Manifestations of Immunodeficiency in the Gastrointestinal Tract
Kay Washington
Chanjuan Shi
Primary Disorders of Immune Deficiency
Many of the primary disorders of immune deficiency (Table 5.1) are associated with gastrointestinal (GI) lesions. Manifestations of immune deficiency in the GI tract may be broadly divided into three categories: (1) increased susceptibility to infection, (2) idiopathic chronic inflammatory conditions, and (3) increased risk of neoplasia. Although many GI lesions are infectious (Table 5.2), chronic inflammatory conditions resembling celiac disease and inflammatory bowel disease (IBD) (Table 5.3) are seen in many patients with antibody deficiencies and are probably the result of the inability of dysfunctional mononuclear cells to suppress unwanted immune responses. All patients with primary immune deficiencies are at increased risk of neoplasia1 (Table 5.4), most commonly non-Hodgkin lymphoma (NHL), and the GI tract is often the primary site of involvement.2 In addition to the risk of lymphoma, some of the primary immune deficiencies are associated with increased risk of gastric adenocarcinoma3 and colorectal carcinoma.4,5
Table 5.1
Molecular Basis of Primary Immunodeficiency Disorders
| Disease | Proposed Cause |
| Selective IgA deficiency | Impaired IgA synthesis; molecular defect unknown in most cases; mutation in TNFRSF13B in ~5% |
| Common variable immunodeficiency | Impaired B cell maturation; molecular defect unknown in most cases; mutation in TNFRSF13B in ~10-15% |
| X-linked agammaglobulinemia | Mutation in BTK results in absence of BTK in B cells |
| Hyper-IgM syndrome | Mutation in CD40L, leading to absence of CD40 ligand on T cells |
| Hyper-IgE syndrome | Mutation in STAT3 in autosomal dominant form; mutation in DOCK8 in autosomal recessive form. |
| Severe combined immunodeficiency | Multiple defects, most commonly in the common γ chain; others include adenosine deaminase deficiency, purine nucleoside deficiency, and T-cell receptor deficiencies |
| Omenn syndrome | Hypomorphic missense mutation in RAG1/2, IL7RA,RMRP, ADA, DNA ligase IV, Artemis, γc |
| Immune dysregulation, polyendocrinopathy, enteropathy, X-linked | Mutation in FOXP3 |
| DiGeorge syndrome | Thymic hypoplasia; microdeletion in 22q11.2 |
| Chronic mucocutaneous candidiasis | Heterogeneous disorder; mutation in AIRE gene in some; mutation in STAT1 in autosomal dominant form |
| Wiskott-Aldrich syndrome | Mutation in WASP gene involved in cell trafficking and motility |
| Chronic granulomatous disease | Mutation in gene for component of NADPH oxidase; CYBB mutation in X-linked form; mutation in CYBA, NCF1, NCF2, or NCF4 in autosomal recessive forms |
Ig, Immunoglobulin; NADPH, reduced nicotinamide adenine dinucleotide phosphate.
From International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies, Notarangelo LD, Fischer A, et al. Primary immunodeficiencies: 2009 update. (Erratum appears in J Allergy Clin Immunol. 2010;125:771-773.) J Allergy Clin Immunol. 2009;124:1161-1178.
Table 5.2
Gastrointestinal Infections in Primary Immunodeficiency
| Disease | Gastrointestinal Infections |
| Selective IgA deficiency | Giardia intestinalis; strongyloidiasis |
| Common variable immunodeficiency | Giardia intestinalis; Cryptosporidium; CMV |
| X-linked agammaglobulinemia | Giardia intestinalis; Cryptosporidium Salmonella, Campylobacter Rotavirus, coxsackievirus, poliovirus |
| Hyper-IgM syndrome | Giardia intestinalis, Cryptosporidium, Entamoeba histolytica, Salmonella, Histoplasma capsulatum |
| Severe combined immunodeficiency | Candida; Salmonella and other bacterial pathogens; CMV, rotavirus, Epstein-Barr virus |
| DiGeorge syndrome | Candida |
| Chronic mucocutaneous candidiasis | Candida; Histoplasma capsulatum |
CMV, Cytomegalovirus; Ig, Immunoglobulin.
Table 5.3
Inflammatory Gastrointestinal Lesions in Primary Immunodeficiency
| Disease | Manifestation |
| IgA deficiency |
GVHD, Graft-versus-host disease; Ig, immunoglobulin.
Table 5.4
Malignancies Involving the Gastrointestinal Tract in Primary Immunodeficiency
| Disease | Gastrointestinal Malignancy |
| Immunoglobulin A deficiency |
Humoral Immunodeficiencies
Selective Immunoglobulin A Deficiency
Defined as a serum immunoglobulin A (IgA) concentration of less than 50 µg/mL, selective IgA deficiency is the most common primary immunodeficiency6; it occurs in 1 of every 600 people of Northern European ancestry.7 The disorder is 20 times more common in white Americans than in African Americans. Defects in antibody production in patients with IgA deficiency represent a continuum with those seen in common variable immunodeficiency (CVID), and 20% to 30% of IgA-deficient patients also have deficits in IgG subclasses. Mutations in the gene TNFRSF13B have been identified and associated with IgA deficiency,8–10 although the mutational defect remains unknown in many cases. This gene encodes a member of the tumor necrosis factor-receptor (TNFR) superfamily, the transmembrane activator and calcium-modulator and cyclophilin-ligand interactor (TACI), which mediates isotype switching in B lymphocytes (see later discussion).
Clinical manifestations of IgA deficiency range from no symptoms to recurrent infections (typically involving mucosal surfaces), autoimmune disorders, allergic diseases, and malignancy, but are typically milder than those seen with CVID. Recurrent upper and lower respiratory tract infections are common in both disorders. Similarly, GI manifestations of IgA deficiency are the same as those associated with CVID. Infections are less common than might be expected, possibly because of compensation for lack of mucosal IgA by transport of IgM across the mucosa into the gut lumen, but include acute diarrheal illnesses caused by bacterial enterocolitis and chronic diarrhea caused by persistent Giardia intestinalis infection. Chronic strongyloidiasis has also been reported.11
Susceptibility to autoimmune disorders such as insulin-dependent diabetes mellitus and celiac disease may be inherited together with IgA deficiency; all three conditions are linked to particular major histocompatibility haplotypes and probably represent genetically linked susceptibilities in certain populations. The prevalence of celiac disease, the most common noninfectious GI complication of IgA deficiency, is 7.7% in children with IgA deficiency, compared with 1 : 500 in the general population,12 and a novel shared risk locus, CTLA4/ICOS, has been described linking CVID, IgA deficiency, and celiac disease.13 Antigliadin IgA and endomysial IgA antibodies cannot be used as screening tools in the IgA-deficient population, but the morphology of celiac disease occurring in the setting of IgA deficiency is similar to that seen in immunocompetent patients. In addition, a sprue-like illness characterized by chronic diarrhea with villous atrophy that does not respond to a gluten-free diet may be seen in IgA deficiency, as in CVID. Pernicious anemia complicating chronic atrophic autoimmune gastritis is seen more commonly in IgA deficiency than in CVID, and nodular lymphoid hyperplasia (NLH) in the small bowel is only rarely reported.14 As with other B cell disorders, the incidence of Crohn disease and gastric adenocarcinoma appears to be increased in IgA deficiency.15
X-Linked Agammaglobulinemia
The typical patient with X-linked agammaglobulinemia (XLAG) is susceptible to bacterial infections because of the absence of all circulating immunoglobulin subtypes; mature circulating B cells are low to absent. This disorder is characterized by an inability to make antibodies to virtually all antigens. The molecular basis of most cases of XLAG was elucidated in 1993, when a defect in the BTK (Bruton tyrosine kinase) gene was found37,38; subsequent studies have identified numerous deleterious mutations.39–41 The BTK gene encodes a nonreceptor tyrosine kinase expressed in B-cell and myelomonocytic cell lineages but not in T cells. BTK functions in intracellular signaling pathways essential for pre–B-cell maturation, but the exact mechanism by which the defects in BTK lead to B-cell maturation arrest remains unclear. XLAG may have more phenotypic diversity than has previously been recognized, because adults with mild or no clinical symptoms but with deficiencies in BTK have been described.42–44 Age at onset, disease severity, and genotype are roughly associated with mutation severity (as classified based on structural and functional consequences) in some but not all cohorts.39,40
GI manifestations are less common in XLAG than in CVID, perhaps because of preserved T-cell function in XLAG,45 although persistent or recurrent diarrhea is reported in 23% to 29% of patients.39,46 Age at onset of GI symptoms is younger than in CVID patients, and autoimmune diseases are less common. Small intestinal and colonic mucosal biopsies in the XLAG patient without GI symptoms are notable only for the lack of plasma cells in the lamina propria, giving the lamina propria an empty appearance. Mucosal architecture is unremarkable, and villous blunting is not seen. Approximately one third of patients are seen initially with GI complaints, most commonly diarrhea or perirectal abscess, and 10% in one study had chronic GI symptoms, either from persistent infection with G. intestinalis, Salmonella, or enteropathic Escherichia coli or secondary to bacterial overgrowth. No cause for chronic diarrhea was found in half of these patients.47 Chronic infection with rotavirus is also reported in this population.48 Because biopsies are not routinely performed for this disorder, few descriptions of histopathologic findings are available, but moderate blunting of duodenal villi with crypt hyperplasia and an increase in lamina propria inflammatory cells are reported in acute infections.49 Degenerative changes may be noted in epithelial cells on the surface of the villus with no increase in crypt apoptosis50; crypt cells are spared, and the crypt zone undergoes a compensatory hyperplasia. The histologic changes of acute rotavirus infection are reported to resemble celiac disease but are patchier and quickly revert to normal with resolution of infection.51
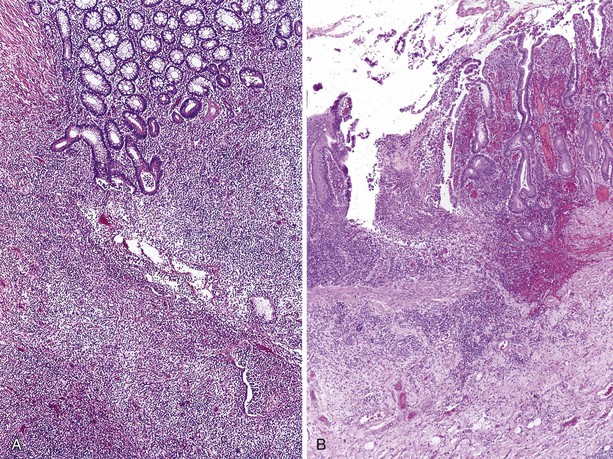
In addition to GI infections, a chronic ulcerating inflammatory condition, clinically similar to Crohn disease, that is manifested by recurrent diarrhea, malabsorption, ulcers, and small bowel strictures (Fig. 5.1). A prominent lymphocytic inflammatory infiltrate without plasma cells or granulomas is seen in the affected areas.23,52 In one case, enterovirus was found by polymerase chain reaction in inflamed ileum and adjacent mesenteric lymph nodes, suggesting that in some XLAG patients infection may be responsible for these lesions.53
Patients with XLAG are at increased risk for malignancy, even in childhood. The most common malignancy in this group is NHL, which often involves the GI tract, and many of these cases occur in children younger than 10 years of age.54 There are rare reported cases of gastric adenocarcinoma54 and colorectal adenocarcinoma.5 The increased incidence of colorectal carcinoma for patients with XLAG was calculated as 30-fold in one study,5 although other registry studies have reported few or no colorectal cancers in their patients with XLAG.46,55 In most of the reported cases, XLAG patients with colorectal carcinoma are young adults in their 20s who are seen with advanced-stage tumors. In one reported case, multiple colorectal adenomas in addition to carcinoma were found.5
X-Linked Hyperimmunoglobuin M Syndrome
X-linked hyperimmunoglobulin M syndrome (XHIM) results from a mutation in the gene for CD40 ligand, which leads to loss of isotype switching. T cells from patients with this disorder lack the CD40 ligand and therefore do not interact with CD40 on the B-cell surface, an event necessary for immunoglobulin class switching. These patients have very low levels of IgG and IgA and normal or elevated levels of IgM.56 They are susceptible to pyogenic infections similar to those encountered in XLAG, and in addition are susceptible to Pneumocystis carinii pneumonia. A variety of intracellular pathogens such as mycobacterial species, fungi, and viruses (e.g., CMV, adenovirus) are implicated in causing disease in these patients. The most common site of infection is the upper or lower respiratory tract (approximately 50% and 80% of cases, respectively).56 Disseminated infection57,58 and esophageal infection with Histoplasma capsulatum are also reported in XHIM.
Diarrhea occurs in one third to one half of patients56,59 and follows a chronic course in most. Chronic watery diarrhea may be caused by Cryptosporidium infection; G. intestinalis, Salmonella, and Entamoeba histolytica have also been implicated, although in most patients no pathogen is identified.56 NLH involving the GI tract is reported in approximately 5% of patients.59 Lymphoid hyperplasia may also result in hepatosplenomegaly, lymphadenopathy, and enlargement of the tonsils.60 IBD was reported in two patients with XHIM and chronic diarrhea; clinical details were not provided.59 Sclerosing cholangitis is a common and serious complication (affecting about 20% of European patients) and is often related to chronic infection with Cryptosporidium; liver transplantation may be necessary.59 In one European series, three of five patients infected with hepatitis B developed hepatocellular carcinoma.59
Patients with XHIM are prone to autoimmune hematologic diseases, including cyclic or chronic neutropenia, and oral and perianal ulcers are common during neutropenic episodes.59 Massive proliferation of IgM-producing plasma cells may involve the GI tract, liver, and gallbladder, usually in the second decade of life, and may prove fatal.61 High-grade neuroendocrine carcinoma of the colon has been reported in this disorder,62–64 and there is an increased incidence of liver and biliary tract tumors.63
Hyperimmunoglobulin E Syndrome
Also known as Job syndrome, hyperimmunoglobulin E syndrome is a rare, multisystem disorder characterized by recurrent elevated serum IgE, eczema, and sinopulmonary infections.65 Three genetic etiologies have been identified: loss of function of DOCK8 in the autosomal recessive form of the disorder,66 mutations in STAT3 in the more common autosomal dominant form,67 and a single reported case of Tyk deficiency.68 In addition to findings related to the immune system, characteristic facial features and dental and skeletal abnormalities occur in the autosomal dominant form, and severe viral cutaneous infections are seen in the autosomal recessive form.65 Chronic diarrhea and disorders resembling IBD are not reported in hyper-IgE syndrome. GI manifestations of the disease appear limited to mucocutaneous candidiasis, tissue-invasive fungal infections with Cryptococcus (reported in the esophagus69 and colon70), and ileocecal histoplasmosis mimicking Crohn disease.71,72 Perforation of the colon, probably related to infection with staphylococcal species, has also been reported,73 as well as diverticulitis in a young patient.74 Patients with hyper-IgE syndrome do not appear to be at increased risk for primary GI malignancy.
Common Variable Immunodeficiency
Although CVID is not a common disorder, it is probably the most common symptomatic primary immunodeficiency. Clinical and immunologic features are heterogeneous, but most patients are seen with recurrent bacterial infections, usually involving the upper and lower respiratory tracts and leading to chronic lung disease and bronchiectasis. Patients may be seen with CVID at any age from infancy to late adult life, and males and females are equally affected. Autoimmune manifestations such as thyroid dysfunction, pernicious anemia, autoimmune hemolytic anemia, autoimmune thrombocytopenia, and rheumatoid arthritis are common, and granulomatous involvement of skin and visceral organs mimicking sarcoidosis may be seen in CVID.16–18 Chronic GI disorders resulting in malabsorption and weight loss occur in approximately 20% of patients with CVID.
CVID is characterized immunologically by hypogammaglobulinemia involving multiple antibody classes. T-cell abnormalities are common; below-normal proliferative responses to mitogens are found in 40% of patients, and 20% have a relative lack of CD4+ T cells.18 The common abnormality shared by IgA deficiency and CVID is failure of terminal maturation of B lymphocytes into plasma cells producing various Ig subtypes. A primary B-cell defect is favored in many patients, but in others defective antigen responsiveness in helper T cells may be the underlying basis for the disorder.
A genetic basis has long been suspected, based on the observation that familial inheritance of CVID occurs in 20% of cases19 and that CVID and IgA deficiency tend to occur in the same family; individual family members may gradually convert from one disorder to the other. In multiple-case families, CVID is often present in the parents and IgA deficiency in the offspring, consistent with the hypothesis that CVID may develop later in life as a more severe manifestation of a common defect involving immunoglobulin class switching. Studies of isotype switching led to the discovery that the gene TNFRSF13B, which encodes the member of the TNFR family, TACI, is mutated in approximately 10% to 15% of patients with CVID and 5% of patients with IgA deficiency.20 TACI has also been shown to induce apoptosis in B cells, and this may be the basis for the susceptibility of patients with TACI mutations to autoimmune and lymphoproliferative disorders.20 New gene defects in BAFFR (TNFRSF13C), CD81, and CD20 (now called MS4A1) resulting in a CVID phenotype have been described in small numbers of patients,21 underscoring the genetic heterogeneity of this disorder, and genome-wide association studies have identified numerous candidate causative genes, as well as a strong association of CVID with the major histocompatibility region.22
Patients with CVID are at particular risk for chronic inflammatory disorders and malignancies affecting the GI tract. The inflammatory disorders may represent a response to acute or chronic infection, but in some patients the GI lesions are probably a manifestation of autoimmunity and are associated with other disorders of autoimmunity.23 In one large clinical study of patients with CVID, 22% had one or more autoimmune diseases, most commonly idiopathic thrombocytopenia purpura (6%) and autoimmune hemolytic anemia (5%).18
Infections in CVID
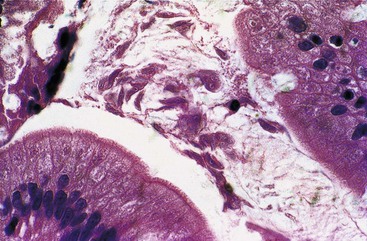
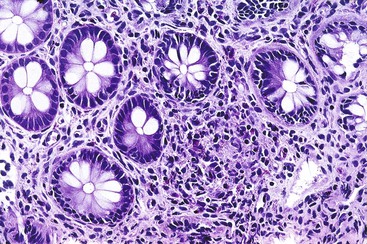
Chronic infection with G. intestinalis is a common problem in patients with CVID and may or may not cause clinical symptoms. In some cases, malabsorption, steatorrhea, and villous abnormalities can be reversed if Giardia is eradicated. Small bowel mucosal abnormalities in giardiasis include villous blunting, increased intraepithelial lymphocytes, and NLH. The trophozoite form of the organism can be identified on small bowel biopsy (Fig. 5.2). The prevalence of Giardia infection in this population appears to be decreasing, but giardiasis remains a significant cause of chronic diarrhea in CVID.15
Other GI infections are less common in CVID. Cryptosporidiosis is occasionally found. The prevalence of common bacterial intestinal infections (e.g., Salmonella, Campylobacter) does not appear to be increased. Although prolonged antibiotic use is common in these patients, an increase in pseudomembranous colitis has not been reported.15 On occasion, viral and fungal organisms infect the GI tract in CVID patients, but such infections are less common in CVID than in acquired immunodeficiency syndrome (AIDS). Cytomegalovirus (CMV) infection involving the esophagus, stomach, jejunum, and ileocecal area and resulting in multiple ulcers and obstructing strictures has been reported in a patient with CVID.15
Inflammatory Disorders and Malignancy in CVID
Stomach
In the stomach, a nonspecific increase in lamina propria lymphocytes is seen in some patients with CVID (Fig. 5.3, A); increased apoptosis of gastric epithelial cells is present in some cases23,24 (see Fig. 5.3, B). In a study of gastric biopsies from 34 patients with CVID and dyspepsia, 41% of patients were infected with Helicobacter pylori. All H. pylori–positive patients and 20% of H. pylori–negative patients had chronic gastritis, and 50% of those infected with H. pylori had multifocal atrophic gastritis. Ten percent of H. pylori–negative patients had multifocal atrophic gastritis.25 Atrophic gastritis resembling autoimmune atrophic gastritis on clinical and morphologic grounds (see Fig. 5.3, C) and resulting in pernicious anemia may occur in the absence of demonstrable anti–parietal cell antibodies in these patents. Atrophic gastritis may develop at a very young age in patients with CVID, and it has been reported in a 6-year-old who developed multifocal gastric adenocarcinoma at age 11.26
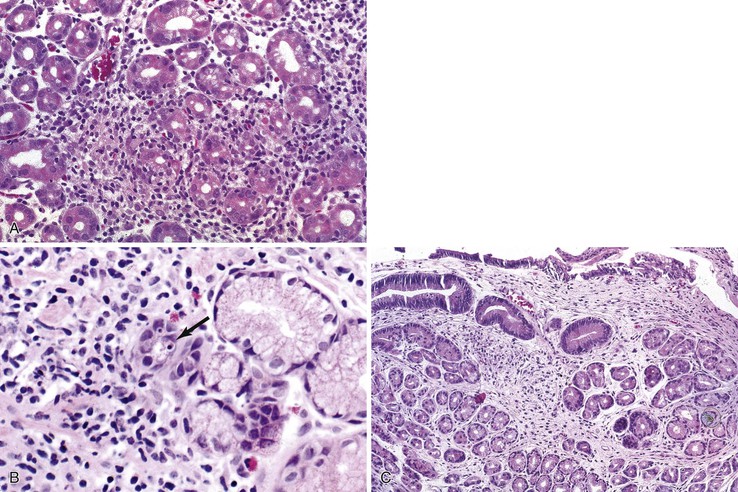
Adults with CVID are also at increased risk for gastric adenocarcinoma. It has been estimated that patients with CVID have a 47-fold increase in gastric carcinoma compared with the general population of Great Britain,27 and gastric carcinoma ultimately develops in 5% to 10% of CVID patients, usually many years after the onset of hypogammaglobulinemia.
Small Bowel
In the small bowel, a sprue like lesion with villous blunting occurs in some patients with CVID and is associated with severe malabsorption, often requiring parenteral nutrition. Villous atrophy associated with CVID generally lacks the degree of crypt hyperplasia seen in celiac disease (Fig. 5.4, A), but may be indistinguishable on biopsy. In general, the lamina propria inflammatory infiltrate is not as prominent as in celiac disease, and enterocyte maturation is normal, with preservation of the brush border.23 Most CVID patients with this small bowel lesion do not respond to a gluten-free diet, although an elemental diet may be beneficial, and most respond to corticosteroids, at least initially.28 Plasma cells are absent or are found only in very small numbers in the lamina propria. Surface intraepithelial lymphocytes are often markedly increased (see Fig. 5.4, B), even in the absence of villous atrophy. In some cases, an increase in apoptotic bodies is found in crypt epithelial cells (see Fig. 5.4, C). Clinical autoimmune enteritis with loss of goblet cells has also been reported.24
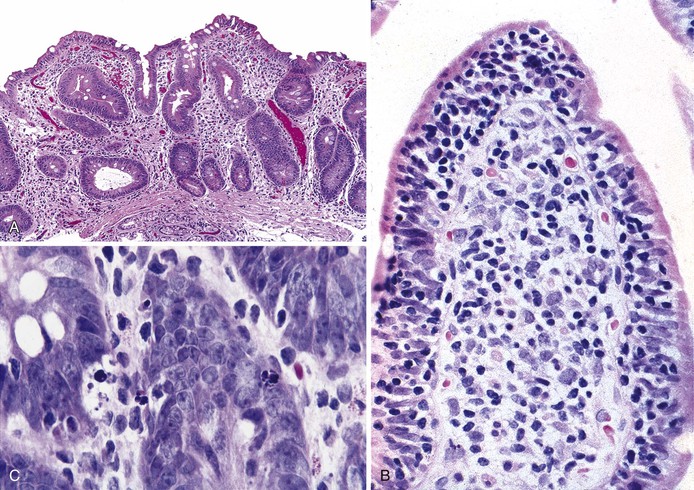
Granulomatous enteropathy has also been reported in patients with CVID and may be associated with protracted diarrhea unresponsive to antibiotic therapy. Poorly formed non-necrotizing granulomas are found in the lamina propria in multiple sites in the GI tract, including the stomach, small intestine, and colon24,29; the diarrhea usually resolves with intravenous immunoglobulin therapy.
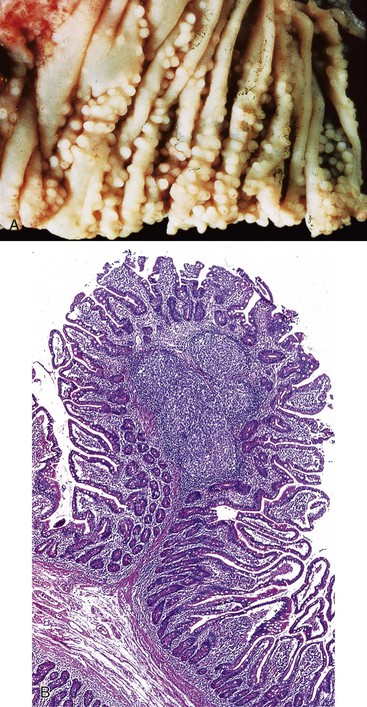
NLH in the GI tract is characterized by multiple discrete hyperplastic lymphoid nodules in the lamina propria and submucosa of the small intestine (Fig. 5.5), large intestine, or both, and is probably a result of chronic antigenic stimulation. The germinal centers of the follicles are composed of proliferating B cells with scattered tingible body macrophages; the mantle zones contain mature and immature B cells, and the extramantle zones contain a mixture of cell types including B cells, T cells, and macrophages. NLH is found in as many as 60% of patients with CVID but may be seen in the setting of giardiasis without antibody deficiency. In contrast to NLH in CVID patients, plasma cells are present in the extramantle zones in nonimmunodeficient patients. NLH is not considered a malignant disorder. However, malignant lymphomas of the GI tract in patients with immunodeficiencies often arise in a background of NLH, and clonal immunoglobulin gene rearrangement has been demonstrated in NLH in the GI tract of a child with CVID.30 Consistent with these observations, the most common malignancy in CVID is NHL, which affects approximately 8% of patients18; these lymphomas often originate in extranodal sites, with small bowel the most common GI site. Most of these lymphomas are of B-cell origin; they include diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma.18
Chronic inflammatory processes involving small or large bowel that are clinically similar to IBD develop in some patients with CVID. In some patients, the small bowel is the primary site of involvement and the lesions resemble Crohn disease, with transmural inflammation and small bowel obstruction. Granulomas usually are not present in these Crohn disease-like disorders.31
Large Bowel
The colitis occurring in CVID is variable in morphology. In some patients, the inflammatory process is limited to the colon and clinically mimics ulcerative colitis. Mucosal architectural distortion with crypt destruction is present, although crypt distortion is less pronounced than in most cases of ulcerative colitis, with less crypt branching (Fig. 5.6, A). Neutrophils are present in the lamina propria and crypt epithelium (see Fig. 5.6, B). In contrast to ulcerative colitis, plasma cells are not present in the lamina propria in CVID-associated colitis, and an increase in CD8+ T cells (compared with normal controls and those with IBD) has been reported in the colon of CVID patients.32 In some cases, the crypt destruction and mucosal distortion is accompanied by increased apoptosis, and the histology is similar to that of colonic graft-versus-host disease (GVHD).23,24 Milder cases of colitis in CVID may resemble lymphocytic colitis, characterized by increased intraepithelial lymphocytes and minimal mucosal distortion,33 and an atypical form of collagenous colitis with pseudomembranes has been reported in one patient.34 The etiology and pathogenesis of colitis in these patients remain largely unknown. The association of chronic GI inflammatory disorders and autoimmune disorders in these patients, and the resemblance of the lesions to other disorders of immune dysregulation, imply that the colitis of CVID may be autoimmune in origin.
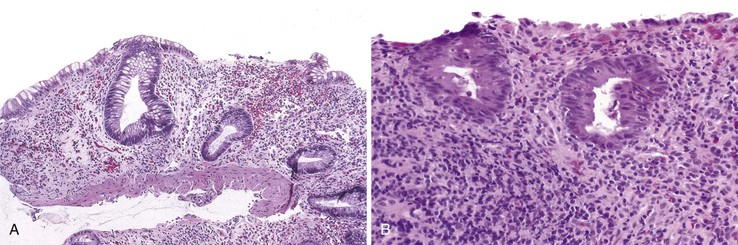
Adenocarcinoma of the colon is reported in young patients with CVID. Small cell neuroendocrine carcinoma of the cecum was reported in a 16-year-old boy, who died of liver metastases 5 months after diagnosis.35 In another case, 9 adenocarcinomas and 20 adenomas were present synchronously in the colon of a 22-year-old man with CVID.36
Combined Cellular/Humoral Immunodeficiencies
Severe Combined Immunodeficiency
Severe combined immunodeficiency (SCID) is a heterogeneous group of congenital disorders characterized by defects in both B- and T-cell function. Children with SCID typically are seen in the first year of life with severe recurrent bacterial or viral infections. A number of molecular defects may result in SCID. Most are autosomal recessive, including adenosine deaminase deficiency, which accounts for 50% of autosomal recessive SCID; purine nucleoside deficiency; T-cell receptor deficiencies; Zap70 deficiency; JAK3 deficiency; and IL-7 receptor deficiency.75 However, X-linked SCID, resulting from a defect in the common γ chain, is the single most common type of SCID in the United States.76 It has a characteristic phenotype of absence of T cells and natural killer (NK) cells but normal B-cell numbers, although the B cells are dysfunctional.
GI disorders in SCID are caused by a variety of infectious pathogens. Oral, esophageal, and perianal candidiasis is common, and profound diarrhea may develop in children with SCID early in life. In general, GI biopsy specimens from these patients show a hypocellular lamina propria without plasma cells or lymphocytes. Because these patients are susceptible to viral infections, examination of stool for viral particles may be indicated. In particular, rotavirus, normally a self-limited infection, may cause chronic diarrhea in these children. Although villous blunting has been described in acute rotavirus infection in normal children51 and in animal models,77 the intestinal pathology of chronic rotavirus infection has not been described in SCID patients. Cytopathic viral infections that may be identified on GI biopsy include CMV and adenovirus (Fig. 5.7). Salmonella may also cause chronic GI infection in SCID patients. Patients receiving nonirradiated blood products or allogeneic bone marrow transplants are susceptible to GVHD. Furthermore, a GVHD-like process affecting the colon and small intestine has been described in patients with SCID who had not undergone bone marrow transplantation.78,79 Children with SCID may be a greater risk for reflux esophagitis than the normal population.80
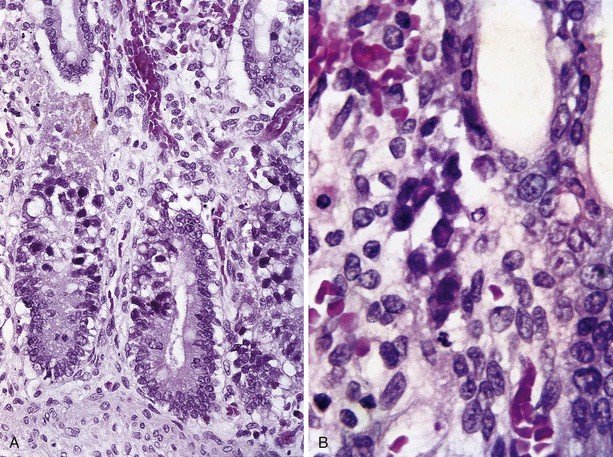
Omenn Syndrome
Omenn syndrome is an autosomal recessive type of SCID with clinical and pathologic features of GVHD. The immunologic hallmark of the disease is expansion of an oligoclonal population of T cells and a near absence of B cells.76 Infants with Omenn syndrome are seen with diffuse erythroderma, hepatosplenomegaly, lymphadenopathy, and failure to thrive81; chronic diarrhea and alopecia are common. Hypereosinophilia and hypogammaglobulinemia are characteristic. Paradoxically, serum IgE levels are increased, although B lymphocytes are not detectable in the circulation, lymph nodes, or skin. Activated circulating T cells are normal to increased in number but constitute an oligoclonal population. The underlying basis for these findings in Omenn syndrome is impairment but not complete loss of the V(D)J recombination process as a result of mutations in RAG1 or RAG2, the recombination activating genes.76 Mutations in these genes were first identified in a subset of SCID patients with T−B− SCID. The occurrence of this type of SCID and Omenn syndrome in the same kindred furnished the clue that Omenn syndrome was caused by mutations in the same genes. Differences between T−B− SCID and Omenn syndrome can be explained by the presence of two entirely defective alleles in SCID and the presence of one marginally functional allele that is capable of establishing the oligoclonal T-cell population in patients with Omenn syndrome. Infants with SCID with maternal T-cell engraftment may exhibit GVHD symptoms indistinguishable on clinical grounds from Omenn syndrome,82 and a diagnosis of Omenn syndrome depends on excluding this possibility by appropriate human leukocyte antigen typing or molecular analysis. Published accounts of the histopathologic changes in Omenn syndrome are scant, but skin changes resemble those of GVHD,83,84 and numerous apoptotic crypt cells are found in colonic biopsies in a pattern similar to that of GVHD (unpublished observations). Crypt injury and an increase in lamina propria eosinophils may also be seen (Fig. 5.8).
Other Primary Immunodeficiencies
DiGeorge Syndrome
DiGeorge syndrome is caused by a microdeletion in chromosome 22q11.2, which leads to a congenital malformation of the third and fourth pharyngeal pouch, resulting in thymic and parathyroid hypoplasia. This disease is the most common microdeletion syndrome in humans,85 and is estimated to affect 1 of every 4000 live births.86 T cells are markedly reduced in number, but B cells are normal in number and functionality. Midline anomalies affecting the GI tract (e.g., esophageal atresia, imperforate anus) are seen in some cases in association with DiGeorge syndrome, and water diarrhea and malabsorption have been described but not well characterized.87 Oral candidiasis is common. Dysphagia and feeding difficulties have been reported in infants with 22q11.2 deletion.88
Chronic Mucocutaneous Candidiasis
Chronic mucocutaneous candidiasis is a heterogeneous group of disorders characterized by persistent Candida infection of the skin, nails, and mucous membranes. Autoimmune disorders and a polyglandular endocrinopathy syndrome including pernicious anemia are common, occurring in more than 50% of patients. There is a high frequency of association with thymoma and systemic lupus erythematosus.89 Mutations in STAT1 have been identified in the autosomal dominant form of the disease,90,91 and the autosomal recessive form has been linked to deficiency in interleukin-17 receptor A.92 Immune defects include disorders of T-cell immunity with variable B-cell involvement. The most common GI manifestation is esophageal candidiasis. Although superficial infection with Candida is a defining characteristic, infections with other fungi (e.g., H. capsulatum) and bacteria are common.93
Wiscott-Aldrich Syndrome
Wiskott-Aldrich syndrome, characterized by early onset of profound thrombocytopenia with small platelets, eczema, and recurrent infections, is inherited as an X-linked recessive disease. Platelets and T cells are most severely affected. The genetic basis of Wiskott-Aldrich syndrome, described in 1994, is a mutation of the WASP gene, which encodes an intracellular protein expressed exclusively in hematopoietic cells.94 This protein is involved in transduction of signals from cell surface receptors to the actin cytoskeleton and is important in cytoskeletal architecture and cell trafficking and motility. Diarrhea is reported in patients with this disorder but has been poorly characterized.87 Bloody diarrhea in these patients is often attributed to thrombocytopenia, and biopsies may not be performed because of the risk of hemorrhage. A Crohn’s disease-like inflammatory process with cobblestone appearance and inflammatory pseudopolyps involving the descending and transverse colon has been reported in Wiskott-Aldrich syndrome.95 Massive hemorrhage from aneurysms involving the liver, small bowel mesentery, and kidney has been reported.96
Chronic Granulomatous Disease
In chronic granulomatous disease (CGD), phagocytic cells are unable to reduce molecular oxygen to create the superoxide anion and its metabolites necessary for eradication of certain catalase-positive intracellular microbes. CGD is genetically heterogeneous, resulting from a mutation in any of four components of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. The most common form of the disease, accounting for 70% of cases, is X-linked recessive; four other forms are autosomal recessive.97 As a result of this defect, patients with CGD suffer from recurrent bacterial and fungal infections; abscesses in a variety of sites and pneumonia are common. They are also prone to develop inflammatory and rheumatic diseases, such as an IBD-like condition and a lupus-like syndrome. GI manifestations are relatively rare in CGD, but reported cases can be broadly grouped into obstructive and inflammatory categories.
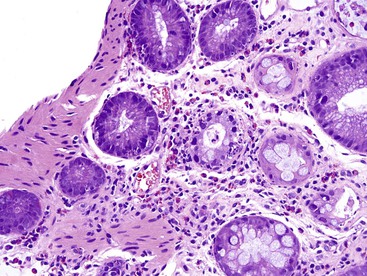
Obstruction can occur in patients with CGD at a number of levels of the GI tract, from esophagus to small bowel. Gastric outlet obstruction is more common in the X-linked form of the disease.97 In some cases, the obstruction is caused by infiltration of the viscus wall by pigment-laden macrophages (the histologic hallmark of CGD) or by granulomatous inflammation. In other cases, the obstruction is reportedly secondary to a functional disturbance in GI motility, although infiltration of the deep layers of the organ by macrophages often cannot be excluded. Esophageal obstruction occurs in 1% of CGD patients97; biopsies of the esophageal mucosa usually show nonspecific findings or reflux esophagitis98 but may demonstrate pigmented macrophages. Involvement of the gastric antrum and pylorus is somewhat more common, occurring in 16% of patients, and gastric outlet obstruction may be the first manifestation of CGD. Granulomas, giant cells, and macrophages laden with brown-yellow fine pigment are commonly present in gastric biopsy specimens,99 but in some cases only nonspecific inflammation is seen.100 Small bowel obstruction is relatively rare in CGD but is occasionally reported in the context of an inflammatory process.101 In a review of small bowel and rectal biopsies from nine patients with CGD, pigment-laden macrophages were found in the lamina propria at both sites. In the small bowel, the macrophages were located deep in the mucosa adjacent to crypts, but when numerous, they also extended into the villus core (Fig. 5.9). In rectal biopsies, the number of pigmented histiocytes was quite variable, ranging from rare scatted cells to large numbers of histiocytes accumulating between the base of the crypts and the muscularis mucosae. Granulomas with giant cells were also present in rectal biopsies from some patients. In one of eight cases, distortion of crypt architecture without crypt abscesses was seen.102
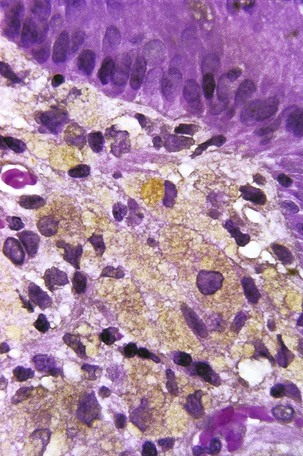
Chronic inflammatory processes that are indistinguishable from IBD affecting the small and large bowel may occur in CGD patients103; as with obstructive lesions of the GI tract, these lesions are more common in the X-linked form of the disease.97 Polymorphisms in genes unrelated to NADPH oxidase may modify the clinical phenotype in CGD; certain polymorphisms in the genes for myeloperoxidase and Fcγ receptors are strongly associated with GI complications.104 Involvement of the small bowel in CGD may produce fistulas, longitudinal ulcers, stenosis, and non-necrotizing granulomatous inflammation that can be mistaken for Crohn disease105; discontinuous inflammation and perianal disease may also contribute to the difficulty in distinguishing the two entities.106 The granulomas seen in intestinal lesions in CGD are often more florid than is usually seen in Crohn disease, but granulomas are not present in all cases. In a study of colitis in one CGD patient, the presence of an acute and chronic inflammatory infiltrate confined to the colonic mucosa, crypt abscesses, and lack of granulomas were more suggestive of ulcerative colitis than Crohn disease. Mucosal architectural distortion and ulceration were not as prominent as is usually the case in ulcerative colitis, however, and pigmented macrophages were present in the lamina propria.103
Miscellaneous Immune Deficiency Syndromes
Other rare disorders of immunity occasionally associated with GI manifestations include leukocyte adhesion deficiency, in which delayed wound healing and susceptibility to bacterial and fungal infection lead to necrotizing enterocolitis.107 A chronic inflammatory process with multiple aphthous ulcers involving the gastric antrum, terminal ileum, cecum, and right colon, which resolved with bone marrow transplantation, has also been reported in leukocyte adhesion deficiency,108 and a case of rectal ulcer resembling Crohn disease has been reported in a pediatric patient.109
Immune dysregulation, polyendocrinopathy, and enteropathy, X-linked (IPEX) syndrome, caused by mutations in FOXP3, usually manifests in affected males with severe watery diarrhea early in life. The entire GI tract may be involved and primarily shows GVHD-like histologic changes with prominent crypt apoptosis (Fig. 5.10). Small bowel disease with villous atrophy mimicking celiac disease has also been reported in IPEX.110,111
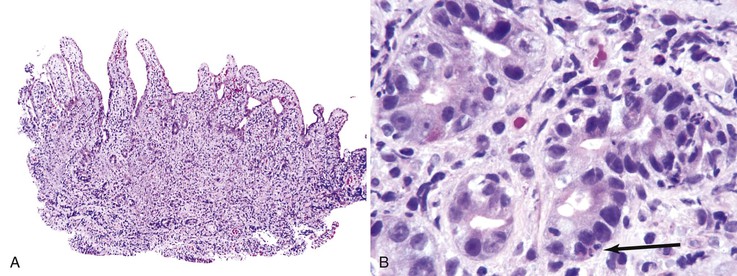
Graft-versus-Host Disease
GVHD, most commonly involving GI tract, skin, or liver, develops in as many as 50% of allogeneic bone marrow transplant recipients.112 Indeed, the most common cause of persistent nausea and anorexia in patients beyond day 20 after transplantation is acute GVHD.113 Changes identical to those seen in GVHD after allogeneic transplantation may be seen in the GI tract and liver after autologous stem cell transplantation and are considered to be a form of GVHD resulting from a lack of regulation of immune mechanisms by the reconstituting immune system. This syndrome of acute GVHD–like changes in the GI tract after autologous transplantation is rare, occurring in gastric biopsies in only 4% of patients with upper GI tract symptoms114 and approximately 5% of all autologous transplantation patients.115 Rarely, GVHD occurs after solid organ transplantation or blood transfusion.116 Symptoms indicative of GI tract involvement by GVHD include profuse diarrhea, crampy abdominal pain, GI hemorrhage, anorexia, nausea, and vomiting. Severe GI bleeding and peritonitis are also reported.117
Historically, acute GVHD has been defined as occurring less than 100 days after myeloablative stem cell transplantation. However, current clinical practices in stem cell transplantation have rendered this classification inadequate, because the use of reduced-intensity regimens, for instance, has led to late occurrence of otherwise typical acute GVHD. Accordingly, recommendations for classification of GVHD as acute or chronic have been developed by an expert panel convened by the National Institutes of Health118 (Table 5.5). Characteristic skin, GI tract, or liver abnormalities are now classified as acute GVHD regardless of time after transplantation. Diagnosis of chronic GVHD requires the presence of at least one diagnostic clinical sign or distinctive clinical manifestation confirmed by biopsy or other relevant tests in the same or another organ. In the GI tract, the presence of an esophageal web, stricture, or concentric rings documented by endoscopy or barium contrast radiography is sufficient to establish the diagnosis of chronic GVHD. Distinctive criteria that are, by themselves, insufficient to establish a diagnosis of chronic GI GVHD include pancreatic exocrine insufficiency, anorexia, nausea, vomiting, diarrhea, weight loss, and wasting syndrome.118
Table 5.5
Classification of Graft-versus-Host Disease
| Classification | Time of Onset | Features |
| “Classic” acute GVHD | ≤100 days after HSCT | Maculopapular rash, nausea, vomiting, anorexia, profuse diarrhea, ileus, or cholestatic hepatitis |
| Persistent, recurrent, or late acute GVHD | >100 days after HSCT | Same as “classic” acute GVHD, without diagnostic or distinctive manifestations of chronic GVHD; often seen after withdrawal of immunosuppression |
| “Classic” chronic GVHD | No time limit | At least one diagnostic or distinctive manifestation of chronic GVHD without features characteristic of acute GVHD |
| Overlap syndrome of acute and chronic GVHD | No time limit | Features of acute and chronic GVHD appear together |
GVHD, Graft-versus-host disease; HSCT, hematopoietic stem cell transplantation.
Acute GVHD
On endoscopic examination, the appearance of the GI mucosa in acute GVHD is variable, ranging from only mucosal edema and erythema to more severe changes such as ulcers and mucosal sloughing.119 The major histologic features of GVHD in the GI tract are epithelial cell apoptosis and a relatively sparse mononuclear inflammatory cell infiltrate (Fig. 5.11, A). Apoptotic epithelial cells are found primarily in the regenerative compartment of the mucosa, such as the crypt in the colon and small intestine and the neck area of gastric glands. In the colon, the apoptotic cells are particularly conspicuous and are termed “exploding crypt cells”; these cells contain intracytoplasmic vacuoles filled with karyorrhectic nuclear debris (see Fig. 5.11B). Apoptotic cells are smaller and less conspicuous in the gastric mucosa (Fig. 5.12). In more severe cases of acute GVHD, crypt abscesses may be seen, and destruction and loss of crypts is seen. In the most severe cases, mucosal sloughing and extensive ulceration occur. In the stomach, granular eosinophilic necrotic cellular debris without neutrophils may be present in the lumina of injured gastric glands.120 Villous blunting is commonly seen in small bowel GVHD. A grading system for acute GVHD affecting the colon has been proposed121 (Fig. 5.13 and Table 5.6); however, correlations with clinical symptoms and patient outcome are weak.
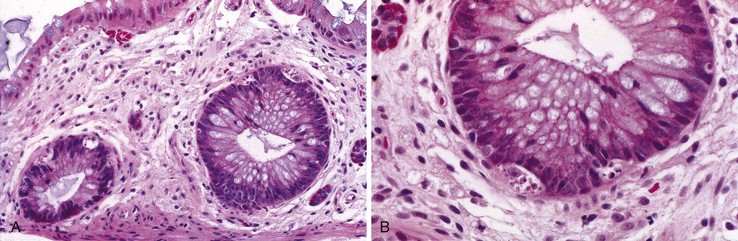
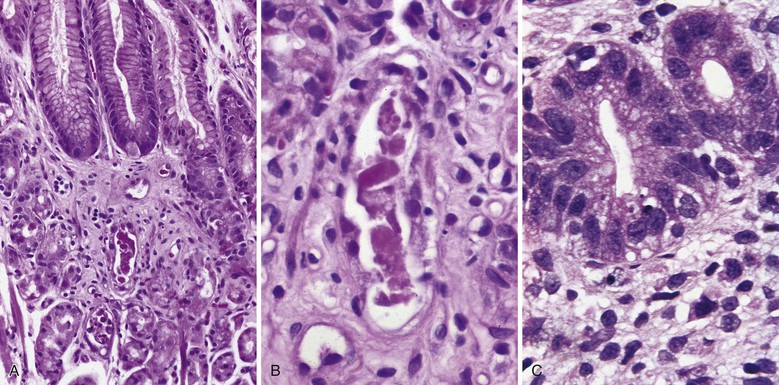
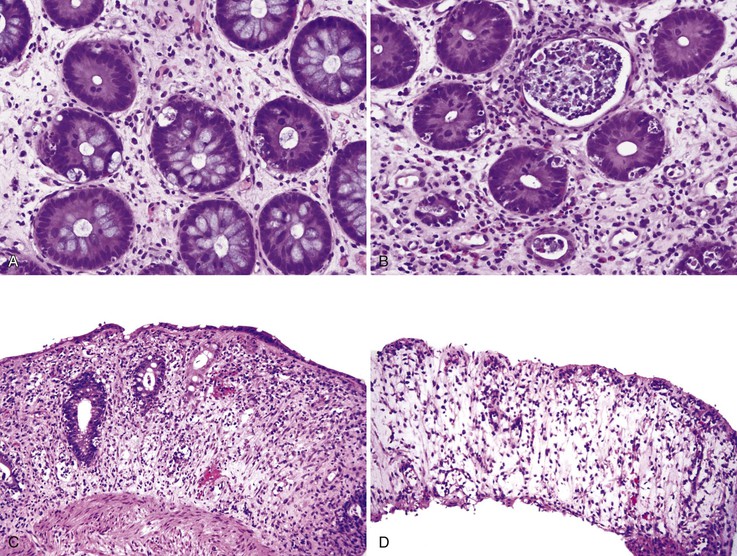
Table 5.6
Grading of Acute Graft-versus-Host Disease of the Colon
| Grade | Histologic Features |
| I | Rare apoptotic cells, without crypt loss |
| II | Loss of individual crypts |
| III | Loss of two or more contiguous crypts |
| IV | No identifiable crypts (mucosal ulceration) |
Data from Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122:1082-1086.
The changes that occur in the GI tract in acute GVHD are not entirely specific. The differential diagnosis includes drug-related injury, infection, recurrent hematologic malignancy, and other causes of immune activation (Table 5.7). In particular, mycophenolate mofetil (MMF) treatment may cause active colitis with ulcers, marked crypt cell apoptosis, and a mixed lamina propria inflammatory infiltrate that can mimic GVHD (Fig. 5.14).122 Other reported patterns of injury include IBD-like changes and, more rarely, ischemic colitis.123 Normal biopsy findings in sites distant from these GVHD-like lesions should raise the question of MMF-associated colitis rather than GVHD124; improvement after discontinuation of MMF is also good evidence that the injury was drug related. Use of proton pump inhibitor (PPI) therapy has been associated with an increase in apoptotic epithelial cells in the gastric mucosa (Fig. 5.15), mimicking the histologic changes seen in GVHD.125
Table 5.7
Differential Diagnosis of Graft-versus-Host Disease
| Diagnosis (Ref. No.) | Features | Comments |
| Conditioning regimen158 | Epithelial cell apoptosis; increased mitotic activity; crypt cell regeneration | Found in early period after transplantation (up to day 20); severe injury at day 20 likely represents GVHD, because effects of chemotherapy and radiation are typically improving by then |
| Cytomegalovirus159 | Increased epithelial cell apoptosis; nuclear inclusions may be sparse | May occur concomitantly with gastrointestinal GVHD |
| Cryptosporidium160 | Increased epithelial cell apoptosis | Rarely encountered in biopsies in HSCT population |
| Mycophenolate mofetil161 | Colitis with increased crypt cell apoptosis, focal ulcers, mixed lamina propria inflammatory infiltrate | Apoptotic bodies in sites other than colon are suggestive of GVHD |
| Proton pump inhibitors162 | Increased epithelial cell apoptosis in gastric antral mucosa, without inflammation | Biopsy of oxyntic mucosa may be more informative than antral mucosa in patients receiving PPI therapy |
| Cord colitis syndrome131 | Chronic active colitis with crypt distortion; granulomas in some cases | Described in patients undergoing umbilical cord blood stem cell transplantation; response to antibiotics suggests infectious etiology |
GVHD, Graft-versus-host disease; HSCT, hematopoietic stem cell transplantation; PPI, proton pump inhibitor.
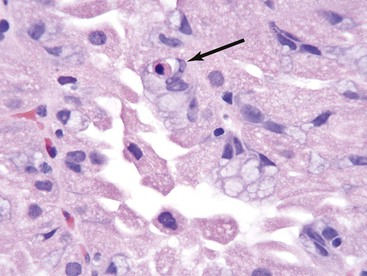
Changes similar to GVHD are reported in colonic biopsies from patients with severe T-cell deficiencies,79 malignant thymoma,126 and primary immune disorders such as CVID.23,24 In the patient who has undergone bone marrow transplantation, the effects of cytoreductive therapy (Fig. 5.16) may resemble changes of GI GVHD in the early posttransplantation period; therefore, a diagnosis of GVHD must be made with caution before day 21 after transplantation. Recurrence of hematologic malignancies, particularly acute lymphoblastic leukemia, may also mimic acute GVHD.127 CMV infection may produce mucosal damage characterized by apoptotic epithelial cells, mimicking GVHD; differentiation from GVHD relies on the demonstration of viral inclusions.120GVHD and CMV infection may occur simultaneously, making it difficult to separate the effects of each process on the GI tract. Infection with Clostridium difficile has also been reported to be associated with GI GVHD and a high nonrelapse mortality rate in this group; it is postulated that C. difficile toxin may predispose to increased severity of GVHD.128
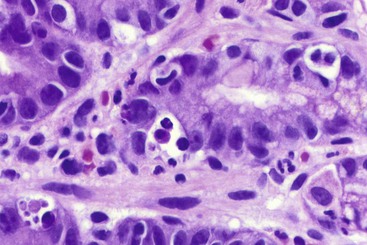
Chronic GVHD
Chronic GVHD is clinically similar in many ways to some of the collagen vascular diseases and has been compared to autoimmune disorders such as scleroderma, Sjögren syndrome, and primary biliary cirrhosis. Chronic GVHD involves multiple organs, such as salivary gland, mouth, eye, and upper respiratory tract, that are not involved by acute GVHD.120
In chronic GVHD, skin changes of dermal fibrosis may resemble scleroderma, and involvement of the oral squamous mucosa leads to painful ulcers and submucosal fibrosis. Involvement of minor salivary glands results in an oral sicca syndrome. In advanced cases, ulcers and submucosal fibrosis occur in the esophagus (Fig. 5.17), the most commonly affected site in the GI tract.124 Small bowel involvement is less common; when present, it is associated with diarrhea. Focal fibrosis of the lamina propria and segmental submucosal fibrosis, with minimal mucosal changes, has been reported.129 In colonic biopsies, chronic colitis similar to that seen in ulcerative colitis has also been reported in allogeneic bone marrow transplant recipients.130 These changes consist of mild to moderate crypt distortion and crypt atrophy, and it is unclear whether the mucosal architectural distortion is caused by chronic GVHD or by other factors. In one study, similar findings of mild chronic active colitis, with granulomas in some cases (Fig. 5.18), were reported in 10% of patients undergoing umbilical cord blood stem cell transplantation, occurring 88 to 314 days after transplantation. Although patients were culture negative, this “cord colitis syndrome” responded to antibiotic therapy, suggesting an infectious etiology.131
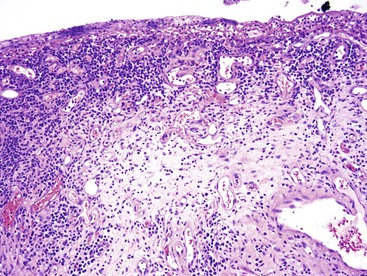
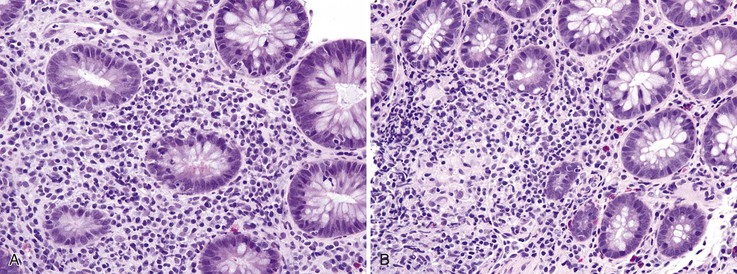
Neutropenic Enterocolitis
Neutropenic enterocolitis (NEC) is a necrotizing inflammatory process that predominantly affects the cecum, terminal ileum, and ascending colon and occurs most commonly in the setting of neutropenia. Hemorrhagic necrosis of the cecum in this setting has also been termed typhlitis. Historically, most patients with NEC have had acute leukemia, although NEC is has also been reported in patients undergoing stem cell or autologous bone marrow transplantation for solid malignancies.132 NEC also occurs in patients with aplastic anemia, renal transplant recipients, and those with other hematologic malignancies. Incidence varies from less than 1% to almost 7% in children with cancer,133,134 with a pooled incidence of about 5% in adult patients.132 Most patients have received chemotherapy in the previous 30 days,135 and absolute neutrophil counts of less than 1500 cells/mm3 are associated with the disease. Patients may be seen with clinical features suggestive of acute appendicitis, such as fever and right lower quadrant pain.136 One third are seen with GI hemorrhage,135 and rarely a palpable right lower quadrant mass is present. The combination of abdominal pain, diarrhea, and fever is the most common presentation; diagnosis is often established by radiographic studies showing a fluid-filled, dilated cecum and inflammatory changes in the abdomen.136
On gross examination, the cecum and other affected portions of the GI tract are dilated, edematous, and congested or hemorrhagic. Pneumatosis intestinalis (Fig. 5.19, A) may be seen but is relatively rare. The mucosa is hemorrhagic, often necrotic, and covered with granular eosinophilic debris; no significant inflammatory reaction is present (see Fig. 5.19, B). The pathogenesis of this disorder is initiated with mucosal injury, primarily related to recent administration of chemotherapeutic agents and augmented by neutropenia. Bacterial invasion of the injured mucosa then occurs, with Clostridium spp. implicated as major offenders; fungi such as Candida spp. have also been implicated as causative or contributing agents. Toxins produced by organisms invading the gut wall lead to edema and necrosis. Distention of the bowel wall leads to decreased blood flow, adding an element of ischemic injury. Most patients become septicemic. If NEC is left untreated, the prognosis is grave, but patients may survive with optimal medical and surgical management; recovery depends on the restoration of adequate neutrophil counts.
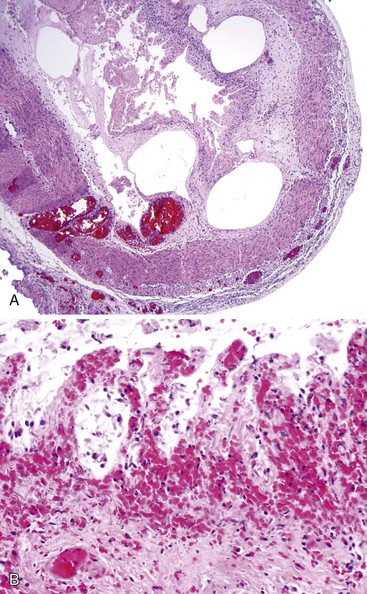
The Gastrointestinal Tract in HIV Infection
GI illnesses are common in human immunodeficiency virus (HIV)-infected patients, with diarrhea, nausea, vomiting, anorexia, and abdominal pain as presenting symptoms. Before the use of new, highly active antiretroviral therapy (HAART), opportunistic infections with such pathogens as Isospora, Mycobacterium avium-intracellulare complex (MAC), microsporidia, Cryptosporidium, and CMV were frequent causes of diarrhea, malabsorption, and wasting (Table 5.8). Although the prevalence of intestinal pathogens has dramatically decreased since then, from a reported 85% (in men with AIDS and diarrhea) to 12% (found almost exclusively in homosexual men), current studies continue to show a high prevalence of GI dysfunction in HIV-infected patients. Chronic diarrhea is reported in approximately 25% of HIV patients and was not associated with degree of immune suppression in one cohort.137
Table 5.8
HIV-Associated Gastrointestinal Diseases
| Infection | Neoplasia |
| Giardia intestinalis | Kaposi sarcoma |
| Cryptosporidium parvum | Burkitt lymphoma |
| Isospora belli | Diffuse large B-cell lymphoma |
| Microsporidia | Plasmablastic lymphoma |
| Cytomegalovirus | Anal squamous cell carcinoma |
| Candida albicans | Hodgkin lymphoma |
| Listeria monocytogenes | |
| Strongyloides stercoralis | |
| Human herpesvirus 8 | |
| Epstein-Barr virus |
HIV, Human immunodeficiency virus.
Infections
CMV remains the most common pathogen in HIV-infected patients, and CMV infection can involve any segment of the GI tract, most commonly the esophagus and colon. CMV esophagitis often manifests with distal esophageal ulceration. CMV primarily infects endothelial cells, followed by macrophages; biopsy specimens obtained from the ulcer bed are, therefore, more likely to demonstrate characteristic CMV cytopathic effects. Endoscopic features in CMV gastritis include patchy erythema, erosions, or multiple small ulcers. Unlike other sites in the GI tract, in the stomach the epithelial cells are preferentially infected. CMV colitis can manifest as colitis with or without ulcers. As in the esophagus, characteristic CMV inclusions can be seen in stromal and endothelial cells.
Although GI herpes simplex virus (HSV) infection occurs less frequently than CMV infection, approximately 5% of esophageal ulcerative lesions in HIV-infected patients are caused by HSV infection.138 HSV esophagitis can manifest as either vesicular or ulcerative lesions. Unlike CMV esophagitis, HSV characteristically infects squamous epithelium.
One of the important differential diagnoses for CMV and HSV esophagitis in HIV-infected patients is idiopathic esophageal ulcer. HIV patients with idiopathic esophageal ulcer always are seen with severe odynophagia and weight loss, which often respond to therapy with corticosteroid or thalidomide or both.139 Endoscopically, HIV-associated idiopathic esophageal ulceration appears as large, irregular ulcers in the middle or distal esophagus. To diagnose idiopathic esophageal ulcer, exclusion of infectious etiologies is required.
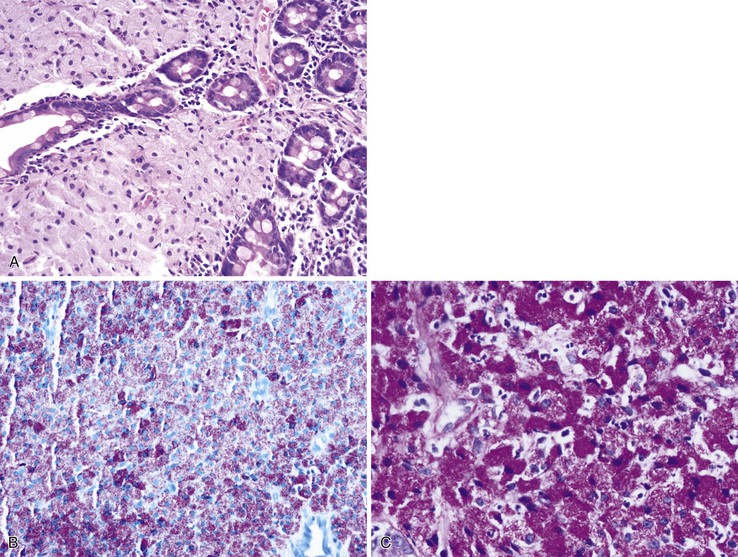
Bacterial pathogens involving the GI tract in the HIV-positive population include C. difficile, mycobacteria, and spirochetes. C. difficile–associated colitis is the most common cause of bacterial diarrhea in HIV-infected patients after HAART.140 GI MAC infection can be seen in severely immunocompromised patients, involving the stomach, the small intestine (Fig. 5.20, A), and, rarely, the colon. Endoscopic presentations include normal-appearing mucosa, friability, multiple raised nodules, or ulceration. Microscopically, the lamina propria of the intestinal mucosa may be stuffed with distended macrophages containing the organisms, which can be confirmed by a special stain for acid-fast bacilli (see Fig. 5.20, B). Periodic acid–Schiff (PAS) stain characteristically reveals abundant delicate bacilli in macrophages (see Fig. 5.20, C).
Human intestinal spirochetosis is a common cause of diarrhea in HIV patients and preferentially infects the surface epithelium of the distal colon and rectum. In addition to HIV-infected individuals, intestinal spirochetosis is also diagnosed in general population without HIV infection.141 The prevalence of intestinal spirochetosis varies considerably from region to region. For example, in the Western countries, the prevalence of intestinal spirochetosis in rectal biopsies is 2% to 7%, whereas in developing countries it is much higher, ranging from 11.4% to 32.6%. Among homosexual and HIV-infected men, the rate is even higher, up to 54%. Intestinal spirochetosis is much more common in men than in women. Two strains of Bachyspira have been identified as being responsible for intestinal spirochetosis—Brachyspira aalborgi and Brachyspira pilosicoli—with B, aalborgi being the major causative agent in Western countries.
Because many cases of intestinal spirochetosis are asymptomatic, there is much debate as to whether intestinal spirochetosis is a pathogen or a commensal. However, accumulating evidence supports the notion that intestinal spirochetosis can be the cause of GI symptoms in a subset of patients, especially in homosexual men, HIV patients, and children.142–146 Association of intestinal spirochetosis with invasive colitis and hepatitis, rectal discharge, rectal bleeding, and even spirochetemia has been reported. Improvement of symptoms after eradication of the spirochetes is seen in individuals with symptomatic intestinal spirochetosis.
For a long time, intestinal spirochetes have been described as noninvasive.147 However, some studies have demonstrated invasion of spirochetes beyond the surface epithelium.148 The presence of spirochetes in epithelial cells, lamina propria, macrophages, and even Schwann cells has been reported. In addition, invasive intestinal spirochetosis is associated with immediate-type immune reaction of the host, featured by a marked increase of IgE-producing plasma cells in the lamina propria and intraepithelial mast cells. Stunting, destruction, and loss of microvilli are seen in invasive cases and can result in reduction of the resorptive surface, leading to diarrhea. Invasive intestinal spirochetosis has been correlated to GI symptoms in the general population, whereas homosexual and HIV-infected men are more likely to be symptomatic regardless of invasion, for unclear reasons.
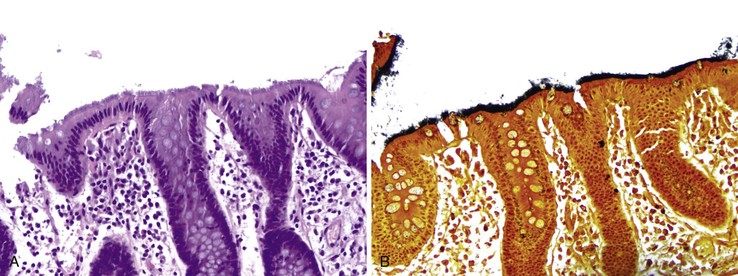
The most common symptoms associated with intestinal spirochetosis include diarrhea, abdominal pain, altered bowel movement habits, and rectal bleeding.142–145 Intestinal spirochetosis can affect the rectum, the colon, or both, with the rectum the most severely involved area. Colonization of appendiceal epithelium by intestinal spirochetosis occurs frequently.141 In one study, spirochetes were found in 7.8% of appendectomy specimens. The endoscopic appearance of the colon is largely normal; however, some subtle changes (e.g., erythema) may be present. Biopsy reveals long, undulating bacteria vertically attached to the brush border, forming a blue bushy layer on hemotoxylin and eosin (H&E) staining (Fig. 5.21, A). The organisms stain dark black with the Warthin-Starry stain (see Fig. 5.21, B). The colonic mucosa is always unremarkable, with no increase in chronic inflammation and no active colitis. However, in a small subset of patients, mild inflammatory changes such as increased intraepithelial lymphoctyes can be seen. A severe acute inflammatory reaction with crypt abscesses and ulcers was reported in two patients with advanced HIV infection.148
In general, no treatment is needed for asymptomatic cases. If intestinal spirochetosis is identified as the sole intestinal pathology in a symptomatic patient, it should be treated with antibiotics to eradicate the spirochetes. Metronidazole is the drug of choice for the treatment of intestinal spirochetosis. Most cases treated with metronidazole show symptom improvement with eradication of the bacteria in follow-up biopsies.142,144
Candida is the most common HIV-associated fungal pathogen, with the esophagus the site most often involved. Esophageal candidiasis is characterized by white or yellow plaques with surrounding mucosal erythema endoscopically. Biopsy of the plaques reveals necroinflammatory exudates, budding yeast forms, and pseudohyphae. In HIV-infected patients, histoplasmosis is most likely to involve the ileocecal region, occasionally leading to GI bleeding, obstruction, perforation, and stricture. Intestinal mucosal biopsies reveal marked infiltration of the lamina propria by macrophages containing budding yeasts.
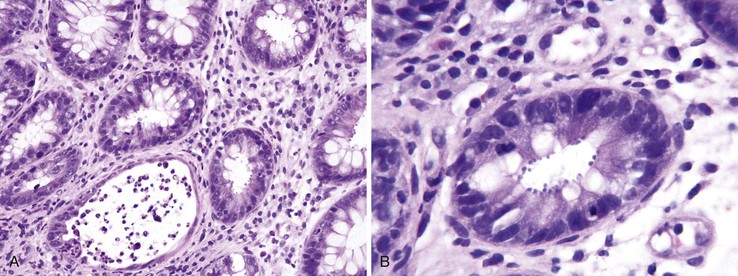
GI parasitic infections are relatively uncommon in HIV-infected patients after HAART. They mainly occur in the small intestine. Infecting agents include Cryptosporidium, Microsporidium, Isospora, and Giardia. In cryptosporidiosis, the parasite inhabits the microvillous brush border of the intestinal epithelium, causing enfacement of the brush border. The intestinal epithelium may be atrophic and in disarray. A relatively mild active colitis may be seen in the colon, with cryptitis and a few crypt abscesses (Fig. 5.22). Microsporidium is difficult to identify on H&E-stained sections; special stains such as Giemsa may be beneficial, and electron microscopy is the gold standard for confirming the presence of the organism in the surface epithelial cells.
HIV Enteropathy
GI dysfunction, as manifested by d-xylose malabsorption, is common, even in early HIV disease. It may be a manifestation of HIV enteropathy, defined as a reduction in small bowel villous surface area associated with chronic diarrhea in the absence of enteric pathogens. The pathogenesis of HIV enteropathy is not well understood. The chronic diarrhea may be caused by an unidentified pathogen, but there is evidence that diarrhea is directly related to local infection by the virus. Studies have shown that HIV-infected patients show improvement in clinical symptoms after initiation of HAART. Studies of intestinal permeability, epithelial cell barrier function, and cytoskeletal integrity have demonstrated changes in HIV-infected subjects after administration of antiviral agents alone.149 These studies indicate that medications should also be considered as a cause of diarrhea in these patients, and they may account for up to 45% of noninfectious cases150; commonly implicated medications are nelfinavir, ritonavir, saquinavir, indinavir, and didanosine.
In small bowel biopsies, HIV enteropathy is characterized by relatively mild villous blunting without well-developed crypt hyperplasia.151 The degree of villous atrophy is typically less than that seen in celiac disease. The histology of the colon in HIV enteropathy is less well understood. Increased epithelial cell apoptosis has been reported in association with HIV infection152; however, it is unclear whether this is related to the presence of the virus, other immune mediators, or antiretroviral therapy. The presence of an opportunistic pathogen, especially Cryptosporidium, Microsporidium, Isospora, or Giardia in the small intestine and Salmonella, Shigella, Mycobacterium, Cryptosporidium, E. histolytica, and CMV in the colon, must always be ruled out in these patients.
Malignancy
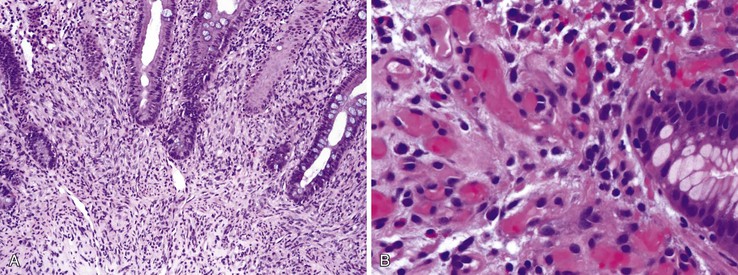
The effective use of HAART in the treatment of HIV has led to a gradual decline in infectious diseases and an increase in HIV-associated malignancies (see Table 5.6). The most prevalent cancers in this population are Kaposi sarcoma and AIDS-related NHL. Kaposi sarcoma is still the most common HIV-associated malignancy, despite a decline in incidence since advent of HAART. Studies have reported GI involvement in 40% of cases at initial presentation and in as much as 80% at autopsy.153 Endoscopically, Kaposi sarcoma appears as flat to raised, purple plaques. Microscopically, the lesion is composed of plump spindle cells, which often form fine vascular lumina containing red blood cells (Fig. 5.23). Hemosiderin and eosinophilic hyaline droplets may be present. Kaposi sarcoma is caused by human herpes virus 8 (HHV-8). Immunohistochemical stain for HHV-8 is useful in confirming the diagnosis.
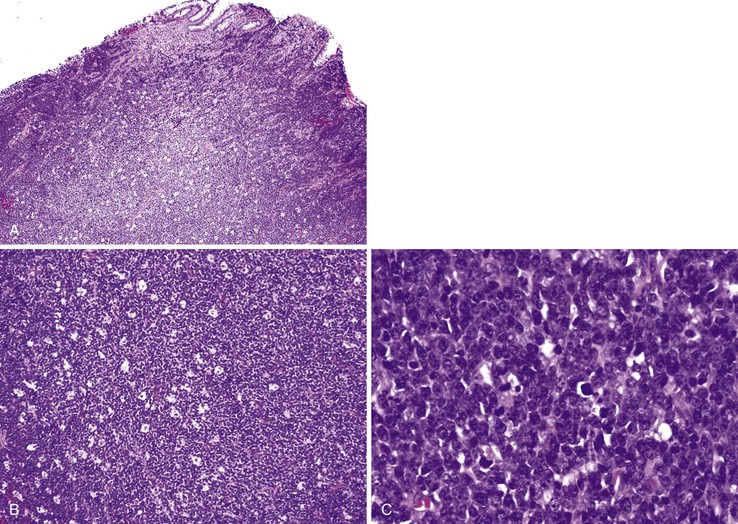
The AIDS-related NHL lymphomas are predominantly of B-cell lineage and include Burkitt lymphoma (Fig. 5.24), DLBCL (immunoblastic, centroblastic, and anaplastic variants), primary effusion lymphoma (PEL), and plasmablastic lymphoma.153,154 The GI tract is the most common site of extranodal NHL, including Burkitt lymphoma and DLBCL.155,156 Classic PEL affects the peritoneal cavity, whereas solid PEL may be seen in extraserous sites such as large intestine and lymph nodes. Plasmablastic lymphoma has been documented in the oral cavity and anorectum.153 The development of these HIV-associated malignancies is often attributable to coinfection by viruses such as HHV-8 (also called Kaposi sarcoma–associated herpesvirus) and Epstein-Barr virus. In addition to these more common HIV-associated cancers, large database studies have shown an association of HIV infection with other malignancies. This list includes Hodgkin lymphoma, multiple myeloma, leukemia, anal squamous cell carcinoma, head and neck squamous cell carcinoma, esophageal carcinoma, and gastric carcinoma.153,157
Related posts:
 Epithelial Neoplasms of the Large Intestine
Epithelial Neoplasms of the Large Intestine
 Inflammatory Disorders of the Large Intestine
Inflammatory Disorders of the Large Intestine
 Inflammatory Disorders of the Small Intestine
Inflammatory Disorders of the Small Intestine
 Polyps of the Stomach
Polyps of the Stomach
 Benign and Malignant Tumors of the Gallbladder and Extrahepatic Biliary Tract
Benign and Malignant Tumors of the Gallbladder and Extrahepatic Biliary Tract
 Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
