[level-membership-for-anesthesiology-category]
Chapter 2 Mechanisms of Spinal Neuromodulation
Chapter Synopsis: Electrical stimulation of the spinal cord (SCS) improves many forms of neuropathic pain; but, contrary to our early understanding, it can also affect some forms of nonneuropathic nociception. Chapter 2 examines the physiology of these indications. The understanding of SCS is rooted in Melzack and Wall’s5 gate control theory of pain transmission. By spinal stimulation of large-fiber neurons, the gate is activated to reduce transmission of neuropathic pain signals from primary small-fiber afferents. The technique generally does not alleviate acute nociception, but it can reduce certain types of peripheral nociception and can even alleviate underlying conditions. SCS has been shown to affect ischemic limb pain caused by peripheral arterial occlusive disorder (PAOD), angina, and gastrointestinal disorders such as irritable bowel syndrome (IBS).
 Transmission of nociceptive information from the site of an injury may generate a perception of pain because an imbalance exists between large and small fiber systems (cf. Head and Thompson1). This concept eventually evolved into Melzack and Wall’s5 gate control theory.
Transmission of nociceptive information from the site of an injury may generate a perception of pain because an imbalance exists between large and small fiber systems (cf. Head and Thompson1). This concept eventually evolved into Melzack and Wall’s5 gate control theory.








Background
Therapeutic effects of neuromodulation are based on the concept that selective excitation of large afferent fibers activates mechanisms that control pain. This fits well with the idea that pain may occur as a result of an imbalance between large and small fiber systems that transmit nociceptive information from the site of injury. Previous investigators have provided a long history of support for this concept. As early as 1906 Head and Thompson1 argued that fine discrimination such as touch normally exerts an inhibitory influence on impulses transmitted in fibers mediating nociception, which results in pain. This inhibition or facilitation of sensory impulses has been proposed to occur in the dorsal horn before nociceptive information is relayed onto secondary neurons. Furthermore, clinical trials performed in the early sixties using sensory thalamic stimulation2,3 were based on the notion that activation of fine discrimination receptors (touch) exerted an inhibitory influence over sensations such as pain, pressure, heat, or cold. It should also be noted that Noordenbos4 used the descriptive phrase “fast blocks slow” to stress the inhibitory influence of fast on slow fibers.
The concept of excitation of large afferent fibers activating pain control mechanisms advanced very rapidly with the publication of the article proposing the gate control theory; it is one of the most cited papers in modern pain literature.5 In this article the authors suggested that the therapeutic implication of their model would be to selectively activate large fibers to control pain. Thus even though the basic idea underlying the gate control theory was not completely unknown, it was built on a foundation of creative experiments using modern electrophysiological techniques. The results of these experiments were clearly synthesized and discussed in a form that postulated a new conceptualization of pain and pain control. Subsequently, numerous studies were conducted to criticize the theory, but nevertheless its simplicity has provided a useful frame of reference to explain mechanisms of pain generation and pain control. As Dickenson6 pointed out in his editorial about the ability of the gate control theory of pain to stand the test of time, the concepts of convergence and modulation changed the focus from destructing pathways for relief of pain to controlling pain by modulation in which excitation is reduced and inhibition is increased. The gate control theory accelerated the pursuit of modern pain research to explore how the pervasive plasticity of the nervous system plays a critical role in the generation, maintenance, and modulation of pain.
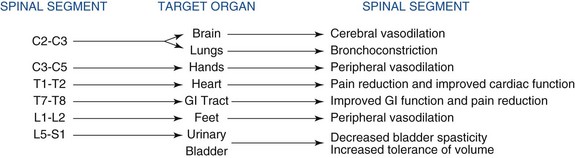
The gate control theory served as a critical catalyst in the clinical arena to spawn the development of various forms of neuromodulation that led to new therapies. The insights gained by Shealy and his colleagues7 and Shealy, Mortimer, and Reswick8 in animal experiments led them to conduct the first human trials with electrical spinal cord stimulation (SCS) as one form of neuromodulation.8 Their experimental studies in conscious cats revealed that stimulating the dorsal aspect of the spinal cord blocked responses to nociceptive peripheral stimuli. On the basis of this study and support of the gate control theory, it was assumed that neuromodulation could be used to treat all forms of nociceptive pain. However, several reports pointed out that SCS is ineffective for treating acute nociceptive conditions in contrast to what was predicted from the gate control theory; but eventually it has become the foremost treatment for neuropathic pain originating from the periphery.9–13 Nevertheless, numerous reports appeared during the eighties to convince clinicians that SCS could also be used to alleviate certain types of nociceptive pain, including selected ischemic pain states such as peripheral arterial occlusive disease (PAOD), vasospastic conditions, and therapy-resistant angina pectoris. The mechanisms of action for SCS are slowly emerging as more solid evidence has revealed some of the underlying physiological mechanisms. Clinical observations coupled with important experimental data clearly demonstrate that SCS applied to different segments of the spinal cord elicits fundamentally different results on various target organs or parts of the body (Fig. 2-1).
Organization and Electrical Properties of the Spinal Cord
The spinal cord is encased within the vertebral canal, which is made up of vertebrae that encircle the spinal cord but limit space for insertion of stimulating electrodes. The spinal cord in an adult human extends from the foramen magnum to the first or second lumbar vertebra and is divided into cervical, thoracic, lumbar, and sacral segments. The naming of the segments is based on the regions of the body innervated by the spinal cord. Examination of a cross section of the spinal cord shows that it is composed of gray matter and surrounded by white matter (Fig. 2-2).
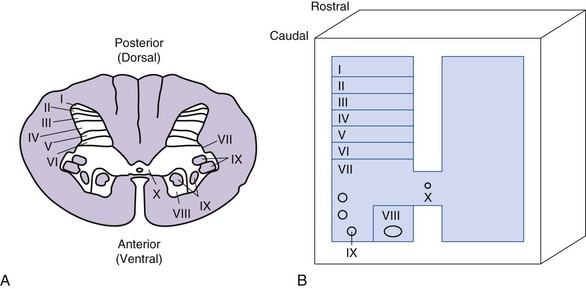
The gray matter is comprised of cell bodies with their dendrites and initial segment of the axon, microglia, and astrocytes. It is divided into a posterior horn, intermediate zone, and the ventral horn. The gray matter is further divided into laminae I to X; these divisions are based on the size, shape, and distribution of neurons located in these laminae.14 The input received by these neurons and the trajectory of the axons from them also help to characterize laminae. Neurons of dorsal and intermediate laminae (I to VII, X) generally receive sensory information originating from peripheral sensory receptors. These neurons integrate this information with input arriving from descending pathways. Some of the cell bodies have short axons and serve as interneurons, whereas others are the cells of origin of ascending sensory pathways. The interneurons may also participate in local reflexes. The ventral laminae (VIII, IX) are generally composed of motoneurons that form the motor nuclei.
The electrical properties, more specifically the electrical conductivity, of white and gray matter of the spinal cord are not homogeneous. For SCS it is important to know that the electrical conductivity of the dorsal column is anisotropic; that is, current can travel in the direction parallel to the axons more easily than in the direction perpendicular to axons.15 The electrical properties within the gray matter also vary because neurons and glia have diverse orientations, ubiquitous dimensions, and different dendritic characteristics.
Neuromodulation using electrical stimulation of the spinal cord depends on the conductivity of the intraspinal elements relative to the position of the electrode.16 If an axon is depolarized or made more electrically positive, it produces an action potential that is transmitted orthodromically and antidromically within the axon. The cathode of an external electrode must be negatively charged to generate the action potential in the axon. In contrast, if an axon is hyperpolarized or made more negatively charged, its ability to generate an action potential is reduced because the threshold for depolarization is increased. A positively charged external electrode or anode produces this effect. Thus the active electrode for electrical stimulation serves as the cathode, whereas the anode or positive electrode may serve as a shield to prevent stimulation of neuronal structures such as dorsal roots that might interfere with effective neuromodulation. For SCS the electrode most commonly is placed on the surface of the dura mater. Activation of the electrode releases electric current that is transmitted through the dura mater and the highly conductive cerebrospinal fluid (CSF) before it reaches the dorsal part of the spinal cord. The dura mater has low conductivity, but it is so thin that the current generally is not impeded significantly as it passes through the dura to the CSF. Furthermore, the vertebral bone has the lowest conductivity so it insulates pelvic structures and visceral organs from the electric field generated by SCS. Once the electric current reaches the spinal cord, several factors may determine the neural structure being stimulated. Jan Holsheimer16 has used computerized models of the spinal cord to study the activation of axons by electrical current. In addition to the fiber diameter, the presence of myelination, and the depth of CSF layer surrounding the cord at the level of an electrode, the axon orientation has important implications for activation thresholds. In general, axons of the dorsal columns have higher activation thresholds than fibers such as the dorsal roots that are oriented laterally or angle as they enter the spinal cord.16
The dorsal column is composed primarily of large-diameter afferent nerve fibers with relatively low thresholds for recruitment when cathodal electrical pulses are generated through the epidural electrode that is attached to a spinal cord stimulator. It is important to note that the electrode for SCS needs to be placed near midline to prevent the activation of dorsal root fibers.17 Stimulation amplitudes are then increased to intensities that recruit large fibers to produce action potentials and produce paresthesias. These action potentials are transmitted orthodromically and antidromically in these axons. The action potentials transmitted antidromically reach the collateral processes that penetrate the gray matter of the spinal cord. Their activation causes the release of transmitters, which activates the “gate.” Activation of the gate sets in motion neural mechanisms that reduce pain and improve organ function. The details of these mechanisms are discussed in subsequent paragraphs.
Neuromodulation Mechanisms in Ischemic Pain
Ischemic painful conditions of the limbs commonly results from PAOD, which is caused by obstruction of blood flow into an arterial tree.18 PAOD is a major cause of disability and loss of work and affects the quality of life.19,20 Morbidity and mortality are relatively high because effective treatments are very limited. Presently SCS is usually implemented only after vascular surgery and medications fail to slow or prevent the progression of PAOD. Surprisingly the success rate of SCS-treated PAOD is greater than 70%.21 Since ischemic pain is characterized generally as essentially nociceptive and several studies have indicated that SCS does not alleviate acute nociceptive pain,9,22,23 SCS-induced pain relief is most likely secondary to attenuation of tissue ischemia that occurs as a result of either increasing/redistributing blood flow to the ischemic area or decreasing tissue oxygen demand.24,25 Cook and associates26 were the first to report that SCS increased peripheral circulation of patients suffering from PAOD. Usually SCS is applied to the dorsal columns of lower thoracic (T10-T12) and higher lumbar spinal segments (L1-L2) to increase peripheral circulation in the legs of PAOD patients.
The mechanisms of SCS-induced vasodilation in the lower limbs and feet are not yet completely understood. Since no animal models of PAOD that generate ischemic pain have emerged, normal anesthetized animal models have been used to investigate the physiologic mechanisms of SCS-induced changes in peripheral blood flow (see reference 23 for review). Cutaneous blood flow and calculated vascular resistance in the glabrous skin of ipsilateral and contralateral hindpaws have been determined most commonly by using laser Doppler flowmetry. A thermistor probe placed next to the laser Doppler probe on the plantar aspect of the foot has been used to measure skin temperature. Various interventions such as injections of hexamethonium, administration of adrenergic agonists and antagonists, sympathetic denervation, dorsal rhizotomies, calcitonin gene-related peptide (CGRP) antagonists, nitric oxide synthetase inhibitors, and local paw cooling have been used to explore the underlying mechanisms of peripheral microcirculation. Studies using Doppler flowmetry and interventions for more than 30 years of clinical and basic science studies have resulted in the evolution of two theories to explain the mechanisms of SCS-induced vasodilation. One theory is that SCS decreases sympathetic outflow and reduces the constriction of arterial vessels27,28; the alternative theory is that SCS antidromically activates sensory fibers, which causes the release of vasodilators29, 30 (Fig. 2-3). The theory for SCS-induced suppression of sympathetic activity was based on results from clinical observations showing that a sympathetic block or sympathectomy produced pain relief and vasodilation imitated effects of treatment with SCS.31,32 This theory was tested in animal models in which SCS-induced cutaneous vasodilation in the rat hindpaw at 66% of motor threshold was abolished by complete surgical sympathectomy.33 SCS-induced vasodilation was markedly attenuated after administrating the ganglionic blocker, hexamethonium, or the neuronal nicotinic ganglionic blocker, chlorisondamine. These results led to the suggestion that efferent sympathetic activity, including nicotinic transmission in the ganglia and the postganglionic α1-adrenergic receptors are suppressed by SCS (see Fig. 2-3). The alternative theory of SCS-induced antidromic activation of sensory fibers was confirmed in studies showing that sensory afferent fibers are important for SCS-induced vasodilation and that at higher, but not painful, SCS intensities C-fibers may also contribute to the response30,34,35 (see Fig. 2-3). Thus SCS applied at the spinal L2-L5 segments excites dorsal column fibers that antidromically activate interneurons, which subsequently stimulate spinal terminals of transient receptor potential V1 (TRPV1) containing sensory fibers, which are primarily made up of C-fiber axons.36,37 These fibers transmit action potentials antidromically to nerve endings in the hindlimb. The action potentials evoke mechanisms that release vasodilators, including the most powerful vasodilator, CGRP, which binds to receptors on endothelial cells. The activation of these receptors leads to production and subsequent release of nitric oxide (NO), which results in relaxation of vascular smooth muscle cells (see reference 30 for review). The overall result is that relaxation of vascular smooth muscle cells decreases vascular resistance and increases peripheral blood flow. It should be noted that SCS applied at 500 Hz significantly increased cutaneous blood flow and decreased vascular resistance when compared to the responses induced at 50 Hz and 200 Hz; the effects at all of these frequencies depend on the activation of TRPV1-containing fibers and release of CGRP.38 The clinical use of such findings remains to be determined.
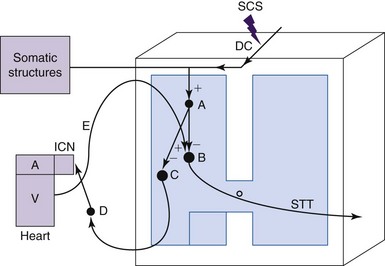
The level of sympathetic nervous system activity may shift the balance between the effects of sympathetic efferent suppression and antidromic activation of sensory afferent fibers. Cooler skin temperatures increase sympathetic activity. A notable observation is that SCS-induced vasodilation of a cooled hindpaw (<25° C) generated an early phase of vasodilation via sensory afferent fibers and a late phase via suppression of the sympathetic efferent activity.39 However, only sensory afferent activation occurred if SCS-induced vasodilation was performed in a warm paw (>28° C). Thus the balance of these two mechanisms most likely depends on the activity level of the sympathetic nervous system. Furthermore, another study showed that preemptive SCS increased the survival rate of skin flaps that were made ischemic by occluding the blood supply to the tissue for as long as 12 hours.40 Concomitant administration of the CGRP-1 receptor antagonist CGRP 8-37 markedly attenuated the cytoprotective effect40; whereas preoperative administration of antiadrenergic drugs such as guanethidine, reserpine, and 6-hydroxydopamine increased experimental flap survival.41 Thus the dual mechanisms of the release of vasodilators by antidromic activation of sensory fibers and suppression of sympathetic activity may all be involved in cytoprotection and prevention of vasospasm.
Neuromodulation Mechanisms of Visceral Organs
This section focuses on the heart and gastrointestinal tract because most of the basic research to determine mechanisms underlying the SCS-induced effects has been performed on these organs. Effects of SCS on spasms of bronchi in lungs were examined, but only an abstract was published.42 To the best of our knowledge the effects of SCS on other visceral organs of animals besides studies of the colon (see section on Gastrointestinal Tract that follows) have not been examined systematically. However, there is abundant evidence of SCS effects on the urinary bladder that were obtained mostly from multiple sclerosis patients.43
Heart
Ischemic heart disease is often presented as shortness of breath and angina pectoris, which is described clinically as an extremely intense pain and severe discomfort that usually radiates to the chest, shoulder, and left arm and occasionally to the neck and jaw.44 This pain usually occurs during episodes of vasospasm or occlusion of the coronary vessels that result in decreased blood flow to the heart. The decreased blood flow generally causes an imbalance between the supply and the demand of oxygen in the heart. Ischemic episodes result in the release of prostaglandins, adenosine, bradykinin, and other substances that activate nociceptive spinal sensory afferent fibers innervating the heart.45 This nociceptive information is transmitted by these sensory afferent fibers, which enter the C7-T5 spinal segments and synapse on spinothalamic tract cells, and cells of other ascending pathways that are also receiving converging cutaneous and muscle input from the overlying somatic structures such as the chest and upper arm.45 Because of this convergent input, angina pectoris is felt as if it is originating from the chest and left arm. On occasion, angina is referred to the neck and jaw because the ischemic episodes can excite nociceptive vagal afferents that converge on spinothalamic tract cells in the upper cervical segments that also receive somatic convergent input from the neck and jaw.45
It is important to note that a large population of patients suffering from chronic angina pectoris does not respond to conventional treatments.46 This group of patients led clinicians to develop alternative strategies such as neuromodulation to provide pain relief. Thus, following a period of using transcutaneous nerve stimulation for various types of visceral pain, including angina pectoris,47 SCS has been used to treat such therapy-resistant angina pectoris since the mid-eighties.48,49 Application of SCS at T1-T2 or higher spinal segments in patients provides pain relief by reducing both the frequency and to some extent the severity of angina attacks; the intake of short-acting nitrates is also reduced.50–53 Thus SCS improves the quality of life in these patients; however, the mechanisms producing pain relief and improved heart function still remain unclear. An early animal study showed that SCS produced antianginal effects by directly inhibiting spinothalamic tract cell activity resulting from cardiac nociception,54 but clinical and animal studies have proven that SCS does not solely relieve pain but also improves cardiac function. However, the primary factor appears to be the resolution of myocardial ischemia. Hautvast and colleagues55 have proposed an SCS-induced flow increase or redistribution of blood supply, whereas Eliasson, Augustinnson, and Mannheimer56 and Mannheimer and associates57 interpret the reduction of coronary ischemia (decreased ST changes; reversal of lactate production) as being mainly caused by decreased cardiomyocyte oxygen demand.
Studies have been conducted to determine if blood flow changes relieve angina pectoris with SCS. In a human experimental study, positron emission tomography (PET) was used to show that SCS appeared to redistribute blood.55 Blood flow and redistribution were also examined in an animal study by determining the distribution of isotope-labeled microspheres in hearts of anesthetized and artificially ventilated adult mongrel dogs.58 A comparison of occluding the left anterior descending coronary artery with and without SCS showed that local blood flow in the myocardium did not increase and no changes occurred in the pressure-volume relationships during SCS. However, this study was limited because it was performed using acute occlusions in animals with a normal heart. It would have been more appropriate to conduct such studies in canine hearts with previous infarctions and long-term ischemic episodes since patients have long-term coronary ischemic disease.
Clinical and animal studies have been done to determine if SCS produces anti-ischemic effects that contribute to improved cardiac function. In patients whose blood supply in the coronary arteries was reduced, SCS applied during standardized heart workloads, analogous to exercise and rapid cardiac pacing, significantly reduced the magnitude of ST segment changes of the electrocardiogram.57,59,60 These results support the idea that SCS may affect cardiac function by improving the working capacity of the heart. A canine animal model of myocardial ischemia was used to resemble the development of chronic ischemic heart disease by implanting an ameroid constrictor ring around the proximal left circumflex coronary artery.61 As the material inside the constrictor ring swells, it gradually reduces arterial blood flow and induces the development of collaterals.62 After 4 to 6 weeks the chest of anesthetized animals was opened, and the exposed heart was paced at a basal rate of 150 beats/min. A plaque containing 191 unipolar contacts was placed on the left ventricle distal to the left coronary artery occluded by the ameroid constrictor. Recordings were obtained from unipolar contact sites to determine changes in the ST segments. The heart was stressed by administering angiotensin II via the coronary artery blood supply to the right atrial ganglionated plexus. Hearts stressed with angiotensin II produced elevations of the ST segments; SCS markedly attenuated this ST segment elevation. These data indicate that SCS may counteract the deleterious effects caused by chemical activation of the intrinsic cardiac nervous system that stressors release in a myocardium with reduced coronary reserve. From these results it was concluded that SCS may produce anti-ischemic effects, which in turn contribute to improved cardiac function. In a more recent study Lopshire and associates63 demonstrated that SCS improved cardiac function in canine heart failure after an experimental myocardial infarction and further stressing the heart by using high-frequency cardiac pacing over 8 weeks.
Further evidence to support the anti-ischemic effects of SCS on the heart is the observation that SCS initiated before the onset of ischemic episodes (preemptive SCS) appears to activate mechanisms that reduce infarct size produced by coronary occlusions.64 This preemptive SCS treatment reduced infarct size produced by coronary occlusions. This reduction in infarct size depends on adrenergic receptors located in the membrane of cardiac myocytes. Thus preemptive SCS appears to provide protection to the heart during periods of critical ischemia. However, protective effects of SCS therapy are ineffective if SCS is initiated after (reactive SCS) the onset of the ischemic episode.
The intrinsic cardiac nervous system is powerfully activated during ischemic episodes.65,66 It is located in the cardiac ganglion plexi of epicardial fat pads adjacent to and within the myocardium.67 This system is composed of interconnecting local circuit neurons and sympathetic efferent, parasympathetic efferent, and sensory afferent fibers. Local circuit neurons have the capacity to produce local interactions and also to connect with neurons arising from other ganglia and higher centers. Thus the intrinsic cardiac nervous system is essential to coordinate regional cardiac function and provide rapid and timely reflex coordination of autonomic neuronal outflow to the heart.68 An important observation in animal studies is that SCS appears to stabilize activity of these intrinsic cardiac neurons during an ischemic challenge resulting from occlusion of a coronary artery. Thus SCS improves cardiac function to a considerable degree by regulating the intrinsic cardiac nervous system23 (Fig. 2-4).
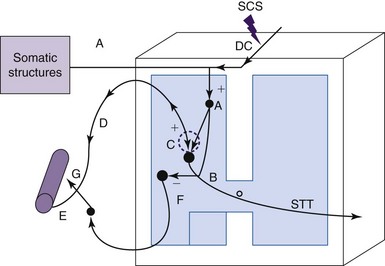
The stabilizing role of the intrinsic nervous system during SCS may also reduce arrhythmias. These effects have been examined in a canine animal model in which mediastinal nerve stimulation can evoke bradycardias and atrial arrhythmias.69 SCS significantly reduces these arrhythmias and those evoked by ischemia,69,70 but bilateral stellectomy eliminated these SCS-induced effects.69 Thus these results provided evidence that SCS prevents the onset of atrial arrhythmias initiated by excessive activation of intrinsic cardiac neurons (mediastinal nerve stimulation), which depends on intact fibers coursing through the stellate ganglion and subclavian ansae.69 Thus modulation of the intrinsic cardiac nervous system may be at least one mechanism that provides protection for the heart during more severe ischemic threats caused by generalized arrhythmias.70 Other mechanisms may also contribute to SCS-induced cardioprotection, including local release of catecholamine in the myocardium64,65 and an α1-PKC pathway and a β-PKA pathway that mediates transient myocardial ischemia-induced apoptosis.64,71 Other neuropeptides such as NO72 and β-endorphin73 may also provide cardioprotection. Some of the pathways and proposed mechanisms contributing to the effects of SCS on cardiac function discussed previously are summarized briefly in Fig. 2-4.
Gastrointestinal Tract—Irritable Bowel Syndrome
Functional bowel disorders, including irritable bowel syndrome (IBS), are common abnormalities of the gastrointestinal tract that are associated with painful abdominal cramps, abnormal bowel habits, and somatic hypersensitivity.74,75 Unfortunately no effective therapy is available because mechanisms that contribute to chronic visceral symptoms of IBS are not well understood. This lack of effective therapy led to speculation that SCS might be a means to treat IBS because it effectively reduces hyperexcitable somatosensory and viscerosomatic (bladder) reflexes in patients experiencing spasticity43 and relieves certain types of visceral pain. This speculation led to the idea of proposing a study that was designed to determine if SCS might be a potential therapy for visceral pain originating from the gastrointestinal tract.76 To simulate the IBS symptoms observed in patients, an animal model of visceral hypersensitivity was adapted by infusing a small concentration of acetic acid into the colon, which produces hypersensitivity but does not damage the mucosa77–79 or by producing postinflammatory colonic hypersensitivity with trinitrobenzenesulfonic acid to create the acute inflammatory insult.80 To quantify the intensity of visceral pain, visceromotor behavioral responses (VMRs) were determined by recording abdominal muscle contractions during noxious colorectal distention.81 In this model a miniature SCS electrode system was implanted chronically with the techniques used in animal studies on neuropathic pain.82 After 1 week, animals were anesthetized briefly with isoflurane so a strain gauge force transducer could be sutured on the right external oblique abdominal muscle. A balloon inserted in the colon was then used to distend normal colons and those irrigated with acetic acid, which sensitizes the colon; the number of abdominal contractions recorded from the strain gauge was determined with and without SCS. The results showed that SCS significantly reduced VMR responses generated with colorectal distention in both normal and acutely sensitized colons. The rat model of postinflammatory colonic hypersensitivity also showed that SCS could significantly reduce VMR responses to innocuous colorectal distention.80 Thus the ability of SCS to reduce colonic sensitivity raises the possibility that SCS may be used therapeutically to treat abdominal cramping and abdominal spasms that result in visceral pain of gastrointestinal origin. The findings from the animal studies were translated from bench to bedside because subsequently a single case study reported that SCS reduced hypersensitivity and produced relief of diarrhea in a patient suffering from severe IBS.83 Further support came from Khan, Raza, and Khan,84 who conducted a retrospective study showing that SCS can be used effectively to treat a variety of visceral pain syndromes such as generalized abdominal pain, chronic nonalcoholic pancreatitis, and pain following posttraumatic splenectomy. Thus the agreement between the clinical reports and animal studies supports the idea that SCS might be used in the future to treat various functional bowel and other visceral disorders. Ongoing randomized cross-over prospective clinical studies indicate that two thirds of patients with IBS can be treated effectively by SCS applied at the T6-T8 segments.85
Neuromodulation Mechanisms for Neuropathic Pain
A limitation of the neural mechanisms used to describe the effects of SCS in the previous sections is that they were based on experiments conducted primarily with normal animals. Although these mechanisms provide clues about the mechanisms, the ability to translate that information to the bedside is reduced. The advantage of neuromodulation mechanisms for neuropathic pain is that the studies from the mid-nineties and onward were performed on different models of nerve injury–induced “painlike behavior.”86 After a nerve lesion is generated by manipulating the sciatic nerve, peripheral branches of the sciatic nerve, or spinal roots, the posture of the animals of the nerve-injured limb soon changes; and the sensitivity of the limb to normally innocuous mechanical and thermal stimuli also increases in many cases. These behavioral changes are the visible results of both peripheral and central sensitization.87 The most common method of evaluating the tactile hypersensitivity is to probe the nerve-injured hindpaw with von Frey filaments and observe the threshold that induces a withdrawal response to innocuous stimuli. This hypersensitivity is the most common behavioral sign in animal models of neuropathy; however, the pathophysiologic mechanisms are still not fully understood.88,89 This measurable sign of hypersensitivity does resemble a “stimulus-evoked painlike reaction,” which can be interpreted as being similar to allodynia observed in patients suffering from painful neuropathic conditions.90 A notable concern in this context is that tactile hypersensitivity occurs in a much larger proportion of nerve-injured rats but only 20% to 40% of neuropathic pain patients present with mechanical allodynia.91 Unfortunately, neuropathic pain animal models almost never express behavioral signs indicating the presence of continuous, spontaneous pain. These issues need to be considered when attempting to translate the results of these animal studies to the bedside.
Spinal Neural Networks of the Dorsal Horn
Tactile hypersensitivity or allodynia primarily results from the involvement of low threshold Aβ fibers and central sensitization of neural networks in the gray matter of the spinal cord.92 The central changes in the spinal cord following peripheral nerve injury depend mainly on altered characteristics primarily of multimodal wide–dynamic range (WDR) neurons. The altered characteristics of these neurons are persistent augmented responses to innocuous somatic stimuli and a marked increase in spontaneous activity. These characteristics are amenable to modulation by SCS. Acute experiments conducted in nerve-lesioned rats have shown that SCS elicits a significant and long-lasting inhibition of the augmented responses to innocuous somatic stimuli and to the after-discharges in WDR cells.93 Furthermore, studies conducted in freely moving, nerve-lesioned rats have shown that in some of the animals SCS may effectively suppress tactile hypersensitivity, similar to the effect on allodynia observed in neuropathic pain patients.94–96 In translating this information to the clinical setting, this SCS suppression of dorsal horn neuronal activity may be related to the beneficial effect of SCS not only on the allodynia but also on the spontaneous neuropathic pain.
The ability of SCS to alter the characteristics of WDR spinal neurons, to affect other components of the spinal neural network, and to reduce tactile hypersensitivity most likely requires activation of multiple transmitter/receptor systems. However, very little data are available from human studies to know about systems that are critically involved in the attenuation of chronic, neuropathic pain by SCS. A series of studies performed in nerve-lesioned animals provide important clues about the transmitters that might contribute to central sensitization and the reduction in tactile hypersensitivity (Fig. 2-5).

The hyperexcitability of WDR cells in the dorsal horns of nerve-lesioned animals93 appears to be correlated with increased basal release of excitatory amino acids such as glutamate, and malfunction of the local spinal γ-aminobutyric acid (GABA) system.97,98 Attenuation of the hyperexcitability of WDR cells by SCS most likely results from an induced release of GABA in the dorsal and simultaneous decrease of the interstitial glutamate concentration.93,97
The GABA-B receptor activation appears to be critical for suppressing release of glutamate.97,99,100 An early study showed that the release of GABA is only observed in animals when SCS reduces tactile sensitivity, but not in a group of nonresponding animals.98 However, an intrathecal injection of the GABA-B receptor agonist baclofen administered in these nonresponding animals could transform them into responders to SCS.100
In addition to the GABA system, the cholinergic system also plays an important role in producing the antinociceptive effects of SCS. The first indication pointing to the involvement of the cholinergic system came from a study showing that subeffective, intrathecal doses of clonidine transformed animals from nonresponders to responders to SCS.101 It has also been shown that SCS releases acetylcholine in the dorsal horn; this effect depends on activation of the muscarinic (M4) receptor.102 Furthermore, a subeffective intrathecal dose of a muscarinic receptor agonist (oxotremorine) could also transform nonresponding animals into responders to SCS.103
An exciting development resulting from these studies is that the findings were translated from bench to bedside. Baclofen was developed into a therapeutic benefit to treat neuropathic pain patients who responded inappropriately or did not experience enough relief from SCS. It is also interesting to note that the beneficial effects in patients who responded to this “drug-enhanced spinal stimulation therapy” have been stable for many years.104,105 In addition to baclofen, intrathecal infusions of clonidine, which depends on the cholinergic system, also proved to be effective as an adjunct to SCS when stimulation alone was ineffective in treating neuropathic pain patients.106,107
SCS-induced release of adenosine, serotonin, and norepinephrine into the dorsal horn may also participate in the relief of neuropathic pain.22,23 In contrast to the cholinergic system that depends on interneurons in the gray matter, serotonin and norepinephrine are released from descending pathways and are involved in inhibition of spinal neuronal activity. El-Khoury and associates108 and Saadé and Jabbur109 have conducted a long series of studies showing that neuropathic pain involves spinal and supraspinal mechanisms and that SCS orthodromically excited dorsal column fibers, which in turn activated neural circuits in the brainstem that transmit information in descending pathways that release these transmitters. These results did not agree with previous observations showing that SCS primarily activated local spinal circuits.110 However, work from the same laboratory showed that serotonin released from the descending tract produces its inhibitory effects via GABA-B receptors in the spinal gray matter.111 Thus more studies need to be done to understand the local and supraspinal mechanisms that produce the relief of neuropathic pain to resolve these differences.
1 Head H, Thompson T. The grouping of afferent impulses within the spinal cord. Brain. 1906;29:537-741.
2 Mazars GJ. Intermittent stimulation of nucleus ventralis posterolateralis for intractable pain. Surg Neurol. 1975;4:93-95.
3 Mazars GJ, Merienne S, Cioloca C. Stimulations thalamiques intermittentes antalgiques. Rev Neurol (Paris). 1973;128:273-279.
4 Noordenbos W, editor. Pain. Amsterdam: Elsevier, 1959.
5 Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150(699):971-979.
6 Dickenson AH. Gate control theory of pain stands the test of time. Br J Anaesth. 2002;88(6):755-757.
7 Shealy CN, et al. Electrical inhibition of pain: experimental evaluation. Anesth Analg. 1967;46:299-305.
8 Shealy CN, Mortimer JT, Reswick JB. Electrical inhibition of pain by stimulation of the dorsal columns: preliminary clinical report. Anesth Analg. 1967;46:489-491.
9 Lindblom U, Meyerson BA. Influence of touch, vibration and cutaneous pain of dorsal column stimulation in man. Pain. 1975;1:257-270.
10 Linderoth B, Meyerson BA. Spinal cord stimulation: techniques, indications and outcome. In: Lozano AM, Gildenberg PL, Tasker RR, editors. Textbook of stereotactic and functional neurosurgery. ed 2. Berlin-Heidelberg: Springer Verlag; 2009:3288. (Chapter 151)
11 Linderoth B, Simpson B, Meyerson BA. Spinal cord and brain stimulation. In McMahon S, Kolzenburg M, editors: Wall and Melzack’s textbook of pain, ed 5, Philadelphia: Elsevier, 2005. (Chapter 37, pp 563-582)
12 Meyerson BA, Linderoth B. Mechanisms of spinal cord stimulation in neuropathic pain, Invited review. Neurol Res. 2000;22:285-292.
13 Meyerson BA, Linderoth B. Therapeutic electrical neurostimulation from a historical perspective. In: Merskey H, Loeser JD, Dubner R, editors. The paths of pain 1975-2005. Seattle: IASP Press, 2005. (Chapter 21, pp 313-327)
14 Rexed B. A cytoarchitectonic atlas of the spinal cord in the cat. J Comp Neurol. 1954;100:297-379.
15 Grill WM. Principles of electric field generation for stimulation of the central nervous system. In: Krames ES, Hunter Peckham P, Rezai AR, editors. Neuromodulation. Amsterdam: Elsevier; 2009:146-154.
16 Holsheimer J. Computer modelling of spinal cord stimulation and its contribution to therapeutic efficacy. Spinal Cord. 1998;36:531-540. Review
17 Barolat G, et al. Mapping of sensory response to epidural stimulation of the intraspinal neural structures in man. J Neurosurg. 1993;78:233-239.
18 Garcia LA. Epidemiology and pathophysiology of lower extremity peripheral arterial disease. J. Endovascular Ther. 2006;13:II3-II9.
19 Golomb BA, Dang TT, Criqui MH. Peripheral arterial disease: morbidity and mortality implications. Circulation. 2006;114:688-699.
20 Hirsch AT, et al. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA. 2001;286:1317-1324. 1
21 Cameron T. Safety and efficacy of spinal cord stimulation for the treatment of chronic pain: a 20-year literature review. J Neurosurg. 2004;100:254-267.
22 Linderoth B, Foreman RD. Physiology of spinal cord stimulation: review and update. Neuromodulation. 1999;2(3):150-164.
23 Linderoth B, Foreman RD. Mechanisms of spinal cord stimulation in painful syndromes: role of animal models. Pain Med. 2006;7:S14-S26.
24 Augustinsson LE, Linderoth B, Mannheimer C. Spinal cord stimulation in different ischemic conditions. In: Illis LS, editor. Spinal cord dysfunction III: functional stimulation. Oxford: Oxford University Press, 1992. Chapter 12, pp 270-293
25 Linderoth B. Spinal cord stimulation in ischemia and ischemic pain. In: Horsch S, Claeys L, editors. Spinal cord stimulation: an innovative method in the treatment of PVD and angina. Darmstadt: Steinkopff Verlag; 1995:19-35.
26 Cook AW, et al. Vascular disease of extremities: electrical stimulation of spinal cord and posterior roots. NY State J Med. 1976;76:366-368.
27 Linderoth B, Gunasekera L, Meyerson BA. Effects of sympathectomy on skin and muscle microcirculation during dorsal column stimulation: animal studies. Neurosurgery. 1991;29:874-879.
28 Linderoth B, Herregodts P, Meyerson BA. Sympathetic mediation of peripheral vasodilation induced by spinal cord stimulation: animal studies of the role of cholinergic and adrenergic receptor subtypes. Neurosurgery. 1994;35:711-719.
29 Croom JE, et al. Cutaneous vasodilation during dorsal column stimulation is mediated by dorsal roots and CGRP. Am J Physiol Heart Circ Physiol. 1997;272:H950-H957.
30 Wu M, Linderoth B, Foreman RD. Putative mechanisms behind effects of spinal cord stimulation on vascular diseases: a review of experimental studies. Auton Neurosci. 2008;138:9-23.
31 Augustinsson L, et al. Epidural electrical stimulation in severe limb ischemia: pain relief, increased blood flow, and a possible limb-saving effect. Ann Surg. 1985;202:104-110.
32 Horsch S, Schulte S, Hess S. Spinal cord stimulation in the treatment of peripheral vascular disease: results of a single-center study of 258 patients. Angiology. 2004;55:111-118.
33 Linderoth B, Fedorcsak I, Meyerson BA. Peripheral vasodilatation after spinal cord stimulation: animal studies of putative effector mechanisms. Neurosurgery. 1991;28:187-195.
34 Tanaka S, et al. Role of primary afferents in spinal cord stimulation-induced vasodilatation: characterization of fiber types. Brain Res. 2003;959:191-198.
35 Tanaka S, et al. Mechanisms of sustained cutaneous vasodilation induced by spinal cord stimulation. Auton Neurosci. 2004;114(1-2):55-60.
36 Wu M, et al. Sensory fibers containing vanilloid receptor-1 (VR-1) participate in spinal cord stimulation-induced vasodilation. Brain Res. 2006;1107:177-184.
37 Wu M. Roles of peripheral terminals of transient receptor potential vanilloid-1 containing sensory fibers in spinal cord stimulation-induced peripheral vasodilation. Brain Res. 2007;1156:80-92.
38 Gao J, et al. Effects of spinal cord stimulation with “standard clinical” and higher frequencies on peripheral blood flow in rats. Brain Res. 2010;1313:53-61.
39 Tanaka S, et al. Local cooling alters neural mechanisms producing changes in peripheral blood flow by spinal cord stimulation. Auton Neurosci. 2003;104:117-127.
40 Gherardini G, et al. Spinal cord stimulation improves survival in ischemic skin flaps: an experimental study of the possible mediation by calcitonin gene-related peptide. Plast Reconstr Surg. 1999;103:1221-1228.
41 Jurell G, Jonsson CE. Increased survival of experimental skin flaps in rats following treatment with antiadrenergic drugs. Scand J Plast Reconstr Surg. 1976;10:169-172.
42 Gersbach P, et al. Influence of high-cervical spinal cord stimulation on antigen-induced bronchospasm in an ovine model (abstract). San Diego: ALA/ATS International Congress; 1999.
43 Illis LS, editor. Spinal cord dysfunction. III: functional stimulation. Oxford: Oxford University Press, 1992.
44 Gibbons RJ, et al. ACC/AHA/ACP-ASIM guidelines for the management of patients with chronic stable angina: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Patients With Chronic Stable Angina). J Am Coll Cardiol. 1999;33:2092-2197.
45 Foreman RD. Mechanisms of cardiac pain. Annu Rev Physiol. 1999;61:143-167.
46 DeJongste MJL, et al. Effects of spinal cord stimulation on daily life myocardial ischemia in patients with severe coronary artery disease: a prospective ambulatory ECG study. Br Heart J. 1994;71:413-418.
47 Mannheimer C, et al. Transcutaneous electrical nerve stimulation in severe angina pectoris. Eur Heart J. 1982;3(4):297-302. Aug
48 Mannheimer C, et al. Epidural spinal electrical stimulation in severe angina pectoris. Br Heart J. 1988;59:56-61.
49 Murphy DF, Giles KE. Dorsal column stimulation for pain relief from intractable angina. Pain. 1987;28:365-368.
50 Buchser E, Durre A, Albrecht E. Spinal cord stimulation for the management of refractory angina pectoris. J Pain Symp Manag. 2006;4(suppl):S36-S42.
51 Chua R, Keogh A. Spinal cord stimulation significantly improves refractory angina pectoris—a local experience spinal cord stimulation in refractory angina. Heart Lung Circ. 2005;14:3-7.
52 Jessurun GA, DeJongste MJ, Blanksma PK. Current views on neurostimulation in the treatment of cardiac ischemic syndromes. Pain. 1996;66:109-116.
53 Yu W, et al. Spinal cord stimulation for refractory angina pectoris: a retrospective analysis of efficacy and cost–benefit. Coron Artery Dis. 2004;15:31-37.
54 Chandler MJ, et al. A mechanism of cardiac pain suppression by spinal cord stimulation: implications for patients with angina pectoris. Eur Heart J. 1993;14:96-105.
55 Hautvast RW, et al. Effect of spinal cord stimulation on myocardial blood flow assessed by positron emission tomography in patients with refractory angina pectoris. Am J Cardiol. 1996;77:462-467.
56 Eliasson T, Augustinnson LE, Mannheimer C. Spinal cord stimulation in severe angina pectoris—presentation of current studies, indications, and clinical experience. Pain. 1996;65:169-179.
57 Mannheimer C, et al. Effects of spinal cord stimulation in angina pectoris induced by pacing and possible mechanism of action. Br Med J. 1993;307:477-480.
58 Kingma J, et al. Neuromodulation therapy does not influence blood flow distribution or left-ventricular dynamics during acute myocardial ischemia. Auton Neurosci. 2001;91(1-2):47-54.
59 Hautvast RW, et al. Effect of spinal cord stimulation on heart rate variability and myocardial ischemia in patients with chronic intractable angina pectoris—a prospective ambulatory electrocardiographic study. Clin Cardiol. 1998;21:33-38.
60 Sanderson JE, et al. Epidural spinal electrical stimulation for severe angina: a study of its effects on symptoms, exercise tolerance and degree of ischaemia. Eur Heart J. 1992;13:628-633.
61 Cardinal R, et al. Spinal cord activation differentially modulates ischemic electrical responses to different stressors in canine ventricles. Auton Neurosci. 2004;111(1):34-47.
62 Tomoike H, et al. Functional significance of collaterals during ameroid–induced coronary stenosis in conscious dogs. Circulation. 1983;67:1001-1008.
63 Lopshire JC, et al. Spinal cord stimulation improves ventricular function and reduces ventricular arrhythmias in a canine postinfarction heart failure model. Circulation. 2009;120:286-294.
64 Southerland EM, et al. Pre-emptive, but not reactive, spinal cord stimulation mitigates transient ischemia induced myocardial infarction via cardiac adrenergic neurons. Am J Physiol Heart Circ Physiol. 2006;292:H311-H317.
65 Armour JA, et al. Long-term modulation of the intrinsic cardiac nervous system by spinal cord neurons in normal and ischaemic hearts. Auton Neurosci. 2002;95:71-79.
66 Foreman RD, et al. Modulation of intrinsic cardiac neurons by spinal cord stimulation: implications for its therapeutic use in angina pectoris. Cardiovasc Res. 2000;47:367-375.
67 Ardell JL. Intrathoracic neuronal regulation of cardiac function. In: Armour JA, Ardell J, editors. Basic and clinical neurocardiology. New York: Oxford University Press; 2004:118-152.
68 Armour JA. Myocardial ischaemia and the cardiac nervous system. Cardiovasc Res. 1999;41:41-54.
69 Cardinal R, et al. Spinal cord stimulation suppresses bradycardias and atrial tachyarrhythmias induced by mediastinal nerve stimulation in dogs. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1369-R1375.
70 Issa ZF, et al. Thoracic spinal cord stimulation reduces the risk of ischemic ventricular arrhythmias in a postinfarction heart failure canine model. Circulation. 2005;111:3217-3220.
71 Broadley KJ, Penson PE. The roles of alpha- and beta-adrenoceptor stimulation in myocardial ischaemia. Auton Autacoid Pharmacol. 2004;24:87-93.
72 Sanada S, Kitakaze M. Ischemic preconditioning: emerging evidence, controversy, and translational trials. Int J Cardiol. 2004;97:263-276.
73 Eliasson T, et al. Myocardial turnover of endogenous opioids and calcitonin-gene-related peptide in the human heart and the effects of spinal cord stimulation on pacing-induced angina pectoris. Cardiology. 1998;89:170-177.
74 Lembo T, et al. Symptom duration in patients with irritable bowel syndrome. Am J Gastroenterol. 1996;91:898-905.
75 Mayer EA, Gebhart GF. Basic and clinical aspects of visceral hyperalgesia. Gastroenterology. 1994;107:271-293.
76 Greenwood-Van Meerveld B, et al. Attenuation by spinal cord stimulation of a nociceptive reflex generated by colorectal distention in a rat model. Auton Neurosci. 2003;104:17-24.
77 Gunter WD, et al. Evidence for visceral hypersensitivity in high-anxiety rats. Physiol Behav. 2000;69:379-382.
78 Langlois A, et al. Response heterogeneity of 5-HT3 receptor antagonists in a rat visceral hypersensitivity model. Eur J Pharmacol. 1995;318:141-144.
79 Plourde V, St-Pierre S, Quirion R. Calcitonin gene-related peptide in viscerosensitive response to colorectal distension in rats. Am J Physiol. 1997;273:G191-G196.
80 Greenwood-Van Meerveld B, et al. Spinal cord stimulation attenuates visceromotor reflexes in a rat model of post-inflammatory colonic hypersensitivity. Auton Neurosci. 2005;122(102):69-76.
81 Ness TJ, Gebhart GF. Colorectal distention as a noxious visceral stimulus: physiologic and pharmacologic characterization of the pseudaffective reflexes in the rat. Brain Res. 1988;450:153-169.
82 Linderoth B, et al. An animal model for the study of brain transmitter release in response to spinal cord stimulation in the awake, freely moving rat: preliminary results from the periaqueductal grey matter. Acta Neurochir. 1993;58(suppl [Wien]):156-160.
83 Krames E, Mousad DG. Spinal cord stimulation reverses pain and diarrheal episodes of irritable bowel syndrome: a case report. Neuromodulation. 2004;7(2):82-88.
84 Khan YN, Raza SS, Khan EA. Application of spinal cord stimulation for the treatment of abdominal visceral pain syndromes: case reports. Neuromodulation. 2005;8:14-27.
85 Lind G et al: A prospective randomized trial of spinal cord stimulation for treatment of irritable bowel syndrome (abstract), Toronto, May 24-27, 2009, WSSFN Quadrennial Meeting. p 164.
86 Campbell JN, Meyer RA. Mechanisms of neuropathic pain (Review). Neuron. 2006;5:77-92.
87 Suzuki R, Dickenson A. Spinal and supraspinal contributions to central sensitization in peripheral neuropathy. Neurosignals. 2005;14(4):175-181.
88 Baba H. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818-830.
89 Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1-32.
90 Harke H, et al. Spinal cord stimulation in sympathetically maintained complex regional pain syndrome type I with severe disability: a prospective clinical study. Eur J Pain. 2005;9:363-373.
91 Hansson P. Difficulties in stratifying neuropathic pain by mechanisms. Eur J Pain. 2003;7(4):353-357.
92 Woolf C, Doubell T. The pathophysiology of chronic pain—increased sensitivity to low threshold A-beta fiber inputs. Curr Opin Neurobiol. 1994;4:525-534.
93 Yakhnitsa V, Linderoth B, Meyerson BA. Spinal cord stimulation attenuates dorsal horn neuronal hyperexcitability in a rat model of mononeuropathy. Pain. 1999;79:223-233.
94 Meyerson BA, Linderoth B. Spinal cord Stimulation: mechanisms of action in neuropathic and ischemic pain. In: Simpson BA, editor. Electrical stimulation and the relief of pain, vol 15. New York: Elsevier; 2003. Chapter 11, pp 161-182
95 Meyerson BA, Linderoth B. Mode of action of spinal cord stimulation in neuropathic pain. J Pain Symptom Manage. 2006;31:6-12.
96 Meyerson BA, et al. Spinal cord stimulation in animal models of mononeuropathy: effects on the withdrawal response and the flexor reflex. Pain. 1995;61:229-243.
97 Cui JG, et al. Spinal cord stimulation attenuates augmented dorsal horn release of excitatory amino acids in mononeuropathy via a GABAergic mechanism. Pain. 1997;73:87-95.
98 Stiller CO, et al. Release of GABA in the dorsal horn and suppression of tactile allodynia by spinal cord stimulation in mononeuropathic rats. Neurosurgery. 1996;39:367-375.
99 Cui JG, Linderoth B, Meyerson BA. Effects of spinal cord stimulation on touch-evoked allodynia involve GABAergic mechanisms: an experimental study in the mononeuropathic rat. Pain. 1996;66:287-295.
100 Cui JG, et al. Effects of spinal cord stimulation on tactile hypersensitivity in mononeuropathic rats is potentiated by GABA-B and adenosine receptor activation. Neurosci Lett. 1998;247:183-186.
101 Schechtmann G, et al. Intrathecal clonidine potentiates suppression of tactile hypersensitivity by spinal cord stimulation in a model of neuropathy. Anesth Analg. 2004;99:135-139.
102 Schechtmann G, et al. Cholinergic mechanisms in the pain relieving effect of spinal cord stimulation in a model of neuropathy. Pain. 2008;139:136-145.
103 Song Z, Meyerson BA, Linderoth B. Muscarinic receptor activation potentiates the effect of spinal cord stimulation on pain-related behaviour in rats with mononeuropathy. Neurosci Lett. 2008;436:7-12.
104 Lind G, et al. Intrathecal baclofen as adjuvant therapy to enhance the effect of spinal cord stimulation in neuropathic pain: a pilot study. Eur J Pain. 2004;8(4):377-383.
105 Lind G, et al. Baclofen-enhanced spinal cord stimulation and intrathecal balofen alone for neuropathic pain: long-term outcome of a pilot study. Eur J Pain. 2008;12:132-136.
106 Schechtmann G, et al. Intrathecal clonidine and baclofen produce enhancement of pain-relieving effects of spinal cord stimulation: a double blind study. Acta Neurochir. 2008;150(9):972. (abstract, ESSFN XVIII Congress, Rimini, Italy October 5-8, 2008)
107 Schechtmann G, et al. Drug-enhanced spinal stimulation intrathecal clonidine VS baclofen: a double-blind clinical pilot study. Pain Pract. 2009;9(Suppl. 1):249-288. (abstract, Fifth Congress World Institute Of Pain, NY, March 13-19, 2009)
108 El-Khoury C, et al. Attenuation of neuropathic pain by segmental and supraspinal activation of the dorsal column system in awake rats. Neuroscience. 2002;112(3):541-553.
109 Saadé NE, Jabbur SJ. Nociceptive behavior in animal models for peripheral neuropathy: spinal and supraspinal mechanisms. Prog Neurobiol. 2008;86:22-47.
110 Yakhnitsa V, Linderoth B, Meyerson BA. Modulation of dorsal horn neuronal activity by spinal cord stimulation in a rat model of neuropathy: the role of the dorsal funicles. Neurophysiology. 1998;30(6):424-427.
111 Song Z, et al. Pain relief by spinal cord stimulation involves serotonergic mechanisms: an experimental study in a rat model of mononeuropathy. Pain. 2009;147:241-248.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 2 Mechanisms of Spinal Neuromodulation
Chapter Synopsis: Electrical stimulation of the spinal cord (SCS) improves many forms of neuropathic pain; but, contrary to our early understanding, it can also affect some forms of nonneuropathic nociception. Chapter 2 examines the physiology of these indications. The understanding of SCS is rooted in Melzack and Wall’s5 gate control theory of pain transmission. By spinal stimulation of large-fiber neurons, the gate is activated to reduce transmission of neuropathic pain signals from primary small-fiber afferents. The technique generally does not alleviate acute nociception, but it can reduce certain types of peripheral nociception and can even alleviate underlying conditions. SCS has been shown to affect ischemic limb pain caused by peripheral arterial occlusive disorder (PAOD), angina, and gastrointestinal disorders such as irritable bowel syndrome (IBS).
Transmission of nociceptive information from the site of an injury may generate a perception of pain because an imbalance exists between large and small fiber systems (cf. Head and Thompson1). This concept eventually evolved into Melzack and Wall’s5 gate control theory.Background
Therapeutic effects of neuromodulation are based on the concept that selective excitation of large afferent fibers activates mechanisms that control pain. This fits well with the idea that pain may occur as a result of an imbalance between large and small fiber systems that transmit nociceptive information from the site of injury. Previous investigators have provided a long history of support for this concept. As early as 1906 Head and Thompson1 argued that fine discrimination such as touch normally exerts an inhibitory influence on impulses transmitted in fibers mediating nociception, which results in pain. This inhibition or facilitation of sensory impulses has been proposed to occur in the dorsal horn before nociceptive information is relayed onto secondary neurons. Furthermore, clinical trials performed in the early sixties using sensory thalamic stimulation2,3 were based on the notion that activation of fine discrimination receptors (touch) exerted an inhibitory influence over sensations such as pain, pressure, heat, or cold. It should also be noted that Noordenbos4 used the descriptive phrase “fast blocks slow” to stress the inhibitory influence of fast on slow fibers.
The concept of excitation of large afferent fibers activating pain control mechanisms advanced very rapidly with the publication of the article proposing the gate control theory; it is one of the most cited papers in modern pain literature.5 In this article the authors suggested that the therapeutic implication of their model would be to selectively activate large fibers to control pain. Thus even though the basic idea underlying the gate control theory was not completely unknown, it was built on a foundation of creative experiments using modern electrophysiological techniques. The results of these experiments were clearly synthesized and discussed in a form that postulated a new conceptualization of pain and pain control. Subsequently, numerous studies were conducted to criticize the theory, but nevertheless its simplicity has provided a useful frame of reference to explain mechanisms of pain generation and pain control. As Dickenson6 pointed out in his editorial about the ability of the gate control theory of pain to stand the test of time, the concepts of convergence and modulation changed the focus from destructing pathways for relief of pain to controlling pain by modulation in which excitation is reduced and inhibition is increased. The gate control theory accelerated the pursuit of modern pain research to explore how the pervasive plasticity of the nervous system plays a critical role in the generation, maintenance, and modulation of pain.
The gate control theory served as a critical catalyst in the clinical arena to spawn the development of various forms of neuromodulation that led to new therapies. The insights gained by Shealy and his colleagues7 and Shealy, Mortimer, and Reswick8 in animal experiments led them to conduct the first human trials with electrical spinal cord stimulation (SCS) as one form of neuromodulation.8 Their experimental studies in conscious cats revealed that stimulating the dorsal aspect of the spinal cord blocked responses to nociceptive peripheral stimuli. On the basis of this study and support of the gate control theory, it was assumed that neuromodulation could be used to treat all forms of nociceptive pain. However, several reports pointed out that SCS is ineffective for treating acute nociceptive conditions in contrast to what was predicted from the gate control theory; but eventually it has become the foremost treatment for neuropathic pain originating from the periphery.9–13 Nevertheless, numerous reports appeared during the eighties to convince clinicians that SCS could also be used to alleviate certain types of nociceptive pain, including selected ischemic pain states such as peripheral arterial occlusive disease (PAOD), vasospastic conditions, and therapy-resistant angina pectoris. The mechanisms of action for SCS are slowly emerging as more solid evidence has revealed some of the underlying physiological mechanisms. Clinical observations coupled with important experimental data clearly demonstrate that SCS applied to different segments of the spinal cord elicits fundamentally different results on various target organs or parts of the body (Fig. 2-1).
Organization and Electrical Properties of the Spinal Cord
The spinal cord is encased within the vertebral canal, which is made up of vertebrae that encircle the spinal cord but limit space for insertion of stimulating electrodes. The spinal cord in an adult human extends from the foramen magnum to the first or second lumbar vertebra and is divided into cervical, thoracic, lumbar, and sacral segments. The naming of the segments is based on the regions of the body innervated by the spinal cord. Examination of a cross section of the spinal cord shows that it is composed of gray matter and surrounded by white matter (Fig. 2-2).
The gray matter is comprised of cell bodies with their dendrites and initial segment of the axon, microglia, and astrocytes. It is divided into a posterior horn, intermediate zone, and the ventral horn. The gray matter is further divided into laminae I to X; these divisions are based on the size, shape, and distribution of neurons located in these laminae.14 The input received by these neurons and the trajectory of the axons from them also help to characterize laminae. Neurons of dorsal and intermediate laminae (I to VII, X) generally receive sensory information originating from peripheral sensory receptors. These neurons integrate this information with input arriving from descending pathways. Some of the cell bodies have short axons and serve as interneurons, whereas others are the cells of origin of ascending sensory pathways. The interneurons may also participate in local reflexes. The ventral laminae (VIII, IX) are generally composed of motoneurons that form the motor nuclei.
The electrical properties, more specifically the electrical conductivity, of white and gray matter of the spinal cord are not homogeneous. For SCS it is important to know that the electrical conductivity of the dorsal column is anisotropic; that is, current can travel in the direction parallel to the axons more easily than in the direction perpendicular to axons.15 The electrical properties within the gray matter also vary because neurons and glia have diverse orientations, ubiquitous dimensions, and different dendritic characteristics.
Neuromodulation using electrical stimulation of the spinal cord depends on the conductivity of the intraspinal elements relative to the position of the electrode.16 If an axon is depolarized or made more electrically positive, it produces an action potential that is transmitted orthodromically and antidromically within the axon. The cathode of an external electrode must be negatively charged to generate the action potential in the axon. In contrast, if an axon is hyperpolarized or made more negatively charged, its ability to generate an action potential is reduced because the threshold for depolarization is increased. A positively charged external electrode or anode produces this effect. Thus the active electrode for electrical stimulation serves as the cathode, whereas the anode or positive electrode may serve as a shield to prevent stimulation of neuronal structures such as dorsal roots that might interfere with effective neuromodulation. For SCS the electrode most commonly is placed on the surface of the dura mater. Activation of the electrode releases electric current that is transmitted through the dura mater and the highly conductive cerebrospinal fluid (CSF) before it reaches the dorsal part of the spinal cord. The dura mater has low conductivity, but it is so thin that the current generally is not impeded significantly as it passes through the dura to the CSF. Furthermore, the vertebral bone has the lowest conductivity so it insulates pelvic structures and visceral organs from the electric field generated by SCS. Once the electric current reaches the spinal cord, several factors may determine the neural structure being stimulated. Jan Holsheimer16 has used computerized models of the spinal cord to study the activation of axons by electrical current. In addition to the fiber diameter, the presence of myelination, and the depth of CSF layer surrounding the cord at the level of an electrode, the axon orientation has important implications for activation thresholds. In general, axons of the dorsal columns have higher activation thresholds than fibers such as the dorsal roots that are oriented laterally or angle as they enter the spinal cord.16
The dorsal column is composed primarily of large-diameter afferent nerve fibers with relatively low thresholds for recruitment when cathodal electrical pulses are generated through the epidural electrode that is attached to a spinal cord stimulator. It is important to note that the electrode for SCS needs to be placed near midline to prevent the activation of dorsal root fibers.17 Stimulation amplitudes are then increased to intensities that recruit large fibers to produce action potentials and produce paresthesias. These action potentials are transmitted orthodromically and antidromically in these axons. The action potentials transmitted antidromically reach the collateral processes that penetrate the gray matter of the spinal cord. Their activation causes the release of transmitters, which activates the “gate.” Activation of the gate sets in motion neural mechanisms that reduce pain and improve organ function. The details of these mechanisms are discussed in subsequent paragraphs.
Neuromodulation Mechanisms in Ischemic Pain
Ischemic painful conditions of the limbs commonly results from PAOD, which is caused by obstruction of blood flow into an arterial tree.18 PAOD is a major cause of disability and loss of work and affects the quality of life.19,20 Morbidity and mortality are relatively high because effective treatments are very limited. Presently SCS is usually implemented only after vascular surgery and medications fail to slow or prevent the progression of PAOD. Surprisingly the success rate of SCS-treated PAOD is greater than 70%.21 Since ischemic pain is characterized generally as essentially nociceptive and several studies have indicated that SCS does not alleviate acute nociceptive pain,9,22,23 SCS-induced pain relief is most likely secondary to attenuation of tissue ischemia that occurs as a result of either increasing/redistributing blood flow to the ischemic area or decreasing tissue oxygen demand.24,25 Cook and associates26 were the first to report that SCS increased peripheral circulation of patients suffering from PAOD. Usually SCS is applied to the dorsal columns of lower thoracic (T10-T12) and higher lumbar spinal segments (L1-L2) to increase peripheral circulation in the legs of PAOD patients.
The mechanisms of SCS-induced vasodilation in the lower limbs and feet are not yet completely understood. Since no animal models of PAOD that generate ischemic pain have emerged, normal anesthetized animal models have been used to investigate the physiologic mechanisms of SCS-induced changes in peripheral blood flow (see reference 23 for review). Cutaneous blood flow and calculated vascular resistance in the glabrous skin of ipsilateral and contralateral hindpaws have been determined most commonly by using laser Doppler flowmetry. A thermistor probe placed next to the laser Doppler probe on the plantar aspect of the foot has been used to measure skin temperature. Various interventions such as injections of hexamethonium, administration of adrenergic agonists and antagonists, sympathetic denervation, dorsal rhizotomies, calcitonin gene-related peptide (CGRP) antagonists, nitric oxide synthetase inhibitors, and local paw cooling have been used to explore the underlying mechanisms of peripheral microcirculation. Studies using Doppler flowmetry and interventions for more than 30 years of clinical and basic science studies have resulted in the evolution of two theories to explain the mechanisms of SCS-induced vasodilation. One theory is that SCS decreases sympathetic outflow and reduces the constriction of arterial vessels27,28; the alternative theory is that SCS antidromically activates sensory fibers, which causes the release of vasodilators29, 30 (Fig. 2-3). The theory for SCS-induced suppression of sympathetic activity was based on results from clinical observations showing that a sympathetic block or sympathectomy produced pain relief and vasodilation imitated effects of treatment with SCS.31,32 This theory was tested in animal models in which SCS-induced cutaneous vasodilation in the rat hindpaw at 66% of motor threshold was abolished by complete surgical sympathectomy.33 SCS-induced vasodilation was markedly attenuated after administrating the ganglionic blocker, hexamethonium, or the neuronal nicotinic ganglionic blocker, chlorisondamine. These results led to the suggestion that efferent sympathetic activity, including nicotinic transmission in the ganglia and the postganglionic α1-adrenergic receptors are suppressed by SCS (see Fig. 2-3). The alternative theory of SCS-induced antidromic activation of sensory fibers was confirmed in studies showing that sensory afferent fibers are important for SCS-induced vasodilation and that at higher, but not painful, SCS intensities C-fibers may also contribute to the response30,34,35 (see Fig. 2-3). Thus SCS applied at the spinal L2-L5 segments excites dorsal column fibers that antidromically activate interneurons, which subsequently stimulate spinal terminals of transient receptor potential V1 (TRPV1) containing sensory fibers, which are primarily made up of C-fiber axons.36,37 These fibers transmit action potentials antidromically to nerve endings in the hindlimb. The action potentials evoke mechanisms that release vasodilators, including the most powerful vasodilator, CGRP, which binds to receptors on endothelial cells. The activation of these receptors leads to production and subsequent release of nitric oxide (NO), which results in relaxation of vascular smooth muscle cells (see reference 30 for review). The overall result is that relaxation of vascular smooth muscle cells decreases vascular resistance and increases peripheral blood flow. It should be noted that SCS applied at 500 Hz significantly increased cutaneous blood flow and decreased vascular resistance when compared to the responses induced at 50 Hz and 200 Hz; the effects at all of these frequencies depend on the activation of TRPV1-containing fibers and release of CGRP.38 The clinical use of such findings remains to be determined.
The level of sympathetic nervous system activity may shift the balance between the effects of sympathetic efferent suppression and antidromic activation of sensory afferent fibers. Cooler skin temperatures increase sympathetic activity. A notable observation is that SCS-induced vasodilation of a cooled hindpaw (<25° C) generated an early phase of vasodilation via sensory afferent fibers and a late phase via suppression of the sympathetic efferent activity.39 However, only sensory afferent activation occurred if SCS-induced vasodilation was performed in a warm paw (>28° C). Thus the balance of these two mechanisms most likely depends on the activity level of the sympathetic nervous system. Furthermore, another study showed that preemptive SCS increased the survival rate of skin flaps that were made ischemic by occluding the blood supply to the tissue for as long as 12 hours.40 Concomitant administration of the CGRP-1 receptor antagonist CGRP 8-37 markedly attenuated the cytoprotective effect40
[/not-level-membership-for-anesthesiology-category]
