5 |
Principles of Clinical Pharmacology |
Drugs are the cornerstone of modern therapeutics. Nevertheless, it is well recognized among physicians and in the lay community that the outcome of drug therapy varies widely among individuals. While this variability has been perceived as an unpredictable, and therefore inevitable, accompaniment of drug therapy, this is not the case. The goal of this chapter is to describe the principles of clinical pharmacology that can be used for the safe and optimal use of available and new drugs.
Drugs interact with specific target molecules to produce their beneficial and adverse effects. The chain of events between administration of a drug and production of these effects in the body can be divided into two components, both of which contribute to variability in drug actions. The first component comprises the processes that determine drug delivery to, and removal from, molecular targets. The resulting description of the relationship between drug concentration and time is termed pharmacokinetics. The second component of variability in drug action comprises the processes that determine variability in drug actions despite equivalent drug delivery to effector drug sites. This description of the relationship between drug concentration and effect is termed pharmacodynamics. As discussed further below, pharmacodynamic variability can arise as a result of variability in function of the target molecule itself or of variability in the broad biologic context in which the drug-target interaction occurs to achieve drug effects.
Two important goals of the discipline of clinical pharmacology are (1) to provide a description of conditions under which drug actions vary among human subjects; and (2) to determine mechanisms underlying this variability, with the goal of improving therapy with available drugs as well as pointing to new drug mechanisms that may be effective in the treatment of human disease. The first steps in the discipline were empirical descriptions of the influence of disease on drug actions and of individuals or families with unusual sensitivities to adverse drug effects. These important descriptive findings are now being replaced by an understanding of the molecular mechanisms underlying variability in drug actions. Thus, the effects of disease, drug coadministration, or familial factors in modulating drug action can now be reinterpreted as variability in expression or function of specific genes whose products determine pharmacokinetics and pharmacodynamics. Nevertheless, it is often the personal interaction of the patient with the physician or other health care provider that first identifies unusual variability in drug actions; maintained alertness to unusual drug responses continues to be a key component of improving drug safety.
Unusual drug responses, segregating in families, have been recognized for decades and initially defined the field of pharmacogenetics. Now, with an increasing appreciation of common and rare polymorphisms across the human genome, comes the opportunity to reinterpret descriptive mechanisms of variability in drug action as a consequence of specific DNA variants, or sets of variants, among individuals. This approach defines the field of pharmacogenomics, which may hold the opportunity of allowing practitioners to integrate a molecular understanding of the basis of disease with an individual’s genomic makeup to prescribe personalized, highly effective, and safe therapies.
IDENTIFYING DRUG TARGETS
Drug therapy is an ancient feature of human culture. The first treatments were plant extracts discovered empirically to be effective for indications like fever, pain, or breathlessness. This symptom-based empiric approach to drug development was supplanted in the twentieth century by identification of compounds targeting more fundamental biologic processes such as bacterial growth or elevated blood pressure; the term “magic bullet,” coined by Paul Ehrlich to describe the search for effective compounds for syphilis, captures the essence of the hope that understanding basic biologic processes will lead to highly effective new therapies. An integral step in modern drug development follows identification of a chemical lead with biologic activity with increasingly sophisticated medicinal chemistry-based structural modifications to develop compounds with specificity for the chosen target, lack of “off-target” effects, and pharmacokinetic properties suitable for human use (e.g., consistent bioavailability, long elimination half-life, no high-risk pharmacokinetic features described further below).
A common starting point for contemporary drug development is basic biologic discovery that implicates potential target molecules: examples of such target molecules include HMG-CoA reductase or the BRAF V600E mutation in many malignant melanomas. The development of compounds targeting these molecules has not only revolutionized treatment for diseases such as hypercholesterolemia or malignant melanoma, but has also revealed new biologic features of disease. Thus, for example, initial spectacular successes with vemurafenib (which targets BRAF V600E) were followed by near-universal tumor relapse, strongly suggesting that inhibition of this pathway alone would be insufficient for tumor control. This reasoning, in turn, supports a view that many complex diseases will not lend themselves to cure by targeting a single magic bullet, but rather single drugs or combinations will need to attack multiple pathways whose perturbation results in disease. The use of combination therapy in settings such as hypertension, tuberculosis, HIV infection, and many cancers highlights potential for such a “systems biology” view of drug therapy.
GLOBAL CONSIDERATIONS
![]() It is true across all cultures and diseases that factors such as compliance, genetic variants affecting pharmacokinetics or pharmacodynamics, and drug interactions contribute to drug responses. In addition, culture- or ancestry-specific factors play a role. For example, the frequency of specific genetic variants modulating drug responses often varies by ancestry, as discussed later. Cost issues or cultural factors may determine the likelihood that specific drugs, drug combinations, or over-the-counter (OTC) remedies are prescribed. The broad principles of clinical pharmacology enunciated here can be used to analyze the mechanisms underlying successful or unsuccessful therapy with any drug.
It is true across all cultures and diseases that factors such as compliance, genetic variants affecting pharmacokinetics or pharmacodynamics, and drug interactions contribute to drug responses. In addition, culture- or ancestry-specific factors play a role. For example, the frequency of specific genetic variants modulating drug responses often varies by ancestry, as discussed later. Cost issues or cultural factors may determine the likelihood that specific drugs, drug combinations, or over-the-counter (OTC) remedies are prescribed. The broad principles of clinical pharmacology enunciated here can be used to analyze the mechanisms underlying successful or unsuccessful therapy with any drug.
INDICATIONS FOR DRUG THERAPY: RISK VERSUS BENEFIT
It is self-evident that the benefits of drug therapy should outweigh the risks. Benefits fall into two broad categories: those designed to alleviate a symptom and those designed to prolong useful life. An increasing emphasis on the principles of evidence-based medicine and techniques such as large clinical trials and meta-analyses have defined benefits of drug therapy in broad patient populations. Establishing the balance between risk and benefit is not always simple. An increasing body of evidence supports the idea, with which practitioners are very familiar, that individual patients may display responses that are not expected from large population studies and often have comorbidities that typically exclude them from large clinical trials. In addition, therapies that provide symptomatic benefits but shorten life may be entertained in patients with serious and highly symptomatic diseases such as heart failure or cancer. These considerations illustrate the continuing, highly personal nature of the relationship between the prescriber and the patient.
Adverse Effects Some adverse effects are so common and so readily associated with drug therapy that they are identified very early during clinical use of a drug. By contrast, serious adverse effects may be sufficiently uncommon that they escape detection for many years after a drug begins to be widely used. The issue of how to identify rare but serious adverse effects (that can profoundly affect the benefit-risk perception in an individual patient) has not been satisfactorily resolved. Potential approaches range from an increased understanding of the molecular and genetic basis of variability in drug actions to expanded postmarketing surveillance mechanisms. None of these have been completely effective, so practitioners must be continuously vigilant to the possibility that unusual symptoms may be related to specific drugs, or combinations of drugs, that their patients receive.
Therapeutic Index Beneficial and adverse reactions to drug therapy can be described by a series of dose-response relations (Fig. 5-1). Well-tolerated drugs demonstrate a wide margin, termed the therapeutic ratio, therapeutic index, or therapeutic window, between the doses required to produce a therapeutic effect and those producing toxicity. In cases where there is a similar relationship between plasma drug concentration and effects, monitoring plasma concentrations can be a highly effective aid in managing drug therapy by enabling concentrations to be maintained above the minimum required to produce an effect and below the concentration range likely to produce toxicity. Such monitoring has been widely used to guide therapy with specific agents, such as certain antiarrhythmics, anticonvulsants, and antibiotics. Many of the principles in clinical pharmacology and examples outlined below, which can be applied broadly to therapeutics, have been developed in these arenas.
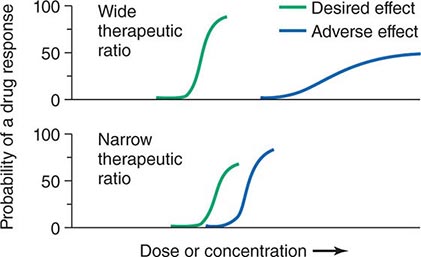
FIGURE 5-1 The concept of a therapeutic ratio. Each panel illustrates the relationship between increasing dose and cumulative probability of a desired or adverse drug effect. Top. A drug with a wide therapeutic ratio, i.e., a wide separation of the two curves. Bottom. A drug with a narrow therapeutic ratio; here, the likelihood of adverse effects at therapeutic doses is increased because the curves are not well separated. Further, a steep dose-response curve for adverse effects is especially undesirable, as it implies that even small dosage increments may sharply increase the likelihood of toxicity. When there is a definable relationship between drug concentration (usually measured in plasma) and desirable and adverse effect curves, concentration may be substituted on the abscissa. Note that not all patients necessarily demonstrate a therapeutic response (or adverse effect) at any dose, and that some effects (notably some adverse effects) may occur in a dose-independent fashion.
PRINCIPLES OF PHARMACOKINETICS
The processes of absorption, distribution, metabolism, and excretion—collectively termed drug disposition—determine the concentration of drug delivered to target effector molecules.
ABSORPTION AND BIOAVAILABILITY
When a drug is administered orally, subcutaneously, intramuscularly, rectally, sublingually, or directly into desired sites of action, the amount of drug actually entering the systemic circulation may be less than with the intravenous route (Fig. 5-2A). The fraction of drug available to the systemic circulation by other routes is termed bioavailability. Bioavailability may be <100% for two main reasons: (1) absorption is reduced, or (2) the drug undergoes metabolism or elimination prior to entering the systemic circulation. Occasionally, the administered drug formulation is inconsistent or has degraded with time; for example, the anticoagulant dabigatran degrades rapidly (over weeks) once exposed to air, so the amount administered may be less than prescribed.
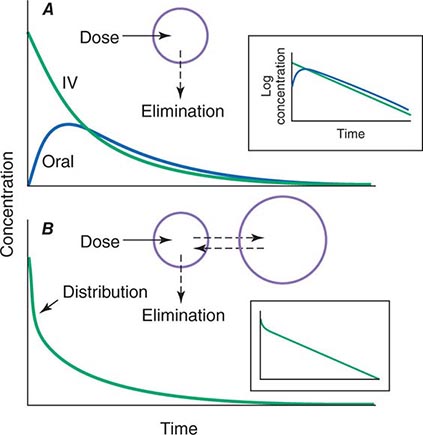
FIGURE 5-2 Idealized time-plasma concentration curves after a single dose of drug. A. The time course of drug concentration after an instantaneous IV bolus or an oral dose in the one-compartment model shown. The area under the time-concentration curve is clearly less with the oral drug than the IV, indicating incomplete bioavailability. Note that despite this incomplete bioavailability, concentration after the oral dose can be higher than after the IV dose at some time points. The inset shows that the decline of concentrations over time is linear on a log-linear plot, characteristic of first-order elimination, and that oral and IV drugs have the same elimination (parallel) time course. B. The decline of central compartment concentration when drug is distributed both to and from a peripheral compartment and eliminated from the central compartment. The rapid initial decline of concentration reflects not drug elimination but distribution.
When a drug is administered by a nonintravenous route, the peak concentration occurs later and is lower than after the same dose given by rapid intravenous injection, reflecting absorption from the site of administration (Fig. 5-2). The extent of absorption may be reduced because a drug is incompletely released from its dosage form, undergoes destruction at its site of administration, or has physicochemical properties such as insolubility that prevent complete absorption from its site of administration. Slow absorption rates are deliberately designed into “slow-release” or “sustained-release” drug formulations in order to minimize variation in plasma concentrations during the interval between doses.
“First-Pass” Effect When a drug is administered orally, it must traverse the intestinal epithelium, the portal venous system, and the liver prior to entering the systemic circulation (Fig. 5-3). Once a drug enters the enterocyte, it may undergo metabolism, be transported into the portal vein, or be excreted back into the intestinal lumen. Both excretion into the intestinal lumen and metabolism decrease systemic bioavailability. Once a drug passes this enterocyte barrier, it may also be taken up into the hepatocyte, where bioavailability can be further limited by metabolism or excretion into the bile. This elimination in intestine and liver, which reduces the amount of drug delivered to the systemic circulation, is termed presystemic elimination, presystemic extraction, or first-pass elimination.
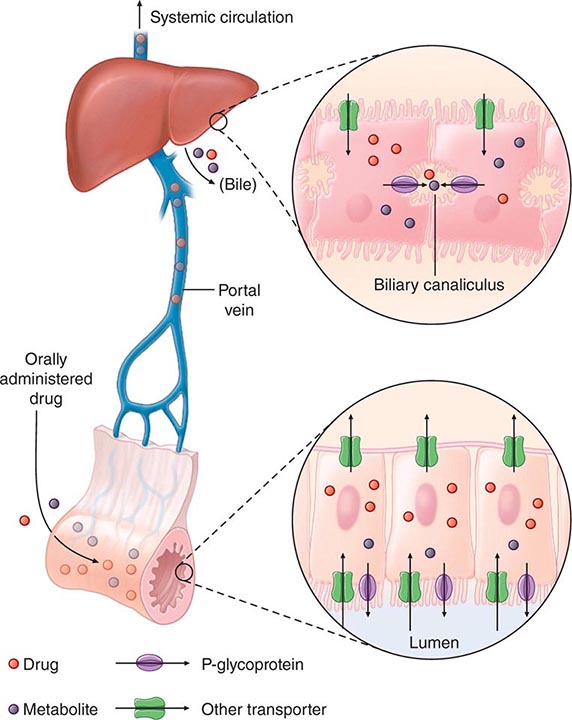
FIGURE 5-3 Mechanism of presystemic clearance. After drug enters the enterocyte, it can undergo metabolism, excretion into the intestinal lumen, or transport into the portal vein. Similarly, the hepatocyte may accomplish metabolism and biliary excretion prior to the entry of drug and metabolites to the systemic circulation. (Adapted by permission from DM Roden, in DP Zipes, J Jalife [eds]: Cardiac Electrophysiology: From Cell to Bedside, 4th ed. Philadelphia, Saunders, 2003. Copyright 2003 with permission from Elsevier.)
Drug movement across the membrane of any cell, including enterocytes and hepatocytes, is a combination of passive diffusion and active transport, mediated by specific drug uptake and efflux molecules. One widely studied drug transport molecule is P-glycoprotein, the product of the MDR1 gene. P-glycoprotein is expressed on the apical aspect of the enterocyte and on the canalicular aspect of the hepatocyte (Fig. 5-3). In both locations, it serves as an efflux pump, limiting availability of drug to the systemic circulation. P-glycoprotein–mediated drug efflux from cerebral capillaries limits drug brain penetration and is an important component of the blood-brain barrier.
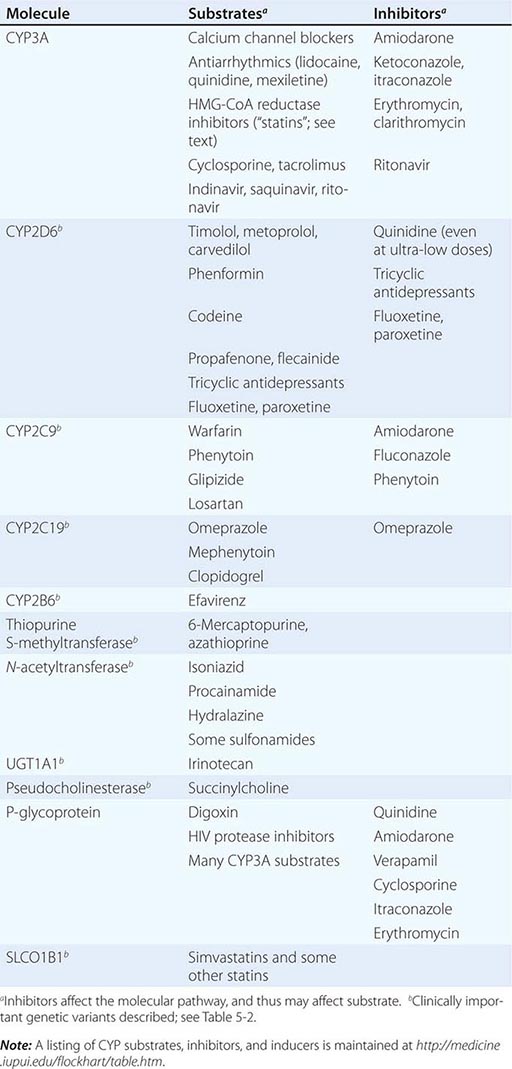
Drug metabolism generates compounds that are usually more polar and, hence, more readily excreted than parent drug. Metabolism takes place predominantly in the liver but can occur at other sites such as kidney, intestinal epithelium, lung, and plasma. “Phase I” metabolism involves chemical modification, most often oxidation accomplished by members of the cytochrome P450 (CYP) monooxygenase superfamily. CYPs that are especially important for drug metabolism are presented in Table 5-1, and each drug may be a substrate for one or more of these enzymes. “Phase II” metabolism involves conjugation of specific endogenous compounds to drugs or their metabolites. The enzymes that accomplish phase II reactions include glucuronyl-, acetyl-, sulfo-, and methyltransferases. Drug metabolites may exert important pharmacologic activity, as discussed further below.
|
MOLECULAR PATHWAYS MEDIATING DRUG DISPOSITION |
Clinical Implications of Altered Bioavailability Some drugs undergo near-complete presystemic metabolism and, thus, cannot be administered orally. Nitroglycerin cannot be used orally because it is completely extracted prior to reaching the systemic circulation. The drug is, therefore, used by the sublingual or transdermal routes, which bypass presystemic metabolism.
Some drugs with very extensive presystemic metabolism can still be administered by the oral route, using much higher doses than those required intravenously. Thus, a typical intravenous dose of verapamil is 1–5 mg, compared to the usual single oral dose of 40–120 mg. Administration of low-dose aspirin can result in exposure of cyclooxygenase in platelets in the portal vein to the drug, but systemic sparing because of first-pass aspirin deacylation in the liver. This is an example of presystemic metabolism being exploited to therapeutic advantage.
PHARMACOKINETIC CONCEPTS
Most pharmacokinetic processes, such as elimination, are first-order; that is, the rate of the process depends on the amount of drug present. Elimination can occasionally be zero-order (fixed amount eliminated per unit time), and this can be clinically important (see “Principles of Dose Selection”). In the simplest pharmacokinetic model (Fig. 5-2A), a drug bolus (D) is administered instantaneously to a central compartment, from which drug elimination occurs as a first-order process. Occasionally, central and other compartments correspond to physiologic spaces (e.g., plasma volume), whereas in others they are simply mathematical functions used to describe drug disposition. The first-order nature of drug elimination leads directly to the relationship describing drug concentration (C) at any time (t) following the bolus:
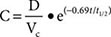
where VC is the volume of the compartment into which drug is delivered and t1/2 is elimination half-life. As a consequence of this relationship, a plot of the logarithm of concentration versus time is a straight line (Fig. 5-2A, inset). Half-life is the time required for 50% of a first-order process to be complete. Thus, 50% of drug elimination is achieved after one drug-elimination half-life, 75% after two, 87.5% after three, etc. In practice, first-order processes such as elimination are near-complete after four–five half-lives.
In some cases, drug is removed from the central compartment not only by elimination but also by distribution into peripheral compartments. In this case, the plot of plasma concentration versus time after a bolus may demonstrate two (or more) exponential components (Fig. 5-2B). In general, the initial rapid drop in drug concentration represents not elimination but drug distribution into and out of peripheral tissues (also first-order processes), while the slower component represents drug elimination; the initial precipitous decline is usually evident with administration by intravenous but not by other routes. Drug concentrations at peripheral sites are determined by a balance between drug distribution to and redistribution from those sites, as well as by elimination. Once distribution is near-complete (four–five distribution half-lives), plasma and tissue concentrations decline in parallel.
Clinical Implications of Half-Life Measurements The elimination half-life not only determines the time required for drug concentrations to fall to near-immeasurable levels after a single bolus, it is also the sole determinant of the time required for steady-state plasma concentrations to be achieved after any change in drug dosing (Fig. 5-4). This applies to the initiation of chronic drug therapy (whether by multiple oral doses or by continuous intravenous infusion), a change in chronic drug dose or dosing interval, or discontinuation of drug.
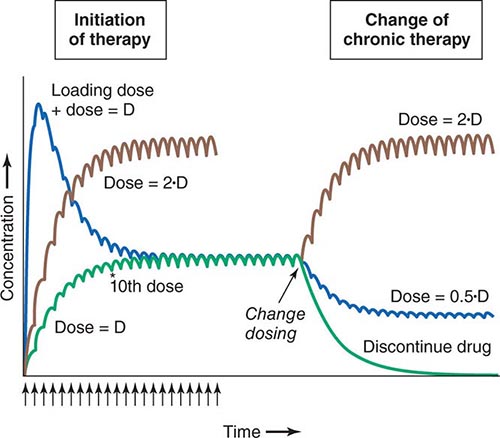
FIGURE 5-4 Drug accumulation to steady state. In this simulation, drug was administered (arrows) at intervals = 50% of the elimination half-life. Steady state is achieved during initiation of therapy after ~5 elimination half-lives, or 10 doses. A loading dose did not alter the eventual steady state achieved. A doubling of the dose resulted in a doubling of the steady state but the same time course of accumulation. Once steady state is achieved, a change in dose (increase, decrease, or drug discontinuation) results in a new steady state in ~5 elimination half-lives. (Adapted by permission from DM Roden, in DP Zipes, J Jalife [eds]: Cardiac Electrophysiology: From Cell to Bedside, 4th ed. Philadelphia, Saunders, 2003. Copyright 2003 with permission from Elsevier.)
Steady state describes the situation during chronic drug administration when the amount of drug administered per unit time equals drug eliminated per unit time. With a continuous intravenous infusion, plasma concentrations at steady state are stable, while with chronic oral drug administration, plasma concentrations vary during the dosing interval but the time-concentration profile between dosing intervals is stable (Fig. 5-4).
DRUG DISTRIBUTION
In a typical 70-kg human, plasma volume is ~3 L, blood volume is ~5.5 L, and extracellular water outside the vasculature is ~20 L. The volume of distribution of drugs extensively bound to plasma proteins but not to tissue components approaches plasma volume; warfarin is one such example. By contrast, for drugs highly bound to tissues, the volume of distribution can be far greater than any physiologic space. For example, the volume of distribution of digoxin and tricyclic antidepressants is hundreds of liters, obviously exceeding total-body volume. Such drugs are not readily removed by dialysis, an important consideration in overdose.
Clinical Implications of Drug Distribution In some cases, pharmacologic effects require drug distribution to peripheral sites. In this instance, the time course of drug delivery to and removal from these sites determines the time course of drug effects; anesthetic uptake into the central nervous system (CNS) is an example.
LOADING DOSES For some drugs, the indication may be so urgent that administration of “loading” dosages is required to achieve rapid elevations of drug concentration and therapeutic effects earlier than with chronic maintenance therapy (Fig. 5-4). Nevertheless, the time required for true steady state to be achieved is still determined only by the elimination half-life.
RATE OF INTRAVENOUS ADMINISTRATION Although the simulations in Fig. 5-2 use a single intravenous bolus, this is usually inappropriate in practice because side effects related to transiently very high concentrations can result. Rather, drugs are more usually administered orally or as a slower intravenous infusion. Some drugs are so predictably lethal when infused too rapidly that special precautions should be taken to prevent accidental boluses. For example, solutions of potassium for intravenous administration >20 mEq/L should be avoided in all but the most exceptional and carefully monitored circumstances. This minimizes the possibility of cardiac arrest due to accidental increases in infusion rates of more concentrated solutions.
Transiently high drug concentrations after rapid intravenous administration can occasionally be used to advantage. The use of midazolam for intravenous sedation, for example, depends upon its rapid uptake by the brain during the distribution phase to produce sedation quickly, with subsequent egress from the brain during the redistribution of the drug as equilibrium is achieved.
Similarly, adenosine must be administered as a rapid bolus in the treatment of reentrant supraventricular tachycardias (Chap. 276) to prevent elimination by very rapid (t1/2 of seconds) uptake into erythrocytes and endothelial cells before the drug can reach its clinical site of action, the atrioventricular node.
Clinical Implications of Altered Protein Binding Many drugs circulate in the plasma partly bound to plasma proteins. Since only unbound (free) drug can distribute to sites of pharmacologic action, drug response is related to the free rather than the total circulating plasma drug concentration. In chronic kidney or liver disease, protein binding may be decreased and thus drug actions increased. In some situations (myocardial infarction, infection, surgery), acute phase reactants transiently increase drug binding and thus decrease efficacy. These changes assume the greatest clinical importance for drugs that are highly protein-bound since even a small change in protein binding can result in large changes in free drug; for example, a decrease in binding from 99% to 98% doubles the free drug concentration from 1% to 2%. For some drugs (e.g., phenytoin), monitoring free rather than total drug concentrations can be useful.
ELIMINATION
Drug elimination reduces the amount of drug in the body over time. An important approach to quantifying this reduction is to consider that drug concentrations at the beginning and end of a time period are unchanged and that a specific volume of the body has been “cleared” of the drug during that time period. This defines clearance as volume/time. Clearance includes both drug metabolism and excretion.
Clinical Implications of Altered Clearance While elimination half-life determines the time required to achieve steady-state plasma concentration (Css), the magnitude of that steady state is determined by clearance (Cl) and dose alone. For a drug administered as an intravenous infusion, this relationship is:
Css = dosing rate/Cl or dosing rate = Cl · Css
When drug is administered orally, the average plasma concentration within a dosing interval (Cavg,ss) replaces Css, and the dosage (dose per unit time) must be increased if bioavailability (F) is less than 1:
Dose/time = Cl · Cavg,ss/F
Genetic variants, drug interactions, or diseases that reduce the activity of drug-metabolizing enzymes or excretory mechanisms lead to decreased clearance and, hence, a requirement for downward dose adjustment to avoid toxicity. Conversely, some drug interactions and genetic variants increase the function of drug elimination pathways, and hence, increased drug dosage is necessary to maintain a therapeutic effect.
ACTIVE DRUG METABOLITES
Metabolites may produce effects similar to, overlapping with, or distinct from those of the parent drug. Accumulation of the major metabolite of procainamide, N-acetylprocainamide (NAPA), likely accounts for marked QT prolongation and torsades des pointes ventricular tachycardia (Chap. 276) during therapy with procainamide. Neurotoxicity during therapy with the opioid analgesic meperidine is likely due to accumulation of normeperidine, especially in renal disease.
Prodrugs are inactive compounds that require metabolism to generate active metabolites that mediate the drug effects. Examples include many angiotensin-converting enzyme (ACE) inhibitors, the angiotensin receptor blocker losartan, the antineoplastic irinotecan, the anti-estrogen tamoxifen, the analgesic codeine (whose active metabolite morphine probably underlies the opioid effect during codeine administration), and the antiplatelet drug clopidogrel. Drug metabolism has also been implicated in bioactivation of procarcinogens and in generation of reactive metabolites that mediate certain adverse drug effects (e.g., acetaminophen hepatotoxicity, discussed below).
THE CONCEPT OF HIGH-RISK PHARMACOKINETICS
When plasma concentrations of active drug depend exclusively on a single metabolic pathway, any condition that inhibits that pathway (be it disease-related, genetic, or due to a drug interaction) can lead to dramatic changes in drug concentrations and marked variability in drug action. This problem of high-risk pharmacokinetics is especially pronounced in two settings. First, variability in bioactivation of a prodrug can lead to striking variability in drug action; examples include decreased CYP2D6 activity, which prevents analgesia by codeine, and decreased CYP2C19 activity, which reduces the antiplatelet effects of clopidogrel. The second setting is drug elimination that relies on a single pathway. In this case, inhibition of the elimination pathway by genetic variants or by administration of inhibiting drugs leads to marked elevation of drug concentration and, for drugs with a narrow therapeutic window, an increased likelihood of dose-related toxicity. Individuals with loss-of-function alleles in CYP2C9, responsible for metabolism of the active S-enantiomer of warfarin, appear to be at increased risk for bleeding. When drugs undergo elimination by multiple-drug metabolizing or excretory pathways, absence of one pathway (due to a genetic variant or drug interaction) is much less likely to have a large impact on drug concentrations or drug actions.
PRINCIPLES OF PHARMACODYNAMICS
The Onset of Drug Action For drugs used in the urgent treatment of acute symptoms, little or no delay is anticipated (or desired) between the drug-target interaction and the development of a clinical effect. Examples of such acute situations include vascular thrombosis, shock, or status epilepticus.
For many conditions, however, the indication for therapy is less urgent, and a delay between the interaction of a drug with its pharmacologic target(s) and a clinical effect is clinically acceptable. Common pharmacokinetic mechanisms that can contribute to such a delay include slow elimination (resulting in slow accumulation to steady state), uptake into peripheral compartments, or accumulation of active metabolites. Another common explanation for such a delay is that the clinical effect develops as a downstream consequence of the initial molecular effect the drug produces. Thus, administration of a proton pump inhibitor or an H2-receptor blocker produces an immediate increase in gastric pH but ulcer healing that is delayed. Cancer chemotherapy similarly produces delayed therapeutic effects.
Drug Effects May Be Disease Specific A drug may produce no action or a different spectrum of actions in unaffected individuals compared to patients with underlying disease. Further, concomitant disease can complicate interpretation of response to drug therapy, especially adverse effects. For example, high doses of anticonvulsants such as phenytoin may cause neurologic symptoms, which may be confused with the underlying neurologic disease. Similarly, increasing dyspnea in a patient with chronic lung disease receiving amiodarone therapy could be due to drug, underlying disease, or an intercurrent cardiopulmonary problem. Thus, the presence of chronic lung disease may argue against the use of amiodarone.
While drugs interact with specific molecular receptors, drug effects may vary over time, even if stable drug and metabolite concentrations are maintained. The drug-receptor interaction occurs in a complex biologic milieu that can vary to modulate the drug effect. For example, ion channel blockade by drugs, an important anticonvulsant and antiarrhythmic effect, is often modulated by membrane potential, itself a function of factors such as extracellular potassium or local ischemia. Receptors may be up- or downregulated by disease or by the drug itself. For example, β-adrenergic blockers upregulate β-receptor density during chronic therapy. While this effect does not usually result in resistance to the therapeutic effect of the drugs, it may produce severe agonist-mediated effects (such as hypertension or tachycardia) if the blocking drug is abruptly withdrawn.
PRINCIPLES OF DOSE SELECTION
The desired goal of therapy with any drug is to maximize the likelihood of a beneficial effect while minimizing the risk of adverse effects. Previous experience with the drug, in controlled clinical trials or in postmarketing use, defines the relationships between dose or plasma concentration and these dual effects (Fig. 5-1) and has important implications for initiation of drug therapy:
1. The target drug effect should be defined when drug treatment is started. With some drugs, the desired effect may be difficult to measure objectively, or the onset of efficacy can be delayed for weeks or months; drugs used in the treatment of cancer and psychiatric disease are examples. Sometimes a drug is used to treat a symptom, such as pain or palpitations, and here it is the patient who will report whether the selected dose is effective. In yet other settings, such as anticoagulation or hypertension, the desired response can be repeatedly and objectively assessed by simple clinical or laboratory tests.
2. The nature of anticipated toxicity often dictates the starting dose. If side effects are minor, it may be acceptable to start chronic therapy at a dose highly likely to achieve efficacy and down-titrate if side effects occur. However, this approach is rarely, if ever, justified if the anticipated toxicity is serious or life-threatening; in this circumstance, it is more appropriate to initiate therapy with the lowest dose that may produce a desired effect. In cancer chemotherapy, it is common practice to use maximum-tolerated doses.
3. The above considerations do not apply if these relationships between dose and effects cannot be defined. This is especially relevant to some adverse drug effects (discussed in further detail below) whose development are not readily related to drug dose.
4. If a drug dose does not achieve its desired effect, a dosage increase is justified only if toxicity is absent and the likelihood of serious toxicity is small.
Failure of Efficacy Assuming the diagnosis is correct and the correct drug is prescribed, explanations for failure of efficacy include drug interactions, noncompliance, or unexpectedly low drug dosage due to administration of expired or degraded drug. These are situations in which measurement of plasma drug concentrations, if available, can be especially useful. Noncompliance is an especially frequent problem in the long-term treatment of diseases such as hypertension and epilepsy, occurring in ≥25% of patients in therapeutic environments in which no special effort is made to involve patients in the responsibility for their own health. Multidrug regimens with multiple doses per day are especially prone to noncompliance.
Monitoring response to therapy, by physiologic measures or by plasma concentration measurements, requires an understanding of the relationships between plasma concentration and anticipated effects. For example, measurement of QT interval is used during treatment with sotalol or dofetilide to avoid marked QT prolongation that can herald serious arrhythmias. In this setting, evaluating the electrocardiogram at the time of anticipated peak plasma concentration and effect (e.g., 1–2 h postdose at steady state) is most appropriate. Maintained high vancomycin levels carry a risk of nephrotoxicity, so dosages should be adjusted on the basis of plasma concentrations measured at trough (predose). Similarly, for dose adjustment of other drugs (e.g., anticonvulsants), concentration should be measured at its lowest during the dosing interval, just prior to a dose at steady state (Fig. 5-4), to ensure a maintained therapeutic effect.
Concentration of Drugs in Plasma as a Guide to Therapy Factors such as interactions with other drugs, disease-induced alterations in elimination and distribution, and genetic variation in drug disposition combine to yield a wide range of plasma levels in patients given the same dose. Hence, if a predictable relationship can be established between plasma drug concentration and beneficial or adverse drug effect, measurement of plasma levels can provide a valuable tool to guide selection of an optimal dose, especially when there is a narrow range between the plasma levels yielding therapeutic and adverse effects. Monitoring is commonly used with certain types of drugs including many anticonvulsants, antirejection agents, antiarrhythmics, and antibiotics. By contrast, if no such relationship can be established (e.g., if drug access to important sites of action outside plasma is highly variable), monitoring plasma concentration may not provide an accurate guide to therapy (Fig. 5-5A).
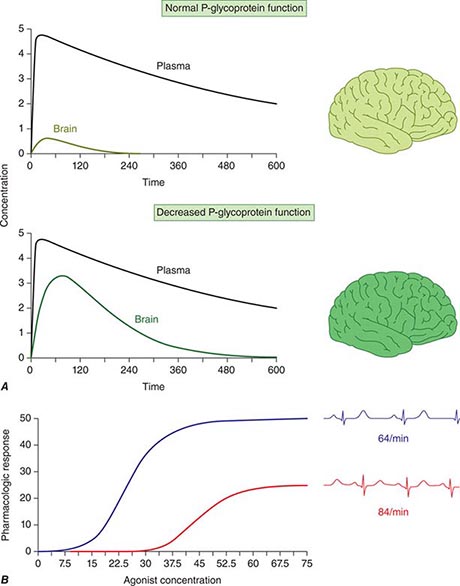
FIGURE 5-5 A. The efflux pump P-glycoprotein excludes drugs from the endothelium of capillaries in the brain and so constitutes a key element of the blood-brain barrier. Thus, reduced P-glycoprotein function (e.g., due to drug interactions or genetically determined variability in gene transcription) increases penetration of substrate drugs into the brain, even when plasma concentrations are unchanged. B. The graph shows an effect of a β1-receptor polymorphism on receptor function in vitro. Patients with the hypofunctional variant (red) may display lesser heart-rate slowing or blood pressure lowering on exposure to a receptor blocking agent.
The common situation of first-order elimination implies that average, maximum, and minimum steady-state concentrations are related linearly to the dosing rate. Accordingly, the maintenance dose may be adjusted on the basis of the ratio between the desired and measured concentrations at steady state; for example, if a doubling of the steady-state plasma concentration is desired, the dose should be doubled. This does not apply to drugs eliminated by zero-order kinetics (fixed amount per unit time), where small dosage increases will produce disproportionate increases in plasma concentration; examples include phenytoin and theophylline.
An increase in dosage is usually best achieved by changing the drug dose but not the dosing interval (e.g., by giving 200 mg every 8 h instead of 100 mg every 8 h). However, this approach is acceptable only if the resulting maximum concentration is not toxic and the trough value does not fall below the minimum effective concentration for an undesirable period of time. Alternatively, the steady state may be changed by altering the frequency of intermittent dosing but not the size of each dose. In this case, the magnitude of the fluctuations around the average steady-state level will change—the shorter the dosing interval, the smaller the difference between peak and trough levels.
EFFECTS OF DISEASE ON DRUG CONCENTRATION AND RESPONSE
RENAL DISEASE
Renal excretion of parent drug and metabolites is generally accomplished by glomerular filtration and by specific drug transporters. If a drug or its metabolites are primarily excreted through the kidneys and increased drug levels are associated with adverse effects, drug dosages must be reduced in patients with renal dysfunction to avoid toxicity. The antiarrhythmics dofetilide and sotalol undergo predominant renal excretion and carry a risk of QT prolongation and arrhythmias if doses are not reduced in renal disease. In end-stage renal disease, sotalol has been given as 40 mg after dialysis (every second day), compared to the usual daily dose, 80–120 mg every 12 h. The narcotic analgesic meperidine undergoes extensive hepatic metabolism, so that renal failure has little effect on its plasma concentration. However, its metabolite, normeperidine, does undergo renal excretion, accumulates in renal failure, and probably accounts for the signs of CNS excitation, such as irritability, twitching, and seizures, that appear when multiple doses of meperidine are administered to patients with renal disease. Protein binding of some drugs (e.g., phenytoin) may be altered in uremia, so measuring free drug concentration may be desirable.
In non-end-stage renal disease, changes in renal drug clearance are generally proportional to those in creatinine clearance, which may be measured directly or estimated from the serum creatinine (Chap. 333e). This estimate, coupled with the knowledge of how much drug is normally excreted renally versus nonrenally, allows an estimate of the dose adjustment required. In practice, most decisions involving dosing adjustment in patients with renal failure use published recommended adjustments in dosage or dosing interval based on the severity of renal dysfunction indicated by creatinine clearance. Any such modification of dose is a first approximation and should be followed by plasma concentration data (if available) and clinical observation to further optimize therapy for the individual patient.
LIVER DISEASE
Standard tests of liver function are not useful in adjusting doses in diseases like hepatitis or cirrhosis. First-pass metabolism may decrease, leading to increased oral bioavailability as a consequence of disrupted hepatocyte function, altered liver architecture, and portacaval shunts. The oral bioavailability for high first-pass drugs such as morphine, meperidine, midazolam, and nifedipine is almost doubled in patients with cirrhosis, compared to those with normal liver function. Therefore, the size of the oral dose of such drugs should be reduced in this setting.
HEART FAILURE AND SHOCK
Under conditions of decreased tissue perfusion, the cardiac output is redistributed to preserve blood flow to the heart and brain at the expense of other tissues (Chap. 279). As a result, drugs may be distributed into a smaller volume of distribution, higher drug concentrations will be present in the plasma, and the tissues that are best perfused (the brain and heart) will be exposed to these higher concentrations, resulting in increased CNS or cardiac effects. As well, decreased perfusion of the kidney and liver may impair drug clearance. Another consequence of severe heart failure is decreased gut perfusion, which may reduce drug absorption and, thus, lead to reduced or absent effects of orally administered therapies.
DRUG USE IN THE ELDERLY
In the elderly, multiple pathologies and medications used to treat them result in more drug interactions and adverse effects. Aging also results in changes in organ function, especially of the organs involved in drug disposition. Initial doses should be less than the usual adult dosage and should be increased slowly. The number of medications, and doses per day, should be kept as low as possible.
Even in the absence of kidney disease, renal clearance may be reduced by 35–50% in elderly patients. Dosages should be adjusted on the basis of creatinine clearance. Aging also results in a decrease in the size of, and blood flow to, the liver and possibly in the activity of hepatic drug-metabolizing enzymes; accordingly, the hepatic clearance of some drugs is impaired in the elderly. As with liver disease, these changes are not readily predicted.
Elderly patients may display altered drug sensitivity. Examples include increased analgesic effects of opioids, increased sedation from benzodiazepines and other CNS depressants, and increased risk of bleeding while receiving anticoagulant therapy, even when clotting parameters are well controlled. Exaggerated responses to cardiovascular drugs are also common because of the impaired responsiveness of normal homeostatic mechanisms. Conversely, the elderly display decreased sensitivity to β-adrenergic receptor blockers.
Adverse drug reactions are especially common in the elderly because of altered pharmacokinetics and pharmacodynamics, the frequent use of multidrug regimens, and concomitant disease. For example, use of long half-life benzodiazepines is linked to the occurrence of hip fractures in elderly patients, perhaps reflecting both a risk of falls from these drugs (due to increased sedation) and the increased incidence of osteoporosis in elderly patients. In population surveys of the noninstitutionalized elderly, as many as 10% had at least one adverse drug reaction in the previous year.
DRUG USE IN CHILDREN
While most drugs used to treat disease in children are the same are those in adults, there are few studies that provide solid data to guide dosing. Drug metabolism pathways mature at different rates after birth, and disease mechanisms may be different in children. In practice, doses are adjusted for size (weight or body surface area) as a first approximation unless age-specific data are available.
GENETIC DETERMINANTS OF THE RESPONSE TO DRUGS
PRINCIPLES OF GENETIC VARIATION AND HUMAN TRAITS (See also CHAPS. 82 AND 84)
The concept that genetically determined variations in drug metabolism might be associated with variable drug levels and hence, effect, was advanced at the end of the nineteenth century, and the examples of familial clustering of unusual drug responses were noted in the mid-twentieth century. A goal of traditional Mendelian genetics is to identify DNA variants associated with a distinct phenotype in multiple related family members (Chap. 84). However, it is unusual for a drug response phenotype to be accurately measured in more than one family member, let alone across a kindred. Thus, non-family-based approaches are generally used to identify and validate DNA variants contributing to variable drug actions.
Candidate Gene Studies in Pharmacogenetics Most studies to date have used an understanding of the molecular mechanisms modulating drug action to identify candidate genes in which variants could explain variable drug responses. One very common scenario is that variable drug actions can be attributed to variability in plasma drug concentrations. When plasma drug concentrations vary widely (e.g., more than an order of magnitude), especially if their distribution is non-unimodal as in Fig. 5-6, variants in single genes controlling drug concentrations often contribute. In this case, the most obvious candidate genes are those responsible for drug metabolism and elimination. Other candidate genes are those encoding the target molecules with which drugs interact to produce their effects or molecules modulating that response, including those involved in disease pathogenesis.
FIGURE 5-6 A. CYP2D6 metabolic activity was assessed in 290 subjects by administration of a test dose of a probe substrate and measurement of urinary formation of the CYP2D6-generated metabolite. The heavy arrow indicates a clear antimode, separating poor metabolizer subjects (PMs, red), with two loss-of-function CYP2D6 alleles, indicated by the intron-exon structures below the chart. Individuals with one or two functional alleles are grouped together as extensive metabolizers (EMs, green). Also shown are ultra-rapid metabolizers (UMs), with 2–12 functional copies of the gene (gray), displaying the greatest enzyme activity. (Adapted from M-L Dahl et al: J Pharmacol Exp Ther 274:516, 1995.) B. These simulations show the predicted effects of CYP2D6 genotype on disposition of a substrate drug. With a single dose (left), there is an inverse “gene-dose” relationship between the number of active alleles and the areas under the time-concentration curves (smallest in UM subjects; highest in PM subjects); this indicates that clearance is greatest in UM subjects. In addition, elimination half-life is longest in PM subjects. The right panel shows that these single dose differences are exaggerated during chronic therapy: steady-state concentration is much higher in PM subjects (decreased clearance), as is the time required to achieve steady state (longer elimination half-life).
Genome-Wide Association Studies in Pharmacogenomics The field has also had some success with “unbiased” approaches such as genome-wide association (GWA) (Chap. 82), particularly in identifying single variants associated with high risk for certain forms of drug toxicity (Table 5-2). GWA studies have identified variants in the HLA-B locus that are associated with high risk for severe skin rashes during treatment with the anticonvulsant carbamazepine and the antiretroviral abacavir. A GWA study of simvastatin-associated myopathy identified a single noncoding single nucleotide polymorphism (SNP) in SLCO1B1, encoding OATP1B1, a drug transporter known to modulate simvastatin uptake into the liver, which accounts for 60% of myopathy risk. GWA approaches have also implicated interferon variants in antileukemic responses and in response to therapy in hepatitis C. Ribavirin, used as therapy in hepatitis C, causes hemolytic anemia, and this has been linked to variants in ITPA, encoding inosine triphosphatase.
|
GENETIC VARIANTS AND DRUG RESPONSES |
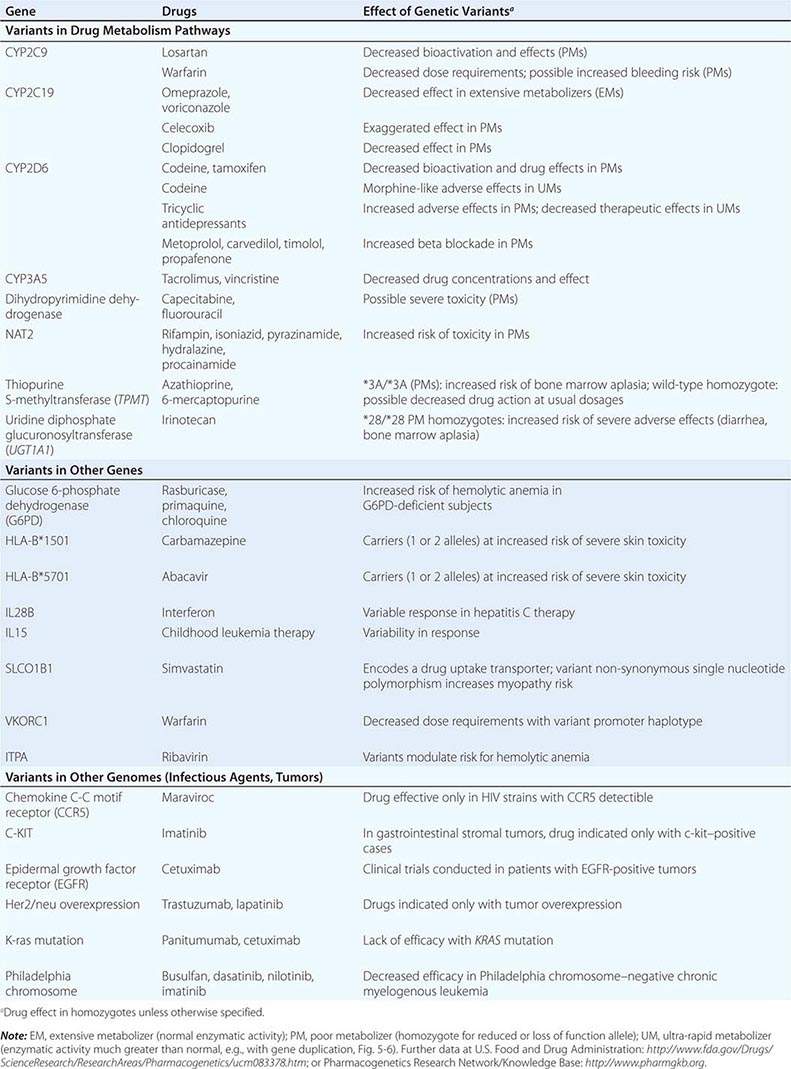
GENETIC VARIANTS AFFECTING PHARMACOKINETICS
Clinically important genetic variants have been described in multiple molecular pathways of drug disposition (Table 5-2). A distinct multimodal distribution of drug disposition (as shown in Fig. 5-6) argues for a predominant effect of variants in a single gene in the metabolism of that substrate. Individuals with two alleles (variants) encoding for nonfunctional protein make up one group, often termed poor metabolizers (PM phenotype); for some genes, many variants can produce such a loss of function, complicating the use of genotyping in clinical practice. Individuals with one functional allele make up a second (intermediate metabolizers) and may or may not be distinguishable from those with two functional alleles (extensive metabolizers, EMs). Ultra-rapid metabolizers with especially high enzymatic activity (occasionally due to gene duplication; Fig. 5-6) have also been described for some traits. Many drugs in widespread use can inhibit specific drug disposition pathways (Table 5-1), and so EM individuals receiving such inhibitors can respond like PM patients (phenocopying). Polymorphisms in genes encoding drug uptake or drug efflux transporters may be other contributors to variability in drug delivery to target sites and, hence, in drug effects.
CYP Variants Members of the CYP3A family (CYP3A4, 3A5) metabolize the greatest number of drugs in therapeutic use. CYP3A4 activity is highly variable (up to an order of magnitude) among individuals, but the underlying mechanisms are not well understood. In whites, but not African Americans, there is a common loss-of-function polymorphism in the closely related CYP3A5 gene. Decreased efficacy of the antirejection agent tacrolimus in African-American subjects has been attributed to more rapid elimination due to relatively greater CYP3A5 activity. A lower risk of vincristine-associated neuropathy has been reported in CYP3A5 “expressers.”
CYP2D6 is second to CYP3A4 in the number of commonly used drugs that it metabolizes. CYP2D6 activity is polymorphically distributed, with about 7% of European- and African-derived populations (but very few Asians) displaying the PM phenotype (Fig. 5-6). Dozens of loss-of-function variants in the CYP2D6 gene have been described; the PM phenotype arises in individuals with two such alleles. In addition, ultra-rapid metabolizers with multiple functional copies of the CYP2D6 gene have been identified.
Codeine is biotransformed by CYP2D6 to the potent active metabolite morphine, so its effects are blunted in PMs and exaggerated in ultra-rapid metabolizers. In the case of drugs with beta-blocking properties metabolized by CYP2D6, greater signs of beta blockade (e.g., bronchospasm, bradycardia) are seen in PM subjects than in EMs. This can be seen not only with orally administered beta blockers such as metoprolol and carvedilol, but also with ophthalmic timolol and with the sodium channel–blocking antiarrhythmic propafenone, a CYP2D6 substrate with beta-blocking properties. Ultra-rapid metabolizers may require very high dosages of tricyclic antidepressants to achieve a therapeutic effect and, with codeine, may display transient euphoria and nausea due to very rapid generation of morphine. Tamoxifen undergoes CYP2D6-mediated biotransformation to an active metabolite, so its efficacy may be in part related to this polymorphism. In addition, the widespread use of selective serotonin reuptake inhibitors (SSRIs) to treat tamoxifen-related hot flashes may also alter the drug’s effects because many SSRIs, notably fluoxetine and paroxetine, are also CYP2D6 inhibitors.
The PM phenotype for CYP2C19 is common (20%) among Asians and rarer (2–3%) in European-derived populations. The impact of polymorphic CYP2C19-mediated metabolism has been demonstrated with the proton pump inhibitor omeprazole, where ulcer cure rates with “standard” dosages were much lower in EM patients (29%) than in PMs (100%). Thus, understanding the importance of this polymorphism would have been important in developing the drug, and knowing a patient’s CYP2C19 genotype should improve therapy. CYP2C19 is responsible for bioactivation of the antiplatelet drug clopidogrel, and several large studies have documented decreased efficacy (e.g., increased myocardial infarction after placement of coronary stents) among Caucasian subjects with reduction of function alleles. In addition, some studies suggest that omeprazole and possibly other proton pump inhibitors phenocopy this effect.
There are common variants of CYP2C9 that encode proteins with loss of catalytic function. These variant alleles are associated with increased rates of neurologic complications with phenytoin, hypoglycemia with glipizide, and reduced warfarin dose required to maintain stable anticoagulation. The angiotensin-receptor blocker losartan is a prodrug that is bioactivated by CYP2C9; as a result, PMs and those receiving inhibitor drugs may display little response to therapy.
Transferase Variants One of the most extensively studied phase II polymorphisms is the PM trait for thiopurine S-methyltransferase (TPMT). TPMT bioinactivates the antileukemic drug 6-mercaptopurine. Further, 6-mercaptopurine is itself an active metabolite of the immunosuppressive azathioprine. Homozygotes for alleles encoding the inactive TPMT (1 in 300 individuals) predictably exhibit severe and potentially fatal pancytopenia on standard doses of azathioprine or 6-mercaptopurine. On the other hand, homozygotes for fully functional alleles may display less anti-inflammatory or antileukemic effect with the drugs.
N-acetylation is catalyzed by hepatic N-acetyl transferase (NAT), which represents the activity of two genes, NAT-1 and NAT-2. Both enzymes transfer an acetyl group from acetyl coenzyme A to the drug; polymorphisms in NAT-2 are thought to underlie individual differences in the rate at which drugs are acetylated and thus define “rapid acetylators” and “slow acetylators.” Slow acetylators make up ~50% of European- and African-derived populations but are less common among Asians. Slow acetylators have an increased incidence of the drug-induced lupus syndrome during procainamide and hydralazine therapy and of hepatitis with isoniazid. Induction of CYPs (e.g., by rifampin) also increases the risk of isoniazid-related hepatitis, likely reflecting generation of reactive metabolites of acetylhydrazine, itself an isoniazid metabolite.
Individuals homozygous for a common promoter polymorphism that reduces transcription of uridine diphosphate glucuronosyltransferase (UGT1A1) have benign hyperbilirubinemia (Gilbert’s syndrome; Chap. 358). This variant has also been associated with diarrhea and increased bone marrow depression with the antineoplastic prodrug irinotecan, whose active metabolite is normally detoxified by UGT1A1-mediated glucuronidation. The antiretroviral atazanavir is a UGT1A1 inhibitor, and individuals with the Gilbert’s variant develop higher bilirubin levels during treatment.
VARIABILITY IN THE MOLECULAR TARGETS WITH WHICH DRUGS INTERACT
Multiple polymorphisms identified in the β2-adrenergic receptor appear to be linked to specific phenotypes in asthma and congestive heart failure, diseases in which β2-receptor function might be expected to determine prognosis. Polymorphisms in the β2-receptor gene have also been associated with response to inhaled β2-receptor agonists, while those in the β1-adrenergic receptor gene have been associated with variability in heart rate slowing and blood pressure lowering (Fig. 5-5B). In addition, in heart failure, a common polymorphism in the β1-adrenergic receptor gene has been implicated in variable clinical outcome during therapy with the investigational beta blocker bucindolol. Response to the 5-lipoxygenase inhibitor zileuton in asthma has been linked to polymorphisms that determine the expression level of the 5-lipoxygenase gene.
Drugs may also interact with genetic pathways of disease to elicit or exacerbate symptoms of the underlying conditions. In the porphyrias, CYP inducers are thought to increase the activity of enzymes proximal to the deficient enzyme, exacerbating or triggering attacks (Chap. 430). Deficiency of glucose-6-phosphate dehydrogenase (G6PD), most often in individuals of African, Mediterranean, or South Asian descent, increases the risk of hemolytic anemia in response to the antimalarial primaquine (Chap. 129) and the uric acid–lowering agent rasburicase, which do not cause hemolysis in patients with normal amounts of the enzyme. Patients with mutations in the ryanodine receptor, which controls intracellular calcium in skeletal muscle and other tissues, may be asymptomatic until exposed to certain general anesthetics, which trigger the rare syndrome of malignant hyperthermia. Certain antiarrhythmics and other drugs can produce marked QT prolongation and torsades des pointes (Chap. 276), and in some patients, this adverse effect represents unmasking of previously subclinical congenital long QT syndrome. Up to 50% of the variability in steady-state warfarin dose requirement is attributable to polymorphisms in the promoter of VKORC1, which encodes the warfarin target, and in the coding region of CYP2C9, which mediates its elimination.
Tumor and Infectious Agent Genomes The actions of drugs used to treat infectious or neoplastic disease may be modulated by variants in these nonhuman germline genomes. Genotyping tumors is a rapidly evolving approach to target therapies to underlying mechanisms and to avoid potentially toxic therapy in patients who would derive no benefit (Chap. 101e). Trastuzumab, which potentiates anthracycline-related cardiotoxicity, is ineffective in breast cancers that do not express the herceptin receptor. Imatinib targets a specific tyrosine kinase, BCR-Abl1, that is generated by the translocation that creates the Philadelphia chromosome typical of chronic myelogenous leukemia (CML). BCR-Abl1 is not only active but may be central to the pathogenesis of CML; its use in BCR-Abl1-positive tumors has resulted in remarkable antitumor efficacy. Similarly, the anti–epidermal growth factor receptor (EGFR) antibodies cetuximab and panitumumab appear especially effective in colon cancers in which K-ras, a G protein in the EGFR pathway, is not mutated. Vemurafenib does not inhibit wild-type BRAF but is active against the V600E mutant form of the kinase.
PROSPECTS FOR INCORPORATING PHARMACOGENETIC INFORMATION INTO CLINICAL PRACTICE
The description of genetic variants linked to variable drug responses naturally raises the question of if and how to use this information in practice. Indeed, the U.S. Food and Drug Administration (FDA) now incorporates pharmacogenetic data into information (“package inserts”) meant to guide prescribing. A decision to adopt pharmacogenetically guided dosing for a given drug depends on multiple factors. The most important are the magnitude and clinical importance of the genetic effect and the strength of evidence linking genetic variation to variable drug effects (e.g., anecdote versus post-hoc analysis of clinical trial data versus randomized prospective clinical trial). The evidence can be strengthened if statistical arguments from clinical trial data are complemented by an understanding of underlying physiologic mechanisms. Cost versus expected benefit may also be a factor.
When the evidence is compelling, alternate therapies are not available, and there are clear recommendations for dosage adjustment in subjects with variants, there is a strong argument for deploying genetic testing as a guide to prescribing. The association between HLA-B*5701 and severe skin toxicity with abacavir is an example. In other situations, the arguments are less compelling: the magnitude of the genetic effect may be smaller, the consequences may be less serious, alternate therapies may be available, or the drug effect may be amenable to monitoring by other approaches. Ongoing clinical trials are addressing the utility of preprescription genotyping in large populations exposed to drugs with known pharmacogenetic variants (e.g., warfarin). Importantly, technological advances are now raising the possibility of inexpensive whole genome sequencing. Incorporating a patient’s whole genome sequence into their electronic medical record would allow the information to be accessed as needed for many genetic and pharmacogenetic applications, and the argument has been put forward that this approach would lower logistic barriers to use of pharmacogenomic variant data in prescribing. There are multiple issues (e.g., economic, technological, and ethical) that need to be addressed if such a paradigm is to be adopted (Chap. 82). While barriers to bringing genomic and pharmacogenomic information to the bedside seem daunting, the field is very young and evolving rapidly. Indeed, one major result of understanding the role of genetics in drug action has been improved screening of drugs during the development process to reduce the likelihood of highly variable metabolism or unanticipated toxicity.
INTERACTIONS BETWEEN DRUGS
Drug interactions can complicate therapy by increasing or decreasing the action of a drug; interactions may be based on changes in drug disposition or in drug response in the absence of changes in drug levels. Interactions must be considered in the differential diagnosis of any unusual response occurring during drug therapy. Prescribers should recognize that patients often come to them with a legacy of drugs acquired during previous medical experiences, often with multiple physicians who may not be aware of all the patient’s medications. A meticulous drug history should include examination of the patient’s medications and, if necessary, calls to the pharmacist to identify prescriptions. It should also address the use of agents not often volunteered during questioning, such as OTC drugs, health food supplements, and topical agents such as eye drops. Lists of interactions are available from a number of electronic sources. While it is unrealistic to expect the practicing physician to memorize these, certain drugs consistently run the risk of generating interactions, often by inhibiting or inducing specific drug elimination pathways. Examples are presented below and in Table 5-3. Accordingly, when these drugs are started or stopped, prescribers must be especially alert to the possibility of interactions.
|
DRUGS WITH A HIGH RISK OF GENERATING PHARMACOKINETIC INTERACTIONS |
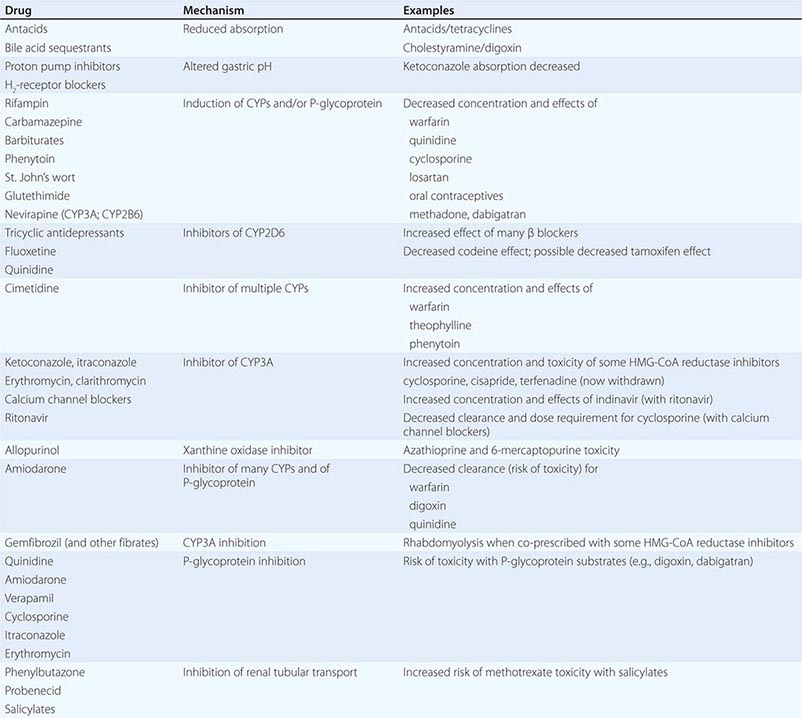
PHARMACOKINETIC INTERACTIONS CAUSING DECREASED DRUG EFFECTS
Gastrointestinal absorption can be reduced if a drug interaction results in drug binding in the gut, as with aluminum-containing antacids, kaolin-pectin suspensions, or bile acid sequestrants. Drugs such as histamine H2-receptor antagonists or proton pump inhibitors that alter gastric pH may decrease the solubility and hence absorption of weak bases such as ketoconazole.
Expression of some genes responsible for drug elimination, notably CYP3A and MDR1, can be markedly increased by inducing drugs, such as rifampin, carbamazepine, phenytoin, St. John’s wort, and glutethimide, and by smoking, exposure to chlorinated insecticides such as DDT (CYP1A2), and chronic alcohol ingestion. Administration of inducing agents lowers plasma levels, and thus effects, over 2–3 weeks as gene expression is increased. If a drug dose is stabilized in the presence of an inducer that is subsequently stopped, major toxicity can occur as clearance returns to preinduction levels and drug concentrations rise. Individuals vary in the extent to which drug metabolism can be induced, likely through genetic mechanisms.
Interactions that inhibit the bioactivation of prodrugs will decrease drug effects (Table 5-1).
Interactions that decrease drug delivery to intracellular sites of action can decrease drug effects: tricyclic antidepressants can blunt the antihypertensive effect of clonidine by decreasing its uptake into adrenergic neurons. Reduced CNS penetration of multiple HIV protease inhibitors (with the attendant risk of facilitating viral replication in a sanctuary site) appears attributable to P-glycoprotein-mediated exclusion of the drug from the CNS; indeed, inhibition of P-glycoprotein has been proposed as a therapeutic approach to enhance drug entry to the CNS (Fig. 5-5A).
PHARMACOKINETIC INTERACTIONS CAUSING INCREASED DRUG EFFECTS
The most common mechanism here is inhibition of drug elimination. In contrast to induction, new protein synthesis is not involved, and the effect develops as drug and any inhibitor metabolites accumulate (a function of their elimination half-lives). Since shared substrates of a single enzyme can compete for access to the active site of the protein, many CYP substrates can also be considered inhibitors. However, some drugs are especially potent as inhibitors (and occasionally may not even be substrates) of specific drug elimination pathways, and so it is in the use of these agents that clinicians must be most alert to the potential for interactions (Table 5-3). Commonly implicated interacting drugs of this type include amiodarone, cimetidine, erythromycin and some other macrolide antibiotics (clarithromycin but not azithromycin), ketoconazole and other azole antifungals, the antiretroviral agent ritonavir, and high concentrations of grapefruit juice (Table 5-3). The consequences of such interactions will depend on the drug whose elimination is being inhibited (see “The Concept of High-Risk Pharmacokinetics,” above). Examples include CYP3A inhibitors increasing the risk of cyclosporine toxicity or of rhabdomyolysis with some HMG-CoA reductase inhibitors (lovastatin, simvastatin, atorvastatin, but not pravastatin), and P-glycoprotein inhibitors increasing the risk of toxicity with digoxin therapy or of bleeding with the thrombin inhibitor dabigatran.
These interactions can occasionally be exploited to therapeutic benefit. The antiviral ritonavir is a very potent CYP3A4 inhibitor that is sometimes added to anti-HIV regimens, not because of its antiviral effects but because it decreases clearance, and hence increases efficacy, of other anti-HIV agents. Similarly, calcium channel blockers have been deliberately coadministered with cyclosporine to reduce its clearance and thus its maintenance dosage and cost.
Phenytoin, an inducer of many systems, including CYP3A, inhibits CYP2C9. CYP2C9 metabolism of losartan to its active metabolite is inhibited by phenytoin, with potential loss of antihypertensive effect.
Grapefruit (but not orange) juice inhibits CYP3A, especially at high doses; patients receiving drugs where even modest CYP3A inhibition may increase the risk of adverse effects (e.g., cyclosporine, some HMG-CoA reductase inhibitors) should therefore avoid grapefruit juice.
CYP2D6 is markedly inhibited by quinidine, a number of neuroleptic drugs (chlorpromazine and haloperidol), and the SSRIs fluoxetine and paroxetine. The clinical consequences of fluoxetine’s interaction with CYP2D6 substrates may not be apparent for weeks after the drug is started, because of its very long half-life and slow generation of a CYP2D6-inhibiting metabolite.
6-Mercaptopurine is metabolized not only by TPMT but also by xanthine oxidase. When allopurinol, an inhibitor of xanthine oxidase, is administered with standard doses of azathioprine or 6-mercaptopurine, life-threatening toxicity (bone marrow suppression) can result.
A number of drugs are secreted by the renal tubular transport systems for organic anions. Inhibition of these systems can cause excessive drug accumulation. Salicylate, for example, reduces the renal clearance of methotrexate, an interaction that may lead to methotrexate toxicity. Renal tubular secretion contributes substantially to the elimination of penicillin, which can be inhibited (to increase its therapeutic effect) by probenecid. Similarly, inhibition of the tubular cation transport system by cimetidine decreases the renal clearance of dofetilide.
DRUG INTERACTIONS NOT MEDIATED BY CHANGES IN DRUG DISPOSITION
Drugs may act on separate components of a common process to generate effects greater than either has alone. Antithrombotic therapy with combinations of antiplatelet agents (glycoprotein IIb/IIIa inhibitors, aspirin, clopidogrel) and anticoagulants (warfarin, heparins) is often used in the treatment of vascular disease, although such combinations carry an increased risk of bleeding.
Nonsteroidal anti-inflammatory drugs (NSAIDs) cause gastric ulcers, and in patients treated with warfarin, the risk of upper gastrointestinal bleeding is increased almost threefold by concomitant use of an NSAID.
Indomethacin, piroxicam, and probably other NSAIDs antagonize the antihypertensive effects of β-adrenergic receptor blockers, diuretics, ACE inhibitors, and other drugs. The resulting elevation in blood pressure ranges from trivial to severe. This effect is not seen with aspirin and sulindac but has been found with the cyclooxygenase 2 (COX-2) inhibitor celecoxib.
Torsades des pointes ventricular tachycardia during administration of QT-prolonging antiarrhythmics (quinidine, sotalol, dofetilide) occurs much more frequently in patients receiving diuretics, probably reflecting hypokalemia. In vitro, hypokalemia not only prolongs the QT interval in the absence of drug but also potentiates drug block of ion channels that results in QT prolongation. Also, some diuretics have direct electrophysiologic actions that prolong QT.
The administration of supplemental potassium leads to more frequent and more severe hyperkalemia when potassium elimination is reduced by concurrent treatment with ACE inhibitors, spironolactone, amiloride, or triamterene.
The pharmacologic effects of sildenafil result from inhibition of the phosphodiesterase type 5 isoform that inactivates cyclic guanosine monophosphate (GMP) in the vasculature. Nitroglycerin and related nitrates used to treat angina produce vasodilation by elevating cyclic GMP. Thus, coadministration of these nitrates with sildenafil can cause profound hypotension, which can be catastrophic in patients with coronary disease.
Sometimes, combining drugs can increase overall efficacy and/or reduce drug-specific toxicity. Such therapeutically useful interactions are described in chapters dealing with specific disease entities.
ADVERSE REACTIONS TO DRUGS
The beneficial effects of drugs are coupled with the inescapable risk of untoward effects. The morbidity and mortality from these adverse effects often present diagnostic problems because they can involve every organ and system of the body and may be mistaken for signs of underlying disease. As well, some surveys have suggested that drug therapy for a range of chronic conditions such as psychiatric disease or hypertension does not achieve its desired goal in up to half of treated patients; thus, the most common “adverse” drug effect may be failure of efficacy.
Adverse reactions can be classified in two broad groups. One type results from exaggeration of an intended pharmacologic action of the drug, such as increased bleeding with anticoagulants or bone marrow suppression with antineoplastics. The second type of adverse reaction ensues from toxic effects unrelated to the intended pharmacologic actions. The latter effects are often unanticipated (especially with new drugs) and frequently severe and may result from recognized as well as previously undescribed mechanisms.
Drugs may increase the frequency of an event that is common in a general population, and this may be especially difficult to recognize; an excellent example is the increase in myocardial infarctions with the COX-2 inhibitor rofecoxib. Drugs can also cause rare and serious adverse effects, such as hematologic abnormalities, arrhythmias, severe skin reactions, or hepatic or renal dysfunction. Prior to regulatory approval and marketing, new drugs are tested in relatively few patients who tend to be less sick and to have fewer concomitant diseases than those patients who subsequently receive the drug therapeutically. Because of the relatively small number of patients studied in clinical trials and the selected nature of these patients, rare adverse effects are generally not detected prior to a drug’s approval; indeed, if they are detected, the new drugs are generally not approved. Therefore, physicians need to be cautious in the prescription of new drugs and alert for the appearance of previously unrecognized adverse events.
Elucidating mechanisms underlying adverse drug effects can assist development of safer compounds or allow a patient subset at especially high risk to be excluded from drug exposure. National adverse reaction reporting systems, such as those operated by the FDA (suspected adverse reactions can be reported online at http://www.fda.gov/safety/medwatch/default.htm) and the Committee on Safety of Medicines in Great Britain, can prove useful. The publication or reporting of a newly recognized adverse reaction can in a short time stimulate many similar such reports of reactions that previously had gone unrecognized.
Occasionally, “adverse” effects may be exploited to develop an entirely new indication for a drug. Unwanted hair growth during minoxidil treatment of severely hypertensive patients led to development of the drug for hair growth. Sildenafil was initially developed as an antianginal, but its effects to alleviate erectile dysfunction not only led to a new drug indication but also to increased understanding of the role of type 5 phosphodiesterase in erectile tissue. These examples further reinforce the concept that prescribers must remain vigilant to the possibility that unusual symptoms may reflect unappreciated drug effects.
Some 25–50% of patients make errors in self-administration of prescribed medicines, and these errors can be responsible for adverse drug effects. Similarly, patients commit errors in taking OTC drugs by not reading or following the directions on the containers. Health care providers must recognize that providing directions with prescriptions does not always guarantee compliance.
In hospitals, drugs are administered in a controlled setting, and patient compliance is, in general, ensured. Errors may occur nevertheless—the wrong drug or dose may be given or the drug may be given to the wrong patient—and improved drug distribution and administration systems are addressing this problem.
SCOPE OF THE PROBLEM
Patients receive, on average, 10 different drugs during each hospitalization. The sicker the patient, the more drugs are given, and there is a corresponding increase in the likelihood of adverse drug reactions. When <6 different drugs are given to hospitalized patients, the probability of an adverse reaction is ~5%, but if >15 drugs are given, the probability is >40%. Retrospective analyses of ambulatory patients have revealed adverse drug effects in 20%. Serious adverse reactions are also well-recognized with “herbal” remedies and OTC compounds; examples include kava-associated hepatotoxicity, L-tryptophan-associated eosinophilia-myalgia, and phenylpropanolamine-associated stroke, each of which has caused fatalities.
A small group of widely used drugs accounts for a disproportionate number of reactions. Aspirin and other NSAIDs, analgesics, digoxin, anticoagulants, diuretics, antimicrobials, glucocorticoids, antineoplastics, and hypoglycemic agents account for 90% of reactions.
TOXICITY UNRELATED TO A DRUG’S PRIMARY PHARMACOLOGIC ACTIVITY
Drugs or more commonly reactive metabolites generated by CYPs can covalently bind to tissue macromolecules (such as proteins or DNA) to cause tissue toxicity. Because of the reactive nature of these metabolites, covalent binding often occurs close to the site of production, typically the liver.
The most common cause of drug-induced hepatotoxicity is acetaminophen overdosage (Chap. 361). Normally, reactive metabolites are detoxified by combining with hepatic glutathione. When glutathione becomes depleted, the metabolites bind instead to hepatic protein, with resultant hepatocyte damage. The hepatic necrosis produced by the ingestion of acetaminophen can be prevented or attenuated by the administration of substances such as N-acetylcysteine that reduce the binding of electrophilic metabolites to hepatic proteins. The risk of acetaminophen-related hepatic necrosis is increased in patients receiving drugs such as phenobarbital or phenytoin, which increase the rate of drug metabolism, or ethanol, which exhausts glutathione stores. Such toxicity has even occurred with therapeutic dosages, so patients at risk through these mechanisms should be warned.
Most pharmacologic agents are small molecules with low molecular weights (<2000) and thus are poor immunogens. Generation of an immune response to a drug therefore usually requires in vivo activation and covalent linkage to protein, carbohydrate, or nucleic acid.
Drug stimulation of antibody production may mediate tissue injury by several mechanisms. The antibody may attack the drug when the drug is covalently attached to a cell and thereby destroy the cell. This occurs in penicillin-induced hemolytic anemia. Antibody-drug-antigen complexes may be passively adsorbed by a bystander cell, which is then destroyed by activation of complement; this occurs in quinine- and quinidine-induced thrombocytopenia. Heparin-induced thrombocytopenia arises when antibodies against complexes of platelet factor 4 peptide and heparin generate immune complexes that activate platelets; thus, the thrombocytopenia is accompanied by “paradoxical” thrombosis and is treated with thrombin inhibitors. Drugs or their reactive metabolites may alter a host tissue, rendering it antigenic and eliciting autoantibodies. For example, hydralazine and procainamide (or their reactive metabolites) can chemically alter nuclear material, stimulating the formation of antinuclear antibodies and occasionally causing lupus erythematosus. Drug-induced pure red cell aplasia (Chap. 130) is due to an immune-based drug reaction.
Serum sickness (Chap. 376) results from the deposition of circulating drug-antibody complexes on endothelial surfaces. Complement activation occurs, chemotactic factors are generated locally, and an inflammatory response develops at the site of complex entrapment. Arthralgias, urticaria, lymphadenopathy, glomerulonephritis, or cerebritis may result. Foreign proteins (vaccines, streptokinase, therapeutic antibodies) and antibiotics are common causes. Many drugs, particularly antimicrobial agents, ACE inhibitors, and aspirin, can elicit anaphylaxis with production of IgE, which binds to mast cell membranes. Contact with a drug antigen initiates a series of biochemical events in the mast cell and results in the release of mediators that can produce the characteristic urticaria, wheezing, flushing, rhinorrhea, and (occasionally) hypotension.
Drugs may also elicit cell-mediated immune responses. Topically administered substances may interact with sulfhydryl or amino groups in the skin and react with sensitized lymphocytes to produce the rash characteristic of contact dermatitis. Other types of rashes may also result from the interaction of serum factors, drugs, and sensitized lymphocytes.
DIAGNOSIS AND TREATMENT OF ADVERSE DRUG REACTIONS
The manifestations of drug-induced diseases frequently resemble those of other diseases, and a given set of manifestations may be produced by different and dissimilar drugs. Recognition of the role of a drug or drugs in an illness depends on appreciation of the possible adverse reactions to drugs in any disease, on identification of the temporal relationship between drug administration and development of the illness, and on familiarity with the common manifestations of the drugs. A suspected adverse drug reaction developing after introduction of a new drug naturally implicates that drug; however, it is also important to remember that a drug interaction may be responsible. Thus, for example, a patient on a chronic stable warfarin dose may develop a bleeding complication after introduction of amiodarone; this does not reflect a direct reaction to amiodarone but rather its effect to inhibit warfarin metabolism. Many associations between particular drugs and specific reactions have been described, but there is always a “first time” for a novel association, and any drug should be suspected of causing an adverse effect if the clinical setting is appropriate.
Illness related to a drug’s intended pharmacologic action is often more easily recognized than illness attributable to immune or other mechanisms. For example, side effects such as cardiac arrhythmias in patients receiving digitalis, hypoglycemia in patients given insulin, or bleeding in patients receiving anticoagulants are more readily related to a specific drug than are symptoms such as fever or rash, which may be caused by many drugs or by other factors.
Electronic listings of adverse drug reactions can be useful. However, exhaustive compilations often provide little sense of perspective in terms of frequency and seriousness, which can vary considerably among patients.
Eliciting a drug history from each patient is important for diagnosis. Attention must be directed to OTC drugs and herbal preparations as well as to prescription drugs. Each type can be responsible for adverse drug effects, and adverse interactions may occur between OTC drugs and prescribed drugs. Loss of efficacy of oral contraceptives or cyclosporine with concurrent use of St. John’s wort (a P-glycoprotein inducer) is an example. In addition, it is common for patients to be cared for by several physicians, and duplicative, additive, antagonistic, or synergistic drug combinations may therefore be administered if the physicians are not aware of the patients’ drug histories. Every physician should determine what drugs a patient has been taking, for the previous month or two ideally, before prescribing any medications. Medications stopped for inefficacy or adverse effects should be documented to avoid pointless and potentially dangerous reexposure. A frequently overlooked source of additional drug exposure is topical therapy; for example, a patient complaining of bronchospasm may not mention that an ophthalmic beta blocker is being used unless specifically asked. A history of previous adverse drug effects in patients is common. Since these patients have shown a predisposition to drug-induced illnesses, such a history should dictate added caution in prescribing new drugs.
Laboratory studies may include demonstration of serum antibody in some persons with drug allergies involving cellular blood elements, as in agranulocytosis, hemolytic anemia, and thrombocytopenia. For example, both quinine and quinidine can produce platelet agglutination in vitro in the presence of complement and the serum from a patient who has developed thrombocytopenia following use of this drug. Biochemical abnormalities such as G6PD deficiency, serum pseudocholinesterase level, or genotyping may also be useful in diagnosis, often after an adverse effect has occurred in the patient or a family member.
Once an adverse reaction is suspected, discontinuation of the suspected drug followed by disappearance of the reaction is presumptive evidence of a drug-induced illness. Confirming evidence may be sought by cautiously reintroducing the drug and seeing if the reaction reappears. However, that should be done only if confirmation would be useful in the future management of the patient and if the attempt would not entail undue risk. With concentration-dependent adverse reactions, lowering the dosage may cause the reaction to disappear, and raising it may cause the reaction to reappear. When the reaction is thought to be allergic, however, readministration of the drug may be hazardous, since anaphylaxis may develop.
If the patient is receiving many drugs when an adverse reaction is suspected, the drugs likeliest to be responsible can usually be identified; this should include both potential culprit agents as well as drugs that alter their elimination. All drugs may be discontinued at once or, if this is not practical, discontinued one at a time, starting with the ones most suspect, and the patient observed for signs of improvement. The time needed for a concentration-dependent adverse effect to disappear depends on the time required for the concentration to fall below the range associated with the adverse effect; that, in turn, depends on the initial blood level and on the rate of elimination or metabolism of the drug. Adverse effects of drugs with long half-lives or those not directly related to serum concentration may take a considerable time to disappear.
SUMMARY
Modern clinical pharmacology aims to replace empiricism in the use of drugs with therapy based on in-depth understanding of factors that determine an individual’s response to drug treatment. Molecular pharmacology, pharmacokinetics, genetics, clinical trials, and the educated prescriber all contribute to this process. No drug response should ever be termed idiosyncratic; all responses have a mechanism whose understanding will help guide further therapy with that drug or successors. This rapidly expanding understanding of variability in drug actions makes the process of prescribing drugs increasingly daunting for the practitioner. However, fundamental principles should guide this process:
• The benefits of drug therapy, however defined, should always outweigh the risk.
• The smallest dosage necessary to produce the desired effect should be used.
• The number of medications and doses per day should be minimized.
• Although the literature is rapidly expanding, accessing it is becoming easier; electronic tools to search databases of literature and unbiased opinion will become increasingly commonplace.
• Genetics play a role in determining variability in drug response and may become a part of clinical practice.
• Electronic medical record and pharmacy systems will increasingly incorporate prescribing advice, such as indicated medications not used; unindicated medications being prescribed; and potential dosing errors, drug interactions, or genetically determined drug responses.
• Prescribers should be particularly wary when adding or stopping specific drugs that are especially liable to provoke interactions and adverse reactions.
• Prescribers should use only a limited number of drugs, with which they are thoroughly familiar.
6e |
Women’s Health |
The National Institutes of Health’s Office of Research on Women’s Health celebrated its twentieth anniversary in 2010 with a new strategic plan recognizing the study of the biologic basis of sex differences as a distinct scientific discipline. It has become clear that both sex chromosomes and sex hormones contribute to these differences. Indeed, it is recommended that the term sex difference be used for biologic processes that differ between males and females and the term gender difference be used for features related to social influences. The clinical discipline of women’s health emphasizes greater attention to patient education and involvement in disease prevention and medical decision-making and has become a model for patient-centered health care.
DISEASE RISK: REALITY AND PERCEPTION
The leading causes of death are the same in women and men: (1) heart disease, and (2) cancer (Table 6e-1; Fig. 6e-1). The leading cause of cancer death, lung cancer, is the same in both sexes. Breast cancer is the second leading cause of cancer death in women, but it causes about 60% fewer deaths than does lung cancer. Men are substantially more likely to die from suicide and accidents than are women.
|
DEATHS AND PERCENTAGE OF TOTAL DEATHS FOR THE LEADING CAUSES OF DEATH BY SEX IN THE UNITED STATES IN 2010 |
FIGURE 6e-1 Death rates per 100,000 population for 2007 by 5-year age groups in U.S. women. Note that the scale of the y axis is increased in the graph on the right compared with that on the left. Accidents and HIV/AIDS are the leading causes of death in young women 20–34 years of age. Accidents, breast cancer, and ischemic heart disease (IHD) are the leading causes of death in women 35–49 years of age. IHD becomes the leading cause of death in women beginning at age 50 years. In older women, IHD remains the leading cause of death, cerebrovascular disease becomes the second leading cause of death, and lung cancer is the leading cause of cancer-related deaths. At age 85 years and beyond, Alzheimer’s disease (AD) becomes the third leading cause of death. Ca, cancer; CLRD, chronic lower respiratory disease; DM, diabetes mellitus. (Data adapted from Centers for Disease Control and Prevention, http://www.cdc.gov/nchs/data/dvs/MortFinal2007_WorkTable210R.pdf.)
Women’s risk for many diseases increases at menopause, which occurs at a median age of 51.4 years. In the industrialized world, women spend one-third of their lives in the postmenopausal period. Estrogen levels fall abruptly at menopause, inducing a variety of physiologic and metabolic responses. Rates of cardiovascular disease (CVD) increase and bone density begins to decrease rapidly after menopause. In the United States, women live on average about 5 years longer than men, with a life expectancy at birth in 2011 of 81.1 years compared with 76.3 years in men. Elderly women outnumber elderly men, so that age-related conditions such as hypertension have a female preponderance. However, the difference in life expectancy between men and women has decreased an average of 0.1 year every year since its peak of 7.8 years in 1979. If this convergence in mortality figures continues, it is projected that mortality rates will be similar by 2054.
Public awareness campaigns have resulted in a marked increase in the percentage of U.S. women knowing that CVD is the leading cause of death in women. In 1997, the majority of U.S. women surveyed thought that cancer (35%) rather than heart disease (30%) was the leading cause of death in women (Fig. 6e-2). In 2012, these perceptions were reversed, with 56% of U.S. women surveyed recognizing that heart disease rather than cancer (24%) was the leading cause of death in women (Fig. 6e-2). Although awareness of heart disease has improved substantially among black and Hispanic women over this time period, these groups were 66% less likely than white women to recognize that heart disease is the leading cause of death in women.
FIGURE 6e-2 Changes in perceived leading causes of death among women surveyed in 1997 compared with those surveyed in 2012. In 1997, cancer was cited as the leading cause of death in women, not heart disease. In 2012, this trend had reversed. The rate of awareness that heart disease is the leading cause of death in women was significantly higher in 2012 (56% vs 30%, p <.001) than in 1997. (Data adapted from L Mosca et al: Circulation 127:1254, 2013.)
Nevertheless, women younger than 65 years still consider breast cancer to be their leading health risk, despite the fact that death rates from breast cancer have been falling since the 1990s. In any specific decade of life, a woman’s risk for breast cancer never exceeds 1 in 34. Although a woman’s lifetime risk of developing breast cancer if she lives past 85 years is about 1 in 9, it is much more likely that she will die from CVD than from breast cancer. In other words, many elderly women have breast cancer but die from other causes. Similarly, a minority of women are aware that lung cancer is the leading cause of cancer death in women. Physicians are also less likely to recognize women’s risk for CVD. Even in 2012, only 21% of U.S. women surveyed reported that their physicians had counseled them about their risk for heart disease. These misconceptions are unfortunate as they perpetuate inadequate attention to modifiable risk factors such as dyslipidemia, hypertension, and cigarette smoking.
SEX DIFFERENCES IN HEALTH AND DISEASE
ALZHEIMER’S DISEASE
(See also Chap. 448) Alzheimer’s disease (AD) affects approximately twice as many women as men. Because the risk for AD increases with age, part of this sex difference is accounted for by the fact that women live longer than men. However, additional factors probably contribute to the increased risk for AD in women, including sex differences in brain size, structure, and functional organization. There is emerging evidence for sex-specific differences in gene expression, not only for genes on the × and Y chromosomes but also for some autosomal genes. Estrogens have pleiotropic genomic and nongenomic effects on the central nervous system, including neurotrophic actions in key areas involved in cognition and memory. Women with AD have lower endogenous estrogen levels than do women without AD. These observations have led to the hypothesis that estrogen is neuroprotective.
Some studies have suggested that estrogen administration improves cognitive function in nondemented postmenopausal women as well as in women with AD, and several observational studies have suggested that postmenopausal hormone therapy (HT) may decrease the risk of AD. However, HT placebo-controlled trials have found no improvement in disease progression or cognitive function in women with AD. Further, the Women’s Health Initiative Memory Study (WHIMS), an ancillary study in the Women’s Health Initiative (WHI), found no benefit compared with placebo of estrogen alone [combined continuous equine estrogen (CEE), 0.625 mg daily] or estrogen with progestin [CEE, 0.625 mg daily, and medroxyprogesterone acetate (MPA), 2.5 mg daily] on cognitive function or the development of dementia in women ≥65 years. Indeed, there was a significantly increased risk for both dementia and mild cognitive impairment in women receiving HT. However, preliminary findings from the Kronos Early Estrogen Prevention Study (KEEPS), a randomized clinical trial of early initiation of HT after menopause that compared CEE 0.45 mg daily, 50 μg of weekly transdermal estradiol (both estrogen arms included cyclic oral micronized progesterone 200 mg daily for 12 days each month), or placebo, found no adverse effects on cognitive function.
CARDIOVASCULAR DISEASE AND STROKE
(See also Chap. 293) There are major sex differences in CVD, the leading cause of death in men and women in developed countries. A greater number of U.S. women than men die annually of CVD and stroke. Deaths from CVD have decreased markedly in men since 1980, whereas CVD deaths only began to decrease substantially in women beginning in 2000. However, in middle-aged women, the prevalence rates of both coronary heart disease (CHD) and stroke have increased in the 1999–2004 National Health and Nutrition Survey (NHANES) compared to the 1988–1994 NHANES, whereas prevalence rates have decreased or remained unchanged, respectively, in men. These increases were paralleled by an increasing prevalence of abdominal obesity and other components of metabolic syndrome in women.
Sex steroids have major effects on the cardiovascular system and lipid metabolism. Estrogen increases high-density lipoprotein (HDL) and lowers low-density lipoprotein (LDL), whereas androgens have the opposite effect. Estrogen has direct vasodilatory effects on the vascular endothelium, enhances insulin sensitivity, and has antioxidant and anti-inflammatory properties. There is a striking increase in CHD after both natural and surgical menopause, suggesting that endogenous estrogens are cardioprotective. Women also have longer QT intervals on electrocardiograms, and this increases their susceptibility to certain arrhythmias.
CHD presents differently in women, who are usually 10–15 years older than their male counterparts and are more likely to have comorbidities such as hypertension, congestive heart failure, and diabetes mellitus (DM). In the Framingham study, angina was the most common initial symptom of CHD in women, whereas myocardial infarction (MI) was the most common initial presentation in men. Women more often have atypical symptoms such as nausea, vomiting, indigestion, and upper back pain. Although awareness that heart disease is the leading cause of death in women has nearly doubled over the last 15 years, women remain less aware that its symptoms are often atypical, and they are less likely to contact 9-1-1 when they experience such symptoms.
Women with MI are more likely to present with cardiac arrest or cardiogenic shock, whereas men are more likely to present with ventricular tachycardia. Further, younger women with MI are more likely to die than are men of similar age. However, this mortality gap has decreased substantially in recent years because younger women have experienced greater improvements in survival after MI than men (Fig. 6e-3). The improvement in survival is due largely to a reduction in comorbidities, suggesting a greater attention to modifiable risk factors in women.
FIGURE 6e-3 Hospital mortality rates in men and women for acute myocardial infarction (MI) in 1994–1995 compared with 2004–2006. Women younger than age 65 years had substantially greater mortality than men of similar age in 1994–1995. Mortality rates declined markedly for both sexes across all age groups in 2004–2006 compared with 1994–1995. However, there was a more striking decrease in mortality in women younger than age 75 years compared with men of similar age. The mortality rate reduction was largest in women less than age 55 years (52.9%) and lowest in men of similar age (33.3%). (Data adapted from V Vaccarino et al: Arch Intern Med 169:1767, 2009.)
Nevertheless, physicians are less likely to suspect heart disease in women with chest pain and less likely to perform diagnostic and therapeutic cardiac procedures in women. Women are less likely to receive therapies such as angioplasty, thrombolytic therapy, coronary artery bypass grafts (CABGs), beta blockers, and aspirin. There are also sex differences in outcomes when women with CHD do receive therapeutic interventions. Women undergoing CABG surgery have more advanced disease, a higher perioperative mortality rate, less relief of angina, and less graft patency; however, 5- and 10-year survival rates are similar. Women undergoing percutaneous transluminal coronary angioplasty have lower rates of initial angiographic and clinical success than men, but they also have a lower rate of restenosis and a better long-term outcome. Women may benefit less and have more frequent serious bleeding complications from thrombolytic therapy compared with men. Factors such as older age, more comorbid conditions, smaller body size, and more severe CHD in women at the time of events or procedures account in part for the observed sex differences.
Elevated cholesterol levels, hypertension, smoking, obesity, low HDL cholesterol levels, DM, and lack of physical activity are important risk factors for CHD in both men and women. Total triglyceride levels are an independent risk factor for CHD in women but not in men. Low HDL cholesterol and DM are more important risk factors for CHD in women than in men. Smoking is an important risk factor for CHD in women—it accelerates atherosclerosis, exerts direct negative effects on cardiac function, and is associated with an earlier age of menopause. Cholesterol-lowering drugs are equally effective in men and women for primary and secondary prevention of CHD. However, because of perceptions that women are at lower risk for CHD, they receive fewer interventions for modifiable risk factors than do men. In contrast to men, randomized trials showed that aspirin was not effective in the primary prevention of CHD in women; it did significantly reduce the risk of ischemic stroke.
The sex differences in CHD prevalence, beneficial biologic effects of estrogen on the cardiovascular system, and reduced risk for CHD in observational studies led to the hypothesis that HT was cardioprotective. However, the WHI, which studied more than 16,000 women on CEE plus MPA or placebo and more than 10,000 women with hysterectomy on CEE alone or placebo, did not demonstrate a benefit of HT for the primary or secondary prevention of CHD. In addition, CEE plus MPA was associated with an increased risk for CHD, particularly in the first year of therapy, whereas CEE alone neither increased nor decreased CHD risk. Both CEE plus MPA and CEE alone were associated with an increased risk for ischemic stroke.
In the WHI, there was a suggestion of a reduction in CHD risk in women who initiated HT closer to menopause. This finding suggests that the time at which HT is initiated is critical for cardioprotection. According to this “timing” hypothesis, HT has differential effects, depending on the stage of atherosclerosis; adverse effects are seen with advanced, unstable lesions. A recent study using data from the Danish Osteoporosis Prevention Study (DOPS), an open-label randomized trial of triphasic oral estradiol compared with no treatment in recently menopausal or perimenopausal women (a cyclic oral synthetic progestin, norethisterone acetate, was added in women who had a uterus), found significantly reduced mortality and CVD after 10 years of HT. However, DOPS was designed to investigate HT for the primary prevention of osteoporotic bone fractures, and CVD outcomes were not prespecified endpoints. Further, there were relatively few CVD events in the study groups.
KEEPS was designed to directly test the “timing” hypothesis. Seven hundred twenty-seven recently menopausal women age 42–58 years (mean 52.7 years) were randomized to oral CEE (lower dose than WHI), transdermal estradiol, or placebo for 4 years; both estrogen arms included oral cyclical micronized progesterone (see above section on AD for dosing details). There were no significant beneficial or deleterious effects on the progression of atherosclerosis by computed tomography assessment of coronary artery calcification in either HT arm. Adverse events including stroke, MI, venous thromboembolism, and breast cancer were not increased in the HT arms compared with the placebo arm. There were improvements in hot flashes, night sweats, mood, sexual function, and bone density in the HT arms. This relatively small study does not suggest that early HT administration, transdermally or orally, reduces atherosclerosis. However, the study suggests that short-term HT may be safely administered for symptom relief in recently menopausal women. HT is discussed further in Chap. 413.
DIABETES MELLITUS
(See also Chap. 417) Women are more sensitive to insulin than men are. Despite this, the prevalence of type 2 DM is similar in men and women. There is a sex difference in the relationship between endogenous androgen levels and DM risk. Higher bioavailable testosterone levels are associated with increased risk in women, whereas lower bioavailable testosterone levels are associated with increased risk in men. Polycystic ovary syndrome and gestational DM—common conditions in premenopausal women—are associated with a significantly increased risk for type 2 DM. Premenopausal women with DM lose the cardioprotective effect of female sex and have rates of CHD identical to those in males. These women have impaired endothelial function and reduced coronary vasodilatory responses, which may predispose to cardiovascular complications. Among individuals with DM, women have a greater risk for MI than do men. Women with DM are more likely to have left ventricular hypertrophy. Women with DM receive less aggressive treatment for modifiable CHD risk factors than men with DM. In the WHI, CEE plus MPA significantly reduced the incidence of DM, whereas with CEE alone, there was only a trend toward decreased DM incidence.
HYPERTENSION
(See also Chap. 298) After age 60, hypertension is more common in U.S. women than in men, largely because of the high prevalence of hypertension in older age groups and the longer survival of women. Isolated systolic hypertension is present in 30% of women >60 years old. Sex hormones affect blood pressure. Both normotensive and hypertensive women have higher blood pressure levels during the follicular phase than during the luteal phase. In the Nurses’ Health Study, the relative risk of hypertension was 1.8 in current users of oral contraceptives, but this risk is lower with the newer low-dose contraceptive preparations. HT is not associated with hypertension. Among secondary causes of hypertension, there is a female preponderance of renal artery fibromuscular dysplasia.
The benefits of treatment for hypertension have been dramatic in both women and men. A meta-analysis of the effects of hypertension treatment, the Individual Data Analysis of Antihypertensive Intervention Trial, found a reduction of risk for stroke and for major cardiovascular events in women. The effectiveness of various antihypertensive drugs appears to be comparable in women and men; however, women may experience more side effects. For example, women are more likely to develop cough with angiotensin-converting enzyme inhibitors.
AUTOIMMUNE DISORDERS
(See also Chap. 377e) Most autoimmune disorders occur more commonly in women than in men; they include autoimmune thyroid and liver diseases, lupus, rheumatoid arthritis (RA), scleroderma, multiple sclerosis (MS), and idiopathic thrombocytopenic purpura. However, there is no sex difference in the incidence of type 1 DM, and ankylosing spondylitis occurs more commonly in men. Women may be more resistant to bacterial infections than men. Sex differences in both immune responses and adverse reactions to vaccines have been reported. For example, there is a female preponderance of postvaccination arthritis.
Adaptive immune responses are more robust in women than in men; this may be explained by the stimulatory actions of estrogens and the inhibitory actions of androgens on the cellular mediators of immunity. Consistent with an important role for sex hormones, there is variation in immune responses during the menstrual cycle, and the activity of certain autoimmune disorders is altered by castration or pregnancy (e.g., RA and MS may remit during pregnancy). Nevertheless, the majority of studies show that exogenous estrogens and progestins in the form of HT or oral contraceptives do not alter autoimmune disease incidence or activity. Exposure to fetal antigens, including circulating fetal cells that persist in certain tissues, has been speculated to increase the risk of autoimmune responses. There is clearly an important genetic component to autoimmunity, as indicated by the familial clustering and HLA association of many such disorders. × chromosome genes also contribute to sex differences in immunity. Indeed, nonrandom × chromosome inactivation may be a risk factor for autoimmune diseases.
HIV INFECTION
(See also Chap. 226) Women account for almost 50% of the 34 million persons infected with HIV-1 worldwide. AIDS is an important cause of death in younger women (Fig. 6e-1). Heterosexual contact with an at-risk partner is the fastest-growing transmission category, and women are more susceptible to HIV infection than are men. This increased susceptibility is accounted for in part by an increased prevalence of sexually transmitted diseases in women. Some studies have suggested that hormonal contraceptives may increase the risk of HIV transmission. Progesterone has been shown to increase susceptibility to infection in nonhuman primate models of HIV. Women are also more likely to be infected by multiple variants of the virus than are men. Women with HIV have more rapid decreases in their CD4 cell counts than do men. Compared with men, HIV-infected women more frequently develop candidiasis, but Kaposi’s sarcoma is less common than it is in men. Women have more adverse reactions, such as lipodystrophy, dyslipidemia, and rash, with antiretroviral therapy than do men. This observation is explained in part by sex differences in the pharmacokinetics of certain antiretroviral drugs, resulting in higher plasma concentrations in women.
OBESITY
(See also Chap. 416) The prevalence of both obesity (body mass index ≥30 kg/m2) and abdominal obesity (waist circumference ≥88 cm in women) is higher in U.S. women than in men. However, between 1999 and 2008, the prevalence of obesity increased significantly in men but not in women. The prevalence of abdominal obesity increased over this time period in both sexes. More than 80% of patients who undergo bariatric surgery are women. Pregnancy and menopause are risk factors for obesity.
There are major sex differences in body fat distribution. Women characteristically have gluteal and femoral or gynoid pattern of fat distribution, whereas men typically have a central or android pattern. Women have more subcutaneous fat than men. In women, endogenous androgen levels are positively associated with abdominal obesity, and androgen administration increases visceral fat. In contrast, there is an inverse relationship between endogenous androgen levels and abdominal obesity in men. Further, androgen administration decreases visceral fat in these obese men. The reasons for these sex differences in the relationship between visceral fat and androgens are unknown. Studies in humans also suggest that sex steroids play a role in modulating food intake and energy expenditure.
In men and women, abdominal obesity characterized by increased visceral fat is associated with an increased risk for CVD and DM. Obesity increases a woman’s risk for certain cancers, in particular postmenopausal breast and endometrial cancer, in part because adipose tissue provides an extragonadal source of estrogen through aromatization of circulating adrenal and ovarian androgens, especially the conversion of androstenedione to estrone. Obesity increases the risk of infertility, miscarriage, and complications of pregnancy.
OSTEOPOROSIS
(See also Chap. 425) Osteoporosis is about five times more common in postmenopausal women than in age-matched men, and osteoporotic hip fractures are a major cause of morbidity in elderly women. Men accumulate more bone mass and lose bone more slowly than do women. Sex differences in bone mass are found as early as infancy. Calcium intake, vitamin D, and estrogen all play important roles in bone formation and bone loss. Particularly during adolescence, calcium intake is an important determinant of peak bone mass. Vitamin D deficiency is surprisingly common in elderly women, occurring in >40% of women living in northern latitudes. Receptors for estrogens and androgens have been identified in bone. Estrogen deficiency is associated with increased osteoclast activity and a decreased number of bone-forming units, leading to net bone loss. The aromatase enzyme, which converts androgens to estrogens, is also present in bone. Estrogen is an important determinant of bone mass in men (derived from the aromatization of androgens) as well as in women.
PHARMACOLOGY
On average, women have lower body weights, smaller organs, a higher percentage of body fat, and lower total-body water than men. There are also important sex differences in drug action and metabolism that are not accounted for by these differences in body size and composition. Sex steroids alter the binding and metabolism of a number of drugs. Further, menstrual cycle phase and pregnancy can alter drug action. Two-thirds of cases of drug-induced torsades des pointes, a rare, life-threatening ventricular arrhythmia, occur in women because they have a longer, more vulnerable QT interval. These drugs, which include certain antihistamines, antibiotics, antiarrhythmics, and antipsychotics, can prolong cardiac repolarization by blocking cardiac voltage-gated potassium channels. Women require lower doses of neuroleptics to control schizophrenia. Women awaken from anesthesia faster than do men given the same doses of anesthetics. Women also take more medications than men, including over-the-counter formulations and supplements. The greater use of medications combined with these biologic differences may account for the reported higher frequency of adverse drug reactions in women than in men.
PSYCHOLOGICAL DISORDERS
(See also Chap. 466) Depression, anxiety, and affective and eating disorders (bulimia and anorexia nervosa) are more common in women than in men. Epidemiologic studies from both developed and developing nations consistently find major depression to be twice as common in women as in men, with the sex difference becoming evident in early adolescence. Depression occurs in 10% of women during pregnancy and in 10–15% of women during the postpartum period. There is a high likelihood of recurrence of postpartum depression with subsequent pregnancies. The incidence of major depression diminishes after age 45 years and does not increase with the onset of menopause. Depression in women appears to have a worse prognosis than does depression in men; episodes last longer, and there is a lower rate of spontaneous remission. Schizophrenia and bipolar disorders occur at equal rates in men and women, although there may be sex differences in symptoms.
Both biologic and social factors account for the greater prevalence of depressive disorders in women. Men have higher levels of the neurotransmitter serotonin. Sex steroids also affect mood, and fluctuations during the menstrual cycle have been linked to symptoms of premenstrual syndrome. Sex hormones differentially affect the hypothalamic-pituitary-adrenal responses to stress. Testosterone appears to blunt cortisol responses to corticotropin-releasing hormone. Both low and high levels of estrogen can activate the hypothalamic-pituitary-adrenal axis.
SLEEP DISORDERS
(See also Chap. 38) There are striking sex differences in sleep and its disorders. During sleep, women have an increased amount of slow-wave activity, differences in timing of delta activity, and an increase in the number of sleep spindles. Testosterone modulates neural control of breathing and upper airway mechanics. Men have a higher prevalence of sleep apnea. Testosterone administration to hypogonadal men as well as to women increases apneic episodes during sleep. Women with the hyperandrogenic disorder polycystic ovary syndrome have an increased prevalence of obstructive sleep apnea, and apneic episodes are positively correlated with their circulating testosterone levels. In contrast, progesterone accelerates breathing, and in the past, progestins were used for treatment of sleep apnea.
SUBSTANCE ABUSE AND TOBACCO
(See also Chaps. 467 and 470) Substance abuse is more common in men than in women. However, one-third of Americans who suffer from alcoholism are women. Women alcoholics are less likely to be diagnosed than men. A greater proportion of men than women seek help for alcohol and drug abuse. Men are more likely to go to an alcohol or drug treatment facility, whereas women tend to approach a primary care physician or mental health professional for help under the guise of a psychosocial problem. Late-life alcoholism is more common in women than in men. On average, alcoholic women drink less than alcoholic men but exhibit the same degree of impairment. Blood alcohol levels are higher in women than in men after drinking equivalent amounts of alcohol, adjusted for body weight. This greater bioavailability of alcohol in women is due to both the smaller volume of distribution and the slower gastric metabolism of alcohol secondary to lower activity of gastric alcohol dehydrogenase than is the case in men. In addition, alcoholic women are more likely to abuse tranquilizers, sedatives, and amphetamines. Women alcoholics have a higher mortality rate than do nonalcoholic women and alcoholic men. Women also appear to develop alcoholic liver disease and other alcohol-related diseases with shorter drinking histories and lower levels of alcohol consumption. Alcohol abuse also poses special risks to a woman, adversely affecting fertility and the health of the baby (fetal alcohol syndrome). Even moderate alcohol use increases the risk of breast cancer, hypertension, and stroke in women.
More men than women smoke tobacco, but this sex difference continues to decrease. Women have a much larger burden of smoking-related disease. Smoking markedly increases the risk of CVD in premenopausal women and is also associated with a decrease in the age of menopause. Women who smoke are more likely to develop chronic obstructive pulmonary disease and lung cancer than men and at lower levels of tobacco exposure. Postmenopausal women who smoke have lower bone density than women who never smoked. Smoking during pregnancy increases the risk of preterm deliveries and low birth weight infants.
VIOLENCE AGAINST WOMEN
More than one in three women in the United States have experienced rape, physical violence, and/or stalking by an intimate partner. Adult women are much more likely to be raped by a spouse, ex-spouse, or acquaintance than by a stranger. Domestic or intimate partner violence is a leading cause of death among young women. Domestic violence may be an unrecognized feature of certain clinical presentations, such as chronic abdominal pain, headaches, and eating disorders, in addition to more obvious manifestations such as trauma. Intimate partner violence is an important risk factor for depression, substance abuse, and suicide in women. Screening instruments can accurately identify women experiencing intimate partner violence. Such screening by health care providers is acceptable to women in settings ensuring adequate privacy and safety.
SUMMARY
Women’s health is now a mature discipline, and the importance of sex differences in biologic processes is well recognized. There has been a striking reduction in the excess mortality rate from MI in younger women. Nevertheless, ongoing misperceptions about disease risk, not only among women but also among their physicians, result in inadequate attention to modifiable risk factors. Research into the fundamental mechanisms of sex differences will provide important biologic insights. Further, those insights will have an impact on both women’s and men’s health.
7e Men’s Health
The emergence of men’s health as a distinct discipline within internal medicine is founded on the evidence that men and women differ across their life span in their susceptibility to disease, in the clinical manifestations of the disease, and in their response to treatment. Furthermore, men and women weigh the health consequences of illness differently and have different motivations for seeking care. Men and women experience different types of disparities in access to health care services and in the manner in which health care is delivered to them because of a complex array of socioeconomic and cultural factors. Attitudinal and institutional barriers to accessing care, fear and embarrassment due to the perception by some that it is not manly to seek medical help, and reticence on the part of patients and physicians to discuss issues related to sexuality, drug use, and aging have heightened the need for programs tailored to address the specific health needs of men.
Sex differences in disease prevalence, susceptibility, and clinical manifestations of disease were discussed in Chap. 6e (“Women’s Health”). It is notable that the two leading causes of death in both men and women—heart disease and cancer—are the same. However, men have a higher prevalence of neurodevelopmental and degenerative disorders; substance abuse disorders, including the use of performance-enhancing drugs and alcohol dependence; diabetes; and cardiovascular disease; and women have a higher prevalence of autoimmune disorders, depression, rheumatologic disorders, and osteoporosis. Men are substantially more likely to die from accidents, suicides, and homicides than women. Among men 15–34 years of age, unintentional injuries, homicides, and suicides account for over three-fourths of all deaths. Among men 35–64 years of age, heart disease, cancer, and unintentional injuries are the leading causes of death. Among men 65 years of age or older, heart disease, cancer, lower respiratory tract infections, and stroke are the major causes of death.
The biologic bases of sex differences in disease susceptibility, progression, and manifestation remain incompletely understood and are likely multifactorial. Undoubtedly, sex-specific differences in the genetic architecture and circulating sex hormones influence disease phenotype; additionally, epigenetic effects of sex hormones during fetal life, early childhood, and pubertal development may imprint sexual and nonsexual behaviors, body composition, and disease susceptibility. Reproductive load and physiologic changes during pregnancy, including profound hormonal and metabolic shifts and microchimerism (transfer of cells from the mother to the fetus and from the fetus to the mother), may affect disease susceptibility and disease severity in women. Sociocultural norms of child-rearing practices, societal expectations of gender roles, and the long-term economic impact of these practices and gender roles also may affect disease risk and its clinical manifestation. The trajectories of age-related changes in sex hormones during the reproductive and postreproductive years vary substantially between men and women and may influence the sex differences in the temporal evolution of age-related conditions such as osteoporosis, breast cancer, and autoimmune disease.
In a reflection of the growing attention to issues related to men’s health, health clinics focused on the health problems of men are being established with increasing frequency. Although the major threats to men’s health have not changed—heart disease, cancer, and unintentional injury continue to dominate the list of major medical causes of morbidity and mortality in men—the men who attend men’s health clinics do so largely for sexual, reproductive, and urologic health concerns involving common conditions such as androgen deficiency syndromes, age-related decline in testosterone levels, sexual dysfunction, muscle dysmorphia and anabolic-androgenic steroid use, lower urinary tract symptoms, and medical complications of prostate cancer therapy, which are the focus of this chapter. Additionally, new categories of body image disorders have emerged in men that had not been recognized until the 1980s, such as body dysmorphia syndrome and the use of performance-enhancing drugs to increase muscularity and lean appearance. Although menopause in women has been the subject of intense investigation for more than five decades, the issues that are specific to men’s health are just beginning to gain the attention that they deserve because of their high prevalence and impact on overall health, well-being, and quality of life.
AGING-RELATED CHANGES IN MALE REPRODUCTIVE FUNCTION (SEE CHAP. 411)
A number of studies have established that testosterone concentrations decrease with advancing age. This age-related decline starts in the third decade of life and progresses thereafter (Fig. 7e-1). Low total and bioavailable testosterone concentrations are associated with decreased skeletal muscle mass and strength, higher visceral fat mass, insulin resistance, and increased risk of coronary artery disease and mortality (Table 7e-1). Most studies suggest that these symptoms and signs develop with total testosterone levels below 320 ng/dL and free testosterone levels below 64 pg/mL in older men. Testing for low testosterone in older men should be limited to those with symptoms or signs attributable to androgen deficiency.
FIGURE 7e-1 Age-related decline in total testosterone levels. Total testosterone levels measured using liquid chromatography tandem mass spectrometry in men of the Framingham Heart Study (FHS), the European Male Aging Study (EMAS), and the Osteoporotic Fractures in Men Study (MrOS). (Reproduced with permission from S Bhasin et al: J Clin Endocrinol Metab 96:2430, 2011.)
|
ASSOCIATION OF TESTOSTERONE LEVELS WITH OUTCOMES IN OLDER MEN |
Testosterone therapy of healthy older men with low testosterone increases lean body mass, grip strength, and self-reported physical function (Fig. 7e-2). Testosterone therapy also increases vertebral but not femoral bone mineral density. In men with sexual dysfunction and low testosterone levels, testosterone therapy improves libido, but effects on erectile function and response to selective phosphodiesterase inhibitors are variable (Chap. 67). As discussed in Chap. 411, there is concern that testosterone therapy may stimulate the growth of prostate cancers.
FIGURE 7e-2 The effects of testosterone therapy on body composition, muscle strength, bone mineral density, and sexual function in intervention trials. The point estimates and the associated 95% confidence intervals are shown. A. The effects of testosterone therapy on lean body mass, grip strength, and fat mass in a meta-analysis of randomized trials. (Data derived from S Bhasin et al: Nat Clin Pract Endocrinol Metab 2:146, 2006.) B. The effects of testosterone therapy on lumbar and femoral bone mineral density in a meta-analysis of randomized trials. (Data derived from a meta-analysis by MJ Tracz et al: J Clin Endocrinol Metab 91:2011, 2006.) C. The effects of testosterone therapy on measures of sexual function in men with baseline testosterone less than 10 nmol/L (290 ng/dL). (Data derived from a meta-analysis by AM Isidori et al: Clin Endocrinol (Oxf) 63:381, 2005.) (Reproduced with permission from M Spitzer et al: Nat Rev Endocrinol 9:414, 2013.)
Sexual Dysfunction (See Chap. 67) Various forms of sexual dysfunction are a major motivating factor for men seeking care at men’s health clinics. The landmark descriptions of the human sexual response cycle by Masters and Johnson, demonstrating that men and women display predictable physiologic responses after sexual stimulation, provided the basis for rational classification of human sexual disorders. Accordingly, sexual disorders have been classified into four categories depending on phase of sexual response cycle in which the abnormality exists:
1. Hypoactive sexual desire disorder
2. Erectile dysfunction
3. Ejaculatory and orgasmic disorders
4. Disorders of pain
Classification of the patient’s disorder into these categories is important because the etiologic factors, diagnostic tests, and therapeutic strategies vary for each class of sexual disorder. Historically, the classification and nomenclature for sexual disorders used criteria identified in the Diagnostic and Statistical Manual of Mental Disorders (DSM), based on the erroneous belief that sexual disorders in men are largely psychogenic in their origin. However, the recognition of erectile dysfunction as a manifestation of systemic disease and the availability of easy-to-use oral selective phosphodiesterase-5 inhibitors have placed sexual disorders in men within the purview of the primary care provider.
MUSCLE DYSMORPHIA SYNDROME IN MEN: A BODY IMAGE DISORDER
Muscle dysmorphia is a form of body image disorder characterized by a pathologic preoccupation with muscularity and leanness. The men with muscle dysmorphia express a strong desire to be more muscular and lean. These men describe shame and embarrassment about their body size and shape and often report adverse symptoms such as dissatisfaction with appearance, preoccupation with bodybuilding and muscularity, and functional impairment. Patients with muscle dysmorphia also report higher rates of mood and anxiety disorders, as well as obsessive and compulsive behaviors. These men often experience impairment of social and occupational functioning.
Patients with muscle dysmorphia syndrome—nearly all men—are almost always engaged in weightlifting and body building and are more likely to use performance-enhancing drugs, especially anabolic-androgenic steroids. Muscle dysmorphia disorder predisposes men to an increased risk of disease due to the combined interactive effects of the intensity of physical exercise, the use of performance-enhancing drugs, and other lifestyle factors associated with weightlifting and the use of performance-enhancing drugs. No randomized trials of any treatment modalities have been conducted; anecdotally, behavioral and cognitive therapies have been tried with varying degrees of success.
Anabolic-Androgenic Steroid Abuse by Athletes and Recreational Body-Builders The illicit use of anabolic-androgenic steroids (AAS) to enhance athletic performance first surfaced in the 1950s among powerlifters and spread rapidly to other sports and to professional as well as high school athletes and recreational bodybuilders. In the early 1980s, the use of AAS spread beyond the athletic community into the general population. As many as 3 million Americans, most of them men, have likely used these compounds. Most AAS users are not athletes, but rather recreational weightlifters who use these drugs to look lean and more muscular.
The most commonly used AAS include testosterone esters, nandrolone, stanozolol, methandienone, and methenolone. AAS users generally use increasing doses of multiple steroids in a practice known as stacking.
The adverse effects of long-term AAS abuse remain poorly understood. Most of the information about the adverse effects of AAS has emerged from case reports, uncontrolled studies, or clinical trials that used replacement doses of testosterone (Table 7e-2). Of note, AAS users may administer 10–100 times the replacement doses of testosterone over many years, making it unjustifiable to extrapolate from trials using replacement doses. A substantial fraction of AAS users also use other drugs that are perceived to be muscle-building or performance-enhancing, such as growth hormone; erythropoiesis-stimulating agents; insulin; and stimulants such as amphetamine, clenbuterol, cocaine, ephedrine, and thyroxine; and drugs perceived to reduce adverse effects such as human chorionic gonadotropin, aromatase inhibitors, or estrogen antagonists. The men who abuse AAS are more likely to engage in other high-risk behaviors than nonusers. The adverse events associated with AAS use may be due to AAS themselves, concomitant use of other drugs, high-risk behaviors, and host characteristics that may render these individuals more susceptible to AAS use or to other high-risk behaviors.
|
POTENTIAL ADVERSE EFFECTS ASSOCIATED WITH THE USE OF ANABOLIC-ANDROGENIC STEROIDS (AAS) |
The high rates of mortality and morbidities observed in AAS users are alarming. The risk of death among elite powerlifters has been reported to be fivefold greater than in age-matched men from the general population. The causes of death among powerlifters included suicides, myocardial infarction, hepatic coma, and non-Hodgkin’s lymphoma.
Numerous reports of cardiac death among young AAS users raise concerns about the adverse cardiovascular effects of AAS. High doses of AAS may induce proatherogenic dyslipidemia, increase thrombosis risk via effects on clotting factors and platelets, induce vasospasm through their effects on vascular nitric oxide, and induce myocardial hypertrophy and fibrosis.
Replacement doses of testosterone, when administered parenterally, are associated with only a small decrease in high-density lipoprotein (HDL) cholesterol and little or no effect on total cholesterol, low-density lipoprotein (LDL) cholesterol, and triglyceride levels. In contrast, supraphysiologic doses of testosterone and orally administered, 17-α-alkylated, nonaromatizable AAS are associated with marked reductions in HDL cholesterol and increases in LDL cholesterol.
Long-term AAS use may be associated with myocardial hypertrophy and fibrosis as well as shortening of QT intervals. AAS use suppresses LH and FSH secretion and inhibits endogenous testosterone production and spermatogenesis. Consequently, stopping AAS may be associated with sexual dysfunction, fatigue, infertility, and depressive symptoms. In some AAS users, hypothalamic-pituitary-testicular axis suppression may last more than a year, and in a few individuals, complete recovery may not occur. The symptoms of androgen deficiency during AAS withdrawal may cause some men to revert back to using AAS, leading to continued use and AAS dependence. As many as 30% of AAS users develop a syndrome of AAS dependence, characterized by long-term AAS use, despite adverse medical and psychiatric effects. Supraphysiologic doses of testosterone may also impair insulin sensitivity, predisposing to diabetes. Elevated liver enzymes, cholestatic jaundice, hepatic neoplasms, and peliosis hepatis have been reported with oral 17-α-alkylated AAS. AAS use may cause muscle hypertrophy without compensatory adaptations in tendons, ligaments, and joints, thus increasing the risk of tendon and joint injuries. AAS use is associated with acne, baldness, and increased body hair.
Unsafe injection practices, high-risk behaviors, and increased rates of incarceration render AAS users at increased risk of HIV and hepatitis B and C. In one survey, nearly 1 in 10 gay men had injected AAS or other substances, and AAS users were more likely to report high-risk unprotected anal sex than other men.
Some AAS users develop hypomanic and manic symptoms during AAS exposure (irritability, aggressiveness, reckless behavior, and occasional psychotic symptoms, sometimes associated with violence) and major depression (sometimes associated with suicidality) during AAS withdrawal. Users may also develop other forms of illicit drug use, which may be potentiated or exacerbated by AAS.
|
DETECTION OF THE USE OF ANABOLIC-ANDROGENIC STEROIDS |
Abbreviations: FSH, follicle-stimulating hormone; LC-MS/MS, liquid chromatography and tandem mass spectrometry; LH, luteinizing hormone.
MALE LOWER URINARY TRACT SYMPTOMS
Lower urinary tract symptoms (LUTS) in men include storage symptoms (urgency, daytime and nighttime frequency, and urgency incontinence), voiding disturbances (slow or intermittent stream, difficulty in initiating micturition, straining to void, pain or discomfort during the passage of urine, and terminal dribbling), or postmicturition symptoms (a sense of incomplete voiding after passing urine and postmicturition dribble). The overactive bladder syndrome refers to urgency with or without urgency incontinence, usually with urinary frequency and nocturia, and is often due to detrusor muscle overactivity. LUTS have historically been attributed to benign prostatic hyperplasia, although it has become apparent that the pathophysiologic mechanisms of LUTS are complex and multifactorial and may include structural or functional abnormalities of the bladder, bladder neck, prostate, distal sphincter mechanism, and urethra, as well as abnormalities in the neural control to the lower urinary tract. A presumptive diagnosis of benign prostatic hyperplasia should be made only in men with LUTS who have demonstrable evidence of prostate enlargement and obstruction based on the size of the prostate. Diuretics, antihistamines, antidepressants, and other medications that have anticholinergic properties can cause or exacerbate LUTS in older men. The intensity of LUTS symptoms tends to fluctuate over time.
LUTS is highly prevalent in older men, affecting nearly 50% of men over the age of 65 and 70% of men over the age of 80. LUTS adversely affects quality of life because of its impact on sleep, ability to perform activities of daily living, and depressive symptoms. LUTS is often associated with erectile dysfunction.
MEDICAL COMPLICATIONS OF PROSTATE CANCER THERAPY
Prostate cancer is the most common malignancy in American men, accounting for 29% of all diagnosed cancers and approximately 13% of all cancer deaths; its incidence is on the rise, partly due to increased screening with PSA. In 2013, approximately 233,000 new cases of prostate cancer were diagnosed in the United States and there were 29,480 deaths related to prostate cancer. The majority of these men have low-grade, organ-confined prostate cancer and excellent prospects of long-term survival. Substantial improvement in survival in men with prostate cancer has focused attention on the high prevalence of sexual dysfunction, physical dysfunction, and low vitality, which are important contributors to poor quality of life among patients treated for prostate cancer. The pathophysiology of these symptoms after radical prostatectomy is multifactorial, but denervation and androgen deficiency are important contributors to these symptoms.
Androgen deficiency is common in men with prostate cancer. Testosterone levels decline with age, and men with prostate cancer are at risk of having low testosterone levels simply by virtue of their age. However, total and free testosterone levels are even lower in men with prostate cancer, who have undergone prostatectomy, when compared with age-matched controls without cancer. Androgen deficiency in men with prostate cancer is associated with distressing symptoms such as fatigue, sexual dysfunction, hot flushes, mobility limitation, and decreased physical function. Even with a bilateral nerve-sparing procedure, more than 50% of men develop sexual dysfunction after surgery. Although there is some recovery of sexual function with passage of time, 40–50% of men undergoing radical prostatectomy find their sexual performance to be problematic 18 months after surgery. Sexual performance problems are a source of psychosocial distress in men with localized prostate cancer. In addition to its causal contribution to distressing symptoms, androgen deficiency in men with prostate cancer increases the risk of bone fractures, diabetes, coronary heart disease, and frailty.
Testosterone Therapy in Men with History of Prostate Cancer A history of prostate cancer has historically been considered a contraindication for testosterone therapy. This guidance is based on observations that testosterone promotes the growth of metastatic prostate cancer. Metastatic prostate cancer generally regresses after orchidectomy and androgen deprivation therapy. Androgen receptor signaling plays a central role in maintaining growth of normal prostate and prostate cancer. PSA levels are lower in hypogonadal men and increase after testosterone therapy. Prostate volume is lower in hypogonadal men and increases after testosterone therapy to levels seen in age-matched controls.
However, the role of testosterone in prostate cancer is complex. Epidemiologic studies have not revealed a consistent relationship between serum testosterone and prostate cancer. In a landmark randomized trial, testosterone therapy of older men with low testosterone did not affect intraprostatic androgen levels or the expression of androgen-dependent prostatic genes. The suppression of circulating testosterone levels by a gonadotropin-releasing hormone (GnRH) antagonist also does not affect intraprostatic androgen concentrations. Open-label trials and retrospective analyses of testosterone therapy in men with prostate cancer, who have undergone radical prostatectomy and have undetectable PSA levels after radical prostatectomy, have found very low rates of PSA recurrence. Even in men with high-grade prostatic intraepithelial neoplasia (HGPIN)—a group at high risk of developing prostate cancer—testosterone therapy for 1 year did not increase PSA or rates of prostate cancer.
After radical prostatectomy, in the absence of residual cancer, PSA becomes undetectable within a month. An undetectable PSA after radical prostatectomy is a good indicator of biochemical recurrence-free survival at 5 years. Therefore, men with organ-confined prostate cancer (pT2), Gleason score ≤6, and a preoperative PSA of <10 ng/mL, who have had undetectable PSA levels (<0.1 ng/mL) for >2 years after radical prostatectomy, have very low risk of disease recurrence (<0.5% at 10 years) and may be considered for testosterone therapy on an individualized basis. If testosterone therapy is instituted, it should be associated with careful monitoring of PSA levels and done in consultation with a urologist.
MEDICAL COMPLICATIONS OF ANDROGEN DEPRIVATION THERAPY
In patients with prostate cancer and distant metastases, androgen deprivation therapy (ADT) improves survival. In patients with locally advanced disease, ADT in combination with external-beam radiation or as an adjuvant therapy (after prostatectomy and pelvic lymphadenectomy) also has been shown to improve survival. However, ADT is being increasingly used as primary therapy in men with localized disease and in men encountering biochemical recurrence without clear evidence of survival advantage. Because most men with prostate cancer die of conditions other than their primary malignancy, recognition and management of these adverse effects is paramount.
Profound hypogonadism resulting from ADT is associated with sexual dysfunction, vasomotor symptoms, gynecomastia, decreased muscle mass and strength, frailty, increased fat mass, anemia, fatigue, bone loss, loss of body hair, depressive symptoms, and reduced quality of life. Diabetes and cardiovascular disease have recently been added to the list of these complications (Fig. 7e-3). Treatment with GnRH agonists in men with prostate cancer is associated with rapid induction of insulin resistance, hyperinsulinemia, and a significant increase in the risk of incident diabetes. Metabolic syndrome is prevalent in over 50% of men undergoing long-term ADT. Some but not all studies have reported an increased risk of cardiovascular events, death due to cardiovascular events, and peripheral vascular disease in men undergoing ADT. Men receiving ADT are also at increased risk of thromboembolic events. The rates of acute kidney injury are higher in men currently receiving ADT than in men not receiving ADT; the increased risk appears to be particularly associated with the use of combined regimens of a GnRH agonist plus an antiandrogen. ADT also is associated with substantially increased risk of osteoporosis and bone fractures.
FIGURE 7e-3 Adverse cardiometabolic and skeletal effects of androgen deprivation therapy (ADT) in men receiving ADT for prostate cancer. Administration of ADT has been associated with increased risk of thromboembolic events, fractures, and diabetes. Some, but not all, studies have reported increased risk of cardiovascular events in men receiving ADT. (Data on relative risk were derived from VB Shahinian et al: N Engl J Med 352:154, 2005; NL Keating et al: J Clin Oncol 24:4448, 2006; and JC Hu et al: Eur Urol 61:1119, 2012.)
|
CHECKLIST FOR MEN UNDERGOING ANDROGEN DEPRIVATION THERAPY (ADT) |
8 Medical Disorders During Pregnancy
Each year, approximately 4 million births occur in the United States, and more than 130 million births occur worldwide. A significant proportion of births are complicated by medical disorders. In the past, many medical disorders were contraindications to pregnancy. Advances in obstetrics, neonatology, obstetric anesthesiology, and medicine have increased the expectation that pregnancy will result in a positive outcome for both mother and fetus despite most of these conditions. A successful pregnancy requires important physiologic adaptations, such as a marked increase in cardiac output. Medical problems that interfere with the physiologic adaptations of pregnancy increase the risk for poor pregnancy outcome; conversely, in some instances, pregnancy may adversely impact an underlying medical disorder.
HYPERTENSION
(See also Chap. 298) In pregnancy, cardiac output increases by 40%, with most of the increase due to an increase in stroke volume. Heart rate increases by ~10 beats/min during the third trimester. In the second trimester, systemic vascular resistance decreases, and this decline is associated with a fall in blood pressure. During pregnancy, a blood pressure of 140/90 mmHg is considered to be abnormally elevated and is associated with an increase in perinatal morbidity and mortality. In all pregnant women, the measurement of blood pressure should be performed in the sitting position, because the lateral recumbent position may result in a blood pressure lower than that recorded in the sitting position. The diagnosis of hypertension requires the measurement of two elevated blood pressures at least 6 h apart. Hypertension during pregnancy is usually caused by preeclampsia, chronic hypertension, gestational hypertension, or renal disease.
PREECLAMPSIA
Approximately 5–7% of all pregnant women develop preeclampsia, the new onset of hypertension (blood pressure >140/90 mmHg) and proteinuria (either a 24 hour urinary protein >300 mg/24 h, or a protein-creatinine ratio ≥0.3) after 20 weeks of gestation. Although the precise pathophysiology of preeclampsia remains unknown, recent studies show excessive placental production of antagonists to both vascular epithelial growth factor (VEGF) and transforming growth factor β (TGF-β). These antagonists to VEGF and TGF-β disrupt endothelial and renal glomerular function resulting in edema, hypertension, and proteinuria. The renal histological feature of preeclampsia is glomerular endotheliosis. Glomerular endothelial cells are swollen and encroach on the vascular lumen. Preeclampsia is associated with abnormalities of cerebral circulatory autoregulation, which increase the risk of stroke at mildly and moderately elevated blood pressures. Risk factors for the development of preeclampsia include nulliparity, diabetes mellitus, a history of renal disease or chronic hypertension, a prior history of preeclampsia, extremes of maternal age (>35 years or <15 years), obesity, antiphospholipid antibody syndrome, and multiple gestation. Low-dose aspirin (81 mg daily, initiated at the end of the first trimester) may reduce the risk of preeclampsia in pregnant women at high risk of developing the disease.
In December, 2013 The American College of Obstetricians and Gynecologists issued a report summarizing the findings and recommendations of their Task Force on Hypertension in Pregnancy. With respect to preeclampsia several pertinent revisions to the diagnostic criteria were made including: proteinuria is no longer an absolute requirement for making the diagnosis; the terms mild and severe preeclampsia have been replaced, and the disease is now termed preeclampsia either with or without severe features; removal of fetal growth restriction as a defining criterion for severe preeclampsia.
Preeclampsia with severe features is the presence of new-onset hypertension and proteinuria accompanied by end-organ damage. Features may include severe elevation of blood pressure (>160/110 mmHg), evidence of central nervous system (CNS) dysfunction (headaches, blurred vision, seizures, coma), renal dysfunction (oliguria or creatinine >1.5 mg/dL), pulmonary edema, hepatocellular injury (serum alanine aminotransferase level more than twofold the upper limit of normal), hematologic dysfunction (platelet count <100,000/L or disseminated intravascular coagulation [DIC]). The HELLP syndrome (hemolysis, elevated liver enzymes, low platelets) is a special subtype of severe preeclampsia and is a major cause of morbidity and mortality in this disease. Platelet dysfunction and coagulation disorders further increase the risk of stroke.
CHRONIC ESSENTIAL HYPERTENSION
Pregnancy complicated by chronic essential hypertension is associated with intrauterine growth restriction and increased perinatal mortality. Pregnant women with chronic hypertension are at increased risk for superimposed preeclampsia and abruptio placentae. Women with chronic hypertension should have a thorough prepregnancy evaluation, both to identify remediable causes of hypertension and to ensure that the prescribed antihypertensive agents (e.g., ACE inhibitors, angiotensin-receptor blockers) are not associated with an adverse outcome of pregnancy. α-Methyldopa, labetalol, and nifedipine are the most commonly used medications for the treatment of chronic hypertension in pregnancy. The target blood pressure is in the range of 130–150 mmHg systolic and 80–100 mmHg diastolic. Should hypertension worsen during pregnancy, baseline evaluation of renal function (see below) is necessary to help differentiate the effects of chronic hypertension from those of superimposed preeclampsia. There are no convincing data that the treatment of mild chronic hypertension improves perinatal outcome.
GESTATIONAL HYPERTENSION
The development of elevated blood pressure during pregnancy or in the first 24 h post-partum in the absence of preexisting chronic hypertension or proteinuria is referred to as gestational hypertension. Mild gestational hypertension that does not progress to preeclampsia has not been associated with adverse pregnancy outcome or adverse long-term prognosis.
RENAL DISEASE
(See also Chaps. 333 and 341) Normal pregnancy is characterized by an increase in glomerular filtration rate and creatinine clearance. This increase occurs secondary to a rise in renal plasma flow and increased glomerular filtration pressures. Patients with underlying renal disease and hypertension may expect a worsening of hypertension during pregnancy. If superimposed preeclampsia develops, the additional endothelial injury results in a capillary leak syndrome that may make management challenging. In general, patients with underlying renal disease and hypertension benefit from aggressive management of blood pressure. Preconception counseling is also essential for these patients so that accurate risk assessment and medication changes can occur prior to pregnancy. In general, a prepregnancy serum creatinine level <133 μmol/L (<1.5 mg/dL) is associated with a favorable prognosis. When renal disease worsens during pregnancy, close collaboration between the internist and the maternal-fetal medicine specialist is essential so that decisions regarding delivery can be weighed to balance the sequelae of prematurity for the neonate versus long-term sequelae for the mother with respect to future renal function.
CARDIAC DISEASE
VALVULAR HEART DISEASE
(See also Chaps. 283–286) Valvular heart disease is the most common cardiac problem complicating pregnancy.
Mitral Stenosis This is the valvular disease most likely to cause death during pregnancy. The pregnancy-induced increase in blood volume, cardiac output, and tachycardia can increase the transmitral pressure gradient and cause pulmonary edema in women with mitral stenosis. Women with moderate to severe mitral stenosis who are planning pregnancy and have either symptomatic disease or pulmonary hypertension should undergo valvuloplasty prior to conception. Pregnancy associated with long-standing mitral stenosis may result in pulmonary hypertension. Sudden death has been reported when hypovolemia occurs. Careful control of heart rate, especially during labor and delivery, minimizes the impact of tachycardia and reduced ventricular filling times on cardiac function. Pregnant women with mitral stenosis are at increased risk for the development of atrial fibrillation and other tachyarrhythmias. Medical management of severe mitral stenosis and atrial fibrillation with digoxin and beta blockers is recommended. Balloon valvulotomy can be carried out during pregnancy. The immediate postpartum period is a time of particular concern secondary to rapid volume shifts. Careful monitoring of cardiac and fluid status should be observed.
Mitral Regurgitation and Aortic Regurgitation and Stenosis The pregnancy-induced decrease in systemic vascular resistance reduces the risk of cardiac failure with these conditions. As a rule, mitral valve prolapse does not present problems for the pregnant patient, and aortic stenosis, unless very severe, is well tolerated. In the most severe cases of aortic stenosis, limitation of activity or balloon valvuloplasty may be indicated.
CONGENITAL HEART DISEASE
(See also Chap. 282) Reparative surgery has markedly increased the number of women with surgically repaired congenital heart disease. Maternal morbidity and mortality are greater among these women than among those without surgical repairs. When pregnant, these patients should be jointly managed by a cardiologist and an obstetrician familiar with these problems. The presence of a congenital cardiac lesion in the mother increases the risk of congenital cardiac disease in the newborn. Prenatal screening of the fetus for congenital cardiac disease with ultrasound is recommended. Atrial or ventricular septal defect is usually well tolerated during pregnancy in the absence of pulmonary hypertension, provided that the woman’s prepregnancy cardiac status is favorable. Use of air filters on IV sets during labor and delivery in patients with intracardiac shunts is recommended.
OTHER CARDIAC DISORDERS
Supraventricular tachycardia (Chap. 276) is a common cardiac complication of pregnancy. Treatment is the same as in the nonpregnant patient, and fetal tolerance of medications such as adenosine and calcium channel blockers is acceptable. When necessary, pharmacologic or electric cardioversion may be performed to improve cardiac performance and reduce symptoms. This intervention is generally well tolerated by mother and fetus.
Peripartum cardiomyopathy (Chap. 287) is an uncommon disorder of pregnancy associated with myocarditis, and its etiology remains unknown. Treatment is directed toward symptomatic relief and improvement of cardiac function. Many patients recover completely; others are left with progressive dilated cardiomyopathy. Recurrence in a subsequent pregnancy has been reported, and women who do not have normal baseline left-ventricular function after an episode of peripartum cardiomyopathy should be counseled to avoid pregnancy.
SPECIFIC HIGH-RISK CARDIAC LESIONS
Marfan Syndrome (See also Chap. 427) This autosomal dominant disease is associated with a high risk of maternal morbidity. Approximately 15% of pregnant women with Marfan syndrome develop a major cardiovascular manifestation during pregnancy, with almost all women surviving. An aortic root diameter <40 mm is associated with a favorable outcome of pregnancy. Prophylactic therapy with beta blockers has been advocated, although large-scale clinical trials in pregnancy have not been performed. Ehlers-Danlos syndrome (EDS) may be associated with premature labor, and in type IV EDS there is increased risk of organ or vascular rupture that may cause death.
Pulmonary Hypertension (See also Chap. 304) Maternal mortality in the setting of severe pulmonary hypertension is high, and primary pulmonary hypertension is a contraindication to pregnancy. Termination of pregnancy may be advisable in these circumstances to preserve the life of the mother. In the Eisenmenger syndrome, i.e., the combination of pulmonary hypertension with right-to-left shunting due to congenital abnormalities (Chap. 282), maternal and fetal deaths occur frequently. Systemic hypotension may occur after blood loss, prolonged Valsalva maneuver, or regional anesthesia; sudden death secondary to hypotension is a dreaded complication. Management of these patients is challenging, and invasive hemodynamic monitoring during labor and delivery is recommended in severe cases.
In patients with pulmonary hypertension, vaginal delivery is less stressful hemodynamically than cesarean section, which should be reserved for accepted obstetric indications.
DEEP VENOUS THROMBOSIS AND PULMONARY EMBOLISM
(See also Chap. 300) A hypercoagulable state is characteristic of pregnancy, and deep venous thrombosis (DVT) occurs in about 1 in 500 pregnancies. In pregnant women, most unilateral DVTs occur in the left leg because the left iliac vein is compressed by the right iliac artery and the uterus compresses the inferior vena cava. Pregnancy is associated with an increase in procoagulants such as factors V and VII and a decrease in anticoagulant activity, including proteins C and S. Pulmonary embolism is one of the most common causes of maternal death in the United States. Activated protein C resistance caused by the factor V Leiden mutation increases the risk for DVT and pulmonary embolism during pregnancy. Approximately 25% of women with DVT during pregnancy carry the factor V Leiden allele. Additional genetic mutations associated with DVT during pregnancy include the prothrombin G20210A mutation (heterozygotes and homozygotes) and the methylenetetrahydrofolate reductase C677T mutation (homozygotes).
ENDOCRINE DISORDERS
DIABETES MELLITUS
(See also Chaps. 417–419) In pregnancy, the fetoplacental unit induces major metabolic changes, the purpose of which is to shunt glucose and amino acids to the fetus while the mother uses ketones and triglycerides to fuel her metabolic needs. These metabolic changes are accompanied by maternal insulin resistance caused in part by placental production of steroids, a growth hormone variant, and placental lactogen. Although pregnancy has been referred to as a state of “accelerated starvation,” it is better characterized as “accelerated ketosis.” In pregnancy, after an overnight fast, plasma glucose is lower by 0.8–1.1 mmol/L (15–20 mg/dL) than in the nonpregnant state. This difference is due to the use of glucose by the fetus. In early pregnancy, fasting may result in circulating glucose concentrations in the range of 2.2 mmol/L (40 mg/dL) and may be associated with symptoms of hypoglycemia. In contrast to the decrease in maternal glucose concentration, plasma hydroxybutyrate and acetoacetate levels rise to two to four times normal after a fast.
GESTATIONAL DIABETES
Gestational diabetes occurs in approximately 4% of pregnancies. All pregnant women should be screened for gestational diabetes unless they are in a low-risk group. Women at low risk for gestational diabetes are those <25 years of age; those with a body mass index <25 kg/m2, no maternal history of macrosomia or gestational diabetes, and no diabetes in a first-degree relative; and those who are not members of a high-risk ethnic group (African American, Hispanic, Native American). A typical two-step strategy for establishing the diagnosis of gestational diabetes involves administration of a 50-g oral glucose challenge with a single serum glucose measurement at 60 min. If the plasma glucose is <7.8 mmol/L (<130 mg/dL), the test is considered normal. Plasma glucose >7.8 mmol/L (>130 mg/dL) warrants administration of a 100-g oral glucose challenge with plasma glucose measurements obtained in the fasting state and at 1, 2, and 3 h. Normal plasma glucose concentrations at these time points are <5.8 mmol/L (<105 mg/dL), 10.5 mmol/L (190 mg/dL), 9.1 mmol/L (165 mg/dL), and 8.0 mmol/L (145 mg/dL), respectively. Some centers have adopted more sensitive criteria, using values of <5.3 mmol/L (<95 mg/dL), <10 mmol/L (<180 mg/dL), <8.6 mmol/L (<155 mg/dL), and <7.8 mmol/L (<140 mg/dL) as the upper norms for a 3-h glucose tolerance test. Two elevated glucose values indicate a positive test. Adverse pregnancy outcomes for mother and fetus appear to increase with glucose as a continuous variable; thus it is challenging to define the optimal threshold for establishing the diagnosis of gestational diabetes.
Pregnant women with gestational diabetes are at increased risk of stillbirth, preeclampsia, and delivery of infants who are large for their gestational age, with resulting birth lacerations, shoulder dystocia, and birth trauma including brachial plexus injury. These fetuses are at risk of hypoglycemia, hyperbilirubinemia, and polycythemia. Tight control of blood sugar during pregnancy and labor can reduce these risks.
OBESITY
(See also Chap. 416) Pregnant women who are obese have an increased risk of stillbirth, congenital fetal malformations, gestational diabetes, preeclampsia, urinary tract infections, post-date delivery, and cesarean delivery. Women contemplating pregnancy should attempt to attain a healthy weight prior to conception. For morbidly obese women who have not been able to lose weight with lifestyle changes, bariatric surgery may result in weight loss and improve pregnancy outcomes. Following bariatric surgery, women should delay conception for 1 year to avoid pregnancy during an interval of rapid metabolic changes.
THYROID DISEASE
(See also Chap. 405) In pregnancy, the estrogen-induced increase in thyroxine-binding globulin increases circulating levels of total T3 and total T4. The normal range of circulating levels of free T4, free T3, and thyroid-stimulating hormone (TSH) remain unaltered by pregnancy.
The thyroid gland normally enlarges during pregnancy. Many physiologic adaptations to pregnancy may mimic subtle signs of hyperthyroidism. Maternal hyperthyroidism occurs at a rate of ~2 per 1000 pregnancies and is generally well tolerated by pregnant women. Clinical signs and symptoms should alert the physician to the occurrence of this condition. Hyperthyroidism in pregnancy is most commonly caused by Graves’ disease, but autonomously functioning nodules and gestational trophoblastic disease should also be considered. Although pregnant women are able to tolerate mild hyperthyroidism without adverse sequelae, more severe hyperthyroidism can cause spontaneous abortion or premature labor, and thyroid storm is associated with a significant risk of maternal death.
Testing for hypothyroidism using TSH measurements before or early in pregnancy may be warranted in symptomatic women and in women with a personal or family history of thyroid disease. With use of this case-finding approach, about 30% of pregnant women with mild hypothyroidism remain undiagnosed, leading some to recommend universal screening. Children born to women with an elevated serum TSH (and a normal total thyroxine) during pregnancy may have impaired performance on neuropsychologic tests.
HEMATOLOGIC DISORDERS
Pregnancy has been described as a state of physiologic anemia. Part of the reduction in hemoglobin concentration is dilutional, but iron and folate deficiencies are major causes of correctable anemia during pregnancy.
In populations at high risk for hemoglobinopathies (Chap. 127), hemoglobin electrophoresis should be performed as part of the prenatal screen. Hemoglobinopathies can be associated with increased maternal and fetal morbidity and mortality. Management is tailored to the specific hemoglobinopathy and is generally the same for both pregnant and nonpregnant women. Prenatal diagnosis of hemoglobinopathies in the fetus is readily available and should be discussed with prospective parents either prior to or early in pregnancy.
Thrombocytopenia occurs commonly during pregnancy. The majority of cases are benign gestational thrombocytopenias, but the differential diagnosis should include immune thrombocytopenia (Chap. 140), thrombotic thrombocytopenic purpura, and preeclampsia. Maternal thrombocytopenia may also be caused by DIC, which is a consumptive coagulopathy characterized by thrombocytopenia, prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT), elevated fibrin degradation products, and a low fibrinogen concentration. Several catastrophic obstetric events are associated with the development of DIC, including retention of a dead fetus, sepsis, abruptio placentae, and amniotic fluid embolism.
NEUROLOGIC DISORDERS
Headache appearing during pregnancy is usually due to migraine (Chap. 21), a condition that may worsen, improve, or be unaffected by pregnancy. A new or worsening headache, particularly if associated with visual blurring, may signal eclampsia (above) or pseudotumor cerebri (benign intracranial hypertension); diplopia due to a sixth-nerve palsy suggests pseudotumor cerebri (Chap. 39). The risk of seizures in patients with epilepsy increases in the postpartum period but not consistently during pregnancy; management is discussed in Chap. 445. The risk of stroke is generally thought to increase during pregnancy because of a hypercoagulable state; however, studies suggest that the period of risk occurs primarily in the postpartum period and that both ischemic and hemorrhagic strokes may occur at this time. Guidelines for use of heparin therapy are summarized above (see “Deep Venous Thrombosis and Pulmonary Embolism”); warfarin is teratogenic and should be avoided.
The onset of a new movement disorder during pregnancy suggests chorea gravidarum, a variant of Sydenham’s chorea associated with rheumatic fever and streptococcal infection (Chap. 381); the chorea may recur with subsequent pregnancies. Patients with preexisting multiple sclerosis (Chap. 458) experience a gradual decrease in the risk of relapses as pregnancy progresses and, conversely, an increase in attack risk during the postpartum period. Disease-modifying agents, including interferon β, should not be administered to pregnant multiple sclerosis patients, but moderate or severe relapses can be safely treated with pulse glucocorticoid therapy. Finally, certain tumors, particularly pituitary adenoma and meningioma (Chap. 403), may manifest during pregnancy because of accelerated growth, possibly driven by hormonal factors.
Peripheral nerve disorders associated with pregnancy include Bell’s palsy (idiopathic facial paralysis) (Chap. 459), which is approximately threefold more likely to occur during the third trimester and immediate postpartum period than in the general population. Therapy with glucocorticoids should follow the guidelines established for nonpregnant patients. Entrapment neuropathies are common in the later stages of pregnancy, presumably as a result of fluid retention. Carpal tunnel syndrome (median nerve) presents first as pain and paresthesia in the hand (often worse at night) and later with weakness in the thenar muscles. Treatment is generally conservative; wrist splints may be helpful, and glucocorticoid injections or surgical section of the carpal tunnel can usually be postponed. Meralgia paresthetica (lateral femoral cutaneous nerve entrapment) consists of pain and numbness in the lateral aspect of the thigh without weakness. Patients are usually reassured to learn that these symptoms are benign and can be expected to remit spontaneously after the pregnancy has been completed. Restless leg syndrome is the most common peripheral nerve and movement disorder in pregnancy. Disordered iron metabolism is the suspected etiology. Management is expectant in most cases.
GASTROINTESTINAL AND LIVER DISEASE
Up to 90% of pregnant women experience nausea and vomiting during the first trimester of pregnancy. Hyperemesis gravidarum is a severe form that prevents adequate fluid and nutritional intake and may require hospitalization to prevent dehydration and malnutrition.
Crohn’s disease may be associated with exacerbations in the second and third trimesters. Ulcerative colitis is associated with disease exacerbations in the first trimester and during the early postpartum period. Medical management of these diseases during pregnancy is similar to management in the nonpregnant state (Chap. 351).
Exacerbation of gallbladder disease is common during pregnancy. In part, this aggravation may be due to pregnancy-induced alteration in the metabolism of bile and fatty acids. Intrahepatic cholestasis of pregnancy is generally a third-trimester event. Profound pruritus may accompany this condition, and it may be associated with increased fetal mortality. Placental bile salt deposition may contribute to progressive uteroplacental insufficiency. Therefore, regular fetal surveillance should be undertaken once the diagnosis of intrahepatic cholestasis is made, and delivery should be planned once the fetus reaches about 37 weeks of gestation. Favorable results with ursodiol have been reported.
Acute fatty liver is a rare complication of pregnancy. Frequently confused with the HELLP syndrome (see “Preeclampsia” above) and severe preeclampsia, the diagnosis of acute fatty liver of pregnancy may be facilitated by imaging studies and laboratory evaluation. Acute fatty liver of pregnancy is generally characterized by markedly increased serum levels of bilirubin and ammonia and by hypoglycemia. Management of acute fatty liver of pregnancy is supportive; recurrence in subsequent pregnancies has been reported.
All pregnant women should be screened for hepatitis B. This information is important for pediatricians after delivery of the infant. All infants receive hepatitis B vaccine. Infants born to mothers who are carriers of hepatitis B surface antigen should also receive hepatitis B immune globulin as soon after birth as possible and preferably within the first 72 h. Screening for hepatitis C is recommended for individuals at high risk for exposure.
INFECTIONS
BACTERIAL INFECTIONS
Other than bacterial vaginosis, the most common bacterial infections during pregnancy involve the urinary tract (Chap. 162). Many pregnant women have asymptomatic bacteriuria, most likely due to stasis caused by progestational effects on ureteral and bladder smooth muscle and later in pregnancy due to compression effects of the enlarging uterus. In itself, this condition is not associated with an adverse outcome of pregnancy. However, if asymptomatic bacteriuria is left untreated, symptomatic pyelonephritis may occur. Indeed, ~75% of pregnancy-associated pyelonephritis cases are the result of untreated asymptomatic bacteriuria. All pregnant women should be screened with a urine culture for asymptomatic bacteriuria at the first prenatal visit. Subsequent screening with nitrite/leukocyte esterase strips is indicated for high-risk women, such as those with sickle cell trait or a history of urinary tract infections. All women with positive screens should be treated. Pregnant women who develop pyelonephritis need careful monitoring, including inpatient IV antibiotic administration due to the elevated risk of urosepsis and acute respiratory distress syndrome in pregnancy.
Abdominal pain and fever during pregnancy create a clinical dilemma. The diagnosis of greatest concern is intrauterine amniotic infection. While amniotic infection most commonly follows rupture of the membranes, this is not always the case. In general, antibiotic therapy is not recommended as a temporizing measure in these circumstances. If intrauterine infection is suspected, induced delivery with concomitant antibiotic therapy is generally indicated. Intrauterine amniotic infection is most often caused by pathogens such as Escherichia coli and group B Streptococcus (GBS). In high-risk patients at term or in preterm patients, routine intrapartum prophylaxis of GBS disease is recommended. Penicillin G and ampicillin are the drugs of choice. In penicillin-allergic patients with a low risk of anaphylaxis, cefazolin is recommended. If the patient is at high risk of anaphylaxis, vancomycin is recommended. If the organism is known to be sensitive to clindamycin, this antibiotic may be used. For the reduction of neonatal morbidity due to GBS, universal screening of pregnant women for GBS between 35 and 37 weeks of gestation, with intrapartum antibiotic treatment of infected women, is recommended.
Postpartum infection is a significant cause of maternal morbidity and mortality. Postpartum endomyometritis is more common after cesarean delivery than vaginal delivery and develops in 2% of women after elective repeat cesarean section and in up to 10% after emergency cesarean section following prolonged labor. To reduce the risk of endomyometritis, prophylactic antibiotics should be given to all patients undergoing cesarean section, and administration 30–60 min prior to skin incision is preferable to administration at the time of umbilical cord clamping. As most cases of postpartum endomyometritis are polymicrobial, broad-spectrum antibiotic coverage with a penicillin, an aminoglycoside, and metronidazole is recommended (Chap. 201). Most cases resolve within 72 h. Women who do not respond to antibiotic treatment for postpartum endomyometritis should be evaluated for septic pelvic thrombophlebitis. Imaging studies may be helpful in establishing the diagnosis, which is primarily a clinical diagnosis of exclusion. Patients with septic pelvic thrombophlebitis generally have tachycardia out of proportion to their fever and respond rapidly to IV administration of heparin.
All pregnant patients are screened prenatally for gonorrhea and chlamydial infections, and the detection of either should result in prompt treatment. Ceftriaxone and azithromycin are the agents of choice (Chaps. 181 and 213).
VIRAL INFECTIONS
Influenza (See also Chap. 224) Pregnant women with influenza are at increased risk of serious complications and death. All women who are pregnant or plan to become pregnant in the near future should receive inactivated influenza vaccine. The prompt initiation of antiviral treatment is recommended for pregnant women in whom influenza is suspected. Treatment can be reconsidered once the results of high-sensitivity tests are available. Prompt initiation of treatment lowers the risk of admission to an intensive care unit and death.
Cytomegalovirus Infection The most common cause of congenital viral infection in the United States is cytomegalovirus (CMV) (Chap. 219). As many as 50–90% of women of childbearing age have antibodies to CMV, but only rarely does CMV reactivation result in neonatal infection. More commonly, primary CMV infection during pregnancy creates a risk of congenital CMV. No currently accepted treatment of CMV infection during pregnancy has been demonstrated to protect the fetus effectively. Moreover, it is difficult to predict which fetus will sustain a life-threatening CMV infection. Severe CMV disease in the newborn is characterized most often by petechiae, hepatosplenomegaly, and jaundice. Chorioretinitis, microcephaly, intracranial calcifications, hepatitis, hemolytic anemia, and purpura may also develop. CNS involvement, resulting in the development of psychomotor, ocular, auditory, and dental abnormalities over time, has been described.
Rubella (See also Chap. 230e) Rubella virus is a known teratogen; first-trimester rubella carries a high risk of fetal anomalies, though the risk significantly decreases later in pregnancy. Congenital rubella may be diagnosed by percutaneous umbilical-blood sampling with the detection of IgM antibodies in fetal blood. All pregnant women and all women of childbearing age should be tested for their immune status to rubella. All nonpregnant women who are not immune to rubella should be vaccinated. The incidence of congenital rubella in the United States is extremely low.
Herpesvirus Infection (See also Chap. 216) The acquisition of genital herpes during pregnancy is associated with spontaneous abortion, prematurity, and congenital and neonatal herpes. A cohort study of pregnant women without evidence of previous herpesvirus infection demonstrated that ~2% acquired a new herpesvirus infection during the pregnancy. Approximately 60% of the newly infected women had no clinical symptoms. Infection occurred with equal frequency in all three trimesters. If herpesvirus seroconversion occurred early in pregnancy, the risk of transmission to the newborn was very low. In women who acquired genital herpes shortly before delivery, the risk of transmission was high. The risk of active genital herpes lesions at term can be reduced by prescribing acyclovir for the last 4 weeks of pregnancy to women who have had their first episode of genital herpes during the pregnancy.
Herpesvirus infection in the newborn can be devastating. Disseminated neonatal herpes carries with it high mortality and morbidity rates from CNS involvement. It is recommended that pregnant women with active genital herpes lesions at the time of presentation in labor be delivered by cesarean section.
Parvovirus Infection (See also Chap. 221) Parvovirus infection (caused by human parvovirus B19) may occur during pregnancy. It rarely causes sequelae, but susceptible women infected during pregnancy may be at risk for fetal hydrops secondary to erythroid aplasia and profound anemia.
HIV Infection (See also Chap. 226) The predominant cause of HIV infection in children is transmission of the virus from mother to newborn during the perinatal period. All pregnant women should be screened for HIV infection. Factors that increase the risk of mother-to-newborn transmission include high maternal viral load, low maternal CD4+ T cell count, prolonged labor, prolonged duration of membrane rupture, and the presence of other genital tract infections, such as syphilis or herpes. Prior to the widespread use of antiretroviral treatment, the perinatal transmission rate was in the range of 20%. In women with a good response to antiretroviral treatment, the transmission rate is about 1%. Measurement of maternal plasma HIV RNA copy number guides the decision for vaginal versus cesarean delivery. For women with <1000 copies of plasma HIV RNA/ml who are receiving combination antiretroviral therapy, the risk of transmission to the newborn is approximately 1% regardless of mode of delivery or duration of membrane rupture. These women may elect to attempt a vaginal birth following the spontaneous onset of labor. For women with a viral load of ≥1000 copies/ml prior to 38 weeks of gestation, a scheduled prelabor cesarean at 38 weeks is recommended to reduce the risk of HIV transmission to the newborn. To reduce the risk of mother-to-newborn transmission, women with >400 copies of HIV RNA/ml should be treated during the intrapartum interval with zidovudine. All newborns of HIV-infected mothers should be treated with zidovudine for 6 months after birth. Women who are HIV-positive may transmit the virus through their breast milk. In developed countries, HIV-infected mothers are advised not to breast-feed.
VACCINATIONS
(See also Chap. 148) For rubella-nonimmune individuals contemplating pregnancy, measles-mumps-rubella vaccine should be administered, ideally at least 3 months prior to conception but otherwise in the immediate postpartum period. In addition, pregnancy is not a contraindication for vaccination against influenza, tetanus, diphtheria, and pertussis (Tdap), and these vaccines are recommended for appropriate individuals.
MATERNAL MORTALITY
Maternal death is defined as death occurring during pregnancy or within 42 days of completion of pregnancy from a cause related to or aggravated by pregnancy, but not due to accident or incidental causes. From 1935 to 2007, the U.S. maternal death rate decreased from nearly 600/100,000 births to 12.7/100,000 births. There are significant health disparities in the maternal mortality rate, with the highest rates among non-Hispanic black women. In 2007, maternal mortality rates (per 100,000) by race were 10.5 among non-Hispanic white women, 8.9 among Hispanic women, and 28.4 among non-Hispanic black women. The most common causes of maternal death in the United States today are pulmonary embolism, obstetric hemorrhage, hypertension, sepsis, cardiovascular conditions (including peripartum cardiomyopathy), and ectopic pregnancy.
![]() As stated above, the maternal mortality rate in the United States is about 12.7/100,00 births. In some countries in sub-Saharan Africa and southern Asia, the maternal mortality rate is about 500/100,000 live births. The most common cause of maternal death in these countries is maternal hemorrhage. The high maternal death rates are due in part to inadequate contraceptive and family-planning services, an insufficient number of skilled birth attendants, and difficulty in accessing birthing centers and emergency obstetrical care units. Maternal death is a global public-health tragedy that could be mitigated with the application of modest resources.
As stated above, the maternal mortality rate in the United States is about 12.7/100,00 births. In some countries in sub-Saharan Africa and southern Asia, the maternal mortality rate is about 500/100,000 live births. The most common cause of maternal death in these countries is maternal hemorrhage. The high maternal death rates are due in part to inadequate contraceptive and family-planning services, an insufficient number of skilled birth attendants, and difficulty in accessing birthing centers and emergency obstetrical care units. Maternal death is a global public-health tragedy that could be mitigated with the application of modest resources.
SUMMARY
With improved diagnostic and therapeutic modalities as well as advances in the treatment of infertility, more patients with medical complications will be seeking and will require complex obstetric care. Improved outcomes of pregnancy in these women will be best attained by a team of internists, maternal-fetal medicine (high-risk obstetrics) specialists, and anesthesiologists assembled to counsel these patients about the risks of pregnancy and to plan their treatment prior to conception. The importance of preconception counseling cannot be overstated. It is the responsibility of all physicians caring for women in the reproductive age group to assess their patients’ reproductive plans as part of their overall health evaluation.





