[level-membership-for-surgery-category]
Chapter 28 Management of Bell’s Palsy and Ramsay Hunt Syndrome
BELL’S PALSY
Bell (1774-1842)1 first described a patient with a facial paralysis in 1818; subsequently, all patients with facial palsy of unknown etiology have come to bear his name. The etiology of this “idiopathic” disorder has become much clearer in recent years. Although first proposed in 1972 by McCormick,2 herpes simplex virus (HSV) has been identified as the disease vector only recently,3–5 and an animal model has been designed.6 Murakami and associates5 identified HSV type 1 (HSV-1) DNA fragments in perineural fluid in 11 of 14 patients undergoing facial nerve decompression. In this study, no control subjects had HSV-1 DNA in perineural fluid. Using polymerase chain reaction to analyze the saliva of patients with Bell’s palsy, Furuta and colleagues3 identified HSV-1 DNA in 50% of patients, which was significantly more often than controls, a finding confirmed by other groups.7 Polymerase chain reaction has also been used to isolate HSV-1 genomic DNA from the geniculate ganglion of a temporal bone in a patient dying during the acute phase of Bell’s palsy.4
Sugita and coworkers6 proposed an animal model of Bell’s palsy. Six days after inoculation of HSV-1 into either the auricle or the tongue of mice, a temporary ipsilateral facial paralysis was identified that recovered spontaneously within 3 to 7 days. Histopathologically, neural edema, vacuolar degeneration, and inflammatory cell infiltrate with associated demyelination or axonal degeneration were shown in the affected facial nerve and nucleus.8 HSV-1 antigens were identified within the facial nerve, geniculate ganglion, and facial nucleus 6 to 20 days after inoculation.6 Similar pathologic findings have been shown in rabbits after HSV-1 inoculation, but without the associated facial paralysis.9
The histopathologic changes seen in autopsy specimens of patients who died with acute idiopathic facial paralysis have provided some insight into the underlying cellular mechanisms in Bell’s palsy. Reddy and coworkers10 found degeneration of the myelin sheath and axons, perivascular inflammation, and a phagocytic cell infiltrate in 10% to 30% of nerve fibers in a patient 17 days after the onset of an acute idiopathic facial paralysis. Fowler11 found diffuse vascular engorgement throughout the intratemporal facial nerve and evidence of acute hemorrhage within the intracanalicular, labyrinthine, and geniculate portions of the facial nerve in a patient who died shortly after the onset of Bell’s palsy. In examining an autopsy specimen of a patient who died 13 days after the onset of Bell’s palsy, McKeever and colleagues12 found a diffuse lymphocytic infiltration of the intratemporal facial nerve with ongoing myelin phagocytosis. They later re-examined the same specimen and noted the most pronounced lymphocytic infiltration of the nerve at the labyrinthine segment, findings that these authors believed were most consistent with an ongoing compression-type injury to the facial nerve.
Multiple reports have found that there seems to be evidence of constriction of the nerve at the meatal foramen in cases of Bell’s palsy. In specimens examined during the acute phase of Bell’s palsy, lymphocytic infiltration, perineural edema, and myelin degeneration have been noted.13,14 In a nerve specimen examined 1 year after the diagnosis of Bell’s Palsy lymphocytic infiltration, perineural edema, and fibrotic changes were noted.15 Intraoperative biopsy specimens of the greater superficial petrosal nerve from patients undergoing nerve decompression procedures for Bell’s palsy have shown axonal demyelination and degeneration with lymphocytic infiltration.16 Together, the histopathologic findings in Bell’s palsy show demyelination and axonal loss with lymphocytic or phagocytic infiltration, findings that seem to be more pronounced at the labyrinthine segment of the facial nerve.
In considering this evidence, the pathogenesis of Bell’s palsy becomes more apparent: a virally induced, inflammatory response that produces edema within the nerve. Fisch and Felix16 first proposed that the facial nerve was entrapped at the meatal foramen as a result of neural edema. Intraoperative conduction studies have shown an electrophysiologic blockage at this site.17 The constriction imposed produces a conduction block at first; however, with prolonged or increased constriction, ischemia results. Subsequently, wallerian degeneration occurs, producing axonotmesis or neurotmesis or both. A spectrum of injury within the nerve from neurapraxia to neurotmesis may occur in Bell’s palsy.11,18 The proportion of each of these determines the amount of facial function that returns when the acute phase of the disease subsides.
Bell’s palsy accounts for nearly three quarters of all acute facial palsies.19 The incidence of Bell’s palsy is 20 to 30 cases per 100,000 per year.20 The median age is 40 years, but it can occur at any age.21 The incidence is highest in patients older than 70 years and lowest in children younger than 10 years. The left and right sides are affected equally. Men and women are equally affected, but the incidence of Bell’s palsy is higher in pregnant women (45 cases per 100,000).
The clinical presentation of Bell’s palsy is well known; however, the clinician must exclude other causes of facial paralysis based on history and physical examination findings. Patients describe an abrupt onset of unilateral paresis that occurs over 24 to 48 hours. The paresis can progress over 1 to 7 days to complete paralysis. Bilateral involvement, either simultaneously or consecutively, has been described.23 A history of progression of weakness over weeks to months, repeated episodes of paralysis, and twitching of the facial muscles should not be considered symptoms of Bell’s palsy. Other associated symptoms of hearing loss, vestibular symptoms, or other cranial nerve neuropathies also rule out the diagnosis of Bell’s palsy.
Audiometric evaluation should reveal symmetric function except for an absent ipsilateral acoustic reflex. If unilateral hearing loss or acoustic reflex decay is present, further evaluation for retrocochlear pathology is necessary. If vestibular complaints are present, an electronystagmogram is performed, and diagnosis of Bell’s palsy should be questioned. If the history and clinical presentation are highly suggestive of Bell’s palsy, magnetic resonance imaging (MRI) and computed tomography (CT) are not performed. High-resolution CT scans and MRI are obtained if patients have associated symptoms of otorrhea, vestibular complaints, and hearing loss. Planned surgical decompression and persistent dense paralysis after 6 months are also indications for imaging. Serologic tests for Lyme disease (IgG, IgM) are an important part of the work-up for unexplained facial paralysis in endemic areas.24 Serologic testing for antibodies to HSV or varicella zoster have provided some correlative evidence for a viral etiology in Bell’s palsy, although laboratory investigations in general have not been found to provide clinically relevant data.
Electrodiagnostic testing is an important element of the diagnostic evaluation of facial paralysis. Testing can determine the extent of facial nerve injury and provide useful prognostic information for the development of management strategies. The technique of electroneuronography (ENoG), developed by Esslen,25 can distinguish the nerve fibers that have undergone wallerian degeneration from fibers that are temporarily blocked (neurapraxia). ENoG testing is not performed until 3 to 4 days after the development of complete unilateral paralysis because wallerian degeneration does not become apparent until 48 to 72 hours after an acute injury to the nerve. Electrodiagnostic testing is not performed when the patient exhibits paresis only. If the paresis progresses to total paralysis, electrodiagnostic testing is performed 3 days after the onset of total paralysis. Presence of voluntary movement 4 to 5 days after the onset of paresis indicates only minor injury, and complete recovery should be anticipated.
ENoG is most accurate when it is performed within 3 weeks of the acute injury. The test is performed using standard electromyography (EMG) equipment, but requires the use of special surface stimulating and recording electrodes.26 The recording electrodes are in a hand-held carrier and are manipulated in the nasolabial fold with the Esslen technique. The recording electrodes are not taped to the skin, as has been described by others.27
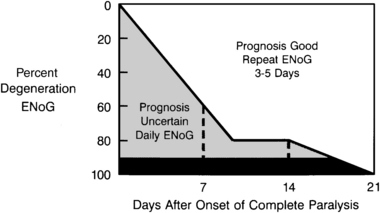
An evoked electric stimulus generates synchronous facial muscle movement that can be recorded from the skin surface (termed the compound muscle action potential [CMAP]). The amplitude of the biphasic CMAP has been found to correlate with the number of blocked or neurapraxic nerve fibers. As the percentage of degenerated fibers within the nerve increases, the amplitude of the CMAP decreases compared with the normal side of the face. Fisch and Esslen28 determined that if 90% or more of the fibers within the facial nerve degenerate within the first 14 days of an acute paralysis, a severe injury has occurred, and the chances of complete recovery are less than 50%. Patients who do not reach the 90% degeneration level by 3 weeks have a very good prognosis and are likely to regain normal facial motion without synkinesis. The time course of degeneration is also important; the more rapid the degeneration, the more severe the injury.29 A patient showing greater than 90% degeneration at 5 days would have a worse prognosis than a patient with 90% degeneration at 14 days.
Topognostic testing is widely reported to be useful in determining the site of injury in acute facial paralysis; however, intraoperative studies have shown that the Schirmer test is not accurate in diagnosing Bell’s palsy.26 The Schirmer test may be used to determine the extent of lacrimation and the need for eye protection.
A review of the natural history of Bell’s palsy shows that approximately 85% of patients begin to display some return of facial movement within 3 weeks of the onset of paresis.30 The remaining 15% begin to improve 3 to 6 months after the onset of the disease. Most patients show a complete return of facial function, but 10% to 15% have residual unilateral weakness and develop secondary deformities of synkinesis, tearing, or contracture. Some motion returns in almost all individuals with Bell’s palsy by 6 months. If no movement returns, a vigorous search for another etiology should begin anew.
The management of patients with Bell’s palsy varies, depending on the type of specialist initially seeing the patient and on the training of the individual specialist. Figure 28-1 presents an overview of our management strategy. Patients presenting within the first week of facial weakness with paresis are placed on steroid therapy (prednisone, 60 to 80 mg/day for 7 days) and an antiviral agent (valacyclovir, 500 mg three times per day for 10 days). If patients are seen 7 to 10 days after onset and motor function is stable or improving, medical treatment is unnecessary. Patients are instructed to return in 1 week for re-evaluation to determine if neural degeneration has occurred. Electrodiagnostic testing is unnecessary as long as voluntary facial movement is present. If total paralysis occurs in the interim, the total paralysis protocol of Figure 28-1 is followed. Stable or improving patients are seen in 1 month.
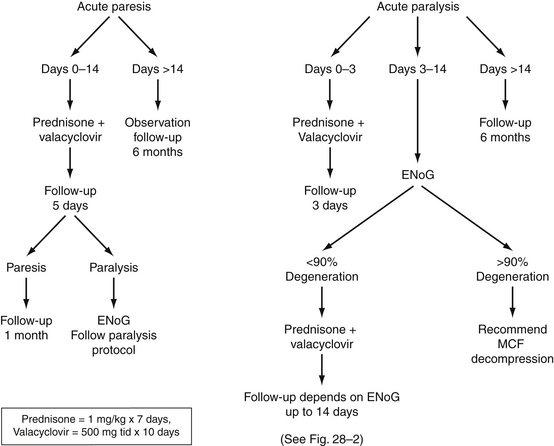
FIGURE 28-1 Algorithm for management of Bell’s palsy. ENoG, electroneuronography; MCF, middle cranial fossa.
The use of steroids in Bell’s palsy has been the subject of much debate. Numerous studies of varying designs in adults have shown better outcomes in patients treated with steroids,21,31,32 especially when initiated early in the course of the disease.33–35 Other randomized studies36 and meta-analyses,37 including many studies in children,38–40 have concluded that steroids did not affect the ultimate facial function outcomes in Bell’s palsy. Grogan and Gronseth,41 in a comprehensive, evidence-based review, concluded that there seemed to be a beneficial effect in the use of steroids in Bell’s palsy. Ramsey and coworkers42 performed a meta-analysis of 47 trials of steroid therapy for Bell’s palsy, and concluded that there seemed to be an improved odds of recovery in patients treated with steroids (49% to 97%) compared with untreated controls (23% to 64%). We use prednisone in a dose of 1 mg/kg daily for 7 days in all cases in which it is not medically contraindicated, in anticipation of speeding recovery, reducing the number of degenerating axons, and reducing the number of patients needing decompression.
The combination of steroids and antivirals may be superior to either one alone. A double-blind, randomized, controlled trial of acyclovir and prednisone versus prednisone alone in the treatment of Bell’s palsy showed better results with the combination therapy.43 This study documented poor facial function recovery in 23% of the prednisone-only group compared with 7% in the acyclovir-plus-prednisone group. Interpreting the results of this study, Grogan and Gronseth41 reported that patients who receive antiviral therapy in addition to steroids were 1.22 times as likely to have a good facial nerve outcome. A multicenter, randomized, placebo-controlled study comparing treatment of patients with Bell’s palsy with steroids and antivirals with treatment with steroids alone concluded that the addition of valacyclovir improved the recovery rate from 75% to 90% in cases of complete palsy, and from 89% to 96% in all cases of facial palsy.44 Many other reviews of the topic have made similar conclusions.45–48
Other studies have found no significant difference between this combination of drugs and the natural history of the disease.24,49 One study found a negative impact of treatment of patients with Bell’s palsy with antivirals alone versus steroids alone.50 Because of the paucity of potential side effects and good patient tolerance of antiviral medications, the addition of antiviral medications to steroids in the treatment of patients with Bell’s palsy seems prudent.
Patients presenting within 1 week of the onset of total unilateral paralysis undergo electrodiagnostic testing (if at least 3 days have passed since the onset of paralysis) and are started on medical therapy. If the patient is seen in the first 3 days after the onset of paralysis, steroid and antiviral therapy are initiated, and follow-up electrodiagnostic studies are arranged. The frequency of follow-up electrodiagnostic examinations is determined by the result of testing and the time interval after paralysis, as suggested by Fisch51 (Fig. 28-2). Patients exhibiting nearly 90% neural degeneration on ENoG examination, or who are degenerating quickly, undergo frequent electrodiagnostic testing (every 1 or 2 days). If greater than 90% degeneration is reached, and there are no motor unit potentials on voluntary EMG testing, the patient is considered a candidate for middle cranial fossa decompression. When 90% degeneration is not reached within 2 weeks (14 days) after the onset of paralysis, no further electrodiagnostic studies need to be performed.
RAMSAY HUNT SYNDROME
Herpes zoster oticus refers to a syndrome of acute otalgia accompanied by a herpetic, vesicular rash. When accompanied by facial paralysis, the syndrome is known as Ramsay Hunt syndrome.52 Ramsay Hunt syndrome is the second most common cause of facial paralysis (after Bell’s palsy), and is induced by the reactivation of the varicella-zoster virus that remains latent in the geniculate ganglion after primary infection with chickenpox.53 Classically, patients present with severe otalgia and unilateral facial paralysis. Vesicular eruptions may or may not be present initially, but usually appear within 3 to 5 days of the paralysis. The vesicular lesions can appear on the concha, ear canal, postauricular skin, and tympanic membrane. Occasionally, the oral cavity, neck, and shoulder are also involved. The disease can affect other cranial nerves, including auditory, vestibular, trigeminal, glossopharyngeal, and vagus, prompting the name herpes zoster cephalicus.54
Also in contrast to Bell’s palsy, the symptoms are more severe, and the prognosis is worse in Ramsay Hunt syndrome. The frequency of complete neural degeneration of the facial nerve is substantially higher than with Bell’s palsy, and complete recovery of facial motor function has ranged from 10% to 31% in several studies.55–57 Patients with auditory and vestibular dysfunction in addition to facial paralysis generally have a worse prognosis. In addition, patients with diabetes, hypertension, and advanced age have been reported to have a poorer prognosis in Ramsay Hunt syndrome.58
The diagnosis of Ramsay Hunt syndrome is based on the history of otalgia, vesicular lesions or eschars, and facial paralysis. MRI shows enhancement of a large portion of the facial nerve, often the vestibular and cochlear nerves, the labyrinth, and the dura lining the internal auditory canal.59 Imaging as part of routine evaluation is unnecessary. Electrodiagnostic studies have been unreliable in herpes zoster oticus.
The management of Ramsay Hunt syndrome has changed with the development of antiviral agents. The natural history of the disease was evaluated by Devriese and Moesker,56 who identified only a 10% rate of complete facial nerve recovery. Many studies have identified a superior recovery rate of 75% using a combination of steroids and antiviral agents.45,60–62 The benefit of early initiation of steroid and antiviral treatment was clearly shown in a study by Murakami and colleagues,60 in which 75% of patients treated within 3 days of the onset had complete recovery, whereas only 30% of patients treated after 7 days experienced a complete recovery. We have experienced similar successful results using intravenous acyclovir (10 mg/kg three times daily), oral acyclovir (800 mg five times daily), or oral valacyclovir (500 mg three times daily) for 10 days, in combination with a 3 week tapering course of prednisone (60 to 80 mg/kg daily). Patients report rapid reduction in pain, and occasionally experience return of facial movement during the medical therapy. Because of the presence of “skip” regions and diffuse neuritis of the facial nerve,63,64 surgical decompression of RHS is not recommended.
FACIAL NERVE DECOMPRESSION FOR BELL’S PALSY
Decompression of the facial nerve for Bell’s palsy has been reported since the 1930s,65 and is recommended in patients who exhibit greater than 90% degeneration on ENoG and no voluntary EMG activity within 14 days of the paralysis. Preliminary results suggested that early decompression of the labyrinthine, geniculate, and tympanic segments of the facial nerve through a middle cranial fossa approach improves recovery of severely degenerated cases,25 whereas decompression of the mastoid segment alone did not alter the natural history of the disease.66 Gantz and associates67 showed a statistically significant difference in facial nerve outcome when decompression is performed within 2 weeks of onset of the paralysis. The patients undergoing decompression exhibited House-Brackmann grade I (n = 14) or II (n = 17) in 91% of cases; two patients had a grade III, and one patient had a grade IV outcome. There were no grade V or VI results in the surgical group. Patients who met surgical criteria but elected not to undergo surgery had a 58% chance of a poor outcome. Within the nonsurgical group, 19 patients had a House-Brackmann grade of either III or IV at 7 month follow-up.
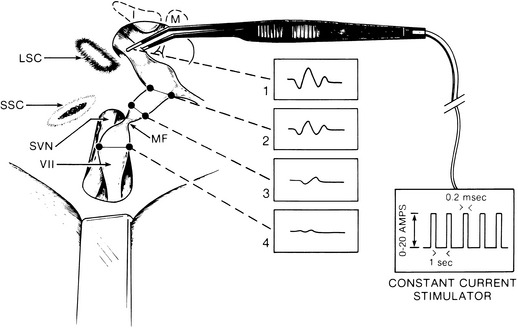
Intraoperative evoked EMG is used to localize the region of the conduction block.17 Intraoperative direct nerve stimulation is useful when the nerve is not completely degenerated, which includes most instances when preoperative ENoG reveals 100% degeneration. This finding suggests that a few nerve fibers remain capable of stimulation even when ENoG shows total nerve degeneration. In most cases, the nerve conduction block is localized between the geniculate ganglion and the internal auditory canal segments of the facial nerve. Failure to generate a motor unit potential when the tympanic segment of the nerve is stimulated indicates that the nerve is 100% degenerated, or that the conduction block is more distal. If the nerve appears normal in the tympanic portion, while there is edema and erythema of the internal auditory canal segment, total degeneration should be suspected. If erythema and edema are apparent in the tympanic segment, a mastoid decompression is added.
Preoperative Preparation
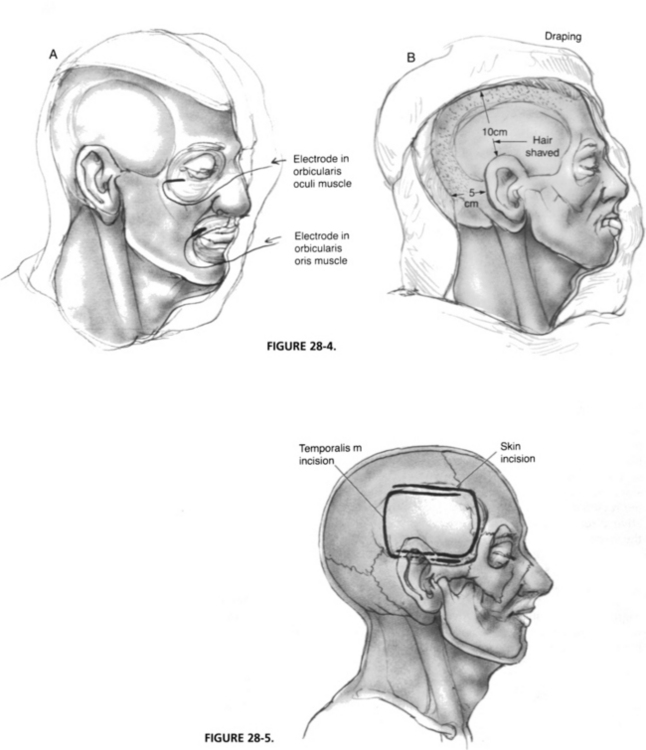
General anesthesia and transoral endotracheal intubation are accomplished; thereafter, the bed is rotated 180 degrees for access (Fig. 28-3). Paralytic agents must be reversed before the skin incision. A urinary catheter is placed to monitor fluids and diuresis. Hair is shaved approximately 10 cm above and 5 cm behind the ear. The entire side of the head and face are prepared, and EMG needles are placed in the orbicularis oculi and oris muscles (Fig. 28-4A) for facial nerve monitoring. A clear drape is placed over the prepared area to allow visualization of the entire side of the face in the case of monitoring equipment failure (Fig. 28-4B). Standard auditory brainstem–evoked recording electrodes are placed in the right and left mastoid tips and the vertex and forehead (ground), and insert headphones are placed in both external auditory canals. The auditory brainstem response is monitored throughout the procedure.
Surgical Technique
The skin incision is marked as shown in Figure 28-5 and carried down to the level of the temporalis fascia. Meanwhile, mannitol (0.5 g/kg body weight) and hyperventilation (partial pressure of carbon dioxide of 25 mm Hg) are initiated by the anesthesiologist to relax the brain. After the posteriorly based skin flap is elevated, a 4 × 6 cm piece of temporalis fascia is harvested and set aside in a moist gauze for use at the time of closure. An anteriorly based, inferiorly staggered muscle flap is elevated down to the level of the linea temporalis and reflected forward with an Adson cerebellar retractor. Staggering the incisions prevents dural exposure if wound dehiscence occurs. The zygomatic root identifies the floor of the middle cranial fossa and is the central landmark of the craniotomy. The skin and muscle flaps should be wrapped with moist sponges and secured with temporary retraction stitches.
The craniotomy should be approximately 4 cm in anteroposterior dimension and 5 cm cephalocaudal (Fig. 28-6A). The anteroposterior margins must be kept parallel for stability of the middle cranial fossa retractor. The craniotomy can be created with either cutting burrs or a craniotomy saw. The bone flap should be elevated with care by use of a blunt dural elevator. Occasionally, the middle meningeal artery is embedded within the bone, requiring bipolar coagulation to free it. The bone flap is wrapped in a moist gauze and set aside for use at closure.
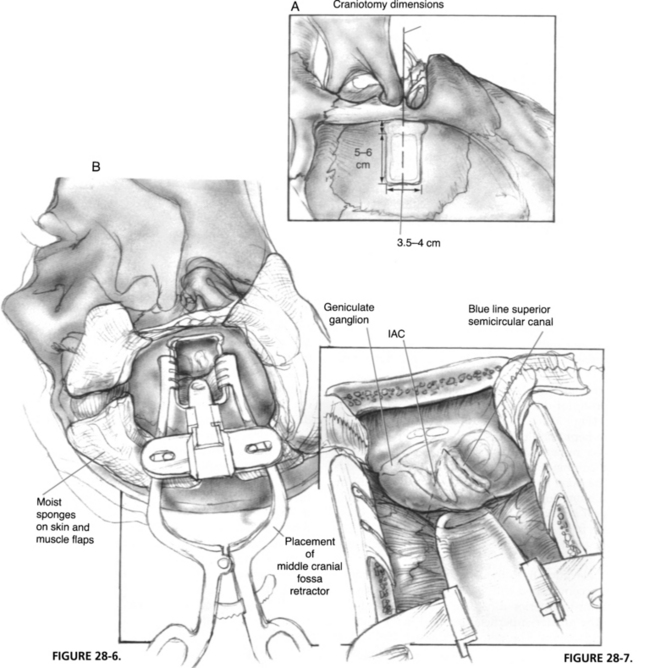
Dural elevation from the floor of the middle cranial fossa is accomplished with a Freer or joker elevator, always in a posterior-to-anterior direction, which prevents injury to the greater superficial petrosal nerve and geniculate ganglion. The petrous ridge is identified at the posterior margin of the craniotomy, and the dura is slowly elevated over the arcuate eminence and meatal plane. Dural reflections are cauterized and sharply transected to allow elevation to the anterior petrous ridge. The hiatus of the facial canal with the greater superficial petrosal nerve and artery is the anterior margin of elevation. Further anterior elevation exposes the foramen spinosum and the pterygoid plexus of veins, which can cause troublesome oozing throughout the procedure. After dural elevation, cottonoid sponges can be placed at the anterior and posterior margins of the elevation to help retract the dura during placement of the self-retaining middle cranial fossa retractor (Fig. 28-6B).
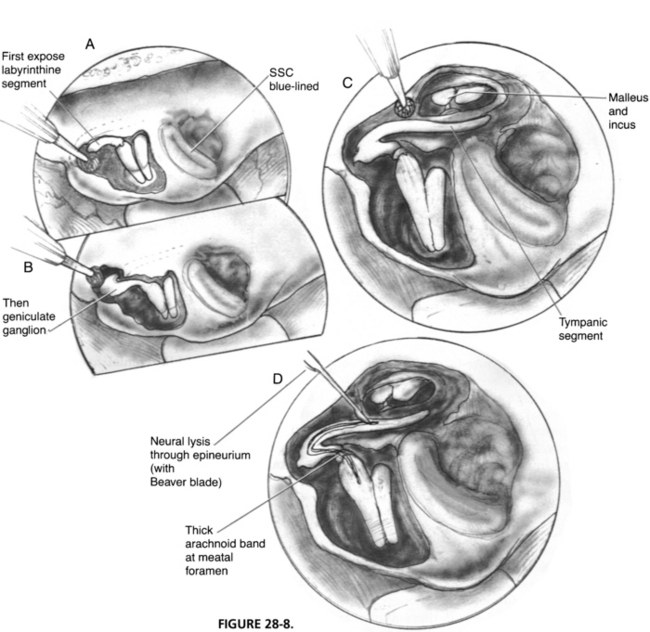
Before the bony exposure of the facial nerve, the Stenvers projection radiograph is re-examined to determine the depth of the superior semicircular canal in the temporal bone. The superior semicircular canal is the first structure to be located. When its blue line is identified, the remaining intratemporal structures have consistent anatomic locations. Landmarks of the middle cranial fossa floor can be quite subtle. The arcuate eminence may not be apparent and, in many instances, is not parallel with the superior semicircular canal. One consistent anatomic feature to remember is that the plane of the superior semicircular canal is almost always perpendicular to the petrous ridge (Fig. 28-7). If the arcuate eminence is not initially apparent, drilling is begun posterior to the semicircular canal, slowly removing the tegmen mastoideum with a moderate-sized diamond burr. The whitish color of the membranous temporal bone can be distinguished from the yellow, dense otic capsule bone of the superior canal. When the superior canal is identified, drilling in a parallel direction with the canal gradually exposes the blue line.
When the location of the superior canal is confirmed, an anterior line 60 degrees to the blue line locates the position of the internal auditory canal. The depth of the internal auditory canal varies, but drilling medially near the petrous ridge provides a safe route to the canal. Drilling laterally near the anterior ampulla of the superior semicircular canal places the geniculate ganglion, labyrinthine segment of the facial nerve, and cochlea at risk. When the blue line of the internal auditory canal is exposed, bone is removed in a lateral direction until the vertical crest is found (Fig. 28-8). Bone can now be removed over the geniculate ganglion and tegmen tympani.
Intraoperative EMG is used to localize the region of the nerve conduction block. Direct stimulation of the most distal exposed tympanic segment of the nerve is performed with monopolar or bipolar microforceps (Fig. 28-9). If the conduction block is medial to the point of stimulation, and the nerve is not completely degenerated, a motor unit potential is observed. Stimulating more medially toward the internal auditory canal fails to elicit a motor unit potential if the conduction block is at the meatal foramen.
Postoperative Care
Postoperative care includes observation in the intensive care unit overnight, limitation of fluids (1500 to 1800 mL/day), dexamethasone (6 mg every 6 hours for 36 hours), cefazolin (1 g every 8 hours for 36 hours), routine neural checks, and limitation of analgesia to codeine. There is little postoperative pain, and narcotics stronger than codeine may mask intracranial complications. The patient is transferred to a routine postoperative floor the next morning, encouraged to begin ambulation, and started on a diet as tolerated. On postoperative day 3, and daily thereafter, observations for cerebrospinal fluid rhinorrhea are made by asking the patient to lean forward with the head between the knees. If cerebrospinal fluid rhinorrhea occurs, a spinal drain must be placed for 4 to 5 days. Following this regimen, only very rarely has a patient required surgical closure of the leak. Patients are usually discharged from the hospital on postoperative days 5 to 7. No intracranial complications, including intracranial hemorrhage, aphasia, or seizures, occurred in the Iowa series.67
1. Bell C. On the nerves, giving an account of some experiments on their structure and function, which led to a new arrangement of the system. Philos Trans. 1821;111:398-424.
2. McCormick D.P. Herpes-simplex virus as a cause of Bell’s palsy. Lancet. 1972;1:937-939.
3. Furuta Y., Fukuda S., Chida E., et al. Reactivation of herpes simplex virus type 1 in patients with Bell’s palsy. J Med Virol. 1998;54:162-166.
4. Burgess R.C., Michaels L., Bale J.F.Jr., et al. Polymerase chain reaction amplification of herpes simplex viral DNA from the geniculate ganglion of a patient with Bell’s palsy. Ann Otol Rhinol Laryngol. 1994;103:775-779.
5. Murakami S., Mizobuchi M., Nakashiro Y., et al. Bell palsy and herpes simplex virus: Identification of viral DNA in endoneurial fluid and muscle. Ann Intern Med. 1996;124(1 Pt 1):27-30.
6. Sugita T., Murakami S., Yanagihara N. Facial nerve paralysis induced by herpes simplex virus in mice: An animal model of acute and transient facial paralysis. Ann Otol Rhinol Laryngol. 1995;104:574-581.
7. Lazarini P.R., Viamna M.F., Alcantara M.P.A., et al. [Herpes simplex virus in the saliva of peripheral Bell’s palsy patients]. Rev Bras Otorrinolaringol (Engl Ed). 2006;72:7-11.
8. Liston S.L., Kleid M.S. Histopathology of Bell’s palsy. Laryngoscope. 1989;99:23-26.
9. Carreno M., Llorente J.L., Hidalgo F., et al. [Application of the polymerase chain reaction to an experimental model of infection by herpes simplex virus type 1]. Acta Otorrinolaringol Esp. 1998;49:15-18.
10. Reddy J.B., Liu J., Balshi S., et al. Histopathology of Bell’s palsy. Eye Ear Nose Throat Mon. 1966;45:62-66.
11. Fowler E.P.Jr. The pathologic findings in a case of facial paralysis. Trans Am Acad Ophthalmol Otolaryngol. 1963;67:187-197.
12. McKeever P., Proctor B., Proud G. Cranial nerve lesions in Bell’s palsy. Otolaryngol Head Neck Surg. 1987;97:326-327.
13. Podvinec M. Facial nerve disorders: Anatomical, histological and clinical aspects. Adv Otorhinolaryngol. 1984;32:124-193.
14. Donoghue O. Histopathologic aspects of Bell’s palsy. Presented at American Academy of Otolaryngology–Head and Neck Surgery. Anaheim, CA: Association for Research in Otolaryngology; 1983.
15. Jackson C.G., et alJohnson G.D., Hyams V.J. Pathologic findings in the labyrinthine segment of the facial nerve in a case of facial paralysis. Ann Otol Rhinol Laryngol. 1990;99(5 Pt 1):327-329.
16. Fisch U., Felix H. On the pathogenesis of Bell’s palsy. Acta Otolaryngol. 1983;95(5-6):532-538.
17. Gantz B.J., Gmur A., Fisch U. Intraoperative evoked electromyography in Bell’s palsy. Am J Otolaryngol. 1982;3:273-278.
18. Proctor B., Corgill D.A., Proud G. The pathology of Bell’s palsy. Trans Sect Otolaryngol Am Acad Ophthalmol Otolaryngol 82:ORL70-ORL80. 1976.
19. Peitersen E. Bell’s palsy: The spontaneous course of 2,500 peripheral facial nerve palsies of different etiologies. Acta Otolaryngol Suppl. 2002;549:4-30.
20. Hauser W.A., et alKarnes W.E., Annis J. Incidence and prognosis of Bell’s palsy in the population of Rochester, Minnesota. Mayo Clin Proc. 1971;46:258-264.
21. Katusic S.K., et alBeard C.M., Wiederholt W.C. Incidence, clinical features, and prognosis in Bell’s palsy, Rochester, Minnesota, 1968-1982. Ann Neurol. 1986;20:622-627.
22. Adour K.K. Current concepts in neurology: Diagnosis and management of facial paralysis. N Engl J Med. 1982;307:348-351.
23. Cwach H., Landis J., Freeman J.W. Bilateral seventh nerve palsy: A report of two cases and a review. S D J Med. 1997;50:99-101.
24. Holland N.J., Weiner G.M. Recent developments in Bell’s palsy. BMJ. 2004;329:553-557.
25. Esslen E. Electromyography and electroneurography. In: Fisch U., editor. Facial Nerve Surgery. Birmingham, AL: Aesculapius; 1977:93-100.
26. Gantz B.J., et alGmuer A.A., Holiday M. Electroneurographic evaluation of the facial nerve: Method and technical problems. Ann Otol Rhinol Laryngol. 1984;93(4 Pt 1):394-398.
27. Blumenthal F., May M. Electrodiagnosis. In: May M., editor. The Facial Nerve. New York: Thieme, 1986.
28. Fisch U., Esslen E. Total intratemporal exposure of the facial nerve: Pathologic findings in Bell’s palsy. Arch Otolaryngol. 1972;95:335-341.
29. Fisch U. Prognostic value of electrical tests in acute facial paralysis. Am J Otol. 1984;5:494-498.
30. Peitersen E. Natural history of Bell’s palsy. Acta Otolaryngol Suppl. 1992;492:122-124.
31. Adour K.K., et alWingerd J., Bell D.N. Prednisone treatment for idiopathic facial paralysis (Bell’s palsy). N Engl J Med. 1972;287:1268-1272.
32. Wolf S.M., et alWagner J.H., Davidson S. Treatment of Bell palsy with prednisone: A prospective, randomized study. Neurology. 1978;28:158-161.
33. Williamson I.G., Whelan T.R. The clinical problem of Bell’s palsy: Is treatment with steroids effective? Br J Gen Pract. 1996;46:743-747.
34. Shafshak T.S., Essa A.Y., Bakey F.A. The possible contributing factors for the success of steroid therapy in Bell’s palsy: A clinical and electrophysiological study. J Laryngol Otol. 1994;108:940-943.
35. Brown J.S. Bell’s palsy: A 5 year review of 174 consecutive cases: An attempted double blind study. Laryngoscope. 1982;92:1369-1373.
36. May M., et alWette R., Hardin W.B.Jr. The use of steroids in Bell’s palsy: A prospective controlled study. Laryngoscope. 1976;86:1111-1122.
37. Salinas R.A., et al. Corticosteroids for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev. (1):2002. CD001942
38. Salman M.S., MacGregor D.L. Should children with Bell’s palsy be treated with corticosteroids? A systematic review. J Child Neurol. 2001;16:565-568.
39. Prescott C.A. Idiopathic facial nerve palsy in children and the effect of treatment with steroids. Int J Pediatr Otorhinolaryngol. 1987;13:257-264.
40. Unuvar E., et alOguz F., Sidal M. Corticosteroid treatment of childhood Bell’s palsy. Pediatr Neurol. 1999;21:814-816.
41. Grogan P.M., Gronseth G.S. Practice parameter: Steroids, acyclovir, and surgery for Bell’s palsy (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:830-836.
42. Ramsey M.J., et alDerSimonian R., Holter M.R. Corticosteroid treatment for idiopathic facial nerve paralysis: A meta-analysis. Laryngoscope. 2000;110(3 Pt 1):335-341.
43. Adour K.K., et alRuboyianes J.M., Von Doersteu P.G. Bell’s palsy treatment with acyclovir and prednisone compared with prednisone alone: A double-blind, randomized, controlled trial. Ann Otol Rhinol Laryngol. 1996;105:371-378.
44. Hato N., et alYamada H., Kohno H. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol. 2007;28(3):408-413.
45. Dickins J.R., Smith J.T., Graham S.S. Herpes zoster oticus: Treatment with intravenous acyclovir. Laryngoscope. 1988;98:776-779.
46. Sweeney C.J., Gilden D.H. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:149-154.
47. Morrow M.J. Bell’s palsy and herpes zoster oticus. Curr Treat Options Neurol. 2000;2:407-416.
48. Axelsson S., Lindberg S., Stjernquist-Desatnik A. Outcome of treatment with valacyclovir and prednisone in patients with Bell’s palsy. Ann Otol Rhinol Laryngol. 2003;112:197-201.
49. Ramos Macias A., de Miguel Martine Z., I Martin Sanchez A.M. Incorporacion del aciclovir en el tratamiento de la paralisis periferica: Un estudio en 45 casos. Acta Otolaryngol Espanola. 1992;43:117-120.
50. De Diego J.I., et alPrim M.P., De Sarriá M.J. Idiopathic facial paralysis: A randomized, prospective, and controlled study using single-dose prednisone versus acyclovir three times daily. Laryngoscope. 1998;108(4 Pt 1):573-575.
51. Fisch U. Surgery for Bell’s palsy. Arch Otolaryngol. 1981;107:1-11.
52. Sachs E.Jr., House R.K. The Ramsay Hunt syndrome, geniculate herpes. Neurology. 1956;6:262-268.
53. Ikeda M., et alHiroshige K., Abiko Y. Impaired specific cellular immunity to the varicella-zoster virus in patients with herpes zoster oticus. J Laryngol Otol. 1996;110:918-921.
54. Adour K.K. Otological complications of herpes zoster. Ann Neurol. 1994;35(Suppl):S62-S64.
55. Devriese P. Herpes zoster causing facial paralysis. In: Fisch U., editor. Facial Nerve Surgery. Birmingham, AL: Aesculapius; 1977:419-420.
56. Devriese P.P., Moesker W.H. The natural history of facial paralysis in herpes zoster. Clin Otolaryngol Allied Sci. 1988;13:289-298.
57. Peitersen E. Spontaneous course of Bell’s palsy. In: Fisch U., editor. Facial Nerve Surgery. Birmingham, AL: Aesculapius; 1977:337-343.
58. Yeo S.W., et alLee D.H., Jun B.C. Analysis of prognostic factors in Bell’s palsy and Ramsay Hunt syndrome. Auris Nasus Larynx. 2007;34:159-164.
59. Brandle P., et alSatoretti-Schefer S., Bohmer A. Correlation of MRI, clinical, and electroneuronographic findings in acute facial nerve palsy. Am J Otol. 1996;17:154-161.
60. Murakami S., et alHato N., Horiuchi J. Treatment of Ramsay Hunt syndrome with acyclovir-prednisone: Significance of early diagnosis and treatment. Ann Neurol. 1997;41:353-357.
61. Stafford F.W., Welch A.R. The use of acyclovir in Ramsay Hunt syndrome. J Laryngol Otol. 1986;100:337-340.
62. Uri N., et alMeyer W., Greenberg E. Herpes zoster oticus: Treatment with acyclovir. Ann Otol Rhinol Laryngol. 1992;101(2 Pt 1):161-162.
63. Denny-Brown D. Pathologic features of herpes zoster: A note on geniculate herpes. Arch Neurol Psychiatry. 1944;20:149-159.
64. Honda N., et alYanagihara N., Hato N. Swelling of the intratemporal facial nerve in Ramsay Hunt syndrome. Acta Otolaryngol. 2002;122:348-352.
65. Balance C. The operative treatment of facial palsy: By the introduction of nerve grafts into the fallopian canal and by other intratemporal methods. Acta Otolaryngol. 1932;15:1-79.
66. May M., Klein S.R., Taylor F.H. Idiopathic (Bell’s) facial palsy: Natural history defies steroid or surgical treatment. Laryngoscope. 1985;95:406-499.
67. Gantz B.J., et alRubinstein J.T., Gidley P. Surgical management of Bell’s palsy. Laryngoscope. 1999;109:1177-1188.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 28 Management of Bell’s Palsy and Ramsay Hunt Syndrome
BELL’S PALSY
Bell (1774-1842)1 first described a patient with a facial paralysis in 1818; subsequently, all patients with facial palsy of unknown etiology have come to bear his name. The etiology of this “idiopathic” disorder has become much clearer in recent years. Although first proposed in 1972 by McCormick,2 herpes simplex virus (HSV) has been identified as the disease vector only recently,3–5 and an animal model has been designed.6 Murakami and associates5 identified HSV type 1 (HSV-1) DNA fragments in perineural fluid in 11 of 14 patients undergoing facial nerve decompression. In this study, no control subjects had HSV-1 DNA in perineural fluid. Using polymerase chain reaction to analyze the saliva of patients with Bell’s palsy, Furuta and colleagues3 identified HSV-1 DNA in 50% of patients, which was significantly more often than controls, a finding confirmed by other groups.7 Polymerase chain reaction has also been used to isolate HSV-1 genomic DNA from the geniculate ganglion of a temporal bone in a patient dying during the acute phase of Bell’s palsy.4
Sugita and coworkers6 proposed an animal model of Bell’s palsy. Six days after inoculation of HSV-1 into either the auricle or the tongue of mice, a temporary ipsilateral facial paralysis was identified that recovered spontaneously within 3 to 7 days. Histopathologically, neural edema, vacuolar degeneration, and inflammatory cell infiltrate with associated demyelination or axonal degeneration were shown in the affected facial nerve and nucleus.8 HSV-1 antigens were identified within the facial nerve, geniculate ganglion, and facial nucleus 6 to 20 days after inoculation.6 Similar pathologic findings have been shown in rabbits after HSV-1 inoculation, but without the associated facial paralysis.9
The histopathologic changes seen in autopsy specimens of patients who died with acute idiopathic facial paralysis have provided some insight into the underlying cellular mechanisms in Bell’s palsy. Reddy and coworkers10 found degeneration of the myelin sheath and axons, perivascular inflammation, and a phagocytic cell infiltrate in 10% to 30% of nerve fibers in a patient 17 days after the onset of an acute idiopathic facial paralysis. Fowler11 found diffuse vascular engorgement throughout the intratemporal facial nerve and evidence of acute hemorrhage within the intracanalicular, labyrinthine, and geniculate portions of the facial nerve in a patient who died shortly after the onset of Bell’s palsy. In examining an autopsy specimen of a patient who died 13 days after the onset of Bell’s palsy, McKeever and colleagues12 found a diffuse lymphocytic infiltration of the intratemporal facial nerve with ongoing myelin phagocytosis. They later re-examined the same specimen and noted the most pronounced lymphocytic infiltration of the nerve at the labyrinthine segment, findings that these authors believed were most consistent with an ongoing compression-type injury to the facial nerve.
Multiple reports have found that there seems to be evidence of constriction of the nerve at the meatal foramen in cases of Bell’s palsy. In specimens examined during the acute phase of Bell’s palsy, lymphocytic infiltration, perineural edema, and myelin degeneration have been noted.13,14 In a nerve specimen examined 1 year after the diagnosis of Bell’s Palsy lymphocytic infiltration, perineural edema, and fibrotic changes were noted.15 Intraoperative biopsy specimens of the greater superficial petrosal nerve from patients undergoing nerve decompression procedures for Bell’s palsy have shown axonal demyelination and degeneration with lymphocytic infiltration.16 Together, the histopathologic findings in Bell’s palsy show demyelination and axonal loss with lymphocytic or phagocytic infiltration, findings that seem to be more pronounced at the labyrinthine segment of the facial nerve.
In considering this evidence, the pathogenesis of Bell’s palsy becomes more apparent: a virally induced, inflammatory response that produces edema within the nerve. Fisch and Felix16 first proposed that the facial nerve was entrapped at the meatal foramen as a result of neural edema. Intraoperative conduction studies have shown an electrophysiologic blockage at this site.17 The constriction imposed produces a conduction block at first; however, with prolonged or increased constriction, ischemia results. Subsequently, wallerian degeneration occurs, producing axonotmesis or neurotmesis or both. A spectrum of injury within the nerve from neurapraxia to neurotmesis may occur in Bell’s palsy.11,18 The proportion of each of these determines the amount of facial function that returns when the acute phase of the disease subsides.
Bell’s palsy accounts for nearly three quarters of all acute facial palsies.19 The incidence of Bell’s palsy is 20 to 30 cases per 100,000 per year.20 The median age is 40 years, but it can occur at any age.21 The incidence is highest in patients older than 70 years and lowest in children younger than 10 years. The left and right sides are affected equally. Men and women are equally affected, but the incidence of Bell’s palsy is higher in pregnant women (45 cases per 100,000).
The clinical presentation of Bell’s palsy is well known; however, the clinician must exclude other causes of facial paralysis based on history and physical examination findings. Patients describe an abrupt onset of unilateral paresis that occurs over 24 to 48 hours. The paresis can progress over 1 to 7 days to complete paralysis. Bilateral involvement, either simultaneously or consecutively, has been described.23 A history of progression of weakness over weeks to months, repeated episodes of paralysis, and twitching of the facial muscles should not be considered symptoms of Bell’s palsy. Other associated symptoms of hearing loss, vestibular symptoms, or other cranial nerve neuropathies also rule out the diagnosis of Bell’s palsy.
Audiometric evaluation should reveal symmetric function except for an absent ipsilateral acoustic reflex. If unilateral hearing loss or acoustic reflex decay is present, further evaluation for retrocochlear pathology is necessary. If vestibular complaints are present, an electronystagmogram is performed, and diagnosis of Bell’s palsy should be questioned. If the history and clinical presentation are highly suggestive of Bell’s palsy, magnetic resonance imaging (MRI) and computed tomography (CT) are not performed. High-resolution CT scans and MRI are obtained if patients have associated symptoms of otorrhea, vestibular complaints, and hearing loss. Planned surgical decompression and persistent dense paralysis after 6 months are also indications for imaging. Serologic tests for Lyme disease (IgG, IgM) are an important part of the work-up for unexplained facial paralysis in endemic areas.24 Serologic testing for antibodies to HSV or varicella zoster have provided some correlative evidence for a viral etiology in Bell’s palsy, although laboratory investigations in general have not been found to provide clinically relevant data.
Electrodiagnostic testing is an important element of the diagnostic evaluation of facial paralysis. Testing can determine the extent of facial nerve injury and provide useful prognostic information for the development of management strategies. The technique of electroneuronography (ENoG), developed by Esslen,25 can distinguish the nerve fibers that have undergone wallerian degeneration from fibers that are temporarily blocked (neurapraxia). ENoG testing is not performed until 3 to 4 days after the development of complete unilateral paralysis because wallerian degeneration does not become apparent until 48 to 72 hours after an acute injury to the nerve. Electrodiagnostic testing is not performed when the patient exhibits paresis only. If the paresis progresses to total paralysis, electrodiagnostic testing is performed 3 days after the onset of total paralysis. Presence of voluntary movement 4 to 5 days after the onset of paresis indicates only minor injury, and complete recovery should be anticipated.
ENoG is most accurate when it is performed within 3 weeks of the acute injury. The test is performed using standard electromyography (EMG) equipment, but requires the use of special surface stimulating and recording electrodes.26 The recording electrodes are in a hand-held carrier and are manipulated in the nasolabial fold with the Esslen technique. The recording electrodes are not taped to the skin, as has been described by others.27
An evoked electric stimulus generates synchronous facial muscle movement that can be recorded from the skin surface (termed the compound muscle action potential [CMAP]). The amplitude of the biphasic CMAP has been found to correlate with the number of blocked or neurapraxic nerve fibers. As the percentage of degenerated fibers within the nerve increases, the amplitude of the CMAP decreases compared with the normal side of the face. Fisch and Esslen28 determined that if 90% or more of the fibers within the facial nerve degenerate within the first 14 days of an acute paralysis, a severe injury has occurred, and the chances of complete recovery are less than 50%. Patients who do not reach the 90% degeneration level by 3 weeks have a very good prognosis and are likely to regain normal facial motion without synkinesis. The time course of degeneration is also important; the more rapid the degeneration, the more severe the injury.29 A patient showing greater than 90% degeneration at 5 days would have a worse prognosis than a patient with 90% degeneration at 14 days.
Topognostic testing is widely reported to be useful in determining the site of injury in acute facial paralysis; however, intraoperative studies have shown that the Schirmer test is not accurate in diagnosing Bell’s palsy.26 The Schirmer test may be used to determine the extent of lacrimation and the need for eye protection.
A review of the natural history of Bell’s palsy shows that approximately 85% of patients begin to display some return of facial movement within 3 weeks of the onset of paresis.30 The remaining 15% begin to improve 3 to 6 months after the onset of the disease. Most patients show a complete return of facial function, but 10% to 15% have residual unilateral weakness and develop secondary deformities of synkinesis, tearing, or contracture. Some motion returns in almost all individuals with Bell’s palsy by 6 months. If no movement returns, a vigorous search for another etiology should begin anew.
The management of patients with Bell’s palsy varies, depending on the type of specialist initially seeing the patient and on the training of the individual specialist. Figure 28-1 presents an overview of our management strategy. Patients presenting within the first week of facial weakness with paresis are placed on steroid therapy (prednisone, 60 to 80 mg/day for 7 days) and an antiviral agent (valacyclovir, 500 mg three times per day for 10 days). If patients are seen 7 to 10 days after onset and motor function is stable or improving, medical treatment is unnecessary. Patients are instructed to return in 1 week for re-evaluation to determine if neural degeneration has occurred. Electrodiagnostic testing is unnecessary as long as voluntary facial movement is present. If total paralysis occurs in the interim, the total paralysis protocol of Figure 28-1 is followed. Stable or improving patients are seen in 1 month.
FIGURE 28-1 Algorithm for management of Bell’s palsy. ENoG, electroneuronography; MCF, middle cranial fossa.
The use of steroids in Bell’s palsy has been the subject of much debate. Numerous studies of varying designs in adults have shown better outcomes in patients treated with steroids,21,31,32 especially when initiated early in the course of the disease.33–35 Other randomized studies36 and meta-analyses,37 including many studies in children,38–40 have concluded that steroids did not affect the ultimate facial function outcomes in Bell’s palsy. Grogan and Gronseth,41 in a comprehensive, evidence-based review, concluded that there seemed to be a beneficial effect in the use of steroids in Bell’s palsy. Ramsey and coworkers42 performed a meta-analysis of 47 trials of steroid therapy for Bell’s palsy, and concluded that there seemed to be an improved odds of recovery in patients treated with steroids (49% to 97%) compared with untreated controls (23% to 64%). We use prednisone in a dose of 1 mg/kg daily for 7 days in all cases in which it is not medically contraindicated, in anticipation of speeding recovery, reducing the number of degenerating axons, and reducing the number of patients needing decompression.
The combination of steroids and antivirals may be superior to either one alone. A double-blind, randomized, controlled trial of acyclovir and prednisone versus prednisone alone in the treatment of Bell’s palsy showed better results with the combination therapy.43 This study documented poor facial function recovery in 23% of the prednisone-only group compared with 7% in the acyclovir-plus-prednisone group. Interpreting the results of this study, Grogan and Gronseth41 reported that patients who receive antiviral therapy in addition to steroids were 1.22 times as likely to have a good facial nerve outcome. A multicenter, randomized, placebo-controlled study comparing treatment of patients with Bell’s palsy with steroids and antivirals with treatment with steroids alone concluded that the addition of valacyclovir improved the recovery rate from 75% to 90% in cases of complete palsy, and from 89% to 96% in all cases of facial palsy.44 Many other reviews of the topic have made similar conclusions.45–48
Other studies have found no significant difference between this combination of drugs and the natural history of the disease.24,49
[/not-level-membership-for-surgery-category]
