40 Malignant Hyperthermia
MH was first described by Denborough and Lovell in 1960, who reported a 21-year-old man with compound fractures of his right tibia, who, with great trepidation, underwent general anesthesia with halothane.1 After 10 minutes of anesthesia, he became hemodynamically unstable with hypotension, tachycardia, and mottled skin that was hot to the touch. The soda lime canister was found to be hot and was changed because it appeared to be exhausted. The anesthetic was discontinued, the patient was packed in ice, and he recovered without sequelae. Postprocedural examination did not reveal any known medical abnormalities. A careful family history disclosed that 10 blood relatives had previously died after ether anesthesia, suggesting an autosomal dominant pattern of inheritance for the disorder. This patient required a subsequent operation and was administered a spinal anesthetic without incident, demonstrating the effectiveness of nontriggering anesthetics.2 Subsequent reports from around the world established this disorder as a familial entity that was potentially fatal.3,4 The term malignant hyperpyrexia (later changed to malignant hyperthermia) was coined in 1967 at the first international meeting on this disorder.
The incidence of MH based on occurrences of MH reactions has been reported to be 1 case per 50,000 to 100,000 adults and 1 case per 3000 to 15,000 children.5,6 Subsequent surveys of suspected MH reactions revealed even greater incidences; the incidence of suspected MH was 1 case per 16,000 anesthetics in adults in Denmark, an incidence that increased to 1 case per 4200 adults when an inhaled anesthetic and succinylcholine were combined.7 However, a survey of anesthesia in a pediatric hospital in the United States during the halothane era revealed an MH incidence of 1 case per 20,000 to 40,000 children, almost one half of that reported previously.8 The incidence of fulminant MH (i.e., rapid increase in temperature accompanied by life-threatening metabolic changes, arrhythmias, and increased serum creatine kinase level) was 1 case per 250,000 general anesthetics in Denmark.7 A similar incidence of 1 case of fulminant MH per 200,000 general anesthetics was reported from the United Kingdom.9 The Danish survey found an incidence of 1 case of masseter muscle spasm per 12,000 anesthetics among children who received succinylcholine, whether in combination with inhalational or intravenous anesthetics.7 Clinically, the demographic data suggested that the incidence or suspicion of MH was greater among children than adults and that the incidence was even greater among children in whom succinylcholine was used.
The frequency of MH reactions is greatest in childhood, with a peak age of 3 years, although recent data suggest that only about 18% of MH reactions occur in childhood.10,11 Reactions have also been reported in neonates and infants, although they have been infrequent.12 Many have perceived a decrease in the incidence of MH reactions in the past 2 decades, although some have challenged this perception.11 Two reasons for the perception that the incidence of MH reactions have decreased are that many families with a genetic predisposition to MH have been identified and bring it to the attention of their surgical and anesthetic care providers preoperatively, and that the routine use of succinylcholine has decreased dramatically as a result of concerns regarding rare complications, such as hyperkalemic cardiac arrest. The latter has resulted in a black box warning admonishing against the routine use of succinylcholine in children, particularly male children younger than 8 years of age who may have unrecognized muscular dystrophy or other myopathy.13,14
It remains certain that all inhalational anesthetics and succinylcholine are potent triggers of MH reactions in susceptible patients,15–19 with the possible exception of xenon based on evidence from MH-sensitive pigs20 and caffeine-halothane contracture tests (CHCTs) from MH-susceptible and normal humans.21 No other drugs used for intravenous or regional anesthesia trigger MH reactions. A comprehensive list of triggering and nontriggering drugs is available from the Malignant Hyperthermia Association of the United States (MHAUS) (http://www.mhaus.org/ [accessed July 2012]).
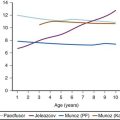
The mortality rate for MH has decreased dramatically from more than 80% in the 1960s to 1.5% to 5% in recent years (Fig. 40-1).11,22–24 For children, the MH mortality rate (0.7%) is 15-fold less than the rate (14%) for adults.11 The overall decrease in mortality may be attributable to several factors: better identification of MH-susceptible individuals; the routine use of capnography and pulse oximetry, facilitating early identification of the signs and symptoms of an acute MH reaction25,26; a better understanding of the pathogenesis of MH; and the widespread availability of dantrolene.10
Clinical Presentation
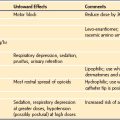
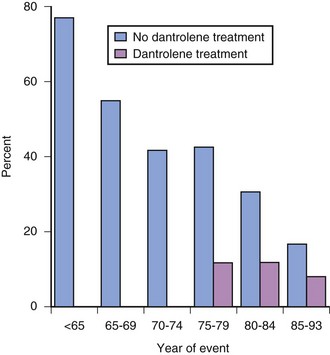
The most common presentation of MH is a hypermetabolic response to the inhalational anesthetics, with or without succinylcholine (Table 40-1). Among the earliest clinical signs is a marked increase in the end-tidal CO2 that resists control by either mechanical ventilation or an increase in minute ventilation when the patient is breathing spontaneously.23,27 Other nonspecific early signs include tachycardia and hemodynamic instability with a trend toward hypertension. Severe masseter muscle spasm (i.e., masseter tetany) refers to the inability to insert a laryngoscope blade into the mouth, the so-called “jaws of steel,” even with no twitches evident on a blockade monitor after administration of succinylcholine, and it strongly indicates MH susceptibility (Fig. 40-2). In vitro live muscle biopsy testing revealed a 28% to 50% incidence of MH susceptibility among children with jaws of steel (Table 40-2).28–30 Generalized muscle rigidity develops as a result of the excessive accumulation of myoplasmic Ca2+ concentrations in MH-susceptible skeletal muscle, causing sustained muscle contractures.27 This occurs even in the presence of neuromuscular blockade with nondepolarizing neuromuscular blocking drugs (NMBDs).
TABLE 40-1 Clinical and Laboratory Findings Associated with Malignant Hyperthermia
| Clinical Findings | Laboratory Findings |
|---|---|
| Tachycardia, tachypnea and hypertension | Increased Paco2 |
| Hypercarbia (etco2) | Acidosis (mixed respiratory and metabolic) |
| Greatly increased minute ventilation | Relative hypoxia, increased alveolar to arterial partial pressure gradient for oxygen |
| Generalized muscle rigidity (unresponsive to nondepolarizing muscle relaxants) | Hyperkalemia |
| Skin mottling | Elevated plasma lactate concentration |
| Hyperthermia (late sign) | Abnormal coagulation studies (late sign) |
| Cardiac arrhythmias (hyperkalemia-induced: PVC, VT, VF) | Myoglobinuria, myoglobinemia |
| Cola-colored urine (late sign) | Increased CPK level (usually a late sign) |
| Disseminated intravascular coagulation (late) |
CPK, Creatinine phosphokinase; etco2, end-tidal carbon dioxide; Paco2, arterial partial pressure of carbon dioxide; PVC, premature ventricular contraction; VF, ventricular fibrillation; VT, ventricular tachycardia.
Data from Malignant Hyperthermia Association of the United States. Available at http://www.mhaus.org (accessed July 2012).
TABLE 40-2 Limited Excursion of the Mandible: Differential Diagnosis
Temporomandibular joint dysfunction: congenital, inflammatory/infectious, trauma, neoplasm, collagen vascular disease (rheumatoid arthritis)
Muscle disease: malignant hyperthermia, Duchenne or Becker muscular dystrophy, myotonia congenita
Integumentary disease: inflammatory or infectious disease, neoplasm, radiation effects, collagen vascular disease (scleroderma)
Ultra-rapid metabolizer of succinylcholine (Nietlich or Cynthiana [C5 enzyme] variant of pseudocholinesterase) (see Chapter 6)
Hyperthermia, often a late sign, results from the greatly increased aerobic and anaerobic metabolic activity of triggered skeletal muscle; the overlying skin soon becomes hot to the touch. In many instances, the large muscle groups such as calf or thigh muscles feel tight or knotted. This is accompanied by exaggerated carbon dioxide (CO2) production, which usually is the first sign of an evolving MH reaction, and the accumulation of lactate fueled by markedly increased glycogenolysis and glycolysis (i.e., mixed respiratory and metabolic acidosis).31,32 If an acute MH reaction is diagnosed and treated early in its evolution, before the body becomes unable to maintain this exaggerated aerobic metabolism, arterial blood gases may show an almost pure respiratory acidosis, which can make the diagnosis of MH difficult (Table 40-3). A mixed venous or peripheral venous blood gas analysis can be helpful in establishing the presence of hypermetabolism because it is more likely to demonstrate an increased CO2 level, significant oxygen desaturation consistent with increased oxygen consumption (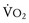 less than 40 mm Hg, despite administering supplemental oxygen for which the expected
less than 40 mm Hg, despite administering supplemental oxygen for which the expected 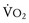 is greater than 60 mm Hg), and a possible increased lactate concentration.
is greater than 60 mm Hg), and a possible increased lactate concentration.
In fulminant cases, untreated MH may increase the temperature as rapidly as 1° C every 10 minutes.27 In one case, the temperature reached 43.8° C (110.8° F) within 18 minutes.33 In addition to the profound hypercarbia and tachycardia, severe hypoxia, skin mottling, exuberant metabolic acidosis, rhabdomyolysis, coagulopathy, and hyperkalemia may follow. Unstable hemodynamics and ventricular arrhythmias inevitably follow. Intractable ventricular arrhythmias, pulmonary edema, disseminated intravascular coagulation, cerebral hypoxia or edema, and renal failure due to myoglobin deposition in the renal tubules are often associated with fatal outcomes.
The presenting signs of MH vary (see Table 40-1). The syndrome may be fulminant or indolent, not all features of a classic MH reaction may be immediately evident, and it may occur intraoperatively or postoperatively, although the frequency of MH reactions occurring postoperatively is only 2%.27,34 The latest that an MH reaction has reportedly occurred postoperatively is 11 hours, although a recent review of reports of postoperative MH in the North American Malignant Hyperthermia Registry has not found any case occurring beyond 40 minutes after the end of the anesthetic.34,35 The likelihood that an MH-susceptible patient will develop MH in the presence of inhalational anesthetics is exasperatingly unpredictable. In one study, 50% of susceptible individuals reported two or more uneventful general anesthesias before an MH reaction was triggered.23,36 Only 6.5% of probands in a retrospective review reported a family or personal history of MH.36 A negative personal or family history is insufficient to conclude that a child is not susceptible to MH. Other disease states may be confused with MH (Table 40-4) and must be distinguished from it to provide correct therapy.
TABLE 40-4 Differential Diagnosis of Malignant Hyperthermia
| Diagnosis | Distinguishing Traits |
|---|---|
| Hyperthyroidism | Patients often present with similar symptoms and physical findings; blood gas abnormalities gradually evolve; creatine phosphokinase value does not increase substantively. |
| Sepsis | Usually, blood gases are normal early, and metabolic acidosis occurs late; creatine phosphokinase remains normal. |
| Pheochromocytoma | Similar to MH, except for marked blood pressure swings |
| Metastatic carcinoid | Flushing, diarrhea, hypotension |
| Cocaine intoxication | Fever, rigidity, rhabdomyolysis similar to NMS |
| Heat stroke | Similar to MH, except that the patient is outside the operating room |
| Masseter muscle rigidity (MMR) | May progress to MH; total body spasm more likely than isolated MMR |
| Neuroleptic malignant syndrome (NMS) | Similar to MH but evolves over weeks; usually associated with the use of antipsychotics |
| Serotonergic toxicity | Similar to MH and NMS; associated with the administration of mood-elevating drugs (e.g., selective serotonin reuptake inhibitors) |
| Nonmalignant hyperthermia syndrome | Reported only once; severe hyperthermia seemingly associated with fentanyl |
Given the variability of the clinical presentation of MH and the dearth of pathognomonic signs for this syndrome, establishing the diagnosis can be difficult. In response to the need for an objective measure to verify a clinical episode of MH, a retrospective, multivariable clinical grading scale was developed.37 This grading scale was devised to clarify the cutoff value for a positive muscle CHCT result. It was not intended to be a clinical guide in the operating room. Despite recommendations not to use this scale to guide treatment and to be more conservative in its application, Tables 40-5 and 40-6 are provided to help clinicians identify true MH reactions. Although this clinical grading scale is somewhat cumbersome and has not been prospectively validated in clinical settings for use by nonexperts, it is a useful guide for the clinician.
TABLE 40-5 Clinical Indicators for Determining the Malignant Hyperthermia Raw Score
| Process | Indicator | Points |
|---|---|---|
| Rigidity | Generalized muscular rigidity | 15 |
| Masseter spasm | 15 | |
| Muscle breakdown | Creatine kinase >20,000 IU after succinylcholine | 15 |
| Creatine kinase >10,000 IU with no succinylcholine | 15 | |
| Cola-colored urine in perioperative period | 10 | |
| Myoglobin in urine >60 µg/L | 5 | |
| Myoglobin in serum >170 µg/L | 5 | |
| Blood, plasma, or serum K+ >6 mEq/L, no renal illness | 3 | |
| Respiratory acidosis | Petco2 >55 mm Hg with controlled ventilation | 15 |
| Arterial Paco2 >60 mm Hg with controlled ventilation | 15 | |
| Petco2 >60 mm Hg with spontaneous ventilation | 15 | |
| Arterial Paco2 >65 mm Hg with spontaneous ventilation | 15 | |
| Inappropriate hypercarbia, anesthesiologist’s call | 15 | |
| Inappropriate tachypnea | 10 | |
| Temperature increase | Inappropriately rapid increase | 15 |
| Inappropriately increased temperature >38.8° C (101.8° F) | 10 | |
| Cardiac involvement | Inappropriate sinus tachycardia | 3 |
| Ventricular tachycardia or fibrillation | 3 | |
| Family history | Positive family history for first-degree relative | 15 |
| Positive family history for more distant relative | 5 | |
| Others | Arterial base excess more negative than −8 mEq/L | 10 |
| Arterial pH <7.25 | 10 | |
| Rapid reversal of maligant hyperthermia (MH) signs after intravenous administration of dantrolene | 5 | |
| Positive MH family history with another indicator from the patient’s anesthesia experience other than increased creatine kinase level | 10 | |
| Elevated creatine kinase level and a family history of MH | 10 |
From Larach MG, Localio AR, Allen GC, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology 1994;80:771-9.
TABLE 40-6 Malignant Hyperthermia Clinical Grading Scale
| Raw Score Range | Rank | Likelihood |
|---|---|---|
| 0 | 1 | Almost never |
| 3-9 | 2 | Unlikely |
| 10-19 | 3 | Somewhat less than likely |
| 20-34 | 4 | Somewhat greater than likely |
| 35-49 | 5 | Very likely |
| ≥50 | 6 | Almost certain |
Modified from Larach MG, Localio AR, Allen GC, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology 1994;80:771-9.
Patient Evaluation and Preparation
Ambulatory surgery has rapidly expanded to include most pediatric surgery. Consequently, the safety of discharging children with a personal or family history of MH after an uneventful, trigger-free anesthesia on the day of surgery has raised concerns. Two retrospective studies concluded that the risk of a child developing an MH reaction after an uneventful, trigger-free anesthesia was exceedingly small.38 Postoperative monitoring for an MH reaction while in the hospital was gradually reduced from 6 hours to 2 hours before discharge.39,40 For MH-susceptible children, parents should be provided with a written description of the signs and symptoms of an MH reaction and a phone number for the on-call anesthesiologist for further advice. Families should contact an anesthesiologist rather than return to the emergency department, because the emergency physician may be unfamiliar with MH, particularly in children. Additional advice for the parents should include the use of an oral antipyretic drug (e.g., acetaminophen) to treat a mild fever. If the fever abates after a dose or two of acetaminophen, the fever was not caused by MH. If the fever persists despite acetaminophen and sponge baths, and is accompanied by tachycardia and tachypnea, the parents should notify the on-call anesthesiologist and return the child to the hospital.
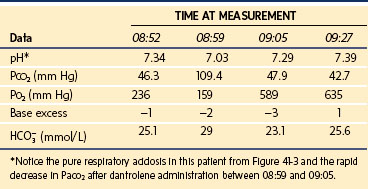
When a child with a known susceptibility to MH is scheduled for general anesthesia, the anesthesia machine (i.e., anesthetic workstation [AWS]) must be prepared to preclude the delivery of triggering agents. First, succinylcholine should be removed from the local vicinity to avoid inadvertent administration. Second, all vaporizers should be physically disengaged from the AWS, which is preferable because they can leak trace concentrations of inhalational anesthetics even in the off state, or if they cannot be removed from the AWS, tape should be placed across them in the off position to avoid accidentally turning them on.41 Third, to accelerate the washout of anesthetics, the CO2 absorbent should be replaced, a new anesthetic breathing circuit installed, and the ventilator bellows flushed and left operating.41–43 Fourth, to eliminate inhalational anesthetics from the AWS, many clinicians follow a standardized protocol of flushing the workstation with 10 L/min of oxygen for 10 to 20 minutes, depending on the manufacturer and age of the machine.44 However, the assumption that one protocol fits all to reduce the anesthetic concentration to less than 10 ppm, which is assumed to be the threshold below which an MH reaction cannot be triggered,45 may not hold true for every AWS, particularly the newer ones, which are more complex in construction and more likely to contain internal working parts made of plastic, which act as sumps for inhalational anesthetics. To address the various types of AWS, the duration of flushing with large fresh gas flows (Table 40-7) and the need to exchange contaminated internal components with clean versions must be determined for each AWS. For some types of AWS, more than 60 minutes may be required to reach anesthetic concentrations less than 10 ppm.44,46,47 To achieve an anesthetic concentration of 10 ppm or less in the Drager Primus, Fabius, and Zeus machines in a timely manner, the ventilator diaphragm and integrated breathing system should be replaced with autoclaved components and then flushed for 20 minutes at a fresh gas flow of 10 L/min.46,47 Table 40-7 lists the published times required to reach an anesthetic concentration of less than 10 ppm without replacing any AWS components.44,47,48 These data support the notion that the previously held protocols to wash out inhalational anesthetics from older AWSs do not hold true for the newer AWSs. The need for a single protocol or intervention that consistently achieves an anesthetic concentration of less than 10 ppm in all AWSs is of even greater importance because none of the available anesthetic agent analyzers is capable of measuring anesthetic concentrations in the 10 ppm range to confirm adequate removal of inhalational anesthetics.
TABLE 40-7 Time to Wash Out Inhalational Anesthetics to Less than 10 ppm from Anesthetic Workstations
After the AWS has been flushed with 10 L/min of an air and oxygen mixture, most anesthesiologists reduce the fresh gas flow during anesthesia. However, evidence has shown that the concentration of inhalational anesthetic surges (≥50 ppm) when the fresh gas flow is reduced, and the magnitude of the rebound directly depends on the fresh gas flow rate.48,49 Those who reduce the fresh gas flow after purging the AWS may be exposing their patients to concentrations of inhalational anesthetics that may trigger an MH reaction, although no MH reactions have been reported in patients in whom a reduced fresh gas flow was used in an AWS that had been purged using a large (>10 lpm) fresh gas flow. To avoid confusion, a single, consistent, effective, and reliable intervention is required to prevent MH reactions in patients exposed to trace gases from a previously contaminated AWS.
A commercially available charcoal filter (Vapor-Clean, Dynasthetics, LLC, Salt Lake City) fitted to the expiratory and inspiratory limbs of the AWS reduces the concentration of inhalational anesthetics to less than 5 ppm within several minutes.50 These filters are sold in pairs. The manufacturer recommends that a filter should be inserted into both limbs of the anesthesia breathing circuit just distal to the valves, which is reasonable during an acute MH reaction. However, when only the machine is contaminated with inhalational anesthetic (e.g., after flushing the AWS for elective MH cases), we apply a single filter in the inspiratory limb and keep the second of the pair to replace the first after 60 to 90 minutes, since it may become expended by that time.50
Monitoring
Capnography and pulse oximetry, key monitors for early signs of an MH reaction, are required for all children who receive general anesthesia, irrespective of the duration of the procedure. Measurement of the body temperature is recommended for all children who undergo general anesthesia when fluctuations may be anticipated, according to the American Society of Anesthesiologists 2011 standards for basic anesthesia monitoring (www.asahq.org/For-Members/Standards-Guidelines-and-Statements.aspx). Monitoring the axillary temperature site (opposite the extremity with the intravenous line) is recommended rather than a core site for early detection of an MH reaction because the axillary region is surrounded by large muscle bulk in the pectoral shoulder girdle. Although most consider an increasing temperature a late sign of an MH reaction, a retrospective review suggested that an increasing body temperature may occur early in an evolving MH reaction.36,51 Evidence also suggests that crystalline skin temperature tapes may not reliably track temperature changes during MH reactions.36
Diagnosis
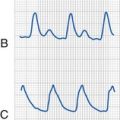
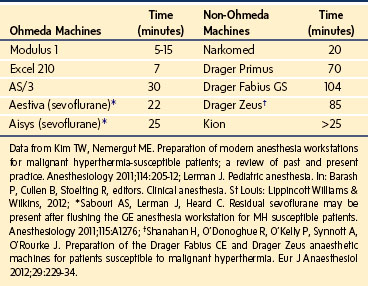
Because the underlying disorder in MH is a hypermetabolic reaction (i.e., increased CO2 production and oxygen consumption), massive volumes of CO2 are released into the circulation, which rapidly increase the partial pressure of CO2 (Pco2) and respiratory rate in the unparalyzed child. The cardiovascular response is an increased cardiac output, heart rate, and in some cases, blood pressure (E-Fig. 40-1). The first clinical signs and symptoms of this hypermetabolic reaction in a spontaneously breathing patient are hypercapnia, tachypnea, and tachycardia (see Table 40-1).36 A steady and relentless increase in the end-tidal CO2 pressure (Petco2) is the earliest sign of a reaction and is usually evident whether respirations are spontaneous or controlled. In some instances, the increase in Petco2 and heart rate occur contemporaneously, alerting the clinician to consider an evolving MH reaction (Fig. 40-3
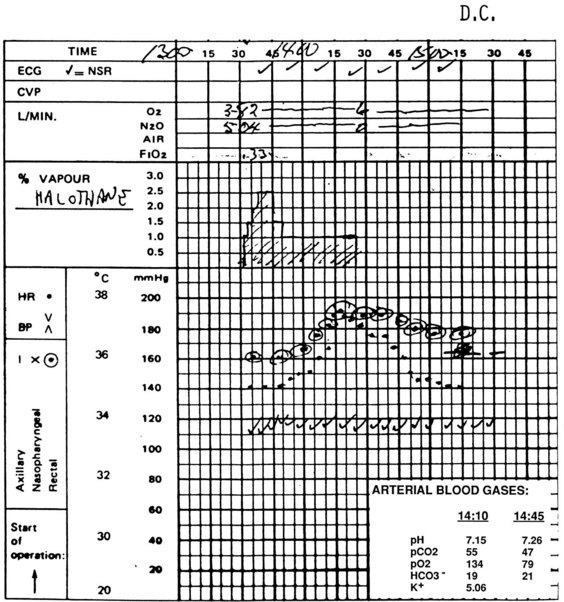
). Sudden unexpected cardiac arrest is a very rare presentation of MH and suggests a disease process other than MH, such as acute rhabdomyolysis and hyperkalemia after succinylcholine in a (male) child with an undiagnosed myopathy.

 ), the elimination of CO2 (i.e., alveolar ventilation ([
), the elimination of CO2 (i.e., alveolar ventilation ([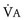 ]), and the inspired concentration of CO2 (F
]), and the inspired concentration of CO2 (F include fever, MH, thyroid storm, and sepsis. Causes of a decreased
include fever, MH, thyroid storm, and sepsis. Causes of a decreased 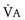 include a deep level of anesthesia, endobronchial intubation, bronchospasm, and a kinked tracheal tube or airway breathing circuit. Causes of increased F
include a deep level of anesthesia, endobronchial intubation, bronchospasm, and a kinked tracheal tube or airway breathing circuit. Causes of increased FDuring airway obstruction and hypermetabolic states such as thyrotoxicosis and sepsis, an increased Petco2 can be readily corrected with mild to moderate hyperventilation. During MH reactions, however, it is very difficult to restore the Petco2 to the normal range, even with vigorous mechanical hyperventilation.26 The CO2 production is sometimes so great that the in-circuit CO2 absorbent rapidly becomes exhausted in an exothermic reaction, and the absorbent container becomes hot to touch.
A child’s response to surgery during light anesthesia often includes tachycardia and may sometimes include bronchoconstriction. However, a dramatic and unexpected increase in heart rate from 120 to 180 beats/min in a healthy, 7-year-old child (or an increase from 70 to 120 beats/min in an adult) strongly suggests a pathologic process, and a differential diagnosis beyond light anesthesia should be seriously considered. Before intervening, it is important to quickly scan all of the monitors to determine whether the aggregate indices point to a specific diagnosis. If tachycardia is associated with an increase in body temperature, a differential diagnosis of fever and tachycardia under anesthesia should be considered. Fever related to sepsis or viral infection usually has a slow onset, whereas fever from an MH reaction typically has a rapid onset. The differential diagnoses include iatrogenic external overheating and an MH reaction (see Table 40-4). A rapid increase in the inspired concentration of desflurane and isoflurane, but not sevoflurane, may cause a sympathetic-based tachycardia that in isolation should not suggest a diagnosis of MH because the Petco2 remains unchanged.52,53 Of the inhalational anesthetics, halothane appears to be the most likely to trigger an MH reaction and the best discriminator for the CHCT. Enflurane provides the smallest trigger, sevoflurane and isoflurane are intermediate, and preliminary data suggest that xenon does not trigger MH reactions.21,45,54–56 If none of these factors appears to be causative and a deeper level of anesthesia (achieved with propofol with or without an opioid) fails to abate the signs, simultaneous venous and arterial blood gases should be analyzed to determine whether the patient has or is developing a hypermetabolic state.
A moderate but gradual increase in body temperature may occur in children excessively draped, those with forced-air warming devices, those with bilateral limb tourniquets, and those covered with plastic occlusive wrap. However, the sudden onset of a high fever must be more thoroughly investigated because it may result from several potentially fatal causes (see Table 40-4).57–60
Management, Susceptibility Screening, and Counseling
Treatment
If the anesthesiologist suspects that a child is experiencing an MH episode, the inhalational anesthetic should be immediately discontinued, 100% oxygen administered at a large fresh gas flow rate (≥10 L/min), and the surgeon informed; if surgery cannot be aborted, it must be completed expeditiously. Charcoal filters should be inserted into both limbs of the breathing circuit until a clean breathing circuit is available (E-Fig. 40-2). They prevent the child from being contaminated by residual anesthetic in the AWS and to prevent the AWS from being contaminated by anesthetic in the patient.50 The MH cart and additional personnel to assist in dissolving the dantrolene should be brought to the operating room immediately (Table 40-8). Minute ventilation should be increased to control the Paco2 and Petco2.


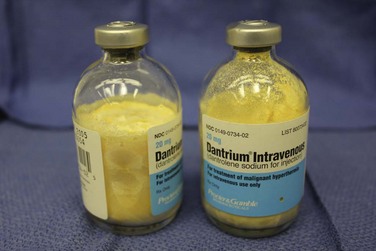
Because native dantrolene is quite insoluble in water, several strategies have been developed to speed its solubility. The current formulation is packaged as a lyophilized yellow powder in 20-mg vials that contain 3 g of mannitol and enough base to maintain a pH of about 9.5 (E-Fig. 40-3). The lyophilized formulation, mannitol, and the alkaline pH all speed the dissolution of dantrolene in water. Sixty milliliters of sterile water should be added to each vial to dissolve the dantrolene, with a resultant dantrolene concentration of 0.33 mg/mL. Warming the sterile water speeds dissolution.61 Most of the dantrolene in a vial dissolves within 60 seconds of adding the water. When the vial is vigorously shaken, any residual crystals dissolve, and the solution turns clear orange. The solution should be withdrawn immediately and administered intravenously as rapidly as possible. Occasionally, 1 or 2 additional minutes may be required to dissolve the last few crystals of dantrolene. Given the extreme alkaline pH of the dantrolene solution, it should be rapidly infused into a large vein to reduce the risk of phlebitis. Extravasation of dantrolene or prolonged continuous infusions of dantrolene may cause thrombophlebitis or thrombosis of large and small veins.62–64 Some have recommended continuous infusions of dantrolene in adults after the initial bolus, although the risk/benefit ratio of this practice is unproved in adults and untested in children.65 A new formulation of nanocrystalline dantrolene that rapidly dissolves and is available in a much greater concentration than the current formulation is undergoing federal review.66

The pharmacokinetics of intravenous dantrolene have been studied in MH-susceptible children 2 to 7 years of age.67 A loading dose of 2.5 mg/kg produced predictable blood concentrations (≥3 µg/mL) for about 6 hours after the loading dose (Fig. 40-4).67 Based on these pharmacokinetic data, if half of the loading dose of dantrolene were repeated at 6 hours after the loading dose, therapeutic blood concentrations of dantrolene would be maintained for a total of 15 hours and possibly prevent a recrudescence.67
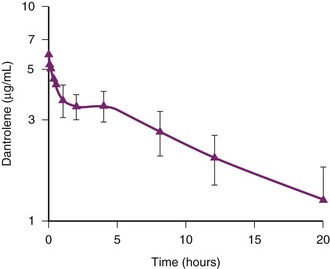
FIGURE 40-4 Pharmacokinetics of dantrolene in children.
(From Lerman J, McLeod ME, Strong HA. Pharmacokinetics of intravenous dantrolene in children. Anesthesiology 1989;70:625-9.)
An initial bolus dose of 2.5 mg/kg can control most MH reactions if the dantrolene is administered as soon after the onset of the reaction as possible (see Fig. 40-3).68 Delay in instituting dantrolene therapy increases the probability of failed therapy and death.36 For a 70-kg patient, this corresponds to 8 vials of dantrolene. The response to dantrolene should be evident within minutes, with a marked reduction in Petco2, heart rate, and respiratory rate (see Fig. 40-3). If there is no response within 3 to 5 minutes, the initial dose should be repeated until signs that the physiologic variables are abating. The clinical end points include resolution of the hypercapnia, tachypnea, and tachycardia; resolution of muscle rigidity; restoration of clear urine output; return of normal consciousness when sedation is discontinued; self-correction of blood gas abnormalities; and resolution of electrolyte disturbances. If these do not occur or are incomplete, additional doses of dantrolene should be administered until all signs and symptoms of the MH reaction have abated or the diagnosis reevaluated.68 There is no upper limit to the amount of dantrolene that can or should be given acutely to stop an MH reaction. In rare instances of persistent MH or recrudescence, a cumulative dose of up to 40 mg/kg has been required.69 Dantrolene is a fairly potent muscle relaxant, and a child with an unintubated airway (particularly one with respiratory disorders) may become weak and require controlled respirations.
Although dantrolene is effective in terminating acute MH reactions, this effect may wane as the blood concentration of dantrolene decreases. In this case, the MH reaction may recrudesce. The prevalence of recrudescence has been reported to be as great as 20% and associated with several predictive factors, including muscular body type and a greater time interval between induction of anesthesia and the development of the initial reaction.70 However, the first episode of recrudescence may occur any time after the initial reaction was successfully treated, up to and as late as 36 hours.71 The possibility of recrudescence must always be considered during the first 2 to 3 days after an MH reaction has occurred. Because recrudescence cannot be predicted, all children who experience an MH reaction must be admitted to the pediatric intensive care unit or a monitored bed until the reaction resolves and the child’s metabolic indices return to and remain normal for 2 to 3 days. MHAUS recommends that 1 mg/kg of dantrolene be administered intravenously every 6 hours for 24 to 48 hours after an MH reaction to prevent recrudescence. This recommendation remains empirical because there are no data to support the effectiveness of dantrolene in preventing recrudescence, although these dosages are consistent with pharmacokinetic data in children.67 We recommend continued vigilance, frequent physical examinations for muscle tightness, and repeated laboratory tests (e.g., blood gas analyses), and monitoring of vital signs, particularly heart rate and expired CO2 tension for evidence of a recrudescence during the first 48 hours after an MH episode. If recrudescence does occur, additional intravenous dantrolene should be administered until the reaction again abates.
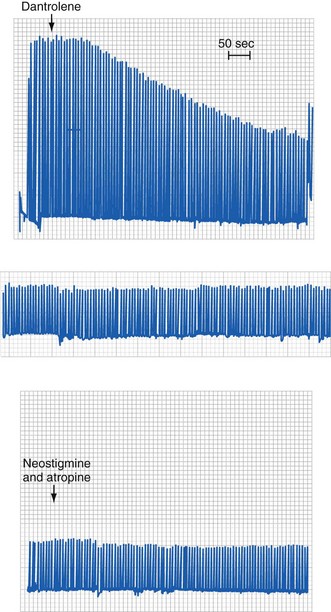
Acute administration of dantrolene causes skeletal muscle weakness that can lead to respiratory embarrassment. Not surprisingly, neostigmine is ineffective in reversing the effects of dantrolene because the latter acts intracellularly, not at the neuromuscular junction (see the section on molecular mechanisms of dantrolene). It may be advisable to maintain control of the airway (combined with intravenous sedation) until there is no further need for dantrolene. Preoperative, orally administered dantrolene, initially recommended in MH-susceptible children 2 decades ago, caused skeletal muscle weakness, dysarthria, sialorrhea, and diplopia and was ineffective in preventing MH.72 As a result, this practice has been discontinued.72 There have been no reports of acute dantrolene toxicity, although reported adverse effects after intravenous administration include muscle weakness (about 22%), phlebitis (9%), gastrointestinal upset (about 4%), and respiratory failure (3.8%).63 Long-term use in the treatment of chronic skeletal muscle spasticity has been reported to cause liver dysfunction and fatal hepatitis in about 1% and 0.1% of patients, respectively.73 Given the risks of an MH reaction, there seems little downside to treating a child with dantrolene before the diagnosis is certain, provided all the necessary blood work and laboratory tests have been collected, because early treatment reduces mortality and morbidity (see Fig. 40-1).36 The probability of developing a complication from an MH reaction increases almost threefold for every 2° C increase in body temperature and 1.6-fold for every 30-minute delay in administering intravenous dantrolene during a reaction.36 Cardiac arrest and death during an MH reaction correlate with a muscular physique and greater time intervals between induction of anesthesia and the maximal value of Petco2.24 Other possible diagnoses should continue to be considered while the treatment for the suspected MH reaction is organized (see Table 40-4). A positive response to dantrolene is not pathognomonic of an MH reaction, because other conditions may respond with resolution of their signs.
The potential for life-threatening acidosis—a pure respiratory acidosis early in the syndrome, followed later by a mixed metabolic/respiratory acidosis—always exists. Metabolic acidosis should be treated with sodium bicarbonate (1 to 2 mEq/kg IV initially), as would be done for any acutely acidotic child, and continue treatment guided by pH and base deficit, although if hypercapnia persists despite aggressive hyperventilation, administration of sodium bicarbonate should be reconsidered because the severity of the respiratory acidosis may increase. Because an acute MH episode is associated with catecholamine stress, hemodynamic instability, particularly due to dysrhythmias, may develop and should be treated according to advanced cardiac life support protocols. Calcium channel blockers should be avoided when treating MH reactions because they may cause cardiovascular collapse or acute hyperkalemia in the presence of dantrolene.74–76 Acute hyperkalemia is common in patients with MH complicated by rhabdomyolysis and acidosis. Glucose and insulin should be immediately available and combined with the judicious use of exogenous calcium for treatment. There is no evidence that the use of calcium in this setting exacerbates an MH reaction.
Other organ systems may be affected after an acute episode of MH. It is common for markers of liver function to increase 12 to 36 hours after the crisis; some liver enzymes, including lactate dehydrogenase, aspartate aminotransferase, and alanine aminotransferase, can also originate from muscle. A concomitant increase in creatine kinase to more than 10,000 IU strongly suggests a severe and acute muscle disorder. Accompanying increases in γ-glutamyltransferase and bilirubin suggest liver involvement. Disseminated intravascular coagulation as part of multisystem organ failure is a common and ominous late complication.77 Coagulation profiles should be followed frequently to guide treatment appropriate to the case.
When necessary, the MH treatment algorithm should be accessed, available on the MHAUS web site (http://www.mhaus.org/). We think that an updated MH treatment algorithm should be attached to every MH cart and anesthesia machine and that operating room personnel should hold practice drills for the treatment of an MH crisis (see “Emergency Therapy for Malignant Hyperthermia” on the inside back cover of this text and at ExpertConsult.com). If the patient experiences what is thought to be an MH reaction, the care provider should call the MH emergency response line (1-800-644-9737 or 001-1-315-464-7079 if calling from outside the United States) for consultation with experienced anesthesiologists who are available 24 hours every day. We strongly encourage completion of an adverse metabolic reaction to anesthesia (AMRA) report by members of the team who provided anesthesia and postoperative care to the child. These forms can be downloaded (www.mhreg.org), sent by the hotline consultant, or obtained from the MHAUS office through regular mail. The data contained in the completed AMRA forms allow the North American Malignant Hyperthermia Registry (NAMHR) and MHAUS to produce better data regarding the variability of MH crises and the effectiveness of treatment. Because anesthesiologists should take a leading role in managing these patients, the families should be strongly encouraged to register the proband before leaving the hospital for MedicAlert identification (www.medicalert.org) that provides critical health information, such as “malignant hyperthermia susceptible, avoid inhaled anesthetics and succinylcholine.”
Stress-Triggered Malignant Hyperthermia
In 1974, Wingard described an MH-susceptible family with a history of exercise- and emotion-induced fevers and sudden death not associated with anesthesia or surgery; he considered the possibility that MH was part of a spectrum of human stress syndromes.78 Likewise, the porcine model of MH, also known as porcine stress syndrome, was first described as an awake, stress-induced syndrome brought about by tightly packing pigs in a train, car, or truck for shipment.79 Dantrolene-responsive cases of awake and heat stroke–induced MH have been reported.80,81 Heat stress–induced MH also seems to be characteristic of susceptible animal models.82–84 A fatal case of exercise-induced MH in a 12-year-old boy who had previously survived a suspected episode of MH during general anesthesia to set a fractured humerus has been reported.85 Eight months after surgery, while playing a game of football, the child became hyperthermic, collapsed, and died. Postmortem DNA testing revealed an MH-associated RYR1 mutation in the child and his surviving father.85 More cases of stress-induced MH have been reported, substantiated by genetic testing and in vitro contracture testing (IVCT).86,87
Screening for MH susceptibility in heat stroke patients and those suffering postexercise cramps or rhabdomyolysis using the in vitro CHCT has led to the laboratory diagnosis of MH susceptibility in some patients.88–92 Although there is laboratory evidence of similarities in skeletal muscle metabolism in exertional heat stroke and MH,93 and one of the mouse MH knock-in models exhibits environmental heat triggering,51 there is as yet little evidence that these are anything more than clinically similar presentations.92 For this reason, dantrolene has rarely been an effective treatment for heat stroke.94 Nonetheless, it seems reasonable to suggest that subsets of MH susceptible patients may be more sensitive to heat stroke. Therefore it may be helpful to test children with exertional heat illness for MH susceptibility.95
MH reactions have been reported infrequently in susceptible patients who received a nontriggering anesthetic.96–98 Of 2214 patients who presented for muscle biopsy for MH susceptibility, 5 (0.46%) of 1082 who had MH-positive biopsy results developed MH reactions in the recovery room. None of the patients with negative biopsy results developed MH reactions. There is a small incidence of MH reactions among susceptible individuals that may occur despite a safe anesthetic regimen. Whether this is caused by stress or trace anesthetic concentrations that were inhaled is unclear. Massive rhabdomyolysis has been reported on rewarming from cardiopulmonary bypass despite the patient having received a nontriggering anesthetic.96,97 The mechanism of these responses may be stress, but evidence is lacking.
Postepisode Counseling
Ideally, after treating a child for MH, the anesthesiologist should arrange for referral of the child and first-degree relatives to an MH diagnostic biopsy center. It is only at such centers that the CHCT can be performed in adults. The CHCT is the only test that can produce a true negative diagnosis (i.e., not MH susceptible),99 but the sensitivity and specificity of this test are less than 100%. False-negative test results may occur, albeit rarely.100 When the anesthetic management of a small number of MH biopsy-negative patients was reviewed, more than one half were given inhalational anesthetics, although the biopsy results for the remainder may not have been known at the time of anesthesia because they were given trigger-free anesthetics.101
Many persons suspected of having MH have normal muscle responses on the CHCT. In the event that an adult who experienced an MH reaction has a normal CHCT result, he or she should be referred to a neurologist with an interest in muscle diseases to determine whether an occult myopathy is responsible for the clinical events. Alternatively, the diagnosis may be incorrect, and other diagnoses should be considered (see Table 40-4). Individuals suspected of being MH susceptible should undergo CHCT; the family should also be evaluated and counseled.
Patients with strongly positive CHCT results should undergo screening for the ryanodine receptor 1 gene (RYR1), because mutations in this gene have been found in about 60% of family members who had an MH episode. If an MH-associated mutation (discussed later) is found in RYR1, first-degree relatives have a 50% probability of having a similar defect. Diagnostic testing for RYR1 is performed on DNA obtained from a blood specimen, obviating the need to travel to an MH diagnostic biopsy center or undergo muscle biopsy for the genetic test. Genetic testing of relatives can be undertaken through the office of the primary care physician or by the anesthesiologist, a process that will simplify evaluation of the family. Failure to identify an MH-causative RYR1 mutation (E-Fig. 40-4) does not confirm a negative diagnosis (i.e., not MH susceptible) because more than one gene is associated with MH susceptibility, and not all are known.

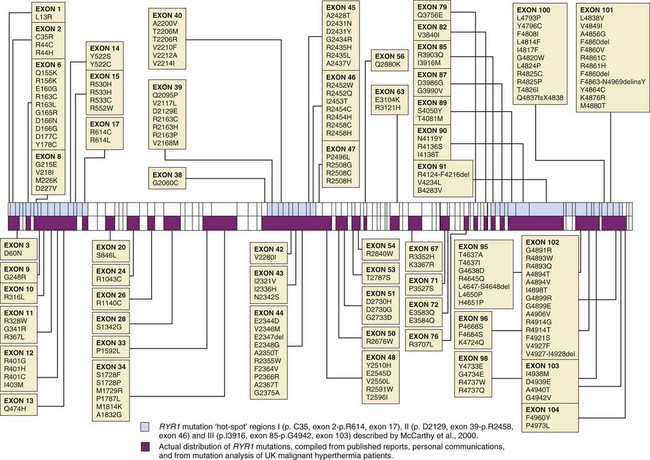
Genetic testing and counseling can be arranged by the anesthesiologist or primary care physician by scheduling an appointment; one such center is located at the Center for Medical Genetics at the University of Pittsburgh. The genetic counselors have extensive experience in counseling patients on the utility of the ryanodine receptor gene test for evaluation of MH susceptibility. The center currently screens 12 exons of genomic RYR1 that commonly contain MH mutations (exons 6, 9, 11, 14, 17, 39, 40, 44, 45, 46, 101, 102). A private commercial laboratory that also offers RYR1 testing is Prevention Genetics (www.preventiongenetics.com), which screens for MH mutations. It has adopted a two-tiered approach: Tier 1 involves bidirectional sequencing of exons 2, 6, 8, 9, 11, 12, 14, 15, 17, 39, 40-41, 44-47, 95, and 100-104. These 22 exons contain most of the conclusively documented MH and central core disease causative mutations in the RYR1 gene (http://www.emhg.org/genetics/). If the first tier is uninformative, their second-tier screen covers the remaining 84 exons of the 106 making up the human RYR1 gene. The Prevention Genetics company corresponds only with physicians and does not provide patient or family counseling.
Genetics
MH in humans follows an autosomal dominant inheritance pattern with incomplete penetrance and variable expressivity.27,102,103 In the context of MH, incomplete penetrance means that there are fewer patients with MH susceptibility than would be predicted by simple autosomal dominant inheritance. Variable expressivity means that the presence of a genetic mutation defining susceptibility is documented, but it does not mean that a patient will have an MH reaction when first exposed to triggering agents. One patient in the NAMHR received 30 anesthetics before an MH reaction was triggered.36 Another was discovered to be MH susceptible by IVCT during screening of a proband’s family and later was inadvertently anesthetized with succinylcholine and isoflurane, but they did not trigger a reaction.104 However, it seems that once an MH reaction has been triggered, the reaction will always be triggered when the patient is exposed to the offending agents.
The molecular and cellular bases of these phenomena remain unknown. Naturally occurring susceptibility to MH seems to follow autosomal dominant inheritance patterns in dogs105 and horses106 but follows a recessive inheritance pattern in pigs.107 This suggests that other genetic and epigenetic factors may play a role in determining the degree and timing of MH susceptibility.
The first breakthrough in finding a gene that predisposed to MH was the serendipitous finding of a similar syndrome in pigs.108,109 When anesthetized with halothane and succinylcholine or with halothane alone, the pigs developed full-blown MH reactions (E-Fig. 40-5). The pig model has been a primary pathophysiologic, genetic, and pharmacologic model for the study of MH over the past 5 decades. A second, serendipitous event introduced a South African anesthesiologist, GG Harrison, to dantrolene, and he successfully tested it in his pig model of MH.110,111 This observation was quickly followed by the successful treatment of a patient with dantrolene.112 An interview about Harrison’s discovery of dantrolene is available from the Wood Library-Museum of the American Society of Anesthesiologists (http://www.woodlibrarymuseum.org/library/media).

One of the most surprising findings was that six separate breeds of pigs at different locations worldwide shared this MH susceptibility to inhalational anesthetics and succinylcholine. All reactions were treated successfully with dantrolene, and all were determined to have an autosomal recessive inheritance pattern. It was established that MH was associated with an uncontrolled increase in intramyoplasmic Ca2+, presumably the result of an exaggerated release of Ca2+ from the sarcoplasmic reticulum (SR).113,114 Much effort has been expended in understanding the physiologic basis of excitation–Ca2+-release coupling (ECRC) as part of the general mechanism of skeletal muscle excitation-contraction coupling.
By the mid-1980s, a large channel in the SR membrane, now known as the ryanodine receptor (RyR1), was identified by its ability to bind a plant toxin, ryanodine. It was discovered to have properties consistent with being a Ca2+ channel.115–117 In 1988, this presumed primary Ca2+-release channel of the SR was found to have gap junction–like channel properties.118 It was hypothesized that the ryanodine receptor might be the site of mutations that caused MH. Within a year, the cDNA for the skeletal muscle ryanodine receptor was cloned.119 Two years after the initial cloning of RyR1 the identical single amino acid mutation (Arg615Cys) was discovered in this channel in all six breeds of MH susceptible pigs.120 RYR1 was immediately acknowledged as a potential target gene in MH susceptibility studies.
Detailed genetic evaluations have linked MH susceptibility to chromosome 19q12-13.2, the location of the human RYR1 gene (19q13.1), in most MH-susceptible families. This is the locus for malignant hyperthermia susceptibility type 1, symbolized by RYR1 (formerly designated MHS1) (Table 40-9).102,121,122 More than 300 variants have been identified in RYR1,123 only about 30 of which have been documented as causing MH (www.emhg.org provides a comprehensive list of known causative mutations in RYR1). Not all MH-susceptible families have disorders linked to this chromosome (see Table 40-9), indicating that this syndrome is genetically heterogeneous.
| Designation | Chromosome Locus | Gene |
|---|---|---|
| Ryanodine receptor 1 (skeletal) (formerly malignant hyperthermia susceptibility 1 [MHS1]) | 19q13.1 | RYR1 |
| Malignant hyperthermia susceptibility 2 | 17q11.2-q24 | MHS2 |
| Malignant hyperthermia susceptibility 4 | 3q13.2 | MHS4 |
| Calcium channel, voltage-dependent, L type, alpha 1S subunit (formerly malignant hyperthermia susceptibility 5 [MHS5]) | 1q32 | CACNA1S |
| Malignant hyperthermia susceptibility 6 | 5p | MHS6 |
The next gene identified, MHS2, was found on chromosome 17q11.2-q24 in North Americans. The previously designated MHS3 locus, a voltage-dependent calcium channel (CACNA2D1), is no longer thought to be linked to MH susceptibility.123 Although the data identify single genes that seem to affect individuals with MH susceptibility, there is discordance between a genetic marker and the likelihood of being MH susceptible.124–126 Linkage analyses of six loci for MH susceptibility and other candidate loci suggest that multiple interacting genes may modify the primary genetic defect and subsequent clinical susceptibility to developing MH.127,128
Complicating the potential of genetic testing for MH even further is the phenomenon of gene silencing. This phenomenon mimics a recessive mutation in heterozygous individuals by allowing expression of only the affected allele while silencing the other, normal, allele.129 Because MH is an autosomal dominant susceptibility, it is conceivable that a mechanism underlying the variability in MH triggering seen in patients with identical, monoallelic RYR1 mutations results from skeletal muscle–specific silencing of the affected gene. It follows that inhalational anesthetics or succinylcholine, after multiple exposures, may release a gene from the silenced state, allowing triggering to occur. This may be an explanation for discordance between genetics, linkage analysis, and trait expressivity.
Although the inheritance of human MH is described as autosomal dominant, there are a few individuals who are allelically homozygous for an RYR1 mutation,130,131 intraallelically heterozygous for two different RYR1 mutations, or compound heterozygotes containing one mutation in RYR1 and a second mutation at another locus for MH susceptibity.132 Surprisingly, no overt myopathies have been reported in these affected individuals.
Physiology
Normal Skeletal Muscle: Excitation-Contraction Coupling
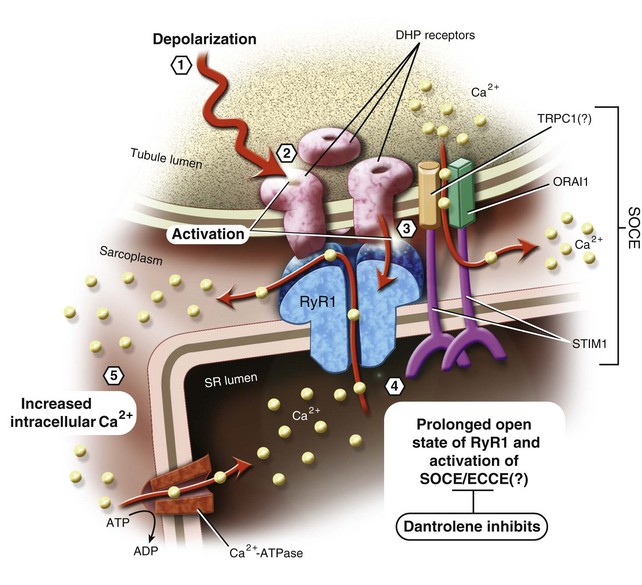
The neurochemical signal that triggers excitation-contraction coupling begins with the release of acetylcholine from the motor nerve terminal at the skeletal muscle nicotinic synapse, resulting in depolarization of the surface membrane, the sarcolemma. Sarcolemmal membrane depolarization is transmitted into the interior of the muscle cell by specialized invaginations of the surface membrane known as transverse tubules (TTs), which occur at regular intervals along the muscle cell (Fig. 40-5). The TT membrane is studded with the skeletal muscle isoform of the voltage-dependent Ca2+ channel known as the dihydropyridine receptor (DHPR). In skeletal muscle, this channel does not transmit Ca2+ in response to sarcolemmal depolarization. Rather, it functions as a sarcolemmal voltage sensor.
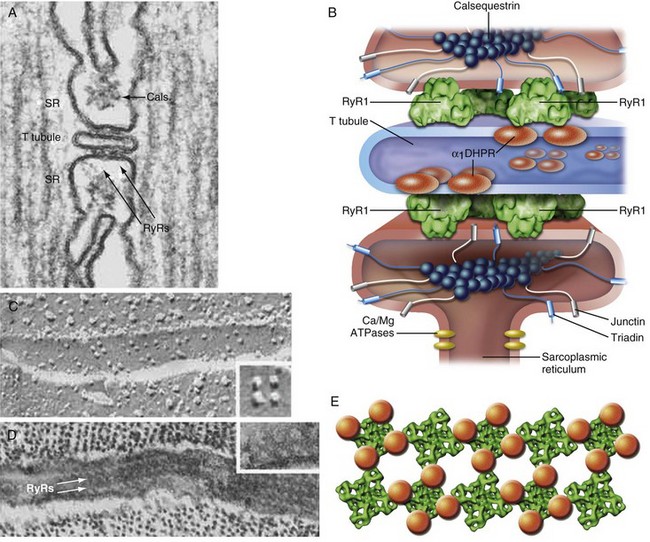
In response to depolarization, intrachannel charge movement across the TT membrane results in a conformational change of the DHPR. The TT is surrounded by specialized portions of the cellular organelle (i.e., SR) that are responsible for maintaining the cellular Ca2+ store, and the SR contains a high-capacity Ca2+ storage protein called calsequestrin, which regulates the ability of the RyR1 channel to open.133 The face of the SR junctional membrane apposing the TT contains a packed, regular array of RyR1 proteins in close apposition to the DHPR. Physically, the DHPR and RyR1 receptor proteins overlie each other in a unique arrangement of four DHPRs (tetrad) per one RyR1, with every other RyR1 lacking a tetrad (Fig. 40-6).134 Physical interaction of the DHPR with RyR1 after depolarization causes the RyR1 channel to open, and SR-stored Ca2+ is released into the myoplasm. Orthograde and anterograde communication between the DHPR and RyR1 results in reciprocal regulation of both entities135,136
In the myoplasm, the troponin C subunit of the troponin complex is bound to tropomyosin, which in the resting state inhibits myosin interaction with actin and maintains a relaxed muscle. Ca2+ binding to troponin C causes a conformational change in troponin and allows the complex to move away from tropomyosin, which rotates along the actin filament in a way that permits myosin head interaction with this fibrous protein. Fiber shortening and muscle contraction then occur in a myosin-ATPase–driven ratcheting reaction. The muscle relaxes when the SR membrane–bound Ca2+-ATPase transports free myoplasmic Ca2+ back into the SR against its concentration gradient in an energy-dependent reaction, thereby driving myoplasmic Ca2+ concentrations down to resting levels. Troponin I, with its bound Ca2+ removed, moves back to block the myosin interaction with actin, thereby preventing muscle contraction and inducing its relaxation. Although a detailed description of the complexity of this process is beyond the scope of this chapter, reviews of excitation-contraction coupling are available.137,138
Pathophysiology of Malignant Hyperthermia
Advances in the pathophysiology of MH have been reviewed in great detail elsewhere.139–142 In brief, at the cellular level, MH is characterized by an inhalational anesthetic–induced, uncontrolled increase in intramyoplasmic Ca2+ levels,143,144 which precedes the metabolic and clinical signs of this syndrome145 (see Fig. 40-6). Increased intramyoplasmic Ca2+ has been demonstrated by directly measuring intracellular Ca2+ in anesthetic-triggered pigs143 and by substantiating the sensitivity of stored Ca2+ release by isolated SR from MH-susceptible pigs.113,114,146,147 There is also a significant loss in the ability of magnesium (Mg2+), the natural inhibitory divalent cation that competes with Ca2+ for binding sites on RyR1, to inhibit Ca2+ release in MH-susceptible skeletal muscle.148–150 RyR1 isolated from MH-susceptible pigs demonstrate greater open probabilities, greater sensitivity to Ca2+ activation, less sensitivity to Ca2+ inactivation, and reduced inhibition by Mg2+. As a result, the affected RyR1 channels spend more time in the open state and less time in the closed state than normal channels.151–155 Although not formally confirmed, this mechanism presumably underlies the sensitivity of RyR1 channels to volatile anesthetics and to the increase in intramyoplasmic Ca2+ seen in MH-susceptible skeletal muscle.
Similar single-channel studies of the Ca2+ responsiveness of human MH-susceptible RyR1 channels have yielded more equivocal results,156 presumably because of the genetic heterogeneity of the human MH-susceptible population. Even within a single individual, heterozygosity for an MH mutation permits a given RyR1 channel that is supposed to be made up of four identical subunits to contain any combination of zero to four MH susceptibility subunits in combination with wild-type, normal RyR1 subunits. When examining single channels from a population of channels isolated from a heterozygous, MH-susceptible patient, the clinician can expect a wider range of channel responses than in the homozygous, inbred, porcine population used for MH models.
Several laboratories have reported the creation of knock-in mice containing one of two known MH-related RyR1 mutations: Tyr522Ser and Arg163Cys.83,84 In contradistinction to the pig model, and like their MH-susceptible human counterparts, the knock-in MH mice are heterozygous. Their homozygous littermates die in utero on day 17. These heterozygous mice become rigid, hyperthermic, and hypermetabolic; die after exposure to inhalational anesthetics or heat stress; and respond therapeutically to dantrolene. They display exaggerated responses to the RyR1 agonists caffeine and 4-chloro-m-cresol and to potassium depolarization. As with MH-susceptible humans and pigs, their muscle is less sensitive to inhibitory Mg2+ and possesses higher resting Ca2+ concentrations than wild-type animals. These experimental animals display a physiologic MH phenotype remarkably similar, if not identical, to the human syndrome and should prove extraordinarily useful in working out the details of MH pathophysiology.
Development of MH seems to require some form of neural input to muscle, because epidural anesthesia in the porcine MH model completely inhibits expression of MH.157 However, complete inhibition of neural input into skeletal muscle in this model by the use of competitive, nondepolarizing, nicotinic cholinergic receptor antagonists such as d-tubocurarine, pancuronium, and vecuronium before a halothane challenge does not inhibit development of MH.158,159 Together, these results suggest that a neurologically significant contribution to MH arises from the central nervous system by means of sympathetic outflow or the neuroendocrine axis, rather than direct skeletal muscle stimulation. This theory is consistent with the awake or stress-related episodes of MH described earlier.
Molecular Mechanisms and Physiologic Effects of Dantrolene
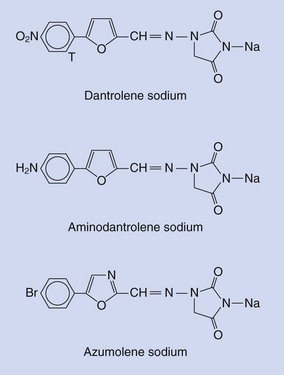
Dantrolene (Fig. 40-7) is a hydantoin derivative originally synthesized as part of an effort to examine the muscle relaxant properties of a series of substituted furan derivatives.160 Thinking that they might have an NMBD, scientists investigated its mechanism of action but found that it was different from the known skeletal muscle relaxants. It affected the intrinsic properties of skeletal muscle without affecting the central nervous system, neuromuscular transmission, electrical properties of the sarcolemma or the T tubule, electromyogram, or train-of-four method for testing neuromuscular blockade. Its action was intracellular (Fig. 40-8).161 Indirect evidence pointed to dantrolene affecting the Ca2+ fluxes that were intrinsic to skeletal muscle contraction.162–164 Direct observations165,166 demonstrated that dantrolene suppressed the rate and amount of Ca2+ released from the SR without completely abolishing it. Subsequently, it was demonstrated that dantrolene inhibited halothane-induced Ca2+ release from the isolated SR of MH-susceptible pigs,146,167,168 while the drug had no effect on calcium reuptake into the SR.169,170
After SR Ca2+ release was identified as the likely target of dantrolene activity, several unsuccessful attempts were made at identifying a dantrolene binding partner in the SR, mainly because of the difficulties of doing detailed pharmacologic receptor analyses with a drug as hydrophobic as dantrolene.171,172 In 1995, an assay for radioactively labeled dantrolene helped to elucidate specific binding sites in porcine skeletal muscle SR.173 Dantrolene inhibits RyR1-dependent cellular Ca2+ fluxes in skeletal muscle, albeit incompletely, but it does not seem to affect RyR1 channel activity or its ability to transport Ca2+.174 With what does dantrolene interact, and how does this interaction relate to reduced Ca2+ fluxes in skeletal muscle and MH?
A second physiologic process, called store-operated Ca2+ entry (SOCE), contributes to the increase in intracellular Ca2+ as a result of the RyR1 channel opening in skeletal muscle.175–177 SOCE is a process by which the SR store of Ca2+ is replenished from the extracellular milieu after significant loss of Ca2+ from the SR.178 Experiments with azumolene, a more water-soluble congener of dantrolene, have demonstrated inhibition of skeletal muscle RyR1-dependent SOCE, not SR Ca2+ release.179 These results raise profound questions. Do the induced Ca2+ fluxes during excitation-contraction coupling, experimental manipulation, and MH that result in an increase in intracellular Ca2+ all result from RyR1-dependent Ca2+ release or RyR1-dependent Ca2+ entry, or both?
Evidence of a dantrolene-sensitive, excitation-coupled Ca2+ entry mechanism of skeletal muscle (ECCE) that is more easily activated in MH skeletal muscle has been described, as well.180–183 ECCE is different from SOCE in that it does not require depletion of the SR Ca2+ store and is activated by high-frequency electrical stimulation of the skeletal muscle membrane. ECCE and SOCE present new physiologic targets of investigation in to the pathophysiology of MH that involve Ca2+ entry rather than Ca2+ release (see Fig. 40-6).
Laboratory Diagnosis
Contracture Testing
The standard for testing susceptibility to MH in the human population is an in vitro contracture test that assesses live human muscle fibers from the vastus lateralis in a physiologic bath. With one end of the muscle tissue attached to a strain gauge, the degree of tension developed at baseline and on electrical stimulation is measured as a function of the concentrations of halothane or caffeine. The test is based on the observation that fresh muscle isolated from MH-susceptible patients behaved abnormally when exposed to halothane or caffeine in vitro (Fig. 40-9).184–187 Two variations of this test have been developed and adopted: the CHCT by the North American Malignant Hyperthermia Group (NAMHG) and the IVCT by the European Malignant Hyperthermia Group (EMHG). The NAMHG test measures the response in separate muscle fascicles to a bolus of 3% halothane or incremental increases in the caffeine concentration,22,188,189 and it requires a positive response to one of the challenges for a diagnosis of MH susceptibility. Those not responding are considered normal. The EMHG test measures the response to graded increases in the halothane concentration up to 2% and incremental increases in the caffeine concentration in different fascicles. The diagnosis of MH susceptibility by the IVCT requires a positive response to both challenges, whereas a positive response to one of the challenging agents results in equivocal diagnostic categorization (i.e., malignant hyperthermia equivocal [MHE]).190–192
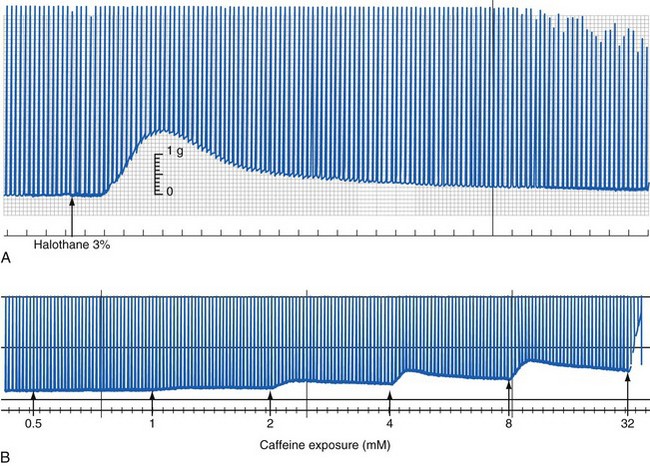
Even though the EMHG protocol may prevent excess false-positive and false-negative results compared to the NAMHG protocol, overall results are comparable.193 The EMHG reports that the sensitivity and specificity of its protocol are 99% and 94%, respectively,191 whereas the NAMHG reports that its sensitivity and specificity are 97% and 78%, respectively.194 Because the North American protocol is less specific and tends to overdiagnose MH susceptibility, it has a very small likelihood of failing to diagnose an MH-susceptible individual. The fact that there are some false-positive findings in the contracture testing demonstrates that other myopathic conditions that are not necessarily concordant with MH have muscle tissue that is also capable of giving an abnormal response to halothane and caffeine exposures. Discordance between the results of an IVCT and the presence of MH mutations in RYR1 can occur in MH families such that 3.1% to 19.4% of family members who do not carry an MH mutation respond positively to an IVCT challenge.75,191 This presumably indicates that there are other factors (e.g., a second MH susceptibility mutation in RYR1, another MH sensitivity locus, a sensitivity to caffeine and halothane that does not reflect MH susceptibility) in these individuals that give a positive IVCT result, but it does not necessarily mean that they are MH susceptible.
Genetic Testing
Limited examination of the RYR1 gene is available for the purpose of diagnosing MH susceptibility at two Clinical Laboratory Improvement Amendments (CLIA)–approved laboratories in North America as discussed earlier. Any physician can write a prescription for the ryanodine receptor gene test for MH susceptibility or fill out the requisition form and send blood for genetic testing. However, the probability of a positive outcome with genetic testing depends in part on the results of the in vitro CHCT because the clinical signs of MH are nonspecific. Current genetic testing indicates that about 60% of those who are positive with the CHCT also have positive genetic test results, whereas only 20% of those without a positive CHCT result yield positive genetic results.75
Other Disorders and Malignant Hyperthermia
Myopathic Syndromes
An incriminatory association between various myopathies and malignant hyperthermia susceptibility, now known to be far more restricted than originally thought, was described in the 1970s and 1980s with much debate over the biochemical and pathophysiologic character of the clinical episodes that resulted from exposure to MH-triggering agents.195,196 Did the clinical episodes represent true MH, did the episodes relate to the instability of myopathic muscle membrane with attendant destruction of muscle cells and release of intracellular proteins, and was there sometimes an associated hyperthermic reaction with these exposures? Although both types of episodes may result in increased creatine kinase levels, hyperkalemia, myoglobinemia, and myoglobinuria, one results from exaggerated ramping of cellular energy and heat production, and the other results from cellular destruction that does not involve abnormal cellular metabolism as a primary cause. Of the congenital myopathies, central core disease, the related multi-minicore disease, and the myopathy of King-Denborough syndrome are the only myopathies known to have a definite relationship with true MH (see Chapter 22).197
Malignant Hyperthermia Mimics
Malignant Hyperthermia–like Syndrome in Pediatric Diabetes Mellitus
Diabetes mellitus has two well-characterized, life-threatening childhood presentations: diabetic ketoacidosis (DKA) and hyperglycemic hyperosmotic nonketotic syndrome (HHNS).198–201 DKA, which is associated with type 1 diabetes, usually manifests as nausea, vomiting, dehydration, and weakness, but shock and coma are uncommon (1% to 2%) in the absence of cerebral edema. Fever is rarely a symptom and usually spurs a search for an underlying infection. HHNS is usually associated with type 2 diabetes and classically manifests with symptoms of increasing polyuria, polydipsia, and lethargy that develop over a few days. The estimated mortality rate is between 12% and 46%, somewhat more dramatic than that seen for DKA (2% to 10%), presumably because most cases of HHNS occur in adults with many other medical problems. The greatest rates of mortality with HHNS occur in adults older than 75 years of age or those with osmolarity values greater than 350 mOsm/L. The incidence of HHNS among U.S. children has been increasing rapidly and appears to be associated with the increase in childhood obesity. Despite this increase, pediatric HHNS is rare, and the presence of fever, as in DKA, usually prompts the search for an underlying infection.
A novel malignant hyperthermia–like syndrome (MHLS) against the background of pediatric diabetes mellitus was described in six adolescent boys between 14 and 18 years of age; the cases were culled from three tertiary care facilities in the United States.202 The features of the syndrome included HHNS with coma, fever, rhabdomyolysis, and severe cardiovascular instability. Among the six adolescents with MHLS, five were obese, five had acanthosis nigricans, four were African American, and four died. Two more cases were later described; one patient died 14 hours after admission due to too rapid a correction of serum osmolarity and resultant cerebral edema and cardiovascular collapse, and a second patient was treated with dantrolene and completely recovered despite developing compartment syndrome in her left upper extremity due to rhabdomyolysis.203 The survivor was tested for metabolic abnormalities, and a deficiency in short-chain acyl-coenzyme A (acyl-CoA) dehydrogenase was found.
Investigators recommend that anyone presenting with symptoms of HHNS and MHLS should be treated with dantrolene as soon as the syndrome is recognized and that fluid and insulin therapy be used for an appropriate rate of correction of serum osmolarity. A search of the literature reveals a similar case described 10 years earlier as fulminant MH associated with DKA in a patient who survived with the addition of dantrolene to his treatment regimen.204 Although it is difficult to ascertain the efficacy of dantrolene in abrogating the deleterious effects on skeletal muscle and in saving critically ill patients with MHLS during HHNS with such a small number of successfully treated patients, it seems prudent to initiate immediate treatment with dantrolene in these cases until the data can inform us about the true efficacy of this drug in MHLS.
Disorders of Fatty Acid Metabolism
Growing numbers of reports have documented cases of rhabdomyolysis in patients with disorders of fatty acid metabolism that in some ways mimic awake MH. These disorders arise from mutations in the enzymes responsible for the metabolism of various fatty acids and for ensuring adequate energy substrate for the mitochondria during periods of reduced glucose availability, such as during stress. Although these disorders have profound effects on energy metabolism, they should not be confused with mitochondrial myopathies, which result from completely different molecular defects and have mutations in the proteins of the mitochondrial respiratory chain (see Chapter 22). In neither case has a real association with MH been established, but there have been case reports of rhabdomyolysis and cardiac arrest in patients with carnitine palmitoyltransferase II deficiency.205–207
Various forms of acyl-CoA dehydrogenase deficiencies (i.e., very-long-chain, long-chain, medium-chain, and short-chain forms) lead to different types of myopathy that can have severe clinical consequences, including hypoglycemia and rhabdomyolysis with attendant multiple-organ dysfunction, which in some deficiencies are brought about by stress, particularly heat and severe exercise.208,209 These diseases can manifest early in childhood or late in adolescence or young adulthood, and they can be a particular problem for individuals in the military or those who participate in intense sports.209 Patients with the very-long-chain acyl-CoA dehydrogenase deficiency can present with acute hypercapnic respiratory failure.210 Dantrolene has been used successfully to treat one case of recurrent rhabdomyolysis in a patient with very-long-chain acyl-CoA dehydrogenase deficiency,211 and it may be useful to treat acute intraoperative rhabdomyolysis in these patients, although there is a dearth of evidence in this regard.
Children may present for surgery without a diagnosis of an inborn error of fatty acid metabolism and develop intraoperative rhabdomyolysis, mimicking aspects of a fulminant MH reaction. This may be the first manifestation of the child’s fatty acid metabolism disorder. Preoperative increases in serum creatine kinase and uric acid levels, presumably due to subclinical rhabdomyolysis, suggest an inborn error of metabolism or myopathy,212 but these tests are not part of the usual preoperative panel of blood tests in normal pediatric anesthesia practice. Perioperative stress that may result from fasting, fear, disease states, and other causes can induce metabolic decompensation and hypoglycemia. As a consequence, an intravenous glucose-electrolyte solution is recommended for affected children.213 Because of a few reports of rhabdomyolysis when these children are anesthetized with inhalational anesthetics, there has been some reluctance to use inhalational anesthetics in them.213,214 It is, however, likely that the number of patients with inborn errors of fatty acid metabolism who undergo surgery is far greater than the paucity of reports of adverse outcomes with inhalational anesthetics. Because these patients vary considerably in their responses to stress, it is not surprising that most do well with any well-managed anesthetic. The available evidence suggests that the perioperative risk is no greater with any particular anesthetic in these children.
If the clinician elects to avoid inhalational anesthetics, two alternatives remain: regional anesthesia and total intravenous anesthesia (TIVA). In children, peripheral limb surgery often allows for the use of intravenous sedation and regional anesthesia that may be suitable depending on the age of the child,191 but most children do not tolerate this technique. However, TIVA often is used in children, most commonly with propofol.215
Propofol infusion syndrome, a rare, usually lethal complication of prolonged infusions of propofol, is diagnosed by cardiovascular collapse associated with lipemic plasma, enlarged fatty liver, severe metabolic acidosis, and rhabdomyolysis or myoglobinuria.216–218 In this syndrome, a large increase in particular fatty acids (i.e., malonylcarnitine and C5-acylcarnitine) that points to impaired entry of long-chain fatty acids into mitochondria and to resultant failure of mitochondrial respiration at complex II has been identified.219 Others suggest that propofol infusion syndrome may uncover medium-chain acyl-CoA dehydrogenase deficiencies, although this remains unproved.216 The notion that propofol can impair fatty acid uptake and subsequent oxidation raises the possibility that the acute administration of propofol to a patient with a defect in fatty acid metabolism can precipitate a metabolic crisis, although this has never been reported. It has also been suggested that the lipid load from a propofol infusion in the absence of adequate carbohydrate intake can expose a carnitine deficiency as a model for propofol infusion syndrome.220 Moreover, propofol itself has been shown to inhibit mitochondrial respiration, possibly compounding the effects of fatty acid oxidation deficiencies.216 In these children, the use of propofol may be associated with an unclear risk of inducing a metabolic crisis. If their metabolic abnormalities are subclinical, there is no easy, inexpensive preoperative screening tool to establish a diagnosis for a rare abnormality. Even if the child is diagnosed with one of the subsets of fatty acid oxidation deficiencies, it is impossible to predict preoperatively which children will be sensitive to propofol and, if they are, how sensitive they are.
Neuroleptic Malignant Syndrome
Neuroleptic malignant syndrome (NMS) is a rare, potentially lethal reaction to neuroleptics (0.1% to 2.5% of patients), which is characterized clinically by the slow onset of fever, muscle rigidity, altered consciousness, and autonomic instability over a protracted period of time.221 Laboratory findings include increased creatine kinase levels, leukocytosis, increased liver enzyme values, and reduced serum iron or potassium concentrations.221 NMS is similar to MH, and the distinction between the two is often difficult to make, except by medication history; neuroleptics are associated with NMS, and inhalational anesthetics and succinylcholine are associated with MH.
Successful therapy for NMS requires early recognition, cessation of offending medications, and intensive medical and nursing care geared toward hydration and restoration of electrolyte balance.221 Specific pharmacologic therapy with dopamine agonists such as bromocriptine or with dantrolene has been advocated, although use of dantrolene is controversial. Despite the fact that textbooks of psychiatry list dantrolene as the first-line pharmacologic treatment for NMS, there are no good data that it has a therapeutic effect in NMS, except for the occasional case report declaring that dantrolene is effective.221–223
Children with psychiatric diagnoses requiring treatment with neuroleptics make up a significant percentage of these pediatric patients. NMS in this pediatric population continues to be a problem, even with the newest of drugs. The perioperative period for the pediatric patient on neuroleptics is one fraught with potential diagnostic dilemmas. For example, one report described postoperative NMS in a child with severe cerebral palsy and seizure disorder who was not taking any neuroleptics and who was successfully treated three times with dantrolene.224 Was this NMS or a mild form of MH? The answer is unclear.
Hirshey Dirksen SJ, Larach MG, Rosenberg H, et al. Special article: Future directions in malignant hyperthermia research and patient care. Anesth Analg. 2011;113:1108–1119.
Hopkins PM. Malignant hyperthermia: pharmacology of triggering. Br J Anaesth. 2011;107:48–56.
Kolb ME, Horne ML, Martz R. Dantrolene in human malignant hyperthermia. Anesthesiology. 1982;56:254–262.
This is a classic report of dantrolene efficacy in treating malignant hyperthermia.
Krause T, Gerbershagen MU, Fiege M, et al. Dantrolene—a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373.
Different aspects of dantrolene pharmacology are well reviewed.
Larach MG, Localio AR, Allen GC, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994;80:771–779.
A retrospective clinical grading scale is used to predict a malignant hyperthermia episode.
Lerman J, McLeod ME, Strong HA. Pharmacokinetics of intravenous dantrolene in children. Anesthesiology. 1989;70:625–629.
The paper describes the first and only treatment of malignant hyperthermia.
Litman RS, Rosenberg H. Malignant hyperthermia: update on susceptibility testing. JAMA. 2005;15:2918–2924.
Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RyR1 in malignant hyperthermia and central core disease. Hum Mutat. 2006;27:977–989.
Rosenberg H, Sambuughin N, Dirksen R. Malignant hyperthermia susceptibility. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews [Internet]. Seattle: University of Washington, 2003. 1993-Dec. 19 [updated Jan. 19, 2010]
The most comprehensive review available in the literature to date.
Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve. 2006;33:715–731.
1 Denborough M, Lovell R. Anesthetic deaths in a family. Lancet. 1960;2:45–46.
2 Denborough MA, Forster JF, Lovell RR, Mapleson PA, Villiers JD. Anaesthetic deaths in a family. Br J Anaesth.. 1962;34:395–396.
3 Saidman LJ, Havard ES, Eger EI. Hyperthermia during anesthesia. JAMA. 1964;190:1029–1032.
4 Cullen WG. Malignant hyperpyrexia during general anaesthesia. Can Anaesth Soc J. 1966;13:415–418.
5 Britt BA. Recent advances in malignant hyperthermia. Anesth Analg. 1972;51:841–850.
6 Relton JE, Britt BA, Steward DJ. Malignant hyperpyrexia. Br J Anaesth. 1973;45:269–275.
7 Ording H. Incidence of malignant hyperthermia in Denmark. Anesth Analg. 1985;64:700–704.
8 Warner LO, Beach TP, Garvin JP, Warner EJ. Halothane and children: the first quarter century. Anesth Analg. 1984;63:838–840.
9 Ellis FR, Halsall PJ. Malignant hyperpyrexia. Br J Hosp Med. 24, 1980. 318–317
10 Strazis KP, Fox AW. Malignant hyperthermia: a review of published cases. Anesth Analg. 1993;77:297–304.
11 Rosero EB, Adesanya AO, Timaran CH, Joshi GP. Trends and outcomes of malignant hyperthermia in the United States, 2000 to 2005. Anesthesiology. 2009;110:89–94.
12 Wilhoit RD, Brown RE, Jr., Bauman LA. Possible malignant hyperthermia in a 7-week-old infant. Anesth Analg. 1989;68:688–691.
13 Goudsouzian NG. Recent changes in the package insert for succinylcholine chloride: should this drug be contraindicated for routine use in children and adolescents? (Summary of the discussions of the anesthetic and life support drug advisory meeting of the Food and Drug Administration, FDA building, Rockville, MD, June 9, 1994.). Anesth Analg. 1995;80:207–208.
14 Larach MG, Rosenberg H, Gronert GA, Allen GC. Hyperkalemic cardiac arrest during anesthesia in infants and children with occult myopathies. Clin Pediatr (Phila). 1997;36:9–16.
15 Britt BA, Gordon RA. Three cases of malignant hyperthermia with special consideration of management. Can Anaesth Soc J. 1969;16:99–105.
16 Chambers FA, Casey W, Dowling F, Moore KP. Malignant hyperthermia during isoflurane anaesthesia. Can J Anaesth. 1994;41:355–356.
17 Ducart A, Adnet P, Renaud B, Riou B, Krivosic-Horber R. Malignant hyperthermia during sevoflurane administration. Anesth Analg. 1995;80:609–611.
18 Otsuka H, Komura Y, Mayumi T, et al. Malignant hyperthermia during sevoflurane anesthesia in a child with central core disease. Anesthesiology. 1991;75:699–701.
19 Fu ES, Scharf JE, Mangar D, Miller WD. Malignant hyperthermia involving the administration of desflurane. Can J Anaesth. 1996;43:687–690.
20 Froeba G, Marx T, Pazhur J, et al. Xenon does not trigger malignant hyperthermia in susceptible swine. Anesthesiology. 1999;91:1047–1052.
21 Baur CP, Klingler W, Jurkat-Rott K, et al. Xenon does not induce contracture in human malignant hyperthermia muscle. Br J Anaesth. 2000;85:712–716.
22 Ali SZ, Taguchi A, Rosenberg H. Malignant hyperthermia. Best Pract Res Clin Anaesthesiol. 2003;17:519–533.
23 Litman RS, Rosenberg H. Malignant hyperthermia: update on susceptibility testing. JAMA. 2005;293:2918–2924.
24 Larach MG, Brandom BW, Allen GC, Gronert GA, Lehman EB. Cardiac arrests and deaths associated with malignant hyperthermia in north america from 1987 to 2006: a report from the north american malignant hyperthermia registry of the malignant hyperthermia association of the United States. Anesthesiology. 2008;108:603–611.
25 Coté CJ, Liu LM, Szyfelbein SK, et al. Intraoperative events diagnosed by expired carbon dioxide monitoring in children. Can Anaesth Soc J. 1986;33:315–320.
26 Baudendistel L, Goudsouzian N, Coté CJ, Strafford M. End-tidal CO2 monitoring. Its use in the diagnosis and management of malignant hyperthermia. Anaesthesia. 1984;39:1000–1003.
27 Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131–1136.
28 Rosenberg H, Fletcher JE. Masseter muscle rigidity and malignant hyperthermia susceptibility. Anesth Analg. 1986;65:161–164.
29 Allen GC, Rosenberg H. Malignant hyperthermia susceptibility in adult patients with masseter muscle rigidity. Can J Anaesth. 1990;37:31–35.
30 O’Flynn RP, Shutack JG, Rosenberg H, Fletcher JE. Masseter muscle rigidity and malignant hyperthermia susceptibility in pediatric patients. An update on management and diagnosis. Anesthesiology. 1994;80:1228–1233.
31 Hall GM, Lucke JN. Porcine malignant hyperthermia. IX: Changes in the concentrations of intramuscular high-energy phosphates, glycogen and glycolytic intermediates. Br J Anaesth. 1983;55:635–640.
32 Decanniere C, Van HP, Vanstapel F, Ville H, Geers R. Metabolic response to halothane in piglets susceptible to malignant hyperthermia: an in vivo 31P-NMR study. J Appl Physiol. 1993;75:955–962.
33 Ryan JF, Papper EM. Malignant fever during and following anesthesia. Anesthesiology. 1970;32:196–201.
34 Litman RS, Flood CD, Kaplan RF, Kim YL, Tobin JR. Postoperative malignant hyperthermia: an analysis of cases from the North American Malignant Hyperthermia Registry. Anesthesiology. 2008;109:825–829.
35 Souliere CR, Jr., Weintraub SJ, Kirchner JC. Markedly delayed postoperative malignant hyperthermia. Arch Otolaryngol Head Neck Surg. 1986;112:564–566.
36 Larach MG, Gronert GA, Allen GC, Brandom BW, Lehman EB. Clinical presentation, treatment, and complications of malignant hyperthermia in North America from 1987 to 2006. Anesth Analg. 2010;110:498–507.
37 Larach MG, Localio AR, Allen GC, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994;80:771–779.
38 Yentis SM, Levine MF, Hartley EJ. Should all children with suspected or confirmed malignant hyperthermia susceptibility be admitted after surgery? A 10-year review. Anesth Analg. 1992;75:345–350.
39 Pollock N, Langton E, McDonnell N, Tiemessen J, Stowell K. Malignant hyperthermia and day stay surgery. Anaesth Intensive Care. 2006;34:40–45.
40 Pollock N, Langtont E, Stowell K, Simpson C, McDonnell N. Safe duration of postoperative monitoring for malignant hyperthermia susceptible patients. Anaesth Intensive Care. 2004;32:502–509.
41 Ritchie PA, Cheshire MA, Pearce NH. Decontamination of halothane from anaesthetic machines achieved by continuous flushing with oxygen. Br J Anaesth. 1988;60:859–863.
42 Beebe JJ, Sessler DI. Preparation of anesthesia machines for patients susceptible to malignant hyperthermia. Anesthesiology. 1988;69:395–400.
43 Gilly H, Weindlmayr-Goettel M, Koberl G, Steinbereithner K. Anaesthetic uptake and washout characteristics of patient circuit tubing with special regard to current decontamination techniques. Acta Anaesthesiol Scand. 1992;36:621–627.
44 Kim TW, Nemergut ME. Preparation of modern anesthesia workstations for malignant hyperthermia-susceptible patients: a review of past and present practice. Anesthesiology. 2011;114:205–212.
45 Hopkins PM. Malignant hyperthermia: pharmacology of triggering. Br J Anaesth. 2011;107:48–56.
46 Crawford MW, Prinzhausen H, Petroz GC. Accelerating the washout of inhalational anesthetics from the Drager Primus anesthetic workstation: effect of exchangeable internal components. Anesthesiology. 2007;106:289–294.
47 Shanahan H, O’Donoghue R, O’Kelly P, Synnott A, O’Rourke J. Preparation of the Drager Fabius CE and Drager Zeus anaesthetic machines for patients susceptible to malignant hyperthermia. Eur J Anaesthesiol. 2012;29:229–234.
48 Sabouri S, Lerman J, Heard C. Residual sevoflurane may be present after flushing the GE Anesthesia Workstation for MH susceptible patients. Anesthesiology. 2011;115:A1276.
49 Petroz GC, Lerman J. Preparation of the Siemens KION anesthetic machine for patients susceptible to malignant hyperthermia. Anesthesiology. 2002;96:941–946.
50 Birgenheier N, Stoker R, Westenskow D, Orr J. Activated charcoal effectively removes inhaled anesthetics from modern anesthesia machines. Anesth Analg. 2011;112:1363–1370.
51 Durham WJ, Aracena-Parks P, Long C, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65.
52 Ebert TJ, Muzi M. Sympathetic hyperactivity during desflurane anesthesia in healthy volunteers. A comparison with isoflurane. Anesthesiology. 1993;79:444–453.
53 Ebert TJ, Muzi M, Lopatka CW. Neurocirculatory responses to sevoflurane in humans. A comparison to desflurane. Anesthesiology. 1995;83:88–95.
54 Britt BA, Endrenyi L, Frodis W, Scott E, Kalow W. Comparison of effects of several inhalation anaesthetics on caffeine-induced contractures of normal and malignant hyperthermic skeletal muscle. Can Anaesth Soc J. 1980;27:12–15.
55 Migita T, Mukaida K, Kobayashi M, Hamada H, Kawamoto M. The severity of sevoflurane-induced malignant hyperthermia. Acta Anaesthesiol Scand. 2012;56:351–356.
56 Metterlein T, Schuster F, Kranke P, Roewer N, Anetseder M. In-vitro contracture testing for susceptibility to malignant hyperthermia: can halothane be replaced? Eur J Anaesthesiol. 2011;28:251–255.
57 Bennett MH, Wainwright AP. Acute thyroid crisis on induction of anaesthesia. Anaesthesia. 1989;44:28–30.
58 Purday JP, Montgomery CJ, Blackstock D. Intraoperative hyperthermia in a paediatric patient with cystinosis. Pediatr Anaesth. 1995;5:389–392.
59 Crowley KJ, Cunningham AJ, Conroy B, O’Connell PR, Collins PG. Phaeochromocytoma—a presentation mimicking malignant hyperthermia. Anaesthesia. 1988;43:1031–1032.
60 Leung WK, Jahr JS, Hotz J, Pollock M. Nonmalignant hyperthermia on induction of anesthesia in a pediatric patient undergoing bidirectional Glenn procedure. J Clin Anesth. 1998;10:427–431.
61 Mitchell LW, Leighton BL. Warmed diluent speeds dantrolene reconstitution. Can J Anaesth. 2003;50:127–130.
62 Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F. Dantrolene—a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373.
63 Brandom BW, Larach MG, Chen MS, Young MC. Complications associated with the administration of dantrolene 1987 to 2006: a report from the North American Malignant Hyperthermia Registry of the Malignant Hyperthermia Association of the United States. Anesth Analg. 2011;112:1115–1123.
64 Britt BA. Dantrolene. Can Anaesth Soc J. 1984;31:61–75.
65 Podranski T, Bouillon T, Schumacher PM, et al. Compartmental pharmacokinetics of dantrolene in adults: do malignant hyperthermia association dosing guidelines work? Anesth Analg. 2005;101:1695–1699.
66 Schutte JK, Becker S, Burmester S, et al. Comparison of the therapeutic effectiveness of a dantrolene sodium solution and a novel nanocrystalline suspension of dantrolene sodium in malignant hyperthermia normal and susceptible pigs. Eur J Anaesthesiol. 2011;28:256–264.
67 Lerman J, McLeod ME, Strong HA. Pharmacokinetics of intravenous dantrolene in children. Anesthesiology. 1989;70:625–629.
68 Kolb ME, Horne ML, Martz R. Dantrolene in human malignant hyperthermia. Anesthesiology. 1982;56:254–262.
69 DeRuyter ML, Wedel DJ, Berge KH. Hyperthermia requiring prolonged administration of high-dose dantrolene in the postoperative period. Anesth Analg. 1995;80:834–836.
70 Burkman JM, Posner KL, Domino KB. Analysis of the clinical variables associated with recrudescence after malignant hyperthermia reactions. Anesthesiology. 2007;106:901–906.
71 Mathieu A, Bogosian AJ, Ryan JF, Crone RK, Crocker D. Recrudescence after survival of an initial episode of malignant hyperthermia. Anesthesiology. 1979;51:454–455.
72 Hackl W, Mauritz W, Winkler M, Sporn P, Steinbereithner K. Anaesthesia in malignant hyperthermia-susceptible patients without dantrolene prophylaxis: a report of 30 cases. Acta Anaesthesiol Scand. 1990;34:534–537.
73 Pinder RM, Brogden RN, Speight TM, Avery GS. Dantrolene sodium: a review of its pharmacological properties and therapeutic efficacy in spasticity. Drugs. 1977;13:3–23.
74 Rubin AS, Zablocki AD. Hyperkalemia, verapamil, and dantrolene. Anesthesiology. 1987;66:246–249.
75 Monnier N, Kozak-Ribbens G, Krivosic-Horber R, et al. Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Hum Mutat. 2005;26:413–425.
76 San Juan AC, Jr., Wong KC, Port JD. Hyperkalemia after dantrolene and verapamil-dantrolene administration in dogs. Anesth Analg. 1988;67:759–762.
77 Jensen AG, Bach V, Werner MU, Nielsen HK, Jensen MH. A fatal case of malignant hyperthermia following isoflurane anaesthesia. Acta Anaesthesiol Scand. 1986;30:293–294.
78 Wingard DW. Malignant hyperthermia: a human stress syndrome [letter]? Lancet. 1974;2:1450–1451.
79 Harrison GG. Porcine malignant hyperthermia—the saga of the “hot” pig. In: Britt BA, ed. Malignant hyperthermia. Boston: Martinus Nijhoff; 1987:103–136.
80 Gronert GA, Thompson RL, Onofrio BM. Human malignant hyperthermia: awake episodes and correction by dantrolene. Anesth Analg. 1980;59:377–378.
81 Denborough MA. Heat stroke and malignant hyperpyrexia. Med J Aust. 1982;1:204–205.
82 Denborough M, Hopkinson KC, O’Brien RO, Foster PS. Overheating alone can trigger malignant hyperthermia in piglets. Anaesth Intensive Care. 1996;24:348–354.
83 Chelu MG, Goonasekera SA, Durham WJ, et al. Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 2006;20:329–330.
84 Yang T, Riehl J, Esteve E, et al. Pharmacologic and functional characterization of malignant hyperthermia in the R163C RyR1 knock-in mouse. Anesthesiology. 2006;105:1164–1175.
85 Tobin JR, Jason DR, Challa VR, Nelson TE, Sambuughin N. Malignant hyperthermia and apparent heat stroke. JAMA. 2001;286:168–169.
86 Gronert GA, Tobin JR, Muldoon S. Malignant hyperthermia—human stress triggering. Biochim Biophys Acta. 2011;1813:2191–2192.
87 Groom L, Muldoon SM, Tang ZZ, et al. Identical de novo mutation in the type 1 ryanodine receptor gene associated with fatal, stress-induced malignant hyperthermia in two unrelated families. Anesthesiology. 2011;115:938–945.
88 Hackl W, Winkler M, Mauritz W, Sporn P, Steinbereithner K. Muscle biopsy for diagnosis of malignant hyperthermia susceptibility in two patients with severe exercise-induced myolysis. Br J Anaesth. 1991;66:138–140.
89 Hopkins PM, Ellis FR, Halsall PJ. Evidence for related myopathies in exertional heat stroke and malignant hyperthermia. Lancet. 1991;338:1491–1492.
90 Ogletree JW, Antognini JF, Gronert GA. Postexercise muscle cramping associated with positive malignant hyperthermia contracture testing. Am J Sports Med. 1996;24:49–51.
91 Köchling A, Wappler F, Winkler G, Schulte am Esch JS. Rhabdomyolysis following severe physical exercise in a patient with predisposition to malignant hyperthermia. Anaesth Intensive Care. 1998;26:315–318.
92 Capacchione JF, Muldoon SM. The relationship between exertional heat illness, exertional rhabdomyolysis, and malignant hyperthermia. Anesth Analg. 2009;109:1065–1069.
93 Bendahan D, Kozak-Ribbens G, Confort-Gouny S, et al. A noninvasive investigation of muscle energetics supports similarities between exertional heat stroke and malignant hyperthermia. Anesth Analg. 2001;93:683–689.
94 Larner AJ. Dantrolene for exertional heatstroke. Lancet. 1992;339:182.
95 Grogan H, Hopkins PM. Heat stroke: implications for critical care and anaesthesia. Br J Anaesth. 2002;88:700–707.
96 Lichtman AD, Oribabor C. Malignant hyperthermia following systemic rewarming after hypothermic cardiopulmonary bypass. Anesth Analg. 2006;102:372–375.
97 Riess FC, Fiege M, Moshar S, et al. Rhabdomyolysis following cardiopulmonary bypass and treatment with enoximone in a patient susceptible to malignant hyperthermia. Anesthesiology. 2001;94:355–357.
98 Carr AS, Lerman J, Cunliffe M, McLeod ME, Britt BA. Incidence of malignant hyperthermia reactions in 2,214 patients undergoing muscle biopsy. Can J Anaesth. 1995;42:281–286.
99 Loke JC, MacLennan DH. Bayesian modeling of muscle biopsy contracture testing for malignant hyperthermia susceptibility. Anesthesiology. 1998;88:589–600.
100 Isaacs H, Badenhorst M. False-negative results with muscle caffeine halothane contracture testing for malignant hyperthermia. Anesthesiology. 1993;79:5–9.
101 Allen GC, Rosenberg H, Fletcher JE. Safety of general anesthesia in patients previously tested negative for malignant hyperthermia susceptibility. Anesthesiology. 1990;72:619–622.
102 Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat. 2006;27:977–989.
103 Harriman DG. Malignant hyperthermia myopathy—a critical review. Br J Anaesth. 1988;60:309–316.
104 Claxton BA, Cross MH, Hopkins PM. No response to trigger agents in a malignant hyperthermia-susceptible patient. Br J Anaesth. 2002;88:870–873.
105 Roberts MC, Mickelson JR, Patterson EE, et al. Autosomal dominant canine malignant hyperthermia is caused by a mutation in the gene encoding the skeletal muscle calcium release channel (RYR1). Anesthesiology. 2001;95:716–725.
106 Aleman M, Riehl J, Aldridge BM, et al. Association of a mutation in the ryanodine receptor 1 gene with equine malignant hyperthermia. Muscle Nerve. 2004;30:356–365.
107 Fletcher JE, Calvo PA, Rosenberg H. Phenotypes associated with malignant hyperthermia susceptibility in swine genotyped as homozygous or heterozygous for the ryanodine receptor mutation. Br J Anaesth. 1993;71:410–417.
108 Hall LW, Woolf N, Bradley JW, Jolly DW. Unusual reaction to suxamethonium chloride. Br Med J. 1966;2:1305.
109 Harrison GG, Biebuyck JF, Terblanche J, et al. Hyperpyrexia during anaesthesia. Br Med J. 1968;3:594–595.
110 Britt BA. A history of malignant hyperthermia. In: Britt BA, ed. Malignant hyperthermia. Boston: Martinus Nijhoff, 1987.
111 Harrison GG. Control of the malignant hyperpyrexic syndrome in MHS swine by dantrolene sodium. Br J Anaesth. 1975;47:62–65.
112 Harrison GG. Dantrolene sodium in the treatment of malignant hyperthermia. A case report. S Afr Med J. 1981;60:909–910.
113 Kim DH, Sreter FA, Ohnishi ST, et al. Kinetic studies of Ca2+ release from sarcoplasmic reticulum of normal and malignant hyperthermia susceptible pig muscles. Biochim Biophys Acta. 1984;775:320–327.
114 Nelson TE. Abnormality in calcium release from skeletal sarcoplasmic reticulum of pigs susceptible to malignant hyperthermia. J Clin Invest. 1983;72:862–870.
115 Pessah IN, Waterhouse AL, Casida JE. The calcium-ryanodine receptor complex of skeletal and cardiac muscle. Biochem Biophys Res Commun. 1985;128:449–456.
116 Pessah IN, Francini AO, Scales DJ, Waterhouse AL, Casida JE. Calcium-ryanodine receptor complex. Solubilization and partial characterization from skeletal muscle junctional sarcoplasmic reticulum vesicles. J Biol Chem. 1986;261:8643–8648.
117 Maurer A, Tanaka M, Ozawa T, Fleischer S. Purification and crystallization of the calcium binding protein of sarcoplasmic reticulum from skeletal muscle. Proc Natl Acad Sci U S A. 1985;82:4036–4040.
118 Ma J, Fill M, Knudson CM, Campbell KP, Coronado R. Ryanodine receptor of skeletal muscle is a gap junction-type channel. Science. 1988;242:99–102.
119 Takeshima H, Nishimura S, Matsumoto T, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–445.
120 Fujii J, Otsu K, Zorzato F, et al. Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science. 1991;253:448–451.
121 Jurkat-Rott K, McCarthy T, Lehmann-Horn F. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve. 2000;23:4–17.
122 Brandom BW. Genetics of malignant hyperthermia. Sci World J. 2006;6:1722–1730.
123 Rosenberg H, Sambuughin N, Dirksen R. Malignant hyperthermia susceptibility. In: Pagon RA, Bird TD, Dolan CR, Adam MP, eds. GeneReviews. Seattle: University of Washington, 2012. Available at http://www.ncbi.nlm.nih.gov/books/NBK1146/ (accessed July 2012)
124 Fagerlund TH, Ording H, Bendixen D, et al. Discordance between malignant hyperthermia susceptibility and RYR1 mutation C1840T in two Scandinavian MH families exhibiting this mutation. Clin Genet. 1997;52:416–421.
125 Galli L, Orrico A, Cozzolino S, et al. Mutations in the RYR1 gene in Italian patients at risk for malignant hyperthermia: evidence for a cluster of novel mutations in the C-terminal region. Cell Calcium. 2002;32:143–151.
126 Robinson RL, Anetseder MJ, Brancadoro V, et al. Recent advances in the diagnosis of malignant hyperthermia susceptibility: how confident can we be of genetic testing? Eur J Hum Genet. 2003;11:342–348.
127 Robinson RL, Curran JL, Ellis FR, et al. Multiple interacting gene products may influence susceptibility to malignant hyperthermia. Ann Hum Genet. 2000;64:307–320.
128 Robinson R, Hopkins P, Carsana A, et al. Several interacting genes influence the malignant hyperthermia phenotype. Hum Genet. 2003;112:217–218.
129 Lu Q, Qiu X, Hu N, et al. Epigenetics, disease, and therapeutic interventions. Ageing Res Rev. 2006;5:449–467.
130 Lynch PJ, Krivosic-Horber R, Reyford H, et al. Identification of heterozygous and homozygous individuals with the novel RYR1 mutation Cys35Arg in a large kindred. Anesthesiology. 1997;86:620–626.
131 Rueffert H, Olthoff D, Deutrich C, Thamm B, Froster UG. Homozygous and heterozygous Arg614Cys mutations (1840C→T) in the ryanodine receptor gene co-segregate with malignant hyperthermia susceptibility in a German family. Br J Anaesth. 2001;87:240–245.
132 Monnier N, Krivosic-Horber R, Payen JF, et al. Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology. 2002;97:1067–1074.
133 Beard NA, Wei L, Dulhunty AF. Control of muscle ryanodine receptor calcium release channels by proteins in the sarcoplasmic reticulum lumen. Clin Exp Pharmacol Physiol. 2009;36:340–345.
134 Paolini C, Protasi F, Franzini-Armstrong C. The relative position of RyR feet and DHPR tetrads in skeletal muscle. J Mol Biol. 2004;342:145–153.
135 Andronache Z, Hamilton SL, Dirksen RT, Melzer W. A retrograde signal from RyR1 alters DHP receptor inactivation and limits window Ca2+ release in muscle fibers of Y522S RyR1 knock-in mice. Proc Natl Acad Sci U S A. 2009;106:4531–4536.
136 Sheridan DC, Takekura H, Franzini-Armstrong C, et al. Bidirectional signaling between calcium channels of skeletal muscle requires multiple direct and indirect interactions. Proc Natl Acad Sci U S A. 2006;103:19760–19765.
137 Dulhunty AF. Excitation-contraction coupling from the 1950s into the new millennium. Clin Exp Pharmacol Physiol. 2006;33:763–772.
138 Protasi F. Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front Biosci. 2002;7:d650–d658.
139 Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve. 2006;33:715–731.
140 Avila G. Intracellular Ca2+ dynamics in malignant hyperthermia and central core disease: established concepts, new cellular mechanisms involved. Cell Calcium. 2005;37:121–127.
141 Nelson TE. Malignant hyperthermia: a pharmacogenetic disease of Ca++ regulating proteins. Curr Mol Med. 2002;2:347–369.
142 Treves S, Anderson AA, Ducreux S, et al. Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul Disord. 2005;15:577–587.
143 Lopez JR, Allen PD, Alamo L, Jones D, Sreter FA. Myoplasmic free [Ca2+] during a malignant hyperthermia episode in swine. Muscle Nerve. 1988;11:82–88.
144 Ohta T, Endo M, Nakano T, et al. Ca-induced Ca release in malignant hyperthermia-susceptible pig skeletal muscle. Am J Physiol. 1989;256:C358–C367.
145 Ryan JF, Lopez JR, Sanchez VB, Sreter FA, Allen PD. Myoplasmic calcium changes precede metabolic and clinical signs of porcine malignant hyperthermia. Anesth Analg. 1994;79:1007–1011.
146 Ohnishi ST, Taylor S, Gronert GA. Calcium-induced Ca2+ release from sarcoplasmic reticulum of pigs susceptible to malignant hyperthermia. The effects of halothane and dantrolene. FEBS Lett. 1983;161:103–107.
147 Mickelson JR, Gallant EM, Litterer LA, et al. Abnormal sarcoplasmic reticulum ryanodine receptor in malignant hyperthermia. J Biol Chem. 1988;263:9310–9315.
148 Lamb GD. Ca2+ inactivation, Mg2+ inhibition and malignant hyperthermia. J Muscle Res Cell Motil. 1993;14:554–556.
149 Duke AM, Hopkins PM, Halsall PJ, Steele DS. Mg2+ dependence of Ca2+ release from the sarcoplasmic reticulum induced by sevoflurane or halothane in skeletal muscle from humans susceptible to malignant hyperthermia. Br J Anaesth. 2006;97:320–328.
150 Owen VJ, Taske NL, Lamb GD. Reduced Mg2+ inhibition of Ca2+ release in muscle fibers of pigs susceptible to malignant hyperthermia. Am J Physiol. 1997;272:C203–C211.
151 Shomer NH, Louis CF, Fill M, Litterer LA, Mickelson JR. Reconstitution of abnormalities in the malignant hyperthermia-susceptible pig ryanodine receptor. Am J Physiol. 1993;264:C125–C135.
152 Fill M, Coronado R, Mickelson JR, et al. Abnormal ryanodine receptor channels in malignant hyperthermia. Biophys J. 1990;57:471–475.
153 Shomer NH, Mickelson JR, Louis CF. Caffeine stimulation of malignant hyperthermia-susceptible sarcoplasmic reticulum Ca2+ release channel. Am J Physiol. 1994;267:C1253–C1261.
154 Carrier L, Villaz M, Dupont Y. Abnormal rapid Ca2+ release from sarcoplasmic reticulum of malignant hyperthermia susceptible pigs. Biochim Biophys Acta. 1991;1064:175–183.
155 Laver DR, Owen VJ, Junankar PR, et al. Reduced inhibitory effect of Mg2+ on ryanodine receptor-Ca2+ release channels in malignant hyperthermia. Biophys J. 1997;73:1913–1924.
156 Nelson TE. Halothane effects on human malignant hyperthermia skeletal muscle single calcium-release channels in planar lipid bilayers. Anesthesiology. 1992;76:588–595.
157 Kerr DD, Wingard DW, Gatz EE. Prevention of porcine malignant hyperthermia by epidural block. Anesthesiology. 1975;42:307–311.
158 Hall GM, Lucke JN, Lister D. Porcine malignant hyperthermia IV: neuromuscular blockade. Br J Anaesth. 1976;48:1135–1141.
159 Buzello W, Williams CH, Chandra P, Watkins ML, Dozier SE. Vecuronium and porcine malignant hyperthermia. Anesth Analg. 1985;64:515–519.
160 Snyder HR, Jr., Davis CS, Bickerton RK, Halliday RP. 1-[(5-arylfurfurylidene)amino]hydantoins. A new class of muscle relaxants. J Med Chem. 1967;10:807–810.
161 Harrison GG. Malignant hyperthermia. Dantrolene—dynamics and kinetics. Br J Anaesth. 1988;60:279–286.
162 Ellis KO, Carpenter JF. Studies on the mechanism of action of dantrolene sodium. A skeletal muscle relaxant. Naunyn Schmiedebergs Arch Pharmacol. 1972;275:83–94.
163 Ellis KO, Bryant SH. Excitation-contraction uncoupling in skeletal muscle by dantrolene sodium. Naunyn Schmiedebergs Arch Pharmacol. 1972;274:107–109.
164 Ellis KO, Carpenter JF. Mechanism of control of skeletal-muscle contraction by dantrolene sodium. Arch Phys Med Rehabil. 1974;55:362–369.
165 Van Winkle WB. Calcium release from skeletal muscle sarcoplasmic reticulum: site of action of dantrolene sodium. Science. 1976;193:1130–1131.
166 Desmedt JE, Hainaut K. Inhibition of the intracellular release of calcium by dantrolene in barnacle giant muscle fibres. J Physiol. 1977;265:565–585.
167 Ohnishi ST, Waring AJ, Fang SR, et al. Abnormal membrane properties of the sarcoplasmic reticulum of pigs susceptible to malignant hyperthermia: modes of action of halothane, caffeine, dantrolene, and two other drugs. Arch Biochem Biophys. 1986;247:294–301.
168 Danko S, Kim DH, Sreter FA, Ikemoto N. Inhibitors of Ca2+ release from the isolated sarcoplasmic reticulum. II. The effects of dantrolene on Ca2+ release induced by caffeine, Ca2+ and depolarization. Biochim Biophys Acta. 1985;816:18–24.
169 Foster PS, Hopkinson KC, Payne N, Denborough MA. The effect of azumolene on hypercontractility and sarcoplasmic reticulum Ca(2+)-dependent ATPase activity of malignant hyperpyrexia-susceptible porcine skeletal muscle. Clin Exp Pharmacol Physiol. 1991;18:489–495.
170 Foster PS, Denborough MA. The effect of calcium channel antagonists and BAY K 8644 on calcium fluxes of malignant hyperpyrexia-susceptible muscle. Int J Biochem. 1993;25:495–504.
171 Dehpour AR, Mofakham S, Mahmoudian M. In vitro binding of dantrolene to sarcoplasmic reticulum of rabbit skeletal muscle. Biochem Pharmacol. 1982;31:965–968.
172 Sengupta C, Meyer UA, Carafoli E. Binding of dantrolene sodium to muscle intracellular membranes. FEBS Lett. 1980;117:37–38.
173 Parness J, Palnitkar SS. Identification of dantrolene binding sites in porcine skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1995;270:18465–18472.
174 Szentesi P, Collet C, Sarkozi S, et al. Effects of dantrolene on steps of excitation-contraction coupling in mammalian skeletal muscle fibers. J Gen Physiol. 2001;118:355–375.
175 Shin DW, Pan Z, Kim EK, et al. A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J Biol Chem. 2003;278:3286–3292.
176 Sampieri A, Diaz-Munoz M, Antaramian A, Vaca L. The foot structure from the type 1 ryanodine receptor is required for functional coupling to store-operated channels. J Biol Chem. 2005;280:24804–24815.
177 Launikonis BS, Rios E. Store-operated Ca2+ entry during intracellular Ca2+ release in mammalian skeletal muscle. J Physiol. 2007;583:81–97.
178 Ambudkar IS, Ong HL, Liu X, Bandyopadhyay BC, Cheng KT. TRPC1: the link between functionally distinct store-operated calcium channels. Cell Calcium. 2007;42:213–223.
179 Zhao X, Weisleder N, Han X, et al. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J Biol Chem. 2006;281:33477–33486.
180 Cherednichenko G, Hurne AM, Fessenden JD, et al. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc Natl Acad Sci U S A. 2004;101:15793–15798.
181 Hurne AM, O’Brien JJ, Wingrove D, et al. Ryanodine receptor type 1 (RyR1) mutations C4958S and C4961S reveal excitation-coupled calcium entry (ECCE) is independent of sarcoplasmic reticulum store depletion. J Biol Chem. 2005;280:36994–37004.
182 Yang T, Allen PD, Pessah IN, Lopez JR. Enhanced excitation-coupled calcium entry in myotubes is associated with expression of RyR1 malignant hyperthermia mutations. J Biol Chem. 2007;282:37471–37478.
183 Cherednichenko G, Ward CW, Feng W, et al. Enhanced excitation-coupled calcium entry in myotubes expressing malignant hyperthermia mutation R163C is attenuated by dantrolene. Mol Pharmacol. 2008;73:1203–1212.
184 Kalow W, Britt BA, Terreau ME, Haist C. Metabolic error of muscle metabolism after recovery from malignant hyperthermia. Lancet. 1970;2:895–898.
185 Ellis FR, Harriman DG, Keaney NP, Kyei-Mensah K, Tyrrell JH. Halothane-induced muscle contracture as a cause of hyperpyrexia. Br J Anaesth. 1971;43:721–722.
186 Anderson IL, Jones EW. Porcine malignant hyperthermia: effect of dantrolene sodium on in-vitro halothane-induced contraction of susceptible muscle. Anesthesiology. 1976;44:57–61.
187 Okumura F, Crocker BD, Denborough MA. Identification of susceptibility to malignant hyperpyrexia in swine. Br J Anaesth. 1979;51:171–176.
188 Larach MG. Standardization of the caffeine halothane muscle contracture test. North American Malignant Hyperthermia Group. Anesth Analg. 1989;69:511–515.
189 Larach MG, Landis JR, Bunn JS, Diaz M. Prediction of malignant hyperthermia susceptibility in low-risk subjects. An epidemiologic investigation of caffeine halothane contracture responses. The North American Malignant Hyperthermia Registry. Anesthesiology. 1992;76:16–27.
190 European Malignant Hyperpyrexia Group. A protocol for the investigation of malignant hyperpyrexia (MH) susceptibility. The European Malignant Hyperpyrexia Group. Br J Anaesth. 1984;56:1267–1269.
191 Ording H, Brancadoro V, Cozzolino S, et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European Malignant Hyperthermia Group. Acta Anaesthesiol Scand. 1997;41:955–966.
192 Islander G, Twetman ER. Comparison between the European and North American protocols for diagnosis of malignant hyperthermia susceptibility in humans. Anesth Analg. 1999;88:1155–1160.
193 Ording H, Bendixen D. Sources of variability in halothane and caffeine contracture tests for susceptibility to malignant hyperthermia. Eur J Anaesthesiol. 1992;9:367–376.
194 Allen GC, Larach MG, Kunselman AR. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. The North American Malignant Hyperthermia Registry of MHAUS. Anesthesiology. 1998;88:579–588.
195 Heiman-Patterson TD, Rosenberg H, Fletcher JE, Tahmoush AJ. Halothane-caffeine contracture testing in neuromuscular diseases. Muscle Nerve. 1988;11:453–457.
196 Benca J, Hogan K. Malignant hyperthermia, coexisting disorders, and enzymopathies: risks and management options. Anesth Analg. 2009;109:1049–1053.
197 Klingler W, Rueffert H, Lehmann-Horn F, Girard T, Hopkins PM. Core myopathies and risk of malignant hyperthermia. Anesth Analg. 2009;109:1167–1173.
198 Pinhas-Hamiel O, Zeitler P. Acute and chronic complications of type 2 diabetes mellitus in children and adolescents. Lancet. 2007;369:1823–1831.
199 Daneman D. Type 1 diabetes. Lancet. 2006;367:847–858.
200 White NH. Diabetic ketoacidosis in children. Endocrinol Metab Clin North Am. 2000;29:657–682.
201 Delaney MF, Zisman A, Kettyle WM. Diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Endocrinol Metab Clin North Am. 2000;29:683–705. V
202 Hollander AS, Olney RC, Blackett PR, Marshall BA. Fatal malignant hyperthermia-like syndrome with rhabdomyolysis complicating the presentation of diabetes mellitus in adolescent males. Pediatrics. 2003;111:1447–1452.
203 Kilbane BJ, Mehta S, Backeljauw PF, Shanley TP, Crimmins NA. Approach to management of malignant hyperthermia–like syndrome in pediatric diabetes mellitus. Pediatr Crit Care Med. 2006;7:169–173.
204 Wappler F, Roewer N, Kochling A, et al. Fulminant malignant hyperthermia associated with ketoacidotic diabetic coma. Intensive Care Med. 1996;22:809–812.
205 Caccia MR, Cerri C, Gravame V, et al. Myopathy with paroxysmal myoglobinuria and focal muscle necrosis following enfluorane anaesthesia. J Neurol Sci. 1978;39:61–69.
206 Vladutiu GD, Bennett MJ, Smail D, et al. A variable myopathy associated with heterozygosity for the R503C mutation in the carnitine palmitoyltransferase II gene. Mol Genet Metab. 2000;70:134–141.
207 Cornelio F, Di DS, Testa D, et al. “Carnitine deficient” myopathy and cardiomyopathy with fatal outcome. Ital J Neurol Sci. 1980;1:95–100.
208 Olpin SE. Fatty acid oxidation defects as a cause of neuromyopathic disease in infants and adults. Clin Lab. 2005;51:289–306.
209 Hoffman JD, Steiner RD, Paradise L, et al. Rhabdomyolysis in the military: recognizing late-onset very long-chain acyl Co-A dehydrogenase deficiency. Mil Med. 2006;171:657–658.
210 Tong MK, Lam CS, Mak TW, et al. Very long-chain acyl-CoA dehydrogenase deficiency presenting as acute hypercapnic respiratory failure. Eur Respir J. 2006;28:447–450.
211 Voermans NC, Poels PJ, Kluijtmans LA, van Engelen BG. The effect of dantrolene sodium in very long chain acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. 2005;15:844–846.
212 Marsden D, Nyhan WL, Barshop BA. Creatine kinase and uric acid: early warning for metabolic imbalance resulting from disorders of fatty acid oxidation. Eur J Pediatr. 2001;160:599–602.
213 Steiner LA, Studer W, Baumgartner ER, Frei FJ. Perioperative management of a child with very-long-chain acyl-coenzyme A dehydrogenase deficiency. Paediatr Anaesth. 2002;12:187–191.
214 Lilker S, Kasodekar S, Goldszmidt E. Anesthetic management of a parturient with carnitine palmitoyltransferase II deficiency. Can J Anaesth. 2006;53:482–486.
215 Hill M, Peat W, Courtman S. A national survey of propofol infusion use by paediatric anaesthetists in Great Britain and Ireland. Paediatr Anaesth. 2008;18:488–493.
216 Fudickar A, Bein B, Tonner PH. Propofol infusion syndrome in anaesthesia and intensive care medicine. Curr Opin Anaesthesiol. 2006;19:404–410.
217 Fodale V, La ME. Propofol infusion syndrome: an overview of a perplexing disease. Drug Saf. 2008;31:293–303.
218 Kam PC, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62:690–701.
219 Wolf A, Weir P, Segar P, Stone J, Shield J. Impaired fatty acid oxidation in propofol infusion syndrome. Lancet. 2001;357:606–607.
220 Uezono S, Hotta Y, Takakuwa Y, Ozaki M. Acquired carnitine deficiency: a clinical model for propofol infusion syndrome [letter]? Anesthesiology. 2005;103:909.
221 Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care. 2007;11:R4.
222 Rosebush PI, Stewart T, Mazurek MF. The treatment of neuroleptic malignant syndrome. Are dantrolene and bromocriptine useful adjuncts to supportive care? Br J Psychiatry. 1991;159:709–712.
223 Silva RR, Munoz DM, Alpert M, Perlmutter IR, Diaz J. Neuroleptic malignant syndrome in children and adolescents. J Am Acad Child Adolesc Psychiatry. 1999;38:187–194.
224 Tsuchiya N, Morimura E, Hanafusa T, Shinomura T. Postoperative neuroleptic malignant syndrome that occurred repeatedly in a patient with cerebral palsy. Paediatr Anaesth. 2007;17:281–284.