[level-membership-for-orthopaedics-category]
CHAPTER 37 Medical Myelopathies
Spinal cord disease may result from a variety of insults. A general classification of these various etiologies is provided in Table 37–1. This chapter addresses the “medical” causes of spinal cord dysfunction.
TABLE 37–1 Etiologies of Myelopathy
Generally, but not invariably, a thorough neurologic examination allows the physician to distinguish spinal cord from other causes of these neurologic complaints. The presence of concomitant neurologic illness attributable to disease of the cerebral hemispheres or brainstem, such as hemianopsia, ophthalmoplegia, and dysarthria, does not rule out the possibility of concomitant spinal cord disease, but should suggest that the findings on neurologic examination may be ascribed to a single or multiple lesions higher in the neuraxis than the spinal cord. The physical examination of patients with spinal cord lesions that have developed gradually or who have acute lesions that have been present for several weeks typically reveals a spastic weakness of the involved extremities with associated hyperreflexia and pathologic reflexes. The latter include Hoffman sign (palmar flexion of the thumb when the distal phalanx of the middle finger of the same hand is rapidly tapped) in the upper extremities and Babinski sign (plantar extension of the hallux when the sole of the foot is stroked) in the lower extremities. Several variants of these signs have been described.1 Superficial reflexes, such as abdominal and cremasteric reflexes, are absent.
Acute Idiopathic Transverse Myelitis and Multiple Sclerosis
Patients with acute idiopathic transverse myelitis most often present with paresthesias of the feet, toes, or fingertips.2 Gradually developing numbness and coincident weakness of the legs follows with subsequent paralysis of the legs. The features of this illness usually evolve over 1 to 3 weeks, but they may develop abruptly. The initial symptoms may be predominantly unilateral, and asymmetrical findings are common. As the illness progresses, upper extremity numbness and weakness and bowel and bladder incontinence may ensue. In some patients, posterior column involvement (decreased vibration and position senses) is spared early but affected later. Occasionally, back (interscapular) pain and, more rarely, calf, arm, or radicular pain may accompany the progressive myelopathy suggesting other pathologic processes, such as an intraspinal neoplasm or epidural abscess. To be considered idiopathic, acute transverse myelitis should be unassociated with a known preceding or concomitant viral infection. The dominant pathologic feature of the spinal cord is demyelination.
Treatment with adrenocorticotropic hormone (ACTH) or corticosteroids has been advocated; the response to therapy may be highly variable, and some studies indicate no benefit.3 A pilot study of high-dose methylprednisolone in children with acute transverse myelitis showed a significant shortening of motor recovery compared with historical controls.4 Approximately one third of patients have a return of a normal gait and bladder function.2 When recovery to a normal or near-normal level of function occurs, it does so within 1 year of the onset of the illness. Of affected patients, 25% become wheelchair-bound or bedridden, and the remainder have varying degrees of lesser disability.2
In some instances, myelopathy develops abruptly. Frequently, a flaccid, areflexic paralysis of the affected limbs accompanies spinal shock. Loss of vision resulting from optic nerve inflammation may accompany this myelopathy. Devic disease (neuromyelitis optica [NMO]) refers to the combination of acute optic neuritis and transverse myelitis. Similar to the more slowly evolving idiopathic transverse myelitis, demyelination is a characteristic neuropathologic hallmark. The pathology has been described as a necrotizing hemorrhagic leukomyelitis. The prognosis of patients with this rapidly evolving myelopathy, particularly when it is accompanied by spinal shock, is worse than the prognosis for the more gradually developing transverse myelitis.5
The differential diagnosis of idiopathic transverse myelitis includes multiple sclerosis; infectious myelitis; postinfectious or postvaccination myelitis; and myelopathy associated with vasculitis and connective tissue diseases, such as Behçet disease, systemic lupus erythematosus (SLE), and Sjögren syndrome. Multiple sclerosis may initially manifest as a transverse myelitis; the likelihood of transverse myelitis being the presenting manifestation of multiple sclerosis was previously reported to be 5% to 15%.2,5–7 Since the advent of more sensitive testing (magnetic resonance imaging [MRI]), this likelihood has been estimated at 42%.8 If cranial MRI is highly suggestive of multiple sclerosis at the time of onset of myelitis, the risk of progression to clinically definite multiple sclerosis is 50% at 2 years, whereas if cranial MRI is negative, the risk is 5%.9 A temporal association with a viral illness (Table 37–2), signs and symptoms that develop over a few days, and a monophasic course should suggest the possibility of a viral myelitis.10
| RNA Viruses | DNA Viruses |
|---|---|
| Nonenveloped | |
| Picornaviruses | Hepatitis B |
| Coxsackieviruses | |
| Echoviruses | |
| Polioviruses | |
| Other enteroviruses | |
| Hepatitis A | |
| Encephalomyocarditis virus | |
| Enveloped | |
| Togaviruses | Herpesviruses |
| Arbovirus | Herpes simplex |
| Rubella | Varicella-zoster |
| Tick-borne encephalitis virus | Epstein-Barr virus |
| Retroviruses | Cytomegalovirus |
| Human immunodeficiency virus type 1 | Herpes simiae |
| Human T-cell lymphotropic virus type I | Poxviruses |
| Orthomyxoviruses | Vaccinia |
| Influenza | Variola |
| Paramyxoviruses | |
| Measles | |
| Mumps | |
| Bunyaviruses | |
| California encephalitis virus | |
| Arenavirus | |
| Lymphocytic choriomeningitis | |
| Rhabdovirus | |
| Rabies | |
Adapted from Tyler KL, Gross RA, Cascino GD: Unusual viral causes of transverse myelitis: Hepatitis A virus and cytomegalovirus. Neurology 36:855-858, 1986.
Spinal cord disease is extremely common in the setting of multiple sclerosis and frequently is responsible for the associated extremity weakness, spasticity, gait abnormalities, and sphincter disturbances. Within 10 to 15 years of the onset of multiple sclerosis, more than 80% of patients exhibit extremity spasticity and weakness, and more than 50% have sphincter disturbances.11,12 Lhermitte sign has been reported in 38% of patients with clinically definite multiple sclerosis.13 Approximately 10% of patients with multiple sclerosis have a primary progressive form of the disease. This disorder often affects the spinal cord predominantly and most commonly develops in women older than 40 years.
NMO has been regarded as a variant of multiple sclerosis; however, the occurrence of a unique IgG antibody to the aquaporin-4 channel (NMO-IgG)14 suggests it is a distinct disorder. NMO may occur as an isolated condition or, more commonly, as recurrent spells of optic neuritis and myelitis. In contrast to the myelitis of multiple sclerosis, which seldom extends beyond two or three segments, the myelitis of NMO is often longitudinally extensive. Optic neuritis or myelitis in isolation may occur with NMO. Treatment recommendations15 include high-dose intravenous methylprednisolone; plasma exchange; intravenous immunoglobulin; and rituximab, an anti-CD20 antibody directed against B cells.
MRI of the involved area of the spinal cord is the best radiographic study. MRI may be normal or reveal cord swelling or hyperintense lesions or both on T2-weighted imaging intrinsic to the spinal cord. Miller and colleagues16 found MRI lesions at the clinically expected level in 64% of patients with a clinical syndrome suggestive of cervical involvement but in only 28% with a suspected thoracic or lumbar lesion. In patients with multiple sclerosis, most lesions are observed in the cervical spinal cord at autopsy and by MRI.17,18 Lesions on MRI are characteristically T2-weighted hyperintense signal abnormalities, generally less than 10 to 15 mm in length,18 and multiple, gapped lesions may be observed. Gadolinium enhancement with active disease is often observed.19 Generally, MRI, if available, negates the need of myelography. If MRI is unavailable, myelography should be performed with water-soluble contrast medium and computed tomography (CT) scan of the affected area. If pain is a significant component of the patient’s illness, a CT scan of the appropriate area and a bone scan may be desirable in eliminating the possibility of an epidural abscess or neoplasm.
Somatosensory evoked potentials can be very helpful in ruling in suspected cord lesions. The presence of abnormalities on brainstem auditory evoked potential and visual evoked potential testing would raise the suspicion of multifocal disease (i.e. multiple sclerosis). Absent F waves on nerve conduction testing can also support evidence of a cord lesion.20
Adrenomyeloneuropathy
Adrenomyeloneuropathy is a phenotypic variant of the genetic disorder adrenoleukodystrophy, in which myelin sheath abnormalities of the white matter are associated with adrenal insufficiency. Although adrenoleukodystrophy is a sex-linked disorder, it has variable expressions, including mild disease in heterozygous women. Pathologically, the white matter abnormality appears to be a diffuse myelinoclastic sclerosis, but it may occasionally appear as a dysmyelinating condition. The neurologic variants are explained by the degree of involvement of brain, spinal cord, and peripheral nerve. Biorefringent material in the adrenal glands and brain observed histopathologically have been shown by sequential extraction methods to be cholesterol esters with high quantities of very long chain fatty acids. Measuring fatty acids in cultured skin fibroblasts from affected individuals, Moser and colleagues21 showed abnormally large amounts of very long chain fatty acids (C24-C30) and a high ratio of C26 to C22 fatty acids. The latter has become the preferred method of diagnosing the disorder.
In patients with the spinal-neuropathic form of this disorder, adrenal insufficiency is usually present since early childhood, and a progressive spastic paraparesis and relatively mild peripheral neuropathy develop in the 3rd decade.22 Other variants affecting the spinal cord have been observed, including a progressive myelopathy in men, a mild spastic paraparesis in women, and combined cerebral and spinal involvement in children and young men. A positive family history is present in approximately 50% of affected individuals. The presence of Addison disease also provides a strong clue to the diagnosis. Addison disease, which is caused by primary adrenal failure, is often accompanied by bronzing of the skin because of excessive secretion of melanocyte-stimulating hormone in association with ACTH.
Infectious Myelopathies
Viral Myelitis
Human Immunodeficiency Virus Infection
Neurologic complications develop in 40% to 60%23–25 of all patients with acquired immunodeficiency syndrome (AIDS), and neurologic disease heralds human immunodeficiency virus (HIV) infection in 10%26 to 20%25 of HIV-infected patients. In retrospective clinical series, spinal cord disease occurring in association with AIDS has been infrequently observed. Levy and colleagues24 found viral myelitis in 3 of 128 AIDS patients with neurologic symptoms, and the collective incidence of viral myelitis in AIDS was 1% when data were pooled from three different hospital series of neurologic complications of AIDS.27 The most common form of myelopathy observed with HIV infection has been a unique degeneration of the spinal cord first described by Petito and colleagues.28 In pathologic series, spinal cord degeneration has been observed in 11%29 to 22%28 of unselected cases. These pathologic series suggest that spinal cord disease occurring in association with HIV infection is common but clinically underrecognized.
The prototypic myelopathy observed with HIV infection is a unique degeneration of the spinal cord.28,30 Petito and colleagues28 observed this myelopathy in 20 of 89 consecutive autopsies of AIDS patients. Although the clinical presentation of this myelopathy may overlap with the presentation of other myelopathies associated with HIV-1 infection, the pathologic appearance is quite distinct. Clinically, patients complain of leg weakness, unsteadiness, and gait impairment. Incontinence of bladder and bowel often supervenes. In one study, incontinence was observed in 60% of patients.28 Patients with this disorder often complain of paresthesias and vague discomfort in their legs. Frequently, these complaints are attributed to the general debilitation of the patient, and the true nature of the illness remains undiagnosed until pathologic examination of the spinal cord at the time of autopsy.
On physical examination, a spastic paraparesis is detected with the degree of weakness exceeding the degree of spasticity. Rarely, marked asymmetry of the leg weakness, a monoparesis, or quadriparesis may be found. Gait ataxia is seen, and the heel-to-knee-to-toe test may reveal dysmetria and dyssynergy. Occasionally, weakness is slight or absent on confrontation testing, but hyperreflexia of the lower extremities and extensor plantar responses are noted. Muscle stretch reflexes may also be diminished or absent in this disorder, however, as a result of concomitant peripheral neuropathy. Sensory examination reveals that vibratory and position sense are disproportionately affected compared with pinprick, temperature, or light touch. A significant impairment of the latter modalities suggests the presence of a concomitant peripheral neuropathy. Electrophysiologic studies may reveal a prolonged latency of cortical evoked responses after tibial nerve stimulation. Typically, this myelopathy is seen late in the course of HIV-1 infection; however, it has been described as the presenting manifestation of infection.31
Gross examination of the spinal cord and dura is generally normal in HIV-associated myelopathy except when the myelopathy is particularly severe.28 The striking finding on histologic examination is the loss of myelin and spongy degeneration. The lateral and posterior columns are more severely affected than the anterior columns. Microvacuolization of the white matter of the spinal cord (Fig. 37–1) associated with lipid-laden macrophages bears an uncanny resemblance to the pathology of subacute combined degeneration of the spinal cord. The vacuolization seems to result from intramyelin swelling. Axons are preserved except in areas of marked vacuolization. Microglial nodules may be detected in the spinal cord gray matter, and 20% of the patients in one series also exhibited central chromatolysis of the anterior horn motor cells.28 Inflammation and intranuclear viral inclusions are generally not seen.
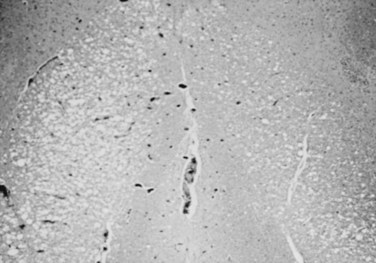
FIGURE 37–1 Microvacuolization of posterior columns of thoracic spinal cord in a patient with HIV-1–related myelopathy.
Although HIV has been cultured from the spinal cord, the specific role of HIV in the causation of this illness is uncertain. A similar clinicopathologic condition has been observed in patients with cancer or other immunosuppressive conditions in the absence of HIV infection.32 The spinal cord pathology is most prominent in the middle and lower thoracic regions. The cord involvement may be asymmetrical and does not seem to be confined to particular tracts.28 Goldstick and colleagues30 described involvement of the posterior columns increasing in intensity with rostral progression, whereas pyramidal tract involvement increased caudally. Petito and colleagues28 were able to correlate the frequency and severity of symptoms to the degree of spinal cord pathology in their series. A potential role of nutritional deficiency as the cause of spinal cord pathology has been suggested.33 Vacuolar myelopathy is not observed in young children with AIDS; however, pathologic abnormalities of the spinal cord are frequently seen at autopsy.34 A loss of myelin and axons in the corticospinal tracts seems to be the most common abnormality.
The diagnosis of this illness is one of exclusion. Numerous other myelopathies have been observed in association with HIV infection. An acute myelopathy of uncertain pathogenesis has been noted at the time of seroconversion.35 Table 37–3 lists other etiologies of spinal cord disease occurring in association with HIV.
TABLE 37–3 Potential Etiologies of the Myelopathies Associated with Human Immunodeficiency Virus (HIV) Infection
| Infectious |
| Viral |
| Bacterial |
| Fungal |
Human T-Cell Lymphotropic Virus Type I
Although thought to be rare in the United States, human T-cell lymphotropic virus type I (HTLV-I) has been observed with increasing frequency in certain subpopulations. A study of volunteer blood donors by the American Red Cross revealed a seropositivity rate of 0.025% among volunteer blood donors.36 Intravenous drug abusers seem to be at particularly high risk for infection with HTLV-I. Seroprevalence rates in this population range from 7% to 49%.37,38 In a study of the seroprevalence among female prostitutes in eight areas of the United States, 6.7% were seropositive for HTLV-I and HTLV-II with prevalence rates ranging from 0% in southern Nevada to 25.4% in Newark, New Jersey.39 A case of transmission of HTLV-I by blood transfusion associated with myelopathy has been confirmed.40 In 10 native-born cases in the United States, 5 had received blood transfusions, and 6 had multiple sex partners (including one who frequented prostitution and had a history of drug abuse).41
In addition to being associated with adult T-cell leukemia/lymphoma, HTLV-I is associated with a chronic progressive myelopathy.42,43 An association between the presence of seropositivity for HTLV-I and multiple sclerosis has also been suggested,44 but this remains controversial.
The myelopathy that occurs with HTLV-I has been referred to as tropical spastic paraparesis (TSP) or HTLV-I associated myelopathy (HAM). This myelopathy is characterized on neuropathologic studies by chronic involvement of the pyramidal tracts chiefly at the thoracic level resulting in spastic lower extremity weakness and a spastic bladder. Paresthesias, pain, and sensory disturbances may also be observed. It is estimated that 1 in 250 individuals infected with HTLV-I develop this progressive myelopathy.45 The major pathologic features of HTLV-I myelopathy are long tract degeneration and demyelination affecting the pyramidal, spinocerebellar, and spinothalamic tracts associated with hyalinoid thickening of the media and adventitia of blood vessels in the brain, spinal cord, and subarachnoid space with perivascular cuffing with leukocytes, astrocytic gliosis, and foamy macrophages.46 These lesions may extend from the upper cervical cord to the lumbar regions. Vacuolization may be observed at the periphery of the lesions.
HTLV-II, a related type C retrovirus in the Oncoviridae subfamily also rarely may result in a myelopathy similar to HAM or TSP.47–53 The epidemiology of this virus is different from HTLV-I because the populations principally affected are American Indians and intravenous drug abusers.54 The mode of transmission parallels that of HTLV-I and HIV. The rarity of these neurologic disorders accompanying HTLV-II has precluded meaningful analysis of treatment options. The same therapies employed in HAM or TSP have been generally recommended, however.
Herpesviruses
Varicella-zoster virus (VZV) is responsible for varicella (chickenpox) and causes shingles in adults. The virus remains latent within the dorsal root ganglia and spreads centrifugally along the corresponding nerves after reactivation resulting in a severely painful, blistering dermatomal eruption. Rarely, when the thoracic dermatomes are involved, the virus may spread centripetally and result in a necrotizing myelopathy.55 The myelitis complicating VZV infection in immunocompetent individuals typically occurs 1 to 2 weeks after the appearance of the dermatomal rash, although VZV myelitis may develop in the absence of a rash. The myelitis occurs at the level of the affected dermatome and results in paraparesis. In immunosuppressed hosts, such as AIDS patients, VZV myelopathy occurs insidiously and progresses. The disease is suspected by the close temporal relationship to the rash and is confirmed by showing the presence of VZV DNA by polymerase chain reaction in the CSF or VZV antibody in the CSF. Treatment with intravenous acyclovir (10 mg/kg every 8 hours) should be initiated.
Rarely, and usually with primary infection, HSV-2, the etiology of genital herpes, may cause a sacral radiculitis56 or an ascending myelitis.57 These neurologic complications are rarely observed with recurrent HSV-2. Epstein-Barr virus,58,59 the etiologic agent of infectious mononucleosis, and cytomegalovirus60 may also result in a transverse myelitis at the time of primary infection. In an immunocompromised host, HSV-1, HSV-2, and cytomegalovirus may result in myelitis.
Other Viruses
Many other viruses have been associated with transverse myelitis (see Table 37–2). Of patients with transverse myelitis, 20% to 40% have evidence of preceding or concurrent viral infection.2,5,7,61,62 Prompted by the increasing availability and efficacy of antiviral therapy and improved diagnostic techniques, in particular, polymerase chain reaction, these percentages are likely to increase.
Myelopathies Resulting from Bacterial Disease
Syphilis
Syphilis may affect the spinal cord in various ways.25 The pathology may be predominantly meningovascular or parenchymatous in nature. Gummas may grow within the substance of the cord or compress the cord by growth from the surrounding meninges. The clinical picture of spinal cord compression in syphilis may also arise as a result of hypertrophic pachymeningitis or vertebral lesions secondary to syphilitic osteitis. Table 37–4 presents a classification based on pathology and modified after the one proposed by Adams and Merritt.63
Adapted from Adams R, Merritt H: Meningeal and vascular diseases of the spinal cord. Medicine 23:181, 1944.
Tabes Dorsalis
Tabes dorsalis is the prototypic spinal cord disorder associated with syphilis. It is characterized by incoordination, pains, anesthesia, and various visceral trophic abnormalities.64 The earliest recognized descriptions of this disorder date to the mid-18th century. By the turn of the 20th century, tabes dorsalis was recognized with increased frequency and, according to Erb, was unequaled in frequency or importance by any other chronic disease of the spinal cord.
The pathology of tabes dorsalis is characterized by changes in the posterior spinal roots and posterior spinal columns (Fig. 37–2). Shrinkage of the spinal cord may be apparent by gross inspection. Leptomeningitis is evidenced by round cell infiltration. The dorsal columns are demyelinated, particularly in the regions of the fasciculus gracilis, root entry zone, and Lissauer tract. Astrocytic proliferation in the posterior columns is accompanied by an increase in connective tissue and thickening of the blood vessel walls. The lower spinal cord bears the brunt of the damage. Nerve fibers in the posterior root are destroyed and replaced by fibrosis. Frequently, lesions are observed in the anterior horns, cranial nerves, and brainstem.
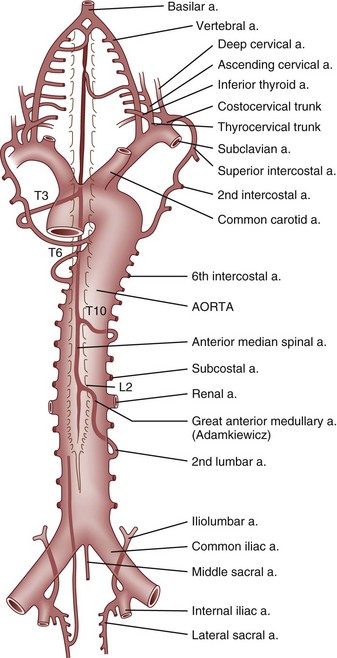
Syphilitic Meningomyelitis
Syphilitic meningomyelitis occurs most commonly in individuals 25 to 40 years old, although it may arise in younger and older age groups. In most series, men predominate. The latency from the onset of the infection to the onset of symptoms with syphilitic meningomyelitis varies from 1 to 30 years with most arising within 6 years of infection.65
Other Forms of Spinal Syphilis
Hypertrophic pachymeningitis66 is an insidious, slowly progressive syphilitic process that results in spinal root and spinal cord dysfunction. A rare but well-recognized complication of tertiary syphilis is a gumma of the spinal cord. The clinical features of a spinal gumma are indistinguishable from an intramedullary glioma if it arises within the cord or may simulate the appearance of an extramedullary tumor if it arises from the meninges and compresses the spinal cord. Spinal cord infarction is a well-recognized complication of syphilis. Another vascular complication of syphilis is aortitis, which may eventuate in an aortic aneurysm resulting rarely in myelopathy by erosion of the vertebrae and ultimately compression of the spinal cord. Progressive spinal muscular atrophy has been observed in association with neurosyphilis, although the relationship may be coincidental. Occasionally, syphilitic caries may affect the vertebrae, particularly vertebrae of the cervical spine, resulting in pain, tenderness to palpation, and loss of mobility, and an abnormal spinal curvature in the involved area is noted. Radicular pains are observed in one third of patients. Compression of the spinal cord, although rare, has been reported.
As with other forms of neurosyphilis, the recommended treatment is 12 to 24 million U of aqueous penicillin daily in divided doses administered every 4 hours for 10 to 14 days.67 Other treatment regimens68 using doxycycline, ceftriaxone, or erythromycin may be considered if the patient cannot tolerate penicillin. These treatment regimens are not well established in treating symptomatic neurosyphilis, however.
Tuberculosis
Neurologic complications of Mycobacterium tuberculosis remain common in some parts of the world but are rare in the developed countries of the Western world. Myelopathy occurring in association with tuberculosis is usually the consequence of tuberculous spondylitis (Pott disease), which accounts for half of all skeletal tuberculosis. Typically, the anterior portion of the vertebral body is affected, with the mycobacterial spread to the vertebrae being hematogenous, lymphatic, or by direct contiguity from the lung.69 The characteristic radiographic defect is anterior wedging of two adjacent vertebrae with loss of the intervening disk space. The spine is enveloped by pus extruding anteriorly from the affected vertebrae. Myelopathy typically results from pressure on the anterior spinal cord by caseous or granulating tissue, inflammatory thrombosis of the anterior spinal artery, or injury to the cord from spinal instability. The last-mentioned may lead to complete spinal cord transection.
Myelopathy occurring in association with tuberculous infection may also occur as a consequence of intraspinal granulomatous tissue unassociated with a bony lesion and intramedullary tuberculomas. In one study of spinal tuberculosis,70 54% of patients had neurologic deficit with bony tuberculous lesions, 39% had neurologic deficit with intraspinal granulomatous tissue occurring in the absence of bony lesions, and 7% had neurologic deficit from intraspinal tuberculomas.
Therapy of patients with spinal tuberculosis requires at least 12 months of antibiotic treatment and surgical decompression in the presence of neurologic abnormalities.70 In the setting of intraspinal granulomatous disease without significant bony destruction, laminectomy and débridement is adequate70; however, more aggressive therapy is warranted when vertebral bodies are involved. A two-stage procedure comprising posterior instrumental stabilization followed by anterior radical decompression permitted earlier mobilization after neurologic recovery.71 Reports of the frequency of neurologic recovery with spinal tuberculosis vary, but functional recovery rates of 90% have been reported.71 Thoracic lesions with severe neurologic deficit show the least improvement, whereas lumbar disease has the best outcome.72
Other Forms of Bacterial Myelopathy
Numerous other bacterial infections have been associated with myelitis. Rarely, the spinal cord may be seeded by bacteria leading to a suppurative myelitis with abscess formation. In a review by Dutton and Alexander,73 direct spread for adjacent infections was most commonly observed; however, hematogenous dissemination from endocarditis, pulmonary infections, and other sites was also frequently observed. Organisms isolated in these cases have included staphylococci, streptococci, Escherichia coli, and Nocardia. Rarely, Whipple disease can manifest as a myelopathy.74,75
More often, myelopathies associated with bacterial infection are parainfectious in nature, similar clinicopathologically to myelopathies occurring after viral infection or vaccination. Potential causes76,77 include scarlet fever, pertussis, whooping cough, mycoplasmal pneumonia, and pneumococcal pneumonia. Myelitis resulting in Brown-Séquard syndrome has also been described with cat-scratch disease.78 Lyme disease, the result of infection with Borrelia burgdorferi, a treponeme, may also result in myelopathy.79
Fungal Myelopathies
Fungal disease of the spinal cord is rare. Certain fungi (Blastomyces, Coccidioides, Aspergillus) may invade the spinal epidural space. Generally, the spinal cord is compromised by lesions arising from a vertebral osteomyelitic focus or by lesions extending through the intervertebral foramina. Certain fungi that result in granulomatous meningitis (e.g., Cryptococcus neoformans) may result in intraspinal or extradural granuloma. Alternatively, these organisms can lead to spinal cord infarction as a result of the associated meningovascular inflammation. Aspergillosis is generally observed only in immunosuppressed patients, with leukemia and lymphoma being common predisposing illnesses. Aspergillosis has been reported to affect the spinal cord in several different ways, including by compromise of the blood supply occurring in association with fungal endarteritis, by direct parenchymal infiltration of the spinal cord, or by cord compression from osteomyelitis and paravertebral mass.80
Parasitic Myelopathies
Among the most common parasites that result in spinal cord disease is Schistosoma,81,82 in particular, Schistosoma haematobium and Schistosoma mansoni. These organisms are seen only in certain geographic regions—the Far East, South America, and Africa. A history of travel to these regions and swimming or bathing in water contaminated with the cercariae that are released from certain aquatic snails may suggest the diagnosis.
Paragonimiases, infection with a lung fluke acquired by eating undercooked freshwater crabs, occurs chiefly in China but may be seen in other parts of the world. Spinal cord disease results from extradural or, more rarely, intradural granuloma formation. Angiostrongylus cantonensis, the most common cause of eosinophilic meningitis and meningoencephalitis in the world, has also been reported to cause spinal cord disease.83 In patients with AIDS, toxoplasmosis has been reported in rare instances to cause an abscess of the spinal cord.
Epidural Abscesses
Spinal epidural abscess may manifest as a surgical emergency evolving rapidly over several days or may arise more indolently. Staphylococcus aureus is the etiologic agent in greater than 50% of acute spinal epidural abscesses, although a broad spectrum of other organisms may be implicated.84 The spread of infection may be directly from a focus of osteomyelitis or hematogenously from a distant site, such as skin furuncles or pulmonary infections. Trauma to the back, typically very minor in nature, has been reported by one third of individuals developing spinal epidural abscess.85 A high degree of suspicion for spinal epidural abscess should be maintained when intravenous drug abusers present with fever and back pain.
Arachnoiditis
Invasion or irritation of the spinal subarachnoid space, as with cases of subarachnoid hemorrhage, meningitis, myelography, spinal or epidural anesthesia, or spinal surgery, can result in arachnoiditis. Arachnoidal inflammation leads to connective tissue proliferation and ultimately to arachnoid thickening, opacification, and adhesion and obliteration of the subarachnoid space. Generally, neurologic complications are the consequence of nerve root involvement; however, in the most severe cases, the nerve roots and the spinal cord may be compressed by bands of connective tissue or cystic loculations of CSF, resulting in myeloradiculopathy. Patients present most frequently with slowly progressive paraparesis, sensory loss, and sphincter dysfunction. Radicular pain of a severe, burning nature may also be present. The pain may not always follow a dermatomal distribution.86 On examination, weakness, hyporeflexia and sensory deficits can be seen. Diagnosis is made by myelography or MRI. Although some patients, particularly patients with mass lesions resulting in cord compression and myelopathic symptoms, may respond to decompression, surgical exploration with resection of adhesions has not been proven to be beneficial in most patients.
Vascular Disease of the Spinal Cord
Paired segmental arteries arising from the aorta and branches of the subclavian and internal iliac arteries supply blood to the spinal cord (Fig. 37–3).87 The most important vascular supply to the cervical spinal cord arises from the vertebral artery, which provides the cephalad origin of the anterior median and posterior lateral spinal arteries. The blood supply to the thoracic and lumbar spinal cord arises from the aorta and internal iliac arteries, and segmental branches of the lateral sacral arteries nourish the sacral spinal cord. The segmental branches that arise from the aorta divide into anterior and posterior rami. A branch of the posterior ramus (see Fig. 37–2), the spinal artery, enters the vertebral foramen and branches at irregular intervals into anterior and posterior medullary arteries, which feed the anterior median spinal artery and the posterior spinal arteries. At regular intervals, the spinal artery also branches into the anterior and posterior radicular arteries, which supply the spinal ganglia and roots.
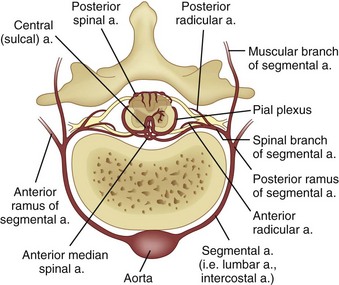
FIGURE 37–3 Anterior view of spinal cord with its segmental blood supply from aorta.
(From Herrick M, Mills PE Jr: Infarction of spinal cord. Arch Neurol 24:228, 1971. Copyright 1971, American Medical Association.)
An occlusion of a dominant medullary artery or, more rarely, the anterior median spinal artery results in an ischemic softening of a variable portion of the anterior two thirds of the spinal cord. This entity is referred to as an anterior spinal artery syndrome and may arise as the consequence of thrombotic atherosclerotic disease, aortic dissection, embolization, or vasculitis (particularly polyarteritis nodosa) or as a complication of aortic angiography.88 Cross clamping of the aorta during cardiac surgery for more than 30 minutes may also result in an infarction in this territory. A clinical hallmark of this entity is a dissociated sensory loss in which position and vibratory sensory perception are maintained, but a sensory level to pinprick is present. The latter is accompanied by paralysis below the level of the lesion, which typically manifests in association with spinal shock. Rarely, anastomotic blood flow allows for the preservation of the white matter of the spinal cord, and the gray matter alone is infarcted. Painful segmental spasm and spinal myoclonus may be observed with this condition.10
An unusual and seldom clinically diagnosed etiology of vascular injury to the spinal cord results from fibrocartilaginous embolization. The embolization typically follows minor trauma to the vertebrae.89 Among the reported precipitants have been lifting heavy weights, prolonged coughing, stooping, or falling.90 Valsalva maneuver has been suggested as a possible contributing factor resulting in the retrograde flow of fibrocartilaginous emboli to the spinal cord. Some investigators have suggested that the fibrocartilaginous debris is forced into the vessels of the spinal leptomeninges and spinal cord through the valveless Batson venous plexus.91 Minor trauma to the vertebrae before onset was noted in most cases with the time to maximum neurologic deficit ranging from minutes to 24 to 48 hours.89 Death typically supervenes within 11 months of onset (median 2.5 to 4 months).89 Multiple emboli have been postulated to occur in some of these instances.92 Involvement of the anterior spinal artery has also been observed.93 Spinal MRI reveals characteristics indicative of vascular lesions.92
Vascular malformations are another etiology of hemorrhage into or around the spinal cord. One type of spinal cord vascular malformation is venous angioma, which is found most often on the dorsal portion of the spinal cord. Middle-aged and elderly men are chiefly affected. The slow and temporally irregular development of symptoms resulting from this lesion is believed to be secondary to ischemic compromise and compression of the spinal cord. Another type of spinal cord vascular malformation is arteriovenous malformation, which predominately affects younger patients. It is most often located on the dorsal thoracic or upper lumbar spinal cord. With this lesion, symptoms may be slowly progressive or may appear suddenly. Rapidly developing symptoms may result from occlusion of a key nutrient vessel or hemorrhage. The association of a cutaneous vascular nevus with a vascular malformation of the spinal cord has been referred to as Klippel-Trenaunay-Weber syndrome. The syndrome of Alajouanine94 or angiodyskinetic myelomalacia is a necrotic myelopathy resulting in a slowly evolving amyotrophic paraplegia in men that has been attributed to spinal venous thrombosis. In the absence of a spinal dural arteriovenous fistula, it is likely to be impossible to distinguish this entity from tumor on radiographic imaging.95 Although its exact nature remains controversial, some investigators have proposed that the lesions are probably acquired rather than congenital.96
Venous infarction of the spinal cord can also occur in the absence of an arteriovenous malformation. This spinal cord disorder may be extremely difficult to identify in light of the absence of specific clinical, laboratory, or radiographic findings short of spinal angiography. For this reason, it is likely seldom recognized and underreported. Kim and colleagues97 classified venous spino-occlusive disease into three types: nonhemorrhagic infarction, hemorrhagic infarction, and embolic infarction. Any region of the spinal cord may be affected, but involvement of the thoracic cord seems to predominate.
Nonhemorrhagic infarction is protracted, seldom accompanied by back pain, evolves slowly, and has survivals averaging approximately 4 years.97 Hemorrhagic infarction is generally sudden in onset and rapidly progressive. Back pain is observed. The full extent of the spinal cord may be affected.98 Survival averages approximately 1 month.97 Hemorrhagic infarction has been reported in association with hypercoagulable syndromes (e.g., Trousseau phenomenon) complicating pancreatic adenocarcinoma.99 Embolic venous infarction was initially reported by Feigin and colleagues91 in 1965. This disorder begins precipitously often with pain in the back and extremities. Asymmetrical and dissociated loss of function occurs more commonly in this form of venous spino-occlusive disease.97
Myelopathy Secondary to Connective Tissue Diseases
Rheumatoid Arthritis
Rheumatoid arthritis may result in myriad complications involving the spinal cord. Among the major abnormalities of the spine and spinal cord in rheumatoid arthritis are vertebral body erosion,100 discitis,101 and spinal cord compression102 that may be secondary to pannus formation. The most dramatic and frequent abnormality occurs at the atlantoaxial region.103 Cervical subluxation may assume many forms in this disorder,103,104 including anterior subluxation, posterior subluxation, vertical subluxation with protrusion of the odontoid, and rotational atlantoaxial subluxation. Other abnormalities include ligamentous calcification, erosion, cystic changes, and spinous process erosion.104 Cervical subluxation may be asymptomatic, although neck pain is common. Lower extremity weakness and spasticity, sensory loss, and sphincter disturbances are seen with lesser degrees of frequency. Hyperextension of the cervical spine, such as may occur with endotracheal intubation, in the face of cervical instability secondary to rheumatoid arthritis may cause severe displacement with rapidly evolving myelopathy.
Unusual causes of myelopathy in a patient with rheumatoid arthritis include “epidural lipomatosis” in patients on high-dose corticosteroid therapy105 and progressive cervical osteomyelitis.106 Rarely, these patients may develop pseudoaneurysm of the vertebral artery or anterior spinal artery occlusion secondary to compression. Marked C1-2 abnormalities seem to be more frequent in young women with severe, long-standing, seropositive rheumatoid arthritis.107 MRI may be useful in identifying pannus formation and craniovertebral involvement in rheumatoid arthritis.108 Many patients stabilize with conservative therapy,109 and the presence of cervical subluxation does not correlate with decreased survival from rheumatoid arthritis. Surgical craniocervical decompression can be helpful in early cases in which the patients remain ambulatory and are medically stable110 but is probably not helpful in advanced nonambulatory cases.111
Sjögren Disease
Sjögren disease is typically a disease of women 30 to 50 years old characterized by dry eyes (xerophthalmia), dry mouth (xerostomia), and noninflammatory arthritis. Neurologic manifestations are diverse and may reflect peripheral nervous system and CNS involvement. Spinal cord involvement includes progressive myelopathy, acute transverse myelitis, and intraspinal hemorrhage.112 Recurrent transverse myelitis may be observed. Pathologic examination revealed angiitis and necrotizing myelitis in one instance.113 Myelopathy related to Sjögren disease can be associated with optic neuropathy or cutaneous vasculopathy.114,115 The evaluation of Sjögren disease includes Schirmer test, serologic testing for anti-Ro (SSa) and anti-La (SSb) antibodies, and labial biopsy. Anecdotal reports have suggested improvement with corticosteroids and plasma exchange.116
Systemic Lupus Erythematosus and Antiphospholipid Antibody Syndrome
Myelopathy is an uncommon manifestation of SLE.117 In one series of 315 patients with SLE, acute transverse myelopathy was observed in 10 (3.2%).118 Most patients have evidence of other systemic disease at the time of diagnosis; however, this and other features of CNS lupus may lead to confusion with multiple sclerosis.119 In a large series of lupus myelopathy, optic neuritis was observed in 48% of patients.120 The association of optic neuritis and myelopathy may be particularly difficult to distinguish from multiple sclerosis. Typically, patients complain of numbness and weakness of the lower extremities manifesting in a subacute fashion, although in one series the cervical cord was involved in 50%.118
SLE-related myelopathy disables two thirds of affected individuals, but the other one third may recover significantly.121 Sedimentation rate and complement levels seem to be insensitive as markers of disease activity for this condition,122 and myelopathy is not associated with anti–ribosomal P, anti-ENA, or antiphospholipid antibodies.118 CSF abnormalities are seen in more than 60% of cases,118 including increased protein, pleocytosis, and even mildly decreased glucose. MRI may reveal prolongation of T1 or T2 signal in the affected cord, spinal cord enlargement, and contrast enhancement.123 The presence of a “longitudinal myelitis,” characterized by increased signal on T2-weighted imaging over many continuous segments has been suggested as a diagnostic clue, particularly for SLE-related myelopathy associated with antiphospholipid antibody.124
Aggressive treatment with intravenous high-dose steroids within 1 week of onset of symptoms has been associated with better outcomes.125 Other treatment modalities have included cyclophosphamide120 and plasmapheresis; however, the relative rarity of the syndrome has precluded rational clinical trials, and the effects of these and other therapeutic options remain uncertain. Subarachnoid spinal hemorrhage may occur in association with SLE.112 Because some cases of SLE-related myelopathy are associated with antiphospholipid antibodies, hypercoagulability or vasculopathy may be partly responsible for the pathogenesis of the disorder.126
Isolated myelopathy127–130 and optic neuropathy associated with myelopathy131 have been reported to accompany antiphospholipid antibody syndrome. Recurrent transverse myelitis has also been reported.132,133 Antiphospholipid antibody syndrome is a disorder in which hypercoagulability is associated with antiphospholipid antibodies (lupus anticoagulant, anticardiolipin, and antibodies directed against β2-glycoprotein I).134 A wide variety of neurologic disorders may be associated with this disorder.135 The myelopathy of antiphospholipid antibody syndrome may be recurrent in nature136 making it difficult to distinguish from multiple sclerosis, particularly when associated with optic neuritis. The disorder may be a variant of SLE. Response to treatment seems to be inconsistent. Recommended therapies have included methylprednisolone, intravenous immunoglobulin, and measures to reduce the antibodies.
Other Autoimmune Myelitides
Transverse myelitis occurring at any level of the spinal cord may complicate polyarteritis nodosa, a disease of small and medium-sized arteries. Cervical spine disease may also be seen with psoriatic arthritis. In 35% of these cases, anklyosing spondylosis occurs with syndesmophytes and ligamentous ossifications. Ankylosing spondylosis may also result in atlantoaxial subluxation in a manner similar to that of rheumatoid arthritis.137
Acute transverse myelitis is also known to occur in mixed connective tissue disorder and ulcerative colitis.138,139 Granulomatous compressive thoracic myelopathy has also been reported as the initial manifestation of Wegener granulomatosis.140 Immunosuppressive therapy seemed to be partially effective in improving the myelopathic features.
Sarcoid Myelopathy
In the study by Junger and colleagues,141 the patients were noted to have paraparesis, urinary bladder dysfunction, radiculopathies, chest wall numbness, gait problems, Brown-Séquard syndrome, or limb numbness or pain. The mean age of onset of neurologic symptoms was 35. Evaluation of the CSF revealed mild or moderate pleocytosis (1 to 200 cells/mm3 [mean 36 cells/mm3]) and elevated protein (52 to 568 mg/dL [mean 162 mg/dL]).
Definitive diagnostic testing requires biopsy showing noncaseating epithelioid granulomas. As intramedullary biopsy is rarely desirable; biopsy material may be obtained from safer sites (e.g., the lungs) if available. MRI can be normal in some suspected cases of spinal sarcoidosis.142 Observed changes in spinal sarcoidosis include gadolinium-enhancing nerve roots, enhancing parenchymal spinal cord masses,143 diffuse spinal cord enlargement, spinal cord atrophy, or focal or diffuse areas of increased T2-weighted signals.141 Gallium scanning may show uptake in the lungs or parotid, salivary, or lacrimal glands. Systemic disease can be detected by increased serum angiotensin-converting enzyme levels. It is unclear at present whether measurements of CSF angiotensin-converting enzyme levels are helpful in diagnosing nervous system involvement. Serum angiotensin-converting enzyme levels are not informative with respect to CNS involvement.
Spontaneous recovery over months or years can occur in 60% to 80% of patients with isolated pulmonary disease, but very little is known about the natural history of CNS disease. Most authorities proceed aggressively with steroids if neurologic involvement becomes symptomatic. If sarcoidosis is refractory to corticosteroids, cyclosporine, cyclophosphamide, chlorambucil, methotrexate, and radiation therapy have been applied in combinations with steroid with some success.144,145
Nutritional Myelopathies
Vitamin B12 Deficiency
The myelopathy that results from vitamin B12 deficiency (subacute combined degeneration) is chiefly characterized neuropathologically by foci of demyelination in the posterior and lateral columns of the cervical and upper thoracic spinal cord. The earliest changes of demyelination are fusiform expansions of the myelin sheaths. Subsequently, the myelin degenerates, and if the process is uninterrupted, gliosis and involvement of the axons eventually ensue. Clinical remissions are anticipated when this myelopathy is treated expeditiously with hydroxycobalamin or cyanocobalamin. The mechanism by which vitamin B12 deficiency results in demyelination is unknown. It is a cofactor in choline synthesis and important in the conversion of methylmalonyl-CoA to succinyl CoA, both important to the myelin sheath.146 Fusiform expansion of the myelin sheaths is followed by myelin degeneration.146,147 If the process is uninterrupted, gliosis and involvement of the axons eventually ensue.146 After effective therapy, the myelopathy of vitamin B12 deficiency not only may resolve clinically, but also radiographically.
Myelopathy Complicating Copper Deficiency
Copper deficiency may result in spinal cord disease that can be clinically reminiscent of the neurologic manifestations of vitamin B12 deficiency.148 Sensory ataxia with a spastic gait and marked ataxia coupled with a distal axonal sensorimotor neuropathy can be observed.149 Causes of copper deficiency include prior gastric surgery, excessive zinc ingestion, and malabsorption; however, an underlying cause is not always uncovered.150 Anemia and neutropenia are often present. Spinal cord disease and peripheral neuropathy developing after gastric bypass surgery should always raise concern about the presence of either vitamin B12 deficiency or copper deficiency.151,152
Metabolic Myelopathies
Myelopathy Resulting from Portosystemic Shunts
A progressive myelopathy has been observed in association with portosystemic shunting.153 Although the illness generally accompanies alcoholic cirrhosis, it may arise secondary to other causes of portosystemic shunting. A selective demyelination predominates in the posterior and lateral funiculi of the spinal cord. Hepatic encephalopathy often accompanies the myelopathy. Myelopathy is manifested by an insidiously developing spastic paraparesis and gait with relative preservation of sensory and sphincter function. Hyperparathyroidism and hyperthyroidism have been rarely associated with cervical myelopathies that remit when the endocrinologic derangement is removed.154–156
Epidural Lipomatosis or Epidural Hibernomas
The prolonged use of large doses of corticosteroids may result in epidural lipomatosis. Epidural lipomatosis, a deposition of epidural fat, causes a compressive myelopathy. Additionally, it has been reported as an etiology of radiculopathy, cauda equina syndrome, and neurogenic claudication.105 Spinal MRI should be highly suggestive of the diagnosis.
Paraneoplastic Myelopathies
Paraneoplastic syndromes are syndromes associated with underlying malignancies not caused by direct tumoral invasion or macroscopic metastatic disease. These remote effects of cancer are usually without an identifiable etiology, although some syndromes are associated with circulating antibodies to nervous system tissue or are the result of a presumptive concomitant viral infection. These syndromes generally are uncommon. Approximately 50% of all paraneoplastic syndromes are associated with small cell carcinoma of the lung.157 The prototypic paraneoplastic syndrome is subacute cerebellar degeneration, which is associated with loss of Purkinje cells in the cerebellar cortex and various circulating antibodies directed against these cells. Paraneoplastic myelopathy is rare158—much less common than myelopathy resulting from metastatic epidural spinal cord compression. Myelopathy occurring in association with malignancy can also be secondary to radiation therapy or the result of the combined effects of radiation therapy and intrathecal chemotherapy, especially methotrexate chemotherapy,159,160 herpes zoster,161 abscess, or hematoma. These myelopathies may occur in isolation or as part of a syndrome in which encephalomyelitis,162 cerebellar degeneration, or peripheral radiculoneuropathies are also observed.
The most typical myelopathy is a necrotizing myelopathy. It is a rare entity associated with lymphoma, leukemia, and small cell lung cancer. Myelopathy may manifest before, concomitant with, or after the initial tumor presentation. Clinically, it is characterized by a rapidly ascending spinal cord dysfunction leading to a flaccid areflexic paraplegia. Myelopathy may manifest asymmetrically and often results in Brown-Séquard syndrome163; however, eventually it becomes bilateral and symmetrical. The brainstem may also be involved. There is no effective treatment. One patient with Hodgkin disease and a paraneoplastic myelopathy was believed to respond favorably to intrathecal corticosteroids.164 CSF usually shows an elevated protein and a mild pleocytosis. Pathologically, there is widespread necrosis of the cord, mostly in the thoracic region.165 Gray and white matter are involved, and lesions may be identified elsewhere in the CNS. In two cases of “paraneoplastic” necrotizing myelopathy, immunohistochemical studies and electron microscopy revealed convincing evidence of infection with HSV-2.166
Other, less common syndromes include a subacute motor neuropathy associated with Hodgkin disease and other malignant lymphomas.167 It is typically diagnosed at a later stage and sometimes after radiation therapy. The motor weakness is clinically of the lower motor neuron type and involves the legs more than the arms. Sensory loss is mild. The course is benign and may stabilize or improve with specific therapy. Pathologically, neuropathy shows degeneration of the anterior horn cells and resembles an indolent poliomyelitis.167
A syndrome of paraneoplastic myelopathy with limbic encephalitis is associated with the antineuronal autoantibody anti-Hu.168 This syndrome is almost always related to small cell lung cancer, and the presence of anti-Hu should lead to aggressive workup for neoplasms. This antibody is also associated with a subacute generalized sensory neuronopathy.
Although not truly a paraneoplastic disorder, intravascular lymphomatosis should be considered in the differential diagnosis of a patient presenting with an unexplained myelopathy. Spinal cord involvement occurs in approximately one third of patients. Although myelopathy, including conus involvement,169 may be the heralding feature of the disorder, it seldom remains the only neurologic manifestation.170 The spinal cord disorder may be indolent or rapidly progressive and may mimic multiple sclerosis or idiopathic transverse myelitis. An initially favorable but incomplete response to corticosteroids may be observed. Damage to the nervous system results from ischemia secondary to occlusion of small-caliber vessels. This disorder is most often the consequence of a clonal expansion of CD20+ B lymphocytes, although T cells and natural killer cells have also been reported. In most cases, the diagnosis remains a conundrum and is established at autopsy.171 Aggressive therapy with CHOP (cyclophosphamide, hydroxydaunomycin, vincristine [Oncovin], and prednisone) has been associated with long-term survival, but the disease is generally fatal.
Myelopathy Secondary to Radiation Therapy
Myelopathy secondary to radiation therapy is an iatrogenic illness. The incidence is affected by the total dose of radiation delivered, the dose per fraction, and the total volume of tissue irradiated.172 In a large study by Kagen and colleagues,173 it was determined that spinal cord injury could be avoided if the total dose delivered was kept to 6000 rads and given over a 30- to 70-day period at a rate not exceeding 200 rads/day or 900 rads/wk.
Two pathophysiologic mechanisms are proposed for this myelopathy: direct damage to the nervous tissue of the spinal cord by irradiation and damage to the vascular supply of the spinal cord. The effects of radiation take the form of an early delayed and late delayed myelopathy. There are no acute effects of radiation on the spinal cord.172 The incidence of this complication is 2% to 3%174 but is substantially higher when radiation therapy is combined with hyperthermia.175
The early delayed radiation myelopathy usually manifests several weeks after radiation therapy as sensory symptoms and paresthesias. These symptoms may be exacerbated by neck flexion, and a typical Lhermitte phenomenon may be detected. These symptoms are believed to be secondary to demyelination and depletion of oligodendrocytes.10 The presence of this early delayed myelopathy does not predict the development of a late delayed radiation injury.176
Late delayed radiation myelopathy may evolve in two distinct forms: (1) a progressive myelopathy that usually occurs 12 to 15 months after radiation therapy and never before 6 months and (2) a progressive lower motor neuron weakness that occurs 3 to 14 months after radiation. The former is characterized by sensory symptoms similar to early delayed myelopathy, but it is accompanied by an asymmetrical weakness. Frequently, the initial picture is that of Brown-Séquard syndrome, which often progresses to complete transverse myelopathy with spastic paraplegia; truncal sensory level; and bowel, bladder, and sexual dysfunction. The CSF profile is unremarkable except for the frequent presence of a slightly elevated protein. Pathologic changes include areas of necrosis that affect gray and white matter, but white matter seems to be preferentially affected. The posterior columns and posterolateral columns may be especially involved.172
Not unexpectedly, the progressive lower motor neuron weakness after radiation therapy is accompanied by pathologic alterations in the anterior horn cells. There is an asymmetrical atrophy with fasciculations and areflexia.177,178 No sensory or sphincter disturbances are noted. This peculiar myelopathy resembles a paraneoplastic subacute motor neuropathy described in patients with lymphoma.167
Toxins
Several agents with industrial, pharmaceutical, and medical applications have been identified as toxins capable of producing myelopathy. Toxic iatrogenic causes of myelopathy include spinal anesthesia and exposure to contrast agents used with myelography and angiography. Spinal anesthesia, with the epidural or intrathecal administration of local anesthetics, has been reported to produce myelopathy.179,180 Although an extremely rare occurrence, myelopathy following spinal anesthesia may be permanent and includes frank paraplegia, sensory loss, and loss of sphincter function.181,182
Some investigators contend that the true incidence of these complications is underreported for medicolegal reasons.183 In a review of 32,718 cases of epidural or spinal anesthesia,184 transient paralysis was observed in 48 (0.1%), and permanent paralysis was observed in 7 (0.02%). Similarly, only 3 of 50,000 patients were reported to have permanent lower extremity weakness in a combined series.185 The first case report of paraplegia after epidural anesthesia was by Davies and colleagues in 1958.186 The mechanisms of spinal anesthesia–induced myelopathy, particularly myelopathy induced by epidural anesthesia, remain elusive. Possible etiologies187 include direct neurotoxicity of these agents, toxicity of drug diluents or contaminants, hypotension leading to spinal cord infarction (typically from anterior spinal artery syndrome), epidural hematoma, epidural abscess, trauma from the epidural needle, and exacerbation of an underlying process. In some instances, a surgical procedure, such as renal transplantation, may be contributory or causative. In the latter example, the artery of Adamkiewicz may be inadvertently traumatized. Cases of myelopathy after spinal anesthesia in the setting of spinal cord tumors or herniated discs have been reported.188
Although oil-soluble and water-soluble myelographic contrast agents may induce arachnoiditis, which may lead to spinal cord dysfunction, these agents may themselves produce a toxic myelopathy. Although the use of modern contrast agents has essentially eliminated this complication, nearly 6% of patients had permanent myelopathic findings after myelography in early reports.189 Transient myelopathic symptoms lasting 24 hours have also been reported after administration of water-soluble contrast agents.77
Myelopathy as a complication of spinal angiography has been well recognized.190 Although this complication is most often ascribed to the induction of vasospasm or embolic thrombosis of spinal vessels resulting in ischemic infarction, a direct neurotoxic effect of the angiographic agents has been implicated in some instances.191,192 Patients may develop pain and spasms immediately on contrast agent injection, or this may be delayed by several hours. Subsequently, patients progress to flaccid paraplegia with frequent sensory and sphincter dysfunction. Partial recovery occurs in about half of the patients, and complete recovery occurs in about 20% within weeks.77 Many patients are left with a spastic paraplegia.
Although an extremely safe agent when used according to recommendations, nitrous oxide can manifest neurotoxicity when abused or under conditions of chronic exposure.193,194 Usually long delayed after nitrous oxide exposure, the resulting myeloneuropathy syndrome may include sensory dysesthesias, leg weakness, spasticity, sphincter dysfunction, and ataxia. Thought to result from a nitrous oxide–induced inhibition of vitamin B12 use, the symptoms tend to resolve after exposure is discontinued.
Intrathecal administration of antineoplastic agents, such as methotrexate or cytosine arabinoside, has been reported to produce transient and permanent myelopathy.195–197 Intrathecal steroid administration may initiate an acute meningeal reaction, probably secondary to polyethylene glycol detergent included in the preparation.198,199 Distinct from problems that are due to a secondary arachnoiditis, this acute syndrome may result in back and leg pain, paresthesias, and sphincter dysfunction. Intravenous heroin administration may also cause an acute transverse myelitis. This syndrome may result from direct drug toxicity, a systemic reaction to the drug itself or to quinine or another drug diluent, a hypersensitivity reaction, or transient spinal cord ischemia.200
Iodochlorohydroxyquinolone, a drug used in the treatment of infectious diarrheas, has been reported to cause a syndrome of myeloneuropathy sometimes accompanied by optic atrophy.201 Seen most frequently in Japan, this syndrome usually follows an episode of abdominal pain or diarrhea and therapy with iodochlorhydroxyquinoline. Although apparently related to the total accumulated dose of the drug, this syndrome might also be related to an enteric virus associated with the patients’ initial abdominal symptoms. Patients initially complain of ascending numbness and paresthesias, which develop into a profound sensory loss. Gait ataxia, leg weakness, and sphincter dysfunction are frequent concomitants; optic atrophy and visual loss are seen in about 25% of patients. After discontinuing the drug, complete or near-complete recovery is the rule.
Triorthocresyl phosphates, used as industrial lubricating oils and solvents, are highly neurotoxic. Although accidental occupational exposure is rare, patients are often exposed by ingesting triorthocresyl phosphates in lieu of ethanol or by ingesting cooking oils contaminated with these compounds. Although the most profound neurologic complication of triorthocresyl phosphate ingestion is an acute peripheral neuritis, clinical signs of spinal cord degeneration can be seen in patients with persistent symptoms. Wallerian degeneration of the pyramidal tract and chromatolytic changes in dorsal and ventral horn cells has been identified.202,203 In patients developing findings of myelopathy, the clinical syndrome is usually permanent.
Electrical Injury
Electrical injury of the nervous system results most frequently from accidental exposure to high-tension currents, although lightning and complication of electroshock therapy have also been reported to cause neurologic injury. Although current sufficient to cause damage to the nervous system is usually fatal, individuals who survive such injuries are subject to damage throughout the neuraxis.77,204 Acutely, patients may have neurologic symptoms referable to cerebral anoxia secondary to cardiac arrhythmias or respiratory arrest. Neural tissue examined shortly after electrical injury shows petechial and perivascular hemorrhages and severe ganglion cell changes. The most abnormal areas of the spinal cord are in the path of the electrical current. Short-term survivors of this acute phase may show focal myelomalacia and mild gliosis on autopsy. Although the pathophysiology is not fully understood, the injury most probably reflects a direct vascular injury to spinal cord vessels, an indirect vascular injury to spinal vasomotor nerves, or a direct effect of current on spinal cord tissue. Pathologic changes noted in long-term survivors postmortem have included demyelination with preservation axons, anterior horn cell loss, and necrosis.
Permanent neurologic manifestations of electrical injury are uncommon. The most characteristic neurologic effect of electrical injury is delayed myelopathy, occurring in 1% to 6% of victims.205,206 Immediately after electrical injury, patients may complain of paresthesias, pain, urinary dysfunction, or impotence. These symptoms tend to improve rapidly. Occasionally, delays in the onset of neurologic symptoms of up to 6 weeks (averaging 1 week) may be observed. Neurologic signs may worsen over 2 to 14 days.207 According to Winkelman,207 one third of patients recover fully, one third have partial recovery, and one third experience no recovery. The last-mentioned is the rule in patients with a complete spinal cord lesion or progression of spinal symptoms over a prolonged period. Rarely, delayed neurologic deficits can appear after a latency of several months.208,209 The symptoms of delayed myelopathy after electrical injury are typically permanent, but rapid recovery has also been reported.210
A cervical myelopathy with atrophic quadriparesis is the most frequent complication; this myelopathy results from current passing from hand to hand.211,212 With low-voltage injuries (<1000 volts), muscular atrophy from anterior horn cell damage is most common.77 With higher voltages, more profound injuries occur to the lateral and posterior columns.77 Pyramidal signs, including spastic weakness, hyperreflexia, and Babinski signs, predominate regardless of the location of the lesion. Sensory findings are less prominent with posterior column dysfunction predominating. Lhermitte phenomenon has been reported rarely.213 Other forms of electrical injury to the spinal cord result in spinal atrophic paralysis, a syndrome of focal muscular atrophy that typically follows low-tension electrical injury that has a predilection to involve gray rather than white matter and, questionably, amyotrophic lateral sclerosis.
Barotrauma
Spinal cord injury following rapid changes in atmospheric pressure, such as can occur in caisson work, scuba diving, and flying, has been well documented.214–216 Spinal cord damage results from too-rapid decompression after exposure to significant increases in atmospheric pressure. At higher atmospheric pressures, increasing amounts of gas are dissolved into tissue. The higher tissue concentration of oxygen is used in oxidative metabolism, whereas nitrogen gas, which is inert, remains dissolved only by virtue of the hyperbaric condition. With decompression, the nitrogen is released; when decompression is too rapid, nitrogen bubbles may form and occlude the spinal cord vasculature.
Therapy for decompression myelopathy is recompression followed by controlled slow decompression. If treatment is instituted rapidly, complete recovery is possible. If it is delayed more than a few hours, the chances of recovery are remote.215,217
Heredofamilial Degenerations
Numerous genetic neurodegenerative diseases can include spasticity attributable in part or total to spinal cord involvement in their complex of multiple signs and symptoms, but two disorders have quite prominent spastic paraparesis. Hereditary spastic paraplegia appears in autosomal recessive, autosomal dominant, and X-linked forms. A locus for the autosomal recessive form has been found on chromosome 8q; loci for the dominant form have been found at 2p, 14q, and 15q; and a locus for the X-linked form has been found at Xq22. In one family with the X-linked form, this has been shown to be due to a mutation in a proteolipoprotein.218 There is spastic weakness in the legs, gait difficulties, hyperactive reflexes, and extensor plantar signs. The course is one of slow but relentless progression. There are rare variants with other associated degenerations, such as optic neuropathy. The diagnosis is made by family history and excluding any other causes.10
Patients with Friedreich ataxia begin having problems in childhood. There is spastic quadriparesis owing to upper motor neuron degeneration, ataxia from cerebellar degeneration, numbness and foot deformity from neuropathy, nystagmus, tremor, and other problems. There is degeneration of the posterior columns, corticospinal tracts, spinocerebellar tracts, dentate nuclei, cranial nerve nuclei, and myocardial muscle fibers. There is no treatment, and death occurs in young adulthood. It has been found more recently that most cases of the illness are due to trinucleotide repeat expansion in the gene X25, which codes for a protein frataxin, on chromosome 9q13. A few cases are due to point mutations in the same gene.219
Pitfalls
Key Points
1 Adams RD, Merritt HH. Meningeal and vascular diseases of the spinal cord. Medicine. 1944;23:181.
2 Kovacs B, Lafferty TL, Brent LH, et al. Transverse myelopathy in systemic lupus erythematosus: An analysis of 14 cases and review of the literature. Ann Rheum Dis. 2000;59:120-124.
3 McLean JM, Palagallo GL, Henderson JP, et al. Myelopathy associated with fibrocartilaginous emboli (FE): Review and two suspected cases. Surg Neurol. 1995;44:228-235.
4 Petito CK, Navia BA, Cho ES, et al. Vacuolar myelopathy pathologically resembling subacute combined degeneration in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1985;312:874.
5 Vernant JC, Maurs L, Gessain A, et al. Endemic tropical spastic paraparesis associated with human T-lymphotropic virus type I: A clinical and seroepidemiological study of 25 cases. Ann Neurol. 1987;21:123.
6 Weinshenker BG, Wingerchuk DM. Neuromyelitis optica: Clinical syndrome and the NMO-IgG autoantibody marker. Curr Top Microbiol Immunol. 2008;318:343-356.
1 DeJong RN. Neurological Examination. New York: Harper & Row; 1979.
2 Ropper AH, Poskanzer DC. The prognosis of acute and subacute transverse myelopathy based on early signs and symptoms. Ann Neurol. 1978;4:51-59.
3 Kalita J, Misra UK. Is methyl prednisolone useful in acute transverse myelitis? Spinal Cord. 2001;39:471-476.
4 Lahat E, Pillar G, Ravid S, et al. Rapid recovery from transverse myelopathy in children treated with methylprednisolone. Pediatr Neurol. 1998;19:279-282.
5 Lipton HL, Teasdall RD. Acute transverse myelopathy in adults: A follow-up study. Arch Neurol. 1973;28:252-257.
6 Altrocchi PH. Acute transverse myelopathy. Arch Neurol. 1963;9:111.
7 Berman M, Feldman S, Alter M, et al. Acute transverse myelitis: Incidence and etiologic considerations. Neurology. 1981;31:966.
8 Miller DH, Ormerod IE, Rudge P, et al. The early risk of multiple sclerosis following isolated acute syndromes of the brainstem and spinal cord. Ann Neurol. 1989;26:635-639.
9 Lee KH, Hashimoto SA, Hooge JP, et al. Magnetic resonance imaging of the head in the diagnosis of multiple sclerosis: A prospective 2-year follow-up with comparison of clinical evaluation, evoked potentials, oligoclonal banding, and CT. Neurology. 1991;41:657-660.
10 Adams R, Victor M. Diseases of the Spinal Cord. Principles of Neurology, 4th ed. New York: McGraw Hill; 1989.
11 Poser S, Wikstrom J, Bauer HJ. Clinical data and the identification of special forms of multiple sclerosis in 1271 cases studied with a standardized documentation system. J Neurol Sci. 1979;40:159-168.
12 Shepherd DI. Clinical features of multiple sclerosis in north-east Scotland. Acta Neurol Scand. 1979;60:218-230.
13 Kanchandani R, Howe JG. Lhermitte’s sign in multiple sclerosis: A clinical survey and review of the literature. J Neurol Neurosurg Psychiatry. 1982;45:308-312.
14 Weinshenker BG, Wingerchuk DM. Neuromyelitis optica: Clinical syndrome and the NMO-IgG autoantibody marker. Curr Top Microbiol Immunol. 2008;318:343-356.
15 Cree B. Neuromyelitis optica: Diagnosis, pathogenesis, and treatment. Curr Neurol Neurosci Rep. 2008;8:427-433.
16 Miller DH, McDonald WI, Blumhardt LD, et al. Magnetic resonance imaging in isolated noncompressive spinal cord syndromes. Ann Neurol. 1987;22:714-723.
17 Oppenheimer DR. The cervical cord in multiple sclerosis. Neuropathol Appl Neurobiol. 1978;4:151-162.
18 Thielen KR, Miller GM. Multiple sclerosis of the spinal cord: Magnetic resonance appearance. J Comput Assist Tomogr. 1996;20:434-438.
19 Thorpe JW, Kidd D, Moseley IF, et al. Serial gadolinium-enhanced MRI of the brain and spinal cord in early relapsing-remitting multiple sclerosis. Neurology. 1996;46:373-378.
20 Syme JAJr, Kelly JJJr. Absent F-waves early in a case of transverse myelitis. Muscle Nerve. 1994;17:462-465.
21 Moser HW, Moser AB. Kawamura N, t al: Adrenoleukodystrophy: Elevated C26 fatty acid in cultured skin fibroblasts. Ann Neurol. 1980;7:542-549.
22 Griffin JW, Goren E, Schaumburg H, et al. Adrenomyeloneuropathy: A probable variant of adrenoleukodystrophy. I. Clinical and endocrinologic aspects. Neurology. 1977;27:1107-1113.
23 Snider WD, Simpson DM, Nielsen S, et al. Neurological complications of acquired immune deficiency syndrome: Analysis of 50 patients. Ann Neurol. 1983;14:403-418.
24 Levy RM, Bredesen DE, Rosenblum ML. Neurological manifestations of the acquired immunodeficiency syndrome (AIDS): Experience at UCSF and review of the literature. J Neurosurg. 1985;62:475-495.
25 Berger JR, Moskowitz L, Fischl M, et al. Neurologic disease as the presenting manifestation of acquired-immunodeficiency-syndrome. South Med J. 1987;80:683-686.
26 Bredesen DE, Messing R. Neurological syndromes heralding the acquired immune-deficiency syndrome. Ann Neurol. 1983;14:141.
27 Helweg-Larsen S, Jakobsen J, Boesen F, et al. Neurological complications and concomitants of AIDS. Acta Neurol Scand. 1986;74:467.
28 Petito CK, Navia BA, Cho ES, et al. Vacuolar myelopathy pathologically resembling subacute combined degeneration in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1985;312:874-879.
29 de la Monte SM, Ho DD, Schooley RT, et al. Subacute encephalomyelitis of AIDS and its relation to HTLV-III infection. Neurology. 1987;37:562-569.
30 Goldstick L, Mandybur TI, Bode R. Spinal cord degeneration in AIDS. Neurology. 1985;35:103-106.
31 Honig LS, Horoupian, DS. Chronic myelopathy as a presenting symptom in HIV infection. Neurology. 1989;39(Suppl 1):419.
32 Kamin SS, Petito CK. Idiopathic myelopathies with white matter vacuolation in non-acquired immunodeficiency syndrome patients. Hum Pathol. 1991;22:816-824.
33 Singh BM, Levine S, Yarrish RL, et al. Spinal cord syndromes in the acquired immune deficiency syndrome. Acta Neurol Scand. 1986;73:590-598.
34 Dickson DW, Belman AL, Kim TS, et al. Spinal cord pathology in pediatric acquired immunodeficiency syndrome. Neurology. 1989;39(2 Pt 1):227-235.
35 Denning DW, Anderson J, Rudge P, et al. Acute myelopathy associated with primary infection with human immunodeficiency virus. BMJ (Clin Res Ed). 1987;294:143-144.
36 Williams AE, Fang CT, Slamon DJ, et al. Seroprevalence and epidemiological correlates of HTLV-I infection in U.S. blood donors. Science. 1988;240:643-646.
37 Robert-Guroff M, Weiss SH, Giron JA, et al. Prevalence of antibodies to HTLV-I, -II, and -III in intravenous drug abusers from an AIDS endemic region. JAMA. 1986;255:3133-3137.
38 Weiss SH, Ginzburg HM, Saxinger WC, et al: Emerging high rates of human T cell lymphotropic virus type 1 (HTLV-I) and HIV infections among US drug abusers. III International Conference on AIDS, Washington, DC, 1987.
39 Khabbaz RF, Darrow WW, Hartley TM, et al. Seroprevalence and risk factors for HTLV-I/II infection among female prostitutes in the United States. JAMA. 1990;263:60-64.
40 Kaplan JE, Litchfield B, Rouault C, et al. HTLV-I-associated myelopathy associated with blood transfusion in the United States: Epidemiologic and molecular evidence linking donor and recipient. Neurology. 1991;41:192-197.
41 Sheremata WA, Berger JR, Harrington WJJr, et al. Human T lymphotropic virus type I-associated myelopathy: A report of 10 patients born in the United States. Arch Neurol. 1992;49:1113-1118.
42 Gessain A, Barin F, Vernant JC, et al. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985;2:407-410.
43 Osame M, Usuku K, Izumo S, et al. HTLV-I associated myelopathy, a new clinical entity. Lancet. 1986;1:1031-1032.
44 Koprowski H, DeFreitas EC, Harper ME, et al. Multiple sclerosis and human T-cell lymphotropic retroviruses. Nature. 1985;318:154-160.
45 Vernant JC, Maurs L, Gessain A, et al. Endemic tropical spastic paraparesis associated with human T-lymphotropic virus type I: A clinical and seroepidemiological study of 25 cases. Ann Neurol. 1987;21:123-130.
46 Akizuki S, Nakazato O, Higuchi Y, et al. Necropsy findings in HTLV-I associated myelopathy. Lancet. 1987;1:156-157.
47 Berger JR, Svenningsson A, Raffanti S, et al. Tropical spastic paraparesis-like illness occurring in a patient dually infected with HIV-1 and HTLV-II. Neurology. 1991;41:85-87.
48 Harrington WJJr, Sheremata W, Hjelle B, et al. Spastic ataxia associated with human T-cell lymphotropic virus type II infection. Ann Neurol. 1993;33:411-414.
49 Jacobson S, Lehky T, Nishimura M, et al. Isolation of HTLV-II from a patient with chronic, progressive neurological disease clinically indistinguishable from HTLV-I-associated myelopathy/tropical spastic paraparesis. Ann Neurol. 1993;33:392-396.
50 Sheremata WA, Harrington WJJr, Bradshaw PA, et al. Association of ‘(tropical) ataxic neuropathy’ with HTLV-II. Virus Res. 1993;29:71-77.
51 Lehky TJ, Flerlage N, Katz D, Houff S, et al. Human T-cell lymphotropic virus type II-associated myelopathy: Clinical and immunologic profiles. Ann Neurol. 1996;40:714-723.
52 Peters AA, Oger JJ, Coulthart MB, et al. An apparent case of human T-cell lymphotropic virus type II (HTLV-II)-associated neurological disease: A clinical, molecular, and phylogenetic characterisation. J Clin Virol. 1999;14:37-50.
53 Silva EA, Otsuki K, Leite AC, et al. HTLV-II infection associated with a chronic neurodegenerative disease: Clinical and molecular analysis. J Med Virol. 2002;66:253-257.
54 Lowis GW, Sheremata WA, Minagar A. Epidemiologic features of HTLV-II: Serologic and molecular evidence. Ann Epidemiol. 2002;12:46-66.
55 Rose FC, Brett EM, Jl Burston. Zoster encephalomyelitis. Arch Neurol. 1964;11:155-172.
56 Caplan LR, Kleeman FJ, Berg S. Urinary retention probably secondary to herpes genitalis. N Engl J Med. 1977;297:920-921.
57 Klatersky J, Cappel R, Snoeck JM, et al. Ascending myelitis in association with herpes simplex virus. N Engl J Med. 1982;287:182-184.
58 Grose C, Feorino PM. Epstein-Barr virus and transverse myelitis. Lancet. 1973;1:892.
59 Silverstein A. Epstein Barr virus infections of the nervous system. In: Vinken P, Bruyn G, editors. Infections of the Nervous System. Amsterdam: North Holland Publishing, 1978.
60 Tyler KL, Gross RA, Cascino GDl. Unusual viral causes of transverse myelitis: Hepatitis A virus and cytomegalovirus. Neurology. 1986;36:855-858.
61 Paine RS, Byers RK. Transverse myelopathy in childhood. AMA Am J Dis Child. 1953;85:151-163.
62 Altrocchi P. Acute transverse myelopathy. Arch Neurol. 1963;9:111.
63 Adams R, Merritt HH. Meningeal and vascular diseases of the spinal cord. Medicine. 1944;23:181.
64 Berger J. Syphilis of the spinal cord. In: Davidoff RA, editor. Handbook of the Spinal Cord. New York: Marcel Dekker, 1987.
65 Fisher M, Poser CM. Syphilitic meningomyelitis: A case report. Arch Neurol. 1977;34:785.
66 Gribble LD. Syphilitic spinal pachymeningitis. S Afr Med J. 1972;46:1326-1328.
67 Syphilis: Recommended treatment schedules, 1976. Recommendations established by the Venereal Disease Control Advisory Committee. Ann Intern Med. 1976;85:94.
68 Berger J. Neurosyphilis. In: Johnson R, editor. Current Therapies in Neurology. Philadelphia: BC Decker, 1990.
69 Griffiths DL. Tuberculosis of the spine: A review. Adv Tuberc Res. 1980;20:92-110.
70 Nussbaum ES, Rockswold GL, Bergman TA, et al. Spinal tuberculosis: A diagnostic and management challenge. J Neurosurg. 1995;83:243-247.
71 Moon MS, Ha KY, Sun DH, et al. Pott’s paraplegia—67 cases. Clin Orthop Relat Res. 1996;323:122-128.
72 Vidyasagar C, Murthy HK. Spinal tuberculosis with neurological deficits. Natl Med J India. 1996;9:25-27.
73 Dutton JE, Alexander GL. Intramedullary spinal abscess. J Neurol Neurosurg Psychiatry. 1954;17:303-307.
74 Clarke CE, Falope ZF, Abdelhadi HA, et al. Cervical myelopathy caused by Whipple’s disease. Neurology. 1998;50:1505-1506.
75 Schroter A, Brinkhoff J, Günthner-Lengsfeld T, et al. Whipple’s disease presenting as an isolated lesion of the cervical spinal cord. Eur J Neurol. 2005;12:276-279.
76 Miller HG, Gibbons JL, Stanton JB. Para-infectious encephalomyelitis and related syndromes: A critical review of the neurological complications of certain specific fevers. QJM. 1956;25:427-505.
77 Kincaid JC. Myelitis and myelopathy. In: Joynt RJ, editor. Clinical Neurology. New York: Harper & Row, 1982.
78 Pickerill RG, Milder JE. Transverse myelitis associated with cat-scratch disease in an adult. JAMA. 1981;246:2840-2841.
79 Reik L, Steere AC, Bartenhagen NH, et al. Neurologic abnormalities of Lyme disease. Medicine (Baltimore). 1979;58:281-294.
80 Koh S, Ross LA, Gilles FH, et al. Myelopathy resulting from invasive aspergillosis. Pediatr Neurol. 1998;19:135-138.
81 Suchet I, Klein C, Horwitz T, et al. Spinal cord schistosomiasis: A case report and review of the literature. Paraplegia. 1987;25:491-496.
82 Ferrari TC. Spinal cord schistosomiasis: A report of 2 cases and review emphasizing clinical aspects. Medicine (Baltimore). 1999;78:176-190.
83 Petjom S, Chaiwun B, Settakorn J, et al. Angiostrongylus cantonensis infection mimicking a spinal cord tumor. Ann Neurol. 2002;52:99-101.
84 Kaufman DM, Kaplan JG, Litman N. Infectious agents in spinal epidural abscesses. Neurology. 1980;30:844-850.
85 Baker AS, Ojemann RG, Swartz MN, et al. Spinal epidural abscess. N Engl J Med. 1975;293:463-468.
86 Aldrete JA. Neurologic deficits and arachnoiditis following neuroaxial anesthesia. Acta Anaesthesiol Scand. 2003;47:3-12.
87 Herrick MK, Mills PEJr. Infarction of spinal cord: Two cases of selective gray matter involvement secondary to asymptomatic aortic disease. Arch Neurol. 1971;24:228-241.
88 Killen DA, Foster JH. Spinal cord injury as a complication of contrast angiography. Surgery. 1966;59:969-981.
89 Bockenek WL, Bach JR. Fibrocartilaginous emboli to the spinal cord: A review of the literature. J Am Paraplegia Soc. 1990;13:18-23.
90 Bots GT, Wattendorff AR, Buruma OJ, et al. Acute myelopathy caused by fibrocartilaginous emboli. Neurology. 1981;31:1250-1256.
91 Feigin I, Popoff N, Adachi M. Fibrocartilaginous venous emboli to the spinal cord with necrotic myelopathy. J Neuropathol Exp Neurol. 1965;24:63-74.
92 McLean JM, Palagallo GL, Henderson JP, et al. Myelopathy associated with fibrocartilaginous emboli (FE): Review and two suspected cases. Surg Neurol. 1995;44:228-234. discussion 234-235
93 Moorhouse DF, Burke M, Keohane C, et al. Spinal-cord infarction caused by cartilage embolus to the anterior spinal artery. Surg Neurol. 1992;37:448-452.
94 Foix C, Alajouanine T. La myelite necrotique subaigue. Rev Neurol. 1926;11:1-42.
95 Mirich DR, Kucharczyk W, Keller MA, et al. Subacute necrotizing myelopathy: MR imaging in four pathologically proved cases. AJNR Am J Neuroradiol. 1991;12:1077-1083.
96 Koeppen AH, Barron KD, Cox JF. Foix-Alajouanine syndrome. Acta Neuropathol (Berl). 1974;29:187-197.
97 Kim RC, Smith HR, Henbest ML, et al. Nonhemorrhagic venous infarction of the spinal cord. Ann Neurol. 1984;15:379-385.
98 Roa KR, Donnenfeld H, Chusid JG, et al. Acute myelopathy secondary to spinal venous thrombosis. J Neurol Sci. 1982;56:107-113.
99 Hughes JT. Venous infarction of the spinal cord. Neurology. 1971;21:794-800.
100 Lorber A, Pearson CM, Rene RMl. Osteolytic vertebral lesions as a manifestation of rheumatoid arthritis and related disorders. Arthritis Rheum. 1961;4:514.
101 Blass J. Rheumatoid arthritis of the cervical spine. Bull Rheum Dis. 1967;18:471.
102 Hopkins JS. Lower cervical rheumatoid subluxation with tetraplegia. J Bone Joint Surg Br. 1967;49:46-51.
103 Shannon KM. Connective tissue diseases and the nervous system. In: Aminoff MJ, editor. Neurology and General Medicine. New York: Churchill Livingstone, 1989.
104 Bundschuk C, Modic MT, Kearney F, et al. Rheumatoid arthritis of the cervical spine: Surface-coil MR imaging. AJR Am J Roentgenol. 1988;151:181.
105 Perling LH, Laurent JP, Cheek WR. Epidural hibernoma as a complication of corticosteroid treatment: Case report. J Neurosurg. 1988;69:613-616.
106 McGrath HJr, McCormick C, Carey ME. Pyogenic cervical osteomyelitis presenting as a massive prevertebral abscess in a patient with rheumatoid arthritis. Am J Med. 1988;84:363-365.
107 Halla JT, Hardin JGJr. The spectrum of atlantoaxial facet joint involvement in rheumatoid arthritis. Arthritis Rheum. 1990;33:325-329.
108 Semble EL, Elster AD, et al. Magnetic-resonance imaging of the craniovertebral junction in rheumatoid-arthritis. J Rheumatol. 1988;15:1367-1375.
109 Smith PH, Sharp J, et al. Natural history of rheumatoid cervical subluxations. Ann Rheum Dis. 1972;31:222-223.
110 Falope ZF, Griffiths ID, Platt PN, Todd NV. Cervical myelopathy and rheumatoid arthritis: a retrospective analysis of management. Clin Rehabil. 2002;16:625-629.
111 Casey AT, et al. Surgery on the rheumatoid cervical spine for the non-ambulant myelopathic patient—too much, too late? Lancet. 1996;347:1004-1007.
112 Fody EP, Netsky MG, et al. Subarachnoid spinal hemorrhage in a case of systemic lupus erythematosus. Arch Neurol. 1980;37:173-174.
113 Rutan G, Martinez AJ, et al. Primary biliary cirrhosis, Sjogren’s syndrome, and transverse myelitis. Gastroenterology. 1986;90:206-210.
114 Harada T, Ohashi T, et al. Optic neuropathy and acute transverse myelopathy in primary Sjogren’s syndrome. Jpn J Ophthalmol. 1995;39:162-165.
115 Lyu RK, Chen ST, et al. Acute transverse myelopathy and cutaneous vasculopathy in primary Sjogren’s syndrome. Eur Neurol. 1995;35:359-362.
116 Konttinen YT, Kinnunen E, et al. Acute transverse myelopathy successfully treated with plasmapheresis and prednisone in a patient with primary Sjogren’s syndrome. Arthritis Rheum. 1987;30:339-344.
117 Ellis SG, Verity MA. Central nervous system involvement in systemic lupus erythematosus: A review of neuropathologic findings in 57 cases, 1955-1977. Semin Arthritis Rheum. 1979;8:212-221.
118 Mok CC, Lau CS, et al. Acute transverse myelopathy in systemic lupus erythematosus: Clinical presentation, treatment, and outcome. J Rheumatol. 1998;25:467-473.
119 Inslicht DV, Stein AB, et al. Three women with lupus transverse myelitis: Case reports and differential diagnosis. Arch Phys Med Rehabil. 1998;79:456-459.
120 Kovacs B, Lafferty TL, et al. Transverse myelopathy in systemic lupus erythematosus: An analysis of 14 cases and review of the literature. Ann Rheum Dis. 2000;59:120-124.
121 Andrianakos AA, Duffy J, et al. Transverse myelopathy in systemic lupus erythematosus: Report of three cases and review of the literature. Ann Intern Med. 1975;83:616-624.
122 Chan KF, Boey ML. Transverse myelopathy in SLE: Clinical features and functional outcomes. Lupus. 1996;5:294-299.
123 Provenzale JM, Barboriak DP, et al. Lupus-related myelitis— serial MR findings. AJNR Am J Neuroradiol. 1994;15:1911-1917.
124 Tellez-Zenteno JF, Remes-Troche JM, Negrete-Pulido RO, et al. Longitudinal myelitis associated with systemic lupus erythematosus: Clinical features and magnetic resonance imaging of six cases. Lupus. 2001;10:851-856.
125 Harisdangkul V, Doorenbos D, et al. Lupus transverse myelopathy: Better outcome with early recognition and aggressive high-dose intravenous corticosteroid pulse treatment. J Neurol. 1995;242:326-331.
126 Cordeiro MF, Lloyd ME, et al. Ischaemic optic neuropathy, transverse myelitis, and epilepsy in an anti-phospholipid positive patient with systemic lupus erythematosus. J Neurol Neurosurg Psychiatry. 1994;57:1142-1143.
127 Hasegawa M, Yamashita J, et al. Spinal cord infarction associated with primary antiphospholipid syndrome in a young child: Case report. J Neurosurg. 1993;79:446-450.
128 Levine SR, Welch KM. The spectrum of neurologic disease associated with antiphospholipid antibodies: Lupus anticoagulants and anticardiolipin antibodies. Arch Neurol. 1987;44:876-883.
129 Quencer RM. Anticardiolipin antibodies and transverse myelopathy: Expanding our understanding of an elusive clinical problem. AJNR Am J Neuroradiol. 1998;19:798-799.
130 Cuadrado MJ, Khamashta MA, et al. Can neurologic manifestations of Hughes (antiphospholipid) syndrome be distinguished from multiple sclerosis? Analysis of 27 patients and review of the literature. Medicine (Baltimore). 2000;79:57-68.
131 Aziz A, Conway MD, et al. Acute optic neuropathy and transverse myelopathy in patients with antiphospholipid antibody syndrome: Favorable outcome after treatment with anticoagulants and glucocorticoids. Lupus. 2000;9:307-310.
132 Kim JH, Lee SI, Park SI, Yoo WH. Recurrent transverse myelitis in primary antiphospholipid antibody syndrome—case report and literature review. Rheumatol Int. 2004;24:244-246.
133 Shaharao V, Bartakke S, Muranjan MN, et al. Recurrent acute transverse myelopathy: association with antiphospholipid antibody syndrome. Indian J Pediatr. 2004;71:559-561.
134 Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346:752-763.
135 Brey RL, Escalante A. Neurological manifestations of antiphospholipid antibody syndrome. Lupus. 1998;7(Suppl 2):S67-S74.
136 Campi A, Filippi M, et al. Recurrent acute transverse myelopathy associated with anticardiolipin antibodies. AJNR Am J Neuroradiol. 1998;19:781-786.
137 Blau RH, Kaufman RL. Erosive and subluxing cervical spine disease in patients with psoriatic arthritis. J Rheumatol. 1987;14:111-117.
138 Mok CC, Lau CS. Transverse myelopathy complicating mixed connective tissue disease. Clin Neurol Neurosurg. 1995;97:259-260.
139 Ray DW, Bridger J, et al. Transverse myelitis as the presentation of Jo-1 antibody syndrome (myositis and fibrosing alveolitis) in long-standing ulcerative colitis. Br J Rheumatol. 1993;32:1105-1108.
140 Kelley PJ, Toker DE, Boyer P, et al. Granulomatous compressive thoracic myelopathy as the initial manifestation of Wegener’s granulomatosis. Neurology. 1998;51:1769-1770.
141 Junger SS, Stern BJ, et al. Intramedullary spinal sarcoidosis: Clinical and magnetic resonance imaging characteristics. Neurology. 1993;43:333-337.
142 Endo T, Koike J, et al. Spinal cord sarcoidosis. Neurology. 1993;43:1059-1060.
143 Lexa FJ, Grossman RI. MR of sarcoidosis in the head and spine: Spectrum of manifestations and radiographic response to steroid therapy. AJNR Am J Neuroradiol. 1994;15:973-982.
144 Agbobu B, et al. Therapeutic considerations in patients with refractory neurosarcoidosis. Arch Neurol. 1995;52:875.
145 Chapelon C, Ziza JM, et al. Neurosarcoidosis: Signs, course and treatment in 35 confirmed cases. Medicine (Baltimore). 1990;69:261-276.
146 Kunze K, Leitenmaier K. Vitamin B12 deficiency and subacute combined degeneration of the spinal cord. Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology, volume 28. 1976:141-198.
147 Smith W. Nutritional deficiencies and disorders. In: Blackwood W, Corsellis J, editors. Greenfield’s Neuropathy. London: Edward Arnold, 1977.
148 Kumar N, Gross JBJr, Ahlskog JE. Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology. 2004;63:33-39.
149 Goodman BP, Mistry DH, Pasha SF, Bosch PE. Copper deficiency myeloneuropathy due to occult celiac disease. Neurologist. 2009;15:355-356.
150 Kumar N. Copper deficiency myelopathy (human swayback). Mayo Clin Proc. 2006;81:1371-1384.
151 Berger JR. The neurological complications of bariatric surgery. Arch Neurol. 2004;61:1185-1189.
152 Juhasz-Pocsine K, Rudnicki SA, Archer RL, Harik SI. Neurologic complications of gastric bypass surgery for morbid obesity. Neurology. 2007;68:1843-1850.
153 Plum F, Hindfeldt B. The neurological complication of liver disease 1976. Vinken P, Bruyn G, editors. Handbook of Clinical Neurology. Amsterdam: North Holland Publishing, 1976.
154 Heyman SN, Michaeli J, et al. Primary hyperparathyroidism presenting as cervical myelopathy. Am J Med Sci. 1986;291:112-114.
155 Juchet H, Ollier S, et al. Neurologic and psychiatric manifestations of primary hyperparathyroidism: Study of 5 cases. Rev Med Int. 1993;14:123-125.
156 Melamed E, Berman M, et al. Posterolateral myelopathy associated with thyrotoxicosis [Letter]. N Engl J Med. 1975;293:778-779.
157 Swash M, Schwartz MS. Paraneoplastic syndromes. In: Johnson RT, editor. Current Therapies in Neurological Diseases—3. Philadelphia: BC Decker, 1990.
158 Norris F. Remote effects of cancer on the spinal cord. In: Vinken P, Bruyn G, editors. Handbook of Clinical Neurology. Amsterdam: North Holland, 1979.
159 Cohen ME, Duffner PK, et al. Myelopathy with severe structural derangement associated with combined modality therapy. Cancer. 1983;52:1590-1596.
160 Gagliano RG, Costanzi JJ. Paraplegia following intrathecal methotrexate: Report of a case and review of the literature. Cancer. 1976;37:1663-1668.
161 Muder RR, Lumish RM, et al. Myelopathy after herpes zoster. Arch Neurol. 1983;40:445-446.
162 Henson RA, Urich H. Cancer and the Nervous System. Oxford: Blackwell Scientific; 1982.
163 Handforth A, Nag S, et al. Paraneoplastic subacute necrotic myelopathy. Can J Neurol Sci. 1983;10:204-207.
164 Dansey RD, Hammond-Tooke GD, et al. Subacute myelopathy: An unusual paraneoplastic complication of Hodgkin’s disease. Med Pediatr Oncol. 1988;16:284-286.
165 Posner JB. Paraneoplastic syndromes involving the nervous system. In: Aminoff MJ, editor. Neurology and General Medicine. New York: Churchill Livingstone, 1989.
166 Iwamasa T, Utsumi Y, et al. Two cases of necrotizing myelopathy associated with malignancy caused by herpes simplex virus type 2. Acta Neuropathol. 1989;78:252-257.
167 Schold SC, Cho ES, et al. Subacute motor neuronopathy: A remote effect of lymphoma. Ann Neurol. 1979;5:271-287.
168 Dalmau J, Graus F, Rosenblum MK, et al. Anti-Hu-associated paraneoplastic encephalomyelitis/sensory neuronopathy: A clinical study of 71 patients. Medicine (Baltimore). 1992;71:59-72.
169 Schwarz S, Zoubaa S, et al. Intravascular lymphomatosis presenting with a conus medullaris syndrome mimicking disseminated encephalomyelitis. Neuro-oncol. 2002;4:187-191.
170 Nakahara T, Saito T, et al. Intravascular lymphomatosis presenting as an ascending cauda equina conus medullaris syndrome: Remission after biweekly CHOP therapy 1999. J Neurol Neurosurg Psychiatry, 67. 1999:403-406.
171 Baumann TP, Hurwitz N, et al. Diagnosis and treatment of intravascular lymphomatosis. Arch Neurol. 2000;57:374-377.
172 Delattre J-Y, Posner J. Paraneoplastic syndromes involving the nervous system. In: Aminoff M, editor. Neurology and General Medicine. New York: Churchill Livingstone, 1989.
173 Kagen AR, Wollin M, Gilbert HA, et al. Comparison of the tolerance of the brain and spinal cord to injury by radiation. In: Gilbert HA, Kagen AR, editors. Radiation Damage to the Nervous System. New York: Raven Press, 1980.
174 Palmer JJ. Radiation myelopathy. Brain. 1972;95:109-122.
175 Douglas MA, Parks LC, et al. Sudden myelopathy secondary to therapeutic total-body hyperthermia after spinal-cord irradiation. N Engl J Med. 1981;304:583-585.
176 Jones A. Transient radiation myelopathy (with reference to Lhermitte’s sign of electrical paraesthesia). Br J Radiol. 1964;37:727-744.
177 Laqueny A, et al. Syndrome de la corne anterieure postradiotherpique. Rev Neurol (Paris). 1985;141:222.
178 Sadowsky CH, Sachs EJr, et al. Postradiation motor neuron syndrome. Arch Neurol. 1976;33:786-787.
179 Steen PA, Michenfelder JD. Neurotoxicity of anesthetics. Anesthesiology. 1979;50:437-453.
180 Usubiaga JE. Neurological complications following epidural anesthesia. Int Anesthesiol Clin. 1975;13:1-153.
181 Dripps RD. Anesthesia. Annu Rev Med. 1954;5:305-322.
182 Phillips OC, Ebner H, et al. Neurologic complications following spinal anesthesia with lidocaine: A prospective review of 10,440 cases. Anesthesiology. 1969;30:284-289.
183 Pujol S, Torrielli R. Neurological accidents after epidural anesthesia in obstetrics. Cah Anesthesiol. 1996;44:341-345.
184 Dawkins CJ. An analysis of the complications of extradural and caudal block. Anaesthesia. 1969;24:554-563.
185 Kane RE. Neurologic deficits following epidural or spinal anesthesia. Anesth Analg. 1981;60:150-161.
186 Davies A, Solomon B, et al. Paraplegia following epidural anaesthesia. BMJ. 1958;2:654-657.
187 Ackerman WE, Juneja MM, et al. Maternal paraparesis after epidural anesthesia and cesarean section. South Med J. 1990;83:695-697.
188 Vandam LD, Dripps RD. Exacerbation of pre-existing neurologic disease after spinal anesthesia. N Engl J Med. 1956;255:843-849.
189 Munro D. Fluorescent screen amplification. Radiography. 1956;22:102-103.
190 Hessel SJ, et al. Complications of angiography. Radiology. 1981;138:273-281.
191 Feigelson HH, Ravin HA. Transverse myelitis following selective bronchial arteriography. Radiology. 1965;85:663-665.
192 Killen DA, Foster JH. Spinal cord injury as a complication of aortography. Ann Surg. 1960;152:211-230.
193 Layzer RB. Myeloneuropathy after prolonged exposure to nitrous oxide. Lancet. 1978;2:1227-1230.
194 Blanco G, Peters HA. Myeloneuropathy and macrocytosis associated with nitrous oxide abuse. Arch Neurol. 1983;40:416-418.
195 Shapiro WR, Young DF. Neurological complications of antineoplastic therapy. Acta Neurol Scand Suppl. 1984;100:125-132.
196 Clark AW, Cohen SR, et al. Paraplegia following intrathecal chemotherapy: Neuropathologic findings and elevation of myelin basic protein. Cancer. 1982;50:42-47.
197 Hahn AF, Feasby TE, et al. Paraparesis following intrathecal chemotherapy. Neurology. 1983;33:1032-1038.
198 Mastaglia FL. Neurology and General Medicine. New York: Churchill Livingstone; 1989.
199 Bernat JL. Intraspinal steroid therapy. Neurology. 1981;31:168-171.
200 Richter R. Drug abuse. In: Rowland L, editor. Merritt’s Textbook of Neurology. Philadelphia: Lea & Febiger, 1984.
201 Sobue I, Ando K, et al. Myeloneuropathy with abdominal disorders in Japan: A clinical study of 752 cases. Neurology. 1971;21:168-173.
202 Chaduri R. Paralytic disease caused by contamination with tricresyl phosphate. Trop Med Hyg. 1965;59:98.
203 Smith HV, Spalding JM. Outbreak of paralysis in Morocco due to ortho-cresyl phosphate poisoning. Lancet. 1959;2:1019-1021.
204 Sprofkin BE. Electrical injuries. In: Rowland L, editor. Merritt’s Textbook of Neurology. Philadelphia: Lea & Febiger, 1984.
205 Varghese G, Mani MM, et al. Spinal cord injuries following electrical accidents. Paraplegia. 1986;24:159-166.
206 Levine NS, Atkins A, et al. Spinal cord injury following electrical accidents: Case reports. J Trauma. 1975;15:459-463.
207 Winkleman M. Complications of thermal and electrical burns. In: Aminoff M, editor. Neurology and General Medicine. New York: Churchill Livingstone; 1995:915-930.
208 Davidson GS, Deck JH. Delayed myelopathy following lightning strike: A demyelinating process. Acta Neuropathol. 1988;77:104-108.
209 Holbrook LA, Beach FX, et al. Delayed myelopathy: A rare complication of severe electrical burns. BMJ. 1970;4:659-660.
210 Clouston PD, Sharpe D. Rapid recovery after delayed myelopathy from electrical burns. J Neurol Neurosurg Psychiatry. 1989;52:1308.
211 Jackson FE, Martin R, et al. Delayed quadriplegia following electrical burn. Milit Med. 1965;130:601-605.
212 Farrell DF, Starr A. Delayed neurological sequelae of electrical injuries. Neurology. 1968;18:601-606.
213 Critchley M. Industrial electrical accidents in their neurological aspect. J State Med. 1932;40:459.
214 Gwozdziewicz J. Changes in the spinal cord of divers connected with chronic forms of caisson disease. Biul Inst Med Morsk Gdansk. 1965;16:171-185.
215 Mastaglia FL, McCallum RI, et al. Myelopathy associated with decompression sickness: A report of six cases. Clin Exp Neurol. 1983;19:54-59.
216 Kim SW, Kim RC, et al. Non-traumatic ischaemic myelopathy: A review of 25 cases. Paraplegia. 1988;26:262-272.
217 Bokeriia LA, Kobaneva RA. Hyperbaric oxygenation in caisson disease. Kilin Med (Mosk). 1973;51:50.
218 Fink JK, Heiman-Patterson T, et al. Hereditary spastic paraplegia: Advances in genetic research. Hereditary Spastic Paraplegia Working group. Neurology. 1996;46:1507-1514.
219 Campuzano V, Montermini L, et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423-1427.
220 Aboulafia DM, Saxton EH, et al. A patient with progressive myelopathy and antibodies to human T-cell leukemia virus type I and human immunodeficiency virus type 1 in serum and cerebrospinal fluid. Arch Neurol. 1990;47:477-479.
221 McArthur JC, Griffin JW, et al. Steroid-responsive myeloneuropathy in a man dually infected with HIV-1 and HTLV-I. Neurology. 1990;40:938-944.
222 Tucker T, Dix RD, et al. Cytomegalovirus and herpes simplex virus ascending myelitis in a patient with acquired immune deficiency syndrome. Ann Neurol. 1985;18:74-79.
223 Britton CB, Mesa-Tejada R, et al. A new complication of AIDS: Thoracic myelitis caused by herpes simplex virus. Neurology. 1985;35:1071-1074.
224 McArthur JC. Neurologic manifestations of AIDS. Medicine (Baltimore). 1987;66:407-437.
225 Doll DC, Yarbro JW, et al. Mycobacterial spinal cord abscess with an ascending polyneuropathy. Ann Intern Med. 1987;106:333-334.
226 Woolsey RM, Chambers TJ, et al. Mycobacterial meningomyelitis associated with human immunodeficiency virus infection. Arch Neurol. 1988;45:691-693.
227 Berger JR. Spinal-cord syphilis associated with human-immunodeficiency-virus infection—a treatable myelopathy. Am J Med. 1992;92:101-103.
228 Herskovitz S, Siegel SE, et al. Spinal cord toxoplasmosis in AIDS. Neurology. 1989;39:1552-1553.
229 Berger JR, Sheremata WA, et al. Multiple sclerosis-like illness occurring with human immunodeficiency virus infection. Neurology. 1989;39:324-329.
230 Weill O, Finaud M, et al. Malignant spinal cord glioma: A new complication of HIV virus infection? Presse Med. 1987;16:1977.
[/level-membership-for-orthopaedics-category][not-level-membership-for-orthopaedics-category]
CHAPTER 37 Medical Myelopathies
Spinal cord disease may result from a variety of insults. A general classification of these various etiologies is provided in Table 37–1. This chapter addresses the “medical” causes of spinal cord dysfunction.
TABLE 37–1 Etiologies of Myelopathy
Generally, but not invariably, a thorough neurologic examination allows the physician to distinguish spinal cord from other causes of these neurologic complaints. The presence of concomitant neurologic illness attributable to disease of the cerebral hemispheres or brainstem, such as hemianopsia, ophthalmoplegia, and dysarthria, does not rule out the possibility of concomitant spinal cord disease, but should suggest that the findings on neurologic examination may be ascribed to a single or multiple lesions higher in the neuraxis than the spinal cord. The physical examination of patients with spinal cord lesions that have developed gradually or who have acute lesions that have been present for several weeks typically reveals a spastic weakness of the involved extremities with associated hyperreflexia and pathologic reflexes. The latter include Hoffman sign (palmar flexion of the thumb when the distal phalanx of the middle finger of the same hand is rapidly tapped) in the upper extremities and Babinski sign (plantar extension of the hallux when the sole of the foot is stroked) in the lower extremities. Several variants of these signs have been described.1 Superficial reflexes, such as abdominal and cremasteric reflexes, are absent.
Acute Idiopathic Transverse Myelitis and Multiple Sclerosis
Patients with acute idiopathic transverse myelitis most often present with paresthesias of the feet, toes, or fingertips.2 Gradually developing numbness and coincident weakness of the legs follows with subsequent paralysis of the legs. The features of this illness usually evolve over 1 to 3 weeks, but they may develop abruptly. The initial symptoms may be predominantly unilateral, and asymmetrical findings are common. As the illness progresses, upper extremity numbness and weakness and bowel and bladder incontinence may ensue. In some patients, posterior column involvement (decreased vibration and position senses) is spared early but affected later. Occasionally, back (interscapular) pain and, more rarely, calf, arm, or radicular pain may accompany the progressive myelopathy suggesting other pathologic processes, such as an intraspinal neoplasm or epidural abscess. To be considered idiopathic, acute transverse myelitis should be unassociated with a known preceding or concomitant viral infection. The dominant pathologic feature of the spinal cord is demyelination.
Treatment with adrenocorticotropic hormone (ACTH) or corticosteroids has been advocated; the response to therapy may be highly variable, and some studies indicate no benefit.3 A pilot study of high-dose methylprednisolone in children with acute transverse myelitis showed a significant shortening of motor recovery compared with historical controls.4 Approximately one third of patients have a return of a normal gait and bladder function.2 When recovery to a normal or near-normal level of function occurs, it does so within 1 year of the onset of the illness. Of affected patients, 25% become wheelchair-bound or bedridden, and the remainder have varying degrees of lesser disability.2
In some instances, myelopathy develops abruptly. Frequently, a flaccid, areflexic paralysis of the affected limbs accompanies spinal shock. Loss of vision resulting from optic nerve inflammation may accompany this myelopathy. Devic disease (neuromyelitis optica [NMO]) refers to the combination of acute optic neuritis and transverse myelitis. Similar to the more slowly evolving idiopathic transverse myelitis, demyelination is a characteristic neuropathologic hallmark. The pathology has been described as a necrotizing hemorrhagic leukomyelitis. The prognosis of patients with this rapidly evolving myelopathy, particularly when it is accompanied by spinal shock, is worse than the prognosis for the more gradually developing transverse myelitis.5
The differential diagnosis of idiopathic transverse myelitis includes multiple sclerosis; infectious myelitis; postinfectious or postvaccination myelitis; and myelopathy associated with vasculitis and connective tissue diseases, such as Behçet disease, systemic lupus erythematosus (SLE), and Sjögren syndrome. Multiple sclerosis may initially manifest as a transverse myelitis; the likelihood of transverse myelitis being the presenting manifestation of multiple sclerosis was previously reported to be 5% to 15%.2,5–7 Since the advent of more sensitive testing (magnetic resonance imaging [MRI]), this likelihood has been estimated at 42%.8 If cranial MRI is highly suggestive of multiple sclerosis at the time of onset of myelitis, the risk of progression to clinically definite multiple sclerosis is 50% at 2 years, whereas if cranial MRI is negative, the risk is 5%.9 A temporal association with a viral illness (Table 37–2), signs and symptoms that develop over a few days, and a monophasic course should suggest the possibility of a viral myelitis.10
| RNA Viruses | DNA Viruses |
|---|---|
| Nonenveloped | |
| Picornaviruses | Hepatitis B |
| Coxsackieviruses | |
| Echoviruses | |
| Polioviruses | |
| Other enteroviruses | |
| Hepatitis A | |
| Encephalomyocarditis virus | |
| Enveloped | |
| Togaviruses | Herpesviruses |
| Arbovirus | Herpes simplex |
| Rubella | Varicella-zoster |
| Tick-borne encephalitis virus | Epstein-Barr virus |
| Retroviruses | Cytomegalovirus |
| Human immunodeficiency virus type 1 | Herpes simiae |
| Human T-cell lymphotropic virus type I | Poxviruses |
| Orthomyxoviruses | Vaccinia |
| Influenza | Variola |
| Paramyxoviruses | |
| Measles | |
| Mumps | |
| Bunyaviruses | |
| California encephalitis virus | |
| Arenavirus | |
| Lymphocytic choriomeningitis | |
| Rhabdovirus | |
| Rabies | |
Adapted from Tyler KL, Gross RA, Cascino GD: Unusual viral causes of transverse myelitis: Hepatitis A virus and cytomegalovirus. Neurology 36:855-858, 1986.
Spinal cord disease is extremely common in the setting of multiple sclerosis and frequently is responsible for the associated extremity weakness, spasticity, gait abnormalities, and sphincter disturbances. Within 10 to 15 years of the onset of multiple sclerosis, more than 80% of patients exhibit extremity spasticity and weakness, and more than 50% have sphincter disturbances.11,12 Lhermitte sign has been reported in 38% of patients with clinically definite multiple sclerosis.13 Approximately 10% of patients with multiple sclerosis have a primary progressive form of the disease. This disorder often affects the spinal cord predominantly and most commonly develops in women older than 40 years.
NMO has been regarded as a variant of multiple sclerosis; however, the occurrence of a unique IgG antibody to the aquaporin-4 channel (NMO-IgG)14 suggests it is a distinct disorder. NMO may occur as an isolated condition or, more commonly, as recurrent spells of optic neuritis and myelitis. In contrast to the myelitis of multiple sclerosis, which seldom extends beyond two or three segments, the myelitis of NMO is often longitudinally extensive. Optic neuritis or myelitis in isolation may occur with NMO. Treatment recommendations15 include high-dose intravenous methylprednisolone; plasma exchange; intravenous immunoglobulin; and rituximab, an anti-CD20 antibody directed against B cells.
MRI of the involved area of the spinal cord is the best radiographic study. MRI may be normal or reveal cord swelling or hyperintense lesions or both on T2-weighted imaging intrinsic to the spinal cord. Miller and colleagues16 found MRI lesions at the clinically expected level in 64% of patients with a clinical syndrome suggestive of cervical involvement but in only 28% with a suspected thoracic or lumbar lesion. In patients with multiple sclerosis, most lesions are observed in the cervical spinal cord at autopsy and by MRI.17,18 Lesions on MRI are characteristically T2-weighted hyperintense signal abnormalities, generally less than 10 to 15 mm in length,18 and multiple, gapped lesions may be observed. Gadolinium enhancement with active disease is often observed.19 Generally, MRI, if available, negates the need of myelography. If MRI is unavailable, myelography should be performed with water-soluble contrast medium and computed tomography (CT) scan of the affected area. If pain is a significant component of the patient’s illness, a CT scan of the appropriate area and a bone scan may be desirable in eliminating the possibility of an epidural abscess or neoplasm.
Somatosensory evoked potentials can be very helpful in ruling in suspected cord lesions. The presence of abnormalities on brainstem auditory evoked potential and visual evoked potential testing would raise the suspicion of multifocal disease (i.e. multiple sclerosis). Absent F waves on nerve conduction testing can also support evidence of a cord lesion.20
Adrenomyeloneuropathy
Adrenomyeloneuropathy is a phenotypic variant of the genetic disorder adrenoleukodystrophy, in which myelin sheath abnormalities of the white matter are associated with adrenal insufficiency. Although adrenoleukodystrophy is a sex-linked disorder, it has variable expressions, including mild disease in heterozygous women. Pathologically, the white matter abnormality appears to be a diffuse myelinoclastic sclerosis, but it may occasionally appear as a dysmyelinating condition. The neurologic variants are explained by the degree of involvement of brain, spinal cord, and peripheral nerve. Biorefringent material in the adrenal glands and brain observed histopathologically have been shown by sequential extraction methods to be cholesterol esters with high quantities of very long chain fatty acids. Measuring fatty acids in cultured skin fibroblasts from affected individuals, Moser and colleagues21 showed abnormally large amounts of very long chain fatty acids (C24-C30) and a high ratio of C26 to C22 fatty acids. The latter has become the preferred method of diagnosing the disorder.
In patients with the spinal-neuropathic form of this disorder, adrenal insufficiency is usually present since early childhood, and a progressive spastic paraparesis and relatively mild peripheral neuropathy develop in the 3rd decade.22 Other variants affecting the spinal cord have been observed, including a progressive myelopathy in men, a mild spastic paraparesis in women, and combined cerebral and spinal involvement in children and young men. A positive family history is present in approximately 50% of affected individuals. The presence of Addison disease also provides a strong clue to the diagnosis. Addison disease, which is caused by primary adrenal failure, is often accompanied by bronzing of the skin because of excessive secretion of melanocyte-stimulating hormone in association with ACTH.
Infectious Myelopathies
Viral Myelitis
Human Immunodeficiency Virus Infection
Neurologic complications develop in 40% to 60%23–25 of all patients with acquired immunodeficiency syndrome (AIDS), and neurologic disease heralds human immunodeficiency virus (HIV) infection in 10%26 to 20%25 of HIV-infected patients. In retrospective clinical series, spinal cord disease occurring in association with AIDS has been infrequently observed. Levy and colleagues24 found viral myelitis in 3 of 128 AIDS patients with neurologic symptoms, and the collective incidence of viral myelitis in AIDS was 1% when data were pooled from three different hospital series of neurologic complications of AIDS.27 The most common form of myelopathy observed with HIV infection has been a unique degeneration of the spinal cord first described by Petito and colleagues.28 In pathologic series, spinal cord degeneration has been observed in 11%29 to 22%28 of unselected cases. These pathologic series suggest that spinal cord disease occurring in association with HIV infection is common but clinically underrecognized.
The prototypic myelopathy observed with HIV infection is a unique degeneration of the spinal cord.28,30 Petito and colleagues28 observed this myelopathy in 20 of 89 consecutive autopsies of AIDS patients. Although the clinical presentation of this myelopathy may overlap with the presentation of other myelopathies associated with HIV-1 infection, the pathologic appearance is quite distinct. Clinically, patients complain of leg weakness, unsteadiness, and gait impairment. Incontinence of bladder and bowel often supervenes. In one study, incontinence was observed in 60% of patients.28 Patients with this disorder often complain of paresthesias and vague discomfort in their legs. Frequently, these complaints are attributed to the general debilitation of the patient, and the true nature of the illness remains undiagnosed until pathologic examination of the spinal cord at the time of autopsy.
On physical examination, a spastic paraparesis is detected with the degree of weakness exceeding the degree of spasticity. Rarely, marked asymmetry of the leg weakness, a monoparesis, or quadriparesis may be found. Gait ataxia is seen, and the heel-to-knee-to-toe test may reveal dysmetria and dyssynergy. Occasionally, weakness is slight or absent on confrontation testing, but hyperreflexia of the lower extremities and extensor plantar responses are noted. Muscle stretch reflexes may also be diminished or absent in this disorder, however, as a result of concomitant peripheral neuropathy. Sensory examination reveals that vibratory and position sense are disproportionately affected compared with pinprick, temperature, or light touch. A significant impairment of the latter modalities suggests the presence of a concomitant peripheral neuropathy. Electrophysiologic studies may reveal a prolonged latency of cortical evoked responses after tibial nerve stimulation. Typically, this myelopathy is seen late in the course of HIV-1 infection; however, it has been described as the presenting manifestation of infection.31
Gross examination of the spinal cord and dura is generally normal in HIV-associated myelopathy except when the myelopathy is particularly severe.28 The striking finding on histologic examination is the loss of myelin and spongy degeneration. The lateral and posterior columns are more severely affected than the anterior columns. Microvacuolization of the white matter of the spinal cord (Fig. 37–1) associated with lipid-laden macrophages bears an uncanny resemblance to the pathology of subacute combined degeneration of the spinal cord. The vacuolization seems to result from intramyelin swelling. Axons are preserved except in areas of marked vacuolization. Microglial nodules may be detected in the spinal cord gray matter, and 20% of the patients in one series also exhibited central chromatolysis of the anterior horn motor cells.28 Inflammation and intranuclear viral inclusions are generally not seen.
FIGURE 37–1 Microvacuolization of posterior columns of thoracic spinal cord in a patient with HIV-1–related myelopathy.
Although HIV has been cultured from the spinal cord, the specific role of HIV in the causation of this illness is uncertain. A similar clinicopathologic condition has been observed in patients with cancer or other immunosuppressive conditions in the absence of HIV infection.32 The spinal cord pathology is most prominent in the middle and lower thoracic regions. The cord involvement may be asymmetrical and does not seem to be confined to particular tracts.28 Goldstick and colleagues30 described involvement of the posterior columns increasing in intensity with rostral progression, whereas pyramidal tract involvement increased caudally. Petito and colleagues28 were able to correlate the frequency and severity of symptoms to the degree of spinal cord pathology in their series. A potential role of nutritional deficiency as the cause of spinal cord pathology has been suggested.33 Vacuolar myelopathy is not observed in young children with AIDS; however, pathologic abnormalities of the spinal cord are frequently seen at autopsy.34 A loss of myelin and axons in the corticospinal tracts seems to be the most common abnormality.
The diagnosis of this illness is one of exclusion. Numerous other myelopathies have been observed in association with HIV infection. An acute myelopathy of uncertain pathogenesis has been noted at the time of seroconversion.35 Table 37–3 lists other etiologies of spinal cord disease occurring in association with HIV.
TABLE 37–3 Potential Etiologies of the Myelopathies Associated with Human Immunodeficiency Virus (HIV) Infection
| Infectious |
| Viral |
| Bacterial |
| Fungal |
Human T-Cell Lymphotropic Virus Type I
Although thought to be rare in the United States, human T-cell lymphotropic virus type I (HTLV-I) has been observed with increasing frequency in certain subpopulations. A study of volunteer blood donors by the American Red Cross revealed a seropositivity rate of 0.025% among volunteer blood donors.36 Intravenous drug abusers seem to be at particularly high risk for infection with HTLV-I. Seroprevalence rates in this population range from 7% to 49%.37,38 In a study of the seroprevalence among female prostitutes in eight areas of the United States, 6.7% were seropositive for HTLV-I and HTLV-II with prevalence rates ranging from 0% in southern Nevada to 25.4% in Newark, New Jersey.39 A case of transmission of HTLV-I by blood transfusion associated with myelopathy has been confirmed.40 In 10 native-born cases in the United States, 5 had received blood transfusions, and 6 had multiple sex partners (including one who frequented prostitution and had a history of drug abuse).41
In addition to being associated with adult T-cell leukemia/lymphoma, HTLV-I is associated with a chronic progressive myelopathy.42,43 An association between the presence of seropositivity for HTLV-I and multiple sclerosis has also been suggested,44 but this remains controversial.
The myelopathy that occurs with HTLV-I has been referred to as tropical spastic paraparesis (TSP) or HTLV-I associated myelopathy (HAM). This myelopathy is characterized on neuropathologic studies by chronic involvement of the pyramidal tracts chiefly at the thoracic level resulting in spastic lower extremity weakness and a spastic bladder. Paresthesias, pain, and sensory disturbances may also be observed. It is estimated that 1 in 250 individuals infected with HTLV-I develop this progressive myelopathy.45 The major pathologic features of HTLV-I myelopathy are long tract degeneration and demyelination affecting the pyramidal, spinocerebellar, and spinothalamic tracts associated with hyalinoid thickening of the media and adventitia of blood vessels in the brain, spinal cord, and subarachnoid space with perivascular cuffing with leukocytes, astrocytic gliosis, and foamy macrophages.46 These lesions may extend from the upper cervical cord to the lumbar regions. Vacuolization may be observed at the periphery of the lesions.
HTLV-II, a related type C retrovirus in the Oncoviridae subfamily also rarely may result in a myelopathy similar to HAM or TSP.47–53 The epidemiology of this virus is different from HTLV-I because the populations principally affected are American Indians and intravenous drug abusers.54 The mode of transmission parallels that of HTLV-I and HIV. The rarity of these neurologic disorders accompanying HTLV-II has precluded meaningful analysis of treatment options. The same therapies employed in HAM or TSP have been generally recommended, however.
Herpesviruses
Varicella-zoster virus (VZV) is responsible for varicella (chickenpox) and causes shingles in adults. The virus remains latent within the dorsal root ganglia and spreads centrifugally along the corresponding nerves after reactivation resulting in a severely painful, blistering dermatomal eruption. Rarely, when the thoracic dermatomes are involved, the virus may spread centripetally and result in a necrotizing myelopathy.55 The myelitis complicating VZV infection in immunocompetent individuals typically occurs 1 to 2 weeks after the appearance of the dermatomal rash, although VZV myelitis may develop in the absence of a rash. The myelitis occurs at the level of the affected dermatome and results in paraparesis. In immunosuppressed hosts, such as AIDS patients, VZV myelopathy occurs insidiously and progresses. The disease is suspected by the close temporal relationship to the rash and is confirmed by showing the presence of VZV DNA by polymerase chain reaction in the CSF or VZV antibody in the CSF. Treatment with intravenous acyclovir (10 mg/kg every 8 hours) should be initiated.
Rarely, and usually with primary infection, HSV-2, the etiology of genital herpes, may cause a sacral radiculitis56 or an ascending myelitis.57 These neurologic complications are rarely observed with recurrent HSV-2. Epstein-Barr virus,58,59 the etiologic agent of infectious mononucleosis, and cytomegalovirus60 may also result in a transverse myelitis at the time of primary infection. In an immunocompromised host, HSV-1, HSV-2, and cytomegalovirus may result in myelitis.
Other Viruses
Many other viruses have been associated with transverse myelitis (see Table 37–2). Of patients with transverse myelitis, 20% to 40% have evidence of preceding or concurrent viral infection.2,5,7,61,62 Prompted by the increasing availability and efficacy of antiviral therapy and improved diagnostic techniques, in particular, polymerase chain reaction, these percentages are likely to increase.
Myelopathies Resulting from Bacterial Disease
Syphilis
Syphilis may affect the spinal cord in various ways.25 The pathology may be predominantly meningovascular or parenchymatous in nature. Gummas may grow within the substance of the cord or compress the cord by growth from the surrounding meninges. The clinical picture of spinal cord compression in syphilis may also arise as a result of hypertrophic pachymeningitis or vertebral lesions secondary to syphilitic osteitis. Table 37–4 presents a classification based on pathology and modified after the one proposed by Adams and Merritt.63
Adapted from Adams R, Merritt H: Meningeal and vascular diseases of the spinal cord. Medicine 23:181, 1944.
Tabes Dorsalis
Tabes dorsalis is the prototypic spinal cord disorder associated with syphilis. It is characterized by incoordination, pains, anesthesia, and various visceral trophic abnormalities.64 The earliest recognized descriptions of this disorder date to the mid-18th century. By the turn of the 20th century, tabes dorsalis was recognized with increased frequency and, according to Erb, was unequaled in frequency or importance by any other chronic disease of the spinal cord.
The pathology of tabes dorsalis is characterized by changes in the posterior spinal roots and posterior spinal columns (Fig. 37–2). Shrinkage of the spinal cord may be apparent by gross inspection. Leptomeningitis is evidenced by round cell infiltration. The dorsal columns are demyelinated, particularly in the regions of the fasciculus gracilis, root entry zone, and Lissauer tract. Astrocytic proliferation in the posterior columns is accompanied by an increase in connective tissue and thickening of the blood vessel walls. The lower spinal cord bears the brunt of the damage. Nerve fibers in the posterior root are destroyed and replaced by fibrosis. Frequently, lesions are observed in the anterior horns, cranial nerves, and brainstem.
Syphilitic Meningomyelitis
Syphilitic meningomyelitis occurs most commonly in individuals 25 to 40 years old, although it may arise in younger and older age groups. In most series, men predominate. The latency from the onset of the infection to the onset of symptoms with syphilitic meningomyelitis varies from 1 to 30 years with most arising within 6 years of infection.65
Other Forms of Spinal Syphilis
Hypertrophic pachymeningitis66 is an insidious, slowly progressive syphilitic process that results in spinal root and spinal cord dysfunction. A rare but well-recognized complication of tertiary syphilis is a gumma of the spinal cord. The clinical features of a spinal gumma are indistinguishable from an intramedullary glioma if it arises within the cord or may simulate the appearance of an extramedullary tumor if it arises from the meninges and compresses the spinal cord. Spinal cord infarction is a well-recognized complication of syphilis. Another vascular complication of syphilis is aortitis, which may eventuate in an aortic aneurysm resulting rarely in myelopathy by erosion of the vertebrae and ultimately compression of the spinal cord. Progressive spinal muscular atrophy has been observed in association with neurosyphilis, although the relationship may be coincidental. Occasionally, syphilitic caries may affect the vertebrae, particularly vertebrae of the cervical spine, resulting in pain, tenderness to palpation, and loss of mobility, and an abnormal spinal curvature in the involved area is noted. Radicular pains are observed in one third of patients. Compression of the spinal cord, although rare, has been reported.
As with other forms of neurosyphilis, the recommended treatment is 12 to 24 million U of aqueous penicillin daily in divided doses administered every 4 hours for 10 to 14 days.67 Other treatment regimens68 using doxycycline, ceftriaxone, or erythromycin may be considered if the patient cannot tolerate penicillin. These treatment regimens are not well established in treating symptomatic neurosyphilis, however.
Tuberculosis
Neurologic complications of Mycobacterium tuberculosis remain common in some parts of the world but are rare in the developed countries of the Western world. Myelopathy occurring in association with tuberculosis is usually the consequence of tuberculous spondylitis (Pott disease), which accounts for half of all skeletal tuberculosis. Typically, the anterior portion of the vertebral body is affected, with the mycobacterial spread to the vertebrae being hematogenous, lymphatic, or by direct contiguity from the lung.69 The characteristic radiographic defect is anterior wedging of two adjacent vertebrae with loss of the intervening disk space. The spine is enveloped by pus extruding anteriorly from the affected vertebrae. Myelopathy typically results from pressure on the anterior spinal cord by caseous or granulating tissue, inflammatory thrombosis of the anterior spinal artery, or injury to the cord from spinal instability. The last-mentioned may lead to complete spinal cord transection.
Myelopathy occurring in association with tuberculous infection may also occur as a consequence of intraspinal granulomatous tissue unassociated with a bony lesion and intramedullary tuberculomas. In one study of spinal tuberculosis,70 54% of patients had neurologic deficit with bony tuberculous lesions, 39% had neurologic deficit with intraspinal granulomatous tissue occurring in the absence of bony lesions, and 7% had neurologic deficit from intraspinal tuberculomas.
Therapy of patients with spinal tuberculosis requires at least 12 months of antibiotic treatment and surgical decompression in the presence of neurologic abnormalities.70 In the setting of intraspinal granulomatous disease without significant bony destruction, laminectomy and débridement is adequate70; however, more aggressive therapy is warranted when vertebral bodies are involved. A two-stage procedure comprising posterior instrumental stabilization followed by anterior radical decompression permitted earlier mobilization after neurologic recovery.71 Reports of the frequency of neurologic recovery with spinal tuberculosis vary, but functional recovery rates of 90% have been reported.71 Thoracic lesions with severe neurologic deficit show the least improvement, whereas lumbar disease has the best outcome.72
Other Forms of Bacterial Myelopathy
Numerous other bacterial infections have been associated with myelitis. Rarely, the spinal cord may be seeded by bacteria leading to a suppurative myelitis with abscess formation. In a review by Dutton and Alexander,73
[/not-level-membership-for-orthopaedics-category]
