Mechanisms of Ventricular Tachycardia and Fibrillation
Sudden cardiac death (SCD) causes an estimated 300,000 deaths in the United States alone.1 Ventricular tachycardia (VT) often precedes the onset of ventricular fibrillation (VF). VF causes approximately one-third of sudden cardiac deaths.2 Patients at high risk for SCD may be implanted with an implanted cardioverter-defibrillator (ICD), but the largest group of victims of SCD does not have risk factors that place them in the high-risk category as candidates for ICD implantation.3 To develop more effective treatments for VT and VF, the mechanisms of VT and VF onset and maintenance must be understood.
Mechanisms of VT Onset and Maintenance
Although cases of idiopathic VF have been reported, most patients with VT and VF have a substrate that increases the probability of reentry. Two primary conditions lead to the onset of VT: ectopic foci and stable reentrant circuits. Ectopic foci due to triggered activity or abnormal automaticity may lead to VT. VT may be hemodyamically stable or unstable and often self-terminates, whereas VF is almost always fatal if not treated by administration of defibrillation shocks within minutes of VF onset. Ventricular tachyarrhythmias often progress from premature ventricular complexes (PVCs) to runs of VT, and finally to VF (Figure 48-1). Ventricular tachycardia and heart failure lead to action potential duration (APD) shortening, and rapid activation rates may lead to intracellular calcium overload. High intracellular calcium and APD shortening promote triggered activity and the initiation of VF.4
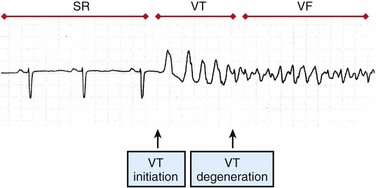
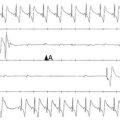
Figure 48-1 Surface ECG recording of the transition from normal sinus rhythm (SR) to ventricular tachycardia (VT) and eventually to ventricular fibrillation (VF). (Reprinted with permission from Weiss JN, Garfinkel A, Karagueuzian HS, et al: Chaos and the transition to ventricular fibrillation: A new approach to antiarrhythmic drug evaluation. Circulation 99:2819-2826, 1999.)
Focal sources that may lead to VT include triggered activity and abnormal automaticity. Focal sources may be the source of repetitive rapid ectopic firing or they may lead to disruption of the normal conduction pathways and the breakup of cohesive wave fronts and establishment of reentrant circuits (Figure 48-2, A). Delayed afterdepolarizations (DADs) cause a rise in resting potential during diastole. If the transmembrane potential rises above the activation threshold, a new action potential may be launched. DADs are traditionally linked to intracellular calcium overload and thus are exacerbated by the rapid heart rates seen in VT and VF. Early afterdepolarizations (EADs) may occur during the plateau (phase 2) or repolarization (phase 3) phase of an action potential. EADs are often associated with bradycardia or slow heart rate and have been linked to numerous ion channel disturbances including L-type calcium, rectifier potassium, and late sodium currents. The sodium/calcium exchanger current is thought to play a critical role in the development of EADs. Chua et al performed optical mapping of tachycardia-induced heart failure and demonstrated that heterogeneous up-regulation of apamin-sensitive K+ current increases sensitivity to intracellular calcium.5 This leads to heterogeneous APD shortening and possibly to late phase 3 EADs in the context of high heart rate or post-defibrillation recovery. Stretch activation may play a role in ventricular arrhythmias, particularly in the context of severe heart failure and volume overload.6 Abnormal automaticity consists of abnormal spontaneous firing action potentials that are not coupled to previous activations.
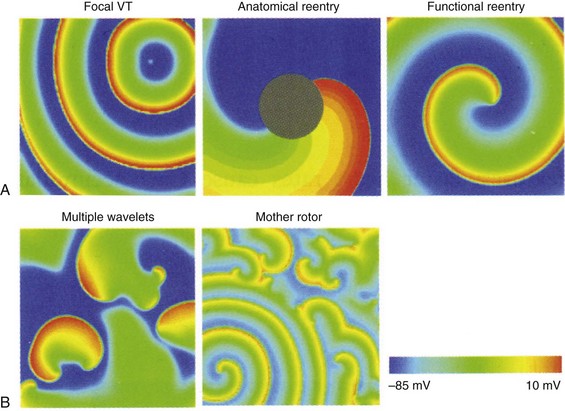
Figure 48-2 The mechanisms of VT and VF are demonstrated in 2D simulations of transmembrane voltage
A, VT may be maintained by a focal source, around an anatomic anchor, or as a reentrant circuit around a functional anchor. B, VF may be maintained by multiple wavelet reentry or by a mother rotor (bottom left of panel) that fractionates and breaks up into multiple daughter wavelets. (Reproduced with permission from Qu Z, Weiss JN: Nonlinear dynamics of excitation and propagation in cardiac muscle. In Zipes DP, Jalife J [eds]: Cardiac Electrophysiology: From Cell to Bedside. Philadelphia, Elsevier, 2009, pp 339-348.)
Stable reentrant circuits around an anatomic anchor such as a scar or a large vessel may lead to sustained VT (see Figure 48-2, A). A section of unexcitable post-infarct scar or a large vessel such as the aorta or the right ventricular (RV) outflow tract may form a pathway for a stable reentrant circuit. Even without an unexcitable core, simulations have shown that a reentrant circuit may be formed around a functional core rather than an anatomic core (see Figure 48-2, A). The center of the circuit may remain excitable but unexcited as the spiral wave circles around the core. Large reentrant circuits that encircle the entire ventricles have been shown to form as well, especially with cardiac dilatation or conduction slowing.
Transition From VT to VF
A primary mechanism for block leading to reentry is nonuniform dispersion of refractoriness. A study by Geizer et al reported that a series of premature stimuli that induced large spatial dispersion of repolarization caused VF in an in vivo dog model.7 This study and others have shown that block can occur even in normal, homogeneous tissue when alternans is caused in APD. Dispersion in APD leads to the development of conduction block and to fractionation of wave fronts, which, in turn, may lead to reentrant circuits and the initiation of VT and VF (Figure 48-3).
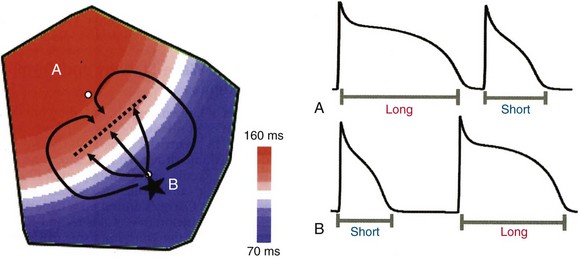
Figure 48-3 The formation of a reentrant circuit due to heterogeneity of APD
An ectopic focus at B initiates in the region of short APD and propagates outward toward the region of long APD. The ectopic beat blocks (dotted line) as it encounters refractory tissue but continues to propagate laterally such that it wraps around the area of block and reenters through the previous region of block and back into the short APD region. (Reproduced with permission from Qu Z, Weiss JN: Nonlinear dynamics of excitation and propagation in cardiac muscle. In Zipes DP, Jalife J [eds]: Cardiac Electrophysiology: From Cell to Bedside. Philadelphia, Elsevier, 2009, pp 339-348.)
Simulations have shown that the restitution curve (the relationship between APD and the diastolic interval) is predictive of the breakdown of a rotor to multiple unstable reentrant circuits. When the slope of the restitution curve (APD/diastolic interval [DI]) is greater than 1, an unstable positive-feedback loop results in increasing oscillations in APD and DI (Figure 48-4). Once the APD and the DI become so shortened that conduction is not possible, the wave front blocks and reentry may occur. A slope of less than 1 of the restitution curve leads to a negative-feedback loop, decreasing alternans, and to convergence of cycle length and DI to an equilibrium.
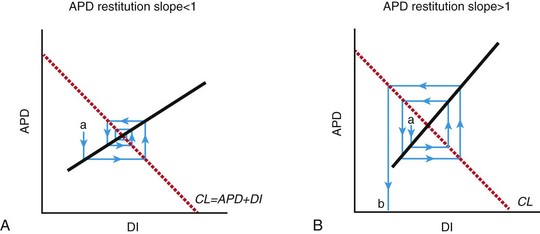
Figure 48-4 Progression of alternans in APD and DI to conduction block and spiral wave breakdown depends on the slope of the restitution curve
A, A spiral wave rotating at a constant cycle length (CL) undergoing a perturbation (a) that decreases DI and will tend to cause a smaller change in APD, which in turn will cause a smaller change in DI. This negative-feedback loop will eventually lead to a return to equilibrium. B, When the restitution slope >1, a perturbation in APD (a) leads to a larger disturbance in DI, which leads to a larger change in APD. This positive-feedback loop leads to increased variation in APD and DI until conduction is no longer sustained, but blocks (b). This leads to spiral wave breakup and chaotic conduction. (Reproduced with permission from Weiss JN, Garfinkel A, Karagueuzian HS, et al: Chaos and the transition to ventricular fibrillation: A new approach to antiarrhythmic drug evaluation. Circulation 99:2819-2826, 1999.)
VF Maintenance
VF changes as global ischemia sets in and the duration of VF increases. Using high-speed cinematography on canine hearts, Wiggers first proposed four stages of VF: (1) an initial tachysystolic stage (<1 s since VF onset), (2) convulsive incoordination (1 to 40 s), (3) tremulous incoordination (40 s to 3 min), and (4) progressive atonic incoordination (>3 min).8 Other groups have proposed 2,9 3,10 4,11 and 512 or more stages to describe the progression of VF over time. Each strategy for classifying VF activation patterns describes a change in VF activation rate and organization at approximately 2 to 3 minutes, with a progressive decline in activation rate and an increased incidence of conduction block thereafter.
Animal studies have shown that the ability of the heart to effectively contract is sufficiently compromised after 3 minutes of VF that even after successful defibrillation, return of spontaneous circulation does not occur without drugs and/or mechanical chest compressions.13,14 With the significant changes in cardiac electrophysiology and function that occur after prolonged VF, it is reasonable to expect that the mechanisms of VF maintenance may change with the progression of VF. Although many studies have investigated the mechanisms of short-duration VF (SDVF; lasting <1 min), relatively few studies have explored the mechanisms of long duration VF (LDVF; lasting >1 min). Although SDVF is of clinical relevance for ICD therapy, the average time from collapse of patients until defibrillation in the prehospital setting ranges from 4 to 10 minutes.15 Therefore, the mechanisms of SDVF and LDVF are of clinical significance and interest. Recent studies have indicated that three different mechanisms may be involved in sustaining VF: (1) mother rotor reentry, (2) wandering wavelet reentry, and (3) focal activity in the Purkinje system. VF mechanisms may vary over time and may switch between mechanisms regularly during LDVF.16
Mother Rotor Reentry
Several groups have proposed that a single, stable reentrant circuit, called a mother rotor, is responsible for maintaining VF. The mother rotor causes a high-frequency, repeatable activation sequence in a single region of the heart, also called the dominant domain. As the distance from the center of the motor rotor increases, the distance traveled by the wave front increases. The far arms of the rotor begin to fractionate and cause daughter reentrant circuits that may be short-lived and meandering. The daughter wave fronts propagate onto the remainder of the working myocardium and do not exhibit repeating activation sequences (Figure 48-2, B). The breakup of the daughter wave fronts and irregular patterns of activation away from the mother rotor give the electrocardiogram (ECG) the chaotic characteristics of VF.
Recent experimental studies and computer simulations have demonstrated that the inward potassium rectifier current plays an important role in rotor stability during VF.17 Increased inward-rectifier current prevents the wave front from colliding with the wave tail and thus prevents breakup of stable rotors. Stable mother rotors have been recorded in isolated sheep ventricles and in guinea pig hearts, and recent canine studies have shown activity that is consistent with mother rotor reentry on the endocardium after 2 minutes of VF.16
Wandering Wavelet Reentry
Wandering wavelet reentry was first proposed as the mechanism of VF maintenance almost 50 years ago. The initial theory proposed that for VF to start, a premature stimulus initiated the arrhythmia, and heterogeneity of refractory periods in the tissue caused wave-front fractionation that facilitated reentry. Wandering wavelet reentry consists of multiple competing wave fronts that constantly undergo collisions, fractionations, and annihilations. Wave fronts meander around refractory tissue and anatomic obstacles such as scar or infarct regions. When a wave front encounters unexcitable tissue, whether anatomic or functional in nature, it may (1) block entirely, (2) proceed around the obstacle, or (3) fractionate and create daughter wave fronts that proceed in different directions from the original wave front (see Figure 48-2, B). Wandering wavelet reentrant pathways are short-lived and transient in nature. Although normal cardiac tissue sustains VF, the onset of VF is facilitated in diseased tissue such as border zones surrounding infarcts and in tissue with anchors such as scar or anatomic boundaries that facilitate reentry. A critical mass of cardiac tissue is required for the wandering wavelets to have sufficient space to collide and create daughter wavelets. The three-dimensional (3D) structure of the ventricles, the fiber orientation, and the tissue anisotropy cause the wave-front propagation to become complicated and difficult to fully map through epicardial or endocardial approaches. The hypothesis of wandering wavelet reentry supposes that wave fronts propagate throughout the cardiac tissue such that there is not a stable high-frequency region that drives the activation rate.
Rogers et al. performed panoramic optical mapping of transmembrane voltage of perfused VF in pig hearts.18 In 17 episodes of mapped VF, they found that 92% of wave fronts were linked through fragmentation and collision. They did not find stable rotors that lasted beyond a few seconds. Although they were able to map only the epicardial surface, investigators were able to account for most of the wave fronts without the need for intramural reentry.
Purkinje Fiber Activity
Studies have shown that following 2 to 3 minutes of VF, activation rates between the endocardial and epicardial surfaces of the heart diverged.19 Although activation rates became slower for both endocardial and epicardial tissue as VF progressed, the epicardial activation rate slowed more dramatically in epicardial tissue. This activation rate gradient develops during LDVF in dogs19 and humans20 but does not develop in pigs19 (Figure 48-5). A study compared VF activation patterns between dogs and pigs using plunge needles distributed throughout the LV wall, and VF was induced electrically.19 Investigators observed that although an activation rate gradient emerged in dogs after a few minutes, this gradient did not develop in pigs. This species-dependent behavior together with the different Purkinje distribution in the two species led investigators to hypothesize that Purkinje fibers could be driving the later VF activation patterns in both dogs and pigs. As is the case with humans, the Purkinje fiber system in dogs is limited primarily to the endocardial surface, and in pigs these fibers arborize from the endocardial surface through the myocardium nearly to the epicardial surface. The development of transmural block and retention of the ability to conduct as LDVF progresses have been shown to represent an irregular and heterogeneous process.21
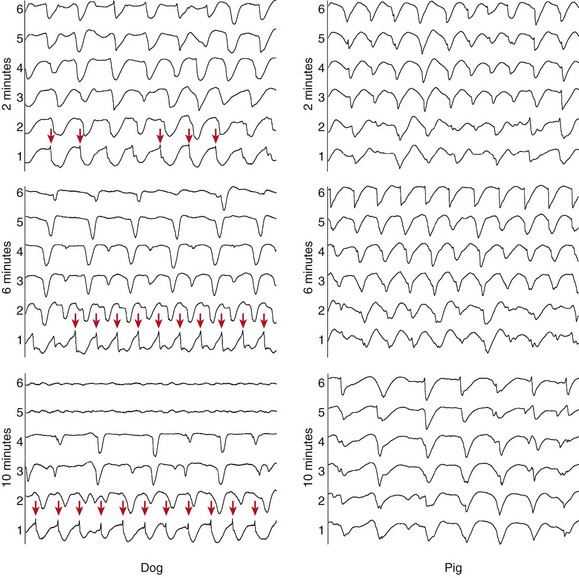
Figure 48-5 Plunge needle recordings from a dog and a pig during LDVF
Electrode 1 was near the endocardium, and electrode 6 was near the epicardium. In both species, the activation rate is similar on the endocardium and the epicardium at 2 minutes of VF. At 6 and 10 minutes of VF, the epicardial activation rate of the dog slows significantly, but an activation rate gradient does not develop in the pig. Purkinje activations (arrows on the dog endocardial recordings) accompany activations in the dog plunge needle recordings. (Reproduced with permission from Allison JS, Qin H, Dosdall DJ, et al: The transmural activation sequence in porcine and canine left ventricle is markedly different during long-duration ventricular fibrillation. J Cardiovasc Electrophysiol 18:1306-1312, 2007.)
To investigate the role of the Purkinje system in LDVF, Tabereaux et al. conducted direct endocardial mapping studies on isolated, perfused dog hearts.22 They recorded endocardial activation maps from the insertion of the anterolateral papillary muscle of electrically induced VF. Purkinje activations were detected separately from myocardial activations, and three distinct types of activations were recorded: (1) wave fronts proceeding from the working myocardium to the Purkinje system, presumably through retrograde conduction pathways; (2) focal activations that could appear in the Purkinje system or in working myocardium; and (3) activations that began outside of the mapped region in the Purkinje system and proceeded to initiate a new waveform in the working myocardium (Figure 48-6). Coupled with the fact that the wave fronts in LDVF tend to propagate from the endocardium to the epicardium, the focal activations appearing in the mapped region suggest that wave fronts are initiated in the Purkinje system near the endocardial surface. The authors concluded that the Purkinje system may play a critical role in both reentrant circuits and focal activations during LDVF.
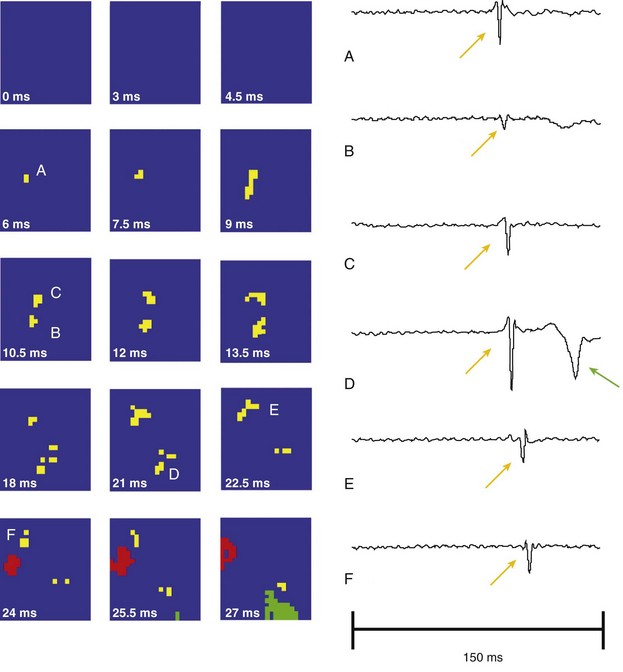
Figure 48-6 A focal activation arising from the center of the array
The focal-appearing PF activation (yellow) is followed by a WVM (red) activation wave front. The temporal derivative of individual electrode recordings shows the wave front initiating at electrode A and spreading outward toward electrodes D and E. The green represents a nonrelated wave front. (Reprinted with permission from Tabereaux PB, Walcott GP, Rogers JM, et al: Activation patterns of Purkinje fibers during long-duration ventricular fibrillation in an isolated canine heart model. Circulation 116:1113-1119, 2007.)
In a subsequent study, Dosdall et al performed mapping of the same anterolateral papillary muscle section of the LV endocardium in dogs.23 They also placed plunge needles on the sides of the plaque. They induced VF in two groups: (1) control hearts and (2) hearts in which the subendocardium, including the Purkinje system, had been chemically ablated by the application of Lugol’s solution. Investigators found that although the endocardial-epicardial rate gradient developed in the control hearts, the rate gradient was eliminated in the ablated hearts. The VF spontaneously terminated after 9.2 ± 3.2 minutes in the control hearts but terminated after 4.9 ± 1.5 minutes in the ablated hearts. This further supports the conclusion that the Purkinje system plays a significant role in maintaining LDVF.
Studies in dogs have demonstrated that periods of highly synchronous activity on the endocardial surface of the LV occur during LDVF. Robichaux et al. inserted a 64-electrode basket into the left ventricular (LV) endocardium and recorded electrically induced VF for 10 minutes.24 They observed periods during which focal activations in the Purkinje system spread rapidly through the entire LV endocardium (Figure 48-7). These activations resulted in rapid synchronous activation of the endocardium and relatively long periods of inactivity between excitations. This activity deviates substantially from wandering wavelet or mother rotor reentry, during which activity would be recorded continuously throughout the endocardium. Another study of LDVF by Li et al. demonstrated that this synchronous activation pattern originated in the Purkinje system on the endocardium, and that these wave fronts blocked at various levels as they proceeded from the endocardium toward the epicardium.25 Researchers found that this type of synchronized activity was present approximately one-third of the time 5 to 10 minutes after the onset of VF. This synchronous pattern was almost entirely eliminated by giving the EAD blocker, pinacidil, but the incidence of this Purkinje-driven activation was not altered by the DAD blocker flunarizine.16
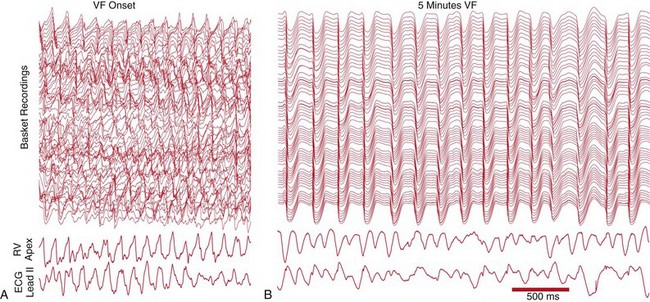
Figure 48-7 Recordings from an LV multi-electrode basket, an RV electrode, and a surface lead II ECG during LDVF
A, Chaotic activation patterns on the LV recordings immediately after VF onset are consistent with wandering wavelet reentry. B, Synchronous, nearly simultaneous activations throughout the LV endocardium demonstrate rapidly spreading activation throughout the LV endocardium. This activity demonstrates that activations spread rapidly throughout the endocardium during periods of LDVF. (Reproduced with permission from Robichaux RP, Dosdall DJ, Osorio J, et al: Periods of highly synchronous, non-reentrant endocardial activation cycles occur during long-duration ventricular fibrillation. J Cardiovasc Electrophysiol 21:1266-1273, 2010.)
Potential mechanisms for focal activity in the Purkinje system during LDVF and after successful defibrillation shocks are beginning to come to light. In a study on perfused rabbit ventricles, Maruyama et al. found spontaneous intracellular calcium levels after defibrillation on the LV endocardial surface but not on the epicardium.26 Triggered activity emerged from the endocardial surface and was correlated with high intracellular calcium levels. Purkinje-like potentials preceded triggered activity on the endocardial surfaces. Spontaneous calcium release and higher diastolic intracellular calcium levels after rapid pacing or VF may lead to triggered activity in the Purkinje system.
Li et al performed high-resolution 3D mapping in pig hearts by placing plunge needles with six electrodes at 2-mm spacing in a 9 × 9 grid with 2-mm spacing on the anterior LV wall.27 They found that although reentrant circuits were common within the mapped region during the first few minutes (Figure 48-8), by 3 minutes after VF the intramural reentry disappeared, and the incidence of focal activations within the mapped region (activations that began de novo in the region rather than as the result of reentry or propagation into the mapped region from outside) increased steadily throughout the first 10 minutes of VF. Although they did not record Purkinje fibers directly, the authors proposed that either focal activity in the Purkinje system or reentrant activity involving the Purkinje system may cause this focal activation, because the Purkinje system permeates the ventricular wall in pigs.
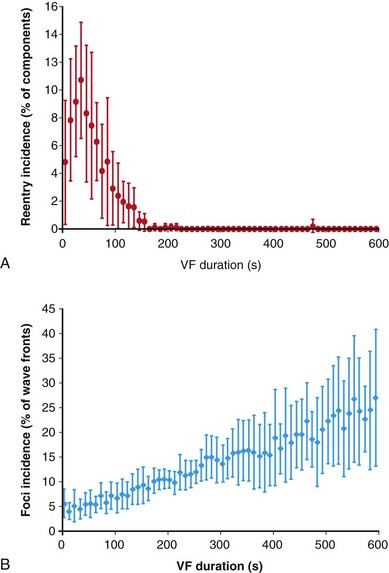
Figure 48-8 Transmural 3D plunge needle mapping in pigs showed that the incidence of intramural reentry within a mapped region went to zero in the first 2 to 3 minutes, and the incidence of focal activations increased during the first 10 minutes of VF. (Reproduced with permission from Li L, Jin Q, Huang J, et al: Intramural foci during long duration fibrillation in the pig ventricle. Circ Res 102:1256-1264, 2008.)
References
1. Zipes, DP, Wellens, HJ. Sudden cardiac death. Circulation. 1998; 98:2334–2351.
2. Cobb, LA, Fahrenbruch, CE, Olsufka, M, et al. Changing incidence of out-of-hospital ventricular fibrillation, 1980–2000. JAMA. 2002; 288:3008–3013.
3. Myerburg, RJ, Kessler, KM, Castellanos, A. Sudden cardiac death: Structure, function, and time-dependence of risk. Circulation. 1992; 85:I2–I10.
4. Ogawa, M, Morita, N, Tang, L, et al. Mechanisms of recurrent ventricular fibrillation in a rabbit model of pacing-induced heart failure. Heart Rhythm. 2009; 6:784–792.
5. Chua, SK, Chang, PC, Maruyama, M, et al. Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res. 2011; 108:971–979.
6. Trayanova, NA, Constantino, J, Gurev, V. Models of stretch-activated ventricular arrhythmias. J Electrocardiol. 2010; 43:479–485.
7. Gelzer, AR, Koller, ML, Otani, NF, et al. Dynamic mechanism for initiation of ventricular fibrillation in vivo. Circulation. 2008; 118:1123–1129.
8. Wiggers, CJ. Studies of ventricular fibrillation caused by electric shock: Cinematographic and electrocardiographic observations of the natural process in the dog’s heart: Its inhibition by potassium and the revival of coordinated beats by calcium. Am Heart J. 1930; 5:351–365.
9. Thurmann, M, Janney, JG, Jr. The diagnostic importance of fibrillatory wave size. Circulation. 1962; 25:991–994.
10. Huizar, JF, Warren, MD, Shvedko, AG, et al. Three distinct phases of vf during global ischemia in the isolated blood-perfused pig heart. Am J Physiol Heart Circ Physiol. 2007; 293:H1617–H1628.
11. Huang, J, Rogers, JM, Killingsworth, CR, et al. Evolution of activation patterns during long-duration ventricular fibrillation in dogs. Am J Physiol Heart Circ Physiol. 2004; 286:H1193–H1200.
12. Cheng, KA, Dosdall, DJ, Li, L, et al. Evolution of activation patterns during long-duration ventricular fibrillation in pigs. Am J Physiol Heart Circ Physiol. 2012; 302:H992–H1002.
13. Menegazzi, JJ, Ramos, R, Wang, HE, et al. Post-resuscitation hemodynamics and relationship to the duration of ventricular fibrillation. Resuscitation. 2008; 78:355–358.
14. Geddes, LA, Roeder, RA, Kemeny, A, et al. The duration of ventricular fibrillation required to produce pulseless electrical activity. Am J Emerg Med. 2005; 23:138–141.
15. Valenzuela, TD, Roe, DJ, Nichol, G, et al. Outcomes of rapid defibrillation by security officers after cardiac arrest in casinos. N Engl J Med. 2000; 343:1206–1209.
16. Li, L, Zheng, X, Dosdall, DJ, et al. Long duration ventricular fibrillation exhibits two distinct, organized, endocardial activation patterns. Heart Rhythm. 2011; 8(5S):S403.
17. Jalife, J. Inward rectifier potassium channels control rotor frequency in ventricular fibrillation. Heart Rhythm. 2009; 6:S44–S48.
18. Rogers, JM, Walcott, GP, Gladden, JD, et al. Panoramic optical mapping reveals continuous epicardial reentry during ventricular fibrillation in the isolated swine heart. Biophys J. 2007; 92:1090–1095.
19. Allison, JS, Qin, H, Dosdall, DJ, et al. The transmural activation sequence in porcine and canine left ventricle is markedly different during long-duration ventricular fibrillation. J Cardiovasc Electrophysiol. 2007; 18:1306–1312.
20. Masse, S, Farid, T, Dorian, P, et al. Effect of global ischemia and reperfusion during ventricular fibrillation in myopathic human hearts. Am J Physiol Heart Circ Physiol. 2009; 297:H1984–H1991.
21. Venable, PW, Taylor, TG, Shibayama, J, et al. Complex structure of electrophysiological gradients emerging during long-duration ventricular fibrillation in the canine heart. Am J Physiol Heart Circ Physiol. 2010; 299:H1405–H1418.
22. Tabereaux, PB, Walcott, GP, Rogers, JM, et al. Activation patterns of Purkinje fibers during long-duration ventricular fibrillation in an isolated canine heart model. Circulation. 2007; 116:1113–1119.
23. Dosdall, DJ, Tabereaux, PB, Kim, JJ, et al. Chemical ablation of the Purkinje system causes early termination and activation rate slowing of long-duration ventricular fibrillation in dogs. Am J Physiol Heart Circ Physiol. 2008; 295:H883–H889.
24. Robichaux, RP, Dosdall, DJ, Osorio, J, et al. Periods of highly synchronous, non-reentrant endocardial activation cycles occur during long-duration ventricular fibrillation. J Cardiovasc Electrophysiol. 2010; 21:1266–1273.
25. Li, L, Jin, Q, Dosdall, DJ, et al. Activation becomes highly organized during long-duration ventricular fibrillation in canine hearts. Am J Physiol Heart Circ Physiol. 2010; 298:H2046–H2053.
26. Maruyama, M, Joung, B, Tang, L, et al. Diastolic intracellular calcium-membrane voltage coupling gain and postshock arrhythmias: Role of Purkinje fibers and triggered activity. Circ Res. 2010; 106:399–408.
27. Li, L, Jin, Q, Huang, J, et al. Intramural foci during long duration fibrillation in the pig ventricle. Circ Res. 2008; 102:1256–1264.