Clinical findings, nonmicrobiologic laboratory tests, and chest radiography are not useful for differentiating M. pneumoniae pneumonia from other types of community-acquired pneumonia. In addition, since M. pneumoniae lacks a cell wall, it is not visible on Gram’s stain. Although of historical interest, the measurement of cold agglutinin titers is no longer recommended for the diagnosis of M. pneumoniae infection because the findings are nonspecific and assays specific for M. pneumoniae are now available.
Acute M. pneumoniae infection can be diagnosed by polymerase chain reaction (PCR) detection of the organism in respiratory tract secretions or by isolation of the organism in culture (Table 212-1). Oropharyngeal, nasopharyngeal, and pulmonary specimens are all acceptable for diagnosing M. pneumoniae pneumonia. Other bodily fluids, such cerebrospinal fluid, are acceptable for extrapulmonary infection. M. pneumoniae culture (which requires special media) is not recommended for routine diagnosis because the organism may take weeks to grow and is often difficult to isolate from clinical specimens. In contrast, PCR allows rapid, specific diagnosis earlier in the course of clinical illness.
|
DIAGNOSTIC TESTS FOR RESPIRATORY MYCOPLASMA PNEUMONIAE INFECTIONa |
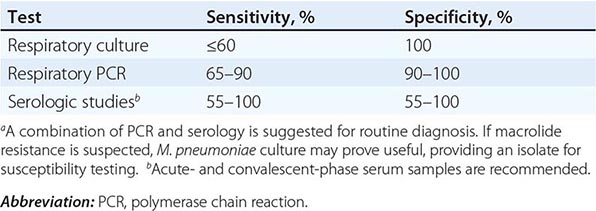
The diagnosis can also be established by serologic tests for IgM and IgG antibodies to M. pneumoniae in paired (acute- and convalescent-phase) serum samples; enzyme-linked immunoassay is the recommended serologic method. An acute-phase sample alone is not adequate for diagnosis, as antibodies to M. pneumoniae may not develop until 2 weeks into the illness; therefore, it is important to test paired samples. In addition, IgM antibody to M. pneumoniae can persist for up to 1 year after acute infection. Thus its presence may indicate recent rather than acute infection.
The combination of PCR of respiratory tract secretions and serologic testing constitutes the most sensitive and rapid approach to the diagnosis of M. pneumoniae infection.
|
TREATMENT |
MYCOPLASMA PNEUMONIAE INFECTIONS |
Although in the majority of untreated cases symptoms resolve within 2–3 weeks without significant associated morbidity, M. pneumoniae pneumonia can be a serious illness that responds to appropriate antimicrobial therapy (Table 212-2). Randomized, double-blind, placebo-controlled trials in adults have demonstrated that antimicrobial treatment significantly decreases the duration of fever, cough, malaise, hospitalization, and radiologic abnormalities in M. pneumoniae pneumonia. Treatment options for acute M. pneumoniae infection include macrolides (e.g., oral azithromycin, 500 mg on day 1, then 250 mg/d on days 2–5), tetracyclines (e.g., oral doxycycline, 100 mg twice daily for 10–14 days), and respiratory fluoroquinolones. However, ciprofloxacin and ofloxacin are not recommended because of their high minimal inhibitory concentrations against M. pneumoniae isolates and their poor performance in experimental studies. A 10- to 14-day course of quinolone therapy appears adequate.
|
ANTIMICROBIAL AGENTS OF CHOICE FOR MYCOPLASMA INFECTIONSa |
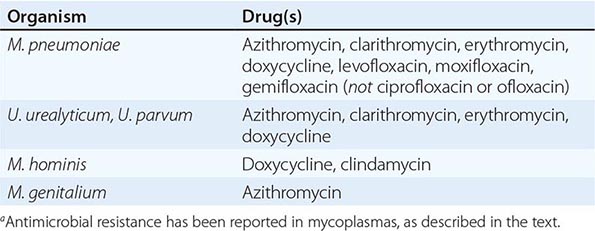
![]() In Japan and China, very high levels (up to ≥90%) of M. pneumoniae resistance to macrolides have been reported. In Europe and to a lesser degree in the United States, macrolide-resistant M. pneumoniae is emerging. In investigated outbreaks of respiratory illness due to M. pneumoniae in the United States, macrolide resistance has been reported in 8–27% of isolates. Clinical studies have demonstrated that, when treated with macrolides, patients with community-acquired pneumonia due to macrolide-resistant M. pneumoniae experience a significantly longer duration of symptoms than do patients infected with macrolide-sensitive organisms; thus macrolide resistance in M. pneumoniae does appear to have clinical significance. If macrolide resistance is prominent in a particular geographic locale or is suspected, then a nonmacrolide antibiotic should be considered for treatment; in addition, culture of M. pneumoniae may prove useful in these instances, providing an isolate for susceptibility testing.
In Japan and China, very high levels (up to ≥90%) of M. pneumoniae resistance to macrolides have been reported. In Europe and to a lesser degree in the United States, macrolide-resistant M. pneumoniae is emerging. In investigated outbreaks of respiratory illness due to M. pneumoniae in the United States, macrolide resistance has been reported in 8–27% of isolates. Clinical studies have demonstrated that, when treated with macrolides, patients with community-acquired pneumonia due to macrolide-resistant M. pneumoniae experience a significantly longer duration of symptoms than do patients infected with macrolide-sensitive organisms; thus macrolide resistance in M. pneumoniae does appear to have clinical significance. If macrolide resistance is prominent in a particular geographic locale or is suspected, then a nonmacrolide antibiotic should be considered for treatment; in addition, culture of M. pneumoniae may prove useful in these instances, providing an isolate for susceptibility testing.
Clinical observations and experimental data suggest that the addition of glucocorticoids to an antibiotic regimen may be of value for the treatment of severe or refractory M. pneumoniae pneumonia. However, relevant clinical experience is limited. Even though appropriate antibiotic therapy significantly reduces the duration of respiratory illness, it does not appear to shorten the duration of detection of M. pneumoniae by culture or PCR; therefore, a test of cure or eradication is not suggested.
The roles of antimicrobial drugs, glucocorticoids, and IV immunoglobulin in the treatment of neurologic disease due to M. pneumoniae remain unknown.
UROGENITAL MYCOPLASMAS (SEE ALSO Chap. 163)
EPIDEMIOLOGY
M. hominis, M. genitalium, U. urealyticum, and U. parvum can cause urogenital tract disease. The significance of isolation of these organisms in a variety of other syndromes is unknown and in some cases is being investigated. M. fermentans has not been shown convincingly to cause human disease.
While urogenital mycoplasmas may be transmitted to a fetus during passage through a colonized birth canal, sexual contact is the major mode of transmission, and the risk of colonization increases dramatically with increasing numbers of sexual partners. In asymptomatic women, these mycoplasmas may be found throughout the lower urogenital tract. The vagina yields the largest number of organisms; next most densely colonized are the periurethral area and the cervix. Ureaplasmas are isolated less often from urine than from the cervix, but M. hominis is found with approximately the same frequency at these two sites. Ureaplasmas are isolated from the vagina of 40–80% of sexually active, asymptomatic women and M. hominis from 21–70%. The two microorganisms are found concurrently in 31–60% of women. In men, colonization with each organism is less prevalent. Mycoplasmas have been isolated from urine, semen, and the distal urethra of asymptomatic men.
CLINICAL MANIFESTATIONS
Urethritis, Pyelonephritis, and Urinary Calculi In many episodes of Chlamydia-negative nongonococcal urethritis, ureaplasmas may be the causative agent. These organisms may also cause chronic voiding symptoms in women. The common presence of ureaplasmas in the urethra of asymptomatic men suggests either that only certain serovars are pathogenic or that predisposing factors, such as lack of immunity, must exist in persons who develop symptomatic infection. Alternatively, disease may develop only upon initial exposure to ureaplasmas. Ureaplasmas have been implicated in epididymitis. M. genitalium also appears to cause urethritis. M. genitalium and ureaplasmas do not have a known role in prostatitis. M. hominis does not appear to play a primary etiologic role in urethritis, epididymitis, or prostatitis.
Evidence suggests that M. hominis causes up to 5% of cases of acute pyelonephritis. Ureaplasmas have not been associated with this disease.
Ureaplasmas play a limited role in the production of urinary calculi. The frequency with which ureaplasmas reach the kidney, the predisposing factors that allow them to do so, and the relative frequency of urinary tract calculi induced by this organism (compared with other organisms) are not known.
Pelvic Inflammatory Disease M. hominis can cause pelvic inflammatory disease. In most episodes, M. hominis occurs as part of a polymicrobial infection, but the organism may play an independent role in a limited number of cases. Some data also support an association of M. genitalium with pelvic inflammatory disease. Ureaplasmas are not thought to cause pelvic inflammatory disease.
Postpartum and Postabortal Infection Studies implicate M. hominis as the primary pathogen in ∼5–10% of women who have postpartum or postabortal fever; ureaplasmas have been implicated to a lesser degree. These infections are generally self-limited; however, if symptoms persist, specific antimicrobial therapy should be given. Ureaplasmas also appear to play a role in occasional postcesarean wound infections.
Non-urogenital Infection In rare instances, M. hominis causes non-urogenital infections, such as brain abscess, wound infection, poststernotomy mediastinitis, endocarditis, and neonatal meningitis. These infections are most common among immunocompromised and hypogammaglobulinemic patients. Ureaplasmas and M. hominis can cause septic arthritis in immunodeficient patients. Ureaplasmas probably cause neonatal pneumonitis; their significant role in the development of bronchopulmonary dysplasia—the chronic lung disease of premature infants—has been documented in a number of studies. It is unclear whether ureaplasmas and M. hominis cause infertility, spontaneous abortion, premature labor, low birth weight, or chorioamnionitis.
DIAGNOSIS
Culture and PCR are both appropriate methods for the isolation of urogenital mycoplasmas. Culture of these organisms, however, requires special techniques and media that generally are available only at larger medical centers and reference laboratories. Serologic testing is not recommended for the clinical diagnosis of urogenital Mycoplasma infections.
|
TREATMENT |
UROGENITAL MYCOPLASMA INFECTIONS |
Because colonization with urogenital mycoplasmas is common, it appears at present that their isolation from the urogenital tract in the absence of disease generally does not warrant treatment. Macrolides and doxycycline are considered the antimicrobial agents of choice for Ureaplasma infections (Table 212-2). Ureaplasma resistance to macrolides, doxycycline, quinolones, and chloramphenicol has been reported. M. hominis is resistant to macrolides. Doxycycline is generally the drug of choice for M. hominis infections, although resistance has been reported. Clindamycin is generally active against M. hominis. Quinolones are active in vitro against M. hominis. For M. genitalium, the agent of choice appears to be azithromycin; treatment failures have been reported with other macrolides as well as with quinolones.
213 |
Chlamydial Infections |
Chlamydiae are obligate intracellular bacteria that cause a wide variety of diseases in humans and animals.
ETIOLOGIC AGENTS
The chlamydiae were originally classified as four species in the genus Chlamydia: C. trachomatis, C. pneumoniae, C. psittaci, and C. pecorum (the last species being found in ruminants). The C. psittaci group has been separated into three species: C. psittaci, C. felis, and C. abortus. The mouse pneumonitis strain (MoPn) is now classified as C. muridarum, and the guinea pig inclusion conjunctivitis strain (GPIC) is now designated C. caviae.
C. trachomatis is divided into two biovars: trachoma and LGV (lymphogranuloma venereum). The trachoma biovar causes two major types of disease in humans: ocular trachoma, the leading infectious cause of preventable blindness in the developing world; and urogenital infections, which are sexually or neonatally transmitted. The 18 serovars of C. trachomatis fall into three groups: the trachoma serovars A, B, Ba, and C; the oculogenital serovars D–K; and the LGV serovars L1–L3. Serovars can be distinguished by serologic typing with monoclonal antibodies or by molecular gene typing. However, serovar identification usually is not important clinically since the antibiotic susceptibility pattern is the same for all three groups. The one exception applies when LGV is suspected on clinical grounds; in this situation, serovar determination is important because a longer treatment duration is required for LGV strains.
BIOLOGY, GROWTH CYCLE, AND PATHOGENESIS
BIOLOGY
During their intracellular growth, chlamydiae produce characteristic intracytoplasmic inclusions that can be visualized by direct fluorescent antibody (DFA) or Giemsa staining of infected clinical material, such as conjunctival scrapings or cervical or urethral epithelial cells. Chlamydiae are nonmotile, gram-negative, obligate intracellular bacteria that replicate within the cytoplasm of host cells, forming the characteristic membrane-bound inclusions that are the basis for some diagnostic tests. Originally considered to be large viruses, chlamydiae differ from viruses in possessing RNA and DNA as well as a cell wall that is quite similar in structure to the cell wall of typical gram-negative bacteria. However, chlamydiae lack peptidoglycan; their structural integrity depends on disulfide binding of outer-membrane proteins.
GROWTH CYCLE
Among the defining characteristics of chlamydiae is a unique growth cycle that involves alternation between two highly specialized morphologic forms (Figs. 213-1 and 213-2): the elementary body (EB), which is the infectious form and is specifically adapted for extracellular survival, and the metabolically active and replicating reticulate body (RB), which is not infectious, is adapted for an intracellular environment, and does not survive well outside the host cell. The biphasic growth cycle begins with attachment of the EB (diameter, 0.25–0.35 μm) at specific sites on the surface of the host cell. The EB enters the cell through a process similar to receptor-mediated endocytosis and resides in an inclusion, where the entire growth cycle is completed. The chlamydiae prevent phagosome-lysosome fusion. The inclusion membrane is modified by insertion of chlamydial antigens. Once the EB has entered the cell, it reorganizes into an RB, which is larger (0.5–1 μm) and contains more RNA. After ~8 h, the RB starts to divide by binary fission. The intracytoplasmic, membrane-bound inclusion body containing the RBs increases in size as the RBs multiply. Approximately 18–24 h after infection of the cell, these RBs begin to become EBs by a reorganization or condensation process that is poorly understood. After rupture of the inclusion body, the EBs are released to initiate another cycle of infection.
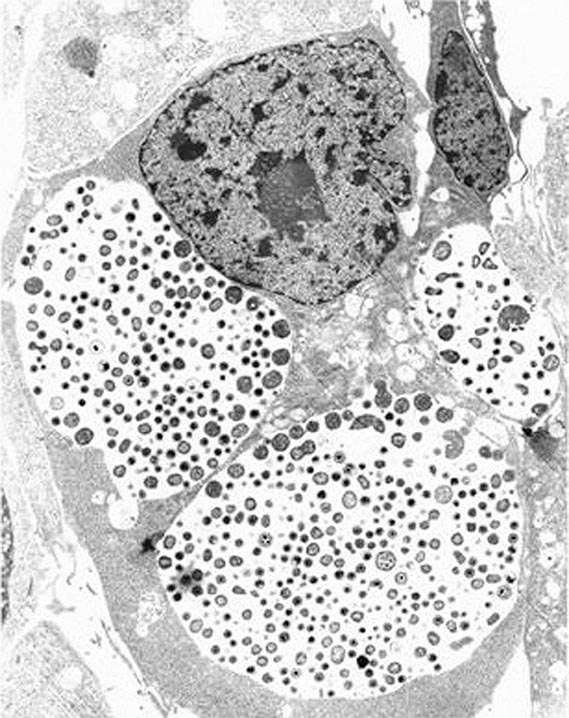
FIGURE 213-1 Chlamydial intracellular inclusions filled with smaller dense elementary bodies and larger reticulate bodies. (Reprinted with permission from WE Stamm: Chlamydial infections, in Harrison’s Principles of Internal Medicine, 17th ed, AS Fauci et al [eds]. New York, McGraw-Hill, 2008, p 1070.)
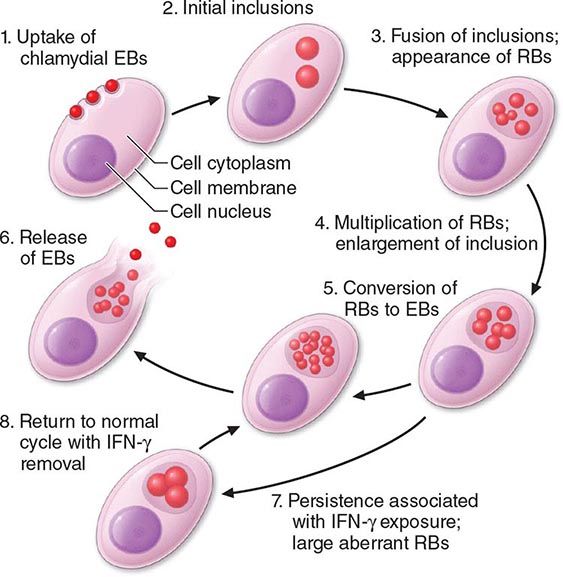
FIGURE 213-2 Chlamydial life cycle. EBs, elementary bodies; RBs, reticulate bodies; IFN-γ, interferon γ. (Reprinted with permission from WE Stamm: Chlamydial infections, in Harrison’s Principles of Internal Medicine, 17th ed, AS Fauci et al [eds]. New York, McGraw-Hill, 2008, p 1071.)
Chlamydiae are susceptible to many broad-spectrum antibiotics and possess a number of enzymes, but they have a very restricted metabolic capacity. None of these metabolic reactions results in the production of energy. Chlamydiae have thus been considered to be energy parasites that use the ATP produced by the host cell for their own metabolic functions. Many aspects of chlamydial molecular biology are not well understood, but the sequencing of several chlamydial genomes and new proteomics research have provided researchers with many relevant tools for elucidating the biology of the life cycle.
PATHOGENESIS
Genital infections are mostly caused by C. trachomatis serovars D–K, with serovars D, E, and F involved most often. Molecular typing of the major outer-membrane protein gene (omp1) from which serovar differences arise has been used to demonstrate that polymorphisms can occur in isolates from patients who are exposed frequently to multiple infections, while less variation is observed in isolates from less sexually active populations. Polymorphisms in the major outer-membrane protein may provide antigenic variation, and the different forms allow persistence in the community because immunity to one is not protective against the others.
The trachoma biovar is essentially a parasite of squamocolumnar epithelial cells; the LGV biovar is more invasive and involves lymphoid cells. As is typical of chlamydiae, C. trachomatis strains are capable of causing chronic, clinically inapparent, asymptomatic infections. Because the duration of the chlamydial growth cycle is ~48–72 h, the incubation period of sexually transmitted chlamydial infections is relatively long—generally 1–3 weeks. C. trachomatis causes cell death as a result of its replicative cycle and can induce cell damage whenever it persists. However, few toxic effects are demonstrated, and cell death because of chlamydial replication is not sufficient to account for disease manifestations, the majority of which are due to immunopathologic mechanisms or nonspecific host responses to the organism or its byproducts.
![]() In recent years, the entire genomes of various chlamydial species have been sequenced, the field of proteomics has become established, host innate immunity has been more precisely delineated, and innovative host cell–chlamydial interaction studies have been conducted. As a result, many insights have been gained into how chlamydiae adapt and replicate in their intracellular environment and produce disease. These insights into pathogenesis include information on the regulation of gene expression, protein localization, the type III secretion system, the roles of CD4+ and CD8+ T lymphocytes in the host response, and T lymphocyte trafficking.
In recent years, the entire genomes of various chlamydial species have been sequenced, the field of proteomics has become established, host innate immunity has been more precisely delineated, and innovative host cell–chlamydial interaction studies have been conducted. As a result, many insights have been gained into how chlamydiae adapt and replicate in their intracellular environment and produce disease. These insights into pathogenesis include information on the regulation of gene expression, protein localization, the type III secretion system, the roles of CD4+ and CD8+ T lymphocytes in the host response, and T lymphocyte trafficking.
The chlamydial heat-shock protein, which shares antigenic epitopes with similar proteins of other bacteria and with human heat-shock protein, may sensitize the host, and repeated infections may cause host cell damage. Persistent or recurrent chlamydial infections are associated with fibrosis, scarring, and complications following simple epithelial infections. A common endpoint of these late consequences is scarring of mucous membranes. Genital complications can lead to pelvic inflammatory disease (PID) and its late consequences of infertility, ectopic pregnancy, and chronic pelvic pain, while ocular infections may lead to blinding trachoma. High levels of antibody to human heat-shock protein have been associated with tubal factor infertility and ectopic pregnancy. Without adequate therapy, chlamydial infections may persist for several years, although symptoms—if present—usually abate.
The pathogenic mechanisms of C. pneumoniae have yet to be completely elucidated. The same is true for C. psittaci, except that this agent infects cells very efficiently and causes disease that may reflect direct cytopathic effects.
CHLAMYDIA TRACHOMATIS INFECTIONS
GENITAL INFECTIONS
Spectrum Although chlamydiae cause a number of human diseases, localized lower genital tract infections caused by C. trachomatis and the sequelae of such infections are the most important in terms of medical and economic impact. Oculogenital infections due to C. trachomatis serovars D–K are transmitted during sexual contact or from mother to baby during childbirth and are associated with many syndromes, including cervicitis, salpingitis, acute urethral syndrome, endometritis, ectopic pregnancy, infertility, and PID in female patients; urethritis, proctitis, and epididymitis in male patients; and conjunctivitis and pneumonia in infants. Women bear the greatest burden of morbidity because of the serious sequelae of these infections. Untreated infections lead to PID, and multiple episodes of PID can lead to tubal factor infertility and chronic pelvic pain. Studies estimate that up to 80–90% of women and >50% of men with C. trachomatis genital infections lack symptoms; other patients have very mild symptoms. Thus a large reservoir of infected persons continues to transmit infection to sexual partners.
As their designations reflect, the LGV serovars (L1, L2, and L3) cause LGV, an invasive sexually transmitted disease (STD) characterized by acute lymphadenitis with bubo formation and/or acute hemorrhagic proctitis (see “Lymphogranuloma Venereum,” below).
![]() Epidemiology C. trachomatis genital infections are global in distribution. The World Health Organization (WHO) estimated in 2008 that >106.4 million cases occur annually worldwide. This figure makes chlamydial infection the most prevalent sexually transmitted bacterial infection in the world. The associated morbidity is substantial, and economic costs are high.
Epidemiology C. trachomatis genital infections are global in distribution. The World Health Organization (WHO) estimated in 2008 that >106.4 million cases occur annually worldwide. This figure makes chlamydial infection the most prevalent sexually transmitted bacterial infection in the world. The associated morbidity is substantial, and economic costs are high.
In the United States, chlamydial infections are the most commonly reported of all infectious diseases. In 2012, 1.3 million cases were reported to the U.S. Centers for Disease Control and Prevention (CDC); however, the CDC estimates that 2–3 million new cases occur per year, with substantial underreporting due to lack of screening in some populations. Rates of infection have increased every year; higher rates among women than among men reflect the focus on expansion of screening programs for women during the past 20 years, the use of increasingly sensitive diagnostic tests, an increased emphasis on case reporting, and improvements in the information systems used for reporting. The CDC and other professional organizations recommend annual screening of all sexually active women ≤25 years of age as well as rescreening of previously infected individuals at 3 months. Young women have the highest infection rates; in 2012, the figures were 3416.5 and 3722.5 cases per 100,000 population at 15–19 and 20–24 years of age, respectively. Age-specific rates among men, while much lower than those among women, were highest in the 20- to 24-year-old age group, at 1343.3 cases per 100,000. In 2012, rates increased for all racial and ethnic groups, with the highest rates among African Americans. For example, the rate of chlamydial infection among African-American girls 15–19 years of age was 7507.1 cases per 100,000—almost six times the rate among Caucasian girls in the same age group (1301.5/100,000). The rate among African-American women 20–24 years old was 4.8 times the rate among Caucasian women in the same age group. Similar racial disparities in reported rates of chlamydial infection exist among men. For boys 15–19 years of age, the rate among African Americans was 11.1 times the rate among Caucasians. The rate among Native Americans/Alaska Natives was more than four times the rate among Caucasians (648.3), and the rate among Latinos (383.6) was two times higher than that among Caucasians. These disparities are important reflections of health inequities in the United States.
The above statistics are based on case reporting. Studies based on screening surveys estimate that the U.S. prevalence of C. trachomatis cervical infection is 5% among asymptomatic female college students and prenatal patients, >10% among women seen in family planning clinics, and >20% among women seen in STD clinics. The prevalence of genital C. trachomatis infections varies substantially by geographic locale, with the highest rates in the southeastern United States. However, asymptomatic infections have been detected in >8–10% of young female military recruits from all parts of the country. The prevalence of C. trachomatis in the cervix of pregnant women is 5–10 times higher than that of Neisseria gonorrhoeae. The prevalence of genital infection with either agent is highest among women who are between the ages of 18 and 24, single, and non-Caucasian (e.g., African-American, Latina, Asian, Pacific Islander). Infections recur frequently in these same risk groups and are often acquired from untreated sexual partners. The use of oral contraception and the presence of cervical ectopy also confer an increased risk. The proportion of infections that are asymptomatic appears to be higher for C. trachomatis than for N. gonorrhoeae, and symptomatic C. trachomatis infections are clinically less severe. Mild or asymptomatic C. trachomatis infections of the fallopian tubes nonetheless cause ongoing tubal damage and infertility. The costs of C. trachomatis infections and their complications to the U.S. health care system have recently been estimated to exceed $516.7 million annually.
Clinical Manifestations • NONGONOCOCCAL AND POSTGONOCOCCAL URETHRITIS C. trachomatis is the most common cause of nongonococcal urethritis (NGU) and postgonococcal urethritis (PGU). The designation PGU refers to NGU developing in men 2–3 weeks after treatment of gonococcal urethritis with single doses of agents such as penicillin or cephalosporins, which lack antimicrobial activity against chlamydiae. Since current treatment regimens for gonorrhea have evolved and now include combination therapy with tetracycline, doxycycline, or azithromycin—all of which are effective against concomitant chlamydial infection—both the incidence of PGU and the causative role of C. trachomatis in this syndrome have declined.
In the United States, most of the estimated 2 million cases of acute urethritis are NGU, and C. trachomatis is implicated in 30–50% of these cases. The cause of most of the remaining cases of NGU is uncertain, but recent evidence suggests that Ureaplasma urealyticum, Mycoplasma genitalium, Trichomonas vaginalis, and herpes simplex virus (HSV) cause some cases. The rate of involvement of C. trachomatis in urethral infection ranges from 3–7% among asymptomatic men to 15–20% among symptomatic men attending STD clinics. A multisite study of men in Baltimore, Seattle, Denver, and San Francisco reported an overall chlamydial prevalence of 7% in urine samples assessed by nucleic acid amplification tests (NAATs). As in women, infection in men is age related, with young age as the greatest risk factor for chlamydial urethritis. The prevalence among men is highest at 20–24 years of age. In STD clinics, urethritis is usually less prevalent among men who have sex with men (MSM) than among heterosexual men and is almost always much more common among African-American men than among Caucasian men. One study reported prevalences of 19% and 9% among nonwhite and white heterosexual men, respectively.
NGU is diagnosed by documentation of a leukocytic urethral exudate and by exclusion of gonorrhea by Gram’s staining or culture. C. trachomatis urethritis is generally less severe than gonococcal urethritis, although in any individual patient these two forms of urethritis cannot reliably be differentiated solely on clinical grounds. Symptoms include urethral discharge (often whitish and mucoid rather than frankly purulent), dysuria, and urethral itching. Physical examination may reveal meatal erythema and tenderness as well as a urethral exudate that is often demonstrable only by stripping of the urethra.
At least one-third of male patients with C. trachomatis urethral infection have no evident signs or symptoms of urethritis. The availability of NAATs for first-void urine specimens has facilitated broader-based testing for asymptomatic infection in male patients. As a result, asymptomatic chlamydial urethritis has been demonstrated in 5–10% of sexually active male adolescents screened at school-based clinics or community centers. Such patients generally have pyuria (≥15 leukocytes per 400× microscopic field in the sediment of first-void urine), a positive leukocyte esterase test, or an increased number of leukocytes on a Gram-stained smear prepared from a urogenital swab inserted 1–2 cm into the anterior urethra. To differentiate between true urethritis and functional symptoms in symptomatic patients or to make a presumptive diagnosis of C. trachomatis infection in high-risk but asymptomatic men (e.g., male patients in STD clinics, sex partners of women with nongonococcal salpingitis or mucopurulent cervicitis, fathers of children with inclusion conjunctivitis), the examination of an endourethral specimen for increased leukocytes is useful if specific diagnostic tests for chlamydiae are not available. Alternatively, urethritis can be assayed noninvasively by examination of a first-void urine sample for pyuria, either by microscopy or by the leukocyte esterase test. Urine (or a urethral swab) can also be tested directly for chlamydiae by DNA amplification methods, as described below (see “Detection Methods”).
EPIDIDYMITIS Chlamydial urethritis may be followed by acute epididymitis, but this condition is rare, generally occurring in sexually active patients <35 years of age; in older men, epididymitis is usually associated with gram-negative bacterial infection and/or instrumentation procedures. It is estimated that 50–70% of cases of acute epididymitis are caused by C. trachomatis. The condition usually presents as unilateral scrotal pain with tenderness, swelling, and fever in a young man, often occurring in association with chlamydial urethritis. The illness may be mild enough to treat with oral antibiotics on an outpatient basis or severe enough to require hospitalization and parenteral therapy. Testicular torsion should be excluded promptly by radionuclide scan, Doppler flow study, or surgical exploration in a teenager or young adult who presents with acute unilateral testicular pain without urethritis. The possibility of testicular tumor or chronic infection (e.g., tuberculosis) should be excluded when a patient with unilateral intrascrotal pain and swelling does not respond to appropriate anti-microbial therapy.
REACTIVE ARTHRITIS Reactive arthritis consists of conjunctivitis, urethritis (or, in female patients, cervicitis), arthritis, and characteristic mucocutaneous lesions. It may develop in 1–2% of cases of NGU and is thought to be the most common type of peripheral inflammatory arthritis in young men. C. trachomatis has been recovered from the urethra of 16–44% of patients with reactive arthritis and from 69% of men who have signs of urogenital inflammation at the time of examination. Antibodies to C. trachomatis have also been detected in 46–67% of patients with reactive arthritis, and Chlamydia-specific cell-mediated immunity has been documented in 72%. In addition, C. trachomatis has been isolated from synovial biopsy samples from 15 of 29 patients in a number of small series and from a smaller proportion of synovial fluid specimens. Chlamydial nucleic acids have been identified in synovial membranes and chlamydial EBs in joint fluid. The pathogenesis of reactive arthritis is unclear, but this condition probably represents an abnormal host response to a number of infectious agents, including those associated with bacterial gastroenteritis (e.g., Salmonella, Shigella, Yersinia, or Campylobacter), or to infection with C. trachomatis or N. gonorrhoeae. Since >80% of affected patients have the HLA-B27 phenotype and since other mucosal infections produce an identical syndrome, chlamydial infection is thought to initiate an aberrant hyperactive immune response that produces inflammation of the involved target organs in these genetically predisposed individuals. Evidence of exaggerated cell-mediated and humoral immune responses to chlamydial antigens in reactive arthritis supports this hypothesis. The finding of chlamydial EBs and DNA in joint fluid and synovial tissue from patients with reactive arthritis suggests that chlamydiae may actually spread from genital to joint tissues in these patients—perhaps in macrophages.
NGU is the initial manifestation of reactive arthritis in 80% of patients, typically occurring within 14 days after sexual exposure. The urethritis may be mild and may even go unnoticed by the patient. Similarly, gonococcal urethritis may precede reactive arthritis, but co-infection with an agent of NGU is difficult to rule out. The urethral discharge may be purulent or mucopurulent, and patients may or may not report dysuria. Accompanying prostatitis, usually asymptomatic, has been described. Arthritis usually begins ~4 weeks after the onset of urethritis but may develop sooner or, in a small percentage of cases, may actually precede urethritis. The knees are most frequently involved; next most commonly affected are the ankles and small joints of the feet. Sacroiliitis, either symmetrical or asymmetrical, is documented in two-thirds of patients. Mild bilateral conjunctivitis, iritis, keratitis, or uveitis is sometimes present but lasts for only a few days. Finally, dermatologic manifestations occur in up to 50% of patients. The initial lesions—usually papules with a central yellow spot—most often involve the soles and palms and, in ~25% of patients, eventually epithelialize and thicken to produce keratoderma blenorrhagicum. Circinate balanitis is usually painless and occurs in fewer than half of patients. The initial episode of reactive arthritis usually lasts 2–6 months.
PROCTITIS Primary anal or rectal infections with C. trachomatis have been described in women and MSM who practice anal intercourse. In these infections, rectal involvement is initially characterized by severe anorectal pain, a bloody mucopurulent discharge, and tenesmus. Oculogenital serovars D–K and LGV serovars L1, L2, and L3 have been found to cause proctitis. The LGV serovars are far more invasive and cause much more severely symptomatic disease, including severe ulcerative proctocolitis that can be clinically confused with HSV proctitis. Histologically, LGV proctitis may resemble Crohn’s disease in that giant cell formation and granulomas are detected. In the United States and Europe, cases of LGV proctitis occur almost exclusively in MSM, many of whom are positive for HIV infection.
The less invasive non-LGV serovars of C. trachomatis cause mild proctitis. Many infected individuals are asymptomatic, and in these cases infection is diagnosed only by routine culture or NAAT of rectal swabs. The number of fecal leukocytes is usually abnormal in both asymptomatic and symptomatic cases. Sigmoidoscopy may yield normal findings or may reveal mild inflammatory changes or small erosions or follicles in the lower 10 cm of the rectum. Histologic examination of rectal biopsies generally shows anal crypts and prominent follicles as well as neutrophilic infiltration of the lamina propria. Chlamydial proctitis is best diagnosed by isolation of C. trachomatis from the rectum and documentation of a response to appropriate therapy. NAATs are reportedly more sensitive than culture for diagnosis and are also specific.
MUCOPURULENT CERVICITIS Although many women with chlamydial infections of the cervix have no symptoms, almost half generally have local signs of infection on examination. Cervicitis is usually characterized by the presence of a mucopurulent discharge, with >20 neutrophils per microscopic field visible in strands of cervical mucus in a thinly smeared, gram-stained preparation of endocervical exudate. Hypertrophic ectopy of the cervix may also be evident as an edematous area near the cervical os that is congested and bleeds easily on minor trauma (e.g., when a specimen is collected with a swab). A Papanicolaou smear shows increased numbers of neutrophils as well as a characteristic pattern of mononuclear inflammatory cells including plasma cells, transformed lymphocytes, and histiocytes. Cervical biopsy shows a predominantly mononuclear cell infiltrate of the subepithelial stroma. Clinical experience and collaborative studies indicate that a cutoff of >30 polymorphonuclear neutrophils (PMNs) per 1000× field in a gram-stained smear of cervical mucus correlates best with chlamydial or gonococcal cervicitis.
Clinical recognition of chlamydial cervicitis depends on a high index of suspicion and careful cervical examination. No genital symptoms are specifically correlated with chlamydial cervical infection. The differential diagnosis of a mucopurulent discharge from the endocervical canal in a young, sexually active woman includes gonococcal endocervicitis, salpingitis, endometritis, and intrauterine contraceptive device–induced inflammation. Diagnosis of cervicitis is based on the presence of PMNs on a cervical swab as noted above; the presence of chlamydiae is confirmed by either culture or NAAT.
PELVIC INFLAMMATORY DISEASE Inflammation of sections of the fallopian tube is often referred to as salpingitis or PID. The proportion of acute salpingitis cases caused by C. trachomatis varies geographically and with the population studied. It has been estimated that C. trachomatis causes up to 50% of PID cases in the United States. PID occurs via ascending intraluminal spread of C. trachomatis or N. gonorrhoeae from the lower genital tract. Mucopurulent cervicitis is often followed by endometritis, endosalpingitis, and finally pelvic peritonitis. Evidence of mucopurulent cervicitis is often found in women with laparoscopically verified salpingitis. Similarly, endometritis, demonstrated by an endometrial biopsy showing plasma cell infiltration of the endometrial epithelium, is documented in most women with laparoscopy-verified chlamydial (or gonococcal) salpingitis. Chlamydial endometritis can also occur in the absence of clinical evidence of salpingitis. Histologic evidence of endometritis has been correlated with a syndrome consisting of vaginal bleeding, lower abdominal pain, and uterine tenderness in the absence of adnexal tenderness. Chlamydial salpingitis produces milder symptoms than gonococcal salpingitis and may be associated with less marked adnexal tenderness. Thus, mild adnexal or uterine tenderness in a sexually active woman with cervicitis suggests chlamydial PID.
Chronic untreated endometrial and tubal inflammation can result in tubal scarring, impaired tubal function, tubal occlusion, and infertility, even among women who report no prior treatment for PID. C. trachomatis has been implicated particularly often in “subclinical” PID on the basis of (1) a lack of history of PID among Chlamydia-seropositive women with tubal damage or (2) detection of chlamydial DNA or antigen among asymptomatic women with tubal infertility. These data suggest that the best method to prevent PID and its sequelae is surveillance and control of lower genital tract infections along with diagnosis and treatment of sex partners and prevention of reinfections. Promotion of early symptom recognition and health care presentation may reduce the frequency and severity of sequelae of PID.
PERIHEPATITIS The Fitz-Hugh–Curtis syndrome was originally described as a complication of gonococcal PID. However, studies over the past several decades have suggested that chlamydial infection is more commonly associated with perihepatitis than is N. gonorrhoeae. Perihepatitis should be suspected in young, sexually active women who develop right-upper-quadrant pain, fever, or nausea. Evidence of salpingitis may or may not be found on examination. Frequently, perihepatitis is strongly associated with extensive tubal scarring, adhesions, and inflammation observed at laparoscopy, and high titers of antibody to the 57-kDa chlamydial heat-shock protein have been documented. Culture and/or serologic evidence of C. trachomatis is found in three-fourths of women with this syndrome.
URETHRAL SYNDROME IN WOMEN In the absence of infection with uropathogens such as coliforms or Staphylococcus saprophyticus, C. trachomatis is the pathogen most commonly isolated from college women with dysuria, frequency, and pyuria. Screening studies can recover C. trachomatis from both the cervix and the urethra; in up to 25% of infected women, the organism is isolated only from the urethra. The urethral syndrome in women consists of dysuria and frequency in conjunction with chlamydial urethritis, pyuria, and no bacteriuria or urinary pathogens. Although symptoms of the urethral syndrome may develop in some women with chlamydial infection, the majority of women attending STD clinics for urethral chlamydial infection do not have dysuria or frequency. Even in women with chlamydial urethritis causing the acute urethral syndrome, signs of urethritis such as urethral discharge, meatal redness, and swelling are uncommon. However, mucopurulent cervicitis in a woman presenting with dysuria and frequency strongly suggests C. trachomatis urethritis. Other correlates of chlamydial urethral syndrome include a duration of dysuria of >7–10 days, lack of hematuria, and lack of suprapubic tenderness. Abnormal urethral Gram’s stains showing >10 PMNs per 1000× field in women with dysuria but without coliform bacteriuria support the diagnosis of chlamydial urethritis. Other possible diagnoses include gonococcal or trichomonal infection of the urethra.
INFECTION IN PREGNANCY AND THE NEONATAL PERIOD Infections during pregnancy can be transmitted to infants during delivery. Approximately 20–30% of infants exposed to C. trachomatis in the birth canal develop conjunctivitis, and 10–15% subsequently develop pneumonia. Consequently, all newborn infants receive ocular prophylaxis at birth to prevent ophthalmia neonatorum. Without treatment, conjunctivitis usually develops at 5–19 days of life and often results in a profuse mucopurulent discharge. Roughly half of infected infants develop clinical evidence of inclusion conjunctivitis. However, it is impossible to differentiate chlamydial conjunctivitis from other forms of neonatal conjunctivitis (e.g., that due to N. gonorrhoeae, Haemophilus influenzae, Streptococcus pneumoniae, or HSV) on clinical grounds; thus laboratory diagnosis is required. Inclusions within epithelial cells are often detected in Giemsa-stained conjunctival smears, but these smears are considerably less sensitive than cultures or NAATs for chlamydiae. Gram-stained smears may show gonococci or occasional small gram-negative coccobacilli in Haemophilus conjunctivitis, but smears should be accompanied by cultures or NAATs for these agents.
C. trachomatis has also been isolated frequently and persistently from the nasopharynx, rectum, and vagina of infected infants—occasionally for >1 year in the absence of treatment. In some cases, otitis media results from perinatally acquired chlamydial infection. Pneumonia may develop in infants from 2 weeks to 4 months of age. C. trachomatis is estimated to cause 20–30% of pneumonia cases in infants <6 months of age. Epidemiologic studies have linked chlamydial pulmonary infection in infants with increased occurrence of subacute lung disease (bronchitis, asthma, wheezing) in later childhood.
![]() LYMPHOGRANULOMA VENEREUM C. trachomatis serovars L1, L2, and L3 cause LGV, an invasive systemic STD. The peak incidence of LGV corresponds with the age of greatest sexual activity: the second and third decades of life. The worldwide incidence of LGV is falling, but the disease is still endemic and a major cause of morbidity in parts of Asia, Africa, South America, and the Caribbean. LGV is rare in industrialized countries; for more than a decade, the reported incidence in the United States has been only 0.1 case per 100,000 population. In the Bahamas, an apparent outbreak of LGV was described in association with a concurrent increase in heterosexual infection with HIV. Reports of outbreaks with the newly identified variant L2b in Europe, Australia, and the United States indicate that LGV is becoming more prevalent among MSM. These cases have usually presented as hemorrhagic proctocolitis in HIV-positive men. More widespread use of NAATs for identification of rectal infections may have enhanced case recognition.
LYMPHOGRANULOMA VENEREUM C. trachomatis serovars L1, L2, and L3 cause LGV, an invasive systemic STD. The peak incidence of LGV corresponds with the age of greatest sexual activity: the second and third decades of life. The worldwide incidence of LGV is falling, but the disease is still endemic and a major cause of morbidity in parts of Asia, Africa, South America, and the Caribbean. LGV is rare in industrialized countries; for more than a decade, the reported incidence in the United States has been only 0.1 case per 100,000 population. In the Bahamas, an apparent outbreak of LGV was described in association with a concurrent increase in heterosexual infection with HIV. Reports of outbreaks with the newly identified variant L2b in Europe, Australia, and the United States indicate that LGV is becoming more prevalent among MSM. These cases have usually presented as hemorrhagic proctocolitis in HIV-positive men. More widespread use of NAATs for identification of rectal infections may have enhanced case recognition.
LGV begins as a small painless papule that tends to ulcerate at the site of inoculation, often escaping attention. This primary lesion heals in a few days without scarring and is usually recognized as LGV only in retrospect. LGV strains of C. trachomatis have occasionally been recovered from genital ulcers and from the urethra of men and the endocervix of women who present with inguinal adenopathy; these areas may be the primary sites of infection in some cases. Proctitis is more common among people who practice receptive anal intercourse, and an elevated white blood cell count in anorectal smears may predict LGV in these patients. Ulcer formation may facilitate transmission of HIV infection and other sexually transmitted and blood-borne diseases.
As NAATs for C. trachomatis are being used more often, increasing numbers of cases of LGV proctitis are being recognized in MSM. Such patients present with anorectal pain and mucopurulent, bloody rectal discharge. Sigmoidoscopy reveals ulcerative proctitis or proctocolitis, with purulent exudate and mucosal bleeding. Histopathologic findings in the rectal mucosa include granulomas with giant cells, crypt abscesses, and extensive inflammation. These clinical, sigmoidoscopic, and histopathologic findings may closely resemble those of Crohn’s disease of the rectum.
The most common presenting picture in heterosexual men and women is the inguinal syndrome, which is characterized by painful inguinal lymphadenopathy beginning 2–6 weeks after presumed exposure; in rare instances, the onset comes after a few months. The inguinal adenopathy is unilateral in two-thirds of cases, and palpable enlargement of the iliac and femoral nodes is often evident on the same side as the enlarged inguinal nodes. The nodes are initially discrete, but progressive periadenitis results in a matted mass of nodes that becomes fluctuant and suppurative. The overlying skin becomes fixed, inflamed, and thin, and multiple draining fistulas finally develop. Extensive enlargement of chains of inguinal nodes above and below the inguinal ligament (“the sign of the groove”) is not specific and, although not uncommon, is documented in only a minority of cases. Spontaneous healing usually takes place after several months; inguinal scars or granulomatous masses of various sizes persist for life. Massive pelvic lymphadenopathy may lead to exploratory laparotomy.
Constitutional symptoms are common during the stage of regional lymphadenopathy and, in cases of proctitis, may include fever, chills, headache, meningismus, anorexia, myalgias, and arthralgias. Other systemic complications are infrequent but include arthritis with sterile effusion, aseptic meningitis, meningoencephalitis, conjunctivitis, hepatitis, and erythema nodosum (Fig. 25e-40). Complications of untreated anorectal infection include perirectal abscess; anal fistulas; and rectovaginal, rectovesical, and ischiorectal fistulas. Secondary bacterial infection probably contributes to these complications. Rectal stricture is a late complication of anorectal infection and usually develops 2–6 cm from the anal orifice—i.e., at a site within reach on digital rectal examination. A small percentage of cases of LGV in men present as chronic progressive infiltrative, ulcerative, or fistular lesions of the penis, urethra, or scrotum. Associated lymphatic obstruction may produce elephantiasis. When urethral stricture occurs, it usually involves the posterior urethra and causes incontinence or difficulty with urination.
Diagnosis • DETECTION METHODS Historically, chlamydiae were cultivated in the yolk sac of embryonated eggs. The organisms can be grown more easily in tissue culture, but cell culture—once considered the diagnostic gold standard—has been replaced by nonculture assays (Table 213-1). In general, culture for chlamydiae in clinical specimens is now performed only in specialized laboratories. The first nonculture assays, such as DFA staining of clinical material and enzyme immunoassay (EIA), have been replaced by NAATS, which are molecular tests that amplify the nucleic acids in clinical specimens. NAATS are currently recommended by the CDC as the diagnostic assays of choice; four or five NAAT assays approved by the U.S. Food and Drug Administration (FDA) are commercially available, some as high-throughput robotic platforms. Point-of-care diagnostic assays (including NAATs), by which patients can be treated before leaving the clinic, are of increasing interest and are becoming available.
|
DIAGNOSTIC TESTS FOR SEXUALLY TRANSMITTED AND PERINATAL CHLAMYDIA TRACHOMATIS INFECTION |
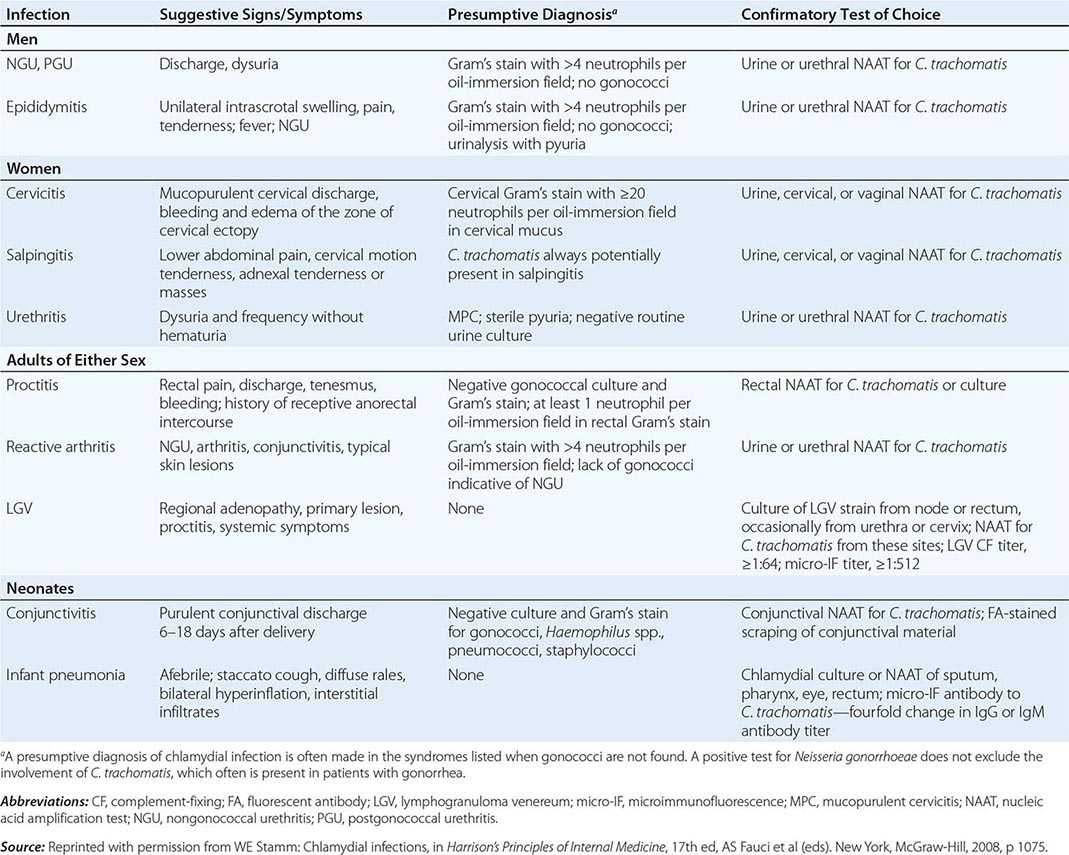
CHOICE OF SPECIMEN Cervical and urethral swabs have traditionally been used for the diagnosis of STDs in female and male patients, respectively. However, given the greatly increased sensitivity and specificity of NAATs, less invasive samples (e.g., urine for both sexes and vaginal swabs for women) can be used. For screening of asymptomatic women, the CDC now recommends that self-collected or clinician-collected vaginal swabs, which are slightly more sensitive than urine, be used. Urine screening tests are often used in outreach screening programs, however. For symptomatic women undergoing a pelvic examination, cervical swab samples are desirable because they have slightly higher chlamydial counts. For male patients, a urine specimen is the sample of choice, but self-collected penile-meatal swabs have been explored.
ALTERNATIVE SPECIMEN TYPES Ocular samples from babies and adults can be assessed by NAATs. However, since commercial NAATs for this purpose have not yet been approved by the FDA, laboratories must perform their own verification studies. Samples from rectal and pharyngeal sites have been used successfully to detect chlamydiae, but laboratories must verify test performance.
OTHER DIAGNOSTIC ISSUES Because NAATs detect nucleic acids instead of live organisms, they should be used with caution as test-of-cure assays. Residual nucleic acid from cells rendered noninfective by antibiotics may continue to yield a positive result in NAATs until as long as 3 weeks after therapy, when viable organisms have actually been eradicated. Therefore, clinicians should not use NAATs for test of cure until after 3 weeks. The CDC currently does not recommend a test of cure after treatment for infection with C. trachomatis. However, because incidence studies have demonstrated that previous chlamydial infection increases the probability of becoming reinfected, the CDC does recommend that previously infected individuals be rescreened 3 months after treatment.
SEROLOGY Serologic testing may be helpful in the diagnosis of LGV and neonatal pneumonia caused by C. trachomatis. The serologic test of choice is the microimmunofluorescence (MIF) test, in which high-titer purified EBs mixed with embryonated chicken yolk-sac material are affixed to a glass microscope slide to which dilutions of serum are applied. After incubation and washing, fluorescein-conjugated IgG or IgM antibody is applied. The test is read with an epifluorescence microscope, with the highest dilution of serum producing visible fluorescence designated as the titer. The MIF test is not widely available and is highly labor intensive. Although the complement fixation (CF) test also can be used, it employs only lipopolysaccharide (LPS) as the antigen and therefore identifies the pathogen only to the genus level. Single-point titers of >1:64 support a diagnosis of LGV, in which it is difficult to demonstrate rising antibody titers; i.e., paired serum samples are difficult to obtain since, by its very nature, the disease results in the patient’s being seen by the physician after the acute stage. Any antibody titer of >1:16 is considered significant evidence of exposure to chlamydiae. However, serologic testing is never recommended for diagnosis of uncomplicated genital infections of the cervix, urethra, and lower genital tract or for C. trachomatis screening of asymptomatic individuals.
|
TREATMENT |
C. TRACHOMATIS GENITAL INFECTIONS |
A 7-day course of tetracycline (500 mg four times daily), doxycycline (100 mg twice daily), erythromycin (500 mg four times daily), or a fluoroquinolone (ofloxacin, 300 mg twice daily; or levofloxacin, 500 mg/d) can be used for treatment of uncomplicated chlamydial infections. A single 1-g oral dose of azithromycin is as effective as a 7-day course of doxycycline for the treatment of uncomplicated genital C. trachomatis infections in adults. Azithromycin causes fewer adverse gastrointestinal reactions than do older macrolides such as erythromycin. The single-dose regimen of azithromycin has great appeal for the treatment of patients with uncomplicated chlamydial infection (especially those without symptoms and those with a likelihood of poor compliance) and of the sexual partners of infected patients. These advantages must be weighed against the considerably greater cost of azithromycin. Whenever possible, the single 1-g dose should be given as directly observed therapy. Although not approved by the FDA for use in pregnancy, this regimen appears to be safe and effective for this purpose. However, amoxicillin (500 mg three times daily for 7 days) also can be given to pregnant women. The fluoroquinolones are contraindicated in pregnancy. A 2-week course of treatment is recommended for complicated chlamydial infections (e.g., PID, epididymitis) and at least a 3-week course of doxycycline (100 mg orally twice daily) or erythromycin base (500 mg orally four times daily) for LGV. Failure of treatment with a tetracycline in genital infections usually indicates poor compliance or reinfection rather than involvement of a drug-resistant strain. To date, clinically significant drug resistance has not been observed in C. trachomatis.
Treatment or testing for chlamydiae should be considered among N. gonorrhoeae–infected patients because of the frequency of co-infection. Systemic treatment with erythromycin has been recommended for ophthalmia neonatorum and for C. trachomatis pneumonia in infants. For the treatment of adult inclusion conjunctivitis, a single 1-g dose of azithromycin is as effective as standard 10-day treatment with doxycycline. Recommended treatment regimens for both bubonic and anogenital LGV include tetracycline, doxycycline, or erythromycin for 21 days.
SEX PARTNERS
The continued high prevalence of chlamydial infections in most parts of the United States is due primarily to the failure to diagnose—and therefore treat—patients with symptomatic or asymptomatic infection and their sex partners. Urethral or cervical infection with C. trachomatis has been well documented in a high proportion of the sex partners of patients with NGU, epididymitis, reactive arthritis, salpingitis, and endocervicitis. If possible, confirmatory laboratory tests for chlamydiae should be undertaken in these individuals, but even those without positive tests or evidence of clinical disease who have recently been exposed to proven or possible chlamydial infection (e.g., NGU) should be offered therapy. A novel approach is partner-delivered therapy, in which infected patients receive treatment and are also provided with single-dose azithromycin to give to their sex partner(s).
NEONATES AND INFANTS
In neonates with conjunctivitis or infants with pneumonia, erythromycin ethylsuccinate or estolate can be given orally at a dosage of 50 mg/kg per day, preferably in four divided doses, for 2 weeks. Careful attention must be given to compliance with therapy—a frequent problem. Relapses of eye infection are common after topical treatment with erythromycin or tetracycline ophthalmic ointment and may also follow oral erythromycin therapy. Thus follow-up cultures should be performed after treatment. Both parents should be examined for C. trachomatis infection and, if diagnostic testing is not readily available, should be treated with doxycycline or azithromycin.
Prevention Since many chlamydial infections are asymptomatic, effective control and prevention must involve periodic screening of individuals at risk. Selective cost-effective screening criteria have been developed. Among women, young age (generally <25 years) is a critical risk factor for chlamydial infections in nearly all studies. Other risk factors include mucopurulent cervicitis; multiple, new, or symptomatic male sex partners; and lack of barrier contraceptive use. In some settings, screening based on young age may be as sensitive as criteria that incorporate behavioral and clinical measures. Another strategy is universal testing of all patients in high-prevalence clinic populations (e.g., STD clinics, juvenile detention facilities, and family planning clinics).
The effectiveness of selective screening in reducing the prevalence of chlamydial infection among women has been demonstrated in several studies. In the Pacific Northwest, where extensive screening began in family planning clinics in 1998 and in STD clinics in 1993, the prevalence declined from 10% in the 1980s to <5% in 2000. Similar trends have occurred in association with screening programs elsewhere. In addition, screening can effect a reduction in upper genital tract disease. In Seattle, women at a large health maintenance organization who were screened for chlamydial infection on a routine basis had a lower incidence of symptomatic PID than did women who received standard care and underwent more selective screening.
In settings with low to moderate prevalence, the prevalence at which selective screening becomes more cost-effective than universal screening must be defined. Most studies have concluded that universal screening is preferable in settings with a chlamydial prevalence of >3–7%. Depending on the criteria used, selective screening is likely to be more cost-effective when prevalence falls below 3%. Nearly all regions of the United States have now initiated screening programs, particularly in family planning and STD clinics. Along with single-dose therapy, the availability of highly sensitive and specific diagnostic NAATs using urine specimens and self-obtained vaginal swabs makes it feasible to mount an effective nationwide Chlamydia control program, with screening of high-risk individuals in traditional health-care settings and in novel outreach and community-based settings. The U.S. Preventive Services Task Force has given C. trachomatis screening a Grade A recommendation, which means that private insurance and Medicare will cover its cost under the Affordable Care Act.
TRACHOMA
![]() Epidemiology Trachoma—a sequela of ocular disease in developing countries—continues to be a leading cause of preventable infectious blindness worldwide. The WHO estimates that ~6 million people have been blinded by trachoma and that ~1.3 million people in developing countries still suffer from preventable blindness due to trachoma; certainly hundreds of millions live in trachoma-endemic areas. Foci of trachoma persist in Australia, the South Pacific, and Latin America. Serovars A, B, Ba, and C are isolated from patients with clinical trachoma in areas of endemicity in developing countries in Africa, the Middle East, Asia, and South America.
Epidemiology Trachoma—a sequela of ocular disease in developing countries—continues to be a leading cause of preventable infectious blindness worldwide. The WHO estimates that ~6 million people have been blinded by trachoma and that ~1.3 million people in developing countries still suffer from preventable blindness due to trachoma; certainly hundreds of millions live in trachoma-endemic areas. Foci of trachoma persist in Australia, the South Pacific, and Latin America. Serovars A, B, Ba, and C are isolated from patients with clinical trachoma in areas of endemicity in developing countries in Africa, the Middle East, Asia, and South America.
The trachoma-hyperendemic areas of the world are in northern and sub-Saharan Africa, the Middle East, drier regions of the Indian subcontinent, and Southeast Asia. In hyperendemic areas, the prevalence of trachoma is essentially 100% by the second or third year of life. Active disease is most common among young children, who are the reservoir for trachoma. By adulthood, active infection is infrequent but sequelae result in blindness. In such areas, trachoma constitutes the major cause of blindness.
Trachoma is transmitted through contact with discharges from the eyes of infected patients. Transmission is most common under poor hygienic conditions and most often takes place between family members or between families with shared facilities. Flies can also transfer the mucopurulent ocular discharges, carrying the organisms on their legs from one person to another. The International Trachoma Initiative founded by the WHO in 1998 aims to eliminate blinding trachoma globally by 2020.
Clinical Manifestations Both endemic trachoma and adult inclusion conjunctivitis present initially as conjunctivitis characterized by small lymphoid follicles in the conjunctiva. In regions with hyperendemic classic blinding trachoma, the disease usually starts insidiously before the age of 2 years. Reinfection is common and probably contributes to the pathogenesis of trachoma. Studies using polymerase chain reaction (PCR) or other NAATs indicate that chlamydial DNA is often present in the ocular secretions of patients with trachoma, even in the absence of positive cultures. Thus persistent infection may be more common than was previously thought.
The cornea becomes involved, with inflammatory leukocytic infiltrations and superficial vascularization (pannus formation). As the inflammation continues, conjunctival scarring eventually distorts the eyelids, causing them to turn inward so that the lashes constantly abrade the eyeball (trichiasis and entropion); eventually the corneal epithelium is abraded and may ulcerate, with subsequent corneal scarring and blindness. Destruction of the conjunctival goblet cells, lacrimal ducts, and lacrimal gland may produce a “dry-eye” syndrome, with resultant corneal opacity due to drying (xerosis) or secondary bacterial corneal ulcers.
Communities with blinding trachoma often experience seasonal epidemics of conjunctivitis due to H. influenzae that contribute to the intensity of the inflammatory process. In such areas, the active infectious process usually resolves spontaneously in affected persons at 10–15 years of age, but conjunctival scars continue to shrink, producing trichiasis and entropion with subsequent corneal scarring in adults. In areas with milder and less prevalent disease, the process may be much slower, with active disease continuing into adulthood; blindness is rare in these cases.
Eye infection with oculogenital C. trachomatis strains in sexually active young adults presents as an acute onset of unilateral follicular conjunctivitis and preauricular lymphadenopathy similar to that seen in acute conjunctivitis caused by adenovirus or HSV. If untreated, the disease may persist for 6 weeks to 2 years. It is frequently associated with corneal inflammation in the form of discrete opacities (“infiltrates”), punctate epithelial erosions, and minor degrees of superficial corneal vascularization. Very rarely, conjunctival scarring and eyelid distortion occur, particularly in patients treated for many months with topical glucocorticoids. Recurrent eye infections develop most often in patients whose sexual partners are not treated with antimicrobial agents.
Diagnosis
The clinical diagnosis of classic trachoma can be made if two of the following signs are present: (1) lymphoid follicles on the upper tarsal conjunctiva; (2) typical conjunctival scarring; (3) vascular pannus; or (4) limbal follicles or their sequelae, Herbert’s pits. The clinical diagnosis of endemic trachoma should be confirmed by laboratory tests in children with relatively marked degrees of inflammation. Intracytoplasmic chlamydial inclusions are found in 10–60% of Giemsa-stained conjunctival smears in such populations, but chlamydial NAATs are more sensitive and are often positive when smears or cultures are negative. Follicular conjunctivitis in European or American adults living in trachomatous regions is rarely due to trachoma.
|
TREATMENT |
TRACHOMA |
Adult inclusion conjunctivitis responds well to treatment with the same regimens used in uncomplicated genital infections—namely, azithromycin (a 1-g single oral dose) or doxycycline (100 mg twice daily for 7 days). Simultaneous treatment of all sexual partners is necessary to prevent ocular reinfection and chlamydial genital disease. Topical antibiotic treatment is not required for patients who receive systemic antibiotics.
PSITTACOSIS
Psittacine birds and many other avian species act as natural reservoirs for C. psittaci–type organisms, which are common pathogens in domestic mammals and birds. The species C. psittaci, which now includes only avian strains, affects humans only as a zoonosis. (The other strains previously included in this species have been placed into different species that generally reflect the animals they infect: C. abortus, C. muridarum, C. suis, C. felis, and C. caviae.) Although all birds are susceptible, pet birds (parrots, parakeets, macaws, and cockatiels) and poultry (turkeys and ducks) are most frequently involved in transmission of C. psittaci to humans. Exposure is greatest among poultry-processing workers and owners of pet birds. Infectious forms of the organisms are shed from both symptomatic and apparently healthy birds and may remain viable for several months. C. psittaci can be transmitted to humans by direct contact with infected birds or by inhalation of aerosols from avian nasal discharges and from infectious avian fecal or feather dust. Transmission from person to person has never been demonstrated.
The diagnosis is usually established serologically. Psittacosis in humans may present as acute primary atypical pneumonia (which can be fatal in up to 10% of untreated cases); as severe chronic pneumonia; or as a mild illness or asymptomatic infection in persons exposed to infected birds.
EPIDEMIOLOGY
Fewer than 50 confirmed cases of psittacosis are reported in the United States each year, although many more cases probably occur than are reported. Control of psittacosis depends on control of avian sources of infection. A pandemic of psittacosis was once stopped by banning shipment or importation of psittacine birds. Birds can receive prophylaxis in the form of a tetracycline-containing feed. Imported birds are currently quarantined for 30 days of treatment.
CLINICAL MANIFESTATIONS
Typical symptoms include fever, chills, muscular aches and pains, severe headache, hepato- and/or splenomegaly, and gastrointestinal symptoms. Cardiac complications may involve endocarditis and myocarditis. Fatal cases were common in the preantibiotic era. As a result of quarantine of imported birds and improved veterinary-hygienic measures, outbreaks and sporadic cases of psittacosis are now rare. Severe pneumonia requiring management in an intensive care unit may develop. Endocarditis, hepatitis, and neurologic complications may occur, and fatal cases have been reported. The incubation period is usually 5–19 days but can last as long as 28 days.
DIAGNOSIS
Previously, the most widely used serologic test for diagnosing chlamydial infections was the genus-specific CF test, in which assay of paired serum specimens often shows fourfold or greater increases in antibody titer. The CF test remains useful, but the gold standard of serologic tests is now the MIF test, which is not widely available (see section on diagnosis of C. trachomatis genital infection, above). Any antibody titer above 1:16 is considered significant evidence of exposure to chlamydiae, and a fourfold titer rise in paired sera in combination with a clinically compatible syndrome can be used to diagnose psittacosis. Some commercially available serologic tests based on measurement of antibodies to LPS can be useful when the clinical diagnosis is consistent with bird exposure; however, since these tests are reactive for all chlamydiae (i.e., all chlamydiae contain LPS), caution must be used in their interpretation.
|
TREATMENT |
PSITTACOSIS |
The antibiotic of choice is tetracycline; the dosage for adults is 250 mg four times a day, continued for at least 3 weeks to avoid relapse. Severely ill patients may need cardiovascular and respiratory support. Erythromycin (500 mg four times a day by mouth) is an alternative therapy.
CHLAMYDIA PNEUMONIAE INFECTIONS
C. pneumoniae is a common cause of human respiratory diseases, such as pneumonia and bronchitis. This organism has been reported to account for as many as 10% of cases of community-acquired pneumonia, most of which are diagnosed by serology. Serologic studies have linked C. pneumoniae to atherosclerosis; isolation and PCR detection in cardiovascular tissues have also been reported. These findings suggest an expanded range of diseases and syndromes for C. pneumoniae. The role of C. pneumoniae in the etiology of atherosclerosis has been discussed since 1988, when Finnish researchers presented serologic evidence of an association of this organism with coronary heart disease and acute myocardial infarction. Subsequently, the organism was identified in atherosclerotic lesions by culture, PCR, immunohistochemistry, and transmission electron microscopy; however, discrepant study results (including those of animal studies) and failure of large-scale treatment studies have raised doubts as to the etiologic role of C. pneumoniae in atherosclerosis. Large-scale case-cohort studies have demonstrated some association of C. pneumoniae with lung cancer, as evaluated by serology.
EPIDEMIOLOGY
![]() Primary infection occurs mainly in school-aged children and reinfection in adults. Seroprevalence rates of 40–70% show that C. pneumoniae is widespread in both industrialized and developing countries. Seropositivity usually is first detected at school age, and rates generally increase by ~10% per decade. About 50% of individuals have detectable antibody at 30 years of age, and most have detectable antibody by the eighth decade of life. Although serologic evidence suggests that C. pneumoniae may be associated with up to 10% of cases of community-acquired pneumonia, most of this evidence is based not on paired serum samples but rather on a single high IgG titer. Some doubt exists about the true prevalence and etiologic role of C. pneumoniae in atypical pneumonia, especially since reports of cross-reactivity have raised questions about the specificity of serology when only a single serum sample is used for diagnosis.
Primary infection occurs mainly in school-aged children and reinfection in adults. Seroprevalence rates of 40–70% show that C. pneumoniae is widespread in both industrialized and developing countries. Seropositivity usually is first detected at school age, and rates generally increase by ~10% per decade. About 50% of individuals have detectable antibody at 30 years of age, and most have detectable antibody by the eighth decade of life. Although serologic evidence suggests that C. pneumoniae may be associated with up to 10% of cases of community-acquired pneumonia, most of this evidence is based not on paired serum samples but rather on a single high IgG titer. Some doubt exists about the true prevalence and etiologic role of C. pneumoniae in atypical pneumonia, especially since reports of cross-reactivity have raised questions about the specificity of serology when only a single serum sample is used for diagnosis.
PATHOGENESIS
Little is known about the pathogenesis of C. pneumoniae infection. It begins in the upper respiratory tract and, in many persons, persists as a prolonged asymptomatic condition of the upper respiratory mucosal surfaces. However, evidence of replication within vascular endothelium and synovial membranes of joints shows that, in at least some individuals, the organism is transported to distant sites, perhaps within macrophages. A C. pneumoniae outer-membrane protein may induce host immune responses whose cross-reactivity with human proteins results in an autoimmune reaction.
As mentioned above, epidemiologic studies have demonstrated an association between serologic evidence of C. pneumoniae infection and atherosclerotic disease of the coronary and other arteries. In addition, C. pneumoniae has been identified in atherosclerotic plaques by electron microscopy, DNA hybridization, and immunocytochemistry. The organism has been recovered in culture from atheromatous plaque—a result indicating the presence of viable replicating bacteria in vessels. Evidence from animal models supports the hypothesis that C. pneumoniae infection of the upper respiratory tract is followed by recovery of the organism from atheromatous lesions in the aorta and that the infection accelerates the process of atherosclerosis, especially in hypercholesterolemic animals. Antimicrobial treatment of the infected animals reverses the increased risk of atherosclerosis. In humans, two small trials in patients with unstable angina or recent myocardial infarction suggested that antibiotics reduce the likelihood of subsequent untoward cardiac events. However, larger-scale trials have not documented an effect of various antichlamydial regimens on the risk of these events.
CLINICAL MANIFESTATIONS
C. pneumoniae was first reported as the etiologic agent of mild atypical pneumonia in military recruits and college students. The clinical spectrum of C. pneumoniae infection includes acute pharyngitis, sinusitis, bronchitis, and pneumonitis, primarily in young adults. The clinical manifestations of primary infection appear to be more severe and prolonged than those of reinfection. The pneumonitis of C. pneumoniae pneumonia resembles that of Mycoplasma pneumonia in that leukocytosis is frequently lacking and patients often have prominent antecedent upper respiratory tract symptoms, fever, nonproductive cough, mild to moderate illness, minimal findings on chest auscultation, and small segmental infiltrates on chest x-ray. In elderly patients, pneumonia due to C. pneumoniae can be especially severe and may necessitate hospitalization and respiratory support.
Chronic infection with C. pneumoniae has been reported among patients with chronic obstructive pulmonary disease and may also play a role in the natural history of asthma, including exacerbations. The clinical symptoms of respiratory infections caused by C. pneumoniae are nonspecific and do not differ from those caused by other agents of atypical pneumonia, such as Mycoplasma pneumoniae.
DIAGNOSIS
Serology, PCR amplification, and culture can be used to diagnose C. pneumoniae infection. Serology has been the traditional method of diagnosing infection by C. pneumoniae. The gold standard serologic test is the MIF test (see section on diagnosis of C. trachomatis genital infection, above). Any antibody titer above 1:16 is considered significant evidence of exposure to chlamydiae. According to a CDC-sponsored expert working group, the diagnosis of acute C. pneumoniae infection requires demonstration of a fourfold rise in titer in paired serum samples. There are no official recommendations for diagnosis of chronic infections, although many research studies have used high titers of IgA as an indicator. The older CF tests and EIAs for LPS are not recommended, as they are not specific for C. pneumoniae but identify the chlamydiae only to the genus level. The organism is very difficult to grow in tissue culture but has been cultivated in HeLa cells, HEp-2 cells, and HL cells. Although NAATs are commercially available for C. trachomatis, only research-based PCR assays are available for C. pneumoniae.
|
TREATMENT |
C. PNEUMONIAE INFECTIONS |
Although few controlled trials of treatment have been reported, C. pneumoniae is inhibited in vitro by erythromycin, tetracycline, azithromycin, clarithromycin, gatifloxacin, and gemifloxacin. Recommended therapy consists of 2 g/d of either tetracycline or erythromycin for 10–14 days. Other macrolides (e.g., azithromycin) and some fluoroquinolones (e.g., levofloxacin and gatifloxacin) also appear to be effective.
ACKNOWLEDGMENT
The authors wish to acknowledge the late Walter E. Stamm, MD, for his significant contributions to the field of Chlamydia research. Dr. Stamm wrote the chapters on chlamydiae for previous editions of Harrison’s Principles of Internal Medicine, and we thank the editors for permission to reproduce Figs. 213-1 and 213-2 as well as Table 213-1 from his chapter in the 17th edition. Dr. Stamm died on December 14, 2009, and this chapter is dedicated to him.
SECTION 11 |
VIRAL DISEASES: GENERAL CONSIDERATIONS |
214e |
Medical Virology |
DEFINING A VIRUS
Viruses are obligate intracellular parasites. They consist of a DNA or RNA genome surrounded by protein. They may also have an outer-membrane lipoprotein envelope. Viruses can replicate only within cells because their nucleic acid does not encode many enzymes necessary for the metabolism of proteins, carbohydrates, or lipids or for the generation of high-energy phosphates. Typically, viral nucleic acids encode messenger RNA (mRNA) and proteins necessary for replicating, packaging, and releasing progeny virus from infected cells.
Viruses differ from virusoids, viroids, and prions. Virusoids are nucleic acids that depend on cells and helper viruses for packaging their nucleic acids into virus-like particles. Viroids are naked, cyclical, mostly double-strand small RNAs that appear to be restricted to plants, spread from cell to cell, and are replicated by cellular RNA polymerase II. Prions (Chap. 453e) are abnormal proteins that propagate and cause disease by altering the structure of a normal cell protein. Prions cause neurodegenerative diseases such as Creutzfeldt-Jakob disease, Gerstmann-Straüssler disease, kuru, and human or bovine spongiform encephalopathy (“mad cow disease”).
VIRUS STRUCTURE
Viral genomes may consist of single- or double-strand DNA, single- or double-strand RNA, single-strand or segmented antisense RNA, or double-strand segmented RNA. Viral nucleic acids may encode only a few genes or more than 100. Sense-strand viral RNA genomes can be translated directly into protein, whereas antisense RNAs must be copied into translatable RNA. Sense and antisense genomes are also referred to as positive-strand and negative-strand genomes, respectively. Viral nucleic acid is usually associated with virus-encoded nucleoprotein(s) in the virus core. Viral nucleic acids and nucleoproteins are almost always enclosed in a protein capsid. Because of the limited genetic complexity of viruses, their capsids are usually composed of multimers of identical capsomeres made up of one or a few proteins. Capsids have icosahedral or helical symmetry. Icosahedral capsid structures approximate spheres and have two-, three-, or fivefold axes of symmetry, whereas helical capsid structures have only a twofold axis of symmetry. The nucleic acid, nucleoprotein(s), and protein capsid together are called a nucleocapsid.
Many viruses are composed of a nucleic acid core and a capsid. For these viruses, the outer capsid surface mediates contact with uninfected cells’ plasma membranes. Other viruses are more complex and have an outer phospholipid, cholesterol, glycoprotein, and glycolipid envelope that is derived from virus-modified infected cell membranes. Cell nuclear, endoplasmic reticulum, Golgi, or plasma membranes that become parts of the viral envelope have usually been modified during infection by the insertion of virus-encoded glycoproteins, which mediate contact of enveloped virus with uninfected cell surfaces. Matrix or tegument proteins may fill the space between the nucleocapsid and the outer envelope of the virus.
Enveloped viruses are usually sensitive to lipid solvents or detergents that can dissolve the envelope, whereas viruses with protein nucleocapsid exteriors may be somewhat detergent resistant. A schematic diagram of large and complex herpesviruses is shown in Fig. 214e-1. Structures of prototypical pathogenic human viruses are described in Table 214e-1. The relative sizes and structures of typical pathogenic human viruses are shown in Fig. 214e-2.
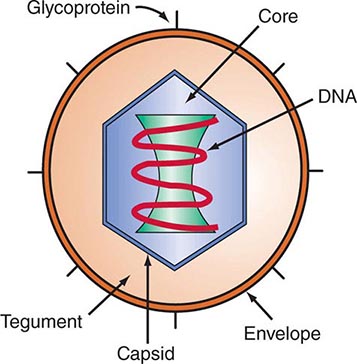
FIGURE 214e-1 Schematic diagram of an enveloped herpesvirus with an icosahedral nucleocapsid. The approximate respective dimensions of the nucleocapsid and the enveloped particles are 110 and 180 nm. The capsid is composed of 162 capsomeres: 150 with sixfold and 12 with fivefold axes of symmetry.
|
VIRUS FAMILIES PATHOGENIC FOR HUMANS |
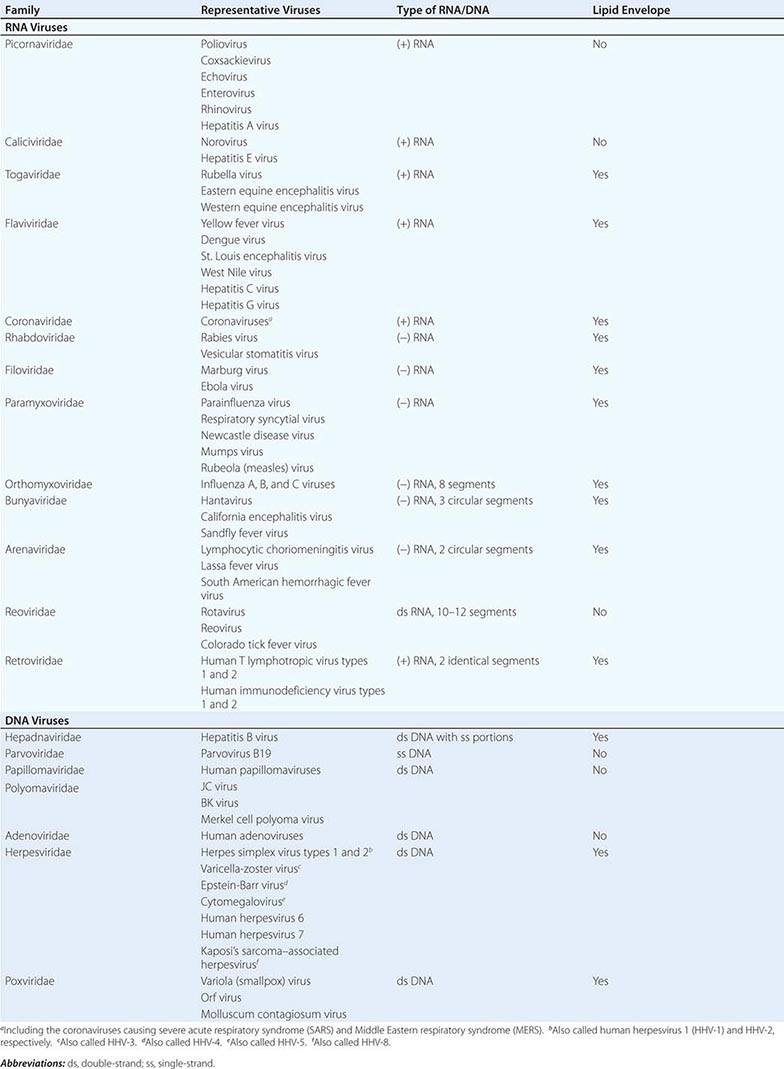
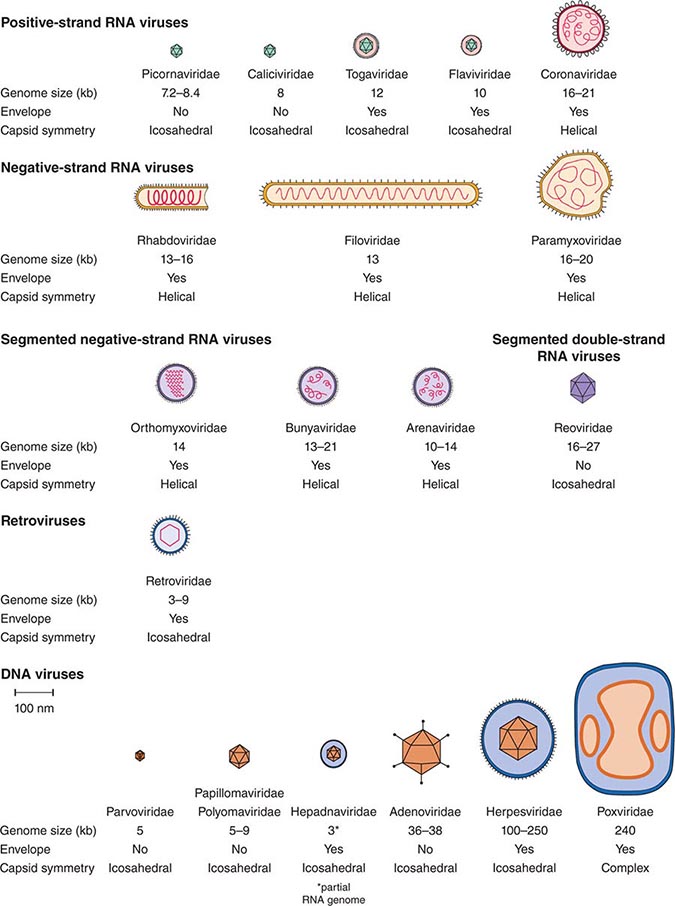
FIGURE 214e-2 Schematic diagrams of the major virus families including species that infect humans. The viruses are grouped by genome type and are drawn approximately to scale. Prototype viruses of each family that cause human disease are listed in Table 214e-1.
TAXONOMY OF PATHOGENIC HUMAN VIRUSES
As is apparent from Table 214e-1 and Fig. 214e-2, the classification of viruses into orders and families is based on nucleic acid composition, nucleocapsid size and symmetry, and presence or absence of an envelope. Viruses of a single family have similar structures and may be morphologically indistinguishable in electron micrographs. Subclassification into genera depends on similarity in epidemiology, biologic effects, and nucleic acid sequence.
Most viruses that infect humans have a common name related to their pathologic effects or the circumstances of their discovery. In addition, formal species names—consisting of the name of the host followed by the family or genus of the virus and a number—have been assigned by the International Committee on Taxonomy of Viruses. This dual terminology can cause confusion when viruses are referred to by either name—e.g., varicella-zoster virus (VZV) or human herpesvirus 3 (HHV-3).
VIRAL INFECTION IN VITRO
STAGES OF VIRAL INFECTION OF CELLS IN CULTURE
Viral Interactions with Cell Surfaces and Cell Entry To deliver its nucleic acid payload to the cell cytoplasm or nucleoplasm, a virus must overcome barriers posed by the cell’s plasma and cytoplasmic membranes. Infection is frequently initiated by weak electrostatic or hydrophobic interactions with the cell surface. Subsequent stronger, more specific attachment to a cell plasma membrane protein, carbohydrate, glycolipid, heparan sulfate proteoglycan, or sialic acid enables stable binding to a specific cell surface “receptor” that mediates fusion with the cell plasma membrane (see Table 145e-1). Receptor binding is often augmented by a viral surface protein interaction with more than one cell surface protein or co-receptor. Receptors and co-receptors are important determinants of the species and cell type that a virus can infect. For example, the HIV envelope glycoprotein binds to the T cell surface protein CD4 and then engages a chemokine receptor that is the definitive co-receptor for the virus and mediates entry into the cell cytoplasm. The Epstein-Barr virus (EBV) glycoprotein gp350 binds to the B lymphocyte complement receptor CD21 and then uses a major histocompatibility complex (MHC) class II molecule as a co-receptor and an integrin for definitive entry.
Viruses have evolved a wide range of strategies to enter cells. Influenza virus has an outer-membrane hemagglutinin glycoprotein that binds to sialic acid on respiratory tract cell plasma membranes. The hemagglutinin mediates adsorption to cell membranes, receptor aggregation, and endocytosis. As the endosome pH decreases in the cell cytoplasm, the influenza hemagglutinin conformation changes, enabling hydrophobic helices, which are initially at the base of the hemagglutinin, to extend, interacting and fusing with the endosome membrane and thereby releasing the viral genome into the cell cytoplasm. The influenza virus M2 membrane channel protein has a key role in lowering endosome pH and permitting virus and cell membrane fusion.
Nonenveloped viruses (e.g., human papillomaviruses [HPVs]) and some enveloped viruses have evolved to partially fuse with cell plasma membrane receptors and be internalized into endosomes. The low pH in an endosome can then trigger virus membrane or capsid fusion with the endocytic membrane, releasing viral DNA into the cytoplasm to initiate infection.
Hydrophobic interactions required for fusion can be susceptible to chemical inhibition or blockade. The HIV envelope glycoprotein gp120 is associated with gp41 on the viral surface. HIV gp120 binding to CD4 and then to specific chemokine receptors results in conformational changes that allow gp41 to initiate cell membrane fusion. The anti-HIV drug enfuvirtide is a small peptide derived from the gp41 structure. Enfuvirtide binds to gp41 and prevents conformational changes required for fusion. In contrast, maraviroc prevents virus entry by binding to the CCR5 receptor, thereby blocking gp120 binding to CCR5 and preventing gp120 fusion with CCR5.
Viral Gene Expression and Replication After uncoating and release of viral nucleoprotein into the cytoplasm, the viral genome is transported to sites of expression and replication. To produce infectious progeny, viruses must produce proteins necessary for replicating their nucleic acids as well as structural proteins necessary for coating their nucleic acids and for assembling nucleic acids and proteins into progeny virus. Different viruses use different strategies and gene repertoires to accomplish these goals. Most DNA viruses, except for poxviruses, replicate their nucleic acid and assemble into nucleocapsids in the cell nucleus. RNA viruses, except for influenza viruses, transcribe and replicate their RNA and assemble in the cytoplasm before envelopment at the cell plasma membrane. The replication strategies of DNA and RNA viruses and of positive- and negative-strand RNA viruses are presented and discussed separately below. Medically important viruses of each group are used for illustrative purposes.
POSITIVE-STRAND RNA VIRUSES RNA viruses of medical importance include positive-strand picornaviruses, flaviviruses, togaviruses, caliciviruses, and coronaviruses. Genome RNA from positive-strand RNA viruses is released into the cytoplasm without associated enzymes. Cell ribosomes recognize and associate with the viral genome’s internal ribosome entry sequence and translate a virus-encoded polyprotein. Proteases within the polyprotein cleave out the viral RNA polymerase and other viral proteins necessary for replication. Antigenomic RNA is next transcribed from the genome RNA template. Positive-strand genomes and mRNAs are then transcribed from the antigenome RNA by the viral RNA polymerase and are translated into capsid proteins. Genomic RNA is encapsidated in the cytoplasm and released as the infected cell undergoes lysis.
NEGATIVE-STRAND RNA VIRUSES Medically important negative-strand RNA viruses include rhabdoviruses, filoviruses, paramyxoviruses, orthomyxoviruses, and bunyaviruses. The genomes of negative-strand viruses are frequently segmented. Negative-strand RNA viral genomes are released into the cytoplasm with an associated RNA polymerase and one or more polymerase accessory proteins. The viral RNA polymerase transcribes mRNAs as well as full-length antigenome RNA, which is the template for genome RNA replication. Viral mRNAs encode the viral RNA polymerase and accessory factors as well as viral structural proteins. Except for influenza virus, which transcribes its mRNAs and antigenome RNAs in the cell nucleus, negative-strand RNA viruses replicate entirely in the cytoplasm. All negative-strand RNA viruses, including influenza viruses, assemble in the cytoplasm.
DOUBLE-STRAND SEGMENTED RNA VIRUSES Double-strand RNA viruses are taxonomically grouped in the family Reoviridae. The medically important viruses in this group are rotaviruses and Colorado tick fever virus. Reovirus genomes have 10–12 RNA segments. Reovirus particles contain an RNA polymerase complex. These viruses replicate and assemble in the cell cytoplasm.
DNA VIRUSES Medically important DNA viruses include parvoviruses, which have small single-strand DNA genomes and cause transient arthritis, and polyomaviruses, including the smaller polyomaviruses such as JC virus, which causes progressive multifocal leukoencephalopathy in immunocompromised patients; BK virus; and Merkel cell polyomavirus. The larger HPVs cause warts as well as cervical, penile, and oral carcinomas. The next larger DNA viruses are adenoviruses, which mostly cause transient respiratory tract and ocular inflammatory disease. The herpesviruses include eight viruses that cause a wide range of inflammatory and malignant diseases in humans. EBV is an important cause of lymphomas and Hodgkin’s disease in both immunocompromised and immunocompetent people and of nasopharyngeal carcinoma in southern Chinese and northern African populations. Cytomegalovirus (CMV) is an important cause of transplacental infections and neonatal neurologic impairment. Poxviruses, the largest DNA viruses and the largest viruses that infect humans (barely visible by light microscopy), cause smallpox, monkeypox, and molluscum contagiosum. Aside from those of poxviruses, other DNA virus genomes enter the cell nucleus and are transcribed by cellular RNA polymerase II.
After receptor binding and fusion with plasma membranes or endocytic vesicle membranes, herpesvirus nucleocapsids are released into the cytoplasm with tegument proteins and are transported along microtubules to a nuclear pore. Capsids then release DNA into the nucleus.
DNA virus transcription and mRNA processing depend on both viral and cellular proteins. For herpes simplex virus (HSV), a viral tegument protein enters the nucleus and activates immediate-early genes, the first genes expressed after infection. Transcription of immediate-early genes requires the viral tegument protein and cell transcription factors. HSV becomes nonreplicating, or latent, in neurons because essential cell transcription factors for expression of viral immediate-early genes are docked in the cytoplasm in neurons. Heat shock or other cell stresses can cause these cell factors to enter the nucleus, activate viral gene expression, and initiate replication. This information explains HSV-1 latency in neurons and activation of replicative infection.
For adenoviruses and herpesviruses, transcription of immediate-early genes results in expression of early proteins necessary for viral DNA replication. Viral DNA synthesis is required to turn on late-gene expression and production of viral structural components. The HPVs, polyomaviruses, and parvoviruses are not dependent on transactivators encoded from the viral genome for early-gene transcription. Instead, their early genes have upstream enhancing elements that bind cell transcription factors. The early genes encode proteins that are necessary for viral DNA synthesis and late-gene transcription. DNA virus late genes encode structural proteins necessary for viral assembly and for viral egress from the infected cell. Late-gene transcription is continuously dependent on DNA replication. Therefore, inhibitors of DNA replication also stop late-gene transcription.
Each DNA virus family uses unique mechanisms for replicating its DNA. Adenovirus and herpesvirus DNAs are linear in the virion. Adenovirus DNA remains linear in infected cells and replicates as a linear genome, using an initiator protein–DNA complex. In contrast, herpesvirus DNA circularizes in the infected cell, and genomes replicate into linear concatemers through a “rolling-circle” mechanism. Full-length DNA genomes are cleaved and packaged into virus. Herpesviruses encode a DNA polymerase and at least six other viral proteins necessary for viral DNA replication. Acyclovir and ganciclovir prevent viral DNA synthesis when they are phosphorylated and incorporated into DNA by the viral polymerase. Herpesviruses also encode enzymes that increase the deoxynucleotide triphosphate pools. HPV and polyomavirus DNAs are circular both within the virus and in infected cells. These genomes are reproduced by cellular DNA replication enzymes and remain circular through replication and packaging. HPV and polyomavirus early proteins are necessary for DNA replication in both latent and viral replicative phases. Early viral proteins stimulate cells to remain in cycle, facilitating viral DNA replication.
Parvoviruses have negative single-strand DNA genomes and are the smallest DNA viruses. Their genomes are half the size of HPV genomes and include only two genes. The replication of autonomous parvoviruses, such as B19, depends on cellular DNA replication and requires the virus-encoded Rep protein. Other parvoviruses, such as adeno-associated virus (AAV), are not autonomous and require helper viruses of the adenovirus or herpesvirus family for their replication. AAV is being used as a potentially safe human gene therapy vector because its replication protein causes integration at a single chromosome site. The small genome size limits the range of proteins that can be expressed from AAV vectors.
As stated above, poxviruses are the largest DNA viruses. They are unique among DNA viruses in replicating and assembling in the cytoplasm. To accomplish cytoplasmic replication, poxviruses encode transcription factors, an RNA polymerase II orthologue, enzymes for RNA capping, enzymes for RNA polyadenylation, and enzymes for viral DNA synthesis. Poxvirus DNA also has a unique structure. The double-strand linear DNA is covalently linked at the ends, making a covalently closed double-strand circular genome. Replication of the circular genomes is initiated by nicking in inverted repeats at the ends of the linear DNA. During DNA replication, the genome is cleaved within the terminal inverted repeats, and the inverted repeats self-prime complementary-strand synthesis by the virus-encoded DNA polymerase. Like herpesviruses, poxviruses encode several enzymes that increase deoxynucleotide triphosphate precursor levels and thus facilitate viral DNA synthesis.
VIRUSES THAT USE BOTH RNA AND DNA GENOMES IN THEIR LIFE CYCLE Retroviruses, including HIV, are RNA viruses that use a DNA intermediate to replicate their genomes. In contrast, hepatitis B virus (HBV) is a DNA virus that uses an RNA intermediate to replicate its genome. Thus these viruses are not purely RNA or DNA viruses. Retroviruses are RNA viruses with two identical sense-strand genomes and associated reverse transcriptase and integrase enzymes. Retroviruses differ from all other viruses in that they reverse-transcribe themselves into partially duplicated double-strand DNA copies and then routinely integrate into the host genome as part of their persistence and replication strategies. Inhibitors of reverse transcriptase (e.g., zidovudine) or integrase (e.g., raltegravir) are now commonly used as antiviral treatments for HIV infection. Integration of remnants and even complete copies of simple retrovirus DNAs into the human genome raises the possibility of replication-competent simple human retroviruses. However, endogenous human retrovirus replication has not been documented or associated with any disease. Integrated, replication-competent retroviral DNAs are also present in many animal species, such as pigs. These porcine retroviruses are a potential cause for concern in xenotransplantation because retrovirus replication could cause disease in humans.
Cellular RNA polymerase II and transcription factors regulate transcription from the integrated provirus DNA genome. Some retroviruses also encode regulators of transcription and RNA processing, such as Tax and Rex in human T lymphotropic virus (HTLV) types 1 and 2. HIV-1 and HIV-2 have orthologous Tat and Rev genes as well as the additional accessory proteins Vpr, Vpu, and Vif, which are important for efficient infection and immune escape. Full-length proviral transcripts are made from a promoter in the viral terminal repeat and serve as both genome RNAs that are packaged in the nucleocapsids and differentially spliced mRNAs that encode for the virus Gag protein, polymerase/integrase protein, and envelope glycoprotein. The Gag protein includes a protease that cleaves it into several components, including a viral matrix protein that coats the viral RNA. Viral RNA polymerase/integrase, matrix protein, and cellular tRNAs are key components in the viral nucleocapsid. Protease inhibitors have been developed as effective agents against infections caused by HIV (e.g., saquinavir) or hepatitis C virus (HCV) (e.g., telaprevir).
HBV replication is unique in several respects. The HBV genome is a partially double-strand DNA genome that is repaired in infected cells to a fully double-strand circular DNA by the virion polymerase. Viral mRNAs are transcribed from the closed circular viral episome by the cellular RNA polymerase II and are translated to yield HBV proteins, including core protein, surface antigen, and polymerase. In addition, a full-genome-length mRNA is packaged into viral core particles in the cytoplasm of infected cells as an intermediate for viral DNA replication. This RNA associates with the viral polymerase, which also has reverse transcriptase activity and converts the full-length encapsidated RNA genome into partially double-strand DNA. Thus, nucleos(t)ide analogs that inhibit reverse transcription (e.g., tenofovir) are commonly used to treat HBV infection. HBV is believed to mature by budding through the cell’s plasma membrane, which has been modified by the insertion of viral surface antigen protein.
Viral Assembly and Egress For most viruses, nucleic acid and structural protein synthesis is accompanied by the assembly of protein and nucleic acid complexes. The assembly and egress of mature infectious virus mark the end of the eclipse phase of infection, during which infectious virus cannot be recovered from the infected cell. Nucleic acids from RNA viruses and poxviruses assemble into nucleocapsids in the cytoplasm. For all DNA viruses except poxviruses, viral DNA assembles into nucleocapsids in the nucleus. In general, the capsid proteins of viruses with icosahedral nucleocapsids can self-assemble into densely packed and highly ordered capsid structures. Herpesviruses require an assemblin protein as a scaffold for capsid assembly. Viral nucleic acid then spools into the assembled capsid. For herpesviruses, a full unit of the viral DNA genome is packaged into the capsid, and a capsid-associated nuclease cleaves the viral DNA at both ends. In the case of viruses with helical nucleocapsids, the protein component appears to assemble around the nucleic acid, which contributes to capsid organization.
Viruses must egress from the infected cell and not bind back to their receptor(s) on the outer surface of the plasma membrane. Viruses can acquire envelopes from cytoplasmic membranes or by budding through the cell’s plasma membrane. Excess viral membrane glycoproteins are synthesized to saturate cell receptors and facilitate separation of the virus from the infected cell. Some viruses encode membrane proteins with enzymatic activity for receptor destruction. Influenza virus, for example, encodes a glycoprotein with neuraminidase activity. Neuraminidase destroys sialic acid on the infected cell’s plasma membrane so that newly released virus does not get stuck to the dying cell. Oseltamivir and zanamivir are neuraminidase inhibitors that are used to treat or provide prophylaxis for influenza virus infection. Herpesvirus nucleocapsids acquire an initial envelope by assembling in the nucleus and then budding through the nuclear membrane into the endoplasmic reticular space. The initially enveloped herpesvirus is then de-enveloped and released from the cell either by exocytosis or by re-envelopment at the plasma membrane. Nonenveloped viruses depend on the death and dissolution of the infected cell for their release.
FIDELITY OF VIRAL REPLICATION
Hundreds or thousands of progeny may be produced from a single virus-infected cell. Many particles partially assemble and never mature into virions. Many mature-appearing virions are imperfect and have only incomplete or nonfunctional genomes. Despite the inefficiency of assembly, a typical virus-infected cell releases 10–1000 infectious progeny. Some of these progeny may contain genomes that differ from those of the virus that infected the cell. Smaller, “defective” viral genomes have been noted with the replication of many RNA and DNA viruses. Virions with defective genomes can be produced in large numbers through packaging of incompletely synthesized nucleic acid. Adenovirus packaging is notoriously inefficient, and a high ratio of particle to infectious virus may limit the amount of recombinant adenovirus that can be administered for gene therapy since the immunogenicity of defective particles may contribute to adverse effects.
Changes in viral genomes can lead to mutant viruses of medical significance. In general, viral nucleic acid replication is more error-prone than cellular nucleic acid replication. RNA polymerases and reverse transcriptases are significantly more error-prone than DNA polymerases. Mutations can also be introduced into the HIV genome by APOBEC3G, a cellular protein that is packaged in the virion. APOBEC3G deaminates cytidine in the virion RNA to uridine. When reverse transcriptase subsequently uses the altered virion RNA as a template in the infected cell, a guanosine-to-adenosine mutation is introduced into the proviral DNA. Mutations resulting in less efficient viral growth, or fitness, may be detrimental to the virus. HIV-encoded Vif blocks APOBEC3G activity in the virion, inhibiting the debilitating effects of hypermutation on genetic integrity. Nevertheless, mutations resulting in evasion of the host immune response or resistance to antiviral drugs are preferentially selected in patients, with the consequent perpetuation of infection. Viral genomes can also be altered by recombination or reassortment between two related viruses in a single infected cell. Although this occurrence is unusual under most circumstances of natural infection, the genome changes can be substantial and can significantly alter virulence or epidemiology. Reassortment of the avian or mammalian influenza A hemagglutinin gene into a human influenza background can result in the emergence of new epidemic or pandemic influenza A strains.
VIRAL GENES NOT REQUIRED FOR VIRAL REPLICATION
Viruses frequently have genes encoding proteins that are not directly involved in replication or packaging of the viral nucleic acid, in virion assembly, or in regulation of the transcription of viral genes involved in those processes. Most of these proteins fall into five classes: (1) proteins that directly or indirectly alter cell growth; (2) proteins that inhibit cellular RNA or protein synthesis so that viral mRNA can be efficiently transcribed or translated; (3) proteins that promote cell survival or inhibit apoptosis so that progeny virus can mature and escape from the infected cell; (4) proteins that inhibit the host interferon response; and (5) proteins that downregulate host inflammatory or immune responses so that viral infection can proceed in an infected person to the extent consistent with the survival of the virus and its efficient transmission to a new host. More complex viruses of the poxvirus or herpesvirus family encode many proteins that serve these functions. Some of these viral proteins have motifs similar to those of cellular proteins, while others are quite novel. Virology has increasingly focused on these more sophisticated strategies evolved by viruses to permit the establishment of long-term infection in humans and other animals. These strategies often provide unique insights into the control of cell growth, cell survival, macromolecular synthesis, proteolytic processing, immune or inflammatory suppression, immune resistance, cytokine mimicry, or cytokine blockade.
MicroRNAs (miRNAs) are small noncoding RNAs that can regulate gene expression at the posttranscriptional level by targeting—and usually silencing—mRNAs. miRNAs were initially discovered in plants and plant viruses, where they alter expression of cell defensins. Herpesviruses are especially rich in miRNAs; for example, at least 23 miRNAs have been identified in EBV and 11 in CMV. Adenovirus and polyomavirus miRNAs have also been described. Increasing data indicate that animal viruses encode miRNAs to alter the growth and survival of host cells and the innate and acquired immune responses.
HOST RANGE
The concept of host range was originally based on the cell types in which a virus replicates in tissue culture. For the most part, the host range is limited by specific cell-surface proteins required for viral adsorption or penetration—i.e., to the cell types that express receptors or co-receptors for a specific virus. Another common basis for host-range limitation is the degree of transcriptional activity from viral promoters in different cell types. Most DNA viruses depend not only on cellular RNA polymerase II and the basal components of the cellular transcription complex but also on activated components and transcriptional accessory factors, both of which differ among differentiated tissues, among cells at various phases of the cell cycle, and between resting and cycling cells.
The importance of host range factors is illustrated by the effects of specific host determinants that limit the replication of influenza virus with avian or porcine hemagglutinins in humans. These viral proteins have adapted to bind avian or porcine sialic acids, and spread of avian or porcine influenza viruses in human populations is limited by their ability to infect human cells.
VIRAL CYTOPATHIC EFFECTS AND INHIBITORS OF APOPTOSIS
The replication of almost all viruses has adverse effects on the infected cell, inhibiting cellular synthesis of DNA, RNA, or proteins through efficient competition for key substrates and enzymatic processes. These general inhibitory effects enable viruses to nonspecifically limit components of innate host resistance, such as interferon (IFN) production. Viruses can specifically inhibit host protein synthesis by attacking a component of the translational initiation complex—frequently, a component that is not required for efficient translation of viral RNAs. Poliovirus protease 2A, for example, cleaves a cellular component of the complex that ordinarily facilitates translation of cellular mRNAs by interacting with their cap structure. Poliovirus RNA is efficiently translated without a cap because it has an internal ribosome entry sequence. Influenza virus inhibits the processing of mRNA by snatching cap structures from nascent cellular RNAs and using them as primers in the synthesis of viral mRNA. HSV has a virion tegument protein that inhibits cellular mRNA translation.
Apoptosis is the expected consequence of virus-induced inhibition of cellular macromolecular synthesis and viral nucleic acid replication. Although the induction of apoptosis may be important for the release of some viruses (particularly nonenveloped viruses), many viruses have acquired genes or parts of genes that enable them to forestall infected-cell death. This delay increases the yield from viral replication. Adenoviruses and herpesviruses encode analogues of the cellular Bc12 protein, which block mitochondrial enhancement of proapoptotic stimuli. Poxviruses and some herpesviruses also encode caspase inhibitors. Many viruses, including HPVs and adenoviruses, encode proteins that inhibit p53 or its downstream proapoptotic effects.
VIRAL INFECTION IN VIVO
TRANSMISSION
The capsid and envelope of a virus protect the genome and enable efficient transmission of the virus from cell to cell and to new prospective hosts. Most common viral infections are spread by direct contact, by ingestion of contaminated water or food, or by inhalation of aerosolized particles. In all these situations, infection begins on an epithelial or mucosal surface and spreads along the mucosa and into deeper tissues. Infection may spread to cells that can enter blood vessels, lymphatics, or neural circuits. HBV, HCV, HTLV, and HIV are dependent on transmission by parenteral inoculation. Some viruses are transmitted only between humans. The dependence of smallpox virus and poliovirus infections on interhuman transmission makes it feasible to eliminate these viruses from human circulation by mass vaccination. Herpesviruses also survive by interhuman transmission but may be more difficult to eliminate because they establish persistent latent infection in humans and continuously reactivate to infect new and naïve generations.
Animals are also important reservoirs and vectors for transmission of viruses causing human disease. Insect vectors can mediate parenteral transfer of viruses that reach high titers in animal or human hosts. Arboviruses are parenterally transmitted from mammalian species to humans by mosquito vectors. Herpes B, monkeypox, rabies, and viral hemorrhagic fevers are other examples of zoonotic infections caused by direct contact with animals, animal tissues, or arthropod vectors.
PRIMARY INFECTION
Initial viral infections usually last for several days or weeks. During this period, the concentration of virus at sites of infection rises and then falls, usually to unmeasurable levels. The rise and fall of viral replication at a given site depend on local innate immune responses and the access of systemic antibody and cell immune effectors to the virus. Typically, primary infections with enteroviruses, mumps virus, measles virus, rubella virus, rotavirus, influenza virus, AAV, adenovirus, HSV, and VZV are cleared from almost all sites within 3–4 weeks. Some viruses are especially proficient in altering or evading innate and acquired immune responses. Primary infection with AAV, EBV, or CMV can last for several months. Characteristically, primary infections due to HBV, HCV, hepatitis D virus (HDV), HIV, HPV, and molluscum contagiosum virus (MCV) extend beyond several weeks. For some of these viruses (e.g., HPV, HBV, HCV, HDV, and MCV), the manifestations of primary infection are almost indistinguishable from the persistent phase.
Disease manifestations usually arise as a consequence of viral replication, infected-cell injury or death, and local inflammatory and innate immune responses. Disease severity may not necessarily correlate with the level of viral replication alone. For example, the clinical manifestations of intense primary infection with poliovirus, enterovirus, rabies virus, measles virus, mumps virus, or HSV at mucosal surfaces may be inapparent or relatively mild, whereas limited replication in neural cells can have dramatic consequences. Similarly, rubella virus or CMV infections in utero or neonatal HSV infections may have much more devastating effects than infections in adults.
Primary infections are cleared by nonspecific innate and specific adaptive immune responses. Thereafter, an immunocompetent host is usually immune to the disease manifestations of reinfection by the same virus. Immunity frequently does not prevent transient surface colonization on reexposure, persistent colonization, or even limited deeper infection.
PERSISTENT AND LATENT INFECTIONS
Relatively few viruses cause persistent or latent infections. HBV, HCV, rabies virus, measles virus, HIV, HTLV, HPV, HHVs, and MCV are notable exceptions. The mechanisms for persistent infection vary. HCV RNA polymerase and HIV reverse transcriptase are error-prone and generate variant genomes. Genome variation can be sufficient to permit evasion of host immune responses, thereby allowing persistent infection. HIV is also directly immunosuppressive, depleting CD4+ T lymphocytes and compromising CD8+ cytotoxic T cell immune responsiveness. Moreover, HIV encodes the Nef protein, which downmodulates MHC class I expression, rendering HIV-infected cells partially resistant to immune CD8+ T cell lysis.
DNA viruses have low mutation rates. Their persistence in human populations usually depends on their ability to establish latent infection in some cells, to reactivate from latency, and then to replicate at epithelial surfaces. Latency is defined as a state of infection in which virus is not replicating, viral genes associated with lytic infection are not expressed, and infectious virus is not made. The complete viral genome is present and may be replicated by cellular DNA polymerase in conjunction with replication of the cell’s genome. HPVs establish latent infection in basal epithelial cells. The latently infected basal cell replicates, along with the HPV episome, by using cellular DNA polymerase. Some of the progeny cells provide new latently infected basal cells, whereas others go on to squamous differentiation. Infected cells that differentiate to squamous cells become permissive for lytic viral infection. Herpesviruses establish latent infection in nonreplicating neural cells (HSV and VZV) or in replicating cells of hematopoietic lineages (EBV, CMV, HHV-6, HHV-7, and Kaposi’s sarcoma–associated herpesvirus [KSHV, also known as HHV-8]). In their latent stage, HPV and herpesvirus genomes are largely hidden from the normal immune response. Reactivated HPV and herpesvirus infections escape immediate and effective immune responses in highly immune hosts by inhibiting host innate immune and inflammatory responses. In addition, HPV, HSV, and VZV are somewhat protected because they replicate in the middle and upper layers of the squamous epithelium—sites not routinely visited by cells that mediate or amplify immune and inflammatory responses. HSV and CMV are also known to encode proteins that downregulate MHC class I expression and antigenic peptide presentation, enabling infected cells to escape recognition by and cytotoxic effects of CD8+ T lymphocytes.
Like other poxviruses, MCV cannot establish latent infection. This virus causes persistent infection in hypertrophic skin lesions that last for months or years. MCV encodes a chemokine homologue that probably blocks inflammatory responses, an MHC class I analogue that blocks cytotoxic T lymphocyte attack, and inhibitors of cell death that prolong infected-cell viability.
PERSISTENT VIRAL INFECTIONS AND CANCER
Persistent viral infection is estimated to be the root cause of as many as 20% of human malignancies. Cancer is an accidental and highly unusual or long-term effect of oncogenic human viral infection. With most “oncogenic viruses,” infection is a critical and ultimately determinative early step in carcinogenesis. Latent HPV infection can block cell death and cause cervical cells to proliferate. A virus-infected cell with an integrated HPV genome overexpressing E6 and E7 undergoes subsequent cellular genetic changes that enhance autonomous malignant cell growth.
Most hepatocellular carcinoma is believed to be caused by chronic inflammatory, immune, and regenerative responses to HBV or HCV infection. Epidemiologic data firmly link HBV and HCV infections to hepatocellular carcinoma. These infections elicit repetitive cycles of virus-induced liver injury followed by tissue repair and regeneration. Over decades, chronic viral infection, repetitive tissue regeneration, and acquired chromosomal changes can result in proliferative nodules. Further chromosomal mutations can lead to the degeneration of cells in a proliferating nodule into hepatocellular carcinoma. In rare instances, HBV DNA integrates into cellular DNA, promoting overexpression of a cell gene that can also contribute to oncogenesis.
Most cervical carcinoma is caused by persistent infection with “high-risk” HPV type 16 or 18. In contrast to HBV and HCV infections, which stimulate cell growth as a consequence of virus-induced cell death, HPV type 16 or 18 proteins E6 and E7 destroy p53 and pRB, respectively. Elimination of these key tumor-suppressive cell proteins increases cell growth, cell survival, and cell genome instability. However, like HBV and HCV infections, HPV infection alone is not sufficient for carcinogenesis. Cervical carcinoma is inevitably associated with persistent HPV infection and integration of the HPV genome into chromosomal DNA. Integrations that result in overexpression of E6 and E7 from HPV type 16 or 18 cause more profound changes in cell growth and survival and permit subsequent chromosomal changes that result in cervical carcinoma.
EBV is the most unusual oncogenic virus in that normal B cell infection results in latency with expression of viral proteins that can cause endless B lymphocyte growth. In almost all humans, strong CD4+ and CD8+ T cell immune responses to the antigenic EBV latent-infection nuclear proteins prevent uncontrolled B cell lymphoproliferation. However, when humans are severely immunosuppressed by transplantation-associated medication, HIV infection, or genetic immune deficiencies, EBV-induced B cell malignancies can emerge.
EBV infection also has a role in the long-term development of B lymphocyte and epithelial cell malignancies. Persistent EBV infection with expression of an EBV latency-associated integral membrane protein (LMP1) in latently infected epithelial cells appears to be a critical early step in the evolution of anaplastic nasopharyngeal carcinoma, a common malignancy in populations in southern China and northern Africa. Genomic instability and chromosomal abnormalities also contribute to the development of EBV-associated nasopharyngeal carcinoma. EBV is an important cause of Hodgkin’s lymphoma. High-level expression of LMP1 or LMP2 in Reed-Sternberg cells is a hallmark in up to 50% of Hodgkin’s lymphoma cases. LMP1-induced nuclear factor-κB (NF-κB) activity may prolong the survival of defective B cells that are normally eliminated by apoptosis, thereby allowing other genetic changes leading to the development of malignant Reed-Sternberg cells.
The HTLV-1 Tax and Rex proteins are critical to the initiation of cutaneous adult T cell lymphoma/leukemias that occur long after primary HTLV-1 infection. Tax-induced NF-κB activation may contribute to cytokine production, infected-cell survival, and eventual outgrowth of malignant cells.
Molecular data confirm the presence of KSHV DNA in all Kaposi’s tumors, including those associated with HIV infection, transplantation, and familial transmission. KSHV infection is also etiologically implicated in pleural-effusion lymphomas and multicentric Castleman’s disease, which are more common among HIV-infected than among HIV-uninfected people. KSHV also has a virus-encoded cyclin, an IFN regulatory factor, and a latency-associated nuclear antigen that are implicated in increased-cell proliferation and survival.
Evidence supporting a causal role for viral infection in all of these malignancies includes (1) epidemiologic data, (2) the presence of viral DNA in all tumor cells, (3) the ability of the viruses to transform human cells in culture, (4) the results of in vitro cell culture–based assays that reveal transforming effects of specific viral genes on cell growth or survival, (5) pathologic data indicating the expression of transforming viral genes in premalignant or malignant cells in vivo, (6) the demonstration in animal models that these viral genes can cause malignant cell growth, and (7) the ability of virus-specific vaccines to reduce the incidence of virus-associated malignancy.
Virus-related malignancies provide an opportunity to expand our understanding of the biologic mechanisms important in the development of cancer. They also offer unique opportunities to develop diagnostics, vaccines, or therapeutics that could prevent or specifically treat cancers associated with viral infection. Widespread immunization against hepatitis B has resulted in a decreased prevalence of HBV-associated hepatitis and will probably prevent most HBV-related liver cancers. Current HPV vaccines can reduce rates of colonization with high-risk HPV strains and thereby decrease the risk of cervical cancer. The successful use of in vitro–expanded EBV-specific T cell populations to treat or prevent EBV-associated posttransplantation lymphoproliferative disease demonstrates the potential of immunoprevention or immunotherapy against virus-associated cancers.
RESISTANCE TO VIRAL INFECTIONS
Resistance to viral infections is initially provided by factors that are not virus-specific. Physical protection is afforded by the cornified layers of the skin and by mucous secretions that continuously sweep over mucosal surfaces. Once the first cell is infected, IFNs are induced and confer resistance to RNA virus replication. Viral infection may also trigger the release of other cytokines from infected cells. These cytokines may be chemotactic to inflammatory and immune cells. Viral protein epitopes expressed on the cell surface in the context of MHC class I and II proteins can stimulate the expansion of T cell populations with receptors that can recognize virus-encoded peptides presented on the cell surface by MHC class I proteins. Cytokines and antigens released by virus-induced cell death further attract inflammatory cells, dendritic cells, granulocytes, natural killer (NK) cells, and B lymphocytes to sites of infection and to draining lymph nodes. IFNs and NK cells are particularly important in containing viral infection for the first several days. Granulocytes and macrophages are also important in the phagocytosis and degradation of viruses, especially after an initial antibody response.
By 7–10 days after infection, virus-specific antibody responses, virus-specific human leukocyte antigen (HLA) class II–restricted CD4+ helper T lymphocyte responses, and virus-specific HLA class I–restricted CD8+ cytotoxic T lymphocyte responses develop. These responses, whose magnitude typically increases over the second and third weeks of infection, are important for rapid recovery. Also between the second and third weeks, the antibody type usually changes from IgM to IgG; IgG or IgA antibody can then be detected at infected mucosal surfaces. Antibody may directly neutralize virus by binding to its surface and preventing cell attachment or penetration. Complement can significantly enhance antibody-mediated virus neutralization. Antibody and complement can also lyse virus-infected cells that express viral membrane proteins on the cell surface. Cells infected with a replicating enveloped virus usually express the virus-envelope glycoproteins on the cell plasma membrane. Specific antibodies can bind to the glycoproteins, fix complement, and lyse the infected cell.
Antibody and CD4+/CD8+ T lymphocyte responses to viral infection can remain at high levels for several months after primary infection but usually wane over time. Low-level persistence of antibody-producing B lymphocytes and CD4+ or CD8+ T lymphocyte responses as memory cells can provide a rapid response to a second infection or an early barrier to reinfection with the same virus. Redevelopment of T cell immunity may take longer than secondary antibody responses, particularly when many years have elapsed between primary infection and reexposure. However, persistent infections or frequent reactivations from latency can result in sustained high-level T cell responses. EBV and CMV typically induce high-level CD4+ and CD8+ T cell responses that are maintained for decades after primary infection.
Some viruses have genes that alter innate and acquired host defenses. Adenoviruses encode small RNAs that inhibit IFN-induced, protein kinase R (PKR)–mediated shutoff of infected-cell protein synthesis. Adenovirus E1A can also directly inhibit IFN-mediated changes in cell gene transcription. Moreover, adenovirus E3 proteins prevent tumor necrosis factor (TNF)–induced cytolysis and block HLA class I synthesis by the infected cell. HSV ICP47 and CMV US11 also block class I antigen presentation. EBV encodes an interleukin (IL) 10 homologue that inhibits NK and T cell responses. Vaccinia virus encodes a soluble receptor for IFN-α and binding proteins for IFN-γ, IL-1, IL-18, and TNF, which inhibit host innate and adaptive immune responses. Vaccinia virus also encodes a caspase inhibitor that inhibits the ability of CD8+ cytotoxic T cells to kill virus-infected cells. Some poxviruses and herpesviruses encode chemokine-binding proteins that inhibit cell inflammatory responses. The adoption of these strategies by viruses highlights the importance of the corresponding host resistance factors in containing viral infection and the importance of redundancy in host resistance.
The host inflammatory and immune responses to viral infection do not come without a price. These responses contribute to the symptoms, signs, and other pathophysiologic manifestations of viral infection. Inflammation at sites of viral infection can subvert an effective immune response and induce tissue death and dysfunction. Moreover, immune responses to viral infection could, in principle, result in immune attack upon cross-reactive epitopes on normal cells, with consequent autoimmunity.
INTERFERONS
All human cells can synthesize IFN-α or IFN-β in response to viral infection. These IFN responses are usually induced by the presence of double-strand viral RNA, which can be made by both RNA and DNA viruses and sensed by double-strand RNA binding proteins (e.g., PKR and RIG-I) in the cell cytoplasm. IFN-γ is not closely related to IFN-α or IFN-β and is produced mainly by NK cells and by immune T lymphocytes responding to IL-12. IFN-α and -β bind to the IFN-α receptor, whereas IFN-γ binds to a different but related receptor. Both receptors signal through receptor-associated JAK kinases and other cytoplasmic proteins, including “STAT” proteins, which are tyrosine-phosphorylated by JAK kinases, translocate to the nucleus, and activate promoters for specific cell genes. Three types of antiviral effects are induced by IFN at the transcriptional level. The first effect is attributable to the induction of 2′-5′ oligo(A) synthetases, which require double-strand RNA for their activation. Activated synthetase polymerizes oligo(A) and thereby activates RNAse L, which in turn degrades single-strand RNA. A second effect results from the induction of PKR, a serine and threonine kinase that is also activated by double-strand RNA. PKR phosphorylates and negatively regulates the translational initiation factor eIF2α, shutting down protein synthesis in the infected cell. A third effect is initiated through the induction of Mx proteins, a family of GTPases that is particularly important in inhibiting the replication of influenza virus and vesicular stomatitis virus. These IFN effects are mostly directed against the infected cell, causing virus and cell dysfunction and thereby limiting viral replication.
DIAGNOSTIC VIROLOGY
A wide variety of methods are used to diagnose viral infection. Serology and virus isolation in tissue culture remain important standards. Acute- and convalescent-phase sera with rising titers of antibody to virus-specific antigens and a shift from IgM to IgG antibodies are generally accepted as diagnostic of acute viral infection. Serologic diagnosis is based on a more than fourfold rise in IgG antibody concentration when acute- and convalescent-phase serum samples are analyzed at the same time.
Immunofluorescence, hemadsorption, and hemagglutination assays for antiviral antibodies are labor-intensive and have been replaced by enzyme-linked immunosorbent assays (ELISAs), which generally use the specific viral proteins most frequently targeted by the antibody response. The proteins are purified from virus-infected cells or produced by recombinant DNA technology and are attached to a solid phase, where they can be incubated with serum, washed to eliminate nonspecific antibodies, and allowed to react with an enzyme-linked reagent to detect human IgG or IgM antibody specifically adhering to the viral antigen. The amount of antibody can then be quantitated by the intensity of a color reaction mediated by the linked enzyme. ELISAs can be sensitive and automated. Western blots can simultaneously confirm the presence of antibody to multiple specific viral proteins. The proteins are separated by size and transferred to an inert membrane, where they are incubated with serum antibodies. Western blots have an internal specificity control because the level of reactivity for viral proteins can be compared with that for cellular proteins in the same sample. Western blots require individual evaluation and are inherently difficult to quantitate or automate.
Isolation of virus in tissue culture depends on infection and replication in susceptible cells. Growth of virus in cell cultures can frequently be identified by effects on cell morphology under light microscopy. For example, HSV produces a typical cytopathic effect in rabbit kidney cells within 3 days. Other viral cytopathic effects may not be as diagnostically distinctive. Identification usually requires confirmation by staining with virus-specific monoclonal antibodies. The efficiency and speed of virus identification can be enhanced by combining short-term culture with immune detection. In assays with “shell vials” of tissue culture cells growing on a coverslip, viral infection can be detected by staining with a monoclonal antibody to a specific viral protein expressed early in viral replication. Thus, virus-infected cells can be detected within hours or days of inoculation, whereas several rounds of infection would be required to produce visible cytopathic effects.
Isolation of virus in tissue culture also depends on the collection of specimens from appropriate sites and the rapid transport of these specimens in appropriate medium to the virology laboratory (Chap. 150e). Rapid transport maintains viral viability and limits bacterial and fungal overgrowth. Enveloped viruses are generally more sensitive to freezing and thawing than nonenveloped viruses. The most appropriate site for culture depends on the pathogenesis of the virus in question. Nasopharyngeal, tracheal, or endobronchial aspirates are most appropriate for the identification of respiratory viruses. Sputum cultures generally are less appropriate because bacterial contamination and viscosity threaten tissue-culture cell viability. Aspirates of vesicular fluid are useful for isolation of HSV and VZV. Nasopharyngeal aspirates and stool specimens may be useful when the patient has fever and a rash and an enteroviral infection is suspected. Adenoviruses can be cultured from the urine of patients with hemorrhagic cystitis. CMV can frequently be isolated from cultures of urine or buffy coat. Biopsy material can be effectively cultured when viruses infect major organs, as in HSV encephalitis or adenovirus pneumonia.
The isolation of a virus does not necessarily establish disease causality. Viruses can persistently or intermittently colonize normal human mucosal surfaces. Saliva can be positive for herpesviruses, and normal urine samples can be positive for CMV. Isolations from blood, cerebrospinal fluid (CSF), or tissue are more often diagnostic of significant viral infection.
Another method aimed at increasing the speed of viral diagnosis is direct testing for antigen or cytopathic effects. Virus-infected cells from the patient may be detected by staining with virus-specific monoclonal antibodies. For example, epithelial cells obtained by nasopharyngeal aspiration can be stained with a variety of specific monoclonal antibodies to identify the specific infecting respiratory virus. Antigen and serologic assays can be multiplexed to detect multiple analytes simultaneously by coupling of reagents to color-coded beads for each analyte and detection by flow cytometry.
Nucleic acid amplification techniques bring speed, sensitivity, and specificity to diagnostic virology. The ability to directly amplify minute amounts of viral nucleic acids in specimens means that detection no longer depends on viable virus and its replication. For example, amplification and detection of HSV nucleic acids in the CSF of patients with HSV encephalitis is a more sensitive detection method than culture of virus from CSF. The extreme sensitivity of these tests can be a problem, because subclinical infection or contamination can lead to false-positive results. Detection of viral nucleic acids does not necessarily indicate virus-induced disease.
Measurement of the amount of viral RNA or DNA in peripheral blood is an important means for determining whether a patient is at increased risk for virus-induced disease and for evaluating clinical responses to antiviral chemotherapy. Nucleic acid technologies for RNA quantification are routinely used in AIDS patients to evaluate responses to antiviral agents and to detect viral resistance or noncompliance with therapy. Virus-load measurements are also useful for evaluating the treatment of patients with HBV and HCV infections. Nucleic acid testing or direct staining with CMV-specific monoclonal antibodies to quantitate virus-infected cells in the peripheral blood (CMV antigenemia) is useful for identifying immunosuppressed patients who may be at risk for CMV-induced disease.
DRUG TREATMENT FOR VIRAL INFECTIONS
Multiple steps in the life cycles of viruses can be effectively targeted by antiviral drugs (Chaps. 215e and 216). Nucleoside and nonnucleoside reverse transcriptase inhibitors prevent HIV provirus synthesis, whereas protease inhibitors block maturation of the HIV and HCV polyprotein after infection of the cell. Enfuvirtide is a small peptide derived from HIV gp41 that acts before cell infection by preventing a conformational change required for initial fusion of the virus with the cell membrane. Raltegravir is an integrase inhibitor that is approved for use with other anti-HIV drugs. Amantadine and rimantadine inhibit the influenza M2 protein, preventing release of viral RNA early during infection, whereas zanamivir and oseltamivir inhibit the influenza neuraminidase, which is necessary for the efficient release of mature virions from infected cells.
Viral genomes can evolve resistance to drugs by mutation and selection, by recombination with a drug-resistant virus, or (in the case of influenza virus and other segmented RNA viral genomes) by reassortment. The emergence of drug-resistant strains can limit the efficacy of antiviral therapy. As in antibacterial therapy, excessive and inappropriate use of antiviral therapy can select for the emergence of drug-resistant strains. HIV genotyping is a rapid method for identifying drug-resistant viruses. Resistance to reverse transcriptase or protease inhibitors has been associated with specific mutations in the reverse transcriptase or protease genes. Identification of these mutations by polymerase chain reaction amplification and nucleic acid sequencing can be clinically useful for determining which antiviral agents may still be effective. Drug resistance also can arise in herpesviruses but is a less common clinical problem.
IMMUNIZATION FOR THE PREVENTION OF VIRAL INFECTIONS
Viral vaccines are among the outstanding accomplishments of medical science. Smallpox has been eradicated except as a potential weapon of biological warfare or bioterrorism (Chap. 261e). Poliovirus eradication may soon follow. Measles can be contained or eliminated. Excess mortality due to influenza virus epidemics can be prevented, and the threat of influenza pandemics can be decreased by contemporary killed or live attenuated influenza vaccines. Mumps, rubella, and chickenpox are well controlled by childhood vaccination in the developed world. Reimmunization of mature adults can be used to control herpes zoster. New rotavirus vaccines can have a major impact on this leading cause of gastroenteritis and prominent cause of childhood death worldwide. Widespread HBV vaccination has dramatically lowered the frequency of acute and chronic hepatitis and is expected to lead to a dramatic decrease in the incidence of hepatocellular carcinoma. The HPV vaccine was the first vaccine specifically licensed to prevent virus-induced cancer. Use of purified proteins, genetically engineered live-virus vaccines, and recombinant DNA–based strategies will make it possible to immunize against severe infections with other viruses. The development of effective HIV and HCV vaccines is complicated by the high mutation rate of viral RNA polymerase and reverse transcriptase, the population-based and individual divergence of HIV or HCV genomes, and repeated high-level exposure in some populations. Concerns about the use of smallpox and other viruses as weapons necessitate maintenance of immunity to agents that are not encountered naturally.
VIRUSES AS NOVEL THERAPEUTIC TOOLS OR AGENTS
Viruses are being used experimentally to deliver biotherapeutic agents or novel vaccines. Foreign genes can be inserted into viral nucleic acids, and the recombinant virus vectors can be used to infect the patient or the patient’s cells ex vivo. Retrovirus integration into the cell genome has been used to functionally replace the abnormal gene in T cells of patients with severe combined immunodeficiency, thereby restoring immune function. Recombinant adenovirus, AAV, and retroviruses are being explored for use in diseases due to single-gene defects, such as cystic fibrosis and hemophilia. AAV carrying a lipoprotein lipase gene is now being used in Europe to treat a rare lipid-processing disease and is the first gene therapy approved for clinical use. Recombinant poxviruses, adenoviruses, and influenza viruses are also being used experimentally as vaccine vectors. Viral vectors are being tested experimentally for the expression of cytokines that can enhance immunity against tumor cells or for the expression of proteins that can increase the sensitivity of tumor cells to chemotherapy. HSV deficient for replication in resting cells is being used to selectively kill proliferating glioblastoma cells after injections into CNS tumors. For improved safety, nonreplicating viruses are frequently used in clinical trials. Potential adverse events associated with virus-mediated gene transfer include the induction of inflammatory and antiviral immune responses. Instances of retrovirus-induced human malignances have raised concerns about the safety of retroviral gene therapy vectors.
215e |
Antiviral Chemotherapy, Excluding Antiretroviral Drugs |
The field of antiviral therapy—both the number of antiviral drugs and our understanding of their optimal use—historically has lagged behind that of antibacterial treatment, but significant progress has been made in recent years on new drugs for several viral infections. The development of antiviral drugs poses several challenges. Viruses replicate intracellularly and often use host cell enzymes, macromolecules, and organelles for synthesis of viral particles. Therefore, useful antiviral compounds must discriminate between host and viral functions with a high degree of specificity; agents without such selectivity are likely to be too toxic for clinical use.
Significant progress has also been made in the development of laboratory assays to assist clinicians in the appropriate use of antiviral drugs. Phenotypic and genotypic assays for resistance to antiviral drugs are becoming more widely available, and correlations of laboratory results with clinical outcomes are being better defined. Of particular note has been the development of highly sensitive and specific methods that measure the concentration of virus in blood (virus load) and permit direct assessment of the antiviral effect of a given drug regimen in that host site. Virus load measurements have been useful in recognizing the risk of disease progression in patients with viral infections and in identifying patients for whom antiviral chemotherapy might be of greatest benefit. As with any in vitro laboratory test, results are highly dependent on and likely vary with the laboratory techniques used.
Information regarding the pharmacodynamics of antiviral drugs, and particularly the relationship of concentration effects to efficacy, has been slow to develop but is also expanding. However, assays to measure concentrations of antiviral drugs, especially of their active moieties within cells, are still primarily research procedures not widely available to clinicians. Thus, there are limited guidelines for adjusting dosages of antiviral agents to maximize antiviral activity and minimize toxicity. Consequently, clinical use of antiviral drugs must be accompanied by particular vigilance for unanticipated adverse effects.
Like that of other infections, the course of viral infections is profoundly affected by interplay between the pathogen and a complex set of host defenses. The presence or absence of preexisting immunity, the ability to mount humoral and/or cell-mediated immune responses, and the stimulation of innate immunity are important determinants of the outcome of viral infections. The state of the host’s defenses needs to be considered when antiviral agents are used or evaluated.
As with any therapy, the optimal use of antiviral compounds requires a specific and timely diagnosis. For some viral infections, such as herpes zoster, the clinical manifestations are so characteristic that a diagnosis can be made on clinical grounds alone. For other viral infections, such as influenza A, epidemiologic information (e.g., the documentation of a community-wide influenza outbreak) can be used to make a presumptive diagnosis with a high degree of accuracy. However, for most of the remaining viral infections, including herpes simplex encephalitis, cytomegaloviral infections other than retinitis, and enteroviral infections, diagnosis on clinical grounds alone cannot be accomplished with certainty. For such infections, rapid viral diagnostic techniques are of great importance. Considerable progress has also been made in recent years in the development of such tests, which are now widely available for a number of viral infections.
Despite these complexities, the efficacy of a number of antiviral compounds has been clearly established in rigorously conducted and controlled studies. As summarized in Table 215e-1, this chapter reviews the antiviral drugs that are currently approved or are likely to be considered for approval in the near future for use against viral infections other than those caused by HIV. Antiretroviral drugs are reviewed in Chap. 226.
|
ANTIVIRAL CHEMOTHERAPY AND CHEMOPROPHYLAXIS |
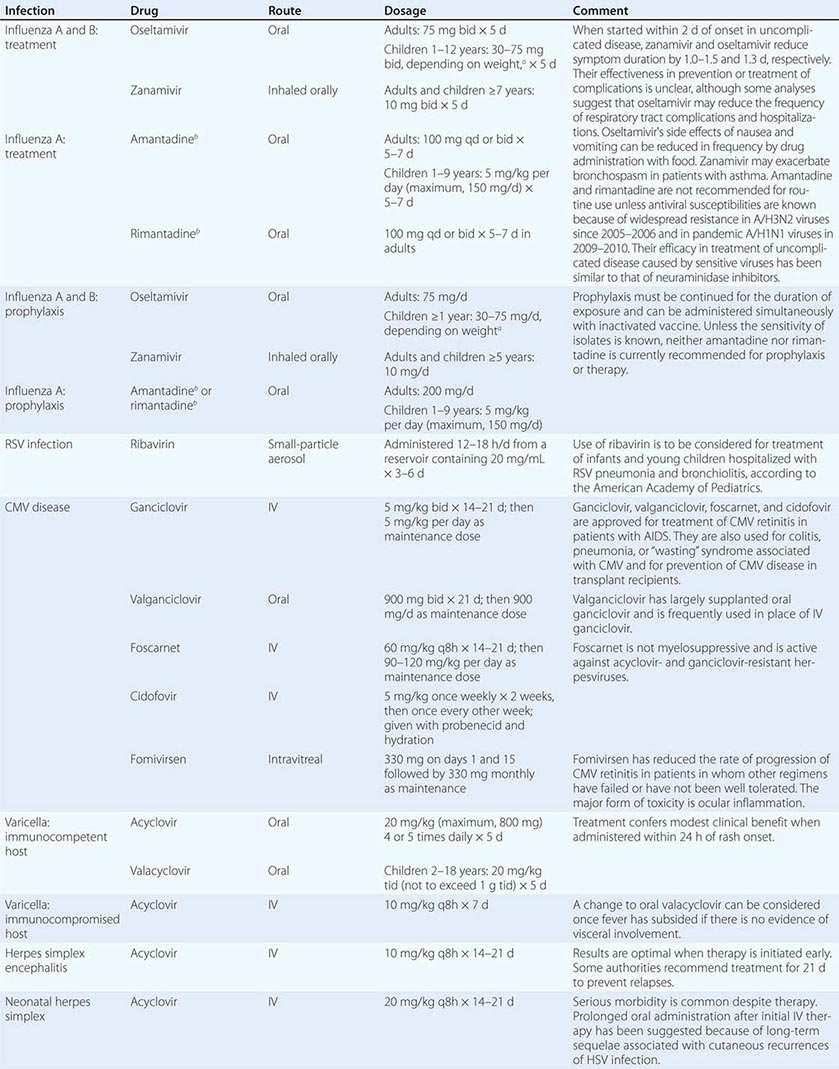
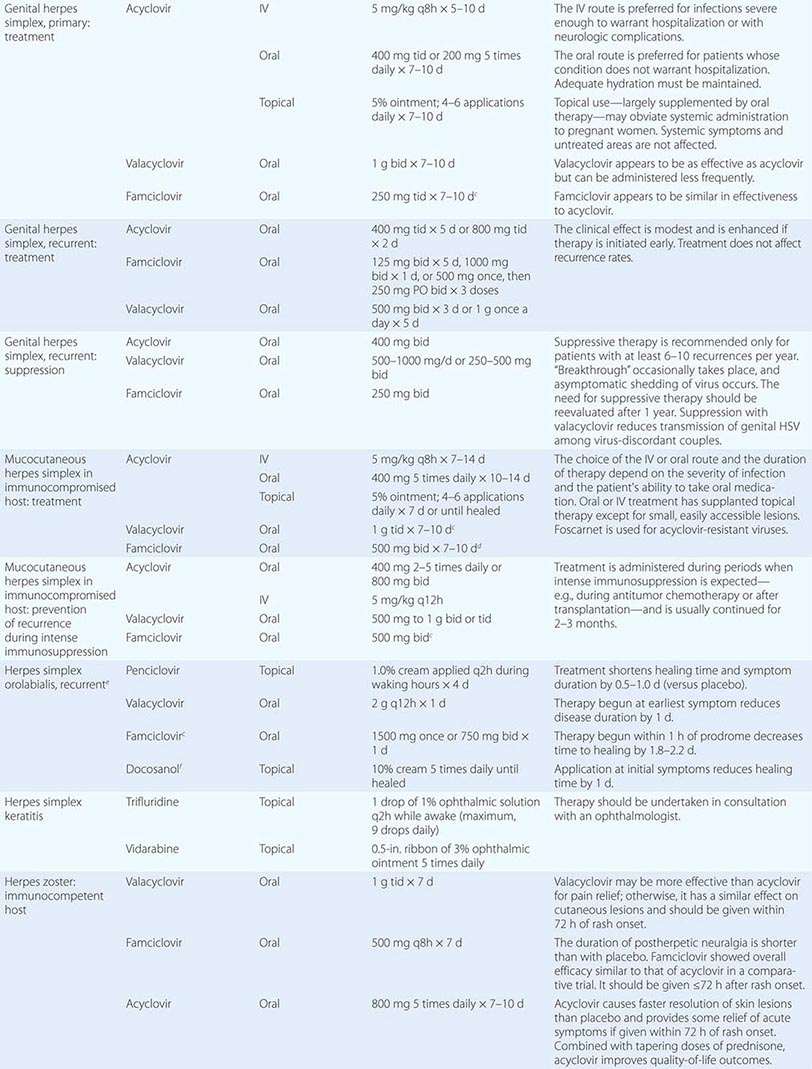
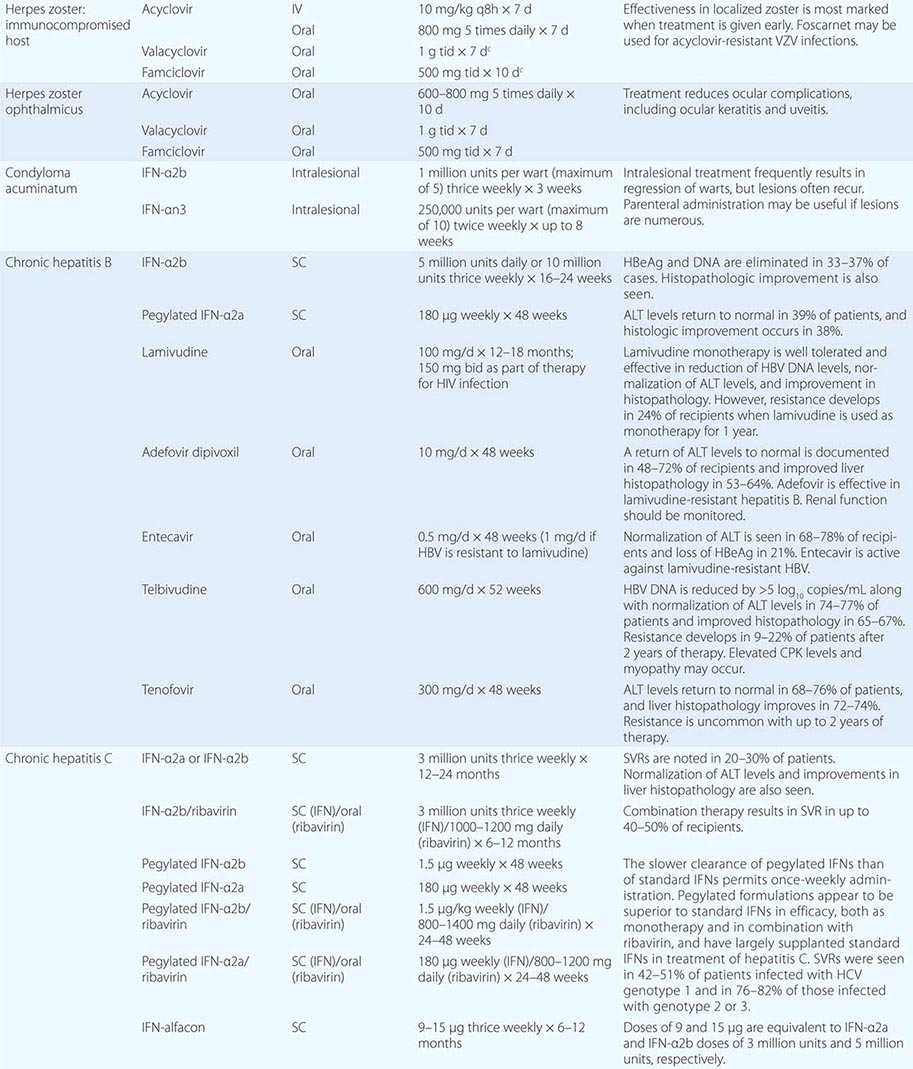
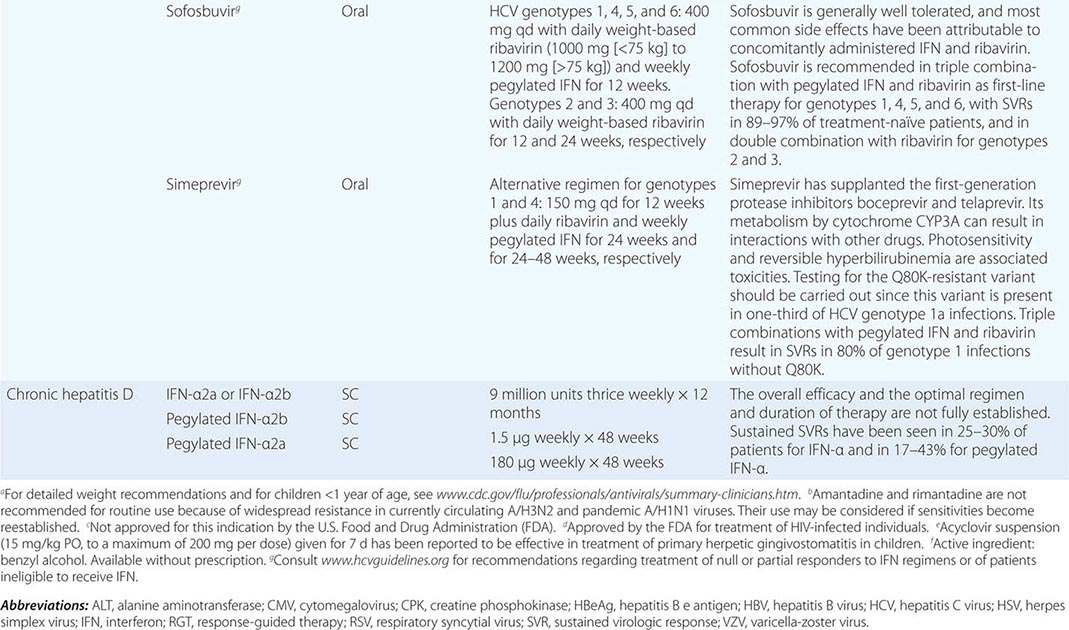
ANTIVIRAL DRUGS ACTIVE AGAINST RESPIRATORY INFECTIONS (SEE ALSO CHAPS. 223 AND 224)
ZANAMIVIR, OSELTAMIVIR, PERAMIVIR, AND LANINAMIVIR
Zanamivir and oseltamivir are inhibitors of the influenza viral neuraminidase enzyme, which is essential for release of virus from infected cells and for its subsequent spread throughout the respiratory tract of the infected host. The enzyme cleaves terminal sialic acid residues and thus destroys the cellular receptors to which the viral hemagglutinin attaches. Zanamivir and oseltamivir are sialic acid transition-state analogues and are highly active and specific inhibitors of the neuraminidases of both influenza A and B viruses. The antineuraminidase activity of the two drugs is similar, although zanamivir has somewhat greater in vitro activity against influenza B virus. Zanamivir may also be active against certain strains of influenza A virus that are resistant to oseltamivir. Both zanamivir and oseltamivir act through competitive and reversible inhibition of the active site of influenza A and B viral neuraminidases and have relatively little effect on mammalian cell enzymes.
Oseltamivir phosphate is an ethyl ester prodrug that is converted to oseltamivir carboxylate by esterases in the liver. Orally administered oseltamivir has a bioavailability of >60% and a plasma half-life of 7–9 h. The drug is excreted unmetabolized, primarily by the kidneys. Zanamivir has low oral bioavailability and is administered orally via a hand-held inhaler. By this route, ~15% of the dose is deposited in the lower respiratory tract, and low plasma levels of the drug are detected. The toxicities most frequently encountered with orally administered oseltamivir are nausea, gastrointestinal discomfort, and (less commonly) vomiting. Gastrointestinal discomfort is usually transient and is less likely if the drug is administered with food. Neuropsychiatric events (delirium, self-injury) have been reported in children who have been taking oseltamivir, primarily in Japan. Zanamivir is orally inhaled and is generally well tolerated, although exacerbations of asthma may occur. An IV formulation of zanamivir is under development and is available from GlaxoSmithKline as part of clinical trials.
Inhaled zanamivir and orally administered oseltamivir have been effective in the treatment of naturally occurring, uncomplicated influenza A or B in otherwise healthy adults. In placebo-controlled studies, illness has been shortened by 1.0–1.5 days of therapy with either of these drugs when treatment is administered within 2 days of onset of symptoms. Pooled analyses of clinical studies of oseltamivir suggest that treatment may reduce the likelihood of hospitalizations and of certain respiratory tract complications associated with influenza, and observational studies suggest that oseltamivir may reduce mortality rates associated with influenza A outbreaks (Chap. 224). Once-daily inhaled zanamivir or once-daily orally administered oseltamivir can provide prophylaxis against laboratory-documented influenza A– and influenza B–associated illness.
Resistance to the neuraminidase inhibitors may develop by changes in the viral neuraminidase enzyme, by changes in the hemagglutinin that make it more resistant to the actions of the neuraminidase, or by both mechanisms. Isolates that are resistant to oseltamivir—most commonly through the H275Y mutation, which leads to a change from histidine to tyrosine at that residue in the neuraminidase—remain sensitive to zanamivir. Certain mutations impart resistance to both oseltamivir and zanamivir (e.g., I223R, which leads to a change from isoleucine to arginine). Because the mechanisms of action of the neuraminidase inhibitors differ from those of the adamantanes (see below), zanamivir and oseltamivir are active against strains of influenza A virus that are resistant to amantadine and rimantadine.
Appropriate use of antiviral agents against influenza viruses depends on a knowledge of the resistance patterns of circulating viruses. As of this writing, currently circulating influenza A/H1N1 and H3N2 viruses (2013–2014) were sensitive to zanamivir and oseltamivir, with a few exceptions for oseltamivir. Up-to-date information on patterns of resistance to antiviral drugs is available from the Centers for Disease Control and Prevention (CDC) at www.cdc.gov/flu.
Zanamivir and oseltamivir have been approved by the U.S. Food and Drug Administration (FDA) for treatment of influenza in adults and in children (those ≥7 years old for zanamivir and those ≥1 year old for oseltamivir) who have been symptomatic for ≤2 days. Oseltamivir is approved for prophylaxis of influenza in individuals ≥1 year of age and zanamivir for those ≥5 years of age (Table 215e-1). Guidelines for the use of oseltamivir in children <1 year of age can be accessed through the CDC website, as noted in the footnote to Table 215e-1.
Peramivir is an investigational neuraminidase inhibitor that can be administered intravenously to patients for whom such an intervention is considered necessary. It has been approved in Japan, China, and South Korea but not in the United States, where it has been available in clinical trials through BioCryst Pharmaceuticals. Oseltamivir-resistant viruses generally exhibit reduced sensitivity to peramivir.
Laninamivir octonoate is an investigational neuraminidase that has been approved in Japan. It is the prodrug of laninamivir, which is administered by oral inhalation and has a prolonged half-life of ~3 days. In limited studies, it has been investigated as single-dose therapy for influenza; its effects were similar to those obtained with multiple dosing of zanamivir or oseltamivir.
AMANTADINE AND RIMANTADINE
Amantadine and the closely related compound rimantadine are primary symmetric amines that have antiviral activity limited to influenza A viruses. Amantadine and rimantadine have a long history of efficacy in the prophylaxis and treatment of influenza A infections in humans. However, high frequencies of resistance to these drugs were noted among influenza A/H3N2 viruses in the 2005–2006 influenza season and continued to be seen in 2013–2014. The pandemic A/H1N1 viruses that circulated in 2009–2010 were also resistant to amantadine and rimantadine, and circulating influenza A/H1N1 viruses in the 2013–2014 season were largely resistant. Therefore, these agents are no longer recommended unless the sensitivity of the particular isolate of influenza A virus is known, in which case their use may be considered. Amantadine and rimantadine act through inhibition of the ion channel function of the influenza A M2 matrix protein, on which uncoating of the virus depends. A substitution of a single amino acid at critical sites in the M2 protein can result in a virus that is resistant to amantadine and rimantadine.
Amantadine and rimantadine have been shown to be effective in the prophylaxis of influenza A in large-scale studies of young adults and in less extensive studies of children and elderly persons. In such studies, efficacy rates of 55–80% in the prevention of influenza-like illness were noted, and even higher rates were reported when virus-specific attack rates were calculated. Amantadine and rimantadine have also been found to be effective in the treatment of influenza A infection in studies involving predominantly young adults and, to a lesser extent, children. Administration of these compounds within 24–72 h after the onset of illness has resulted in a reduction of the duration of signs and symptoms by ~50% compared with that in placebo recipients. The effect on signs and symptoms of illness is superior to that of commonly used antipyretic-analgesic agents. Only anecdotal reports are available concerning the efficacy of amantadine or rimantadine in the prevention or treatment of complications of influenza (e.g., pneumonia).
Amantadine and rimantadine are available only in oral formulations and are ordinarily administered to adults once or twice daily, with a dosage of 100–200 mg/d. Despite their structural similarities, the two compounds have different pharmacokinetics. Amantadine is not metabolized and is excreted almost entirely by the kidneys, with a half-life of 12–17 h and peak plasma concentrations of 0.4 μg/mL. In contrast, rimantadine is extensively metabolized to hydroxylated derivatives and has a half-life of 30 h. Only 30–40% of an orally administered dose of rimantadine is recovered in the urine. The peak plasma levels of rimantadine are approximately half those of amantadine, but rimantadine is concentrated in respiratory secretions to a greater extent than amantadine. For prophylaxis, the compounds must be administered daily for the period at risk (i.e., duration of the exposure). For therapy, amantadine or rimantadine is generally administered for 5–7 days.
Although these compounds are generally well tolerated, 5–10% of amantadine recipients experience mild central nervous system side effects consisting primarily of dizziness, anxiety, insomnia, and difficulty in concentrating. These effects are rapidly reversible upon cessation of the drug’s administration. At a dose of 200 mg/d, rimantadine is better tolerated than amantadine; in a large-scale study of young adults, adverse effects were no more frequent among rimantadine recipients than among placebo recipients. Seizures and worsening of congestive heart failure have also been reported in patients treated with amantadine, although a causal relationship has not been established. The dosage of amantadine should be reduced to 100 mg/d in patients with renal insufficiency—i.e., a creatinine clearance rate (CrCl) of <50 mL/min—and in the elderly. A rimantadine dose of 100 mg/d should be used for patients with a CrCl of <10 mL/min and for the elderly.
RIBAVIRIN
Ribavirin is a synthetic nucleoside analogue that inhibits a wide range of RNA and DNA viruses. The mechanism of action of ribavirin is not completely defined and may be different for different groups of viruses. Ribavirin-5′-monophosphate blocks the conversion of inosine-5′-monophosphate to xanthosine-5′-monophosphate and interferes with the synthesis of guanine nucleotides as well as with that of both RNA and DNA. Ribavirin-5′-monophosphate also inhibits capping of virus-specific messenger RNA in certain viral systems.
Ribavirin administered as a small-particle aerosol to young children hospitalized with respiratory syncytial virus (RSV) infection has been clinically beneficial and has improved oxygenation in some studies (7 of 11). Although ribavirin has been approved for treatment of infants hospitalized with RSV infection, the American Academy of Pediatrics has recommended that it be considered on an individual basis rather than used routinely in that setting. Aerosolized ribavirin has also been administered to older children and adults (including immunosuppressed patients) with severe RSV and parainfluenza virus infections and to older children and adults with influenza A or B infection, but the benefit of this treatment, if any, is unclear. In RSV infections in immunosuppressed patients, ribavirin has been given in combination with anti-RSV immunoglobulins.
Orally administered ribavirin has not been effective in the treatment of influenza A virus infections. IV or oral ribavirin has reduced mortality rates among patients with Lassa fever; it has been particularly effective in this regard when given within the first 6 days of illness. IV ribavirin has been reported to be of clinical benefit in the treatment of hemorrhagic fever with renal syndrome caused by Hantaan virus and as therapy for Argentinean hemorrhagic fever. Oral ribavirin has also been recommended for the treatment and prophylaxis of Congo-Crimean hemorrhagic fever. Use of IV ribavirin in patients with hantavirus pulmonary syndrome in the United States has not been associated with clear-cut benefits.
Oral administration of ribavirin reduces serum aminotransferase levels in patients with chronic hepatitis C virus (HCV) infection; because it appears not to reduce serum HCV RNA levels, the mechanism of this effect is unclear. The drug provides added benefit when given by mouth in doses of 800–1200 mg/d in combination with interferon (IFN) α2b or α2a (see below), and the triple combination of ribavirin, IFN, and sofosbuvir or simeprevir has been approved for the treatment of patients with chronic HCV infection (see below). Recent data suggest that oral ribavirin may be beneficial in resolution of chronic hepatitis E infection associated with organ transplantation. Large oral doses of ribavirin (800–1000 mg/d) have been associated with reversible hematopoietic toxicity. This effect has not been observed with aerosolized ribavirin, apparently because little drug is absorbed systemically. Aerosolized administration of ribavirin is generally well tolerated but occasionally is associated with bronchospasm, rash, or conjunctival irritation. It should be administered under close supervision—particularly in the setting of mechanical ventilation, where precipitation of the drug is possible. Health care workers exposed to the drug have experienced minor toxicity, including eye and respiratory tract irritation. Because ribavirin is mutagenic, teratogenic, and embryotoxic, its use is generally contraindicated in pregnancy. Its administration as an aerosol poses a risk to pregnant health care workers. Because clearance of ribavirin is primarily renal, dose reduction is required in the setting of significant renal dysfunction.
AGENTS OF INVESTIGATIVE INTEREST
DAS181 is an investigational antiviral agent with activity against influenza A and B and parainfluenza viruses. It is a sialidase linked to a respiratory epithelium–anchoring domain. This agent cleaves the terminal sialic acid residues on human respiratory cells, reducing the binding of the aforementioned respiratory viruses. DAS181 is administered by oral inhalation and is being evaluated in the treatment of parainfluenza type 3 infections in recipients of lung and stem cell transplants.
ANTIVIRAL DRUGS ACTIVE AGAINST HERPESVIRUS INFECTIONS
ACYCLOVIR AND VALACYCLOVIR
Acyclovir is a highly potent and selective inhibitor of the replication of certain herpesviruses, including herpes simplex virus (HSV) types 1 and 2, varicella-zoster virus (VZV), and Epstein-Barr virus (EBV). It is relatively ineffective in the treatment of human cytomegalovirus (CMV) infections; however, some studies have indicated effectiveness in the prevention of CMV-associated disease in immunosuppressed patients. Valacyclovir, the L-valyl ester of acyclovir, is converted almost entirely to acyclovir by intestinal and hepatic hydrolysis after oral administration. Valacyclovir offers pharmacokinetic advantages over orally administered acyclovir: it exhibits significantly greater oral bioavailability, results in higher blood levels, and can be given less frequently than acyclovir (two or three rather than five times daily).
The high degree of selectivity of acyclovir is related to its mechanism of action, which requires that the compound first be phosphorylated to acyclovir monophosphate. This phosphorylation occurs efficiently in herpesvirus-infected cells by means of a virus-coded thymidine kinase. In uninfected mammalian cells, little phosphorylation of acyclovir occurs, and the drug is therefore concentrated in herpesvirus-infected cells. Acyclovir monophosphate is subsequently converted by host cell kinases to a triphosphate that is a potent inhibitor of virus-induced DNA polymerase but has relatively little effect on host cell DNA polymerase. Acyclovir triphosphate can also be incorporated into viral DNA, with early chain termination.
Acyclovir is available in IV, oral, and topical forms, while valacyclovir is available in an oral formulation. IV acyclovir is effective in the treatment of mucocutaneous HSV infections in immunocompromised hosts, in whom it reduces time to healing, duration of pain, and virus shedding. When administered prophylactically during periods of intense immunosuppression (e.g., related to chemotherapy for leukemia or transplantation) and before the development of lesions, IV acyclovir reduces the frequency of HSV-associated disease. After prophylaxis is discontinued, HSV lesions recur. IV acyclovir is also effective in the treatment of HSV encephalitis.
Because VZV is generally less sensitive to acyclovir than is HSV, higher doses of acyclovir must be used to treat VZV infections. In immunocompromised patients with herpes zoster, IV acyclovir reduces the frequency of cutaneous dissemination and visceral complications and—in one comparative trial—was more effective than vidarabine. Acyclovir, administered at oral doses of 800 mg five times a day, had a modest beneficial effect on localized herpes zoster lesions in both immunocompromised and immunocompetent patients. Combination of acyclovir with a tapering regimen of prednisone appeared to be more effective than acyclovir alone in terms of quality-of-life outcomes in immunocompetent patients over age 50 with herpes zoster. A comparative study of acyclovir (800 mg PO five times daily) and valacyclovir (1 g PO three times daily) in immunocompetent patients with herpes zoster indicated that the latter drug may be more effective in eliciting the resolution of zoster-associated pain. Orally administered acyclovir (600 mg five times a day) reduced complications of herpes zoster ophthalmicus in a placebo-controlled trial.
In chickenpox, a modest overall clinical benefit is attained when oral acyclovir therapy is begun within 24 h of the onset of rash in otherwise healthy children (20 mg/kg, up to a maximum of 800 mg, four times a day) or adults (800 mg five times a day). IV acyclovir has also been reported to be effective in the treatment of immunocompromised children with chickenpox.
The most widespread use of acyclovir is in the treatment of genital HSV infections. IV or oral acyclovir or oral valacyclovir has shortened the duration of symptoms, reduced virus shedding, and accelerated healing when used for the treatment of primary genital HSV infections. Oral acyclovir and valacyclovir have also had a modest effect in treatment of recurrent genital HSV infections. However, the failure of treatment of either primary or recurrent disease to reduce the frequency of subsequent recurrences has indicated that acyclovir is ineffective in eliminating latent infection. Documented chronic oral administration of acyclovir for up to 6 years or of valacyclovir for up to 1 year has reduced the frequency of recurrences markedly during therapy; once the drug is discontinued, lesions recur. In one study, suppressive therapy with valacyclovir (500 mg once daily for 8 months) reduced transmission of HSV-2 genital infections among discordant couples by 50%. A modest effect on herpes labialis (i.e., a reduction of disease duration by 1 day) was seen when valacyclovir was administered upon detection of the first symptom of a lesion at a dose of 2 g every 12 h for 1 day. In AIDS patients, chronic or intermittent administration of acyclovir has been associated with the development of HSV and VZV strains resistant to the action of the drug and with clinical failures. The most common mechanism of resistance is a deficiency of the virus-induced thymidine kinase. Patients with HSV or VZV infections resistant to acyclovir have frequently responded to foscarnet.
With the availability of the oral and IV forms, there are few indications for topical acyclovir, although treatment with this formulation has been modestly beneficial in primary genital HSV infections and in mucocutaneous HSV infections in immunocompromised hosts.
Overall, acyclovir is remarkably well tolerated and is generally free of toxicity. The most frequently encountered form of toxicity is renal dysfunction because of drug crystallization, particularly after rapid IV administration or with inadequate hydration. Central nervous system changes, including lethargy and tremors, are occasionally reported, primarily in immunosuppressed patients. However, whether these changes are related to acyclovir, to concurrent administration of other therapy, or to underlying infection remains unclear. Acyclovir is excreted primarily unmetabolized by the kidneys via both glomerular filtration and tubular secretion. Approximately 15% of a dose of acyclovir is metabolized to 9-[(carboxymethoxy)methyl]guanine or other minor metabolites. Reduction in dosage is indicated in patients with a CrCl of <50 mL/min. The half-life of acyclovir is ~3 h in normal adults, and the peak plasma concentration after a 1-h infusion of a dose of 5 mg/kg is 9.8 μg/mL. Approximately 22% of an orally administered acyclovir dose is absorbed, and peak plasma concentrations of 0.3–0.9 μg/mL are attained after administration of a 200-mg dose. Acyclovir penetrates relatively well into the cerebrospinal fluid (CSF), with concentrations approaching half of those found in plasma.
Acyclovir causes chromosomal breakage at high doses, but its administration to pregnant women has not been associated with fetal abnormalities. Nonetheless, the potential risks and benefits of acyclovir should be carefully assessed before the drug is used in pregnancy.
Valacyclovir exhibits three to five times greater bioavailability than acyclovir. The concentration-time curve for valacyclovir, given as 1 g PO three times daily, is similar to that for acyclovir, given as 5 mg/kg IV every 8 h. The safety profiles of valacyclovir and acyclovir are similar, although thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome has been reported in immunocompromised patients who have received high doses of valacyclovir (8 g/d). Valacyclovir is approved for the treatment of herpes zoster, of initial and recurrent episodes of genital HSV infection, and of herpes labialis in immunocompetent adults as well as for suppressive treatment of genital herpes. Although it has not been extensively studied in other clinical settings involving HSV or VZV infections, many consultants use valacyclovir rather than oral acyclovir in settings where only the latter has been approved because of valacyclovir’s superior pharmacokinetics and more convenient dosing schedule.
CIDOFOVIR
Cidofovir is a phosphonate nucleotide analogue of cytosine. Its major use is in CMV infections, but it is active against a broad range of herpesviruses, including HSV, human herpesvirus (HHV) types 6A and 6B, HHV-8, and certain other DNA viruses such as polyomaviruses, papillomaviruses, adenoviruses, and poxviruses, including variola (smallpox) and vaccinia. Cidofovir does not require initial phosphorylation by virus-induced kinases; the drug is phosphorylated by host cell enzymes to cidofovir diphosphate, which is a competitive inhibitor of viral DNA polymerases and, to a lesser extent, of host cell DNA polymerases. Incorporation of cidofovir diphosphate slows or terminates nascent DNA chain elongation. Cidofovir is active against HSV isolates that are resistant to acyclovir because of absent or altered thymidine kinase and against CMV isolates that are resistant to ganciclovir because of UL97 phosphotransferase mutations. CMV isolates resistant to ganciclovir on the basis of UL54 mutations are usually resistant to cidofovir as well. Cidofovir is usually active against foscarnet-resistant CMV, although cross-resistance to foscarnet has been described.
Cidofovir has poor oral availability and is administered intravenously. It is excreted primarily by the kidney and has a plasma half-life of 2.6 h. Cidofovir diphosphate’s intracellular half-life of >48 h is the basis for the recommended dosing regimen of 5 mg/kg once a week for the initial 2 weeks and then 5 mg/kg every other week. The major toxic effect of cidofovir is proximal renal tubular injury, as manifested by elevated serum creatinine levels and proteinuria. The risk of nephrotoxicity can be reduced by vigorous saline hydration and by concomitant oral administration of probenecid. Neutropenia, rashes, and gastrointestinal tolerance may also occur.
IV cidofovir has been approved for the treatment of CMV retinitis in AIDS patients who are intolerant of ganciclovir or foscarnet or in whom those drugs have failed. In a controlled study, a maintenance dosage of 5 mg/kg per week administered to AIDS patients reduced the progression of CMV retinitis from that seen at 3 mg/kg. Intravitreal cidofovir has been used to treat CMV retinitis but has been associated with significant toxicity. IV cidofovir has been reported anecdotally to be effective for treatment of acyclovir-resistant mucocutaneous HSV infections. Likewise, topically administered cidofovir is reportedly beneficial against mucocutaneous HSV infections in HIV-infected patients. Anecdotal use of IV cidofovir has been described in disseminated adenoviral infections in immunosuppressed patients and in genitourinary infections with BK virus in renal transplant recipients; however, its efficacy, if any, in these circumstances is not established.
CMX-001 (brincidofovir) is an ester prodrug of cidofovir that can be administered orally and may be less nephrotoxic than IV cidofovir. It is being evaluated for prevention of CMV infection in stem cell transplant recipients and for treatment of BK nephropathy and adenovirus infections.
FOMIVIRSEN
Fomivirsen is the first antisense oligonucleotide approved by the FDA for therapy in humans. This phosphorothioate oligonucleotide, 21 nucleotides in length, inhibits CMV replication through interaction with CMV messenger RNA. Fomivirsen is complementary to messenger transcripts of the major immediate early region 2 (IE2) of CMV, which codes for proteins regulating viral gene expression. In addition to its antisense mechanism of action, fomivirsen may exert activity against CMV through inhibition of viral adsorption to cells as well as direct inhibition of viral replication. Because of its different mechanism of action, fomivirsen is active against CMV isolates that are resistant to nucleoside or nucleotide analogues, such as ganciclovir, foscarnet, or cidofovir.
Fomivirsen has been approved for intravitreal administration in the treatment of CMV retinitis in AIDS patients who have failed to respond to other treatments or cannot tolerate them. Injections of 330 mg for two doses 2 weeks apart, followed by maintenance doses of 330 mg monthly, significantly reduce the rate of progression of CMV retinitis. The major toxicity is ocular inflammation, including vitritis and iritis, which usually responds to topically administered glucocorticoids.
GANCICLOVIR AND VALGANCICLOVIR
An analogue of acyclovir, ganciclovir is active against HSV and VZV and is markedly more active than acyclovir against CMV. Ganciclovir triphosphate inhibits CMV DNA polymerase and can be incorporated into CMV DNA, whose elongation it eventually terminates. In HSV- and VZV-infected cells, ganciclovir is phosphorylated by virus-encoded thymidine kinases; in CMV-infected cells, it is phosphorylated by a viral kinase encoded by the UL97 gene. Ganciclovir triphosphate is present in tenfold higher concentrations in CMV-infected cells than in uninfected cells. Ganciclovir is approved for the treatment of CMV retinitis in immunosuppressed patients and for the prevention of CMV disease in transplant recipients. It is widely used for the treatment of other CMV-associated syndromes, including pneumonia, esophagogastrointestinal infections, hepatitis, and “wasting” illness.
Ganciclovir is available for IV or oral administration. Because its oral bioavailability is low (5–9%), relatively large doses (1 g three times daily) must be administered by this route. Oral ganciclovir has largely been supplanted by valganciclovir, which is the L-valyl ester of ganciclovir. Valganciclovir is well absorbed orally, with a bioavailability of 60%, and is rapidly hydrolyzed to ganciclovir in the intestine and liver. The area under the curve for a 900-mg dose of valganciclovir is equivalent to that for 5 mg/kg of IV ganciclovir, although peak serum concentrations are ~40% lower for valganciclovir. The serum half-life is 3.5 h after IV administration of ganciclovir and 4.0 h after PO administration of valganciclovir. Ganciclovir is excreted primarily by the kidneys in an unmetabolized form, and its dosage should be reduced in cases of renal failure. Ganciclovir therapy at the most commonly used initial IV dosage—i.e., 5 mg/kg every 12 h for 14–21 days—can be changed to valganciclovir (900 mg PO twice daily) when the patient can tolerate oral therapy. The maintenance dose is 5 mg/kg IV daily or five times per week for ganciclovir and 900 mg by mouth once a day for valganciclovir. Dose adjustment in patients with renal dysfunction is required. Intraocular ganciclovir, given by either intravitreal injection or intraocular implantation, has also been used to treat CMV retinitis.
Ganciclovir is effective as prophylaxis against CMV-associated disease in organ and bone marrow transplant recipients. Oral ganciclovir administered prophylactically to AIDS patients with CD4+ T cell counts of <100/μL has provided protection against the development of CMV retinitis. However, the long-term benefits of this approach to prophylaxis in AIDS patients have not been established, and most experts do not recommend the use of oral ganciclovir for this purpose. As already mentioned, valganciclovir has supplanted oral ganciclovir in settings where oral prophylaxis or therapy is considered.
The administration of ganciclovir has been associated with profound bone marrow suppression, particularly neutropenia, which significantly limits the drug’s use in many patients. Bone marrow toxicity is potentiated in the setting of renal dysfunction and when other bone marrow suppressants, such as zidovudine or mycophenolate mofetil, are used concomitantly.
Resistance has been noted in CMV isolates obtained after therapy with ganciclovir, especially those from patients with AIDS or from patients receiving prolonged ganciclovir therapy after organ transplantation. Such resistance may develop through a mutation in either the viral UL97 gene or the viral DNA polymerase. Ganciclovir-resistant isolates are usually sensitive to foscarnet (see below) or may be sensitive to cidofovir, depending on the mechanism of resistance (see above).
FAMCICLOVIR AND PENCICLOVIR
Famciclovir is the diacetyl 6-deoxyester of the guanosine analogue penciclovir. This agent is well absorbed orally, has a bioavailability of 77%, and is rapidly converted to penciclovir by deacetylation and oxidation in the intestine and liver. Penciclovir’s spectrum of activity and mechanism of action are similar to those of acyclovir. Thus, penciclovir usually is not active against acyclovir-resistant viruses. However, some acyclovir-resistant viruses with altered thymidine kinase or DNA polymerase substrate specificity may be sensitive to penciclovir. This drug is phosphorylated initially by a virus-encoded thymidine kinase and subsequently by cellular kinases to penciclovir triphosphate, which inhibits HSV-1, HSV-2, VZV, and EBV as well as hepatitis B virus (HBV). The serum half-life of penciclovir is 2 h, but the intracellular half-life of penciclovir triphosphate is 7–20 h—markedly longer than that of acyclovir triphosphate. The latter is the basis for the less frequent (twice-daily) dosing schedule for famciclovir than for acyclovir. Penciclovir is eliminated primarily in the urine by both glomerular filtration and tubular secretion. The usually recommended dosage interval should be adjusted for renal insufficiency.
Clinical trials involving immunocompetent adults with herpes zoster showed that famciclovir was superior to placebo in eliciting the resolution of skin lesions and virus shedding and in shortening the duration of postherpetic neuralgia; moreover, administered at 500 mg every 8 h, famciclovir was at least as effective as acyclovir administered at an oral dose of 800 mg five times daily. Famciclovir was also effective in the treatment of herpes zoster in immunosuppressed patients. Clinical trials have demonstrated its effectiveness in the suppression of genital HSV infections for up to 1 year and in the treatment of initial and recurrent episodes of genital herpes. Famciclovir is effective as therapy for mucocutaneous HSV infections in HIV-infected patients. Application of a 1% penciclovir cream reduces the duration of signs and symptoms of herpes labialis in immunocompetent patients (by 0.5–1 day) and has been approved for that purpose by the FDA. Famciclovir is generally well tolerated, with occasional headache, nausea, and diarrhea reported in frequencies similar to those among placebo recipients. The administration of high doses of famciclovir for 2 years was associated with an increased incidence of mammary adenocarcinomas in female rats, but the clinical significance of this effect is unknown.
FOSCARNET
Foscarnet (phosphonoformic acid) is a pyrophosphate-containing compound that potently inhibits herpesviruses, including CMV. This drug inhibits DNA polymerases at the pyrophosphate binding site at concentrations that have relatively little effect on cellular polymerases. Foscarnet does not require phosphorylation to exert its antiviral activity and is therefore active against HSV and VZV isolates that are resistant to acyclovir because of deficiencies in thymidine kinase as well as against most ganciclovir-resistant strains of CMV. Foscarnet also inhibits the reverse transcriptase of HIV and is active against HIV in vivo.
Foscarnet is poorly soluble and must be administered intravenously via an infusion pump in a dilute solution over 1–2 h. The plasma half-life of foscarnet is 3–5 h and increases with decreasing renal function because the drug is eliminated primarily by the kidneys. It has been estimated that 10–28% of a dose may be deposited in bone, where it can persist for months. The most common initial dosage of foscarnet—60 mg/kg every 8 h for 14–21 days—is followed by a maintenance dose of 90–120 mg/kg once a day.
Foscarnet is approved for the treatment of CMV retinitis in patients with AIDS and of acyclovir-resistant mucocutaneous HSV infections. In a comparative clinical trial, the drug appeared to be about as efficacious as ganciclovir against CMV retinitis but was associated with a longer survival period, possibly because of its activity against HIV. Intraocular foscarnet has been used to treat CMV retinitis. In addition, foscarnet has been employed to treat acyclovir-resistant HSV and VZV infections as well as ganciclovir-resistant CMV infections, although resistance to foscarnet has been reported in CMV isolates obtained during therapy. Foscarnet has also been used to treat HHV-6 infections in immunosuppressed patients.
The major form of toxicity associated with foscarnet is renal impairment. Thus renal function should be monitored closely, particularly during the initial phase of therapy. Because foscarnet binds divalent metal ions, hypocalcemia, hypomagnesemia, hypokalemia, and hypo- or hyperphosphatemia can develop. Saline hydration and slow infusion appear to protect the patient against nephrotoxicity and electrolyte disturbances. Although hematologic abnormalities have been documented (most commonly anemia), foscarnet is not generally myelosuppressive and can be administered concomitantly with myelosuppressive medications.
TRIFLURIDINE
Trifluridine is a pyrimidine nucleoside active against HSV-1, HSV-2, and CMV. Trifluridine monophosphate irreversibly inhibits thymidylate synthetase, and trifluridine triphosphate inhibits viral and, to a lesser extent, cellular DNA polymerases. Because of systemic toxicity, trifluridine’s use is limited to topical therapy. Trifluridine is approved for treatment of HSV keratitis, against which trials have shown that it is more effective than topical idoxuridine but similar in efficacy to topical vidarabine. The drug has benefited some patients with HSV keratitis who have failed to respond to idoxuridine or vidarabine. Topical application of trifluridine to sites of acyclovir-resistant HSV mucocutaneous infection has also been beneficial in some cases.
VIDARABINE
Vidarabine is a purine nucleoside analogue with activity against HSV-1, HSV-2, VZV, and EBV. Vidarabine inhibits viral DNA synthesis through its 5′-triphosphorylated metabolite, although its precise molecular mechanisms of action are not completely understood. IV-administered vidarabine has been shown to be effective in the treatment of herpes simplex encephalitis, mucocutaneous HSV infections, herpes zoster in immunocompromised patients, and neonatal HSV infections. Its use has been supplanted by that of IV acyclovir, which is more effective and easier to administer. Production of the IV preparation has been discontinued by the manufacturer, but vidarabine is available as an ophthalmic ointment, which is effective in the treatment of HSV keratitis.
AGENTS OF INVESTIGATIVE INTEREST
Maribavir is a benzimidazole that inhibits CMV and EBV. This drug inhibits the CMV UL97 kinase and does not require intracellular phosphorylation for its antiviral activity. Its mechanism of action involves blocking viral DNA synthesis and virion egress. Maribavir is orally administered and has been associated with taste disturbance and diarrhea. In phase 3 studies, it was not efficacious in the prevention of CMV infection in recipients of hematopoietic stem cell and adult liver transplants. However, when used at somewhat higher doses, it may be efficacious for the treatment of refractory or resistant CMV infections in transplant recipients.
Letermovir is an investigational drug with activity against CMV. It is a dihydroquinozoline that acts through inhibition of the viral terminase enzyme complex. This mechanism of action differs from that of ganciclovir, foscarnet, and cidofovir, which inhibit viral DNA polymerase; therefore, letermovir is active against CMV isolates that are resistant to those drugs. It is orally administered and is reportedly well tolerated. Letermovir is being evaluated as prophylaxis against CMV in hematopoietic stem cell recipients.
Inhibition of the helicase-primase heterotrimeric complex of HSV-1 and HSV-2 represents a novel mechanism of action of amenamevir and pritelivir. These drugs are being assessed for prevention and treatment of HSV genital infection. The efficacy of amenamevir, administered as a single oral dose of 1200 mg for recurrent genital herpes, was comparable to that of valacyclovir given for 3 days. Pritelivir has a long half-life (up to 80 h) and was studied in a placebo-controlled trial of suppression of genital HSV infections. Compared with placebo, pritelivir—a loading dose followed by either a daily oral dose of 75 mg for 4 weeks or a weekly dose of 400 mg for 4 weeks—reduced HSV shedding and days of genital lesions. Additional clinical studies of the helicase-primase inhibitors of HSV are planned.
ANTIVIRAL DRUGS ACTIVE AGAINST HEPATITIS VIRUSES
LAMIVUDINE
Lamivudine is a pyrimidine nucleoside analogue that is used primarily in combination therapy against HIV infection (Chap. 226). Its activity against HBV is attributable to inhibition of the viral DNA polymerase. This drug has also been approved for the treatment of chronic HBV infection. At doses of 100 mg/d given for 1 year to patients positive for hepatitis B e antigen (HBeAg), lamivudine is well tolerated and results in suppression of HBV DNA levels, normalization of serum amino-transferase levels in 40–75% of patients, and reduction of hepatic inflammation and fibrosis in 50–60% of patients. Loss of HBeAg occurs in 30% of patients. Lamivudine also appears to be useful in the prevention or suppression of HBV infection associated with liver transplantation. Resistance to lamivudine develops in 24% of patients treated for 1 year and is associated with changes in the YMDD motif of HBV DNA polymerase. Because of the frequency of development of resistance, lamivudine has been largely supplanted by less-resistance-prone drugs for the treatment of HBV infection.
ADEFOVIR DIPIVOXIL
Adefovir dipivoxil is the oral prodrug of adefovir, an acyclic nucleotide analogue of adenosine monophosphate that is active against HBV, HIV, HSV, CMV, and poxviruses. It is phosphorylated by cellular kinases to the active triphosphate moiety, which is a competitive inhibitor of HBV DNA polymerase and results in chain termination after incorporation into nascent viral DNA. Adefovir is administered orally and is eliminated primarily by the kidneys, with a plasma half-life of 5–7.5 h. In clinical studies, therapy with adefovir at a dose of 10 mg/d for 48 weeks resulted in normalization of serum alanine aminotransferase (ALT) levels in 48–72% of patients and improved liver histology in 53–64%; it also resulted in a 3.5- to 3.9-log10 reduction in the number of HBV DNA copies per milliliter of plasma. Adefovir was effective in treatment-naïve patients as well as in those infected with lamivudine-resistant HBV. Resistance to adefovir appears to develop less readily than that to lamivudine, but adefovir resistance rates of 15–18% have been reported after 192 weeks of treatment and may reach 30% after 5 years. This agent is generally well tolerated. Significant nephrotoxicity attributable to adefovir is uncommon at the dose used in the treatment of HBV infections (10 mg/d) but is a treatment-limiting adverse effect at the higher doses used in therapy for HIV infections (30–120 mg/d). In any case, renal function should be monitored in patients taking adefovir, even at the lower dose. Adefovir is approved only for treatment of chronic HBV infection.
TENOFOVIR DISOPROXIL FUMARATE
Tenofovir disoproxil fumarate is a prodrug of tenofovir, a nucleotide analogue of adenosine monophosphate with activity against both retroviruses and hepadnaviruses. In both immunocompetent and immunocompromised patients (including those co-infected with HIV and HBV), tenofovir given at a dose of 300 mg/d for 48 weeks reduced HBV replication by 4.6–6 log10, normalized ALT levels in 68–76% of patients, and improved liver histopathology in 72–74% of patients. Resistance develops uncommonly during ≥2 years of therapy, and tenofovir is active against lamivudine-resistant HBV. The safety profile of tenofovir is similar to that of adefovir, but nephrotoxicity has not been encountered at the dose used for HBV therapy. Tenofovir is approved for the treatment of HIV and chronic HBV infections. For a more detailed discussion of tenofovir, see Chap. 226.
ENTECAVIR
Entecavir is a cyclopentyl 2′-deoxyguanosine analogue that inhibits HBV through interaction of entecavir triphosphate with several HBV DNA polymerase functions. At a dose of 0.5 mg/d given for 48 weeks, entecavir reduced HBV DNA copies by 5.0–6.9 log10, normalized serum aminotransferase levels in 68–78% of patients, and improved histopathology in 70–72% of patients. Entecavir inhibits lamivudine-resistant viruses that have M550I or M550V/L526M mutations but only at serum concentrations 20- or 30-fold higher than those obtained with the 0.5-mg/d dose. Thus, higher doses of entecavir (1 mg/d) are recommended for the treatment of patients infected with lamivudine-resistant HBV. Development of resistance to entecavir is uncommon in treatment-naïve patients but does occur at unacceptably high rates (43% after 4 years) in patients previously infected with lamivudine-resistant virus. Entecavir-resistant strains appear to be sensitive to adefovir and tenofovir.
Entecavir is highly bioavailable but should be taken on an empty stomach because food interferes with its absorption. The drug is eliminated primarily in unchanged form by the kidneys, and its dosage should be adjusted for patients with CrCl values of <50 mL/min. Overall, entecavir is well tolerated, with a safety profile similar to that of lamivudine. As with other anti-HBV treatments, exacerbation of hepatitis may occur when entecavir therapy is stopped. Entecavir is approved for treatment of chronic hepatitis B, including infection with lamivudine-resistant viruses, in adults. Entecavir has some activity against HIV-1 (median effective concentration, 0.026 to >10 μM) but should not be used as monotherapy in HIV-positive patients because of the potential for development of HIV resistance due to the M184V mutation.
TELBIVUDINE
Telbivudine is a β-L enantiomer of thymidine and is a potent, selective inhibitor of HBV. Its active form is telbivudine triphosphate, which inhibits HBV DNA polymerase and causes chain termination but has little or no activity against human DNA polymerase. Administration of telbivudine at an oral dose of 600 mg/d for 52 weeks to patients with chronic hepatitis B resulted in reduction of HBV DNA by 5.2–6.4 log10 copies/mL along with normalization of ALT levels in 74–77% of recipients and improved histopathology in 65–67% of patients. Telbivudine-resistant HBV is generally cross-resistant with lamivudine-resistant virus but is usually susceptible to adefovir. After 2 years of therapy, resistance to telbivudine was noted in isolates from 22% of HBeAg-positive patients and in those from 9% of HBeAg-negative patients.
Orally administered telbivudine is rapidly absorbed; because it is eliminated primarily by the kidneys, its dosage should be reduced in patients with a CrCl value of <50 mL/min. Telbivudine is generally well tolerated, but increases in serum levels of creatinine kinases as well as fatigue and myalgias have been observed. As with other anti-HBV drugs, hepatitis may be exacerbated in patients who discontinue telbivudine therapy. Telbivudine has been approved for the treatment of adults with chronic hepatitis B who have evidence of viral replication and either persistently elevated serum aminotransferase levels or histopathologically active disease, but it has not been widely used because of the frequency of development of resistance, as noted above.
INTERFERONS
IFNs are cytokines that exhibit a broad spectrum of antiviral activities as well as immunomodulating and antiproliferative properties. IFNs are not available for oral administration but must be given IM, SC, or IV. Early studies with human leukocyte IFN demonstrated an effect in the prophylaxis of experimentally induced rhinovirus infections in humans and in the treatment of VZV infections in immunosuppressed patients. DNA recombinant technology has made available highly purified α, β, γ, and λ IFNs that have been evaluated in a variety of viral infections. Results of such trials have confirmed the effectiveness of intranasally administered IFN in the prophylaxis of rhinovirus infections, although its use has been associated with nasal mucosal irritation. Studies have also demonstrated a beneficial effect of intralesionally or systemically administered IFNs on genital warts. The effect of systemic administration consists primarily of a reduction in the size of the warts, and this mode of therapy may be useful in persons who have numerous warts that cannot easily be treated by individual intralesional injections. However, lesions frequently recur after either intralesional or systemic IFN therapy is discontinued.
IFNs have undergone extensive study in the treatment of chronic HBV infection. The administration of standard IFN-α2b (5 million units daily or 10 million units three times a week for 16–24 weeks) to patients with stable chronic HBV infection resulted in loss of markers of HBV replication, such as HBeAg and HBV DNA, in 33–37% of cases; 8% of patients also became negative for hepatitis B surface antigen. In most patients who lose HBeAg and HBV DNA markers, serum aminotransferases return to normal levels, and both short- and long-term improvements in liver histopathology have been described. Predictors of a favorable response to standard IFN therapy include low pretherapy levels of HBV DNA, high pretherapy serum levels of ALT, a short duration of chronic HBV infection, and active inflammation in liver histopathology. Poor responses are seen in immunosuppressed patients, including those with HIV infection.
In pegylated IFNs, IFN alphas are linked to polyethylene glycol. This linkage results in slower absorption, decreased clearance, and more sustained serum concentrations, thereby permitting a more convenient, once-weekly dosing schedule; in many instances, pegylated IFN has supplanted standard IFN. After 48 weeks of treatment with 180 μg of pegylated IFN-α2a, HBV DNA was reduced by 4.1–4.5 log10 copies/mL, with normalization of serum ALT levels in 39% of patients and improved histology in 38%. Response rates were somewhat higher when lamivudine was administered with pegylated IFN-α2a. Adverse effects of IFN are common and include fever, chills, myalgia, fatigue, neurotoxicity (manifested primarily as somnolence, depression, anxiety, and confusion), and leukopenia. Autoantibodies (e.g., antithyroid antibodies) can also develop. IFN-α2b and pegylated IFN-α2a are approved for the treatment of patients with chronic hepatitis B. Data supporting the therapeutic efficacy of pegylated interferon-α2b in HBV infection have been published; the drug has not been approved for this indication in the United States but has been approved for treatment of chronic HBV infection in other countries.
Several IFN preparations, including IFN-α2a, IFN-α2b, IFN-alfacon-1, and IFN-αm1 (lymphoblastoid), have been studied as therapy for chronic HCV infections. A variety of monotherapy regimens have been studied, of which the most common for standard IFN is IFN-α2b or -α2a at 3 million units three times per week for 12–18 months. The addition of oral ribavirin to IFN-α2b—either as initial therapy or after failure of IFN therapy alone—results in significantly higher rates of sustained virologic and/or serum ALT responses (40–50%) than are obtained with monotherapy. Comparative studies indicate that pegylated IFN-α2b or -α2a therapy is more effective than standard IFN treatment against chronic HCV infection. The combination of SC pegylated IFN and oral ribavirin results in sustained virologic responses (SVRs) in 42–51% of patients with HCV genotype 1 infection and in 76–82% of patients with genotype 2 or 3 infection. Ribavirin appears to have a small antiviral effect in HCV infection but may also be working through an immunomodulatory effect in combination with IFN. Optimal results with ribavirin appear to be associated with weight-based dosing. Prognostic factors for a favorable response include an age of <40 years, a short duration of infection, low levels of HCV RNA, a lesser degree of liver histopathology, and infection with HCV genotypes other than 1. IFN-alfacon, a synthetic “consensus” α interferon, appears to produce response rates similar to those elicited by standard IFN-α2a or -α2b alone. In 2014, the approval of a polymerase inhibitor, sofosbuvir, and a second-generation protease inhibitor, simeprevir, led to revised recommendations for treatment of hepatitis C with triple combinations of pegylated IFN, ribavirin, and one of these two drugs, depending on the viral genotype (see below and Table 215e-1).
IFN-α and pegylated IFN-α are active against hepatitis D, but high doses are required (9 million units three times per week for 48 weeks). IFN-α elicited an SVR in 25–30% of patients, whereas pegylated IFN-α had a variable effect, evoking an SVR in 17–43% of patients. However, long-term biochemical and histologic improvements have been seen, even in the absence of sustained inhibition of viral replication.
POLYMERASE INHIBITORS
SOFOSBUVIR
Sofosbuvir is the prodrug of a uridine nucleoside inhibitor of the HCV RNA NS5B polymerase. Its metabolism to the active uridine nucleoside triphosphate results in chain termination. Sofosbuvir is active against all HCV genotypes (1–6) and has a median effective concentration (EC50) of 0.7–2.6 μM against NS5B. Resistance to sofosbuvir is conferred by an S282T substitution in NS5B, but clinically expressed resistance to treatment has only rarely been encountered in patients who receive sofosbuvir.
Sofosbuvir is administered orally and is unaffected by food. After oral administration, plasma concentrations of sofosbuvir and of its active metabolite peak in 0.5–2 h and 2–4 h, respectively. Approximately 61–65% of sofosbuvir is bound in plasma proteins, but very little of the active metabolite is bound. Both sofosbuvir and its active metabolite are cleared renally, with t1/2 values of 0.4 h and 27 h, respectively. Sofosbuvir is relatively free from clinically significant drug interactions, although P′glycoprotein inducers can reduce sofosbuvir concentrations.
Sofosbuvir is generally well tolerated and has not been associated with significant toxicity. The most common side effects in recipients of sofosbuvir have been attributable to concomitant administration of IFN and ribavirin in combination clinical trials (see below).
Sofosbuvir has been studied in a variety of controlled and open-label clinical trials. In late 2013, the results of these trials led to its recommendation—in triple combination with pegylated IFN and ribavirin—as first-line treatment for chronic hepatitis due to HCV genotypes 1, 4, 5, and 6, in which SVRs among treatment-naïve patients were 89–97%. For HCV genotypes 2 and 3, IFN-free regimens consisting of sofosbuvir and ribavirin have been recommended, with SVRs among treatment-naïve patients of 93% for genotype 2 and 61% for genotype 3.
PROTEASE INHIBITORS
BOCEPREVIR, TELAPREVIR
This drug class is specifically designed to inhibit the 3/4A (NS3/4A) HCV protease. These agents resemble the HCV polypeptide and, when processed by the viral protease, form a covalent bond with the catalytic NS3 serine residues, block further activity, and prevent proteolytic cleavage of the HCV polyprotein into NS4A, NS4B, NS5A, and NS5B proteins. Boceprevir and telaprevir are linear ketoamide compounds that are active against HCV genotype 1 (1b > 1a) and much less so against genotypes 2 and 3. These first-generation protease inhibitors received approval for combination therapy (with IFN and ribavirin) for genotype 1 infection. Neither boceprevir nor telaprevir is now recommended for the treatment of hepatitis C. These drugs have been supplanted by sofosbuvir and by simeprevir, a second-generation protease inhibitor with improved pharmacokinetic properties, fewer drug–drug interactions, and less overall toxicity (see below).
SIMEPREVIR
Simeprevir is a second-generation NS3/4A protease inhibitor with antiviral activity against genotype 1 (1b > 1a); the EC50 is 9.4 nM in an HCV genotype 1b replicon. The NS3 polymorphism Q80K, which is present in approximately one-third of patients carrying HCV genotype 1b, increases the EC50 by elevenfold and results in clinical resistance to simeprevir. Thus, testing for Q80K should be carried out if treatment with simeprevir is being considered. Cross-resistance occurs between simeprevir and the first-generation protease inhibitors boceprevir and telaprevir.
Simeprevir is orally administered as a 150-mg capsule, and its bioavailability is increased by administration with food. The serum concentration peaks 4–6 h after oral administration. The drug’s elimination half-life is 10–13 h in healthy individuals and 41 h in patients with hepatitis C. Simeprevir is nearly entirely bound by plasma proteins and cleared by biliary excretion. Because there is no renal excretion, dose adjustments are not required in the presence of renal dysfunction. Simeprevir is metabolized by hepatic CYP3A and therefore should not be used in patients with decompensated liver function.
Because of its metabolism by cytochrome P450 3A (CYP3A), simeprevir interacts with drugs that induce or inhibit CYP3A, and these interactions may concomitantly increase or reduce plasma concentrations of simeprevir. Administration of simeprevir may also increase plasma concentrations of drugs that are substrates for hepatic organic anion-transporting polypeptide 1B1 or 1B3 or for P glycoprotein transporters.
Toxicity observed during clinical trials with simeprevir included photosensitivity (usually mild or moderate) in 28% of recipients and reversible hyperbilirubinemia (both conjugated and unconjugated), which was generally mild to moderate. Most of the other adverse effects that were seen in clinical trials with simeprevir were attributable to concomitant administration of IFN and ribavirin.
Simeprevir has been recommended as a component of alternative treatment—in combination with pegylated IFN and ribavirin—of chronic infection with HCV genotypes 1 and 4. Daily simeprevir, daily ribavirin, and weekly pegylated IFN for 12 weeks followed by another 12 weeks of pegylated IFN and ribavirin resulted in an SVR of 80% in the absence of the Q80K variant. In general, simeprevir-based triple therapy appeared to be 10% less likely to yield an SVR than sofosbuvir-based therapy and more likely to cause adverse effects. However, for prior nonresponders or partial responders to pegylated IFN, the IFN-free regimen of simeprevir, sofosbuvir, and ribavirin shows promise.
INVESTIGATIVE AGENTS OF INTEREST
Next-generation direct-acting antivirals against HCV inhibitors are under active development. These agents include second-generation inhibitors of NS3/4, NS5B polymerase inhibitors, and inhibitors of NS5A (a membrane-associated phosphoprotein that is part of the HCV RNA replication complex). These investigational agents are making progress toward IFN-free regimens, shorter courses of therapy, improved tolerability, and reduction of resistance. For updated information, readers should consult http://www.hcvguidelines.org/.
SECTION 12 |
INFECTIONS DUE TO DNA VIRUSES |
216 |
Herpes Simplex Virus Infections |
DEFINITION
Herpes simplex viruses (HSV-1, HSV-2; Herpesvirus hominis) produce a variety of infections involving mucocutaneous surfaces, the central nervous system (CNS), and—on occasion—visceral organs. Prompt recognition and treatment reduce the morbidity and mortality rates associated with HSV infections.
ETIOLOGIC AGENT
![]() The genome of HSV is a linear, double-strand DNA molecule (molecular weight, ~100 × 106) that encodes >90 transcription units with 84 identified proteins. The genomic structures of the two HSV subtypes are similar. The overall genomic sequence homology between HSV-1 and HSV-2 is ~50%, whereas the proteome homology is >80%. The homologous sequences are distributed over the entire genome map, and most of the polypeptides specified by one viral type are antigenically related to polypeptides of the other viral type. Many type-specific regions unique to HSV-1 and HSV-2 proteins do exist, however, and a number of them appear to be important in host immunity. These type-specific regions have been used to develop serologic assays that distinguish between the two viral subtypes. Either restriction endonuclease analysis or sequencing of viral DNA can be used to distinguish between the two subtypes and among strains of each subtype. The variability of nucleotide sequences from clinical strains of HSV-1 and HSV-2 is such that HSV isolates obtained from two individuals can be differentiated by restriction enzyme patterns or genomic sequences. Moreover, epidemiologically related sources, such as sexual partners, mother-infant pairs, or persons involved in a common-source outbreak, can be inferred from such patterns.
The genome of HSV is a linear, double-strand DNA molecule (molecular weight, ~100 × 106) that encodes >90 transcription units with 84 identified proteins. The genomic structures of the two HSV subtypes are similar. The overall genomic sequence homology between HSV-1 and HSV-2 is ~50%, whereas the proteome homology is >80%. The homologous sequences are distributed over the entire genome map, and most of the polypeptides specified by one viral type are antigenically related to polypeptides of the other viral type. Many type-specific regions unique to HSV-1 and HSV-2 proteins do exist, however, and a number of them appear to be important in host immunity. These type-specific regions have been used to develop serologic assays that distinguish between the two viral subtypes. Either restriction endonuclease analysis or sequencing of viral DNA can be used to distinguish between the two subtypes and among strains of each subtype. The variability of nucleotide sequences from clinical strains of HSV-1 and HSV-2 is such that HSV isolates obtained from two individuals can be differentiated by restriction enzyme patterns or genomic sequences. Moreover, epidemiologically related sources, such as sexual partners, mother-infant pairs, or persons involved in a common-source outbreak, can be inferred from such patterns.
The viral genome is packaged in a regular icosahedral protein shell (capsid) composed of 162 capsomeres (see Fig. 214e-1). The outer covering of the virus is a lipid-containing membrane (envelope) acquired as the DNA-containing capsid buds through the inner nuclear membrane of the host cell. Between the capsid and lipid bilayer of the envelope is the tegument. Viral replication has both nuclear and cytoplasmic phases. Initial attachment to the cell membrane involves interactions of viral glycoproteins C and B with several cellular heparan sulfate–like surface receptors. Subsequently, viral glycoprotein D binds to cellular co-receptors that belong to the tumor necrosis factor receptor family of proteins, the immunoglobulin superfamily (nectin family), or both. The ubiquity of these receptors contributes to the wide host range of herpesviruses. HSV replication is highly regulated. After fusion and entry, the nucleocapsid enters the cytoplasm and several viral proteins are released from the virion. Some of these viral proteins shut off host protein synthesis (by increasing cellular RNA degradation), whereas others “turn on” the transcription of early genes of HSV replication. These early gene products, designated α genes, are required for synthesis of the subsequent polypeptide group, the β polypeptides, many of which are regulatory proteins and enzymes required for DNA replication. Most current antiviral drugs interfere with β proteins, such as viral DNA polymerase. The third (γ) class of HSV genes requires viral DNA replication for expression and encodes most structural proteins specified by the virus.
After viral genome replication and structural protein synthesis, nucleocapsids are assembled in the cell’s nucleus. Envelopment occurs as the nucleocapsids bud through the inner nuclear membrane into the perinuclear space. In some cells, viral replication in the nucleus forms two types of inclusion bodies: type A basophilic Feulgen-positive bodies that contain viral DNA and eosinophilic inclusion bodies that are devoid of viral nucleic acid or protein and represent a “scar” of viral infection. Enveloped virions are then transported via the endoplasmic reticulum and the Golgi apparatus to the cell surface.
Viral genomes are maintained by some neuronal cells in a repressed state called latency. Latency, which is associated with transcription of only a limited number of virus-encoded RNAs, accounts for the presence of viral DNA and RNA in neural tissue at times when infectious virus cannot be isolated. Maintenance and growth of neural cells from latently infected ganglia in tissue culture result in production of infectious virions (explantation) and in subsequent permissive infection of susceptible cells (co-cultivation). Activation of the viral genome may then occur, resulting in reactivation—the normal pattern of regulated viral gene expression and replication and HSV release. The release of virions from the neuron follows a complex process of anterograde transport down the length of neuronal axons. In experimental animals, ultraviolet light, systemic and local immunosuppression, and trauma to the skin or ganglia are associated with reactivation.
Three noncoding RNA latency-associated transcripts (LATs) are found in the nuclei of latently infected neurons. Deletion mutants of the LAT region exhibit reduced efficiency in their later reactivation. Substitution of HSV-1 LATs for HSV-2 LATs induces an HSV-1 reactivation pattern. These data indicate that LATs apparently maintain—rather than establish—latency. HSV-1 LATs promote the survival of acutely infected neurons, perhaps by inhibiting apoptotic pathways. LAT transcript abundance and low genome-copy number correlate with subnuclear positioning of HSV genomes around the centromere. Indeed, chromatization of HSV DNA appears to play a vital role in silencing expression of lytic replication genes. Highly expressed during latency, LAT-derived micro-RNA appears to silence expression of the key neurovirulence factor infected-cell protein 34.5 (ICP34.5) and to bind in an antisense configuration to the immediate-early protein ICP0 messenger RNA to prevent expression, which is vital to HSV reactivation. Although certain viral transcripts are known to be necessary for reactivation from latency, the molecular mechanisms of HSV latency are not fully understood, and strategies to interrupt or maintain latency in neurons are in developmental stages.
While latency is the predominant state of virus on a per-neuron basis, the high frequency of oral and genital tract reactivation for HSV-1 and HSV-2 suggests that the viruses are rarely quiescent within the entire biomass of ganglionic tissue. Recent data indicate that HSV-2 antigen is often shed: most persons infected with HSV-2 have frequent subclinical bursts of reactivation lasting 2–4 h, and the host mucosal immune system can contain viral reactivation in the mucosa before the development of clinical reactivation. Supporting this clinical observation, recent work using microdissection plus real-time polymerase chain reaction (PCR) of individual neurons from cadaveric trigeminal ganglia explants revealed that many more neurons (2–10%) harbor HSV than would be predicted by in situ hybridization studies for LATs. Viral copy number is highly variable between neurons, with extremely high levels in certain neurons, and HSV DNA copy numbers are similar in LAT-positive and LAT-negative neurons; these findings add to the uncertainty about the role that LATs play in preventing reactivation.
PATHOGENESIS
Exposure to HSV at mucosal surfaces or abraded skin sites permits entry of the virus into cells of the epidermis and dermis and initiation of viral replication therein. HSV infections are usually acquired subclinically. Whether clinical or subclinical, HSV acquisition is associated with sufficient viral replication to permit infection of either sensory or autonomic nerve endings. On entry into the neuronal cell, the virus—or, more likely, the nucleocapsid—is transported intra-axonally to the nerve cell bodies in ganglia. In humans, the transit interval of spread to the ganglia after virus inoculation into peripheral tissue is unknown. During the initial phase of infection, viral replication occurs in ganglia and contiguous neural tissue. Virus then spreads to other mucocutaneous surfaces through centrifugal migration of infectious virions via peripheral sensory nerves. This mode of spread helps explain the large surface area involved, the high frequency of new lesions distant from the initial crop of vesicles that is characteristic in patients with primary genital or oral-labial HSV infection, and the ability to recover virus from neural tissue distant from neurons innervating the inoculation site. Contiguous spread of locally inoculated virus also may take place and allow further mucosal extension of disease. Recent studies have demonstrated HSV viremia—another mechanism for extension of infection throughout the body—in ~30–40% of persons with primary HSV-2 infection. Latent infection with both viral subtypes in both sensory and autonomic ganglia has been demonstrated. For HSV-1 infection, trigeminal ganglia are most commonly infected, although extension to the inferior and superior cervical ganglia also occurs. With genital infection, sacral nerve root ganglia (S2–S5) are most commonly affected.
After resolution of primary disease, infectious HSV can no longer be cultured from the ganglia; however, latent infection, as defined by the presence of viral DNA, persists in 2–11% of ganglionic cells in the anatomic region of the initial infection. The mechanism of reactivation from latency is unknown. Increasingly, studies indicate that host T cell responses at the ganglionic and peripheral mucosal levels influence the frequency and severity of HSV reactivation. HSV-specific T cells have been recovered from peripheral-nerve root ganglia. Many of these resident CD8+ T cells are juxtaposed with latently HSV-1-infected neurons in the trigeminal ganglia and can block reactivation with both interferon (IFN) γ release and granzyme B degradation of the immediate-early protein ICP4. In addition, there appears to be a latent viral load in the ganglia that correlates positively with the number of neurons infected and the rate of reactivation but inversely with the number of CD8+ cells present. It is not known whether reactivating stimuli transiently suppress these immune cells, independently upregulate transcription of lytic genes, or both. Moreover, host containment in the mucosa has been demonstrated. Once virus reaches the dermal-epidermal junction, there are three possible outcomes: rapid host containment of infection near the site of reactivation; spread of virus into the epidermis, with a micro-ulceration associated with low-titer subclinical shedding; and subsequent rapid (within hours) containment of virus with widespread replication and necrosis of epithelial cells and subsequent clinical recurrence (the latter defined clinically by a skin blister and ulceration). Histologically, herpetic lesions involve a thin-walled vesicle or ulceration in the basal region, multinucleated cells that may include intranuclear inclusions, necrosis, and an acute inflammatory infection. Re-epithelialization occurs once viral replication is restricted, almost always in the absence of a scar.
Analysis of the DNA from sequential isolates of HSV or from isolates from multiple infected ganglia in any one individual has revealed similar, if not identical, restriction endonuclease or DNA sequence patterns in most persons. As more sensitive genomic technologies are developed, evidence of multiple strains of the same subtype is increasingly being reported. For example, infection of individual neurons with multiple strains of drug-susceptible and drug-resistant virus in severely immunosuppressed patients indicates that ganglia can be reseeded during chronic infection. Because exposure to mucosal shedding is relatively common during a person’s lifetime, current data suggest that exogenous infection with different strains of the same subtype, while possible, is uncommon.
IMMUNITY
Host responses influence the acquisition of HSV disease, the severity of infection, resistance to the development of latency, the maintenance of latency, and the frequency of recurrences. Both antibody-mediated and cell-mediated reactions are clinically important. Immunocompromised patients with defects in cell-mediated immunity experience more severe and more extensive HSV infections than those with deficits in humoral immunity, such as agammaglobulinemia. Experimental ablation of lymphocytes indicates that T cells play a major role in preventing lethal disseminated disease, although antibodies help reduce titers of virus in neural tissue. Some clinical manifestations of HSV appear to be related to the host immune response (e.g., stromal opacities associated with recurrent herpetic keratitis). The surface viral glycoproteins have been shown to be targets of antibodies that mediate neutralization and immune-mediated cytolysis (antibody-dependent cell-mediated cytotoxicity). Monoclonal antibodies specific for each of the known viral glycoproteins have, in experimental infections, conferred protection against subsequent neurologic disease or ganglionic latency. In humans, however, subunit glycoprotein vaccines have been largely ineffective in reducing acquisition of infection. Multiple cell populations, including natural killer cells, macrophages, and a variety of T lymphocytes, play a role in host defenses against HSV infections, as do lymphokines generated by T lymphocytes. In animals, passive transfer of primed lymphocytes confers protection from subsequent HSV challenge. Maximal protection usually requires the activation of multiple T cell subpopulations, including cytotoxic T cells and T cells responsible for delayed hypersensitivity. The latter may confer protection by the antigen-stimulated release of lymphokines (e.g., IFNs), which in turn have a direct antiviral effect and both activate and enhance a variety of specific and nonspecific effector cells. The HSV virion contains a variety of genes that are directed at the inhibition of host responses. These include gene no. 12 (US-12), which can bind to the cellular transporter-activating protein TAP-1 and reduce the ability of this protein to bind HSV peptides to human leukocyte antigen (HLA) class I, thereby reducing recognition of viral proteins by cytotoxic T cells of the host. This effect can be overcome by the addition of IFN-γ, but this reversal requires 24–48 h; thus, the virus has time to replicate and invade other host cells. Entry of infectious HSV-1 and HSV-2 inhibits several signaling pathways of both CD4+ and CD8+ T cells, leading to their functional impairment in killing and influencing the spectrum of their cytokine secretion.
Increasing evidence suggests that HSV-specific CD8+ T cell responses are critical for clearance of virus from lesions. Immunosuppressed patients with frequent and prolonged HSV lesions have fewer functional CD8+ T cells directed at HSV. HSV-specific CD8+ T cells have been shown to persist in the genital skin at the dermal–epidermal junction contiguous to neuronal endings. Even during clinical quiescence, these CD8+ T cells make both antiviral and cytotoxic proteins indicative of immune surveillance. These resident memory CD8+ T cells appear to be “first responders” capable of controlling viral reactivation at the site of viral release into the dermis. This rapid “on and off” interplay between the virus and host helps explain the variability in clinical disease severity between episodes in any single individual. Differences of 30–60 min in host responses can result in 100- to 1000-fold differences in viral levels and can determine whether an episode of disease is subclinical or clinical.
There is a strong association between the magnitude of the CD8+ T lymphocyte response and the clearance of virus from genital lesions. The location, effectiveness, and longevity of the T lymphocytes (and perhaps of other immune effector cells) may be important in the expression of disease and the likelihood of transmission over time.
EPIDEMIOLOGY
![]() Seroepidemiologic studies have documented HSV infections worldwide. The past 15 years have shown that the prevalence of HSV-2 is even higher in the developing than in the developed world. In sub-Saharan Africa, HSV-2 seroprevalence among pregnant women may approach 60%, and annual acquisition rates among teenage girls may verge on 20%. The global incidence has been estimated at ~23.6 million infections per year. As in the developed world, the rate of HSV-2 coital acquisition as well as the serologic prevalence is higher among women than among men. Most of this HSV-2 acquisition is preceded by acquisition of HSV-1; the frequency of genital HSV-1 in the developing world is low at present.
Seroepidemiologic studies have documented HSV infections worldwide. The past 15 years have shown that the prevalence of HSV-2 is even higher in the developing than in the developed world. In sub-Saharan Africa, HSV-2 seroprevalence among pregnant women may approach 60%, and annual acquisition rates among teenage girls may verge on 20%. The global incidence has been estimated at ~23.6 million infections per year. As in the developed world, the rate of HSV-2 coital acquisition as well as the serologic prevalence is higher among women than among men. Most of this HSV-2 acquisition is preceded by acquisition of HSV-1; the frequency of genital HSV-1 in the developing world is low at present.
Infection with HSV-1 is acquired more frequently and earlier than infection with HSV-2. More than 90% of adults have antibodies to HSV-1 by the fifth decade of life. In populations of low socioeconomic status, most persons acquire HSV-1 infection before the third decade of life. Antibodies to HSV-2 are not detected routinely until puberty. Antibody prevalence rates correlate with past sexual activity and vary greatly among different population groups. There is evidence that the prevalence of HSV-2 has decreased slightly over the past decade in the United States. Serosurveys indicate that 15–20% of the U.S. population has antibodies to HSV-2. In most routine obstetric and family planning clinics, 25% of women have HSV-2 antibodies, although only 10% of those who are seropositive for HSV-2 report a history of genital lesions. As many as 50% of heterosexual adults attending sexually transmitted disease clinics have antibodies to HSV-2.
Many studies continue to show that both incident and—more important—prevalent HSV-2 infection enhances the acquisition rate of HIV-1. More specifically, HSV-2 infection is associated with a two- to fourfold increase in HIV-1 acquisition. This association has been amply demonstrated in heterosexual men and women in both the developed and the developing worlds. Epidemiologically, regions of the world with high HSV-2 prevalence and selected populations within such regions have a higher population-based incidence of HIV-1. One study indicated that approximately one-quarter of HIV infections in the high-prevalence city of Kisumu, Kenya, were directly attributable to HSV-2.
In addition, HSV-2 facilitates the spread of HIV into low-risk populations on a per-coital basis, and prevalent HSV-2 appears to increase the risk of HIV infection by seven- to ninefold. Mathematical models suggest that ~33–50% of HIV-1 infections may be attributable to HSV-2 both in men who have sex with men (MSM) and in sub-Saharan Africa. In addition, HSV-2 is more frequently reactivated in and transmitted by persons co-infected with HIV-1 as opposed to persons not co-infected. Thus, most areas of the world with a high HIV-1 prevalence also have a high HSV-2 prevalence. A wide variety of serologic surveys have indicated a similar or even higher seroprevalence of HSV-2 in most parts of Central America, South America, and Africa. In Africa, HSV-2 seroprevalence has ranged from 40% to 70% in obstetric and other sexually experienced populations. Antibody prevalence rates average ~5–10% higher among women than among men.
Several studies suggest that many cases of “asymptomatic” genital HSV-2 infection are, in fact, simply unrecognized or confined to anatomic regions of the genital tract that are not easily visualized. Asymptomatic seropositive persons shed virus on mucosal surfaces almost as frequently as do those with symptomatic disease. This large reservoir of unidentified carriers of HSV-2 and the frequent asymptomatic reactivation of the virus from the genital tract have fostered the continued spread of genital herpes throughout the world. HSV-2 infection is an independent risk factor for the acquisition and transmission of infection with HIV-1. Among co-infected persons, HIV-1 virions can be shed from herpetic lesions of the genital region. This shedding may facilitate the spread of HIV through sexual contact. HSV-2 reactivation is associated with a localized persistent inflammatory response consisting of high concentrations of CCR5-enriched CD4+ T cells as well as inflammatory dendritic cells in the submucosa of the genital skin. These cells can support HIV infection and replication and hence are likely to account for the almost threefold increase in HIV acquisition among persons with genital herpes. Unfortunately, antiviral therapy does not reduce this subclinical postreactivation inflammation, probably because of the inability of current antiviral agents to prevent the release of small amounts of HSV antigen into the genital mucosa.
HSV infections occur throughout the year. Transmission can result from contact with persons who have active ulcerative lesions or with persons who have no clinical manifestations of infection but who are shedding HSV from mucocutaneous surfaces. HSV reactivation on genital skin and mucosal surfaces is common. The frequency of sampling influences the frequency of detection. Recent studies indicate that most HSV-1 and HSV-2 episodes last <4–6 h; thus, replication of virus and clearance by the host are rapid. Even with once-daily sampling, HSV DNA can be detected on 20–30% of days by PCR. Corresponding figures for HSV-1 in oral secretions are similar. Rates of shedding are highest during the initial years after acquisition, with viral shedding occurring on as many as 30–50% of days during this period. Immunosuppressed patients shed HSV from mucosal sites at an even higher frequency (20–80% of days). These high rates of mucocutaneous reactivation suggest that exposure to HSV from sexual or other close contact (kissing, sharing of glasses or silverware) is common and help explain the continuing spread and high seroprevalence of HSV infections worldwide. Reactivation rates vary widely among individuals. Among HIV-positive patients, a low CD4+ T cell count and a high HIV-1 load are associated with increased rates of HSV reactivation. Daily antiviral chemotherapy for HSV-2 infection can reduce shedding rates but does not eliminate shedding, as measured by PCR or culture.
CLINICAL SPECTRUM
HSV has been isolated from nearly all visceral and mucocutaneous sites. The clinical manifestations and course of HSV infection depend on the anatomic site involved, the age and immune status of the host, and the antigenic type of the virus. Primary HSV infections (i.e., first infections with either HSV-1 or HSV-2 in which the host lacks HSV antibodies in acute-phase serum) are frequently accompanied by systemic signs and symptoms. Compared with recurrent episodes, primary infections, which involve both mucosal and extramucosal sites, are characterized by a longer duration of symptoms and virus isolation from lesions. The incubation period ranges from 1 to 26 days (median, 6–8 days). Both viral subtypes can cause genital and oral-facial infections, and the infections caused by the two subtypes are clinically indistinguishable. However, the frequency of reactivation of infection is influenced by anatomic site and virus type. Genital HSV-2 infection is twice as likely to reactivate and recurs 8–10 times more frequently than genital HSV-1 infection. Conversely, oral-labial HSV-1 infection recurs more frequently than oral-labial HSV-2 infection. Asymptomatic shedding rates follow the same pattern.
Oral-Facial Infections Gingivostomatitis and pharyngitis are the most common clinical manifestations of first-episode HSV-1 infection, whereas recurrent herpes labialis is the most common clinical manifestation of reactivation HSV-1 infection. HSV pharyngitis and gingivostomatitis usually result from primary infection and are most common among children and young adults. Clinical symptoms and signs, which include fever, malaise, myalgias, inability to eat, irritability, and cervical adenopathy, may last 3–14 days. Lesions may involve the hard and soft palate, gingiva, tongue, lip, and facial area. HSV-1 or HSV-2 infection of the pharynx usually results in exudative or ulcerative lesions of the posterior pharynx and/or tonsillar pillars. Lesions of the tongue, buccal mucosa, or gingiva may occur later in the course in one-third of cases. Fever lasting 2–7 days and cervical adenopathy are common. It can be difficult to differentiate HSV pharyngitis clinically from bacterial pharyngitis, Mycoplasma pneumoniae infections, and pharyngeal ulcerations of noninfectious etiologies (e.g., Stevens-Johnson syndrome). No substantial evidence suggests that reactivation of oral-labial HSV infection is associated with symptomatic recurrent pharyngitis.
Reactivation of HSV from the trigeminal ganglia may be associated with asymptomatic virus excretion in the saliva, development of intraoral mucosal ulcerations, or herpetic ulcerations on the vermilion border of the lip or external facial skin. About 50–70% of seropositive patients undergoing trigeminal nerve-root decompression and 10–15% of those undergoing dental extraction develop oral-labial HSV infection a median of 3 days after these procedures. Clinical differentiation of intraoral mucosal ulcerations due to HSV from aphthous, traumatic, or drug-induced ulcerations is difficult.
In immunosuppressed patients, HSV infection may extend into mucosal and deep cutaneous layers. Friability, necrosis, bleeding, severe pain, and inability to eat or drink may result. The lesions of HSV mucositis are clinically similar to mucosal lesions caused by cytotoxic drug therapy, trauma, or fungal or bacterial infections. Persistent ulcerative HSV infections are among the most common infections in patients with AIDS. HSV and Candida infections often occur concurrently. Systemic antiviral therapy speeds the rate of healing and relieves the pain of mucosal HSV infections in immunosuppressed patients. The frequency of HSV reactivation during the early phases of transplantation or induction chemotherapy is high (50–90%), and prophylactic systemic antiviral agents such as IV acyclovir and penciclovir or the oral congeners of these drugs are used to reduce reactivation rates. Patients with atopic eczema may also develop severe oral-facial HSV infections (eczema herpeticum), which may rapidly involve extensive areas of skin and occasionally disseminate to visceral organs. Extensive eczema herpeticum has resolved promptly with the administration of IV acyclovir. Erythema multiforme may also be associated with HSV infections (see Figs. 70-9 and 25e-25); some evidence suggests that HSV infection is the precipitating event in ~75% of cases of cutaneous erythema multiforme. HSV antigen has been demonstrated both in circulatory immune complexes and in skin lesion biopsy samples from these cases. Patients with severe HSV-associated erythema multiforme are candidates for chronic suppressive oral antiviral therapy.
HSV-1 and varicella-zoster virus (VZV) have been implicated in the etiology of Bell’s palsy (flaccid paralysis of the mandibular portion of the facial nerve). Some but not all trials have documented quicker resolution of facial paralysis with the prompt initiation of antiviral therapy, with or without glucocorticoids. However, other trials have shown little benefit. Thus there is no consensus on the relative value of antiviral drugs alone, glucocorticoids alone, and the two modalities combined for the treatment of Bell’s palsy.
Genital Infections First-episode primary genital herpes is characterized by fever, headache, malaise, and myalgias. Pain, itching, dysuria, vaginal and urethral discharge, and tender inguinal lymphadenopathy are the predominant local symptoms. Widely spaced bilateral lesions of the external genitalia are characteristic (Fig. 216-1). Lesions may be present in varying stages, including vesicles, pustules, or painful erythematous ulcers. The cervix and urethra are involved in >80% of women with first-episode infections. First episodes of genital herpes in patients who have had prior HSV-1 infection are associated with systemic symptoms in a few patients and with faster healing than primary genital herpes. Subclinical DNAemia has been found in ~30% of cases of true primary genital herpes. The clinical courses of acute first-episode genital herpes are similar for HSV-1 and HSV-2 infection. However, the recurrence rates of genital disease differ with the viral subtype: the 12-month recurrence rates among patients with first-episode HSV-2 and HSV-1 infections are ~90% and ~55%, respectively (median number of recurrences, 4 and <1, respectively). Recurrence rates for genital HSV-2 infections vary greatly among individuals and over time within the same individual. HSV has been isolated from the urethra and urine of men and women without external genital lesions. A clear mucoid discharge and dysuria are characteristics of symptomatic HSV urethritis. HSV has been isolated from the urethra of 5% of women with the dysuria-frequency syndrome. Occasionally, HSV genital tract disease is manifested by endometritis and salpingitis in women and by prostatitis in men. About 15% of cases of HSV-2 acquisition are associated with nonlesional clinical syndromes, such as aseptic meningitis, cervicitis, or urethritis. A more complete discussion of the differential diagnosis of genital herpes is presented in Chap. 163.
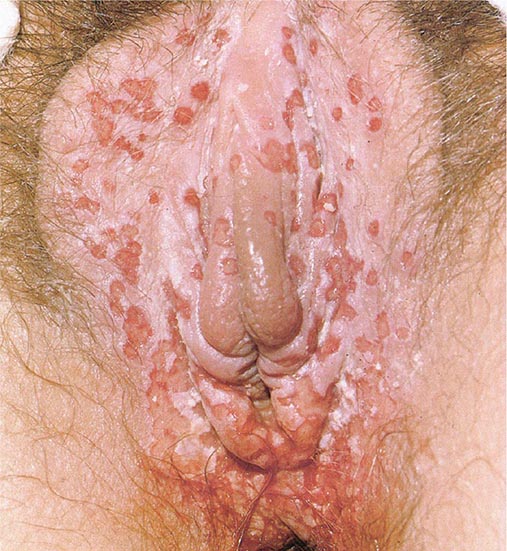
FIGURE 216-1 Genital herpes: primary vulvar infection, with multiple, extremely painful, punched-out, confluent, shallow ulcers on the edematous vulva and perineum. Micturition is often very painful. Associated inguinal lymphadenopathy is common. (Reprinted with permission from K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.)
Both HSV-1 and HSV-2 can cause symptomatic or asymptomatic rectal and perianal infections. HSV proctitis is usually associated with rectal intercourse. However, subclinical perianal shedding of HSV is detected in women and men who report no rectal intercourse. This phenomenon is due to the establishment of latency in the sacral dermatome from prior genital tract infection, with subsequent reactivation in epithelial cells in the perianal region. Such reactivations are often subclinical. Symptoms of HSV proctitis include anorectal pain, anorectal discharge, tenesmus, and constipation. Sigmoidoscopy reveals ulcerative lesions of the distal 10 cm of the rectal mucosa. Rectal biopsies show mucosal ulceration, necrosis, polymorphonuclear and lymphocytic infiltration of the lamina propria, and (in occasional cases) multinucleated intranuclear inclusion-bearing cells. Perianal herpetic lesions are also found in immunosuppressed patients receiving cytotoxic therapy. Extensive perianal herpetic lesions and/or HSV proctitis is common among patients with HIV infection.
Herpetic Whitlow Herpetic whitlow—HSV infection of the finger—may occur as a complication of primary oral or genital herpes by inoculation of virus through a break in the epidermal surface or by direct introduction of virus into the hand through occupational or some other type of exposure. Clinical signs and symptoms include abrupt-onset edema, erythema, and localized tenderness of the infected finger. Vesicular or pustular lesions of the fingertip that are indistinguishable from lesions of pyogenic bacterial infection are seen. Fever, lymphadenitis, and epitrochlear and axillary lymphadenopathy are common. The infection may recur. Prompt diagnosis (to avoid unnecessary and potentially exacerbating surgical therapy and/or transmission) is essential. Antiviral chemotherapy is usually recommended (see below).
Herpes Gladiatorum HSV may infect almost any area of skin. Mucocutaneous HSV infections of the thorax, ears, face, and hands have been described among wrestlers. Transmission of these infections is facilitated by trauma to the skin sustained during wrestling. Several recent outbreaks have illustrated the importance of prompt diagnosis and therapy to contain the spread of this infection.
Eye Infections HSV infection of the eye is the most common cause of corneal blindness in the United States. HSV keratitis presents as an acute onset of pain, blurred vision, chemosis, conjunctivitis, and characteristic dendritic lesions of the cornea. Use of topical glucocorticoids may exacerbate symptoms and lead to involvement of deep structures of the eye. Debridement, topical antiviral treatment, and/or IFN therapy hastens healing. However, recurrences are common, and the deeper structures of the eye may sustain immunopathologic injury. Stromal keratitis due to HSV appears to be related to T cell–dependent destruction of deep corneal tissue. An HSV-1 epitope that is autoreactive with T cell–targeting corneal antigens has been postulated to be a factor in this infection. Chorioretinitis, usually a manifestation of disseminated HSV infection, may occur in neonates or in patients with HIV infection. HSV and VZV can cause acute necrotizing retinitis as an uncommon but severe manifestation.
Central and Peripheral Nervous System Infections HSV accounts for 10–20% of all cases of sporadic viral encephalitis in the United States. The estimated incidence is ~2.3 cases per 1 million persons per year. Cases are distributed throughout the year, and the age distribution appears to be biphasic, with peaks at 5–30 and >50 years of age. HSV-1 causes >95% of cases.
The pathogenesis of HSV encephalitis varies. In children and young adults, primary HSV infection may result in encephalitis; presumably, exogenously acquired virus enters the CNS by neurotropic spread from the periphery via the olfactory bulb. However, most adults with HSV encephalitis have clinical or serologic evidence of mucocutaneous HSV-1 infection before the onset of CNS symptoms. In ~25% of the cases examined, the HSV-1 strains from the oropharynx and brain tissue of the same patient differ; thus some cases may result from reinfection with another strain of HSV-1 that reaches the CNS. Two theories have been proposed to explain the development of actively replicating HSV in localized areas of the CNS in persons whose ganglionic and CNS isolates are similar. Reactivation of latent HSV-1 infection in trigeminal or autonomic nerve roots may be associated with extension of virus into the CNS via nerves innervating the middle cranial fossa. HSV DNA has been demonstrated by DNA hybridization in brain tissue obtained at autopsy—even from healthy adults. Thus, reactivation of long-standing latent CNS infection may be another mechanism for the development of HSV encephalitis.
![]() Recent studies have identified genetic polymorphisms in two separate genes among families with a high frequency of HSV encephalitis. Peripheral-blood mononuclear cells from these patients (predominantly children) appear to secrete reduced levels of IFN in response to HSV. These observations suggest that some cases of sporadic HSV encephalitis may be related to host genetic determinants.
Recent studies have identified genetic polymorphisms in two separate genes among families with a high frequency of HSV encephalitis. Peripheral-blood mononuclear cells from these patients (predominantly children) appear to secrete reduced levels of IFN in response to HSV. These observations suggest that some cases of sporadic HSV encephalitis may be related to host genetic determinants.
The clinical hallmark of HSV encephalitis has been the acute onset of fever and focal neurologic symptoms and signs, especially in the temporal lobe (Fig. 216-2). Clinical differentiation of HSV encephalitis from other viral encephalitides, focal infections, or noninfectious processes is difficult. Elevated cerebrospinal fluid (CSF) protein levels, leukocytosis (predominantly lymphocytes), and red blood cell counts due to hemorrhagic necrosis are common. While brain biopsy has been the gold standard for defining HSV encephalitis, a highly sensitive and specific PCR for detection of HSV DNA in CSF has largely replaced biopsy for defining CNS infection. Although titers of antibody to HSV in CSF and serum increase in most cases of HSV encephalitis, they rarely do so earlier than 10 days into the illness and therefore, although useful in retrospect, generally are not helpful in establishing an early clinical diagnosis. In rare cases, demonstration of HSV antigen, HSV DNA, or HSV replication in brain tissue obtained by biopsy is highly sensitive; examination of such tissue also provides the opportunity to identify alternative, potentially treatable causes of encephalitis. Antiviral chemotherapy with acyclovir reduces the rate of death from HSV encephalitis. Most authorities recommend the administration of IV acyclovir to patients with presumed HSV encephalitis until the diagnosis is confirmed or an alternative diagnosis is made. All confirmed cases should be treated with IV acyclovir (30 mg/kg per day in three divided doses for 14–21 days). After the completion of therapy, the clinical recurrence of encephalitis requiring more treatment has been reported. For this reason, some authorities prefer to treat initially for 21 days, and many continue therapy until HSV DNA has been eliminated from the CSF. Even with therapy, neurologic sequelae are common, especially among persons >50 years of age.
FIGURE 216-2 Computed tomography and diffusion-weighted magnetic resonance imaging scans of the brain of a patient with left-temporal-lobe herpes simplex virus encephalitis.
HSV DNA has been detected in CSF from 3–15% of persons presenting to the hospital with aseptic meningitis. HSV meningitis, which is usually seen in association with primary genital HSV infection, is an acute, self-limited disease manifested by headache, fever, and mild photophobia and lasting 2–7 days. Lymphocytic pleocytosis in the CSF is characteristic. Neurologic sequelae of HSV meningitis are rare. HSV is the most commonly identified cause of recurrent lymphocytic meningitis (Mollaret’s meningitis). Demonstration of HSV antibodies in CSF or persistence of HSV DNA in CSF can establish the diagnosis. For persons with frequent recurrences of HSV meningitis, daily antiviral therapy has reduced the occurrence of such episodes.
Autonomic nervous system dysfunction, especially of the sacral region, has been reported in association with both HSV and VZV infections. Numbness, tingling of the buttocks or perineal areas, urinary retention, constipation, CSF pleocytosis, and (in males) impotence may occur. Symptoms appear to resolve slowly over days or weeks. Occasionally, hypoesthesia and/or weakness of the lower extremities persists for many months. Transitory hypoesthesia of the area of skin innervated by the trigeminal nerve and vestibular system dysfunction (as measured by electronystagmography) are the predominant signs of disease. Whether antiviral chemotherapy can abort these signs or reduce their frequency and severity is not yet known. Rarely, transverse myelitis, manifested by a rapidly progressive symmetric paralysis of the lower extremities or Guillain-Barré syndrome, follows HSV infection. Similarly, peripheral nervous system involvement (Bell’s palsy) or cranial polyneuritis may be related to reactivation of HSV-1 infection.
Visceral Infections HSV infection of visceral organs usually results from viremia, and multiple-organ involvement is common. Occasionally, however, the clinical manifestations of HSV infection involve only the esophagus, lung, or liver. HSV esophagitis may result from direct extension of oral-pharyngeal HSV infection into the esophagus or may occur de novo by reactivation and spread of HSV to the esophageal mucosa via the vagus nerve. The predominant symptoms of HSV esophagitis are odynophagia, dysphagia, substernal pain, and weight loss. Multiple oval ulcerations appear on an erythematous base with or without a patchy white pseudomembrane. The distal esophagus is most commonly involved. With extensive disease, diffuse friability may spread to the entire esophagus. Neither endoscopic nor barium examination can reliably differentiate HSV esophagitis from Candida esophagitis or from esophageal ulcerations due to thermal injury, radiation, or corrosives. Endoscopically obtained secretions for cytologic examination and culture or DNA detection by PCR provide the most useful material for diagnosis. Systemic antiviral chemotherapy usually reduces the severity and duration of symptoms and heals esophageal ulcerations.
HSV pneumonitis is uncommon except in severely immunosuppressed patients and may result from extension of herpetic tracheobronchitis into lung parenchyma. Focal necrotizing pneumonitis usually ensues. Hematogenous dissemination of virus from sites of oral or genital mucocutaneous disease may also occur, producing bilateral interstitial pneumonitis. Bacterial, fungal, and parasitic pathogens are commonly present in HSV pneumonitis. The mortality rate from untreated HSV pneumonia in immunosuppressed patients is high (>80%). HSV has also been isolated from the lower respiratory tract of persons with acute respiratory distress syndrome and prolonged intubation. Most authorities believe that the presence of HSV in tracheal aspirates in such settings is due to reactivation of HSV in the tracheal region and localized tracheitis in persons with long-term intubation. Such patients should be evaluated for extension of HSV infection into the lung parenchyma. Controlled trials assessing the role of antiviral agents used against HSV in morbidity and mortality associated with acute respiratory distress syndrome have not been conducted. The role of lower respiratory tract HSV infection in overall rates of morbidity and mortality associated with these conditions is unclear. HSV is an uncommon cause of hepatitis in immunocompetent patients. HSV infection of the liver is associated with fever, abrupt elevations of bilirubin and serum aminotransferase levels, and leukopenia (<4000 white blood cells/μL). Disseminated intravascular coagulation may also develop.
Other reported complications of HSV infection include monarticular arthritis, adrenal necrosis, idiopathic thrombocytopenia, and glomerulonephritis. Disseminated HSV infection in immunocompetent patients is rare. In immunocompromised patients, burn patients, or malnourished individuals, HSV occasionally disseminates to other visceral organs, such as the adrenal glands, pancreas, small and large intestines, and bone marrow. Rarely, primary HSV infection in pregnancy disseminates and may be associated with the death of both mother and fetus. This uncommon event is usually related to the acquisition of primary infection in the third trimester. Disseminated HSV infection is best detected by the presence of HSV DNA in plasma or blood.
Neonatal HSV Infections Of all HSV-infected populations, neonates (infants younger than 6 weeks) have the highest frequency of visceral and/or CNS infection. Without therapy, the overall rate of death from neonatal herpes is 65%; <10% of neonates with CNS infection develop normally. Although skin lesions are the most commonly recognized features of disease, many infants do not develop lesions at all or do so only well into the course of disease. Neonatal infection is usually acquired perinatally from contact with infected genital secretions at delivery. Congenitally infected infants have been reported. Of neonatal HSV infections, 30–50% are due to HSV-1 and 50–70% to HSV-2. The risk of developing neonatal HSV infection is 10 times higher for an infant born to a mother who has recently acquired HSV than for other infants. Neonatal HSV-1 infections may also be acquired through postnatal contact with immediate family members who have symptomatic or asymptomatic oral-labial HSV-1 infection or through nosocomial transmission within the hospital. All neonates with presumed herpes should be treated with IV acyclovir and then placed on maintenance oral antiviral therapy for the first 6–12 months of life. Antiviral chemotherapy with high-dose IV acyclovir (60 mg/kg per day) has reduced the mortality rate from neonatal herpes to ~15%. However, rates of morbidity, especially among infants with HSV-2 infection involving the CNS, are still very high.
HSV in Pregnancy In the United States, 22% of all pregnant women and 55% of non-Hispanic black pregnant women are seropositive for HSV-2. However, the risk of mother-to-child transmission of HSV in the perinatal period is highest when the infection is acquired near the time of labor—that is, in previously HSV-seronegative women. The clinical manifestations of recurrent genital herpes—including the frequency of subclinical versus clinical infection, duration of lesions, pain, and constitutional symptoms—are similar in pregnant and nonpregnant women. Recurrences increase in frequency over the course of pregnancy. However, when women are seropositive for HSV-2 at the outset of pregnancy, no effect on neonatal outcomes (including birth weight and gestational age) is seen. First-episode infections in pregnancy have more severe consequences for mother and infant. Maternal visceral dissemination during the third trimester occasionally occurs, as does premature birth or intrauterine growth retardation. The acquisition of primary disease in pregnancy, whether related to HSV-1 or HSV-2, carries the risk of transplacental transmission of virus to the neonate and can result in spontaneous abortion, although this outcome is relatively uncommon. For newly acquired genital HSV infection during pregnancy, most authorities recommend treatment with acyclovir (400 mg three times daily) or valacyclovir (500–1000 mg twice daily) for 7–10 days. However, the impact of this intervention on transmission is unknown. The high HSV-2 prevalence rate in pregnancy and the low incidence of neonatal disease (1 case per 6000–20,000 live births) indicate that only a few infants are at risk of acquiring HSV. Therefore, cesarean section is not warranted for all women with recurrent genital disease. Because intrapartum transmission of infection accounts for the majority of cases, abdominal delivery need be considered only for women who are shedding HSV at delivery. Several studies have shown no correlation between recurrence of viral shedding before delivery and viral shedding at term. Hence, weekly virologic monitoring and amniocentesis are not recommended.
The frequency of transmission from mother to infant is markedly higher among women who acquire HSV near term (30–50%) than among those in whom HSV-2 infection is reactivated at delivery (<1%). Although maternal antibody to HSV-2 is protective, antibody to HSV-1 offers little or no protection against neonatal HSV-2 infection. Primary genital infection with HSV-1 leads to a particularly high risk of transmission during pregnancy and accounts for an increasing proportion of neonatal HSV cases. Moreover, during reactivation, HSV-1 appears more transmissible to the neonate than HSV-2. Only 2% of women who are seropositive for HSV-2 have HSV-2 isolated from cervical secretions at delivery, and only 1% of infants exposed in this manner develop infection, presumably because of the protective effects of maternally transferred antibodies and perhaps lower viral titers during reactivation. Despite the low frequency of transmission of HSV in this setting, 30–50% of infants with neonatal HSV are born to mothers with established genital herpes.
Isolation of HSV by cervicovaginal swab at the time of delivery is the greatest risk factor for intrapartum HSV transmission (relative risk = 346); however, culture-negative, PCR-positive cases of intrapartum transmission are well described. New acquisition of HSV (odds ratio [OR] = 49), isolation of HSV-1 versus HSV-2 (OR = 35), cervical versus vulvar HSV detection (OR = 15), use of fetal scalp electrodes (OR = 3.5), and young age confer further risk of transmission, whereas abdominal delivery is protective (OR = 0.14). Physical examination poorly predicts the absence of shedding, and PCR far exceeds culture in terms of sensitivity and speed. Therefore, PCR detection at the onset of labor should be used to aid clinical decision-making for women with HSV-2 antibody. Because cesarean section appears to be an effective means of reducing maternal-fetal transmission, patients with recurrent genital herpes should be encouraged to come to the hospital early at the time of delivery for careful examination of the external genitalia and cervix as well as collection of a swab sample for viral isolation. Women who have no evidence of lesions can have a vaginal delivery. The presence of active lesions on the cervix or external genitalia is an indication for cesarean delivery.
If first-episode exposure has occurred (e.g., if HSV serologies show that the mother is seronegative or if the mother is HSV-1-seropositive and the isolate at delivery is found to be HSV-2), many authorities would initiate antiviral therapy for the infant with IV acyclovir. At a minimum, samples for viral cultures and PCR should be obtained from the throat, nasopharynx, eyes, and rectum of these infants immediately and at 5- to 10-day intervals. Lethargy, skin lesions, or fever should be evaluated promptly. All infants from whom HSV is isolated 24 h after delivery should be treated with IV acyclovir at recommended doses.
DIAGNOSIS
Both clinical and laboratory criteria are useful for diagnosing HSV infections. A clinical diagnosis can be made accurately when characteristic multiple vesicular lesions on an erythematous base are present. However, herpetic ulcerations may resemble skin ulcerations of other etiologies. Mucosal HSV infections may also present as urethritis or pharyngitis without cutaneous lesions. Thus, laboratory studies to confirm the diagnosis and to guide therapy are recommended. While staining of scrapings from the base of the lesions with Wright’s, Giemsa’s (Tzanck preparation), or Papanicolaou’s stain to detect giant cells or intranuclear inclusions of Herpesvirus infection is a well-described procedure, few clinicians are skilled in this technique, the sensitivity of staining is low (<30% for mucosal swabs), and these cytologic methods do not differentiate between HSV and VZV infections.
HSV infection is best confirmed in the laboratory by detection of virus, viral antigen, or viral DNA in scrapings from lesions. HSV DNA detection by PCR is the most sensitive laboratory technique for detecting mucosal or visceral HSV infections and should be used when available. HSV causes a discernible cytopathic effect in a variety of cell culture systems, and this effect can be identified within 48–96 h after inoculation. Spin-amplified culture with subsequent staining for HSV antigen has shortened the time needed to identify HSV to <24 h. The sensitivity of all detection methods depends on the stage of the lesions (with higher sensitivity for vesicular than for ulcerative lesions), on whether the patient has a first or a recurrent episode of the disease (with higher sensitivity in first than in recurrent episodes), and on whether the sample is from an immunosuppressed or an immunocompetent patient (with more antigen or DNA in immunosuppressed patients). Laboratory confirmation permits subtyping of the virus; information on subtype may be useful epidemiologically and may help to predict the frequency of reactivation after first-episode oral-labial or genital HSV infection.
Serologic assays with whole-virus antigen preparations, such as complement fixation, neutralization, indirect immunofluorescence, passive hemagglutination, radioimmunoassay, and enzyme-linked immunosorbent assay, are useful for differentiating uninfected (seronegative) persons from those with past HSV-1 or HSV-2 infection, but they do not reliably distinguish between the two viral subtypes. Serologic assays that identify antibodies to type-specific surface proteins (epitopes) of the two viral subtypes have been developed and can distinguish reliably between the human antibody responses to HSV-1 and HSV-2. The most commonly used assays are those that measure antibodies to glycoprotein G of HSV-1 (gG1) and HSV-2 (gG2). A western blot assay that can detect several HSV type-specific proteins can also be used.
Acute- and convalescent-phase serum samples can be useful in demonstrating seroconversion during primary HSV-1 or HSV-2 infection. However, few available tests report titers, and increases in index values do not reflect first episodes in all patients. Serologic assays based on type-specific proteins should be used to identify asymptomatic carriers of HSV-1 or HSV-2. No reliable IgM method for defining acute HSV infection is available.
Several studies have shown that persons with previously unrecognized HSV-2 infection can be taught to identify symptomatic reactivations. Individuals seropositive for HSV-2 should be told about the high frequency of subclinical reactivation on mucosal surfaces that are not visible to the eye (e.g., cervix, urethra, perianal skin) or in microscopic ulcerations that may not be clinically symptomatic. Transmission of infection during such episodes is well established. HSV-2-seropositive persons should be educated about the high likelihood of subclinical shedding and the role condoms (male or female) may play in reducing transmission. Antiviral therapy with valacyclovir (500 mg once daily) has been shown to reduce the transmission of HSV-2 between sexual partners.
|
TREATMENT |
HERPES SIMPLEX VIRUS INFECTIONS |
Many aspects of mucocutaneous and visceral HSV infections are amenable to antiviral chemotherapy. For mucocutaneous infections, acyclovir and its congeners famciclovir and valacyclovir have been the mainstays of therapy. Several antiviral agents are available for topical use in HSV eye infections: idoxuridine, trifluorothymidine, topical vidarabine, and cidofovir. For HSV encephalitis and neonatal herpes, IV acyclovir is the treatment of choice.
All licensed antiviral agents for use against HSV inhibit the viral DNA polymerase. One class of drugs, typified by the drug acyclovir, is made up of substrates for the HSV enzyme thymidine kinase (TK). Acyclovir, ganciclovir, famciclovir, and valacyclovir are all selectively phosphorylated to the monophosphate form in virus-infected cells. Cellular enzymes convert the monophosphate form of the drug to the triphosphate, which is then incorporated into the viral DNA chain. Acyclovir is the agent most frequently used for the treatment of HSV infections and is available in IV, oral, and topical formulations. Valacyclovir, the valyl ester of acyclovir, offers greater bioavailability than acyclovir and thus can be administered less frequently. Famciclovir, the oral formulation of penciclovir, is clinically effective in the treatment of a variety of HSV-1 and HSV-2 infections. Ganciclovir is active against both HSV-1 and HSV-2; however, it is more toxic than acyclovir, valacyclovir, and famciclovir and generally is not recommended for the treatment of HSV infections. Anecdotal case reports suggest that ganciclovir may also be less effective than acyclovir for treatment of HSV infections. All three recommended compounds—acyclovir, valacyclovir, and famciclovir—have proved effective in shortening the duration of symptoms and lesions of mucocutaneous HSV infections in both immunocompromised and immunocompetent patients (Table 216-1). IV and oral formulations prevent reactivation of HSV in seropositive immunocompromised patients during induction chemotherapy or in the period immediately after bone marrow or solid organ transplantation. Chronic daily suppressive therapy reduces the frequency of reactivation disease among patients with frequent genital or oral-labial herpes. Only valacyclovir has been subjected to clinical trials that demonstrated reduced transmission of HSV-2 infection between sexual partners. IV acyclovir (30 mg/kg per day, given as a 10-mg/kg infusion over 1 h at 8-h intervals) is effective in reducing rates of death and morbidity from HSV encephalitis. Early initiation of therapy is a critical factor in outcome. The major side effect associated with IV acyclovir is transient renal insufficiency, usually due to crystallization of the compound in the renal parenchyma. This adverse reaction can be avoided if the medication is given slowly over 1 h and the patient is well hydrated. Because CSF levels of acyclovir average only 30–50% of plasma levels, the dosage of acyclovir used for treatment of CNS infection (30 mg/kg per day) is double that used for treatment of mucocutaneous or visceral disease (15 mg/kg per day). Even higher doses of IV acyclovir are used for neonatal HSV infection (60 mg/kg per day in three divided doses).




