Meconium Disease
Intestinal obstruction is one of the most common admitting diagnoses to the neonatal intensive care unit, accounting for as many as one-third of all admissions.1 Failure to pass meconium within the first 24 to 48 hours of life, feeding intolerance, abdominal distension, and bilious emesis are hallmarks of intestinal obstruction in the newborn, and evoke a differential diagnosis of obstruction based on anatomic, metabolic, and functional considerations. The term meconium disease refers to meconium ileus and meconium plug syndrome. These conditions are considered separately from functional or anatomic causes of neonatal intestinal obstruction, such as Hirschsprung disease, intestinal atresia, and anorectal malformations.
Meconium Ileus
Meconium ileus (MI) is one of the most common causes of intestinal obstruction in the newborn, accounting for 9–33% of neonatal intestinal obstructions.2 It is characterized by extremely viscid, protein-rich, inspissated meconium causing an intraluminal obstruction in the distal ileum, usually at the ileocecal valve. It is often the earliest clinical manifestation of cystic fibrosis (CF), occurring in approximately 16% of patients with CF.3 Although MI can occur with other uncommon conditions such as pancreatic aplasia and total colonic aganglionosis, it is often considered pathognomonic for CF.4,5 MI may be an early indication of a more severe phenotype of cystic fibrosis, as suggested by significantly diminished pulmonary function found in children with a history of MI compared to age- and gender-matched children with CF who did not have MI.6
Due to abnormalities of exocrine mucus secretion and pancreatic enzyme deficiency, the meconium in MI differs from normal meconium. Meconium in MI has less water content (65% vs 75%) when compared to normal meconium, lower sucrase and lactase levels, increased albumin, and decreased pancreatic enzymes.7–9 Additionally, concentrations of sodium, potassium, magnesium, heavy metals, and carbohydrates in MI meconium are reduced in CF. Concentrations of protein nitrogen are increased and composed of abnormal mucoproteins.10–12 Therefore, more viscous intestinal mucus in the absence of degrading enzymes results in thick, dehydrated meconium that obstructs the intestine.13
Cystic Fibrosis
An understanding of CF is important for all clinicians involved in the management of MI patients. CF is the most common, potentially lethal genetic defect affecting Caucasians. Each year 1,200 infants are born with CF (1 : 2500 live births), and 30,000 children and young adults live with CF in the USA.4 It is an inherited autosomal recessive disease with a 4–5% carrier rate.14 The incidence of CF is much lower in non-Caucasian populations: 1 in 10,500 Native American Aleut (Eskimo) births, 1 in 13,500 in Hispanic–Caucasian births, 1 in 15,000 African–American births (much lower in native Africans), and 1 in 31,000 in Asian–American births.
Genetics
In 1989, the CF locus was localized through linkage analysis to human chromosome 7q31, and it was discovered that mutations in the CF transmembrane (conductance) regulator (CFTR) gene result in CF.15–18 The cell membrane protein coded by CFTR is a 3′–5′-cyclic adenosine monophosphate (cAMP)-induced chloride channel, which also regulates the flow of other ions across the apical surface of epithelial cells. The alteration in CFTR results in an abnormal electrolyte content in the environment external to the apical surface of epithelial membranes. This leads to desiccation and reduced clearance of secretions from tubular structures lined by affected epithelia.
The most common mutation of the CFTR gene, F508del (previously known as ΔF508), is a three base-pair deletion that results in the removal of a phenylalanine residue at amino acid position 508 of the CFTR. Although there are currently 1,903 mutations listed in the CFTR database, the F508del mutation is responsible for approximately 70% of abnormal CF genes.15,16,18,19 In families with MI, there is a significantly higher occurrence rate than the expected 25% for an autosomal recessive genetic disorder.20,21 In one series, 79% of CF patients with the F508del mutation presented with abdominal complaints (including MI) rather than pulmonary complaints.4 However, there is no evidence of distinct allelic frequencies or haplotypic variants in CF patients with MI compared with those without22 or in CF patients with significant liver disease.23,24
Gastrointestinal Pathophysiology
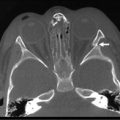
Cystic fibrosis is characterized by mucoviscidosis of exocrine secretions throughout the body resulting from abnormal transport of chloride ions across apical membranes of epithelial cells.4,25–27 Abnormal bicarbonate transport also affects mucin formation in CF.28 The clinical result is chronic obstruction and infection of the respiratory tract, insufficiency of the exocrine pancreas, and elevated sweat chloride levels.29 Other clinical variants, such as patients with chronic sinusitis or adult males with congenital bilateral absence of the vas deferens (CBAVD), who typically have little other clinical involvement, have been described (Fig. 32-1).30–32 In patients with CBAVD, the CFTR genotype usually includes at least one mild mutation not typical of CF patients. The mild-mutation allele is frequently associated with a severe mutation on the other allele, such as the F508del mutation.6,33 CBAVD has been described in a patient with F508del and G551D mutations, both of which were categorized as severe.34 The allele G551D is the third most common CF-associated mutation, and patients affected by this mutation may have pancreatic insufficiency, pulmonary symptoms, and an episode of MI equivalent, indicating CBAVD may be associated with a more severe CF phenotype.7,35
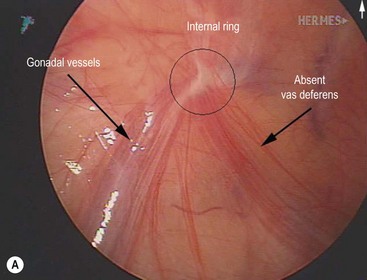
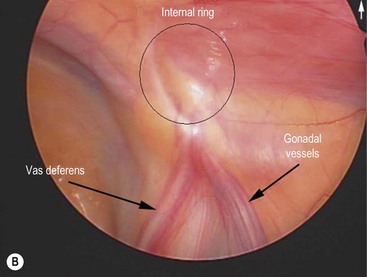
FIGURE 32-1 Congenital bilateral absence of the vas deferens (CBAVD). (A) Laparoscopic view of a patient’s left internal ring. (B) Compare with a laparoscopic view of a similar-aged patient’s right internal ring with a normal vas deferens. (From Escobar MA, Lau ST, Glick PL. Congenital bilateral absence of the vas deferens. J Pediatr Surg 2008;43:1222–3.)
Development of both the pancreas and intestinal tract in fetuses with CF is abnormal. In patients with CF, abnormal pancreatic secretions obstruct the ductal system leading to autodigestion of the acinar cells, fatty replacement of pancreatic parenchyma, and fibrosis. Although this process begins in utero, it occurs variably over time. Regardless, pancreatic insufficiency is prevalent in young infants with CF and has a significant impact on growth and nutrition.23
Pancreatic insufficiency plays a central role in the pathogenesis of MI. Congenital stenosis of the pancreatic ducts is associated with meconium-induced bowel obstruction.36 This is further supported by the fact that two-thirds of infants found to have CF by neonatal screening are pancreatic insufficient at birth.37 However, approximately 10% of patients with CF are pancreatic sufficient and tend to have a milder course. Also, pancreatic lesions are variable at birth and become more severe in CF children older than age 1.38 This finding suggests that pancreatic insufficiency is not the leading cause of abnormal meconium in MI. It appears that a prevalence of intestinal glandular abnormalities contribute more significantly to the production of abnormal meconium.39 The lack of concordance between MI and the severity of pancreatic disease and the preponderance of intestinal glandular lesions implies that intraluminal intestinal factors contribute more to the development of MI than the absence of pancreatic secretions.9,11,12,36–43
Abnormal intestinal motility may also contribute to the development of MI. Some patients with CF have prolonged small intestinal transit times.44,45 Also, the CFTR ion channel defect results in an exocrine secretion that is rich in sodium and chloride which can lead to further dehydration of the intraluminal contents, resulting in impaired clearance.6 Non-CF diseases associated with abnormal gut motility, such as Hirschsprung disease and chronic intestinal pseudo-obstruction, have been associated with MI-like disease, signifying that decreased peristalsis may allow for increased reabsorption of water, thus favoring the development of abnormal meconium.46–48
Prenatal Diagnosis and Screening
The American College of Obstetrics and Gynecology recommends all women of reproductive age should be offered CF carrier screening.14 Based on the results of CF screening, the antenatal diagnosis of MI can be made in two different groups: a high-risk group and a low-risk group. In the low-risk group, the diagnosis is suspected when the sonographic appearances of MI are found on routine prenatal ultrasound in a mother with a negative CF carrier screen. Sonographic findings consistent with MI in a fetus with parents who are known carriers of CF, and pregnancies subsequent to the birth of a CF-affected child, are considered high risk. Parents of a child with CF are considered to be obligate carriers of a CF mutation.
An algorithm has been established which may be useful in decision making and management of the fetus suspected of having MI (Fig. 32-2).49–51 If both parents are carriers, evaluation of the fetus should be made by chorionic villus sampling or amniocentesis. In a pregnancy where CF is suspected, sonographic examinations are performed monthly until delivery. This evaluation allows the early detection of potential complications and prepares the clinicians for special or urgent medical or surgical needs upon delivery.
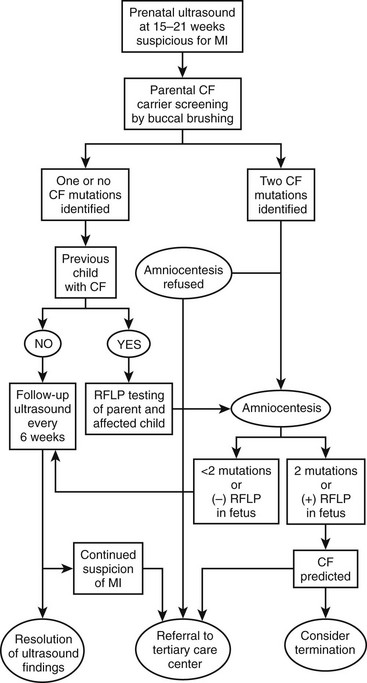
FIGURE 32-2 Suggested algorithm for antenatal management of suspected meconium ileus (MI) and cystic fibrosis (CF). US, ultrasonography; RFLP, restriction fragment length polymorphism. (Adapted from Irish MS, Ragi JM, Karamanoukian H, et al. Prenatal diagnosis of the fetus with cystic fibrosis and meconium ileus. Pediatr Surg Int 1997;12:434–6.)
Sonographic Evaluation
Sonographic characteristics associated with MI include a hyperechoic, intra-abdominal mass (inspissated meconium) (Fig. 32-3), dilated bowel, and nonvisualization of the gallbladder.52 Normal fetal meconium, when visualized in the second and third trimesters, is usually hypoechoic or isoechoic to adjacent abdominal structures.52–57 The sensitivity of intra-abdominal echogenic masses in the detection of MI/CF is reported to be between 30–70%.57 In addition to MI, hyperechoic bowel has been reported with Down syndrome, intrauterine growth retardation, prematurity, in utero cytomegalovirus infection, intestinal atresia, abruptio placenta, and fetal demise.52–55,58–64 The importance of hyperechoic fetal bowel is related to gestational age at detection, ascites, calcification, volume of amniotic fluid, and the presence of other fetal anomalies.57 The positive predictive value of hyperechoic masses in a high-risk fetus is estimated to be 52%, but is only 6.4% in the low-risk fetus.52 It is important to note that hyperechoic bowel has been found to be a normal variant in both the second and third trimesters.56,57,65
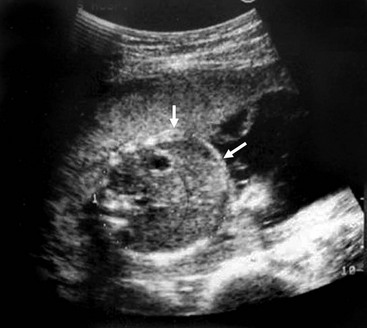
FIGURE 32-3 Ultrasound image of a 22-week gestation demonstrating a 2 cm × 3 cm intraluminal (distal ileum) mass (arrows) consistent with meconium inspissation (meconium ileus). (From Irish MS, Ragi JM, Karamanoukian H, et al. Prenatal diagnosis of the fetus with cystic fibrosis and meconium ileus. Pediatr Surg Int 1997;12:434–6.)
The finding of dilated bowel on prenatal ultrasound (US), in association with a family history of CF, has been reported less frequently than that of hyperechoic bowel. In MI, bowel dilation is caused by obstruction from meconium, but mimics findings in midgut volvulus, congenital bands, intestinal atresia, intestinal duplication, internal hernia, meconium plug syndrome, and Hirschsprung disease.66 The correlation of dilated fetal bowel and MI suggests that dilated fetal bowel warrants parental testing for CF and continued sonographic surveillance of the fetus.
The inability to visualize the gallbladder on fetal ultrasound has also been associated with CF.67 Combined with other sonographic features, nonvisualization of the gallbladder can be useful in the prenatal detection of the disease. However, caution should be exercised in the interpretation of an absent gallbladder as the differential diagnosis also includes biliary atresia, omphalocele, diaphragmatic hernia, chromosomal abnormalities, and a normal pregnancy.
Clinical Presentation
MI is categorized as either simple or complicated. The thickened meconium begins to form in utero. As it obstructs the mid-ileum, proximal bowel dilatation and thickening, along with congestion, occur. Approximately one-half of these neonates present with simple uncomplicated obstruction.4 The remaining patients present with complications of MI, including volvulus, gangrene, atresia, and/or perforation, which may result in meconium peritonitis and giant cystic meconium peritonitis.68–73
Simple Meconium Ileus
In simple MI, the terminal ileum is filled with firm concretions. The bowel in this area is small in diameter and molds around the inspissated lumps of meconium. The ileum becomes dilated and is filled with thick sticky meconium with gas and fluid found within the small bowel proximal to this area.4 Newborns with uncomplicated MI often appear healthy immediately after birth. However, within 1 to 2 days, they develop abdominal distension and bilious emesis. Normal meconium will not be passed. Eventually, dilated loops of bowel become visible on exam and have a ‘doughy’ character that indent on palpation. The rectum and anus are often narrow, a finding that may be misinterpreted as anal stenosis. The presentation of the baby with MI is similar to many types of neonatal small bowel obstruction. Therefore, the clinician should simultaneously consider malrotation, small intestinal atresia, colonic atresia, and meconium plug syndrome. The history, physical examination, and contrast enema help distinguish between these entities.
Complicated Meconium Ileus
Infants with complicated MI present with symptoms within 24 hours of birth. Some newborns are symptomatic immediately after birth as a result of in utero perforation or bowel compromise. Signs of peritonitis, including distension, tenderness, abdominal wall edema and erythema, and clinical evidence of sepsis may be found on the initial neonatal exam. Abdominal distension can be so severe as to cause immediate respiratory distress. A palpable mass suggests pseudocyst formation, which results from in utero bowel perforation.74,75 The neonate may present in extremis and need urgent resuscitation and operative exploration.
Historically, segmental volvulus was reported to be the most common complication of MI.68,69 Prenatal volvulus of the meconium-distended segment of ileum may lead to interruption of the mesenteric blood flow, which can result in ischemic necrosis, intestinal atresia with an associated mesenteric defect, or perforation. When an in utero perforation occurs, most of the sterile meconium is reabsorbed with trace amounts becoming calcified. Atretic segments are common in MI and the affected bowel may appear viable, showing no evidence of perforation or gangrene. 12–17% of neonates born with jejunoileal atresia have CF.4,76,77 Therefore, all neonates with jejunoileal atresia and an abnormal meconium presentation (MI, meconium plug syndrome, giant cystic meconium peritonitis, etc.) should undergo a sweat chloride test.4
The incidence of CF in neonates with meconium peritonitis is reported to be 15–40%.78 Four types of meconium peritonitis have been recognized including: adhesive meconium peritonitis, giant cystic meconium peritonitis or pseudocyst, meconium ascites, and infected meconium peritonitis.79 In addition to MI, other causes of in utero bowel perforation must also be considered (atresia, stenosis, colonic disorders, imperforate anus) in this clinical setting. The differences in clinical presentation are secondary to the timing of the perforation and whether or not the perforation sealed spontaneously. The site of perforation is usually closed by birth. Not surprisingly, mortality is increased in cases where the perforation remains open.79 Initially, meconium peritonitis is a nonbacterial, chemical, and foreign body peritonitis occurring during gestation. As meconium escapes the obstructed bowel, a sterile chemical peritonitis ensues. After delivery, bacterial superinfection may occur with colonization of the gastrointestinal tract. It is important to note that meconium peritonitis can also occur without MI and is not pathognomonic for CF.4,75,79
Radiographic Features
Simple MI is characterized by a pattern of unevenly dilated loops of bowel on an abdominal radiograph with the variable presence of air–fluid levels.69,80,81 The absence of air–fluid levels is due to the viscosity of the meconium not allowing an air interface with the fluid. As swallowed air mixes with the tenacious meconium, bubbles of gas may be seen. This soap bubble appearance (Fig. 32-4) depends on the viscosity of the meconium and is not a constant feature.71,80,81 While each of these features alone is not diagnostic of MI, collectively with a family history of CF, they strongly suggest the diagnosis.
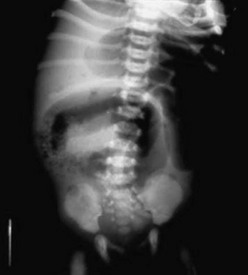
FIGURE 32-4 This abdominal radiograph in a neonate with meconium ileus shows the typical ground-glass appearance in the right lower abdomen. Also note the different-sized loops of distended small bowel.
Radiographic findings in complicated MI vary with the complication. Prenatal ultrasound findings include ascites, intra-abdominal cystic masses, dilated bowel, and calcification.82 Neonatal radiographic findings may include peritoneal calcifications, free air, and/or air-fluid levels (related to atresia).4 Air–fluid levels may be minimally present or absent, misleading the clinician to make an incorrect diagnosis of uncomplicated MI. Speckled calcification on abdominal plain films is highly suggestive of intrauterine intestinal perforation and meconium peritonitis. Radiographic findings of obstruction and a large dense mass with a rim of calcification imply a pseudocyst (Fig. 32-5). These calcium deposits are linear and course along the parietal peritoneum and serosal surface of the visceral organs.83 Interestingly, one-third of cases of complicated MI have no radiologic findings that suggest a complication.84
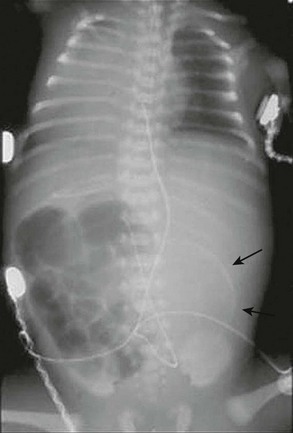
FIGURE 32-5 This neonate presented with evidence of meconium peritonitis. A mass effect in the left abdomen with a rim of calcification (arrows) implies in utero perforation and a pseudocyst.
A contrast enema should be performed in all cases of low intestinal obstruction in the newborn. We advocate an initial water-soluble contrast enema for both diagnosis and treatment. In MI, contrast instillation is monitored fluoroscopically and demonstrates a colon of small caliber, described as the ‘microcolon of disuse,’ often containing small, inspissated rabbit pellets (scybala) of meconium (Fig. 32-6). The enema also identifies cecal position, indicating whether malrotation is present. In complicated cases, such as atresia, a microcolon with reflux into a decompressed terminal ileum may be noted.4 If contrast cannot be refluxed into the dilated small bowel, operative exploration is required for diagnosis and therapy.
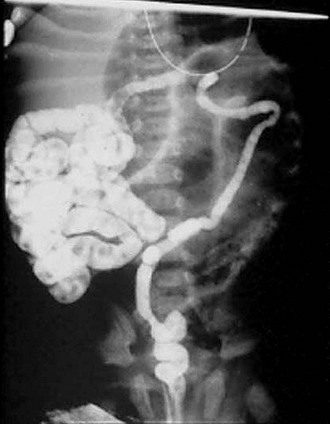
FIGURE 32-6 Classic radiographic findings of meconium ileus are seen on this retrograde contrast study. First, a ‘microcolon of disuse’ is seen. The colon is extremely small and unused. Second, inspissated pellets (filling defects) of meconium are seen in the more proximal small bowel. Third, note there is a small bowel obstruction as the contrast material has not reached the markedly dilated loops of small bowel.
Diagnostic Testing
The diagnosis of CF is established with a sweat test. A sodium concentration of 60 mmol/L in 100 mg of sweat is diagnostic of CF with 40–60 mmol/L being intermediate (but more likely to be diagnostic in infants) and less than 40 mmol/L being normal.85 The test is typically performed at several weeks of life to obtain an adequate sample size. Neonatal CF screening programs using the Guthrie blood spot test for raised concentrations of immunoreactive trypsinogen is available in many countries, but must be confirmed in a two-stage approach incorporating CFTR mutation analysis.86,87 Genetic testing for CFTR mutations is available, however, commercial assays test for a limited number of mutations. Most regional laboratories will provide the results for the four or five most common mutations for the relevant ethnic group or geographical region in their area using the amplification refractory mutation system (ARMS) technique. Stool analyses for albumin, trypsin, and chymotrypsin are available, and abnormal values coupled with operative findings suggest CF.37
Neonates with MI who fail to respond to nonoperative measures may be treated by appendectomy and irrigation with water-soluble contrast into the small bowel via the small bowel or appendiceal stump.88 The appendix (or other intestinal biopsy) may be sent for histologic analysis. Pathognomonic findings or histology for CF include goblet cell hyperplasia and accumulated secretions within crypts or lumen.89
Nonoperative Management of Simple Meconium Ileus
Neonates should initially be managed as any other newborn with intestinal obstruction. This management should include volume resuscitation and ventilator support as necessary. Gastric decompression to prevent progressive abdominal distension, aspiration, and pulmonary compromise is important. In addition, correction of any coagulation disorders and empiric broad-spectrum antibiotic coverage should be initiated.
Under fluoroscopic control, the water-soluble contrast material is slowly infused at low hydrostatic pressure through a catheter inserted into the rectum. Inflation of the catheter balloon should be avoided to minimize the risk of perforation. Upon completion, the catheter is withdrawn and an abdominal radiograph is obtained to evaluate for perforation. The infant is then returned to the neonatal care unit for intensive monitoring and fluid resuscitation. Usually there is rapid passage of meconium pellets followed by semi-liquid meconium, which continues in the ensuing 24–48 hours. Upon instillation of the enema, extraluminal fluid is drawn into the intestinal lumen, hydrating and softening the meconium mass. Warm saline enemas containing 1% N-acetylcysteine (Mucomyst; Apothecon, Princeton, New Jersey) may be given to help complete the evacuation.73 Radiographs should be obtained as clinically indicated to confirm evacuation of the obstruction and to exclude late perforation. If evacuation is incomplete, or if the first attempt at contrast enema evacuation does not reflux contrast into dilated bowel, a second enema may be necessary. However, if progressive distension, signs of peritonitis, or clinical deterioration occur, operative exploration is indicated. After two failed attempts at nonoperative water-soluble enemas, operative intervention is likely warranted.
Following successful evacuation and resuscitation, 5 mL of a 10% N-acetylcysteine solution may be administered every six hours through a nasogastric tube to liquefy the upper gastrointestinal secretions. Feedings with supplemental pancreatic enzymes for those infants confirmed with CF may be initiated when signs of obstruction have subsided. In the past, the success rate of patients with uncomplicated MI, treated with Gastrografin® enemas, has ranged between 63–83%.90,91 However, more recent reports indicate a much lower success rate likely secondary to the use of isotonic enema fluid.4,92
Several potential complications exist with the use of enemas in treating MI. The risk of rectal perforation can be avoided by careful placement of the catheter under fluoroscopic guidance and by not inflating the balloon-tipped catheter. A 23% perforation rate has been demonstrated in patients when inflated balloon catheters were used, and the risk of perforation increases with repeated enemas.93,94 Late perforation, occurring between 12 and 48 hours following the enema, can occur as well. Potential causes for late perforation include severe bowel distension by fluid osmotically drawn into the intestine or by injury to the bowel mucosa by the contrast medium.94 Lower perforation rates have been reported more recently, possibly related to less aggressive enema attempts and isotonic enema agents.4,92 Hypovolemic shock is a risk when delivering hypertonic enemas. Ischemia caused by overdistension is worsened by hypoperfusion caused by hypovolemia due to inadequate fluid resuscitation.95
Operative Management
Simple Meconium Ileus
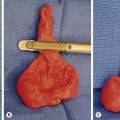
At laparotomy, manual evacuation of the inspissated meconium can be aided by intraoperative instillation of 2% or 4% N-acetylcysteine or saline solutions. These fluids can be passed antegrade through a nasogastric tube, retrograde through the appendiceal stump, or directly into the meconium through an enterotomy. A purse-string suture is placed in the antimesenteric wall of the bowel and a red rubber catheter is inserted through a small incision within the purse-string. This is followed by gentle instillation of the solution into the proximal bowel and terminal ileum to avoid perforation. Often the thick tenacious meconium can be removed directly through the enterotomy (Fig. 32-7). The dissolved meconium and pellets can be either removed directly or milked into the colon. It is important that the surgeon avoids exposure of the meconium to the peritoneum. Once the meconium is cleared, the enterotomy or appendiceal stump is closed. If necessary, an indwelling intestinal catheter or a T-tube may be left through the enterotomy for the purpose of postoperative bowel irrigation, decompression, pancreatic enzyme instillation, and/or feeding.96 The enterostomy tube should be positioned at the junction of the proximal dilated bowel and collapsed distal ileum. The irrigations are begun in the early postoperative period and after successful clearance of meconium, the tubes are removed and the enterocutaneous fistula is allowed to close spontaneously.96–100
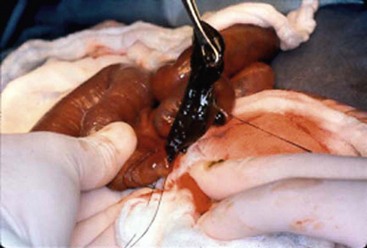
FIGURE 32-7 At operation, the meconium in a neonate with cystic fibrosis is very thick and tenacious.
Although uncommon, resection with primary anastomosis is occasionally required and was first described in 1962.101 Anastomotic leakage complicated early attempts with this approach, but improved results have been recently reported.102–104 Successful outcome following resection with primary anastomosis depends on adequate resection of compromised bowel, complete proximal and distal evacuation of meconium, and preservation of an adequate blood supply.
Other surgical approaches involve resection, anastomosis, and temporary enterostomy through which postoperative irrigations can be delivered (Fig. 32-8). Several stomas have been used: the Mikulicz double-barreled enterostomy, the Bishop-Koop distal chimney enterostomy, and the Santulli and Blanc proximal enterostomy.98,104 Disadvantages of these and other procedures employing resection and stoma(s) include potential high-volume stoma output, bowel length loss due to resection, and the need for a second procedure to re-establish intestinal continuity.
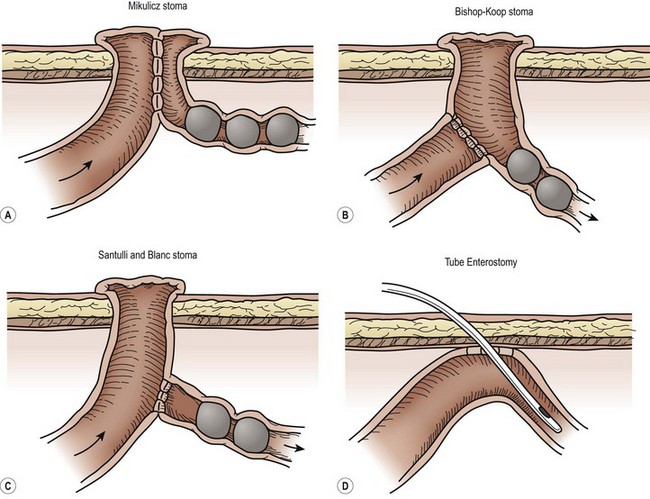
FIGURE 32-8 There are a number of options for surgical management of a neonate with meconium ileus. Options for creation of an ileostomy include the (A) double-barrel ileostomy (Mikulicz enterostomy), (B) the Bishop-Koop ileostomy, and (C) the Santulli ileostomy. Both the Bishop-Koop and Santulli ileostomies require an intra-abdominal anastomosis. (D) Another option is to place a red rubber catheter into the uninvolved proximal small bowel for postoperative irrigations.
Complicated Meconium Ileus
Operative management is almost always required in cases of complicated MI. One exception is the rare in utero spontaneously sealed perforation with intact intestinal continuity and extraluminal intraperitoneal calcified meconium.68 Late findings include calcified meconium identified in a patent processus vaginalis during herniorrhaphy or incidentally on abdominal radiographs. Indications for operation include peritonitis, persistent intestinal obstruction, enlarging abdominal mass, and ongoing sepsis. Surgical management includes debridement of necrotic material, pseudocyst resection, diverting stoma(s), antibiotics, and meticulous postoperative care.73 Creation of an ostomy is usually the fastest and safest operative course, alleviating concern over bowel discrepancy, anastomotic leak/obstruction, and return of bowel activity. In cases with pseudocyst formation, decortication of the cyst wall is recommended if possible.74
Nutritional Management
Enteral feeds in infants with uncomplicated MI and CF may be initiated with breast milk or infant formula, along with supplemental pancreatic enzymes and vitamins.105,106 Caution must be used when prescribing enteric enzyme medication to patients with MI/CF. Treatment failures and complications include fibrosing colonopathy from excessive enzyme doses and MI equivalent, or distal intestinal obstruction syndrome (DIOS) from inadequate enzyme therapy or generic substitutions for proprietary medications.48,105,107–109 Often patients with a complicated postoperative course will require either continuous enteral feeds or total parenteral nutrition (TPN). Dilation of the small bowel by the obstructing meconium may lead to mucosal damage that could contribute to poor peristalsis or malabsorption. In patients with complicated MI or in those with significant loss of intestinal length, initiating the enteral feeding with a predigested, diluted formula at low continuous volumes is best. If this is well tolerated, the concentration should be increased followed by the volume. Pancreatic enzymes should be given with enteral feeds (even with predigested formula) starting at 2,000–4,000 lipase units per 120 mL of full strength formula. Capsules containing enteric-coated microspheres can be opened and the contents mixed with formula or applesauce in older infants. The microcapsules should not be crushed as this will expose the enzymes to the acid of the stomach where they will be destroyed. Uncrushed pancreatic enzymes should be given even with MCT-oil containing formulas.110
Gastric acid hypersecretion is seen in patients who have short bowel syndrome.111 An acidic intestinal environment inactivates pancreatic enzymes and prevents dissolution of enteric-coated microcapsules. H2-receptor antagonists or proton pump inhibitors may be used as an adjunct with pancreatic enzyme therapy in patients who have had significant bowel resections. Patients with excessive sweat and intestinal sodium losses may develop a total body sodium deficit. Urine sodium should be measured in infants with ileostomies, especially when there is growth failure, even if serum sodium levels are normal. Those with a urine sodium less than 10 mEq/L will need sodium (and possibly bicarbonate) supplementation.107,112
Prognosis
The prognosis for infants with MI was uniformly poor, despite operative treatment, prior to the mid-1900s. Early series reported mortality rates of 50–67%.98,99 The improved survival in infants with MI can be attributed to many factors. Advances in prenatal diagnosis, pulmonary and neonatal intensive care, nutrition, antibiotics, anesthesia, operative management, and an improved understanding of the pathophysiology and treatment of the CF complications have resulted in dramatic prognostic improvement for infants with both complicated and simple MI.22,113 Survival rates of 85 to 100% have been reported in uncomplicated MI, and up to 93% in complicated cases.4,84,113
Previously, it was thought that patients with CF presenting with MI have worse outcomes than those without MI. However, it is no longer clear if this is accurate. Several long-term follow-up studies of patients with MI report pulmonary function at age 13 years to be no different between those born with and those without MI.114,115 However, a recent prospective study found children with MI have worse lung function and more obstructive lung disease than those with CF but without MI.111 Furthermore, comparison of the nutritional status of a similar population of patients with CF suggests that those who presented with MI suffer long-term nutritional complications and other problems.116
Meconium Plug Syndrome
The meconium plug syndrome (MPS) was first described in 1956.117 It was hypothesized initially that either colonic motility or the character of the meconium was altered, thereby preventing its normal passage and subsequent decompression of the colon in the newborn period. Under normal conditions, the terminal two centimeters of neonatal meconium is firm in texture, forming a whitish cap. Most newborns pass this meconium cap before, during, or shortly after delivery. One in 500 newborns will have a longer, more tenacious obstructive plug. Failure to pass this plug results in MPS, and the term ‘plugged-up babies’ was coined.
Pathologic causes of MPS include CF, small left colon syndrome, and Hirschsprung disease.117–120 Less common causes include congenital hypothyroidism, maternal narcotic addiction, and neuronal intestinal dysplasia. A contrast enema may be therapeutic as well as diagnostic. Following resolution, a sweat test should be performed to exclude CF and a thryroid-stimulating hormone level should be obtained. A rectal biopsy should be performed to evaluate for Hirschsprung disease if there is a dysfunctional stooling pattern after resolution of the plug.73,118,119
Complications of Meconium Ileus and Cystic Fibrosis
Gastroesophageal Reflux Disease
GER occurs with increased prevalence in patients with CF. Aspiration in CF children may aggravate failure to thrive, adversely affect pulmonary function, and may account for the predilection of CF lung disease in the right upper lobe.121,122 Pathological reflux with endoscopic and histological esophagitis is present in more than 50% of CF patients and the incidence of GER in patients with CF is approximately 80% in patients younger than 5 years.123 It is clear that early diagnosis and treatment of GER is of prime importance if its complications are to be minimized and respiratory function maximized.
Antireflux medications, modification of chest physiotherapy, and eliminating the 30° head-down tilt may all decrease the incidence of GER in this population.124 Children unresponsive to medical management should undergo evaluation for an antireflux procedure. Our preferred approach is the laparoscopic Nissen fundoplication. Recent data suggests that a fundoplication may improve respiratory function (improved FEV1 slope) in CF children with mild versus moderate disease.125 Patients with symptomatic GER requiring an antireflux procedure may benefit from concurrent placement of a gastrostomy if inadequate caloric intake is problematic.
Barrett esophagus, a rare finding in children, has been reported in older children with CF.4,126 Although an antireflux procedure may halt the advancement of metaplasia, if dysplasia is present, the malignant potential remains.127 In cases with metaplasia, endoscopic monitoring is the same for patients with CF as those without.127 In adults, if high-grade dysplasia is confirmed by two pathologists and aggressive medical therapy fails to eliminate the dysplasia, esophagectomy is recommended. With so little data available in children with CF, at a minimum, it is reasonable that patients who have dysplastic esophageal changes should be evaluated for an antireflux procedure.
Biliary Tract Disease
Multiple macroscopic cysts may replace the pancreas in CF.27 Although it has been thought that hepatic and pancreatic dysfunction occurred together, hepatic dysfunction may occur in patients with normal pancreatic function.128 The most common hepatic complications of CF are steatosis, fibrosis, biliary cirrhosis, atretic gallbladder, cholelithiasis, sclerosing cholangitis, and biliary dyskinesia. Obstruction of intrahepatic biliary ductules by abnormal mucoid secretions or inspissated bile, resulting from the absence of functional CFTR in bile duct epithelial cells, results in the development of cirrhosis in patients with CF.128 When biopsied, the classic liver histology in CF is focal biliary fibrosis with progression to multilobular, biliary cirrhosis. Prolonged cholestatic liver disease in CF patients may lead to cirrhosis, portal hypertension, and ultimately liver failure and death without liver transplantation.
Though more common in older patients with CF, intrahepatic cholestasis can be seen in the neonate. In extreme forms, this process can be associated with a marked decrease in ductal diameter, varying from hypoplasia to atresia. Additionally, these neonates are at increased risk for cholestatic jaundice when they are not being fed enterally. Cholestatic jaundice is suggested by prolonged jaundice unresponsive to choleretics, nondilated bile ducts and gallbladder on ultrasound, absent biliary excretion on nuclear scan, and characteristic liver biopsy.129–131
End-stage liver disease (ESLD) is manifest by loss of synthetic function, growth failure, or portal hypertension presenting as variceal hemorrhage.132 Although abnormal liver function tests have been noted in 13% of CF patients, only 4.2% manifest overt liver disease (although the prevalence is as high as 37% depending on the definition of liver disease).133 Portosystemic shunts, transjugular intrahepatic portosystemic shunts (TIPS), partial splenectomy, and endoscopic injection sclerotherapy have been advocated in treating CF patients with portal hypertension.134 Other surgical options for these patients are direct ligation of the varices, esophageal transection, or the Sugiura procedure (gastric devascularization).135,136 These procedures are all palliative, with the only curative treatment for portal hypertension and end-stage liver disease being orthotopic liver transplantation (OLT).
Liver transplantation has been successfully carried out in CF patients with ESLD who did not have respiratory failure.137,138 There are several successful reports of combined liver and intestinal transplantation, combined liver and pancreas transplant, kidney transplant after combined heart and lung transplants, and triple organ transplant (pancreas, liver and kidney) in patients with exocrine pancreatic insufficiency and insulin-dependent diabetes related to CF.4,139,140 Long-term studies are demonstrating preservation or maintenance of respiratory function and nutritional status following OLT in patients with CF.137,138
Gallbladder disease is prevalent in the CF population, including cholelithiasis in up to 24%, and abnormal cholecystograms in 46%.136,141,142 Other abnormalities include microgallbladder, atretic cystic duct, and hyperviscous mucus. Many CF patients with gallstones are asymptomatic, with the incidence of symptomatic gallbladder disease in CF reported to be approximately 4%.143 Because the stones are radiolucent, ultrasound rather than computed tomography (CT) is recommended in patients with CF.136,144 Bile in patients with CF is not cholesterol supersaturated; hence the stones are composed of protein and calcium bilirubinate.141
CF patients with symptomatic gallbladder disease (symptomatic cholelithiasis and/or acute cholecystitis) should undergo prompt cholecystectomy.144,145 Although of historical interest only, the complication rate with open cholecystectomy was quite low with aggressive pulmonary toilet.146,147 The laparoscopic approach is now the accepted standard.4 Due to the low incidence of common bile duct (CBD) stones in CF patients, routine intraoperative cholangiograms or preoperative endoscopic retrograde cholangiopancreatography (ERCP) are not needed.4,147 In fact, the biliary tract abnormalities often encountered in patients with CF make penetration of radiocontrast dye into the biliary tract during ERCP difficult.136 Intraoperative cholangiography is recommended if jaundice, pancreatitis, cholangitis, dilated CBD, or palpable stones in the CBD are present.4
Distal Intestinal Obstruction Syndrome
DIOS (formerly called MI equivalent) is a recurrent, partial or complete intestinal obstruction unique to teenage and young adult patients with CF that occurs secondary to abnormally viscid mucofeculent material in the distal ileum and right colon.148–151 The etiology of DIOS is unclear, but these patients are more likely to have a history of steatorrhea from pancreatic exocrine insufficiency despite adequate enzyme therapy. A number of aspects particular to gastrointestinal function in the CF patient may help to explain this syndrome. In addition to inherently slow intestinal motility, other contributing factors may include thickening of chyme secondary to the presence of undigested protein and fat, precipitation of undigested protein and bile acids in duodenal fluid with reduced pH, lower water content of pancreatic and duodenal secretions, hyperviscosity of mucus resulting from abnormal ion and water transport, abnormal regulation of mucin secretion, and altered biochemical properties of mucus glycoprotein.152–154 Precipitating factors include sudden withdrawal of (or noncompliance with) enzyme supplementation, immobilization, dehydration, respiratory tract infections, and recovery from surgery. However, in the majority of cases, no identifiable cause will be found.4
DIOS occurs in 15–37% of patients with CF, and is seen particularly in those with associated pancreatic insufficiency with malabsorption and severe pulmonary limitation.148,149,151 One study noted a 12% incidence in children with CF, with the majority (63%) having MI as an infant.4 Children with normal fat absorption are rarely affected.
A supine and erect abdominal film is the most useful initial investigation when DIOS is suspected (Fig. 32-9). This will show distended small bowel with scattered air-fluid levels and a granular, bubbly pattern of intestinal gas representing the mixing of air and inspissated meconium in the right lower quadrant, similar to infants with MI.
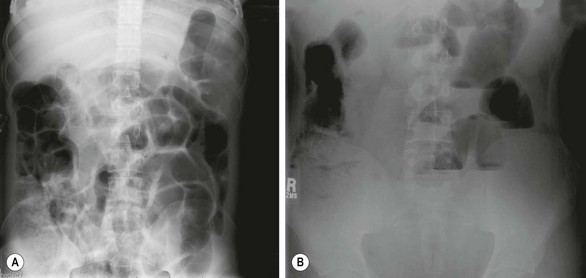
FIGURE 32-9 This 18-year-old with cystic fibrosis presented with crampy abdominal pain that was localized to the right lower abdomen. In addition, he also had a decreased frequency of defecation. (A) The ground-glass appearance in the loops of bowel on the right side are typical findings in a patient with distal intestinal obstruction syndrome. (B) The upright abdominal film shows air-fluid levels. This patient responded well to a contrast enema with relief of his symptoms.
In the absence of mechanical small bowel obstruction due to adhesions, intussusception, or appendiceal disease, a trial of medical management aimed at relieving the inspissated distal bowel obstruction is suggested. After adequate volume resuscitation and colonic enema washout, a balanced polyethylene glycol-electrolyte solution, such as GoLytely® or Colyte®, can be given orally or by nasogastric tube.155 The dose is 20–40 mL/kg/h with a maximum of 1200 mL/h. Alternatively, ingestion of a nonabsorbable intestinal lavage solution may produce the most striking results.156 Younger patients will usually require nasogastric tube placement whereas older children may be able to ingest sufficient volumes of lavage solution to relieve the impacted material.
The passage of stool, resolution of symptoms, and the disappearance of a previously palpable right iliac fossa mass imply successful treatment. Sequential abdominal radiographs will help to document the resolution of DIOS, but if symptoms persist then the differential diagnosis must be re-considered. Some authors have recommended DIOS prophylaxis with use of scheduled laxatives and high dietary fiber.148
When there is complete obstruction or evidence of peritonitis, an operation is necessary and oral or rectal therapies are contraindicated. A nasogastric tube should be passed for decompression and adequate resuscitative measures initiated. At laparotomy, the bowel wall will feel thickened and filled with tenacious material. It can be decompressed and irrigated via a small catheter placed through the appendiceal stump, as previously described for uncomplicated MI. It is also possible to leave an irrigating tube in situ to irrigate the bowel postoperatively. Some children may require lysis of adhesions and/or bowel resections with either primary anastomosis or creation of an ostomy.4
Intussusception
Intussusception occurs in approximately 1% of children with CF with the average age of onset of 9.5 years.151 In contrast, the average age of onset in children with idiopathic intussusception in the general pediatric population is 6–18 months.121 Toddlers and older children presenting with intussusception and a history of recurrent pulmonary infections should be tested for CF. The most common site for intussusception is ileocolic, but it may be ileoileal, cecocolic, or colocolic.122 The abnormally thick stool adheres to the bowel wall and acts as a lead point.157 The appendix may also be a lead point.158 Controversy exists over conservative management of intussusception in CF patients. Some report high rates of successful hydrostatic reduction, while others report less optimal results.157,159 If the intussusception is unable to be reduced operatively, a bowel resection with anastomosis is required. The appendix should be removed at operation in patients with an intussusception.
Fibrosing Colonopathy
Fibrosing colonopathy is a result of colonic strictures, and presents with signs and symptoms of DIOS.105–107,160,161 Histologic findings include colonic strictures with histopathologic changes of post-ischemic ulcer repair, erythematous cobblestone appearance to the mucosa, mucosal and submucosal fibrosis, and destruction of the muscularis mucosa. A change from conventional enteric-coated pancreatic enzymes to high-strength products 12–15 months before presentation has been described.105 In the largest case-control study reported, the absolute dose of pancreatic enzymes, rather than the type of enzyme, was the strongest predictor of fibrosing colonopathy.105
The diagnosis of fibrosing colonopathy should be considered in CF patients who have been exposed to high doses of pancreatic enzymes and present with symptoms of abdominal pain, distension, chylous ascites, change in bowel habit, or failure to thrive. Continued diarrhea may also be a prominent feature, which unfortunately may prompt the family to increase supplemental enzymes further. On occasion, the diarrhea may be bloody. A barium enema may reveal mucosal irregularity, loss of haustral markings with a foreshortened colon, and varying degrees of stricture formation. In some cases, the entire colon is involved. Colonoscopy may show an erythematous mucosa with areas of narrowing, from which it is advisable to take multiple biopsies.73
Outlook
The operative mortality in patients with CF has decreased considerably in the past three decades. The mortality rate for meconium ileus and peritonitis was 55% in the 1960s and 1970s.159 Moreover, there was a significant decrease in survival in the first year of life for these infants compared to those patients with CF who did not have MI.121 After the first year of life, survival among infants with MI approached that of other infants with CF. Recently, these statistics have improved dramatically. There are now reports documenting a 100% early survival and 86% late survival in patients with MI, and a 91.6% survival for uncomplicated MI and 85% for complicated cases at 1 year.113,114 Recently, survival for patients with simple MI was found to be 93% (early 100%, late 93%) and complicated 89% (early 96%, late 93%).4
Discussing the long-term needs of the patient with MI means discussing the long-term needs of the patient with CF. A multidisciplinary approach to the management of the surgical patient with CF including respiratory care, nutrition support, and pancreatic enzyme therapy allows for a low operative morbidity and mortality. Children with MI need long-term follow-up as they are prone to develop DIOS and fibrosing colonopathy. Furthermore, patients may be more prone to develop mechanical bowel obstructions later in life if operated on as an infant for MI.4 Other late complications of MI such as gallstones, cirrhosis, and male sterility may also be viewed as late complications of CF in general. Many patients with CF are now surviving into the third and even fourth decades of life. Hence, many of the surgical complications of CF occur later in life.
References
1. Lister, J. Intestinal obstruction: General considerations. Neonatal Surgery; vol. 3. Butterworth, London, 1990.
2. DeLorimier, AA, Fonkalsrud, EW, Hays, DM. Congenital atresia and stenosis of the jejunum and ileum. Surgery. 1969; 65:819–827.
3. FitzSimmons, SC. The changing epidemiology of cystic fibrosis. J Pediatr. 1993; 122:1–9.
4. Escobar, MA, Grosfeld, JL, Burdick, JJ, et al. Surgical considerations in Cystic Fibrosis: A 32-year evaluation of outcomes. Surgery. 2005; 138:560–572.
5. Stringer, MD, Brereton, RJ, Drake, DP, et al. Meconium ileus due to extensive intestinal aganglionosis. J Pediatr Surg. 1994; 23:501–503.
6. Evans, AK, Fitzgerald, DA, McKay, KO. The impact of meconium ileus on the clinical course of children with cystic fibrosis. Eur Respir J. 2001; 18:784–789.
7. Antonowicz, I, Lebenthal, E, Schwachman, H. Disaccharidase activities in small intestinal mucosa in patients with cystic fibrosis. J Pediatr. 1978; 92:214–219.
8. Schwachman, H, Antionowicz, I. Studies on meconium. In: E Lebenthal, ed. Gastroenterology and Nutrition in Infancy. New York: Raven Press; 1981:83–93.
9. Emery, J. Laboratory observations of the viscidity of meconium. Arch Dis Child. 1954; 29:34–37.
10. Di Sant’Agnese, PA, Dische, Z, Danilczenko, A. Physiochemical differences of mucoproteins in duodenal fluid of patients with cystic fibrosis of the pancreas and controls. Pediatrics. 1957; 19:252–260.
11. Green, M, Clarke, J, Shwachman, H. Studies in cystic fibrosis of the pancreas: Protein pattern in meconium ileus. Pediatrics. 1958; 21:635–641.
12. Stephan, U, Busch, EW, Kollberg, H, et al. Cystic fibrosis detection by means of a test-strip. Pediatrics. 1975; 55:35–38.
13. Eggermont, E, De Boeck, K. Small-intestinal abnormalities in cystic fibrosis patients. Eur J Pediatr Surg. 1991; 150:824–828.
14. American College of Obstetricians and Gynecologists Committee on Genetics. ACOG Committee Opinion No. 486: Update on carrier screening for cystic fibrosis. Obstet Gynecol. 2011; 117:1028–1031.
15. Rommens, JM, Iannuzzi, MC, Kerem, BS, et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science. 1989; 245:1059–1065.
16. Kerem, BS, Rommens, JM, Buchanan, JA, et al. Identification of the cystic fibrosis gene: Genetic analysis. Science. 1989; 245:1073–1080.
17. Welsh, MJ, Anderson, MP, Rich, DP, et al. Cystic fibrosis transmembrane conductance regulator: A chloride channel with novel regulation. Neuron. 1992; 8:821–829.
18. Riordan, JR, Rommens, JM, Kerem, B, et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA [published erratum appears in Science 1989; 245:1437. Science. 1989; 245:1066–1073.
19. Cystic fibrosis mutation database statistics [Internet]. Toronto: Cystic Fibrosis Centre at the Hospital for Sick Children in Toronto; 2012—[cited February 8, 2012]. Available from http://www.genet.sickkids.on.ca/StatisticsPage.html.
20. Mornet, E, Simon-Bouy, B, Serre, JL, et al. Genetic differences between cystic fibrosis with and without meconium ileus. Lancet. 1988; 1:376–378.
21. Fanconi, G, Uehlinger, E, Knauer, C. Das Coeliakiesyndrom bei angeborener zystischer pancreasfibromatose und bronchiektasien. Wien Med Wochenschr. 1936; 27/28:753–756.
22. Kerem, E, Corey, M, Kerem, B, et al. Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J Pediatr. 1989; 114:767–773.
23. Bronstein, MN, Sokol, RJ, Abman, SH, et al. Pancreatic insufficiency, growth, and nutrition in infants identified by newborn screening as having cystic fibrosis. J Pediatr. 1992; 120:533–540.
24. Duthie, A, Doherty, DG, Williams, C, et al. Genotype analysis for delta F508, G551D and R553X mutations in children and young adults with cystic fibrosis with and without chronic liver disease. Hepatology. 1992; 15:660–664.
25. Wilmott, RW, Tyson, SL, Dinwiddie, R, et al. Survival rates in cystic fibrosis. Arch Dis Child. 1983; 38:835–836.
26. Rescorla, FJ, Grosfeld, JL. Contemporary management of meconium ileus. World J Surg. 1993; 17:318–325.
27. Westwood, ATR, Ireland, JD, Bowie, MD. Surgery in cystic fibrosis—a 20-year review. S Afr J Surg. 1997; 35:181–184.
28. Quinton, PM. Cystic fibrosis: Impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008; 372:415–417.
29. Boat, T, Welsh, M, Beaudet, A. Cystic Fibrosis. In: Scriver C, Beaudet A, Sly W, Valle D, eds. The Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 1989:2649–2680.
30. Dohle, GR, Veeze, HJ, Overbeek, SE, et al. The complex relationships between cystic fibrosis and congenital bilateral absence of the vas deferens: Clinical, electrophysiological and genetic data. Hum Reprod. 1999; 14:371–374.
31. Jarvi, K, McCallum, S, Zielenski, J, et al. Heterogeneity of reproductive tract abnormalities in men with absence of the vas deferens: Role of cystic fibrosis transmembrane conductance regulator gene mutations. Fertil Steril. 1998; 70:724–728.
32. Shin, D, Gilbert, F, Goldstein, M, et al. Congenital absence of the vas deferens: Incomplete penetrance of cystic fibrosis gene mutations. J Urol. 1997; 158:1794–1799.
33. Daudin, M, Bieth, E, Bujan, L, et al. Congenital bilateral absence of the vas deferens: Clinical characteristics, biological parameters, cystic fibrosis transmembrane conductance regulator gene mutations, and implications for genetic counseling. Fertil Steril. 2000; 74:1164–1174.
34. Escobar, MA, Lau, ST, Glick, PL. Congenital bilateral absence of the vas deferens. J Pediatr Surg. 2008; 43:1222–1223.
35. Bompadre, SG, Sohma, Y, Li, M, Hwang, TC. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J Gen Physiol. 2007; 129:285–298.
36. Farber, S. The relation of pancreatic achylia to meconium ileus. J Pediatr. 1944; 24:387–392.
37. Kerem, E, Corey, M, Kerem, BS, et al. The relation between genotype and phenotype in cystic fibrosis–analysis of the most common mutation (delta F508). N Engl J Med. 1990; 323:1517–1522.
38. Hinen, E. Meconium ileus with no pancreatic abnormality. Arch Dis Child. 1950; 25:99–100.
39. Thomaidis, TS, Arey, JB. The intestinal lesions in cystic fibrosis of the pancreas. J Pediatr. 1963; 63:444–453.
40. Glanzmann, E. Dysporia entero-bronco-pancreatica congenita familiaris. Ann Paediat. 1946; 166:289.
41. Buchanan, D, Rapoport, S. Chemical comparison of normal meconium and meconium from patients with meconium ileus. Pediatrics. 1952; 9:304–310.
42. Bodian M, ed. Fibrocystic Disease of the Pancrease: Congenital disorder of mucous production-mucosis. New York: Grune and Stratton, Inc., 1953.
43. Foulkes, AG, Harris, A. Localization of expression of the cystic fibrosis gene in human pancreatic development. Pancreas. 1993; 8:3–6.
44. Bali, A, Stableforth, DE, Asquith, P. Prolonged small-intestinal transit time in cystic fibrosis. Br Med J (Clin Res Ed). 1983; 287:1011–1013.
45. Dalzell, AM, Freestone, NS, Billington, D, et al. Small intestinal permeability and orocaecal transit time in cystic fibrosis. Arch Dis Child. 1990; 65:585–588.
46. Toyosaka, A, Tomimoto, Y, Nose, K, et al. Immaturity of the myenteric plexus is the aetiology of meconium ileus without mucoviscidosis: A histopathologic study. Clin Auton Res. 1994; 4:175–184.
47. Emery, JL. Colonic retention syndrome (megacolon) associated with immaturity of intestinal intramural plexus. Proc R Soc Med. 1973; 66:222–223.
48. Wilcox, DT, Borowitz, DS, Stovroff, MC, et al. Chronic intestinal pseudo-obstruction with meconium ileus at onset. J Pediatr. 1993; 123:751–752.
49. Irish, MS, Gollin, Y, Borowitz, DS, et al. Meconium Ileus: Antenatal diagnosis and perinatal care. Fetal Matern Med Rev. 1996; 8:79–83.
50. Irish, MS, Ragi, JM, Karamanoukian, H, et al. Prenatal diagnosis of the fetus with cystic fibrosis and meconium ileus. Pediatr Surg Int. 1997; 12:434–436.
51. Irish, M, Boriwitz, DS, Glick, PL. Meconium Ileus. In: Ziegler MM, Azizkhan RG, Gauderer MWL, Weber TR, eds. Operative Pediatric Surgery. Stamford: Appleton & Lange, Co., 2003.
52. Dicke, JM, Crane, JP. Sonographically detected hyperechoic fetal bowel: Significance and implications for pregnancy management. Obstet Gynecol. 1992; 80:778–782.
53. Denholm, TA, Crow, HC, Edwards, WH, et al. Prenatal sonographic appearance of meconium ileus in twins. AJR Am J Roentgenol. 1984; 143:371–372.
54. Caspi, B, Elchalal, U, Lancet, M, et al. Prenatal diagnosis of cystic fibrosis: Ultrasonographic appearance of meconium ileus in the fetus. Prenat Diagn. 1988; 8:379–382.
55. Benacerraf, BR, Chaudhury, AK. Echogenic fetal bowel in the third trimester associated with meconium ileus secondary to cystic fibrosis. A case report. J Reprod Med. 1989; 34:299–300.
56. Fakhry, J, Reiser, M, Shapiro, LR, et al. Increased echogenicity in the lower fetal abdomen: A common normal variant in the second trimester. J Ultrasound Med. 1986; 5:489–492.
57. Lince, DM, Pretorius, DH, Manco-Johnson, ML, et al. The clinical significance of increased echogenicity in the fetal abdomen. AJR Am J Roentgenol. 1985; 145:683–686.
58. Goldstein, RB, Filly, RA, Callen, PW. Sonographic diagnosis of meconium ileus in utero. J Ultrasound Med. 1987; 6:663–666.
59. Boue, A, Muller, F, Nezelof, C, et al. Prenatal diagnosis in 200 pregnancies with a 1-in-4 risk of cystic fibrosis. Hum Genet. 1986; 74:288–297.
60. Bromley, B, Doubilet, P, Frigoletto, FD, Jr., et al. Is fetal hyperechoic bowel on second-trimester sonogram an indication for amniocentesis? Obstet Gynecol. 1994; 83:647–651.
61. Nyberg, DA, Resta, RG, Luthy, DA, et al. Prenatal sonographic findings of Down syndrome: Review of 94 cases. Obstet Gynecol. 1990; 76:370–377.
62. Gollin, Y, Shaffer, W, Gollin, G, et al. Increased abdominal echogenicity in utero: A marker for intestinal obstruction. Am J Obstet Gynecol. 1993; 168:349.
63. Bahado-Singh, R, Morotti, R, Copel, JA, Mahoney, MJ. Hyperechoic fetal bowel: The perinatal consequences. Prenat Diagn. 1994; 14:981–987.
64. Forouzan, I. Fetal abdominal echogenic mass: An early sign of intrauterine cytomegalovirus infection. Obstet Gynecol. 1992; 80:535–537.
65. Paulson, EK, Hertzberg, BS. Hyperechoic meconium in the third trimester fetus: An uncommon normal variant. J Ultrasound Med. 1991; 10:677–680.
66. De Backer, AI, De Schepper, AM, Deprettere, A, et al. Radiographic manifestations of intestinal obstruction in the newborn. JBR-BTR. 1999; 82:159–166.
67. Duchatel, F, Muller, F, Oury, JF, et al. Prenatal diagnosis of cystic fibrosis: Ultrasonography of the gallbladder at 17–19 weeks of gestation. Fetal Diagn Ther. 1993; 8:28–36.
68. Rescorla, FJ, Grosfeld, JL, West, KJ, et al. Changing patterns of treatment and survival in neonates with meconium ileus. Arch Surg. 1989; 124:837–840.
69. Donnison, AB, Shwachman, H, Gross, RE. A review of 164 children with meconium ileus seen at the Children’s Hospital Medical Center, Boston. Pediatrics. 1966; 37:833–850.
70. Holsclaw, DS, Eckstein, JB, Nixon, HH. Meconium ileus: 20 year review of 109 cases. AMA M J Dis Child. 1965; 109:101–113.
71. Leonidas, JC, Berdon, WE, Baker, DH, et al. Meconium ileus and its complications. A reappraisal of plain film roentgen diagnostic criteria. Am J Roentgenol Radium Ther Nucl Med. 1970; 108:598–609.
72. Santulli, T, Blanc, W. Congenital atresia of the intestine: Pathogenesis and treatment. Ann Surg. 1961; 154:939–948.
73. Rescorla, FJ, Grosfeld, JL. Contemporary management of meconium ileus. World J Surg. 1993; 17:318–325.
74. Careskey, JM, Grosfeld, JL, Weber, TR, et al. Giant cystic meconium peritonitis (GCMP): Improved management based on clinical and laboratory observations. J Pediatr Surg. 1982; 17:482–489.
75. Andrassy, R, Nirgiotis, J. Meconium disease of infancy: Meconium ileus, meconium plug syndrome,and meconium peritonitis. In: Holder T, Ashcraft K, eds. Pediatric Surgery. Philadelphia: W. B. Saunders; 1990:331–340.
76. Rescorla, FJ, Grosfeld, JL. Intestinal atresia and stensosis: Analysis of survival in 120 cases. Surgery. 1985; 98:668–676.
77. Dalla Vecchia, LK, Grosfeld, JL, West, KW, et al. Intestinal atresia and stenosis: a 25-year experience with 277 cases. Arch Surg. 1998; 1333:490–497.
78. Lloyd, D. Meconium ileus. In: Welch K, Randolph J, Ravitch M, et al, eds. Pediatric Surgery. 4th ed. Chicago: Year Book Medical Publishers, Inc.; 1986:849–858.
79. Martin, LW. Meconium peritonitis. In: Ravitch MM, Welch KJ, Benson CD, et al, eds. Pediatric Surgery. Chicago: Year Book Medical Publishers; 1979:952–955.
80. Speck, CR, Moore, TC, Stout, FE. Antenatal roentgen diagnosis of meconium peritonitis. Am J Radiol. 1962; 88:566–570.
81. Herson, R. Meconium ileus. Radiology. 1957; 68:568–571.
82. Foster, MA, Nyberg, DA, Mahony, BS, et al. Meconium peritonitis: Prenatal sonographic findings and their clinical significance. Radiology. 1987; 165:665–668.
83. White, H. Meconium ileus: A new roentgen sign. Radiology. 1956; 66:567–571.
84. Ziegler, MM. Meconium ileus. Curr Probl Surg. 1994; 31:731–777.
85. Gibson, LE, Cooke, RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics. 1959; 23:545–549.
86. Wallis, C. Diagnosing cystic fibrosis: Blood, sweat, and tears. Arch Dis Child. 1997; 76:85–88.
87. Southern, KW, Munck, A, Pollitt, R, et al. A survey of newborn screening for cystic fibrosis in Europe. J Cyst Fibros. 2007; 6:57–65.
88. Fitzgerald, R, Conlon, K. Use of the appendix stump in the treatment of meconium ileus. J Pediatr Surg. 1989; 24:899–900.
89. Oppenheimer, EH, Easterly, JR. Pathological evidence of cystic fibrosis patients with meconium ileus. Pediatr Res. 1973; 7:339.
90. Noblett, H. Meconium Ileus. In: Ravtch M, Welch K, Benson C, et al, eds. Pediatric Surgery. 3rd ed. Chicago: Year Book Medical Publishers; 1979:943–951.
91. Rowe, MI, Furst, AJ, Altman, DH, et al. The neonatal response to gastrografin enema. Pediatrics. 1971; 48:29–35.
92. Copeland, DR, St Peter, SD, Sharp, SW, et al. Diminishing role of contrast enema in simple meconium ileus. J Pediatr Surg. 2009; 44:2130–2132.
93. Ein, S, Shandling, B, Reilly, B, et al. Bowel perforation with nonoperative treatment of meconium ileus. J Pediatr Surg. 1987; 22:146–147.
94. Rowe, MI, Seagram, G, Weinberger, M. Gastrografin-induced hypertonicity. The pathogenesis of a neonatal hazard. Am J Surg. 1973; 125:185–188.
95. Lutzger, LG, Factor, SM. Effects of some water-soluble contrast media on the colonic mucosa. Radiology. 1976; 118:545–548.
96. Steiner, Z, Mogilner, J, Siplovich, L, et al. T-tubes in the management of meconium ileus. Pediatr Surg Int. 1997; 12:140–141.
97. Jawaheer, J, Khalil, B, Plummer, T, et al. Primary resection and anastomosis for complicated meconium ileus: A safe procedure? Pediatr Surg Int. 2007; 23:1091–1093.
98. Bishop, H, Koop, C. Management of meconium ileus: Resection, Roux-en-Y anastomosis and ileostomy irrigation with pancreatic enzymes. Ann Surg. 1957; 145:410–414.
99. Gross, R. The surgery of infants and childhood. Philadelphia: W. B. Saunders Co.; 1953.
100. Harberg, FJ, Senekjian, EK, Pokorny, WJ. Treatment of uncomplicated meconium ileus via T-tube ileostomy. J Pediatr Surg. 1981; 16:61–63.
101. Swenson, O. Pediatric Surgery, 2nd ed. New York: Appelton-Century-Crofts; 1962.
102. Mabogunje, OA, Wang, CI, Mahour, H. Improved survival of neonates with meconium ileus. Arch Surg. 1982; 117:37–40.
103. Chappell, JS. Management of meconium ileus by resection and end-to-end anastomosis. S Afr Med J. 1977; 52:1093–1094.
104. Santulli, T. Meconium ileus. In: Holder T, Ashcraft K, eds. Pediatric Surgery. Philadelphia: W. B. Saunders, 1980.
105. FitzSimmons, SC, Burkhart, GA, Borowitz, D, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Engl J Med. 1997; 336:1283–1289.
106. Borowitz, DS, Grand, RJ, Durie, PR. Use of pancreatic enzyme supplements for patients with cystic fibrosis in the context of fibrosing colonopathy. Consensus Committee. J Pediatr. 1995; 127:681–684.
107. Coates, AJ, Crofton, PM, Marshall, T. Evaluation of salt supplementation in CF infants. J Cystic Fibrosis. 2009; 8:382–385.
108. Hendeles, L. Use bioequivalency rating to select generics [letter]. Am Pharm. 1989; NS29:6.
109. Hendeles, L, Dorf, A, Stecenko, A, et al. Treatment failure after substitution of generic pancrelipase capsules. Correlation with in vitro lipase activity. JAMA. 1990; 263:2459–2461.
110. Durie, PR, Newth, CJ, Forstner, GG, et al. Malabsorption of medium-chain triglycerides in infants with cystic fibrosis: Correction with pancreatic enzyme supplement. J Pediatr. 1980; 96:862–864.
111. Li, Z, Lai, HJ, Kosorok, MR, et al. Longitudinal pulmonary status of cystic fibrosis children with meconium ileus. Pediatr Pulmonol. 2004; 38:277–284.
112. Bower, TR, Pringle, KC, Soper, RT. Sodium deficit causing decreased weight gain and metabolic acidosis in infants with ileostomy. J Pediatr Surg. 1988; 23:567–572.
113. McPartlin, JF, Dickson, JA, Swain, VA. Meconium ileus. Immediate and long-term survival. Arch Dis Child. 1972; 47:207–210.
114. Efrati, O, Nir, J, Fraser, D, et al. Meconium ileus in patients with cystic fibrosis is not a risk factor for clinical deterioration and survival: The Israeli Multicenter Study. JPGN. 2010; 50:173–178.
115. Johnson, J, Bush, A, Buchdahl, R. Does presenting with meconium ileus affect the prognosis of children with cystic fibrosis? Pediatr Pulmonol. 2010; 45:951–958.
116. Lai, HC, Kosorok, MR, Laxova, A, et al. Nutritional status of patients with cystic fibrosis with meconium ileus: A comparison with patients without meconium ileus and diagnosed early through neonatal screening. Pediatrics. 2000; 105:53–61.
117. Clatworthy, H, Howard, W, Lloyd, J. The meconium plug syndrome. Surgery. 1956; 39:131–142.
118. Flake, AW, Ryckman, FC. Meconium plug syndrome. In: Fanaroff AA, Martin RJ, eds. Neonatal-Perinatal Medicine, Disease of the Fetus and Infant. 5th ed. St. Louis: Mosby-Year Book; 1992:1054–1055.
119. Keckler, SJ, St Peter, SD, Spilde, TL, et al. Current significance of meconium plug syndrome. J Pediatr Surg. 2008; 43:896–898.
120. Stewart, DR, Mixon, GW, Johnson, DG, et al. Neonatal small left colon syndrome. Ann Surg. 1977; 186:741–795.
121. Gross, K, Desanto, A, Grosfeld, JL, et al. Intra-abdominal complications of cystic fibrosis. J Pediatr Surg. 1985; 20:431–435.
122. Beierle, EA, Vinocur, CD. Gastrointestinal surgery in cystic fibrosis. Curr Opin Pulm Med. 1998; 4:319–325.
123. Malfroot, A, Dab, I. New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up. Arch Dis Child. 1991; 66:1339–1345.
124. Button, BM, Heine, RG, Catto-Smith, AG, et al. Postural drainage and gastro-oesophageal reflux in infants with cystic fibrosis. Arch Dis Child. 1997; 76:148–150.
125. Boesch, RP, Acton, JD. Outcomes of fundoplication in children with cystic fibrosis. J Pediatr Surg. 2007; 42:1341–1344.
126. Hassall, E, Isreal, DM, Davidson, AG, et al. Barrett’s esophagus in children with cystic fibrosis: Not a coincidental association. Am J Gastroenterol. 1993; 88:1934–1938.
127. McDonald, ML, Trastek, VF, Allen, MS, et al. Barrett’s esophagus: Does an antireflux procedure reduce the need for endoscopic surveillance? J Thorac Cardiovasc Surg. 1996; 111:1135–1138.
128. Cohn, JA, Strong, TV, Picciotto, MR, et al. Localization of the cystic fibrosis transmembrane conductance regulator in human bile duct epithelial cells. Gastroenterol. 1993; 105:1857–1864.
129. Shapira, R, Hadzic, N, Francavilla, R, et al. Retrospective review of cystic fibrosis presenting as infantile liver disease. Arch Dis Child. 1999; 81:125–128.
130. Greenholz, SK, Krishnadasan, B, Marr, C, et al. Biliary obstruction in infants with cystic fibrosis requiring Kasai portoenterostomy. J Pediatr Surg. 1997; 32:175–180.
131. Oppenheimer, EH, Esterly, JR. Hepatic changes in young infants with cystic fibrosis: possible relation to focal biliary cirrhosis. J Pediatr. 1975; 86:683–689.
132. Scott-Jupp, R, Lama, M, Tanner, MS. Prevalence of liver disease in cystic fibrosis. Arch Dis Child. 1991; 66:698–701.
133. Roy, CC, Weber, AM, Morin, CL, et al. Hepatobiliary disease in cystic fibrosis: A survey of current issues and concepts. J Pediatr Gastroenterol Nutr. 1982; 1:469–478.
134. Debray, D, Lykavieris, P, Frédéric, G, et al. Outcome of cystic fibrosis-associated liver cirrhosis: Management of portal hypertension. J Hepatol. 1999; 31:77–83.
135. Karrer, FM. Portal hypertension. Semin Pediatr Surg. 1992; 1:134–144.
136. Williams, SGJ, Westaby, D, Tanner, MS, et al. Liver and biliary problems in cystic fibrosis. Br Med Bull. 1992; 48:877–892.
137. Dowman, JK, Watson, D, Loganathan, S, et al. Long-term impact of liver transplantation on respiratory function and nutritional status in children and adults with cystic fibrosis. Am J Transplant. 2012; 12:954–964.
138. Miller, MR, Sokol, RJ, Narkewicz, MR, et al. Pulmonary function in individuals with cystic fibrosis from the USA cystic fibrosis foundation registry who had undergone liver transplant. Liver Transpl. 2012; 18:585–593.
139. Fridell, JA, Mazariegos, GV, Orenstein, RS, et al. Liver and intestinal transplantation in a child with cystic fibrosis: A case report. Pediatr Transplant. 2003; 7:240–242.
140. Stern, RC, Mayes, JT, Weber, FL, Jr. Restoration of exocrine pancreatic function following pancreas-liver-kidney transplantation in a cystic fibrosis patient. Clin Transplant. 1994; 8:1–4.
141. Angelico, M, Gandin, C, Canuzzi, P, et al. Gallstones in cystic fibrosis: A critical reappraisal. Hepatology. 1991; 14:768–775.
142. Rovsing, H, Sloth, K. Micro-gallbladder and biliary calculi in mucoviscidosis. Acta Radiol. 1973; 14:588–592.
143. L’heureux, PR, Isenberg, JN, Sharp, HL, et al. Gallbladder disease in cystic fibrosis. AJR Am J Roentgenol. 1977; 128:953–956.
144. Stern, RC, Rothstein, FC, Doershunk, CF. Treatment and prognosis of symptomatic gallbladder disease in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 1986; 5:35–40.
145. Snyder, CL, Ferrell, KL, Saltzman, DA, et al. Operative therapy of gallbladder disease in patients with cystic fibrosis. Am J Surg. 1989; 157:557–561.
146. Anagnostopoulos, D, Tsagari, N, Noussia-Arvanitaki, S, et al. Gallbladder disease in patients with cystic fibrosis. Eur J Pediatr Surg. 1993; 3:348–351.
147. Saltzman, DA, Johnson, EM, Feltis, BA, et al. Surgical experience in patients with cystic fibrosis: A 25-year perspective. Pediatr Pulmonol. 2002; 33:106–110.
148. Hanly, JG, Ritzgerald, MX. Meconium ileus equivalent in older patients with cystic fibrosis. Br Med J. 1983; 286:1411–1413.
149. Dalzell, AM, Heaf, DP, Carty, H. Pathology mimicking distal intestinal obstruction syndrome in cystic fibrosis. Arch Dis Child. 1990; 65:540–541.
150. Matseshe, JW, Go, VLW, Dimagno, E. Meconium ileus equivalent complicating cystic fibrosis in post-neonatal children and young adults. Gastroenterology. 1972; 72:732–736.
151. Van der Doef, HP, Kokke, FT, van der Ent, CK, et al. Intestinal obstruction syndromes in cystic fibrosis: Meconium ileus, distal intestinal obstruction syndrome, and constipation. Curr Gastroenterol Rep. 2011; 12:265–270.
152. Wilschanski, M, Rivlin, J, Cohen, S, et al. Clinical and genetic risk factors for cystic fibrosis-related liver disease. Pediatrics. 1999; 103:52–57.
153. Marino, CR, Gorelick, FS. Scientific advances in cystic fibrosis. Gastroenterology. 1992; 103:681–693.
154. Kopelman, H, Corey, M, Gaskin, K, et al. Impaired chloride secretion, as well as bicarbonate secretion, underlies the fluid secretory defect in the cystic fibrosis pancreas. Gastroenterology. 1988; 95:349–355.
155. O’Halloran, SM, Gilbert, J, McKendrick, OM, et al. Gastrografin in acute meconium ileus equivalent. Arch Dis Child. 1986; 61:1128–1130.
156. Cleghorn, GJ, Forstner, GG, Stringer, DA, et al. Treatment of distal intestinal obstruction syndrome in cystic fibrosis with a balanced intestinal lavage solution. Lancet. 1986; 1:8–11.
157. Holsclaw, DS, Rocmans, C, Shwachman, H. Intussusception in patients with cystic fibrosis. Pediatrics. 1971; 48:51–58.
158. Coughlin, JP, Gauderer, MWL, Stern, RC, et al. The spectrum of appendiceal disease in cystic fibrosis. J Pediatr Surg. 1990; 25:835–839.
159. Olsen, MM, Gauderer, MWL, Girz, MK, et al. Surgery in patients with cystic fibrosis. J Pediatr Surg. 1987; 22:613–618.
160. Lloyd-Still, JD, Beno, DW, Kimura, RM. Cystic fibrosis colonopathy. Curr Gastroenterol Rep. 1999; 1:231–237.
161. Serban, DE, Florescu, P, Miu, N. Fibrosing colonopathy revealing cystic fibrosis in a neonate before any pancreatic enzyme supplementation. J Pediatr Gastroenterol Nutr. 2002; 35:356–359.