[level-membership-for-neurosurgery-category]
Chapter 12 Management of Primary Central Nervous System Lymphomas
Pathogenesis and Molecular Pathology
PCNSLs are almost exclusively of B-cell origin with only 2% of T-cell origin. The most common histologic subtype is the CD20-positive, diffuse, large-cell, B-cell, non-Hodgkin’s lymphoma (NHL), with a smattering of other more indolent B-cell lymphomas reported. The disease is more common in the immunocompromised than in the immunocompetent, but the pathogenesis of these disorders is uncertain regardless of the immunocompetency of the patient.1,2
Normal B cells arise from the hematopoietic stem cell and initially undergo antigen-independent differentiation, with immunoglobulin rearrangement in the bone marrow prior to emerging from the marrow as mature but naive B cells. These B cells move to secondary lymphoid organs where, upon encountering antigen, they undergo somatic hypermutation of the immunoglobulin variable region in the germinal center microenvironment. The presence of T cells, antigen presenting cells and the appropriate cytokine milieu are generally considered a requirement for somatic hypermutation with subsequent affinity maturation.3 B cells displaying the highest affinity for antigen are rescued from apoptosis and become either a memory cell or the terminally differentiated plasma cell.
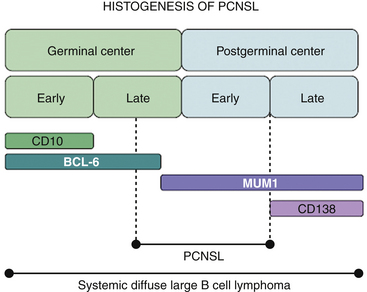
Malignant B cells can be viewed as B cells arrested at a certain stage of differentiation. The developmental state of the cell will be reflected in its morphologic attributes, the degree of immunoglobulin rearrangement, the expression of surface molecules including CD10, BCL-6, MUM-1, and CD138 (which serve as markers of the B cell’s transition through the germinal center), and the presence of intraclonal heterogeneity. In the majority of PCNSLs, the malignant cells are BCL-6 positive (60%–100%), MUM-1 positive (90%–100%), and have undergone immunoglobulin rearrangement with somatic hypermutation. These findings suggest that the malignant cell has seen antigen, passed through the germinal center but has not yet become a plasma B cell, that is, the cell of origin in most PCNSLs is a B cell on the verge of exiting the germinal center (Fig. 12-1).4–6 Because there are no germinal centers in the brain, the B cell has, in all likelihood, migrated from a node to the CNS, probably in response to an antigen.7 It is unclear, however, whether the malignant transformation of B cell occurs prior to or following migration into the CNS.
A rat model of the disease, developed by Knopf and colleagues,8 suggests that PCNSL arises in response to an antigenic stimulus, an infection perhaps, where the antigen has moved into a draining lymph node and serves to recruit naive B cells. Presumably, antigen retained in the CNS as well as the expression of specific chemokines prompts trafficking of B cells back into the CNS. Although this hypothetical scenario is compatible with the pathologic stage of development and differentiation of the malignant cell, several questions remain unanswered, including identification of the site of malignant transformation as mentioned above, the complete lack of involved lymph nodes, and the identification of the intracerebral antigen driving the process.
Certainly, there is abundant data that Epstein-Barr virus (EBV) is involved in the pathogenesis of PCNSL in the HIV-positive patient. There are, however, equally compelling data that EBV positivity is a rarity in PCNSL occurring in the immunocompetent individual.3,9,10 Thus, it is quite possible that the underlying pathogenesis differs, depending on the immunocompetency of the patient.
Epidemiology
PCNSL was considered a rare tumor occurring in a few immunosuppressed organ transplant recipients until the early 1980s when, coinciding with the AIDS epidemic, there was a marked increase in its frequency. The increase in incidence was seen in all age groups but was more evident in men than in women.11 At its peak, prior to the highly active antiretroviral therapy (HAART) era, the relative-risk in HIV-infected individuals was 1000- to 3600-fold higher than in immunocompetent individuals; this has declined dramatically, essentially by an order of magnitude, since the introduction of HAART.12,13 However, a definite and persistent rise in incidence in the immunocompetent population has been observed.14,15 Patients without immune suppression are usually older, and the male-to-female ratio is 1.2 to 1.7:1.11 Most studies corroborate that this change in the incidence of PCNSL is independent of trends in the incidence of brain tumors and in NHL.16
Clinical Manifestations
The clinical effects of PCNSL are indistinguishable from those associated with other brain tumors. In addition to the routine medical history, special care should be taken to elicit information about the possibility of immune suppression, especially HIV infection. PCNSL usually occurs several years after the diagnosis of HIV infection has been made.17 Because PCNSL only occurs in about 3% of all AIDS patients,18 infections like toxoplasmosis are a more likely diagnosis in this setting. It should be noted, however, that as a cause of intracranial mass lesions in an individual with AIDS, PCNSL is second only to toxoplasmosis.12
Approximately 8% of immunocompetent patients will have a history of successful treatment of a non–nervous system malignancy prior to the diagnosis of PCNSL.19 In these cases, the diagnosis is even more challenging and might be delayed because of the concern of secondary nervous system involvement from the previous malignancy. When the previous malignancy is an extraneural NHL, absence of systemic disease on diagnostic workup and comparison of gene rearrangement studies on the biopsy specimens of both lymphomas can demonstrate that they are separate entities. Whether these patients have an increased predisposition to multiple neoplastic processes, or the PCNSL is the result of the antineoplastic treatment for the first tumor, is unknown.19
The relative frequency of clinical manifestations of PCNSL does not differ greatly between immunosuppressed and immunocompetent individuals. Nevertheless, there are some differences that might be of clinical relevance when considering the diagnosis. In immunocompetent patients, the median age at presentation is in the sixth decade of life, whereas the median age for AIDS patients is in the fourth decade of life. Patients with AIDS more often have multiple lesions than do immunocompetent individuals, making the clinical topographic diagnosis difficult, and the latency between the onset of symptoms and diagnosis seems to be shorter.18
The clinical course is usually subacute, with a few months elapsing between the onset of symptoms and the diagnosis of a mass lesion by imaging studies. There are several reports of spontaneous transient remission of symptoms associated with PCNSL.20 The most common symptoms associated with PCNSL are focal neurologic deficit, increased intracranial pressure, alteration of mental function, or a combination of these manifestations. At the time of initial presentation, approximately one third will have symptoms of increased intracranial pressure, about 50% will have behavioral changes, and approximately 10% will have seizures.18 Between 30% and 42% of patients will experience a combination of focal and nonfocal symptoms at the time of diagnosis. The neurologic examination will yield a variety of signs that can be localizing or nonlocalizing (e.g., increased intracranial pressure or alteration in the mental function). Hemiparesis and ataxia are the most common focal neurologic signs, but aphasia, acalculia, visual field defects, and cranial nerve palsies are also common.18 Cranial-spinal nerve palsies and hydrocephalus might be secondary to lymphomatous meningeal infiltration, which is present in up to 42% of all patients with PCNSL.18 Visual symptoms might precede or follow the diagnosis of PCNSL and will depend on the ocular structure affected by the tumor. However, 50% of the patients with PCNSL and ocular involvement detected by slit-lamp examination are asymptomatic.21 Clinically apparent ocular involvement at presentation is found in about 8% to 10% of PCNSL patients,18 but vitreous involvement of the eye occurring prior to or during the course of CNS lymphoma has been noted in up to 25% of patients.22 In about half of the cases with ocular involvement, visual symptoms can be the first manifestation of PCNSL, preceding neurologic symptoms by several months. Decreased visual acuity or floaters may prompt the patient to seek medical attention, and any nonspecific uveitis refractory to topical or systemic steroids should bring ocular PCNSL to mind.21
Rare clinical syndromes that sometimes are associated with PCNSL include those where the tumor location is restricted to the spinal cord, the leptomeninges, and the hypothalamus. These are especially challenging cases because, in addition to other neoplastic diseases, benign inflammatory entities can have a similar clinical and radiologic presentation. Isolated spinal cord involvement occurs in 1% to 2% of all PCNSL18 and can be associated with syringomyelia.23 The level and extent of myelopathic involvement will guide the clinical presentation. Secondary involvement of the spinal cord in patients with cerebral lesions is not a rare occurrence.24 PCNSL may also present as hypothalamic dysfunction causing diabetes insipidus,18 as pituitary apoplexy with bitemporal hemianopsia or as isolated third nerve palsy.25
A variant of PCNSL, clinically presenting with progressive cognitive decline and gait disorder and associated with diffuse white matter abnormality without enhancement on MRI was initially described in 1999.26 The term “lymphomatosis cerebri” has been used to describe this uncommon condition, which can be difficult to diagnose because of the nonspecific clinical and imaging findings;26–30 it can be erroneously diagnosed as vascular leukoencephalopathy, multiple sclerosis (MS), or gliomatosis cerebri.
Diagnosis
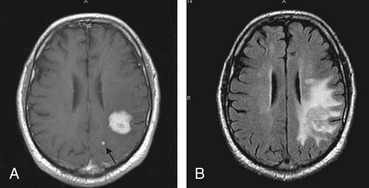
A definitive diagnosis of PCNSL cannot be made on clinical or imaging grounds, and histologic confirmation is essential. CT scan or MRI will initially establish the presence of a mass lesion with characteristics that, although suggestive of PCNSL, are not specific for this entity (Fig. 12-2). CSF analysis helps in the differential diagnosis and in some cases makes the diagnosis by the demonstration of malignant B lymphocytes.18 HIV testing is required in all patients suspected of having PCNSL. In spite of the information obtained from these studies, tissue diagnosis is required in most circumstances.
Imaging Studies
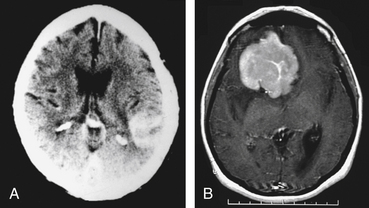
There are some characteristics on imaging studies that, although not pathognomonic, would strongly suggest that the mass lesion identified might be of lymphomatous origin. In the immunocompetent host a higher level of suspicion is required. Head CT scan shows a hyperdense or isodense mass, solitary in 86% of the cases,31 that usually exhibits homogenous enhancement after the administration of iodinated contrast.32 Lesions are usually supratentorial and localized in the deep periventricular areas.31 Because of their infiltrative nature, the lesions might have indistinct borders and result in minimal surrounding edema or compressive effect (see Fig. 12-2). Occasionally it can appear as a ring-enhancing lesion with a hypodense, necrotic core, indistinguishable from a high-grade glioma. Lesions can be multiple, suggestive of metastatic disease or infection, but the scarce perilesional edema should raise the suspicion of PCNSL. In about 10% of the cases the lesions are localized in the posterior fossa.
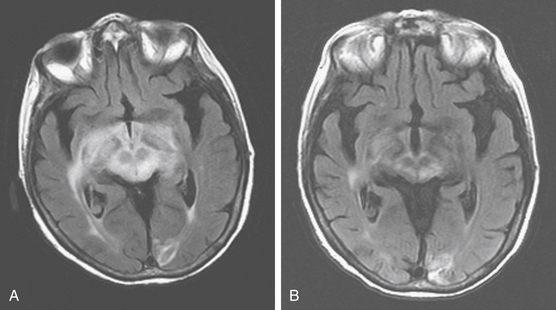
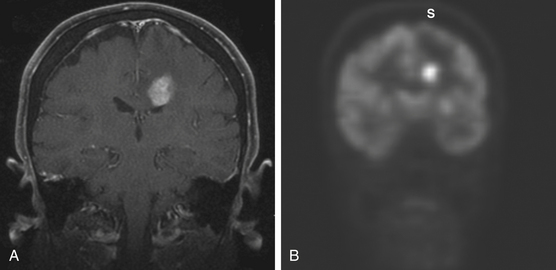
On MRI, most of the lesions are hypointense or isointense on T1-weighted images, and only about 40% are hyperintense on T2-weighted images.32 Although most lesions enhance after the administration of gadolinium exceptions include cases when the study follows the administration of steroids or when there is a diffuse infiltrating lymphoma (lymphomatosis cerebri) (Fig. 12-3). The appearance of PCNSL is of such heterogeneity that it should be considered in the differential diagnosis of any mass lesions detected on imaging studies (Fig. 12-4).
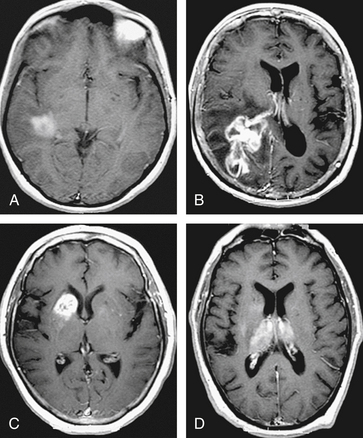
PCNSL in the immunocompromised patient may have a more variable appearance on imaging studies.33 Infection is more likely to be responsible for a mass or enhancing lesion on imaging studies than PCNSL in this population, but no specific pattern has been established that can be used to distinguish between CNS lymphoma, toxoplasmosis, or other CNS diseases that occur in patients with AIDS.18 Unlike immunocompetent patients, AIDS-associated PCNSL has been reported to present with multiple lesions in 71% to 80% of cases, to show ring-like enhancement in 50% of cases, and to lack enhancement in about 10% to 27% of the lesions.33 Spontaneous hemorrhage, a nonenhancing lesion, or diffuse white matter changes do not exclude lymphoma in an immunocompromised patient.
Advanced MRI techniques may be helpful to pre-operatively suggest PCNSL. Magnetic resonance spectroscopy (MRS) shows massively elevated lipid resonances in PCNSL; although this may also be present in glioblastoma, the finding of elevated lipid resonances in combination with a markedly elevated choline/creatine ratio, may improve the preoperative differentiation of PCNSL and glioma.34 Dynamic susceptibility contrast-enhanced perfusion MRI reveals that the relative cerebral blood volume ratio is lower for PCNSL, correlating with the lower microvessel density by immunohistochemistry, than high grade gliomas.35 Using diffusion tensor imaging, with the enhancing lesion as the region of interest and comparing to the contralateral normal-appearing white matter, the fractional anisotropy and the apparent diffusion coefficient (ADC) ratios can be measured. Both parameters are significantly lower in lymphomas than glioblastoma and may assist in differentiating between these two entities.36 The ADC ratios, however, do not allow a reliable distinction between toxoplasmosis and lymphoma.37
Nuclear medicine has also been suggested as a possible method to discriminate between PCNSL and other types of malignant as well as nonmalignant pathologies without resorting to histologic diagnosis. In a study comparing N-isopropyl-p-(123I)-iodoamphetamine (123I-IMP) single photon emission computerized tomography (SPECT) in patients with malignant glioma, PCNSL and meningioma, the 123I-IMP retention uptake in the 6-hour to 24-hour SPECT images were significantly higher in PCNSL than in those of both malignant gliomas and meningiomas.38 In patients with AIDS, the thallium201-SPECT delayed retention index may be useful to discriminate PCNSL from infectious lesions with high sensitivity and specificity.39 The diagnostic utility of these techniques has yet to be determined in larger series, and histologic confirmation is still considered the gold standard for diagnosis of PCNSL.
Tumor infiltration of the nervous system can be more diffuse than is appreciated on imaging studies. Correlation of autopsy and MRI findings in 10 patients who died with PCNSL showed that all had tumor infiltration in CNS regions that were normal radiographically, including T2 sequences.40 Therefore, the infiltrative microscopic tumor burden of PCNSL renders futile any attempt to resect these lesions. The surgical role is restricted to a tissue biopsy for histologic diagnosis.
Tissue Diagnosis and Staging
While the diagnosis of PCNSL is usually suggested by the appearance of a focal lesion on CT or MRI, confirmation of the diagnosis of PCNSL in the immunocompetent requires tissue to definitively differentiate PCNSL from metastatic disease, glioma, sarcoidosis, and inflammatory lesions (Table 12-1). Because the CSF is involved at diagnosis 20% of the time, a brain biopsy can be avoided if malignant cells can be obtained from the CSF.41 Similarly, malignant (though often asymptomatic) uveitis is apparent in 10% to 20% of patients with PCNSL at presentation and may serve as the source of cells on which the diagnosis may be based.42,43 Cytologic examination of cells, as the sole parameter by which the diagnosis is made, is not optimal because it cannot determine monoclonality, a condition that is necessary though not sufficient for the diagnosis of lymphoma. In the case of B-cell lymphoma, monoclonality can be determined by flow cytometric detection of light-chain restriction if there are sufficient cells available. In T-cell disease, and when there are inadequate numbers of malignant B cells for flow cytometry, the diagnosis can be established by polymerase chain reaction (PCR) studies on the basis of T-cell receptor or immunoglobulin rearrangement, respectively. This technique is susceptible to false positives if too few cells are present and to false negatives if the DNA is highly degraded.44 It is important to underscore the fact that most lymphomas, including PCNSL, are quite sensitive to steroids. Cell death and tumor regression may occur as early as 24 hours after initiation of therapy. If tissue or a specimen for cytology is obtained following initiation of steroids, the result may be nondiagnostic because of cell death and tissue necrosis. Unless the patient is showing evidence of rapid neurologic deterioration or impending herniation, tissue should be obtained prior to starting any steroid therapy.45,46 Although a retrospective study exists challenging this concept,47 it seems prudent to avoid the unnecessary use of corticosteroids prior to diagnostic biopsy for PCNSL.
| Disease | Diagnostic Studies |
|---|---|
| Multiple sclerosis | Past medical history, CSF |
| High-grade glioma | SPECT-MRS |
| Infection | HIV infection, CSF |
| Sarcoidosis | ACE level, calcium |
| Meningioma | MRS |
| Vascular | Cerebral angiogram, MRI DWIs |
ACE, angiotensin-converting enzyme; CSF, cerebrospinal fluid; DWI, diffusion-weighted images; HIV, human immunodeficiency virus; MRS, magnetic resonance spectroscopy; SPECT, single photon emission computerized tomography.
Once the diagnosis is made, patients generally undergo staging to determine the extent of involvement. Baseline evaluations include a physical and a neurologic exam, as well as cognitive function assessment. Because systemic involvement tends to occur more commonly at relapse, and because evidence of systemic disease is found in less than 5% at diagnosis, the necessity for a full lymphoma staging has been called into question.48 There is general agreement that, besides a complete history and physical, a CBC, standard chemistries with an LDH, HIV status, chest x-ray, CSF examination, and slit-lamp examination are absolutely required. A full staging would also include CT of the chest, abdomen, and pelvis, as well as bilateral bone marrow biopsies. Finally, testicular ultrasound should be considered in all elderly men. Full staging is inevitably required for any patient enrolled in a clinical trial and is recommended for all patients with PCNSL in the published guidelines for standardization of baseline evaluations.49
The role of 18F-fluorodeoxyglucose (FDG) PET to rule out systemic disease in the initial evaluation of patients presenting with PCNSL is uncertain. Two retrospective studies using PET in the initial staging of immunocompetent patients have revealed abnormalities (e.g., other malignancies, systemic sites of lymphoma as well as concurrent sites within the nervous system) undetected by other diagnostic studies in 19% and 28% of patients respectively.50,51 It remains to be determined if FDG PET is needed in all patients with the presumptive diagnosis of PCNSL (Fig. 12-5).
The approach to a focal brain lesion in the HIV population is slightly different. The detection of EBV DNA by PCR in the CSF was initially found to be a sensitive (80% to 84%) and highly specific (100%) diagnostic marker of AIDS-related PCNSL, thus potentially obviating the need for a biopsy; these results, however, could not be confirmed in a later study.52–54 A brain biopsy in the AIDS patient carries a significant morbidity (8.4%) and mortality (2.9%),55 but is recommended when the CSF is negative for lymphoma, the focal lesion is atypical for toxoplasmosis, there is progression on a brief trial of antitoxoplasmosis therapy, toxoplasma serologies are negative, there is a rapid neurologic decline, or there are discordant CSF EBV and thallium201-SPECT results.12,17 It is noteworthy that a pre-HAART retrospective study of presumed or confirmed PCNSL in HIV-positive patients revealed there was no difference in the overall survival (1.2 months) between the two groups, presumptive and biopsy-confirmed, leading the authors to conclude there may be little benefit in subjecting the patient to the diagnostic procedure given the dismal outcome.56 The use of HAART and its impact on the CD4 count has allowed the use of more aggressive chemotherapeutic regimens without undue toxicity, and resulted in improved life expectancy for these patients. These changes will undoubtedly influence the algorithm, and they suggest that earlier diagnostic procedures may be in order for this patient population.
Differential Diagnosis
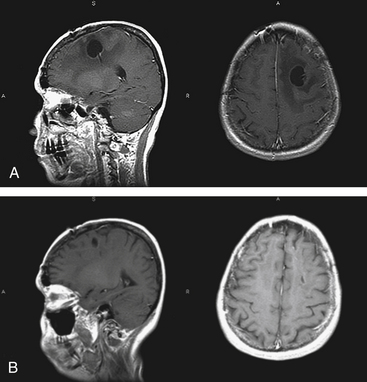
PCNSL may present with a variety of signs and symptoms and has a capacity to mimic many other nonmalignant neurologic conditions.57 It can be confused with multiple sclerosis (MS) in patients who present with neurologic dysfunction, a nonenhancing periventricular lesion, and CSF pleocytosis. Administration of corticosteroid causes clinical improvement and regression of PCNSL in some patients; this may be interpreted as a steroid-induced remission from an exacerbation of MS. Sustained clinical dependence on corticosteroid is unusual in MS, and should lead to consideration of PCNSL. Repeat CSF examination and gadolinium-enhanced MRI scan obtained off corticosteroid should differentiate between the two diagnostic possibilities.58 PCNSL cortical lesions may be difficult to distinguish from extra-axial masses such as meningioma on imaging studies (Fig. 12-6). In the case of immunocompromised individuals, infections such as Toxoplasma gondii should always be included in the differential diagnosis.
Treatment
Steroids
Steroids induce apoptosis of lymphoid cells and can result in complete disappearance of the clinical and imaging manifestations associated with PCNSL.59 Although reduced edema plays a role, most of the rapid and dramatic responsiveness to corticosteroids is mediated by their cytotoxic activity. Because of the exquisite sensitivity of lymphoma cells to steroids, their administration to the patient with PCNSL carries important diagnostic and therapeutic implications. In 40% of patients, a rapid and dramatic regression of PCNSL follows their administration.60 Further, there can be a complete response after steroids in about 15% of the cases, resulting in the complete disappearance of the target for the biopsy.59 In another series, however, the majority of immunocompetent PCNSL patients received steroids before biopsy and had neither significant MRI change nor required of second biopsy for diagnosis.47 A dramatic clinical and MRI improvement would make the diagnosis of PCNSL very likely, although as previously mentioned there are other entities that exhibit similar steroid-responsiveness.
Even when a complete response is obtained on steroids alone, the disease will recur either at the same or in a remote site within the nervous system. Due to the potential development of steroid resistance, their use as monotherapy for PCNSL is not recommended, though they are usually used in combination with chemotherapy as part of the therapeutic regimen. It has been shown that the addition of corticosteroids to the first course of methotrexate chemotherapy was associated with a higher rate of complete responses.61
Surgery
PCNSL is an infiltrative tumor, usually localized in the deep periventricular regions, and is highly responsive to radiation and chemotherapy. Therefore, surgical resection is of limited benefit and can carry significant morbidity. In a retrospective study including 248 immunocompetent patients with PCNSL, 132 underwent stereotactic biopsy for diagnosis of PCNSL, resulting in a procedure-related morbidity of 3.7% and no mortality. The remaining 116 underwent surgical resection (because imaging studies were not suggestive of lymphoma) with a mortality rate of 3.4%. Multivariate analysis revealed that surgical resection was an unfavorable prognostic factor31 and may increase functional deficit.18 When there is the suspicion of PCNSL, the role of surgery is restricted to a stereotactic biopsy to provide tissue for diagnostic purposes.
In AIDS patients, the diagnosis of PCNSL is extremely likely in patients with hyperactive lesions on thallium201-SPECT and a CSF positive for EBV-DNA. If the lesion is hypoactive on SPECT, with negative CSF EBV-DNA, the recommendation is empiric antitoxoplasma therapy. If there is disagreement between the SPECT and CSF PCR results, a brain biopsy is advisable.53
Despite common contiguity with the ventricular system, only 7% of the patients have hydrocephalus requiring shunting.31 When leptomeningeal lymphomatous spread is documented, intraventricular chemotherapy is often recommended. An intraventricular catheter with a subgaleal reservoir for ease of administration is usually placed. Before therapeutic use of the reservoir, a nuclear medicine flow study is performed to ensure correct catheter placement as well as good distribution of the chemotherapy agent. Reservoirs have been associated with complications necessitating removal of the device (Fig. 12-7). Infection is the most common complication, occurring in about 15% of patients with PCNSL.
Radiation Therapy
Radiotherapy was the first intervention to have a significant impact on PCNSL survival, with an increased median survival of 12 to 18 months.31,46,62 Like systemic lymphomas, PCNSLs are considered to be extremely radiosensitive as was exemplified in a study in which a 40-Gy dose to the whole brain (WB) followed by a 20-Gy boost to the lesion(s) yielded an 83% complete remission rate. Unfortunately, the remissions were not durable; the median survival from the start of radiation therapy (RT) was only 11.6 months.62 Unlike other tumors, there is no classic dose–response curve for radiation in PCNSL, but there does appear to be a threshold dose of 50 Gy. A review of the literature, including patients receiving only RT, found a 42.3% 5-year survival for patients receiving greater than 50 Gy, as compared with a 12.8% 5-year survival for those receiving less than 50 Gy.62
A devastating and irreversible complication, treatment-related delayed neurotoxicity occurs most commonly following radiation or combination chemoradiation therapy (particularly when radiation precedes the chemotherapy) and only rarely following chemotherapy alone. The elderly are particularly vulnerable with an incidence of 80% in those surviving more than 5 years. It manifests as cognitive dysfunction early on followed by motor and autonomic dysfunction.63 While consolidation with WBRT after chemotherapy does improve the failure-free survival, it does not improve overall survival.64 For patients who are 60 years or older, current clinical trials rarely include radiation as initial treatment because of the high risk of severe cognitive impairment and neurotoxicity-related mortality. Recent trial designs have deferred RT or recommended the use of low-dose RT consolidation after chemotherapy. RT deferred until relapse yields a response rate (79%) and overall survival rate (32 months from diagnosis) comparable to those obtained when RT was included in the initial treatment plan.65 Reduced-dose RT (40 to 45 Gy) in patients younger than 60 years who achieve a complete response with aggressive chemotherapy may decrease the occurrence of severe cognitive impairment, but the cost is an extremely high incidence of chemotherapy-related toxicity and lower response rates.66 Recently, response-adapted therapy adjusting the radiation dose to less than 24 Gy for patients who achieved a complete remission with immunochemotherapy has resulted in excellent disease control (the increased incidence of neutropenia notwithstanding) and no decline in cognitive testing though median follow-up is only 37 months.67
Radiosurgery
Although PCNSL is a diffuse disease frequently with multiple enhancing lesions, CSF dissemination and ocular involvement, treatment with radiosurgery has been utilized. Two small studies have reported excellent initial local control of disease with gamma knife radiosurgery but, as expected, the majority of patients develop new lesions in other locations.68,69 Therefore, the potential benefit and long-term neurotoxicity of this therapeutic modality in the management of PCNSL is uncertain and possibly limited for symptom control of localized recurrent disease.
Methotrexate-Based Chemotherapy
The use of combined modality chemoradiation therapy is generally considered to yield improved outcomes over radiation therapy alone, although this consensus is based on retrospective studies only (Fig. 12-8).60 Further, regimens containing either high-dose methotrexate (defined as more than 1.5 gm/m2 per cycle) or high-dose cytarabine are associated with improved survival though only the use of high-dose methotrexate appears to be an independent favorable prognostic factor.70 These two chemotherapeutic agents are commonly used in lymphoma therapy and have excellent penetration into the CNS, even in the presence of an intact blood-brain barrier (BBB). Importantly, early studies established that neither route of administration (intrathecal versus intravenous) nor chemotherapeutic agent, regardless of age, is correlated with increased neurotoxicity.70 Multiple studies have confirmed these findings over the past decade, with median survivals ranging from 33 months to 60 months when a high-dose methotrexate regimen was used with or without subsequent WBRT (Table 12-2).70–82
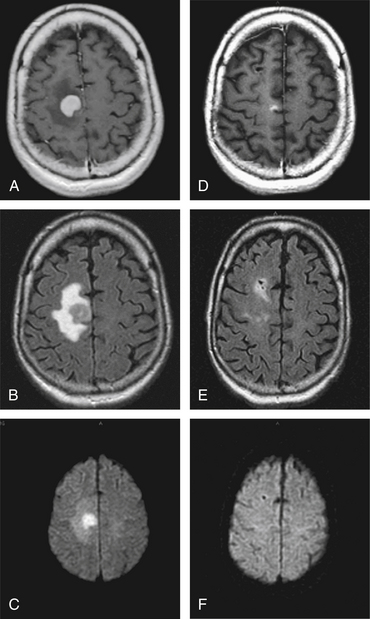
Table 12-2 Response Rates, Overall and Progression-Free Survival, and Toxicity to PCNSL Treatment Regimens with and without Radiation, for Patients of All Ages Including Elderly
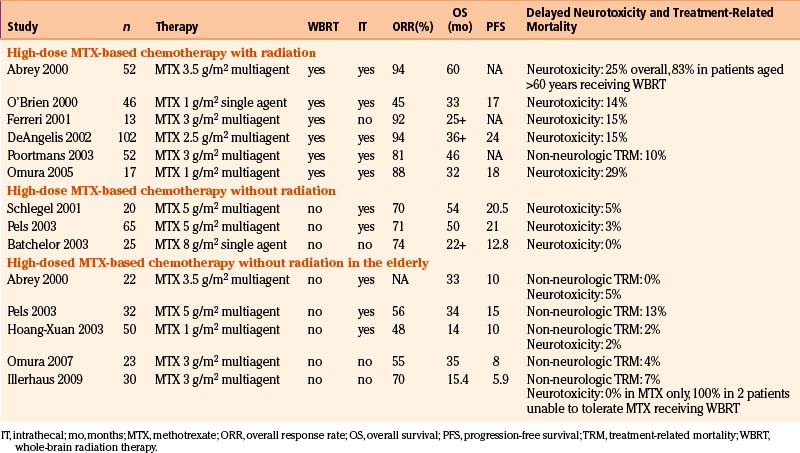
In the elderly, studies using high-dose methotrexate-based regimens without RT have resulted in lower response rates and shorter durability of remission than in younger patients with PCNSL. The event-free survival of 5.9 to 15 months and the overall survival of 14 to 34 months are disappointing but the low incidence of both treatment-related mortality and neurotoxicity as well as a plateau in the survival curves suggests that, even in this population usually exhibiting both poor prognostic features (age and poor performance status), durable remissions with acceptable toxicity are possible.73,74,79,82
Intrathecal Therapy
High-dose intravenous (IV) methotrexate, which penetrates an intact BBB and results in therapeutic levels of drug in the CSF, has brought into question the routine use of intrathecal (IT) therapy for patients with PCNSL. Based on retrospective data, there is a growing consensus that outside of a clinical trial, IT therapy should be used only in the setting of a positive CSF cytology, if at all, in up-front treatment.83 Of note, however, a phase II trial of patients treated without IT therapy was stopped following interim analysis due to a high rate of early relapse when compared to the results of a previous trial of comparable PCNSL patients who received the same chemotherapy regimen but with the addition of IT therapy84 further supporting the need for randomized trials to answer this pressing question. IT therapy is routinely used at relapse or if leptomeningeal disease persists after initiation of systemic high-dose methotrexate therapy.83,85 Methotrexate, cytarabine, and hydrocortisone have all been used singly or in combination for IT therapy. Rituximab, an anti-CD20 monoclonal antibody beneficial in systemic large-cell B-cell lymphoma but which does not penetrate the BBB when given IV, has been shown to be well tolerated and effective when given intrathecally.86–88
Other Therapeutic Options
Durable remissions and cure rates are distinctly lower in diffuse large-cell lymphoma of the CNS than in systemic diffuse large-cell, B-cell NHL. This is, in part, because of the difficulty in obtaining adequate drug levels when given systemically across an intact BBB. The chemotherapeutic regimens for systemic diffuse large-cell lymphoma routinely utilize only a few drugs, specifically those of the alkylator class, that are known to penetrate the CNS. Within this class, penetration varies from drug to drug (Table 12-3). Other strategies to increase the uptake of drugs include the use of a lipid-soluble drug (i.e., temozolomide), increased plasma concentrations, intra-arterial drug injection, and osmotic disruption of the BBB.89 The results of a multi-institutional trial using intra-arterial delivery following osmotic BBB disruption has resulted in an excellent overall response rate (81%) but significant toxicity was incurred with a combined procedural and chemotherapeutic complication rate of 33%.90 High-dose IV therapy, which results in better penetration, is limited by systemic toxicity. However, if the dose-limiting toxicity of the agents is bone marrow suppression or ablation, stem cell rescue can be used to overcome that particular toxicity. On the assumption that there is no significant difference in the biologic behavior between systemic and CNS-isolated large-cell B-cell lymphoma, stem cell transplant, which is routinely used in systemic lymphoma,91,92 has been proposed as a potentially curative intervention in PCNSL. Initially trialed in young PCNSL patients with poor prognostic indicators or relapsed/refractory disease,93–95 the results were so encouraging that high-dose therapy with autologous stem cell rescue has been moved up to first-line therapy by several groups.96–100 Results in newly diagnosed patients using high-dose methotrexate, ablative chemotherapy with stem cell rescue, and, finally, WBRT post-transplant yielded OS of 87% at 5 years, but unacceptable neurotoxicity rates of 24%.96 A revised protocol using radiation in the post-transplant period only when a complete response was not obtained following chemotherapy, has given very encouraging results: the disease free and overall survival at 3 years is 77% without evidence of significant neurotoxicity to date.96
Table 12-3 Systemic Antineoplastic Agents Used in Lymphoma Therapy That Penetrate the Blood–Brain Barrier
| Methotrexate |
| Cytosine arabinoside |
| Procarbazine |
| BCNU |
| CCNU |
| Thiotepa |
| Temozolomide |
| Mercaptopurine (low concentrations) |
| Melphalan (low concentrations) |
| Cyclophosphamide (low concentrations) |
Therapy for Intraocular Lymphoma
The therapy for intraocular lymphoma, whether it occurs in isolation or in the presence of other CNS lesions, deserves separate mention. Neither involved field irradiation nor systemic chemotherapy alone results in durable remissions in the eye, but the addition of orbital radiation to a high-dose, methotrexate-based regimen has been associated with both a higher response rate and decreased intraocular relapse rate.21 No difference in outcome has been found between patients treated with local (RT or intravitreal chemotherapy) as opposed to systemic chemo- or chemoradiation therapy.101 When used, radiation to both orbits is recommended due to the fact that bilateral eye involvement occurs in almost 80% of cases.21 Ocular RT carries a substantial morbidity including radiation retinitis, dry eye syndrome, corneal erosions, glaucoma, and cataracts. Intravitreal methotrexate (400 μg on a once or twice weekly basis with or without monthly maintenance) has been used with success.102–104 A retrospective interinstitutional study of intravitreal methotrexate in patients with intraocular lymphoma revealed resolution of intraocular disease in all patients for whom there is complete information. No patient in this study relapsed in the eye; complications included keratopathy, cataract formation or acceleration and rarely, neovascularization.102 The optimal therapy for PCNSL with intraocular involvement remains to be defined. At present, combined chemotherapy followed by ocular irradiation appears to give the best results.
Treatment for AIDS-Related PCNSL
Treatment of AIDS-related lymphomas in general has been discouraging. Full-dose chemotherapy in the immunocompromised host results in an unacceptable morbidity and mortality, whereas dose reduction results in inadequate therapy, with tumor progression and drug resistance. Standard therapy has been palliative, using corticosteroids and WBRT, with only 10% of patients surviving more than 1 year. Less aggressive chemotherapy (procarbazine, CCNU, vincristine [PCV]) does appear to increase the median survival from 4 months with radiation alone to 13 months in one small study.105 The benefit of combined modality therapy over radiotherapy has been confirmed in other studies.56 The decision to treat with combined modality, as opposed to radiation alone, should be based on the patient’s performance status, extent of disease, comorbid conditions, projected prognosis, and the patient’s desire for aggressive therapy.105 However, a linked SEER-Medicare database search of AIDS patients with PCNSL revealed that 46% of patients received RT alone and 40% received no treatment at all with the result that the prognosis for AIDS-related PCNSL remains dismal.106 Given that AIDS-associated PCNSL is uniformly EBV+, hydroxyurea, which at a low dose can deplete deoxyribonucleotide reserves exerting an antiviral effect, has been trialed in this population.107 To date there are only case reports of regression of PCNSL following initiation of HAART and improvement in the CD4+ cell count.108 Further study will be needed to establish a causal link between lymphoma remission and restoration of immunocompetency.
Treatment of Recurrent PCNSL
In patients who only received chemotherapy initially, WBRT has been used as salvage therapy for refractory/relapsing PCNSL with a 58% complete response rate reported in one study, though neurotoxicity occurred in 29% within a median of 7 months following RT.65 As noted above, high-dose chemotherapy with hematopoietic stem cell rescue was initially studied in relapsed or refractory patients younger than 65 years and has yielded encouraging results.109
Other chemotherapy options that have been used include topotecan, PCV, high-dose cytarabine and rechallenge with high-dose methotrexate-based regimen with variable results.110–113 Temozolomide, an oral alkylating agent with good nervous system penetration and a favorable side effect profile, has been examined in a trial of recurrent PCNSL resulting in a 31% overall response rate and minimal toxicity.114 A retrospective analysis of patients receiving IV rituximab in conjunction with temozolomide, followed by single agent temozolomide, revealed an objective response rate of 53% with an acceptable toxicity profile.115 Nonetheless, the majority of responders relapsed with a median survival of only 14 months.
Prognosis
The ability to determine which patients will likely do well would be of great benefit and would aid in tailoring therapy to the patient. There is general agreement that age and performance status are the most important predictors of outcome.77,79,116 In a multicenter retrospective study of 378 immunocompetent patients, Ferreri and colleagues have attempted to identify other factors that may predict response. Besides age and performance status, serum LDH, CSF protein concentration, and involvement of the deep structures of the brain were independent predictors of survival.117 Survival, of course, is not the only endpoint worthy of measure, and chronic toxicity to the CNS, resulting in an unacceptable quality of life, must be factored into the equation. The opportunity to study, for the first time, PCNSL patients surviving for more than 5 years, has revealed that in patients under 60 years of age, combination chemoradiation therapy yields a superior overall survival to chemotherapy alone but the price is a 26% incidence of debilitating and irreversible neurotoxicity. In patients over 60 years of age, overall survival was identical in the two groups (with and without WBRT) but the cause of death in those receiving WBRT was neurotoxicity while in those who did not, the cause of death was relapse.63 Although the optimal therapy for PCNSL has not yet been determined, it does appear that PCNSL may, like its systemic counterpart, be a potentially curable malignancy. Current therapies hold the hope that the price of cure will not include unacceptable cognitive dysfunction.
Abrey L.E., Batchelor T.T., Ferreri A.J., et al. Report of an international workshop to standardize baseline evaluation and response criteria for primary CNS lymphoma. J Clin Oncol. 2005;23:5034-5043.
Abrey L.E., Yahalom J., DeAngelis L.M. Treatment for primary CNS lymphoma: the next step. J Clin Oncol. 2000;18:3144-3150.
Angelov L., Doolittle N.D., Kraemer D.F., et al. Blood-brain barrier disruption and intra-arterial methotrexate-based therapy for newly diagnosed primary CNS lymphoma: a multi-institutional experience. J Clin Oncol. 2009;27:3503-3509.
Antinori A., De Rossi G., Ammassari A., et al. Value of combined approach with thallium-201 single-photon emission computed tomography and Epstein-Barr virus DNA polymerase chain reaction in CSF for the diagnosis of AIDS-related primary CNS lymphoma. J Clin Oncol. 1999;17:554-560.
Bataille B., Delwail V., Menet E., et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg. 2000;92:261-266.
DeAngelis L.M., Seiferheld W., Schold S.C., et al. Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: radiation Therapy Oncology Group Study 93-10. J Clin Oncol. 2002;20:4643-4648.
Ekenel M., Iwamoto F.M., Ben-Porat L.S., et al. Primary central nervous system lymphoma: the role of consolidation treatment after a complete response to high-dose methotrexate-based chemotherapy. Cancer. 2008;113:1025-1031.
Ferreri A.J., Blay J.Y., Reni M., et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol. 2003;21:266-272.
Ferreri A.J., Abrey L.E., Blay J.Y., et al. Summary statement on primary central nervous system lymphomas from the Eighth International Conference on Malignant Lymphoma, Lugano, Switzerland, June 12 to 15, 2002. J Clin Oncol. 2003;21:2407-2414.
Gavrilovic I.T., Hormigo A., Yahalom J., et al. Long-term follow-up of high-dose methotrexate-based therapy with and without whole brain irradiation for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2006;24:4570-4574.
Grimm S.A., Pulido J.S., Jahnke K., et al. Primary intraocular lymphoma: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann Oncol. 2007;18:1851-1855.
Herrlinger U., Schabet M., Bitzer M., et al. Primary central nervous system lymphoma: from clinical presentation to diagnosis. J Neurooncol. 1999;43:219-226.
Hoang-Xuan K., Taillandier L., Chinot O., et al. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: a multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21:2726-2731.
Hottinger A.F., DeAngelis L.M., Yahalom J., Abrey L.E. Salvage whole brain radiotherapy for recurrent or refractory primary CNS lymphoma. Neurology. 2007;69:1178-1182.
Jahnke K., Thiel E., Martus P., et al. Relapse of primary central nervous system lymphoma: clinical features, outcome and prognostic factors. J Neurooncol. 2006;80:159-165.
Kasamon Y.L., Ambinder R.F. AIDS-related primary central nervous system lymphoma. Hematol Oncol Clin North Am. 2005;19:665-687. vi-vii
Knopf P.M., Harling-Berg C.J., Cserr H.F., et al. Antigen-dependent intrathecal antibody synthesis in the normal rat brain: tissue entry and local retention of antigen-specific B cells. J Immunol. 1998;161:692-701.
Kreisl T.N., Panageas K.S., Elkin E.B., et al. Treatment patterns and prognosis in patients with human immunodeficiency virus and primary central system lymphoma. Leuk Lymphoma. 2008;49:1710-1716.
Nelson D.F. Radiotherapy in the treatment of primary central nervous system lymphoma (PCNSL). J Neurooncol. 1999;43:241-247.
Omuro A.M., DeAngelis L.M., Yahalom J., Abrey L.E. Chemoradiotherapy for primary CNS lymphoma: an intent-to-treat analysis with complete follow-up. Neurology. 2005;64:69-74.
Schlegel U., Schmidt-Wolf I.G., Deckert M. Primary CNS lymphoma: clinical presentation, pathological classification, molecular pathogenesis and treatment. J Neurol Sci. 2000;181:1-12.
Shah G.D., Yahalom J., Correa D.D., et al. Combined immunochemotherapy with reduced whole-brain radiotherapy for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2007;25:4730-4735.
Siegal T., Zylber-Katz E. Strategies for increasing drug delivery to the brain: focus on brain lymphoma. Clin Pharmacokinet. 2002;41:171-186.
Soussain C., Hoang-Xuan K., Taillandier L., et al. Intensive chemotherapy followed by hematopoietic stem-cell rescue for refractory and recurrent primary CNS and intraocular lymphoma: societe Francaise de Greffe de Moelle Osseuse-Therapie Cellulaire. J Clin Oncol. 2008;26:2512-2518.
Sparano J.A. Clinical aspects and management of AIDS-related lymphoma. Eur J Cancer. 2001;37:1296-1305.
1. Schlegel U., Schmidt-Wolf I.G., Deckert M. Primary CNS lymphoma: clinical presentation, pathological classification, molecular pathogenesis and treatment. J Neurol Sci.. 2000;181:1-12.
2. Paulus W. Classification, pathogenesis and molecular pathology of primary CNS lymphomas. J Neurooncol. 1999;43:203-208.
3. MacLennan I.C. Germinal centers. Annu Rev Immunol. 1994;12:117-139.
4. Larocca L.M., Capello D., Rinelli A., et al. The molecular and phenotypic profile of primary central nervous system lymphoma identifies distinct categories of the disease and is consistent with histogenetic derivation from germinal center-related B cells. Blood. 1998;92:1011-1019.
5. Montesinos-Rongen M., Siebert R., Deckert M. Primary lymphoma of the central nervous system: just DLBCL or not? Blood. 2009;113:7-10.
6. Camilleri-Broet S., Criniere E., Broet P., et al. A uniform activated B-cell-like immunophenotype might explain the poor prognosis of primary central nervous system lymphomas: analysis of 83 cases. Blood. 2006;107:190-196.
7. Thompsett A.R., Ellison D.W., Stevenson F.K., Zhu D. V(H) gene sequences from primary central nervous system lymphomas indicate derivation from highly mutated germinal center B cells with ongoing mutational activity. Blood. 1999;94:1738-1746.
8. Knopf P.M., Harling-Berg C.J., Cserr H.F., et al. Antigen-dependent intrathecal antibody synthesis in the normal rat brain: tissue entry and local retention of antigen-specific B cells. J Immunol. 1998;161:692-701.
9. Anthony I.C., Crawford D.H., Bell J.E. B lymphocytes in the normal brain: contrasts with HIV-associated lymphoid infiltrates and lymphomas. Brain. 2003;126:1058-1067.
10. Krogh-Jensen M., Johansen P., D’Amore F. Primary central nervous system lymphomas in immunocompetent individuals: histology, Epstein-Barr virus genome, Ki-67 proliferation index, p53 and bcl-2 gene expression. Leuk Lymphoma. 1998;30:131-142.
11. Schabet M. Epidemiology of primary CNS lymphoma. J Neurooncol. 1999;43:199-201.
12. Kasamon Y.L., Ambinder R.F. AIDS-related primary central nervous system lymphoma. Hematol Oncol Clin North Am. 2005;19:665-687. vi-vii
13. Newell M.E., Hoy J.F., Cooper S.G., et al. Human immunodeficiency virus-related primary central nervous system lymphoma: factors influencing survival in 111 patients. Cancer. 2004;100:2627-2636.
14. Haldorsen I.S., Krossnes B.K., Aarseth J.H., et al. Increasing incidence and continued dismal outcome of primary central nervous system lymphoma in Norway 1989-2003: time trends in a 15-year national survey. Cancer. 2007;110:1803-1814.
15. Rubenstein J., Ferreri A.J., Pittaluga S. Primary lymphoma of the central nervous system: epidemiology, pathology and current approaches to diagnosis, prognosis and treatment. Leuk Lymphoma. 2008;49(suppl 1):43-51.
16. Olson J.E., Janney C.A., Rao R.D., et al. The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: a surveillance, epidemiology, and end results analysis. Cancer. 2002;95:1504-1510.
17. Sparano J.A. Clinical aspects and management of AIDS-related lymphoma. Eur J Cancer. 2001;37:1296-1305.
18. Herrlinger U., Schabet M., Bitzer M., et al. Primary central nervous system lymphoma: from clinical presentation to diagnosis. J Neurooncol. 1999;43:219-226.
19. Reni M., Ferreri A.J., Zoldan M.C., Villa E. Primary brain lymphomas in patients with a prior or concomitant malignancy. J Neurooncol. 1997;32:135-142.
20. Al-Yamany M., Lozano A., Nag S., et al. Spontaneous remission of primary central nervous system lymphoma: report of 3 cases and discussion of pathophysiology. J Neurooncol. 1999;42:151-159.
21. Ferreri A.J., Blay J.Y., Reni M., et al. Relevance of intraocular involvement in the management of primary central nervous system lymphomas. Ann Oncol. 2002;13:531-538.
22. Hochberg F.H., Miller D.C. Primary central nervous system lymphoma. J Neurosurg. 1988;68:835-853.
23. Landan I., Gilroy J., Wolfe D.E. Syringomyelia affecting the entire spinal cord secondary to primary spinal intramedullary central nervous system lymphoma. J Neurol Neurosurg Psychiatry. 1987;50:1533-1535.
24. Kawasaki K., Wakabayashi K., Koizumi T., et al. Spinal cord involvement of primary central nervous system lymphomas: histopathological examination of 14 autopsy cases. Neuropathology. 2002;22:13-18.
25. Quintero Wolfe S., Hood B., Barker J., Benveniste R.J. Primary central nervous system lymphoma mimicking pituitary apoplexy: case report. Pituitary. 2009;12:76-79.
26. Bakshi R., Mazziotta J.C., Mischel P.S., et al. Lymphomatosis cerebri presenting as a rapidly progressive dementia: clinical, neuroimaging and pathologic findings. Dement Geriatr Cogn Disord. 1999;10:152-157.
27. Kanai R., Shibuya M., Hata T., et al. A case of “lymphomatosis cerebri” diagnosed in an early phase and treated by whole brain radiation: case report and literature review. J Neurooncol. 2008;86:83-88.
28. Lewerenz J., Ding X., Matschke J., et al. Dementia and leukoencephalopathy due to lymphomatosis cerebri. J Neurol Neurosurg Psychiatry. 2007;78:777-778.
29. Rollins K.E., Kleinschmidt-DeMasters B.K., Corboy J.R., Damek D.M., Filley C.M. Lymphomatosis cerebri as a cause of white matter dementia. Hum Pathol. 2005;36:282-290.
30. Sugie M., Ishihara K., Kato H., et al. Primary central nervous system lymphoma initially mimicking lymphomatosis cerebri: an autopsy case report. Neuropathology. 2009.
31. Bataille B., Delwail V., Menet E., et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg. 2000;92:261-266.
32. Coulon A., Lafitte F., Hoang-Xuan K., et al. Radiographic findings in 37 cases of primary CNS lymphoma in immunocompetent patients. Eur Radiol. 2002;12:329-340.
33. Thurnher M.M., Rieger A., Kleibl-Popov C., et al. Primary central nervous system lymphoma in AIDS: a wider spectrum of CT and MRI findings. Neuroradiology. 2001;43:29-35.
34. Harting I., Hartmann M., Jost G., et al. Differentiating primary central nervous system lymphoma from glioma in humans using localised proton magnetic resonance spectroscopy. Neurosci Lett. 2003;342:163-166.
35. Liao W., Liu Y., Wang X., et al. Differentiation of primary central nervous system lymphoma and high-grade glioma with dynamic susceptibility contrast-enhanced perfusion magnetic resonance imaging. Acta Radiol. 2009;50:217-225.
36. Toh C.H., Castillo M., Wong A.M., et al. Primary cerebral lymphoma and glioblastoma multiforme: differences in diffusion characteristics evaluated with diffusion tensor imaging. AJNR Am J Neuroradiol. 2008;29:471-475.
37. Schroeder P.C., Post M.J., Oschatz E., et al. Analysis of the utility of diffusion-weighted MRI and apparent diffusion coefficient values in distinguishing central nervous system toxoplasmosis from lymphoma. Neuroradiology. 2006;48:715-720.
38. Shinoda J., Yano H., Murase S., et al. High 123I-IMP retention on SPECT image in primary central nervous system lymphoma. J Neurooncol. 2003;61:261-265.
39. Lorberboym M., Estok L., Machac J., et al. Rapid differential diagnosis of cerebral toxoplasmosis and primary central nervous system lymphoma by thallium-201 SPECT. J Nucl Med. 1996;37:1150-1154.
40. Lai R., Rosenblum M.K., DeAngelis L.M. Primary CNS lymphoma: a whole-brain disease? Neurology. 2002;59:1557-1562.
41. Behin A., Hoang-Xuan K., Carpentier A.F., Delattre J.Y. Primary brain tumours in adults. Lancet. 2003;361:323-331.
42. Verbraeken H.E., Hanssens M., Priem H., Lafaut B.A., De Laey J.J. Ocular non-Hodgkin’s lymphoma: a clinical study of nine cases. Br J Ophthalmol. 1997;81:31-36.
43. Merchant A., Foster C.S. Primary intraocular lymphoma. Int Ophthalmol Clin. 1997;37:101-115.
44. Gleissner B., Siehl J., Korfel A., et al. CSF evaluation in primary CNS lymphoma patients by PCR of the CDR III IgH genes. Neurology. 2002;58:390-396.
45. Nasir S., DeAngelis L.M. Update on the management of primary CNS lymphoma. Oncology (Williston Park). 2000;14:228-234. discussion 37-42, 44
46. Plasswilm L., Herrlinger U., Korfel A., et al. Primary central nervous system (CNS) lymphoma in immunocompetent patients. Ann Hematol. 2002;81:415-423.
47. Porter A.B., Giannini C., Kaufmann T., et al. Primary central nervous system lymphoma can be histologically diagnosed after previous corticosteroid use: a pilot study to determine whether corticosteroids prevent the diagnosis of primary central nervous system lymphoma. Ann Neurol. 2008;63:662-667.
48. Herrlinger U. Primary CNS lymphoma: findings outside the brain. J Neurooncol. 1999;43:227-230.
49. Abrey L.E., Batchelor T.T., Ferreri A.J., et al. Report of an international workshop to standardize baseline evaluation and response criteria for primary CNS lymphoma. J Clin Oncol. 2005;23:5034-5043.
50. Mohile N.A., Deangelis L.M., Abrey L.E. The utility of body FDG PET in staging primary central nervous system lymphoma. Neuro Oncol. 2008;10:223-228.
51. Karantanis D., O’Eill B.P., Subramaniam R.M., et al. 18F-FDG PET/CT in primary central nervous system lymphoma in HIV-negative patients. Nucl Med Commun. 2007;28:834-841.
52. Cingolani A., De Luca A., Larocca L.M., et al. Minimally invasive diagnosis of acquired immunodeficiency syndrome-related primary central nervous system lymphoma. J Natl Cancer Inst. 1998;90:364-369.
53. Antinori A., De Rossi G., Ammassari A., et al. Value of combined approach with thallium-201 single-photon emission computed tomography and Epstein-Barr virus DNA polymerase chain reaction in CSF for the diagnosis of AIDS-related primary CNS lymphoma. J Clin Oncol. 1999;17:554-560.
54. Ivers L.C., Kim A.Y., Sax P.E. Predictive value of polymerase chain reaction of cerebrospinal fluid for detection of Epstein-Barr virus to establish the diagnosis of HIV-related primary central nervous system lymphoma. Clin Infect Dis. 2004;38:1629-1632.
55. Skolasky R.L., Dal Pan G.J., Olivi A., et al. HIV-associated primary CNS morbidity and utility of brain biopsy. J Neurol Sci. 1999;163:32-38.
56. Bower M., Fife K., Sullivan A., et al. Treatment outcome in presumed and confirmed AIDS-related primary cerebral lymphoma. Eur J Cancer. 1999;35:601-604.
57. Grant J.W., Isaacson P.G. Primary central nervous system lymphoma. Brain Pathol. 1992;2:97-109.
58. DeAngelis L.M. Primary central nervous system lymphoma imitates multiple sclerosis. J Neurooncol. 1990;9:177-181.
59. Weller M. Glucocorticoid treatment of primary CNS lymphoma. J Neurooncol. 1999;43:237-239.
60. Ferreri A.J., Abrey L.E., Blay J.Y., et al. Summary statement on primary central nervous system lymphomas from the Eighth International Conference on Malignant Lymphoma, Lugano, Switzerland, June 12 to 15, 2002. J Clin Oncol. 2003;21:2407-2414.
61. Herrlinger U., Kuker W., Platten M., et al. First-line therapy with temozolomide induces regression of primary CNS lymphoma. Neurology. 2002;58:1573-1574.
62. Nelson D.F. Radiotherapy in the treatment of primary central nervous system lymphoma (PCNSL). J Neurooncol. 1999;43:241-247.
63. Gavrilovic I.T., Hormigo A., Yahalom J., et al. Long-term follow-up of high-dose methotrexate-based therapy with and without whole brain irradiation for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2006;24:4570-4574.
64. Ekenel M., Iwamoto F.M., Ben-Porat L.S., et al. Primary central nervous system lymphoma: the role of consolidation treatment after a complete response to high-dose methotrexate-based chemotherapy. Cancer. 2008;113:1025-1031.
65. Hottinger A.F., DeAngelis L.M., Yahalom J., Abrey L.E. Salvage whole brain radiotherapy for recurrent or refractory primary CNS lymphoma. Neurology. 2007;69:1178-1182.
66. DeAngelis L.M. Primary central nervous system lymphoma: a curable brain tumor. J Clin Oncol. 2003;21:4471-4473.
67. Shah G.D., Yahalom J., Correa D.D., et al. Combined immunochemotherapy with reduced whole-brain radiotherapy for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2007;25:4730-4735.
68. Dong Y., Pan L., Wang B., et al. Stereotactic radiosurgery in the treatment of primary central nervous system lymphoma. Chin Med J (Engl). 2003;116:1166-1170.
69. Kenai H., Yamashita M., Nakamura T., et al. Gamma knife surgery for primary central nervous system lymphoma: usefulness as palliative local tumor control. J Neurosurg. 2006;105(suppl):133-138.
70. Blay J.Y., Conroy T., Chevreau C., et al. High-dose methotrexate for the treatment of primary cerebral lymphomas: analysis of survival and late neurologic toxicity in a retrospective series. J Clin Oncol. 1998;16:864-871.
71. Omuro A.M., DeAngelis L.M., Yahalom J., Abrey L.E. Chemoradiotherapy for primary CNS lymphoma: an intent-to-treat analysis with complete follow-up. Neurology. 2005;64:69-74.
72. Ferreri A.J., Reni M., Dell’Oro S., et al. Combined treatment with high-dose methotrexate, vincristine and procarbazine, without intrathecal chemotherapy, followed by consolidation radiotherapy for primary central nervous system lymphoma in immunocompetent patients. Oncology. 2001;60:134-140.
73. Illerhaus G., Marks R., Muller F., et al. High-dose methotrexate combined with procarbazine and CCNU for primary CNS lymphoma in the elderly: results of a prospective pilot and phase II study. Ann Oncol. 2009;20:319-325.
74. Omuro A.M. Is single-agent temozolomide the treatment of choice for recurrent primary central nervous system lymphoma? Nat Clin Pract Oncol. 2007;4:514-515.
75. O’Brien P., Roos D., Pratt G., et al. Phase II multicenter study of brief single-agent methotrexate followed by irradiation in primary CNS lymphoma. J Clin Oncol. 2000;18:519-526.
76. Abrey L.E., Yahalom J., DeAngelis L.M. Treatment for primary CNS lymphoma: the next step. J Clin Oncol. 2000;18:3144-3150.
77. DeAngelis L.M., Seiferheld W., Schold S.C., et al. Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: radiation Therapy Oncology Group Study 93-10. J Clin Oncol. 2002;20:4643-4648.
78. Schlegel U., Pels H., Glasmacher A., et al. Combined systemic and intraventricular chemotherapy in primary CNS lymphoma: a pilot study. J Neurol Neurosurg Psychiatry. 2001;71:118-122.
79. Hoang-Xuan K., Taillandier L., Chinot O., et al. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: a multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21:2726-2731.
80. Batchelor T., Carson K., O’Neill A., et al. Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07. J Clin Oncol. 2003;21:1044-1049.
81. Poortmans P.M., Kluin-Nelemans H.C., Haaxma-Reiche H., et al. High-dose methotrexate-based chemotherapy followed by consolidating radiotherapy in non–AIDS-related primary central nervous system lymphoma: european Organization for Research and Treatment of Cancer Lymphoma Group Phase II Trial 20962. J Clin Oncol. 2003;21:4483-4488.
82. Pels H., Schmidt-Wolf I.G., Glasmacher A., et al. Primary central nervous system lymphoma: results of a pilot and phase II study of systemic and intraventricular chemotherapy with deferred radiotherapy. J Clin Oncol. 2003;21:4489-4495.
83. Khan R.B., Shi W., Thaler H.T., et al. Is intrathecal methotrexate necessary in the treatment of primary CNS lymphoma? J Neurooncol. 2002;58:175-178.
84. Pels H., Juergens A., Glasmacher A., et al. Early relapses in primary CNS lymphoma after response to polychemotherapy without intraventricular treatment: results of a phase II study. J Neurooncol. 2009;91:299-305.
85. Plotkin S.R., Batchelor T.T. Advances in the therapy of primary central nervous system lymphoma. Clin Lymphoma. 2001;1:263-275. discussion 76–77
86. Rubenstein J.L., Combs D., Rosenberg J., et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood. 2003;101:466-468.
87. Rubenstein J.L., Fridlyand J., Abrey L., et al. Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol. 2007;25:1350-1356.
88. Schulz H., Pels H., Schmidt-Wolf I., et al. Intraventricular treatment of relapsed central nervous system lymphoma with the anti-CD20 antibody rituximab. Haematologica. 2004;89:753-754.
89. Siegal T., Zylber-Katz E. Strategies for increasing drug delivery to the brain: focus on brain lymphoma. Clin Pharmacokinet. 2002;41:171-186.
90. Angelov L., Doolittle N.D., Kraemer D.F., et al. Blood-brain barrier disruption and intra-arterial methotrexate-based therapy for newly diagnosed primary CNS lymphoma: a multi-institutional experience. J Clin Oncol. 2009;27:3503-3509.
91. Perry A.R., Goldstone A.H. High-dose therapy for diffuse large-cell lymphoma in first remission. Ann Oncol. 1998;9(suppl 1):S9-S14.
92. Shipp M.A., Abeloff M.D., Antman K.H., et al. International Consensus Conference on High-Dose Therapy with Hematopoietic Stem Cell Transplantation in Aggressive Non-Hodgkin’s Lymphomas: report of the jury. J Clin Oncol. 1999;17:423-429.
93. Chang C.C., Kampalath B., Schultz C., et al. Expression of p53, c-Myc, or Bcl-6 suggests a poor prognosis in primary central nervous system diffuse large B-cell lymphoma among immunocompetent individuals. Arch Pathol Lab Med. 2003;127:208-212.
94. Soussain C., Suzan F., Hoang-Xuan K., et al. Results of intensive chemotherapy followed by hematopoietic stem-cell rescue in 22 patients with refractory or recurrent primary CNS lymphoma or intraocular lymphoma. J Clin Oncol. 2001;19:742-749.
95. Abrey L.E., Moskowitz C.H., Mason W.P., et al. Intensive methotrexate and cytarabine followed by high-dose chemotherapy with autologous stem-cell rescue in patients with newly diagnosed primary CNS lymphoma: an intent-to-treat analysis. J Clin Oncol. 2003;21:4151-4156.
96. Illerhaus G., Marks R., Ihorst G., et al. High-dose chemotherapy with autologous stem-cell transplantation and hyperfractionated radiotherapy as first-line treatment of primary CNS lymphoma. J Clin Oncol. 2006;24:3865-3870.
97. Illerhaus G., Muller F., Feuerhake F., et al. High-dose chemotherapy and autologous stem-cell transplantation without consolidating radiotherapy as first-line treatment for primary lymphoma of the central nervous system. Haematologica. 2008;93:147-148.
98. Colombat P., Lemevel A., Bertrand P., et al. High-dose chemotherapy with autologous stem cell transplantation as first-line therapy for primary CNS lymphoma in patients younger than 60 years: a multicenter phase II study of the GOELAMS group. Bone Marrow Transplant. 2006;38:417-420.
99. Makino K., Nakamura H., Kino T., et al. Rising incidence of primary central nervous system lymphoma in Kumamoto, Japan. Surg Neurol. 2006;66:503-506.
100. Montemurro M., Kiefer T., Schuler F., et al. Primary central nervous system lymphoma treated with high-dose methotrexate, high-dose busulfan/thiotepa, autologous stem-cell transplantation and response-adapted whole-brain radiotherapy: results of the multicenter Ostdeutsche Studiengruppe Hamato-Onkologie OSHO-53 phase II study. Ann Oncol. 2007;18:665-671.
101. Grimm S.A., Pulido J.S., Jahnke K., et al. Primary intraocular lymphoma: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann Oncol. 2007;18:1851-1855.
102. Frenkel S., Hendler K., Siegal T., et al. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Br J Ophthalmol. 2008;92:383-388.
103. de Smet M.D., Vancs V.S., Kohler D., et al. Intravitreal chemotherapy for the treatment of recurrent intraocular lymphoma. Br J Ophthalmol. 1999;83:448-451.
104. Fishburne B.C., Wilson D.J., Rosenbaum J.T., Neuwelt E.A. Intravitreal methotrexate as an adjunctive treatment of intraocular lymphoma. Arch Ophthalmol. 1997;115:1152-1156.
105. Chamberlain M.C., Kormanik P.A. AIDS-related central nervous system lymphomas. J Neurooncol. 1999;43:269-276.
106. Kreisl T.N., Panageas K.S., Elkin E.B., et al. Treatment patterns and prognosis in patients with human immunodeficiency virus and primary central system lymphoma. Leuk Lymphoma. 2008;49:1710-1716.
107. Slobod K.S., Taylor G.H., Sandlund J.T., et al. Epstein-Barr virus-targeted therapy for AIDS-related primary lymphoma of the central nervous system. Lancet. 2000;356:1493-1494.
108. Taiwo B.O. AIDS-related primary CNS lymphoma: a brief review. AIDS Read. 2000;10:486-491.
109. Soussain C., Hoang-Xuan K., Taillandier L., et al. Intensive chemotherapy followed by hematopoietic stem-cell rescue for refractory and recurrent primary CNS and intraocular lymphoma: societe Francaise de Greffe de Moelle Osseuse-Therapie Cellulaire. J Clin Oncol. 2008;26:2512-2518.
110. Herrlinger U., Brugger W., Bamberg M., et al. PCV salvage chemotherapy for recurrent primary CNS lymphoma. Neurology. 2000;54:1707-1708.
111. Doolittle N.D., Jahnke K., Belanger R., et al. Potential of chemo-immunotherapy and radioimmunotherapy in relapsed primary central nervous system (CNS) lymphoma. Leuk Lymphoma. 2007;48:1712-1720.
112. Jahnke K., Thiel E., Martus P., et al. Relapse of primary central nervous system lymphoma: clinical features, outcome and prognostic factors. J Neurooncol. 2006;80:159-165.
113. Voloschin A.D., Betensky R., Wen P.Y., et al. Topotecan as salvage therapy for relapsed or refractory primary central nervous system lymphoma. J Neurooncol. 2008;86:211-215.
114. Reni M., Zaja F., Mason W., et al. Temozolomide as salvage treatment in primary brain lymphomas. Br J Cancer. 2007;96:864-867.
115. Enting R.H., Demopoulos A., DeAngelis L.M., Abrey L.E. Salvage therapy for primary CNS lymphoma with a combination of rituximab and temozolomide. Neurology. 2004;63:901-903.
116. Corry J., Smith J.G., Wirth A., et al. Primary central nervous system lymphoma: age and performance status are more important than treatment modality. Int J Radiat Oncol Biol Phys. 1998;41:615-620.
117. Ferreri A.J., Blay J.Y., Reni M., et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol. 2003;21:266-272.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 12 Management of Primary Central Nervous System Lymphomas
Pathogenesis and Molecular Pathology
PCNSLs are almost exclusively of B-cell origin with only 2% of T-cell origin. The most common histologic subtype is the CD20-positive, diffuse, large-cell, B-cell, non-Hodgkin’s lymphoma (NHL), with a smattering of other more indolent B-cell lymphomas reported. The disease is more common in the immunocompromised than in the immunocompetent, but the pathogenesis of these disorders is uncertain regardless of the immunocompetency of the patient.1,2
Normal B cells arise from the hematopoietic stem cell and initially undergo antigen-independent differentiation, with immunoglobulin rearrangement in the bone marrow prior to emerging from the marrow as mature but naive B cells. These B cells move to secondary lymphoid organs where, upon encountering antigen, they undergo somatic hypermutation of the immunoglobulin variable region in the germinal center microenvironment. The presence of T cells, antigen presenting cells and the appropriate cytokine milieu are generally considered a requirement for somatic hypermutation with subsequent affinity maturation.3 B cells displaying the highest affinity for antigen are rescued from apoptosis and become either a memory cell or the terminally differentiated plasma cell.
Malignant B cells can be viewed as B cells arrested at a certain stage of differentiation. The developmental state of the cell will be reflected in its morphologic attributes, the degree of immunoglobulin rearrangement, the expression of surface molecules including CD10, BCL-6, MUM-1, and CD138 (which serve as markers of the B cell’s transition through the germinal center), and the presence of intraclonal heterogeneity. In the majority of PCNSLs, the malignant cells are BCL-6 positive (60%–100%), MUM-1 positive (90%–100%), and have undergone immunoglobulin rearrangement with somatic hypermutation. These findings suggest that the malignant cell has seen antigen, passed through the germinal center but has not yet become a plasma B cell, that is, the cell of origin in most PCNSLs is a B cell on the verge of exiting the germinal center (Fig. 12-1).4–6 Because there are no germinal centers in the brain, the B cell has, in all likelihood, migrated from a node to the CNS, probably in response to an antigen.7 It is unclear, however, whether the malignant transformation of B cell occurs prior to or following migration into the CNS.
A rat model of the disease, developed by Knopf and colleagues,8 suggests that PCNSL arises in response to an antigenic stimulus, an infection perhaps, where the antigen has moved into a draining lymph node and serves to recruit naive B cells. Presumably, antigen retained in the CNS as well as the expression of specific chemokines prompts trafficking of B cells back into the CNS. Although this hypothetical scenario is compatible with the pathologic stage of development and differentiation of the malignant cell, several questions remain unanswered, including identification of the site of malignant transformation as mentioned above, the complete lack of involved lymph nodes, and the identification of the intracerebral antigen driving the process.
Certainly, there is abundant data that Epstein-Barr virus (EBV) is involved in the pathogenesis of PCNSL in the HIV-positive patient. There are, however, equally compelling data that EBV positivity is a rarity in PCNSL occurring in the immunocompetent individual.3,9,10 Thus, it is quite possible that the underlying pathogenesis differs, depending on the immunocompetency of the patient.
Epidemiology
PCNSL was considered a rare tumor occurring in a few immunosuppressed organ transplant recipients until the early 1980s when, coinciding with the AIDS epidemic, there was a marked increase in its frequency. The increase in incidence was seen in all age groups but was more evident in men than in women.11 At its peak, prior to the highly active antiretroviral therapy (HAART) era, the relative-risk in HIV-infected individuals was 1000- to 3600-fold higher than in immunocompetent individuals; this has declined dramatically, essentially by an order of magnitude, since the introduction of HAART.12,13 However, a definite and persistent rise in incidence in the immunocompetent population has been observed.14,15 Patients without immune suppression are usually older, and the male-to-female ratio is 1.2 to 1.7:1.11 Most studies corroborate that this change in the incidence of PCNSL is independent of trends in the incidence of brain tumors and in NHL.16
Clinical Manifestations
The clinical effects of PCNSL are indistinguishable from those associated with other brain tumors. In addition to the routine medical history, special care should be taken to elicit information about the possibility of immune suppression, especially HIV infection. PCNSL usually occurs several years after the diagnosis of HIV infection has been made.17 Because PCNSL only occurs in about 3% of all AIDS patients,18 infections like toxoplasmosis are a more likely diagnosis in this setting. It should be noted, however, that as a cause of intracranial mass lesions in an individual with AIDS, PCNSL is second only to toxoplasmosis.12
Approximately 8% of immunocompetent patients will have a history of successful treatment of a non–nervous system malignancy prior to the diagnosis of PCNSL.19 In these cases, the diagnosis is even more challenging and might be delayed because of the concern of secondary nervous system involvement from the previous malignancy. When the previous malignancy is an extraneural NHL, absence of systemic disease on diagnostic workup and comparison of gene rearrangement studies on the biopsy specimens of both lymphomas can demonstrate that they are separate entities. Whether these patients have an increased predisposition to multiple neoplastic processes, or the PCNSL is the result of the antineoplastic treatment for the first tumor, is unknown.19
The relative frequency of clinical manifestations of PCNSL does not differ greatly between immunosuppressed and immunocompetent individuals. Nevertheless, there are some differences that might be of clinical relevance when considering the diagnosis. In immunocompetent patients, the median age at presentation is in the sixth decade of life, whereas the median age for AIDS patients is in the fourth decade of life. Patients with AIDS more often have multiple lesions than do immunocompetent individuals, making the clinical topographic diagnosis difficult, and the latency between the onset of symptoms and diagnosis seems to be shorter.18
The clinical course is usually subacute, with a few months elapsing between the onset of symptoms and the diagnosis of a mass lesion by imaging studies. There are several reports of spontaneous transient remission of symptoms associated with PCNSL.20 The most common symptoms associated with PCNSL are focal neurologic deficit, increased intracranial pressure, alteration of mental function, or a combination of these manifestations. At the time of initial presentation, approximately one third will have symptoms of increased intracranial pressure, about 50% will have behavioral changes, and approximately 10% will have seizures.18 Between 30% and 42% of patients will experience a combination of focal and nonfocal symptoms at the time of diagnosis. The neurologic examination will yield a variety of signs that can be localizing or nonlocalizing (e.g., increased intracranial pressure or alteration in the mental function). Hemiparesis and ataxia are the most common focal neurologic signs, but aphasia, acalculia, visual field defects, and cranial nerve palsies are also common.18 Cranial-spinal nerve palsies and hydrocephalus might be secondary to lymphomatous meningeal infiltration, which is present in up to 42% of all patients with PCNSL.18 Visual symptoms might precede or follow the diagnosis of PCNSL and will depend on the ocular structure affected by the tumor. However, 50% of the patients with PCNSL and ocular involvement detected by slit-lamp examination are asymptomatic.21 Clinically apparent ocular involvement at presentation is found in about 8% to 10% of PCNSL patients,18 but vitreous involvement of the eye occurring prior to or during the course of CNS lymphoma has been noted in up to 25% of patients.22 In about half of the cases with ocular involvement, visual symptoms can be the first manifestation of PCNSL, preceding neurologic symptoms by several months. Decreased visual acuity or floaters may prompt the patient to seek medical attention, and any nonspecific uveitis refractory to topical or systemic steroids should bring ocular PCNSL to mind.21
Rare clinical syndromes that sometimes are associated with PCNSL include those where the tumor location is restricted to the spinal cord, the leptomeninges, and the hypothalamus. These are especially challenging cases because, in addition to other neoplastic diseases, benign inflammatory entities can have a similar clinical and radiologic presentation. Isolated spinal cord involvement occurs in 1% to 2% of all PCNSL18 and can be associated with syringomyelia.23 The level and extent of myelopathic involvement will guide the clinical presentation. Secondary involvement of the spinal cord in patients with cerebral lesions is not a rare occurrence.24 PCNSL may also present as hypothalamic dysfunction causing diabetes insipidus,18 as pituitary apoplexy with bitemporal hemianopsia or as isolated third nerve palsy.25
A variant of PCNSL, clinically presenting with progressive cognitive decline and gait disorder and associated with diffuse white matter abnormality without enhancement on MRI was initially described in 1999.26 The term “lymphomatosis cerebri” has been used to describe this uncommon condition, which can be difficult to diagnose because of the nonspecific clinical and imaging findings;26–30 it can be erroneously diagnosed as vascular leukoencephalopathy, multiple sclerosis (MS), or gliomatosis cerebri.
Diagnosis
A definitive diagnosis of PCNSL cannot be made on clinical or imaging grounds, and histologic confirmation is essential. CT scan or MRI will initially establish the presence of a mass lesion with characteristics that, although suggestive of PCNSL, are not specific for this entity (Fig. 12-2). CSF analysis helps in the differential diagnosis and in some cases makes the diagnosis by the demonstration of malignant B lymphocytes.18 HIV testing is required in all patients suspected of having PCNSL. In spite of the information obtained from these studies, tissue diagnosis is required in most circumstances.
Imaging Studies
There are some characteristics on imaging studies that, although not pathognomonic, would strongly suggest that the mass lesion identified might be of lymphomatous origin. In the immunocompetent host a higher level of suspicion is required. Head CT scan shows a hyperdense or isodense mass, solitary in 86% of the cases,31 that usually exhibits homogenous enhancement after the administration of iodinated contrast.32 Lesions are usually supratentorial and localized in the deep periventricular areas.31 Because of their infiltrative nature, the lesions might have indistinct borders and result in minimal surrounding edema or compressive effect (see Fig. 12-2). Occasionally it can appear as a ring-enhancing lesion with a hypodense, necrotic core, indistinguishable from a high-grade glioma. Lesions can be multiple, suggestive of metastatic disease or infection, but the scarce perilesional edema should raise the suspicion of PCNSL. In about 10% of the cases the lesions are localized in the posterior fossa.
On MRI, most of the lesions are hypointense or isointense on T1-weighted images, and only about 40% are hyperintense on T2-weighted images.32 Although most lesions enhance after the administration of gadolinium exceptions include cases when the study follows the administration of steroids or when there is a diffuse infiltrating lymphoma (lymphomatosis cerebri) (Fig. 12-3). The appearance of PCNSL is of such heterogeneity that it should be considered in the differential diagnosis of any mass lesions detected on imaging studies (Fig. 12-4).
PCNSL in the immunocompromised patient may have a more variable appearance on imaging studies.33 Infection is more likely to be responsible for a mass or enhancing lesion on imaging studies than PCNSL in this population, but no specific pattern has been established that can be used to distinguish between CNS lymphoma, toxoplasmosis, or other CNS diseases that occur in patients with AIDS.18 Unlike immunocompetent patients, AIDS-associated PCNSL has been reported to present with multiple lesions in 71% to 80% of cases, to show ring-like enhancement in 50% of cases, and to lack enhancement in about 10% to 27% of the lesions.33 Spontaneous hemorrhage, a nonenhancing lesion, or diffuse white matter changes do not exclude lymphoma in an immunocompromised patient.
Advanced MRI techniques may be helpful to pre-operatively suggest PCNSL. Magnetic resonance spectroscopy (MRS) shows massively elevated lipid resonances in PCNSL; although this may also be present in glioblastoma, the finding of elevated lipid resonances in combination with a markedly elevated choline/creatine ratio, may improve the preoperative differentiation of PCNSL and glioma.34 Dynamic susceptibility contrast-enhanced perfusion MRI reveals that the relative cerebral blood volume ratio is lower for PCNSL, correlating with the lower microvessel density by immunohistochemistry, than high grade gliomas.35 Using diffusion tensor imaging, with the enhancing lesion as the region of interest and comparing to the contralateral normal-appearing white matter, the fractional anisotropy and the apparent diffusion coefficient (ADC) ratios can be measured. Both parameters are significantly lower in lymphomas than glioblastoma and may assist in differentiating between these two entities.36 The ADC ratios, however, do not allow a reliable distinction between toxoplasmosis and lymphoma.37
Nuclear medicine has also been suggested as a possible method to discriminate between PCNSL and other types of malignant as well as nonmalignant pathologies without resorting to histologic diagnosis. In a study comparing N-isopropyl-p-(123I)-iodoamphetamine (123I-IMP) single photon emission computerized tomography (SPECT) in patients with malignant glioma, PCNSL and meningioma, the 123I-IMP retention uptake in the 6-hour to 24-hour SPECT images were significantly higher in PCNSL than in those of both malignant gliomas and meningiomas.38 In patients with AIDS, the thallium201-SPECT delayed retention index may be useful to discriminate PCNSL from infectious lesions with high sensitivity and specificity.39 The diagnostic utility of these techniques has yet to be determined in larger series, and histologic confirmation is still considered the gold standard for diagnosis of PCNSL.
Tumor infiltration of the nervous system can be more diffuse than is appreciated on imaging studies. Correlation of autopsy and MRI findings in 10 patients who died with PCNSL showed that all had tumor infiltration in CNS regions that were normal radiographically, including T2 sequences.40 Therefore, the infiltrative microscopic tumor burden of PCNSL renders futile any attempt to resect these lesions. The surgical role is restricted to a tissue biopsy for histologic diagnosis.
Tissue Diagnosis and Staging
While the diagnosis of PCNSL is usually suggested by the appearance of a focal lesion on CT or MRI, confirmation of the diagnosis of PCNSL in the immunocompetent requires tissue to definitively differentiate PCNSL from metastatic disease, glioma, sarcoidosis, and inflammatory lesions (Table 12-1). Because the CSF is involved at diagnosis 20% of the time, a brain biopsy can be avoided if malignant cells can be obtained from the CSF.41 Similarly, malignant (though often asymptomatic) uveitis is apparent in 10% to 20% of patients with PCNSL at presentation and may serve as the source of cells on which the diagnosis may be based.42,43 Cytologic examination of cells, as the sole parameter by which the diagnosis is made, is not optimal because it cannot determine monoclonality, a condition that is necessary though not sufficient for the diagnosis of lymphoma. In the case of B-cell lymphoma, monoclonality can be determined by flow cytometric detection of light-chain restriction if there are sufficient cells available. In T-cell disease, and when there are inadequate numbers of malignant B cells for flow cytometry, the diagnosis can be established by polymerase chain reaction (PCR) studies on the basis of T-cell receptor or immunoglobulin rearrangement, respectively. This technique is susceptible to false positives if too few cells are present and to false negatives if the DNA is highly degraded.44 It is important to underscore the fact that most lymphomas, including PCNSL, are quite sensitive to steroids. Cell death and tumor regression may occur as early as 24 hours after initiation of therapy. If tissue or a specimen for cytology is obtained following initiation of steroids, the result may be nondiagnostic because of cell death and tissue necrosis. Unless the patient is showing evidence of rapid neurologic deterioration or impending herniation, tissue should be obtained prior to starting any steroid therapy.45,46 Although a retrospective study exists challenging this concept,47 it seems prudent to avoid the unnecessary use of corticosteroids prior to diagnostic biopsy for PCNSL.
| Disease | Diagnostic Studies |
|---|---|
| Multiple sclerosis | Past medical history, CSF |
| High-grade glioma | SPECT-MRS |
| Infection | HIV infection, CSF |
| Sarcoidosis | ACE level, calcium |
| Meningioma | MRS |
| Vascular | Cerebral angiogram, MRI DWIs |
ACE, angiotensin-converting enzyme; CSF, cerebrospinal fluid; DWI, diffusion-weighted images; HIV, human immunodeficiency virus; MRS, magnetic resonance spectroscopy; SPECT, single photon emission computerized tomography.
Once the diagnosis is made, patients generally undergo staging to determine the extent of involvement. Baseline evaluations include a physical and a neurologic exam, as well as cognitive function assessment. Because systemic involvement tends to occur more commonly at relapse, and because evidence of systemic disease is found in less than 5% at diagnosis, the necessity for a full lymphoma staging has been called into question.48 There is general agreement that, besides a complete history and physical, a CBC, standard chemistries with an LDH, HIV status, chest x-ray, CSF examination, and slit-lamp examination are absolutely required. A full staging would also include CT of the chest, abdomen, and pelvis, as well as bilateral bone marrow biopsies. Finally, testicular ultrasound should be considered in all elderly men. Full staging is inevitably required for any patient enrolled in a clinical trial and is recommended for all patients with PCNSL in the published guidelines for standardization of baseline evaluations.49
The role of 18F-fluorodeoxyglucose (FDG) PET to rule out systemic disease in the initial evaluation of patients presenting with PCNSL is uncertain. Two retrospective studies using PET in the initial staging of immunocompetent patients have revealed abnormalities (e.g., other malignancies, systemic sites of lymphoma as well as concurrent sites within the nervous system) undetected by other diagnostic studies in 19% and 28% of patients respectively.50,51 It remains to be determined if FDG PET is needed in all patients with the presumptive diagnosis of PCNSL (Fig. 12-5).
The approach to a focal brain lesion in the HIV population is slightly different. The detection of EBV DNA by PCR in the CSF was initially found to be a sensitive (80% to 84%) and highly specific (100%) diagnostic marker of AIDS-related PCNSL, thus potentially obviating the need for a biopsy; these results, however, could not be confirmed in a later study.52–54 A brain biopsy in the AIDS patient carries a significant morbidity (8.4%) and mortality (2.9%),55 but is recommended when the CSF is negative for lymphoma, the focal lesion is atypical for toxoplasmosis, there is progression on a brief trial of antitoxoplasmosis therapy, toxoplasma serologies are negative, there is a rapid neurologic decline, or there are discordant CSF EBV and thallium201-SPECT results.12,17 It is noteworthy that a pre-HAART retrospective study of presumed or confirmed PCNSL in HIV-positive patients revealed there was no difference in the overall survival (1.2 months) between the two groups, presumptive and biopsy-confirmed, leading the authors to conclude there may be little benefit in subjecting the patient to the diagnostic procedure given the dismal outcome.56 The use of HAART and its impact on the CD4 count has allowed the use of more aggressive chemotherapeutic regimens without undue toxicity, and resulted in improved life expectancy for these patients. These changes will undoubtedly influence the algorithm, and they suggest that earlier diagnostic procedures may be in order for this patient population.
