under synergic interaction with endothelial cells. Acta Anaesthesiol Scand. 2002;46:987–993.
61 Makris M, van Veen JJ, Maclean R. Warfarin anticoagulation reversal: management of the asymptomatic and bleeding patient. J Thromb Thrombolysis. 2010;29:171–181.
62 Tempe DK, Virmani S. Coagulation abnormalities in patients with cyanotic congenital heart disease. J Cardiothorac Vasc Anesth. 2002;16:752–765.
63 Ekert H, Gilchrist GS, Stanton R, Hammond D. Hemostasis in cyanotic congenital heart disease. J Pediatr. 1970;76:221–230.
64 Weber TP, Grosse Hartlage MA, Van Aken H, Booke M. Anaesthetic strategies to reduce perioperative blood loss in paediatric surgery. Eur J Anaesthesiol. 2003;20:175–181.
65 Arnold P, Jackson S, Bolton D, et al. Coagulation and neonatal bypass: an assessment of changes in coagulation factor concentration during cardiopulmonary bypass in neonates, with modified ultrafiltration. Critical Care. 1999;4(Suppl A):5–66.
66 Davidson SJ, Burman JF, Philips SM, et al. Correlation between thrombin potential and bleeding after cardiac surgery in adults. Blood Coagul Fibrinolysis. 2003;14:175–179.
67 Woodman RC, Harker LA. Bleeding complications associated with cardiopulmonary bypass. Blood. 1990;76:1680–1697.
68 Weerasinghe A, Taylor KM. The platelet in cardiopulmonary bypass. Ann Thorac Surg. 1998;66:2145–2152.
69 Guay J, Ruest P, Lortie L. Cardiopulmonary bypass induces significant platelet activation in children undergoing open-heart surgery. Eur J Anaesthesiol. 2004;21:953–956.
70 Williams GD, Bratton SL, Riley EC, Ramamoorthy C. Coagulation tests during cardiopulmonary bypass correlate with blood loss in children undergoing cardiac surgery. J Cardiothorac Vasc Anesth. 1999;13:398–404.
71 Mossad EB, Machado S, Apostolakis J. Bleeding following deep hypothermia and circulatory arrest in children. Semin Cardiothorac Vasc Anesth. 2007;11:34–46.
72 Chung JH, Gikakis N, Rao AK, et al. Pericardial blood activates the extrinsic coagulation pathway during clinical cardiopulmonary bypass. Circulation. 1996;93:2014–2018.
73 Boisclair MD, Lane DA, Philippou H, et al. Mechanisms of thrombin generation during surgery and cardiopulmonary bypass. Blood. 1993;82:3350–3357.
74 Williams GD, Bratton SL, Riley EC, Ramamoorthy C. Association between age and blood loss in children undergoing open heart operations. Ann Thorac Surg. 1998;66:870–875. discussion 75-6
75 Yan Q, Chang AC. Pharmacologic therapy for postoperative bleeding in children after cardiac surgery: when will the bleeding stop? Pediatr Crit Care Med. 2004;5:297–298.
76 Arnold P. Treatment and monitoring of coagulation abnormalities in children undergoing heart surgery. Paediatr Anaesth. 2011;21:494–503.
77 Afshari A, Wikkelso A, Brok J, Moller AM, Wetterslev J. Thrombelastography (TEG) or thromboelastometry (ROTEM) to monitor haemotherapy versus usual care in patients with massive transfusion. Cochrane Database Syst Rev. 2011. CD007871
78 Ak K, Isbir CS, Tetik S, et al. Thromboelastography-based transfusion algorithm reduces blood product use after elective CABG: a prospective randomized study. J Card Surg. 2009;24:404–410.
79 Shore-Lesserson L, Manspeizer HE, DePerio M, et al. Thromboelastography-guided transfusion algorithm reduces transfusions in complex cardiac surgery. Anesth Analg. 1999;88:312–319.
80 Spalding GJ, Hartrumpf M, Sierig T, et al. Cost reduction of perioperative coagulation management in cardiac surgery: value of “bedside” thrombelastography (ROTEM). Eur J Cardiothorac Surg. 2007;31:1052–1057.
81 Spiess BD, Gillies BS, Chandler W, Verrier E. Changes in transfusion therapy and reexploration rate after institution of a blood management program in cardiac surgical patients. J Cardiothorac Vasc Anesth. 1995;9:168–173.
82 Romlin BS, Wahlander H, Berggren H, et al. Intraoperative thromboelastometry is associated with reduced transfusion prevalence in pediatric cardiac surgery. Anesth Analg. 2011;112:30–36.
83 Holland LL, Brooks JP. Toward rational fresh frozen plasma transfusion: the effect of plasma transfusion on coagulation test results. Am J Clin Pathol. 2006;126:133–139.
84 Stanworth SJ, Grant-Casey J, Lowe D, et al. The use of fresh-frozen plasma in England: high levels of inappropriate use in adults and children. Transfusion. 2011;51:62–70.
85 Stanworth SJ, Brunskill SJ, Hyde CJ, McClelland DB, Murphy MF. Is fresh frozen plasma clinically effective? A systematic review of randomized controlled trials. Br J Haematol. 2004;126:139–152.
86 Kakkar N, Kaur R, Dhanoa J. Improvement in fresh frozen plasma transfusion practice: results of an outcome audit. Transfus Med. 2004;14:231–235.
87 Tavares M, DiQuattro P, Nolette N, Conti G, Sweeney J. Reduction in plasma transfusion after enforcement of transfusion guidelines. Transfusion. 2011;51:754–761.
88 Alam HB, Bice LM, Butt MU, et al. Testing of blood products in a polytrauma model: results of a multi-institutional randomized preclinical trial. J Trauma. 2009;67:856–864.
89 Oliver WC, Jr., Beynen FM, Nuttall GA, et al. Blood loss in infants and children for open heart operations: albumin 5% versus fresh-frozen plasma in the prime. Ann Thorac Surg. 2003;75:1506–1512.
90 McCall MM, Blackwell MM, Smyre JT, et al. Fresh frozen plasma in the pediatric pump prime: a prospective, randomized trial. Ann Thorac Surg. 2004;77:983–987. discussion 987
91 Moganasundram S, Hunt BJ, Sykes K, et al. The relationship among thromboelastography, hemostatic variables, and bleeding after cardiopulmonary bypass surgery in children. Anesth Analg. 2010;110:995–1002.
92 Bevan DH. Cryoprecipitate: no longer the best therapeutic choice in congenital fibrinogen disorders? Thromb Res. 2009;124(Suppl 2):S12–S16.
93 Hellstern P. Production and composition of prothrombin complex concentrates: correlation between composition and therapeutic efficiency. Thromb Res. 1999;95(4 Suppl 1):S7–12.
94 Fraser TA, Corke CF, Mohajeri M, Stevenson L, Campbell PJ. A retrospective audit of the use of Prothrombinex-HT for refractory bleeding following adult cardiac surgery. Crit Care Resusc. 2006;8:141–145.
95 Bruce D, Nokes TJ. Prothrombin complex concentrate (Beriplex P/N) in severe bleeding: experience in a large tertiary hospital. Crit Care. 2008;12:R105.
96 Carling MS, Jeppsson A, Wessberg P, et al. Preoperative fibrinogen plasma concentration is associated with perioperative bleeding and transfusion requirements in scoliosis surgery. Spine (Phila Pa 1976). 2011;36:549–555.
97 Rossaint R, Bouillon B, Cerny V, et al. Management of bleeding following major trauma: an updated European guideline. Crit Care. 2010;14:R52.
98 Weinkove R, Rangarajan S. Fibrinogen concentrate for acquired hypofibrinogenaemic states. Transfus Med. 2008;18:151–157.
99 Karlsson M, Ternstrom L, Hyllner M, et al. Prophylactic fibrinogen infusion reduces bleeding after coronary artery bypass surgery. A prospective randomised pilot study. Thromb Haemost. 2009;102:137–144.
100 Rahe-Meyer N, Solomon C, Winterhalter M, et al. Thromboelastometry-guided administration of fibrinogen concentrate for the treatment of excessive intraoperative bleeding in thoracoabdominal aortic aneurysm surgery. J Thorac Cardiovasc Surg. 2009;138:694–702.
101 Velik-Salchner C, Haas T, Innerhofer P, et al. The effect of fibrinogen concentrate on thrombocytopenia. J Thromb Haemost. 2007;5:1019–1025.
102 Bishop P, Lawson J. Recombinant biologics for treatment of bleeding disorders. Nat Rev Drug Discov. 2004;3:684–694.
103 Brody JI, Pickering NJ, Fink GB. Concentrations of factor VIII-related antigen and factor XIII during open heart surgery. Transfusion. 1986;26:478–480.
104 Godje O, Haushofer M, Lamm P, Reichart B. The effect of factor XIII on bleeding in coronary surgery. Thorac Cardiovasc Surg. 1998;46:263–267.
105 Blome M, Isgro F, Kiessling AH, et al. Relationship between factor XIII activity, fibrinogen, haemostasis screening tests and postoperative bleeding in cardiopulmonary bypass surgery. Thromb Haemost. 2005;93:1101–1107.
106 Godje O, Gallmeier U, Schelian M, Grunewald M, Mair H. Coagulation factor XIII reduces postoperative bleeding after coronary surgery with extracorporeal circulation. Thorac Cardiovasc Surg. 2006;54:26–33.
107 Levy JH, Gill R, Nussmeier NA, et al. Repletion of factor XIII following cardiopulmonary bypass using a recombinant A-subunit homodimer. A preliminary report. Thromb Haemost. 2009;102:765–771.
108 Schroth M, Meissner U, Cesnjevar R, et al. Plasmatic [corrected] factor XIII reduces severe pleural effusion in children after open-heart surgery. Pediatr Cardiol. 2006;27:56–60.
109 Bulutcu FS, Ozbek U, Polat B, et al. Which may be effective to reduce blood loss after cardiac operations in cyanotic children: tranexamic acid, aprotinin or a combination? Paediatr Anaesth. 2005;15:41–46.
110 Chauhan S, Kumar BA, Rao BH, et al. Efficacy of aprotinin, epsilon aminocaproic acid, or combination in cyanotic heart disease. Ann Thorac Surg. 2000;70:1308–1312.
111 Chauhan S, Bisoi A, Modi R, Gharde P, Rajesh MR. Tranexamic acid in paediatric cardiac surgery. Indian J Med Res. 2003;118:86–89.
112 Chauhan S, Das SN, Bisoi A, Kale S, Kiran U. Comparison of epsilon aminocaproic acid and tranexamic acid in pediatric cardiac surgery. J Cardiothorac Vasc Anesth. 2004;18:141–143.
113 Chauhan S, Bisoi A, Kumar N, et al. Dose comparison of tranexamic acid in pediatric cardiac surgery. Asian Cardiovasc Thorac Ann. 2004;12:121–124.
114 Rao BH, Saxena N, Chauhan S, Bisoi AK, Venugopal P. Epsilon aminocaproic acid in paediatric cardiac surgery to reduce postoperative blood loss. Indian J Med Res. 2000;111:57–61.
115 Reid RW, Zimmerman AA, Laussen PC, et al. The efficacy of tranexamic acid versus placebo in decreasing blood loss in pediatric patients undergoing repeat cardiac surgery. Anesth Analg. 1997;84:990–996.
116 Zonis Z, Seear M, Reichert C, Sett S, Allen C. The effect of preoperative tranexamic acid on blood loss after cardiac operations in children. J Thorac Cardiovasc Surg. 1996;111:982–987.
117 Toda Y, Kazuyoshi S, Iwasaki T, Takeuchi M, Morita K. Tranexamic acid reduces blood loss during and after pediatric cardiac surgery. Anesthesiology. 2008;109:A153.
118 Ries M, Zenker M, Gaffney PJ. Differences between neonates and adults in the urokinase-plasminogen activator (u-PA) pathway of the fibrinolytic system. Thromb Res. 2000;100:341–351.
119 Ries M, Easton RL, Longstaff C, et al. Differences between neonates and adults in tissue-type-plasminogen activator (t-PA)-catalyzed plasminogen activation with various effectors and in carbohydrate sequences of fibrinogen chains. Thromb Res. 2001;103:173–184.
120 Henry DA, Carless PA, Moxey AJ, et al. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev. 3, 2011. CD001886
121 European Medicines Agency, ICH: E 11: Clinical investigation of medicinal products in the paediatric population—Step 5. Jan. 1, 2001. Available at http://www.ema.europa.eu/docs/en_GB/ document_library/Scientific_guideline/2009/09/WC500002926.pdf (accessed June 2012)
122 Syed S, Arnold P. Use of antifibrinolytics in pediatric heart surgery: a survey of UK practice. Abstract to Association of Paediatric Anaesthetists meeting, 2010.
123 Franck M, Sladen R. Drugs to prevent and reverse anticoagulation. Anesthesiol Clin North Am. 1999;17:799–811.
124 Dowd NP, Karski JM, Cheng DC, et al. Pharmacokinetics of tranexamic acid during cardiopulmonary bypass. Anesthesiology. 2002;97:390–399.
125 Logan AC, Yank V, Stafford RS. Off-label use of recombinant factor VIIa in U.S. hospitals: analysis of hospital records. Ann Intern Med. 2011;154:516–522.
126 Toda Y, Shimizu K, Iwasaki T, Takeuchi M, Morita K. Serum tranexamic acid level and blood loss in pediatric cardiac surgery patients. Abstract A1186 in Proceedings of the 2009 Annual Meeting of the American Society Anesthesiologists.
127 Brodsky I, Gill DN, Lusch CJ. Fibrinolysis in congenital heart disease. Preoperative treatment with epsilon-aminocaproic acid. Am J Clin Pathol. 1969;51:51–57.
128 Ririe DG, James RL, O’Brien JJ, et al. The pharmacokinetics of epsilon-aminocaproic acid in children undergoing surgical repair of congenital heart defects. Anesth Analg. 2002;94:44–49.
129 Porte RJ, Leebeek FW. Pharmacological strategies to decrease transfusion requirements in patients undergoing surgery. Drugs. 2002;62:2193–2211.
130 Mangano DT, Tudor IC, Dietzel C. The risk associated with aprotinin in cardiac surgery. N Engl J Med. 2006;354:353–365.
131 Levi M, Cromheecke ME, de Jonge E, et al. Pharmacological strategies to decrease excessive blood loss in cardiac surgery: a meta-analysis of clinically relevant endpoints. Lancet. 1999;354:1940–1947.
132 Fremes SE, Wong BI, Lee E, et al. Metaanalysis of prophylactic drug treatment in the prevention of postoperative bleeding. Ann Thorac Surg. 1994;58:1580–1588.
133 Dunn CJ, Goa KL. Tranexamic acid: a review of its use in surgery and other indications. Drugs. 1999;57:1005–1032.
134 Casati V, Bellotti F, Gerli C, et al. Tranexamic acid administration after cardiac surgery: a prospective, randomized, double-blind, placebo-controlled study. Anesthesiology. 2001;94:8–14.
135 Abou-Diwan C, Sniecinski RM, Szlam F, et al. Plasma and cerebral spinal fluid tranexamic acid quantitation in cardiopulmonary bypass patients. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:553–556.
136 Sander M, Spies C, Martiny V, et al. Increased incidence of convulsive seizures following administration of tranexamic acid. [abstract]. J Cardiothorac Vasc Anesth. 23(Suppl), 2009.
137 Breuer T, Martin K, Wilhelm M, et al. The blood sparing effect and the safety of aprotinin compared to tranexamic acid in paediatric cardiac surgery. Eur J Cardiothorac Surg. 2009;35:167–171. author reply 71
138 Arnold DM, Fergusson DA, Chan AK, et al. Avoiding transfusions in children undergoing cardiac surgery: a meta-analysis of randomized trials of aprotinin. Anesth Analg. 2006;102:731–737.
139 Murugesan C, Banakal SK, Garg R, Keshavamurthy S, Muralidhar K. The efficacy of aprotinin in arterial switch operations in infants. Anesth Analg. 2008;107:783–787.
140 Williams GD, Ramamoorthy C, Pentcheva K, et al. A randomized, controlled trial of aprotinin in neonates undergoing open-heart surgery. Paediatr Anaesth. 2008;18:812–819.
141 Brown JR, Toler AW, Kramer RS, Landis RC. Anti-inflammatory effect of aprotinin: a meta-analysis. J Extra Corpor Technol. 2009;41:79–86.
142 Hsia TY, McQuinn TC, Mukherjee R, et al. Effects of aprotinin or tranexamic acid on proteolytic/cytokine profiles in infants after cardiac surgery. Ann Thorac Surg. 2010;89:1843–1852. discussion 52
143 Ferreira CA, Vicente WV, Evora PR, et al. Assessment of aprotinin in the reduction of inflammatory systemic response in children undergoing surgery with cardiopulmonary bypass. Rev Bras Cir Cardiovasc. 2010;25:85–98.
144 Fergusson DA, Hebert PC, Mazer CD, et al. A comparison of aprotinin and lysine analogues in high-risk cardiac surgery. N Engl J Med. 2008;358:2319–2331.
145 Pasquali SK, Hall M, Li JS, et al. Safety of aprotinin in congenital heart operations: results from a large multicenter database. Ann Thorac Surg. 2010;90:14–21.
146 Guzzetta NA, Evans FM, Rosenberg ES, et al. The impact of aprotinin on postoperative renal dysfunction in neonates undergoing cardiopulmonary bypass: a retrospective analysis. Anesth Analg. 2009;108:448–455.
147 Monroe DM, Hoffman M, Oliver JA, Roberts HR. Platelet activity of high-dose factor VIIa is independent of tissue factor. Br J Haematol. 1997;99:542–547.
148 Kheirabadi BS, Delgado AV, Dubick MA, et al. In vitro effect of activated recombinant factor VII (rFVIIa) on coagulation properties of human blood at hypothermic temperatures. J Trauma. 2007;63:1079–1086.
149 Eaton MP. Antifibrinolytic therapy in surgery for congenital heart disease. Anesth Analg. 2008;106:1087–1100.
150 Sachs B, Delacy D, Green J, et al. Recombinant activated factor VII in spinal surgery: a multicenter, randomized, double-blind, placebo-controlled, dose-escalation trial. Spine (Phila Pa 1976). 2007;32:2285–2293.
151 Oliver WC, Jr., Fass DN, Nuttall GA, et al. Variability of plasma aprotinin concentrations in pediatric patients undergoing cardiac surgery. J Thorac Cardiovasc Surg. 2004;127:1670–1677.
152 Martin K, Gertler R, Sterner A, et al. Comparison of blood-sparing efficacy of ε-aminocaproic acid and tranexamic acid in newborns undergoing cardiac surgery. Thorac Cardiovasc Surg. 2011;59:276–280.
153 Schindler E, Photiadis J, Sinzobahamvya N, et al. Tranexamic acid: an alternative to aprotinin as antifibrinolytic therapy in pediatric congenital heart surgery. Eur J Cardiothorac Surg. 2011;39:495–499.
154 Urban MK, Beckman J, Gordon M, Urquhart B, Boachie-Adjei O. The efficacy of antifibrinolytics in the reduction of blood loss during complex adult reconstructive spine surgery. Spine (Phila Pa 1976). 2001;26:1152–1156.
155 Bird S, McGill N. Blood conservation and pain control in scoliosis corrective surgery: an online survey of UK practice. Paediatr Anaesth. 2011;21:50–53.
156 Palmer GM, Pirakalathanan P, Skinner AV. A multi-centre multi-national survey of anaesthetists regarding the range of anaesthetic and surgical practices for paediatric scoliosis surgery. Anaesth Intensive Care. 2010;38:1077–1084.
157 Tzortzopoulou A, Cepeda MS, Schumann R, Carr DB. Antifibrinolytic agents for reducing blood loss in scoliosis surgery in children. Cochrane Database Syst Rev. 2008. CD006883
158 Dhawale AA, Shah SA, Sponseller PD, et al. Are antifibrinolytics helpful in decreasing blood loss and transfusions during spinal fusion surgery in children with cerebral palsy scoliosis? Spine (Phila Pa 1976). 2012;37:E549–E555.
159 Shapiro F, Zurakowski D, Sethna NF. Tranexamic acid diminishes intraoperative blood loss and transfusion in spinal fusions for Duchenne muscular dystrophy scoliosis. Spine. 2007;32:2278–2283.
160 Khoshhal K, Mukhtar I, Clark P, et al. Efficacy of aprotinin in reducing blood loss in spinal fusion for idiopathic scoliosis. J Pediatr Orthop. 2003;23:661–664.
161 Stricker PA, Cladis FP, Fiadjoe JE, McCloskey JJ, Maxwell LG. Perioperative management of children undergoing craniofacial reconstruction surgery: a practice survey. Paediatr Anaesth. 2011.
162 Dadure C, Sauter M, Bringuier S, et al. Intraoperative tranexamic acid reduces blood transfusion in children undergoing craniosynostosis surgery: a randomized double-blind study. Anesthesiology. 2011;114:856–861.
163 Goobie SM, Meier PM, Pereira LM, et al. Efficacy of tranexamic acid in pediatric craniosynostosis surgery: a double-blind, placebo-controlled trial. Anesthesiology. 2011;114:862–871.
164 Duran de la Fuente P, Garcia-Fernandez J, Perez-Lopez C, Carceller F, Gilsanz Rodriguez F. [Usefulness of tranexamic acid in cranial remodeling surgery]. Rev Esp Anestesiol Reanim. 2003;50:388–394.
165 D’Errico CC, Munro HM, Buchman SR, Wagner D, Muraszko KM. Efficacy of aprotinin in children undergoing craniofacial surgery. J Neurosurg. 2003;99:287–290.
166 Shakur H, Roberts I, Bautista R, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376:23–32.
167 Roberts I, Shakur H, Afolabi A, et al. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet. 2011;377:1096–1101.
168 D’Errico CC, Shayevitz JR, Martindale SJ, Mosca RS, Bove EL. The efficacy and cost of aprotinin in children undergoing reoperative open heart surgery. Anesth Analg. 1996;83:1193–1199.
169 Seear MD, Wadsworth LD, Rogers PC, Sheps S, Ashmore PG. The effect of desmopressin acetate (DDAVP) on postoperative blood loss after cardiac operations in children. J Thorac Cardiovasc Surg. 1989;98:217–219.
170 Reynolds LM, Nicolson SC, Jobes DR, et al. Desmopressin does not decrease bleeding after cardiac operation in young children. J Thorac Cardiovasc Surg. 1993;106:954–958.
171 Gill JC, Montgomery RR. Failure of response to DDAVP in children less than 1 year of age. Pediatr Res. 1986;20:279.
172 Sutor AH. Desmopressin (DDAVP) in bleeding disorders of childhood. Semin Thromb Hemost. 1998;24:555–566.
173 Miller BE, Williams GD. Bleeding and coagulation: monitoring and management. In: Andropoulos DB, Stayer SA, Russell IA, eds. Anesthesia for congenital heart disease. Oxford, England: Blackwell; 2005:157–172.
174 Brady KM, Easley RB, Tobias JD. Recombinant activated factor VII (rFVIIa) treatment in infants with hemorrhage. Paediatr Anaesth. 2006;16:1042–1046.
175 Pychynska-Pokorska M, Moll JJ, Krajewski W, Jarosik P. The use of recombinant coagulation factor VIIa in uncontrolled postoperative bleeding in children undergoing cardiac surgery with cardiopulmonary bypass. Pediatr Crit Care Med. 2004;5:246–250.
176 Dominguez TE, Mitchell M, Friess SH, et al. Use of recombinant factor VIIa for refractory hemorrhage during extracorporeal membrane oxygenation. Pediatr Crit Care Med. 2005;6:348–351.
177 Filan PM, Mills JF, Clarnette TD, Ekert H, Ekert P. Spontaneous liver hemorrhage during laparotomy for necrotizing enterocolitis: a potential role for recombinant factor VIIa. J Pediatr. 2005;147:857–859.
178 Kulkarni R, Daneshmand A, Guertin S, et al. Successful use of activated recombinant factor VII in traumatic liver injuries in children. J Trauma. 2004;56:1348–1352.
179 Mathew P, Winter SS, Frost JD, et al. Novel applications of recombinant factor VIIa for the management of pediatric coagulopathic diseases. J Pediatr Hematol Oncol. 2003;25:499–502.
180 Fontaine MJ, Lazarchick J, Taylor S, Annibale D. Use of recombinant factor VIIa in infants with severe coagulopathy. J Perinatol. 2004;24:310–311.
181 Leibovitch L, Kenet G, Mazor K, et al. Recombinant activated factor VII for life-threatening pulmonary hemorrhage after pediatric cardiac surgery. Pediatr Crit Care Med. 2003;4:444–446.
182 Atkison PR, Jardine L, Williams S, et al. Use of recombinant factor VIIa in pediatric patients with liver failure and severe coagulopathy. Transplant Proc. 2005;37:1091–1093.
183 Brown JB, Emerick KM, Brown DL, Whitington PF, Alonso EM. Recombinant factor VIIa improves coagulopathy caused by liver failure. J Pediatr Gastroenterol Nutr. 2003;37:268–272.
184 Markiewicz M, Kalicinski P, Kaminski A, et al. Acute coagulopathy after reperfusion of the liver graft in children correction with recombinant activated factor VII. Transplant Proc. 2003;35:2318–2319.
185 Morenski JD, Tobias JD, Jimenez DF. Recombinant activated factor VII for cerebral injury-induced coagulopathy in pediatric patients. Report of three cases and review of the literature. J Neurosurg. 2003;98:611–616.
186 Kalenka A, Munch E, Fiedler F. Recombinant factor VIIa to treat traumatic bleeding in a children. Paediatr Anaesth. 2005;15:1025–1027.
187 Lisman T, De Groot PG. Mechanism of action of recombinant factor VIIa. J Thromb Haemost. 2003;1:1138–1139.
188 Warren OJ, Rogers PL, Watret AL, et al. Defining the role of recombinant activated factor VII in pediatric cardiac surgery: where should we go from here? Pediatr Crit Care Med. 2009;10:572–582.
189 Diprose P, Herbertson MJ, O’Shaughnessy D, Gill RS. Activated recombinant factor VII after cardiopulmonary bypass reduces allogeneic transfusion in complex non-coronary cardiac surgery: randomized double-blind placebo-controlled pilot study. Br J Anaesth. 2005;95:596–602.
190 Gill R, Herbertson M, Vuylsteke A, et al. Safety and efficacy of recombinant activated factor VII: a randomized placebo-controlled trial in the setting of bleeding after cardiac surgery. Circulation. 2009;120:21–27.
191 Ekert H, Brizard C, Eyers R, Cochrane A, Henning R. Elective administration in infants of low-dose recombinant activated factor VII (rFVIIa) in cardiopulmonary bypass surgery for congenital heart disease does not shorten time to chest closure or reduce blood loss and need for transfusions: a randomized, double-blind, parallel group, placebo-controlled study of rFVIIa and standard haemostatic replacement therapy versus standard haemostatic replacement therapy. Blood Coagul Fibrinolysis. 2006;17:389–395.
192 Friederich PW, Henny CP, Messelink EJ, et al. Effect of recombinant activated factor VII on perioperative blood loss in patients undergoing retropubic prostatectomy: a double-blind placebo-controlled randomised trial. Lancet. 2003;361:201–205.
193 Lin Y, Stanworth S, Birchall J, Doree C, Hyde C. Recombinant factor VIIa for the prevention and treatment of bleeding in patients without haemophilia. Cochrane Database Syst Rev. 2011. CD005011
194 Yank V, Tuohy CV, Logan AC, et al. Systematic review: benefits and harms of in-hospital use of recombinant factor VIIa for off-label indications. Ann Intern Med. 2011;154:529–540.
195 Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical trials. N Engl J Med. 2010;363:1791–1800.
196 Puetz J, Darling G, Brabec P, Blatny J, Mathew P. Thrombotic events in neonates receiving recombinant factor VIIa or fresh frozen plasma. Pediatr Blood Cancer. 2009;53:1074–1078.
197 Witmer CM, Huang YS, Lynch K, Raffini LJ, Shah SS. Off-label recombinant factor VIIa use and thrombosis in children: a multi-center cohort study. J Pediatr. 2011;158:820–825.
198 Welsby IJ, Monroe DM, Lawson JH, Hoffmann M. Recombinant activated factor VII and the anaesthetist. Anaesthesia. 2005;60:1203–1212.
199 Chin C. Recombinant activated factor VII. Paediatr Anaesth. 2006;16:907–909.
200 Santagostino E, Morfini M, Rocino A, et al. Relationship between factor VII activity and clinical efficacy of recombinant factor VIIa given by continuous infusion to patients with factor VIII inhibitors. Thromb Haemost. 2001;86:954–958.
201 Razon Y, Erez E, Vidne B, et al. Recombinant factor VIIa (NovoSeven) as a hemostatic agent after surgery for congenital heart disease. Paediatr Anaesth. 2005;15:235–240.
202 Erhardtsen E. Pharmacokinetics of recombinant activated factor VII (rFVIIa). Semin Thromb Hemost. 2000;26:385–391.
203 Villar A, Aronis S, Morfini M, et al. Pharmacokinetics of activated recombinant coagulation factor VII (NovoSeven) in children vs. adults with haemophilia A. Haemophilia. 2004;10:352–359.
204 Veldman A, Josef J, Fischer D, Volk WR. A prospective pilot study of prophylactic treatment of preterm neonates with recombinant activated factor VII during the first 72 hours of life. Pediatr Crit Care Med. 2006;7:34–39.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
18 Medications for Hemostasis
This chapter will examine specific treatments aimed at correcting coagulopathy and at reducing the requirement for transfusion of blood products by encouraging hemostasis. Although similar, these objectives are not the same. During many types of surgery, in vitro tests may only demonstrate mild coagulation defects, and yet the use of hemostatic medications may reduce blood loss and obviate the need to transfuse red cells.1,2 In the absence of these medications, bleeding itself is often unlikely to be life-threatening. In comparison, severe coagulopathic bleeding is, by its nature, life-threatening. Transfusion of blood products cannot be avoided and the hemostatic medications used are often the blood products themselves. Fundamentally, the equation of risk and benefit is very different in these two situations. In the former case, benefit will occur if the risk reduction associated with the avoidance of blood products is greater than any risk from the medication itself. In the latter case, the use of medications that carry a high risk of causing adverse effects is more acceptable if the aim is to prevent death from uncontrolled bleeding. As an example of this, aprotinin has been largely rejected for use during adult cardiac surgery as a strategy to reduce transfusion,3 whereas use of recombinant factor VIIa (almost certainly a more “hazardous” drug) is still considered appropriate for the treatment of life-threatening bleeding.
Avoidance of transfusion of human blood products seems to be a fundamentally good notion. However, many common ideas about the potential harm of transfusion may be mistaken. A national audit of the serious hazards of transfusion (SHOT) in the United Kingdom reported 3239 adverse events over a 10-year period; this is only 0.013% of all blood transfusions.4 These adverse events were more common in infants, but were only reported in 0.037% of infants given transfusions. The true incidence of events is likely to be more common because of underreporting, though many of these “events” were procedural errors not associated with actual harm. It is clear that the risk of immediate harm directly attributable to transfusion is exceptionally low. The risk of direct transmission of infection (the most feared risk in the public perception) is particularly small; the risk of transmission of HIV from transfusion of blood in the UK is 1 in 5 million, and no child has contracted a viral infection from transfusion in the last 4 years.4 The greatest hazard of immediate harm is of a mistake leading to transfusion of incompatible blood.5 However, subtle negative effects of transfusion on children’s outcomes may be considerably more important.6 Transfusion of blood may lead to deterioration in pulmonary function and immunologic effects that predispose children to infection. In addition, if the objective of transfused red cells is to boost tissue oxygenation by improving oxygen carrying capacity, then transfused blood is less effective than the child’s own blood for this function.7 Additional considerations include the increasing costs of transfused blood products, the logistical difficulties of maintaining a secure blood supply (in both well-resourced and less well developed health systems), substantially greater risks of transfusion (in poorly developed health systems), and the possibility of new, unrecognized infective agents entering the blood supply.
There are 2.3 million transfusions each year (37 per 1000 population) in the UK; 4.2% are given to children under 18 years of age (7.1 per 1000) and 1.7% to infants (52 per 1000). An audit of pediatric blood transfusion from Australia revealed 41% of blood transfused was given perioperatively,8 and blood was transfused during 6.3% of instances of anesthesia. The majority of units were used during heart surgery (58% of perioperative use) while a very small minority, 4% were used during major trauma surgery. Heart surgery (on and off bypass), craniosynostosis surgery, and liver transplantation were all commonly associated with blood transfusion. The epidemiology of major bleeding is less certain. Reports of blood use may be misleading (e.g., neonates undergoing cardiac surgery to correct congenital defects may receive few blood units despite significant bleeding), and blood is also used for purposes such as priming the bypass circuit. Observational studies have reported very heavy blood loss associated with heart surgery in children, with increased (per kg) blood loss in smaller children and more complex surgery.9,10 Cardiac surgery can probably be considered the main cause of major blood loss in children and will account for most severe bleeding events in children less than 1 year of age.
Physiology of Coagulation
The understanding of coagulation has shifted greatly in the last few years. The “traditional” model, which emphasizes the importance of a cascade of proteolytic enzymes has given way to the “cell based” model of coagulation that emphasizes the importance of cellular elements in coagulation and presents coagulation as a complex web of interactions rather than a linear process.11,12
To achieve effective hemostasis, a platelet plug must form at the site of vessel injury. In addition, the procoagulant factors need to remain localized to the injured site to avoid widespread clotting activation. This is achieved through changes at the cell surface and localization of the procoagulant reactions on the surfaces of specific cells. Different cells possess different procoagulant and anticoagulant properties; these are incompletely understood, but platelets and cells bearing tissue factor (TF) are central to the process. Intact endothelium is also vital to normal control of coagulation, as it keeps these different procoagulant components apart and modifies coagulation through expression of inhibitory proteins such as thrombomodulin. The described phases of coagulation are overlapping and involve initiation, amplification, and propagation.12
Initiation
Coagulation is initiated by a membrane-bound lipoprotein called tissue factor. This is usually expressed on subendothelial TF-bearing cells, such as stromal fibroblasts; in the absence of vessel injury it is separated from the vessel lumen. After injury, a complex is formed between TF and factor VIIa (TF/VIIa), which activates factors IX and X. Factor Xa, in association with cofactor Va, forms “prothrombinase” complexes on the surface of the TF-bearing cell, which activates a small amount of thrombin (factor IIa)12 (Fig. 18-1). The purpose of this small generation of thrombin and Xa is to activate platelets and factors V and VIII.
Low levels of IX and X activation occur in the absence of tissue injury without causing clot formation. The process only leads to amplification when damage to the vasculature allows intravascular platelets and a complex formed by factor VIII and von Willebrand factor (VIII/vWF) to adhere to the extravascular TF-bearing cells.11
Amplification
Small quantities of thrombin are generated on the TF-bearing cells. This sets up the subsequent propagation phase, during which thrombin is generated in large quantities (Fig. 18-2). This thrombin has many major functions:
Activation of platelets, exposing receptors and binding sites for clotting factors
Activation of factor XI to XIa13
Activation of factor XIII (fibrin stabilizing factor) and promotion of fibrin crosslinking
Cleaving fibrinopeptides A and B from fibrinogen (forming fibrin)
Propagation
Propagation occurs on the surface of activated platelets that are recruited to the site in large numbers. Activated factor IX (from both initiation phase and provided by factor XI on the platelet) binds to VIIIa. The resultant IXa/VIIIa complex activates factor X on the platelet surface. This factor Xa associates with factor Va and forms the prothrombinase complex. The prothrombinase complex causes a “burst” of thrombin generation to cause clotting via fibrinogen (Fig. 18-3).11
This sequence explains why children with hemophilia bleed despite having a normal TF/VIIa complex; the factor Xa during the initiation phase is broken down by ATIII and TFPI if it dissociates from the TF-bearing cell. It is therefore unable to activate the “burst” of thrombin generation that normally occurs in this propagation phase. The factor Xa needs to be generated on the platelet itself via the IXa/VIIIa complex.11
Of note, an alternative pathway is initiated by contact factors (XII, XI, prekallikrein, and high-molecular-weight kininogen [HMWK]). It is of no physiologic importance in terms of coagulation activation; however, it provides important acceleration loops through feedback activation of factors VIII, IX, and XI14 and is important in fibrinolytic and inflammatory pathways.15
Clot Inhibition
Several indirect systems inhibit thrombin, such as the protein C–protein S–thrombomodulin (TM) and the TFPI systems. Thrombin binds to TM on the surface of intact endothelial cells and can no longer cleave fibrinogen to form fibrin. The TM/thrombin complex is neither able to activate platelets nor activate factors V and VIII. Instead, this complex activates protein C, which binds to the cofactor protein S and inactivates factors Va (on the surface of endothelial cells and platelets) and VIIIa.11,14 TFPI bound to endothelial surfaces can form complexes with factor Xa that inhibit factor VIIa. Thrombin generation is therefore inhibited.14
Fibrinolysis
Fibrinolysis (the breakdown of fibrin into soluble degradation products) is mediated by the proteolytic enzyme, plasmin. Plasmin is formed from an inactive zymogen, plasminogen, which is produced in the liver. This process is controlled by activators and inhibitors. The principal plasminogen activators are tissue plasminogen activator (tPA) and urokinase (uPA). Although overlap exists between the functions of these activators, they have distinct physiologic roles. uPA is primarily involved in a wide variety of extracellular processes, including remodeling of tissues. tPA is the major intravascular activator of fibrinolysis and is discussed in more detail later.16 The main inhibitory proteins are plasminogen activator inhibitor-1 (PAI-1), antiplasmins (α2-PI and α2-macroglobulin), and thrombin activated fibrinolysis inhibitor (TAFI)17 (Fig. 18-4).
As well as cleaving fibrin, plasmin metabolizes a number of other proteins, including the platelet receptor for fibrinogen (glycoprotein IIb/IIIa) and fibrinogen.18 In addition, plasmin accelerates its own production by metabolizing the conversion of single chain plasminogen activators to more active two-chain versions. The action of plasmin on fibrin produces a series of degradation products some of which convey anticoagulant properties; this effect is achieved by preventing polymerization of fibrinogen and by inhibition of platelet function.
tPA is released by vascular endothelium of small blood vessels. Release is increased in the presence of stimuli such as trauma, endotoxins, ischemia, or normal exercise. This effect is mediated via contact activation (through the kallikrein system) and also by a series of other substances, including thrombin. Once released, tPA is rapidly metabolized by the liver with a half-life of approximately 5 minutes.17 Fibrin binds both plasminogen and tPA and greatly accelerates the conversion of plasminogen to plasmin (facilitating its own degradation, but also localizing the process to areas of clot). An alternative mechanism for plasminogen activation by tPA exists through binding to receptors expressed by certain cells (endothelium, white cells and some tumor cells); the importance of this in health or disease is unclear.
Excessive fibrinolysis can directly result from excess production of fibrin, as in disseminated intravascular coagulation. This is termed secondary hyperfibrinolysis and in this context the fibrinolysis is considered beneficial because it prevents widespread vascular occlusion. Therapy is directed at replacement of consumed clotting factors, inhibition of excessive coagulation, and treatment of the underlying cause. Primary hyperfibrinolysis can occur during cardiac bypass, massive blood loss, trauma, and liver transplantation.19 During the anhepatic stage of liver transplant surgery, there is hyperfibrinolysis because of failure to metabolize tPA. On reperfusion of the liver, a further surge of tPA occurs that can take several hours to return to normal. In coagulopathic patients, reduced thrombin formation may lead to reduced production of TAFI (important in inhibition of membrane bound plasmin), whereas conversion of single- to two-strand tPA by plasmin may further sustain the process. Individual susceptibility is likely to be important and may have a genetic component.20,21
Two groups of drugs are used clinically to inhibit fibrinolysis:
Synthetic lysine analogues are fairly specific inhibitors of plasminogen activation, working by competitively binding to lysine-binding sites on the plasminogen molecule (Fig. 18-5). This blocks the binding of plasminogen to fibrin; a required step for the conversion of plasminogen to plasmin by plasminogen activators.22 At larger doses, they may have additional effects through direct inhibition of plasmin; this includes inhibition of the plasmin mediated effects on platelets.
Aprotinin is a less specific inhibitor of proteolytic enzymes; it has actions on the kallikrein–kinin (contact) system, as well as enzymes involved in coagulation and fibrinolysis. In addition, aprotinin may be associated with greater preservation of platelet function, as well as an antiinflammatory effect. This wider spectrum of effects from aprotinin may have additional benefits over the antifibrinolytic lysine analogues. Some additional effects may not necessarily be of benefit; for example, the contact system may have protective effects against ischemic reperfusion injury that are inhibited by aprotinin.15
Developmental Coagulation
The hemostatic system in the neonate rapidly matures towards that of the adult (see also Chapter 9).23–25 One must consider how the coagulation system matures in the child to interpret coagulation tests and use appropriate modalities to manipulate hemostasis in vivo.
All fetal coagulation factors are produced independently of the mother; fibrinogen starts to be formed as early as 5.5 weeks gestation, and blood can clot at 11 weeks. The introduction of microassays in the 1980s allowed for the determination of reference ranges for the coagulation factors beginning at 19 weeks gestational age.14,23,24 In general, there are four fundamental differences between the coagulation systems in the infant and the adult26:
Concentrations of components of the hemostatic system
The turnover rate of various components of the coagulation cascade
Differences in the overall ability to generate and regulate the key enzymes: thrombin and plasmin
Despite the upregulation of coagulation factors at birth, the vitamin K–dependent factors II, VII, IX, and X in the neonate are only 50% of adult values; this leads to a slightly prolonged prothrombin time (PT) or international normalized ratio (INR).14 The contact factors HMWK, prekallikrein, and factors XI and XII are also approximately 50% of adult values.14,26 The reduced contact factors account for a disproportionally prolonged activated partial thromboplastin time (aPTT). The reduced concentration of factors at birth is probably explained by the reduced synthesis of factors by the liver; however, concentrations increase rapidly, reaching approximately 80% of adult values by 6 months of age.14,27
In contrast, the plasma concentrations of fibrinogen and factors V and VIII at birth are similar to those in adults, although the fetal form of fibrinogen differs in structure from that of the adult. The physiologic significance of this is not clear.28 Concentrations of vWF in the first 2 months of life are greater than those in adults.29
The inhibitor systems of coagulation also differ from adults. At birth, plasma protein C and S are 35% of adult values, although the fetal forms of the proteins differ from the adult forms. The concentration of protein C does not reach adult values until adolescence. Neonatal concentrations of ATIII and HCII are 50% of adult values; they reach adult concentrations by 6 months of age. The α2-macroglobulin value, however, is increased at birth and remains increased throughout childhood; it is postulated that this may be one of the mechanisms that protects young children from thromboembolic complications.30
Finally, thrombin generation in vitro is reduced in children to approximately 75% of adult values,30 but there is difficulty in interpreting the apparent discrepancy between the sufficient hemostasis seen in vivo and these in vitro laboratory thrombin generation assays. It still remains to be clarified whether neonates and children are more susceptible to bleeding14; however, the risk of thromboembolic complications appears to increase with age.30
Despite reduced plasma concentrations of many procoagulant and anticoagulant proteins in infants, there still appears to be an effective hemostatic balance; healthy fetuses, neonates, and children do not suffer excessive hemorrhage in the presence of minor challenges. This is consistent with the thromboelastogram (TEG) studies of healthy children younger than 2 years of age; no defects in coagulation were noted using this test compared with adults, indicating an intact hemostatic system.31 Another TEG study reported that infants younger than 1 year of age with complex congenital heart disease have an intact and balanced coagulation–fibrinolytic system but at a “lower level” than healthy children. This has been interpreted as a reduction in hemostatic potential with less reserve.32
Genetics of Bleeding
The hemophilias are a group of genetic diseases that cause excessive bleeding, often in response to minor trauma. von Willebrand disease and hemophilia A are the most common variants (see also Chapter 9), associated with low levels of von Willebrand factor and factor VIII respectively. A wide range of single gene defects resulting in deficiencies of single clotting proteins or regulatory proteins have now been described. A further group of single gene disorders may result in thrombophilic disorders, associated with abnormalities of inhibitory proteins.
Unexplained variation has been observed in bleeding between apparently similar patients in the absence of specific factor deficiency. The causes of this variation are likely to be multifold according to nuances of surgical technique and subtle difference in disease process and therapy. It might appear counterintuitive that genetic factors have a significant role in acquired bleeding resulting from surgery; however, recently there has been increased interest in the interplay of genetic and environmental factors in the progress of acquired diseases. The genetics of most clotting proteins has been described, and common variations within populations have been revealed for some. Of these, a common polymorphism of PAI-1 has been well described. PAI-1 is an important endogenous inhibitor of fibrinolysis, and deficiency is associated with increased bleeding.33 The G5/G5 polymorphism is common (about 20% in European populations) and is associated with lower levels of PAI-1. It has been linked (though not consistently) to bleeding after heart surgery and to increased benefit from use of antifibrinolytics.20,21 Should these findings be substantiated, then PAI-1 may still be an exceptional example. In general, the influence and consequences of genetic determinants of bleeding are likely to be more subtle. In infancy an additional factor in the mix may be the relationship between developmental and genetic factors. Many clotting proteins are present in infants as isoforms distinct from those in adults. This implies a different gene expression in the young. It is possible that understanding genetically determined variations in bleeding will increase our understanding of bleeding and, perhaps, allow us to guide therapy for the individual patient. The practical applications of such techniques, however, remain speculative.
Coagulopathy and Major Surgery
Coagulation changes during major surgery and bleeding are complex34 and depend on the clinical context in which bleeding occurs. Coagulation changes that occur in surgical patients have some similarities to those who present after severe trauma; however, the balance of pathophysiologic factors is likely to be very different. The factors underlying these coagulation changes include:
Dilution. Components of the coagulation system are lost in shed blood. The volume of blood lost is then replaced by crystalloid, colloid, or blood products lacking these components, leading to progressively smaller concentrations of these coagulation components. To some degree such changes are balanced, as the concentration of coagulation inhibitors also falls. In addition, coagulation components may be produced or released in response to trauma, which limits the reduction in concentration. To complicate this issue, a reduction in the concentration of one component of the coagulation system may not have the same clinical effect as the same reduction of another component. For example, substantial decreases in the concentration of many clotting proteins will not result in severe bleeding, while even modest decreases in platelet numbers or in the fibrinogen concentration may be significant (see also Chapter 10).
Physiologic derangement associated with blood loss. Acidosis, hypothermia, and hypocalcemia are associated with excess bleeding.35 Hypothermia will result in a general slowing of proteolytic enzyme activity, reduced fibrin synthesis, and reduced platelet function. These effects are largely reversible on rewarming. Acidosis is associated with a marked reduction in activity of coagulation proteins.36 These effects are not fully reversed by correcting acidosis. The degree to which coagulation changes reflect acidosis itself or reflect other underlying factors is unclear. During major bleeding, calcium ion concentration should be monitored and replaced as needed.
Effects of treatment. The use of some synthetic colloids may worsen bleeding to an extent greater than might be expected by dilution. The use of hydroxyethyl starch leads to increased risk of coagulation abnormalities and of acute kidney injury when compared to albumin, gelatins, or crystalloids. Whether differences exist in coagulation effects between different starches is controversial.37–39 Caution should be exercised in the use of starches in children at risk of coagulation problems, and in larger volumes.
Use of specific techniques during surgery. The important effects of cardiac bypass and anticoagulation are discussed later. Liver transplant surgery (see also Chapter 29) and major trauma (see also Chapter 38) are discussed in detail elsewhere in this book.
Alterations in Hemostasis during Pediatric Cardiac Surgery
Routine Anticoagulation
Heparin
Heparin remains the most effective anticoagulant used to facilitate cardiopulmonary bypass (CPB).40 The binding of heparin to lysine sites on ATIII causes a conformational change in ATIII. This results in an increase in ATIII potency; the inhibition of thrombin and factors IXa, Xa, XIa, and XIIa are increased by a factor of 1000.41,42 In infants ATIII levels are low until 3 to 6 months of age and other heparin cofactors, in particular, α2-macroglobulin may have greater importance. However neonates requiring surgery for congenital heart disease have unusually reduced concentrations of all major heparin cofactors, and this may explain the greater concentrations of thrombin production in these patients.43
There are limitations to the use of the ACT; first, the ACT is altered by hypothermia, hemodilution, platelet activation, activation of the hemostatic system, and aprotinin therapy.44,45 Accordingly, it does not accurately reflect the heparin concentrations. One study found that as soon as children go on CPB, the heparin concentrations decreased by 50% as a result of hemodilution, even though the ACT doubled.46 Second, in the bleeding child, the ACT is unable to differentiate between bleeding resulting from excess heparin or from other acquired hemostatic defects.46 The gold standard for measurement of heparin concentration is considered to be the antifactor Xa assay; however, this test remains too cumbersome for routine clinical use. A further method is protamine titration, which has been available as a point-of-care test for some years (Medtronic Hepcon HMS Plus, Medtronic, Minneapolis, USA). In adult patients, use of this system has been shown to lead to reduced thrombin formation47; however, the accuracy of the device has been questioned.48 Reduced bleeding and reduced thrombin generation in children has been demonstrated with the use of this system, whereas other studies have demonstrated reduced thrombin formation22,49 in infants. A trial in small infants was terminated early when increased bleeding and increased length of stay was demonstrated in children in whom the Hepcon device was used.49 The device underestimated heparin concentration in this group, leading to excess dosing of heparin and inadequate reversal with protamine. After modification of their protocol, use of the device demonstrated reduced bleeding, length of stay, and reduced thrombin formation compared to standard treatment.49 Agreement between protamine titration, measures of heparin concentration, and laboratory measures has been demonstrated, but the Hepcon devices tended to underestimate heparin concentrations in infants.50
A common feature of the pediatric studies using the Hepcon device is increased heparin use compared to regimes based on units/kg dosing or ACT. This is consistent with other studies of traditional dosing regimes. Using common pediatric heparin regimens (300 units/kg before CPB, then 100 units/kg to keep the ACT above 450 sec), 50% of children on CPB had low levels of heparin (less than 2 units/mL).46 It is suggested that reduced heparin concentrations during CPB is a major factor responsible for activation of coagulation and fibrinolysis. It is likely that widely used regimens for dosing of heparin in children lead to inadequate dosing, and that units/kg dosing fails to allow for important pharmacokinetic (PK) and pharmacodynamic (PD) differences in children.
Even effective dosing of heparin does not completely abolish the production of thrombin. Low-grade ongoing thrombin production leads to ongoing activation of the coagulation cascade, platelets, fibrinolysis, and the endothelium. The continued thrombin generation and activity during CPB reflects the inability of the heparin–ATIII complex to inactivate fibrin-bound thrombin or to inhibit thrombin-induced platelet activation.51 Theoretically, direct thrombin inhibition may be free of these limitations. Practically, the use of the current generation of thrombin inhibitors (such as hirudin and bivalirudin) is limited because of a lack of effective monitoring and reversal. Currently few reports exist of the use of hirudin in children, although it would be indicated in children in whom heparin use is not possible.52
Reversal of Anticoagulation with Protamine
Protamine is a positively charged polypeptide derived from salmon sperm. It neutralizes heparin by forming an ionic bond with heparin. The resultant complex is removed by the reticuloendothelial system. The most appropriate dosing regimen has yet to be determined. Current dosing used in pediatric practice fails to take into account the range of concentrations of heparin that occurs in infants and children.40,49 The administered heparin dose is often used to guide the dose of protamine; however, it is unclear how this should be modified by various factors, such as additional doses of heparin administered (to prime or during bypass), duration of bypass, techniques (e.g., ultrafiltration), or developmental coagulation differences in children.53
Excessive protamine has been associated with catastrophic pulmonary hypertension and hemorrhagic pulmonary edema. It is also known that protamine can be associated with coagulation abnormalities; an increasing ACT occurs at a protamine-heparin ratio of 2.6 : 1, and platelet aggregation occurs with a minimal excess in protamine.54 Although some studies on titrating protamine regimens in adults demonstrated encouraging results in terms of reduced bleeding,55 others failed to demonstrate differences in transfusion requirements.56
The clearance of protamine is greater than that of heparin, and “heparin rebound” is described as tissue-bound heparin redistributes.40 The diagnosis of residual heparin effect or heparin rebound is challenging. The ACT is not a specific measure of excessive heparin and is also poor at detecting heparin at low concentrations (less than 0.5 unit/mL).57 The aPTT and PT are similarly nonspecific and may be increased after CPB in the absence of heparin.58 An unmodified TEG does not reliably detect heparin rebound if the heparin concentration is small.59 The sensitivity of these tests may be improved by performing similar tests in parallel and comparing the results, such as a reptilase time (unaffected by heparin), or by eliminating residual heparin in vitro with heparinase or protamine. Protamine titration can be used to guide protamine dose in children; however, in infants protocols will require modification (50% higher than calculated dose).49 In practice most anesthesiologists continue to give protamine empirically at a protamine-heparin ratio of between 1 and 1.3 to 1.
Failure of Hemostasis Associated with Cardiac Surgery
Complex abnormalities occur in the coagulation system in cardiac surgery, owing to the profound surgical insult, hypothermia, acid-base disturbance, blood transfusion, anticoagulants, CPB (contact activation, platelet dysfunction, and hemodilution), and in some cases, deep hypothermic cardiorespiratory arrest. In addition children and infants with congenital heart disease may have preexisting coagulation defects or be taking medications that affect coagulation before surgery (Table 18-1).
TABLE 18-1 Causes of Excessive Bleeding after Pediatric Cardiac Surgery
Antiplatelet drugs such as aspirin or clopidogrel may exacerbate bleeding. The clinician must balance the risk of perioperative bleeding against the risk of drug discontinuation when deciding if and when to withhold the drug before surgery. In most cases aspirin can be safely discontinued 5 days before surgery. Prostaglandin E1 can inhibit platelet aggregation, acting in synergy with endothelial cell–derived factors (nitric oxide and prostacyclin) at clinically relevant concentrations,60 although this effect was too subtle to detect in vitro using the TEG.32 It is usually not possible to stop the prostaglandin E1 infusions, because neonates are dependent on its use to maintain ductal patency. For elective surgery in children on oral anticoagulant therapy, it is usually possible to transfer to heparin before surgery. An INR of less than 1.5 is usually considered acceptable for surgery. If urgent correction of anticoagulants is required, prothrombin complex concentrates, together with vitamin K, are more effective than FFP.61 FFP should only be used if prothrombin complex concentrates are not available.
In the child with cyanotic congenital heart disease, hemostasis is further impaired because of polycythemia, low platelet count and altered function, reduced factors V, VII, and VIII, and increased fibrinolysis.32,62 The degree of derangement is related to the degree of cyanosis.63 Preexisting coagulopathy may also occur in children with severe underlying illness or poor nutritional status.
Despite improvements in design and materials of the CPB circuit, abnormal activation of the coagulation and fibrinolytic systems persist. The normal balance of coagulation and fibrinolysis is particularly delicate in children, and more susceptible to exogenous perturbations.64 Shortly after CPB is established in children, all hemostatic protein concentrations are decreased (to a variable degree) as a result of dilution.65 Because of the relative volume of the circuit in relation to the size of the child, it is not surprising that the magnitude of the effect of hemodilution is greater than in adults.46
Reductions in coagulation factor concentrations after cardiac surgery contribute to bleeding in adult cardiac patients,66 and a reduction in platelet count is well described on initiation of bypass. In adults, the platelet count is reduced on CPB by approximately 50%.67 This is also attributed primarily to dilution, although other factors include organ sequestration, mechanical disruption, and adhesion to the circuit.68 A relatively greater reduction will occur in smaller children, although newer combined filter and oxygenators should considerably reduce the circuit volume and, hence, the degree of dilution. Significant platelet dysfunction also occurs.46,69 Low platelet counts have been strongly associated with increased bleeding.10,31,70
Further activation of platelets and denaturing of plasma proteins will occur at interfaces between blood and air, or on contact with extravascular tissue. CPB pump suction and spilling of blood into the pericardium will further contribute to coagulopathy. Greater coagulopathy is also seen during prolonged bypass and when deep hypothermic circulatory arrest is employed.71
The major mechanism for activation of the coagulation cascade during CPB is thought to be the “extrinsic” TF pathway, which is activated as a result of surgical trauma and inflammation.42,72,73 Inflammatory mediators such as tumor necrosis factor (TNF) and interleukin-1 induce expression of TF on endothelial cells and monocytes. The “intrinsic” coagulation system is also activated when factor XII is adsorbed onto the surface of the CPB circuit, causing activation of complement, neutrophils, and the fibrinolytic system via kallikrein.42 Although the intrinsic system has little role in initiating coagulation, activations of kinins will lead to increased fibrinolysis and inflammation.15
The blood loss in the first 24 hours after cardiac surgery can vary between 15 and 110 mL/kg, although the risk of excessive blood loss is greatest among those weighing less than 8 kg, younger than 1 year of age, and children undergoing complex surgery.10 The common preoperative and intraoperative risk factors for excessive bleeding are summarized in Table 18-2.
| Preoperative | Intraoperative |
|---|---|
| Age <1 year or weight <8 kg | Individual surgeon |
| High hematocrit | Complex surgery |
| Congestive heart failure | Low platelet count during CPB |
| Repeat sternotomy | Prolonged CPB |
| Congenital and preoperative acquired coagulopathy | Duration of hypothermia on CPB |
| Cyanotic congenital heart disease | Deep hypothermic cardiac arrest |
CPB, Cardiopulmonary bypass.
Williams GD, Bratton SL, Ramamoorthy C. Factors associated with blood loss and blood product transfusions: a multivariate analysis in children after open-heart surgery. Anesth Analg 1999;89:57-64.
Blood loss and transfusion requirements in pediatric cardiac surgery vary inversely with age; neonates bleed more and receive more products per kilogram than any other age group.74 There are some correlations between coagulation tests before and during CPB and postoperative blood component transfusion requirements, although the sensitivity and specificity of the tests are not sufficient to justify routine coagulation tests while on CPB. Of all the tests, a platelet count of less than 108,000/mm3 while on CPB yielded the greatest sensitivity (83%) and specificity (58%) for predicting excessive blood loss.70
Medications Used for Hemostasis
The pathophysiology of coagulopathies and bleeding in children during surgery are complex, and the etiology is multifactorial (see Table 18-1). No single blood component or drug treatment can reverse the abnormal clotting profile. Initially an attempt should be made to identify causes of bleeding that can be remedied by surgical interventions. The anesthesiologist should attempt to ensure adequate reversal of heparin and restore normal physiologic variables, such as body temperature, serum [Ca2+], and acid-base balance. In the postoperative period, platelets, and other blood products, such as fresh frozen plasma (FFP) and cryoprecipitate, remain the mainstay of treatment for excessive bleeding.75 Blood products (see also Chapter 10) are briefly discussed here, primarily within the context of pediatric heart surgery.
Blood Products
Frequently it is necessary to administer blood products on a largely empirical basis; laboratory tests describe only parts of the coagulation process32 and are practically too slow to direct the anesthesiologist in real time as to the requirement for blood components. The TEG is a dynamic whole-blood test that is frequently used as a near-patient test to assess clot elasticity properties and, hence, more precisely delineate the bleeding and homeostasis profile. The role of TEG during pediatric heart surgery has been recently reviewed.76 The use of treatment algorithms based on TEG can limit transfusion of blood products in adults77–81 and children.82 In children, it may not be appropriate to use protocols originally designed for adults. An alternative approach has recently been proposed.76
