Chapter 9 Malignant disease
Introduction
The term ‘malignant disease’ encompasses a wide range of illnesses, including common ones such as lung, breast and colorectal cancer (Table 9.1) as well as rare ones, like the acute leukaemias. Malignant disease is widely prevalent and, in the West, almost one-third of the population will develop cancer at some time during their life. It is second only to cardiovascular disease as the cause of death. Although the mortality of cancer is high, many advances have been made, both in terms of treatment and in understanding the biology of the disease at the molecular level.
Table 9.1 Relative 5-year survival estimates based on survival probabilities observed during 2000–2001, by sex and site, England and Wales
| 5-year survival (%) | ||
|---|---|---|
| Men | Women | |
|
Pancreas |
3 |
2 |
|
Lung |
6 |
6 |
|
Oesophagus |
7 |
8 |
|
Stomach |
12 |
13 |
|
Brain |
13 |
15 |
|
Multiple myeloma |
24 |
22 |
|
Ovary |
|
34 |
|
Leukaemia |
38 |
36 |
|
Kidney |
45 |
43 |
|
Colon |
46 |
45 |
|
Rectum |
45 |
48 |
|
Non-Hodgkin’s lymphoma |
51 |
52 |
|
Prostate |
61 |
|
|
Larynx |
67 |
|
|
Bladder |
71 |
61 |
|
Cervix |
|
68 |
|
Melanoma |
78 |
90 |
|
Breast |
|
79 |
|
Hodgkin’s lymphoma |
84 |
83 |
|
Testis |
95 |
|
The biology of cancer
Most human neoplasms are clonal in origin, i.e. they arise from a single population of precursor or cancer stem cells. This process is typically initiated by genetic aberrations within this precursor cell. Cancer is increasingly common the older we get and can be related to a time dependent accumulation of DNA damage that is not repaired by the normal mechanisms of genome maintenance, damage tolerance and checkpoint pathways. Malignant transformation may result from a gain in function as cellular proto-oncogenes become mutated (e.g. ras), amplified (e.g. HER2) or translocated (e.g. BCR-ABL). However, these mutations are insufficient to cause malignant transformation by themselves. Alternatively, there may be a loss of function of tumour suppressor genes such as P53 that normally suppress growth. Loss or gain of function may also involve alterations in the genes controlling the transcription of the oncogenes or tumour suppressor genes (p. 46). Over subsequent cell divisions, heterogeneity develops with the accumulation of further genetic abnormalities (Fig. 9.1).
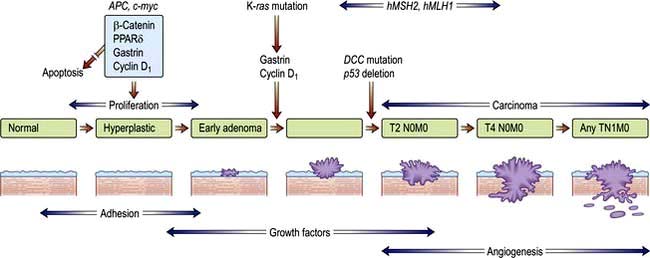
The genes most commonly affected can be characterized as those controlling cell cycle checkpoints, DNA repair and DNA damage recognition, apoptosis, differentiation, growth factor receptors and signalling pathways and tumour suppressor genes (Table 9.2). Recognition of critical genetic alterations has enabled extensive development of new targeted drugs such as imatinib that inhibits the growth signals of the abnormal tyrosine kinase BCR/ABL. Proliferation may continue at the expense of differentiation which, together with the failure of apoptosis, leads to tumour formation with the accumulation of morphologically abnormal cells varying in size, shape and cytoplasmic or nuclear maturity.
| Gene | Example |
|---|---|
|
Control cell cycle checkpoints |
Cyclin D1, p15, p16 |
|
DNA repair |
FANCA, ATM |
|
Apoptosis |
Bcl2 |
|
Differentiation |
PML/RARA |
|
Growth factor receptors |
EGF, VEGF, FGF, BCR/ABL, TGF-B, KIT, L-FLT3 |
|
Signalling pathways |
RAS, BRAF, JAK2, NF1, PTCH |
|
Hedgehog signalling pathway |
See p. 26 |
|
Tumour suppressor genes |
P53, Rb, WT1, VHL |
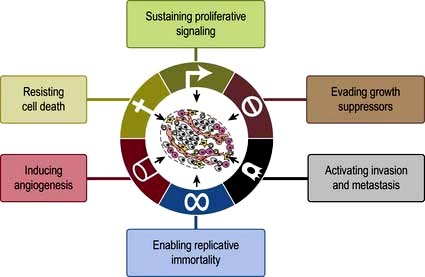
The hallmarks in developing cancer are shown in Figure 9.2.
Tumour immunology
Tumour cells are usually not recognized and killed by the immune system. There are two main reasons. The first is failure to express molecules such as HLA and co-stimulatory B7 molecules that are required for activation of cytotoxic, or ‘killer’, T lymphocytes. Second, tumours may also actively secrete immunosuppressive cytokines and cause a generalized immunosuppression. Successful strategies for tumour vaccines that overcome these obstacles are developing in renal cancer and prostate cancer. The monoclonal antibody ipilimumab against the inhibitory cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) molecule that is expressed after T-cell activation, is used in melanoma (p. 479)
Invasion and metastasis
Solid cancers spread by both local invasion and by distant metastasis through the vessels of the blood and lymphatic systems. Infiltration into surrounding tissues is associated with loss of cell–cell cohesion, which is mediated by active homotypic cell adhesion molecules (CAMs). Epithelial cadherin (E-cadherin) is expressed by many carcinomas and mutated in some such as familial gastric carcinoma (see p. 252).
Aetiology and epidemiology
Genetic factors
Rather than occurring by somatic mutation in response to mutagens, germline mutations in the genes that predispose to the development of cancer may be inherited and therefore present in all tissues. Examples of such cancer syndromes are given in Table 9.3. Expression of the mutation and hence carcinogenesis, will depend upon the penetrance (due to the level of expression and the presence of other genetic events) of the gene and whether the mutated allele has a dominant or recessive effect. There is a small group of autosomal dominant inherited mutations such as RB (in retinoblastoma), and a small group of recessive mutations (Table 9.3). Carriers of the recessive mutations are at risk of developing cancer if the second allele becomes mutated, leading to ‘loss of heterozygosity’ within the tumour, although this is seldom sufficient as carcinogenesis is a multistep process.
| Gene | Neoplasms | |
|---|---|---|
|
Autosomal dominant |
|
|
|
Retinoblastoma |
RB1 |
Eye |
|
Wilms’ tumour |
WT1 |
Kidney |
|
Li–Fraumeni |
p53 |
Sarcoma/brain/leukaemia |
|
Neurofibromatosis type 1 |
NF1 |
Neurofibromas/ leukaemia |
|
Familial adenomatous polyposis (FAP) |
APC |
Colon |
|
Hereditary non-polyposis colon cancer (HNPCC) |
MLH1 and MSH2 |
Colon, endometrium |
|
Hereditary diffuse gastric cancer syndrome |
E-cadherin |
Stomach |
|
Breast ovary families |
BRCA1 |
Breast/ovary |
|
BRCA2 |
||
|
p53 |
||
|
Melanoma |
p16 |
Skin |
|
Von Hippel–Lindau |
VHL |
Renal cell carcinoma and haemangioblastoma |
|
Multiple endocrine neoplasia type 1 |
MEN1 |
Pituitary, pancreas, parathyroid |
|
Multiple endocrine neoplasia type 2 |
RET |
Thyroid, adrenal medulla |
|
Autosomal recessive |
|
|
|
Xeroderma pigmentosa |
XP |
Skin |
|
Ataxia telangiectasia |
AT |
Leukaemia, lymphoma |
|
Fanconi’s anaemia |
FA |
Leukaemia, lymphoma |
|
Bloom’s syndrome |
BS |
Leukaemia, lymphoma |
Environmental factors
A wide range of environmental factors have been identified as being associated with the development of malignancy (Table 9.4) and may be amenable to preventative action such as smoking cessation, dietary modification and antiviral immunization (Box 9.1). Environmental factors interact with genetic predisposition. For example, subsequent generations of people moving from countries with a low incidence to those with a high incidence of breast or colon cancer acquire the cancer incidence of the country to which they have moved while northern European people exposed to strong UV radiation have the highest risk of developing melanoma.
Table 9.4 Some causative factors associated with the development of cancer
|
Smoking |
Mouth, pharynx, oesophagus, larynx, lung, bladder, lip |
|
Alcohol |
Mouth, pharynx, larynx, oesophagus, colorectal |
|
Iatrogenic |
|
|
Alkylating agents |
Bladder, bone marrow |
|
Oestrogens |
Endometrium, vagina, breast, cervix |
|
Androgens |
Prostate |
|
Radiotherapy (e.g. mantle radiotherapy) |
Carcinoma of breast and bronchus |
|
Diet |
|
|
High-fat diet |
Colorectal cancer |
|
Environmental/occupation |
|
|
Vinyl chloride |
Liver (angiosarcoma) |
|
Polycyclic hydrocarbons |
Skin, lung, bladder, myeloid leukaemia |
|
Aromatic amines |
Bladder |
|
Asbestos |
Lung, mesothelium |
|
Ultraviolet light |
Skin, lip |
|
Radiation |
e.g. leukaemia, thyroid cancer |
|
Aflatoxin |
Liver |
|
Biological agents |
|
|
Hepatitis B virus |
Liver (hepatocellular carcinoma) |
|
Hepatitis C virus |
Liver (hepatocellular carcinoma) |
|
Human T-cell leukaemia virus |
Leukaemia/lymphoma |
|
Epstein–Barr virus |
Burkitt’s lymphoma |
|
Hodgkin’s lymphoma |
|
|
Nasopharyngeal carcinoma |
|
|
Human papillomavirus types 16, 18 |
Cervix |
|
Oral cancer (type 16) |
|
|
Schistosoma japonicum |
Bladder |
|
Helicobacter pylori |
Stomach |
 Box 9.1
Box 9.1








Tobacco
The incidence of lung cancer in both men and women increased dramatically in the last 25 years worldwide, but is now falling in many developed countries. The association of smoking with lung cancer is indisputable and causative mechanisms have been identified: cigarette tobacco is responsible for one-third of all deaths from cancer in the UK. Smoking not only causes lung cancer, it is also associated with cancer of the mouth, larynx, oesophagus and bladder. Smoking is discussed on page 806.
Diet
Dietary factors have been attributed to account for one-third of cancer deaths, although it is often difficult to differentiate these from other epidemiological factors. For example, the incidence of stomach cancer is particularly high in the Far East, while breast and colon cancers are more common in the Western, economically more developed countries. Many associations have been observed without a causative mechanism being identified between the incidence of cancer and the consumption of dietary fibre, red meat, saturated fats, salted fish, vitamin E, vitamin A and many others. Food and its role in the causation of gastrointestinal cancer is discussed in Chapter 5 (see p. 218). Increasing levels of obesity in the developed world have been associated with increases in women of cancers associated with oestrogenic stimulation of the breast and endometrium.
Infectious agents
Hepatocellular carcinoma occurs in patients with hepatitis B and C virus infections and Burkitt’s lymphoma and nasopharyngeal carcinoma are associated with the Epstein–Barr virus. EBV is also linked with Hodgkin’s lymphoma (see p. 459).
Radiation
Diagnostic. Imaging procedures involving radiation exposure are associated with an increased risk of cancer. This risk is cumulative, dose dependent and time dependent, i.e. children are at higher risk than adults. The cancer risk of various common investigations is shown in Table 9.5. All doctors should strive to minimize diagnostic exposure to radiation where possible using alternative modalities such as ultrasound or MRI. Good documentation of radiation doses is required. This is particularly so in children and pregnant women.
Table 9.5 Radiation exposure from common diagnostic radiological procedures
| Procedure | mSv |
|---|---|
|
CXR |
0.02 |
|
IVU |
3 |
|
CT chest |
7 |
|
CT abdomen |
8–10 |
|
Whole body CT |
20 |
|
Percutaneous coronary intervention |
15 |
|
Myocardial perfusion imaging |
15.6 |
UK background radiation is 2.6 mSv per year. 1 mSv carries a lifetime cancer risk of 1 in 17 500 and 5 mSv a risk of 1 in 3500.
Modified from: Smith-Bindman R, Lipson J, Marcus R et al. Archives of Internal Medicine 2009; 169:2078–2086 and Fazel R, Krumholz HM, Wang Y et al. New England Journal of Medicine 2009; 361:849–857.
Geographical distribution
The incidence of cancer across the world is dependent on the local environmental factors, the diet and the genetics of the population (see above) (Figs 9.3, 9.4). Age is also a factor as most cancers occur in those over the age of 65 who comprise 3.3% of the population in Africa compared with 15.2% in Europe. Reproductive patterns also influence breast cancer. Migrating individuals often take on the risks of the local environmental factors.
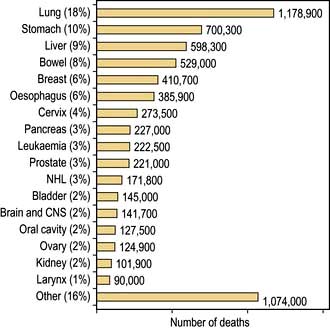
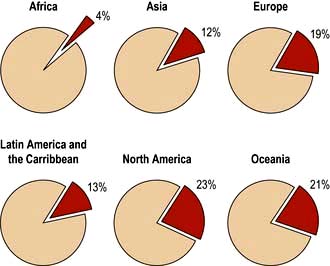
Figure 9.4 Percentage of all deaths due to cancer in the different regions of the world.
(From: http://info.canceresearchuk.org/cancerstats/world/the-global-picture/)
The clinical presentation of malignant disease
Asymptomatic detection through screening
Most common cancers start as focal microscopic clones of transformed cells and diagnosis only becomes likely once sufficient tumour bulk has accumulated to cause symptoms or signs. In order to try to make an earlier diagnosis and increase the curative possibilities, an increasing number of screening programmes are being developed which target the asymptomatic or preinvasive stages of the cancer as in cervix, breast and colon or use serum tumour markers as in prostate and ovarian cancers. Genetic screening can be used to target screening to groups at most risk of developing cancer, e.g. BRCA1 positive and breast cancer (see Table 9.3). The aim of screening programmes is to improve individual and/or population survival by detecting cancer at its very early stages when the patient is asymptomatic. This strategy is dependent upon finding tests that are sufficiently sensitive and specific, using detection methods that identify cancer before it has spread and having curative treatments that are practical and consistent with maintenance of a normal lifestyle and quality of life.
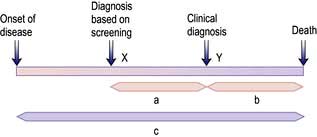
Unfortunately, earlier diagnosis does not necessarily mean longer survival and randomized trials are necessary to prove benefit. With lead time bias, the patient is merely treated at an earlier date and hence the survival appears longer; death still occurs at the same time from the point of genesis of the cancer (Fig. 9.5). With length time bias, a greater number of slowly growing tumours are detected when screening asymptomatic individuals leading to a false impression of an improvement in survival.
An effective screening procedure should:
 be affordable to the healthcare system
be affordable to the healthcare system
 be acceptable to all social groups so that they attend for screening
be acceptable to all social groups so that they attend for screening
 have a good discriminatory index between benign and malignant lesions
have a good discriminatory index between benign and malignant lesions

Colorectal cancer (CRC). Faecal occult blood is a cheap test for the detection of CRC. Large randomized studies have shown a reduction in cancer-related mortality of 15–33%. However, the false-positive rates are high, meaning many unnecessary colonoscopies (see p. 291). The UK has recently introduced a national screening programme using faecal occult blood in patients aged 60–64 years, in which positive tests have identified that 10% have cancer and 40% adenomas. A randomized trial in Norway has found an increased number of early stage cancers in the screened population but a high incidence of interval cancers between biennial screens.
Colonoscopy is the ‘gold-standard’ technique for the examination of the colon and rectum and is the investigation of choice for high-risk patients. Universal screening strategies have been recommended in the USA, but the shortage of skilled endoscopists, the expense, the need for full bowel preparation and the small risk of perforation make colonoscopy impractical as a population screening tool at present and CT colonography (‘virtual colonoscopy’) (see Fig. 6.5) may become an alternative along with genetic testing and stool DNA tests.
Other population-based screening programmes that are being used or are in trials are:
FURTHER READING
Menon U, Gentry-Maharaj A, Hallett R et al. Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer and stage distribution of detected cancers: results of the prevalence screen of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS). Lancet. Oncology 2009; 10:327–340.
Paimela H, Malila N, Palva T et al. Early detection of colorectal cancer with faecal occult blood test screening. British Journal of Surgery 2010; 97:1567–1571.
Schroder FH et al. Prostate cancer mortality at 11 years of follow-up. N Engl J Med 2012; 366:981–990.
The symptomatic patient with cancer
Patients may offer information of predisposing conditions and family history that alerts the clinician to the likelihood of a cancer diagnosis. Many present with a history of tumour site-specific symptoms, e.g. pain, and physical signs, e.g. a mass, which readily identify the primary site of the cancer. However, some only seek medical attention when more systemic and nonspecific symptoms occur such as weight loss, night sweats, fever, fatigue, recurrent infections and anorexia. These usually indicate a more advanced stage of the disease, except in some paraneoplastic and ectopic endocrine syndromes (see below). Other patients are only diagnosed upon the discovery of established metastases such as the abdominal distension of ovarian cancer, the back pain of metastatic prostatic cancer or the liver enlargement of metastatic gastrointestinal cancer (Table 9.6).
| Degree of spread | Anatomical location | Examples of clinical problems |
|---|---|---|
|
Local |
Mass |
Thyroid nodule, pigmented naevus, breast lump, abdominal mass, testicular mass |
|
Local infiltration of skin |
Dermal nodules, peau d’orange, ulceration |
|
|
Local infiltration of nerve |
Neuropathic pain and loss of function |
|
|
Local infiltration of vessel |
Venous thrombosis, tumour emboli, haemorrhage, e.g. GI |
|
|
Obstruction of viscera or duct |
Small or large bowel obstruction, dysphagia, SVC obstruction |
|
|
Nodal |
Peripheral |
Supraclavicular fossa, Virchow’s node, lymphoedema |
|
Central |
Mediastinum – SVC obstruction, porta hepatis – obstructive jaundice, para-aortic nodes and back pain |
|
|
Metastatic |
Lung |
Pleuritic pain, cough, shortness of breath, lymphangitis and respiratory failure, recurrent pneumonia |
|
Liver |
RUQ pain, anorexia, fever, raised serum liver enzymes, jaundice |
|
|
Brain |
Headache and vomiting of raised intracranial pressure, focal deficit, coma, seizure |
|
|
Bone |
Bone pain, cord compression, fracture, hypercalcaemia |
|
|
Pleura |
Effusion, pain, shortness of breath |
|
|
Peritoneum |
Ascites, Krukenberg tumours |
|
|
Adrenal |
Addison’s disease (hypoadrenalism) |
|
|
Umbilicus |
Sister Mary Joseph’s nodule |
Paraneoplastic syndromes are indirect effects of cancer (Box 9.2, Fig. 9.6) that are often associated with specific types of cancer and may be reversible with treatment of the cancer. The effects and mechanisms can be very variable. For example in the Lambert–Eaton syndrome (see p. 1152), there is cross-reactivity between tumour antigens and the normal tissues, e.g. the acetylcholine receptors at neuromuscular junctions.
 Box 9.2
Box 9.2
Paraneoplastic syndromes
| Syndrome | Tumour | Serum antibodies |
|---|---|---|
|
Neurological |
|
|
|
Lambert–Eaton syndrome |
Lung (small-cell) lymphoma |
Anti-VGLC |
|
Peripheral sensory neuropathy |
Lung (small-cell), breast and ovary lymphoma |
Anti-Hu |
|
Cerebellar degeneration |
Lung (particularly small-cell) lymphoma |
Anti-Yo |
|
Opsoclonus/myoclonus |
Breast, lung (small-cell) |
Anti-Ri |
|
Stiff person syndrome |
Breast, lung (small-cell) |
Anti-amphiphysin |
|
Limbic, hypothalamic, brain stem encephalitis |
Lung |
Anti-Ma protein |
|
|
Testicular |
Anti-NMDAR |
|
Endocrine/metabolic |
|
|
|
SIADH |
Lung (small-cell) |
|
|
Ectopic ACTH secretion |
Lung (small-cell) |
|
|
Hypercalcaemia |
Renal, breast, myeloma, lymphoma |
|
|
Fever |
Lymphoma, renal |
|
|
Musculoskeletal |
|
|
|
Hypertrophic pulmonary osteoarthropathy |
Lung (non-small-cell) |
|
|
Clubbing |
Lung |
|
|
Skin |
|
|
|
Dermatomyositis/polymyositis |
Lung and upper GI |
|
|
Acanthosis nigricans |
Mainly gastric |
|
|
Velvet palms |
Gastric, lung (non-small cell) |
|
|
Hyperpigmentation |
Lung (small-cell) |
|
|
Pemphigus |
Non-Hodgkin’s lymphoma, CLL |
|
|
Haematological |
|
|
|
Erythrocytosis |
Renal cell carcinoma, hepatocellular carcinoma, cerebellar haemangioblastoma |
|
|
Thrombocytosis |
Ovarian cancer |
|
|
Migratory thrombophlebitis |
Pancreatic adenocarcinoma |
|
|
DVT |
Adenocarcinoma |
|
|
DIC |
Adenocarcinoma |
|
|
Renal |
|
|
|
Nephrotic syndrome |
Myeloma, amyloidosis |
|
|
Membranous glomerulonephritis |
Lymphoma |
|
SIADH, syndrome of inappropriate antidiuretic hormone secretion; ACTH, adrenocorticotrophic hormone; CLL, chronic lymphocytic leukaemia; DIC, disseminated intravascular coagulation; NMDAR, N-methyl-D-aspartate receptors.
The coagulopathy of cancer may present with thrombophlebitis, deep venous thrombosis and pulmonary emboli, particularly in association with cancers of pancreas, stomach and breast. Some 18% of patients with recurrent pulmonary embolus will be found to have an underlying cancer and many cancer patients are at increased risk of venous thromboembolism (VTE) following diagnosis. Trousseau’s syndrome – superficial thrombophlebitis migrans – refers to this process in the superficial venous system. All patients with active cancer admitted to hospital are at high risk of VTE and should be given prophylaxis with subcutaneous LMW heparin in the absence of any contraindications (see p. 429). Dabigatran, an oral direct thrombin inhibitor, is an alternative therapy.
Other symptoms are related to peptide or hormone release, e.g. carcinoid or Cushing’s syndrome.
Serum tumour markers
Tumour markers are intracellular proteins or cell surface glycoproteins released into the circulation and detected by immunoassays. Examples are given in Table 9.7. Values in the normal range do not necessarily equate with the absence of disease and a positive result must be corroborated by histology as these markers can be seen in many benign conditions. They are most useful in the serial monitoring of response to treatment. As discussed in subsequent sections, a proportion of low-grade B-cell lymphomas and a majority of cases of myeloma will produce a monoclonal paraprotein of intact immunoglobulin molecule or light chains. This acts as a valuable tumour marker in the diagnosis and assessment of response.
|
α-Fetoprotein |
Hepatocellular carcinoma and non-seminomatous germ cell tumours of the gonads |
|
β-Human chorionic gonadotrophin (β-hCG) |
Choriocarcinomas, germ cell tumours (testicular) and lung cancers |
|
Prostate-specific antigen (PSA) |
Carcinoma of prostate |
|
Carcinoma embryonic antigen (CEA) |
Gastrointestinal cancers |
|
CA125 |
Ovarian cancer |
|
CA19–9 |
Gastrointestinal cancers, particularly pancreatic cancer |
|
CA15–3 |
Breast cancer |
|
Osteopontin |
Many cancers including mesothelioma |
|
M-band (Ig or light chain) |
Myeloma, chronic lymphocytic leukaemia, small lymphocytic lymphoma, lymphoplasmacytic lymphoma, amyloid |
Biopsy and histological examination
Molecular markers of genetic abnormalities have long been available in the haematological cancers and are increasingly available in solid cancers. For example, fluorescent in situ hybridization (FISH, see p. 40) can be used to look for characteristic chromosomal translocations, e.g. in lymphoma and leukaemia, as well as deletions or amplifications, e.g. in breast cancer (see genetic basis of cancer, p. 45). Tissue microarrays can identify patterns of multiple genomic alterations and single nucleotide polymorphisms (SNPs), e.g. in breast cancer and lymphoma (see p. 35), and RNA assays with RT-PCR can be used to identify tissue of origin with prognostic and predictive relevance.
Cancer treatment
Aims of treatment
The organization across multiple departments and coordination from primary to secondary and tertiary care has become known as a patient pathway. Establishment of agreed patient pathways has enabled more effective and timely delivery of care and post-treatment rehabilitation. The aim is to provide optimal treatment and for the patient to experience seamless and high quality care and to allow audit and continuing improvement against agreed standards. Central to this endeavour is the involvement of the patient, through education as to the nature of their disease and the treatment options available. An informed choice can then be made, even if in the end it is simply to abide by the decisions made by the professionals. Good communication embodies a humane approach which preserves hope at an appropriate level through empathy and understanding of the patient’s position (see p. 14).
Assessments before treatment
Staging
The staging systems vary according to the type of tumour and may be site specific (see Hodgkin’s lymphoma, p. 461), or the TNM (tumour, node, metastases) classification shown in Table 15.29, which can be adapted for application to most common cancers.
Performance status
In addition to anatomical staging, the person’s age and general state of health need to be taken into account when planning treatment. The latter has been called ‘performance status’ and is of great prognostic significance for all tumour types (Table 9.8). Performance status reflects the effects of the cancer on the patient’s functional capacity. An alternative performance rating scale is by Karnowsky. With a performance status of 2, response to and survival following treatment are greatly reduced for most tumour types.
Table 9.8 Eastern Cooperative Oncology Group (ECOG) performance status scale
| Status | Description |
|---|---|
|
0 |
Asymptomatic, fully active and able to carry out all predisease performance without restrictions |
|
1 |
Symptomatic, fully ambulatory but restricted in physically strenuous activity and able to carry out performance of a light or sedentary nature, e.g. light housework, office work |
|
2 |
Symptomatic, ambulatory and capable of all self-care but unable to carry out any work activities. Up and about >50% of waking hours: in bed <50% of day |
|
3 |
Symptomatic, capable of only limited self-care, confined to bed or chair >50% of waking hours, but not bedridden |
|
4 |
Completely disabled. Cannot carry out any self-care. Totally bedridden |
Assessing the benefits of treatment
Response to treatment can be subjective or objective.
An objective response to treatment is assessed clinically and radiologically. The term ‘remission’ is often used synonymously with ‘response’, which if complete, means an absence of detectable disease without necessarily implying a cure of the cancer. The terms used to evaluate the responses of tumours are given in Box 9.3. For a complete response all previous clinical abnormalities should have resolved and this needs to be confirmed by clinical examination or sampling of the primary disease site, e.g. by bone marrow examination in leukaemia. Where a tumour marker exists, such as a paraprotein in myeloma or β-hCG in testicular cancer, reductions in the level of tumour markers are useful surrogates for evidence of tumour response. They are also useful predictors for disease recurrence. Radiologically, a complete response, is the complete disappearance of all detectable disease and a partial response, defined since 1999 by the Response Evaluation Criteria in Solid Tumors (RECIST) convention, is a ≥30% reduction in the sum of all measurable lesion diameters.
 Box 9.3
Box 9.3
RECIST criteria for assessing response to treatment
|
Complete response |
Disappearance of all target lesions |
|
Partial response |
≥30% decrease in the baseline sum of the longest diameters of all target lesions |
|
Progression |
≥20% increase in the sum of the longest diameters of all target lesions compared with the smallest recorded sum since treatment started, or the appearance of any new lesions |
|
Stable disease |
All values between partial response and progressive disease |
|
Target lesions |
Measurable lesions up to 5 per organ and 10 in total, selected on size and replicable measurement |
|
Non-target lesions |
Non-measurable lesions recorded as present or absent at baseline and on follow-up. |
|
Measurable |
Lesions with the longest diameter in one dimension ≥2.0 cm or ≥1.0 cm if assessed by spiral CT scan |
|
Non-measurable |
e.g. bone, meningeal, ascites, pleural effusion, inflammatory breast cancer, lymphangitis cutis and pulmonis, cystic lesions |
Principles of chemotherapy
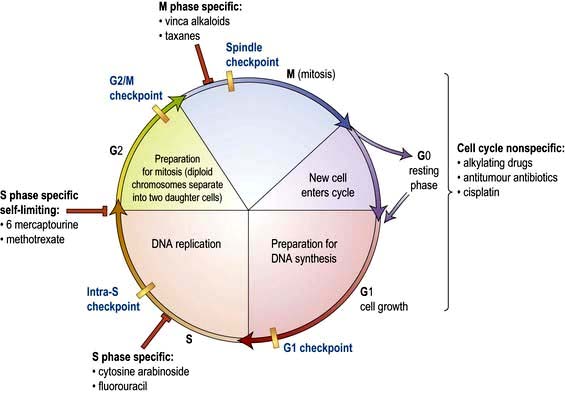
Cytotoxic chemotherapy employs systemically administered drugs that directly damage cellular DNA (and RNA). It kills cells by promoting apoptosis and sometimes frank necrosis. Different cytotoxic drugs work at different stages in the cell cycle (Fig. 9.7; see also Fig. 2.13).
There is a narrow therapeutic window between effective treatment of the cancer and normal tissue toxicity, because cytoxic drugs are not cancer specific (unlike some of the targeted biological agents) and the increased proliferation in cancers is not much greater than in normal tissues (see tumour growth and failure of apoptosis, p. 32, 33). The dose and schedule of the chemotherapy is limited by the normal tissue tolerance, especially in those more proliferative tissues of the bone marrow and gastrointestinal tract mucosa. All tissues can be affected, however, depending upon the pharmacokinetics of the drug and affinity for particular tissues (e.g. heavy metal compounds for kidneys and nerves).
The therapeutic effect on the cancer is achieved by a variety of mechanisms which seek to exploit differences between normal and transformed cells. While most of the drugs have been derived in the past by empirical testing of many different compounds, e.g. alkylating agents, the new molecular biology is leading to targeting of particular genetic defects in the cancer (see tyrosine kinase inhibitors, p. 445).
Toxicity to normal tissue can be limited in some instances by supplying growth factors such as granulocyte colony-stimulating factor (G-CSF) or by the infusion of stem cell preparations to diminish myelotoxicity. The use of more specific targeted biological agents with relatively weak pro-apoptotic effects in combination with the general cytotoxics has also improved the therapeutic ratio (see trastuzumab and breast cancer, p. 475). Certain cytotoxic therapies may also be administered into the pleural space, the peritoneum, the CSF or into the arterial supply of a tumour.
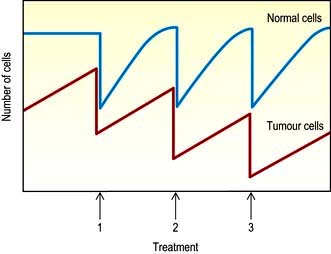
Most tumours rapidly develop resistance to single agents given on their own through changes in membrane transport and DNA repair pathways. For this reason, the principle of intermittent combination chemotherapy was developed. Several drugs are combined together, chosen on the basis of differing mechanisms of action and non-overlapping toxicities. These drugs are given over a period of a few days, followed by a rest of a few weeks, during which time the normal tissues have the opportunity for regrowth. If the normal tissues are more proficient at DNA repair than the cancer cells, it may be possible to deplete the tumour while allowing the restoration of normal tissues between chemotherapy cycles (Fig. 9.8).
Classification of cytotoxic drugs (Table 9.9)
Antitubulin agents
Vinca alkaloids. Drugs such as vincristine, vinblastine and vinorelbine act by binding to tubulin and inhibiting microtubule formation during mitosis (see p. 20). They are used in the treatment of haematological (vincristine and vinblastine) and non-haematological cancers (vinorelbine). They are associated with neurotoxicity due to their anti-microtubule effect and must never be given intrathecally as this is lethal.
Side-effects of chemotherapy
Chemotherapy carries many potentially serious side-effects and should be used only by trained practitioners; however, an appreciation of its common potential side-effects is necessary for any general physician who is called to see a cancer patient on chemotherapy. The five most common side-effects are vomiting, hair loss, tiredness, myelosuppression and mucositis (Table 9.10). Side-effects are much more directly dose related than anticancer effects and it has been the practice to give drugs at doses close to their maximum tolerated dose, although this is not always necessary to achieve their maximum anticancer effect. Common combination chemotherapeutic regimens are shown in Table 9.11.
|
|
Table 9.11 Some common chemotherapy regimens
|
Hodgkin’s lymphoma |
ABVD |
Doxorubicin, bleomycin, vinblastine, dacarbazine |
|
Non-Hodgkin’s lymphoma |
CHOP |
Cyclophosphamide, hydroxy-doxorubicin, vincristine, prednisolone |
|
Breast |
AC |
Adriamycin and cyclophosphamide |
|
Lung |
PE |
Cisplatin, etoposide |
|
Stomach |
ECF |
Epirubicin, cisplatin, 5-fluorouracil |
|
Colorectal |
FolFOx |
Oxaliplatin, 5FU, folinic acid |
Note: Some abbreviations are related to trade names.
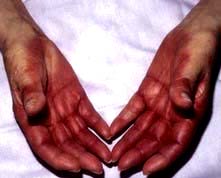
Hair, skin and nails. Many but not all cytotoxic drugs are capable of causing hair loss. Scalp cooling can sometimes be used to reduce hair loss but in general this side-effect can only be avoided by selection of drugs where this is possible. Hair regrows on completion of chemotherapy. Nails will demonstrate banding reflecting periods of cessation of growth during each chemotherapy cycle and skin toxicity may be particularly pronounced with 5FU, capecitabine and docetaxel (Fig. 9.9).

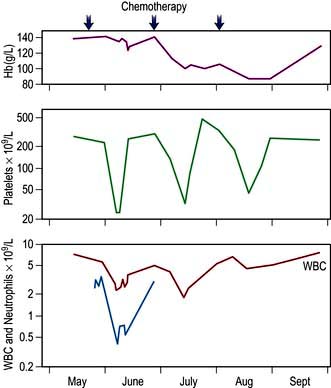
Bone marrow suppression and immunosuppression. Suppression of the production of red blood cells, white blood cells and platelets occurs with most cytotoxic drugs and is a dose-related phenomenon (Fig. 9.10). Severely myelosuppressive chemotherapy may be required if treatment is to be given with curative intent despite the potential for rare but fatal infection or bleeding. Anaemia and thrombocytopenia are managed by red cell or platelet transfusions. (Neutropenic infection is discussed on p. 448.) The risk of infective problems can be ameliorated by the use of prophylactic antimicrobials, such as ciprofloxacin, or the use of GCSF as primary prophylaxis in those chemotherapy regimens with a significant risk of febrile neutropenia or those patients on less intensive therapies who are at higher risk due to age or co-morbidity.
Haemopoietic stem cell transplantation (HSCT)
HSCT is used in a range of malignancies and some non-malignant disorders such as sickle cell disease. It relies on the ability of transfused haemopoietic stem cells to repopulate the marrow niche that has been rendered temporarily or permanently hypoplastic from chemotherapy with or without additional radiotherapy. Such procedures vary in the source of the stem cells and in the type and intensity of the preparatory conditioning regimen (Box 9.4).
 Box 9.4
Box 9.4



Principles of endocrine therapy
Oestrogens are capable of stimulating the growth of breast and endometrial cancers and androgens the growth of prostate cancer. Removal of these growth factors by manipulation of the hormonal environment may result in apoptosis and regression of the cancer. Endocrine therapy can be curative in a proportion of patients treated for micrometastatic disease in the adjuvant setting for breast and prostate cancer and provides a minimally toxic non-curative (palliative) treatment in advanced/metastatic disease. The presence of detectable cellular receptors for the hormone is strongly predictive of response. However, this is also modified by the many molecular interactions between the activation pathways of, for example, EGFR and oestrogen receptor (ER). (See breast cancer p. 475 and prostate cancer p. 481.)
Principles of biological and targeted therapy
Biological therapies
Interferons
Alpha-interferon (IFN-α) has been used to treat advanced melanoma and renal cell carcinoma.
Treatment with IFN has side-effects (see p. 321), most commonly flu-like symptoms, which tend to diminish with time, and fatigue, which generally does not, and can be treatment limiting. IFN was given as a daily subcutaneous injection, but conjugation with polyethylene glycol (PEG interferon) has led to a reduction in frequency of injection and severity of side-effects.
Targeted therapies
Monoclonal antibodies



Intracellular signal inhibitors (Table 9.12)
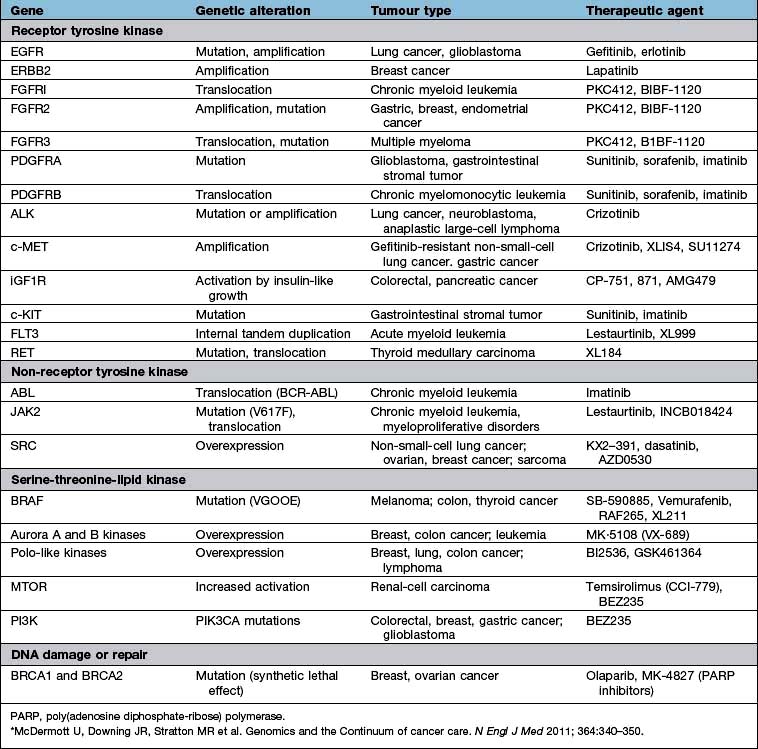
Many cancer cells are transformed by the activity of the protein products of oncogenes that signal growth by phosphorylation of tyrosine residues on the intracellular portion of growth factor receptors. Small molecule inhibitors have many pharmacokinetic advantages over the MAb inhibitors. The first example was the tyrosine kinase inhibitor (TKi) imatinib, which specifically inhibits the BCR-ABL fusion oncoprotein and c-Kit. This compound is an extremely effective treatment for chronic myeloid leukaemia and gastrointestinal stromal tumours (GIST), which are characterized by the presence of the c-Kit target. Lapatinib, which inhibits Her2, has increased survival in breast cancer. The less specific TKIs sunitinib and sorafenib which inhibit signalling by EGFR and VEGFR have proved effective in metastatic renal cancer, while erlotinib and gefitinib have shown activity in lung cancer. Vemurafenib is a kinase inhibitor and has a specificity for BRAF-V600 as used in malignant melanoma ( p. 1226). Many other similar molecules are in preclinical or early clinical development but heterogeneity within a single tumour occurs and genomic anomalies may not all be represented in a single biopsy specimen.
Principles of radiation therapy



Clinical application of radiation therapy
The cancers for which radiotherapy is usually employed in a primary curative approach, when the tumour is anatomically localized, are listed in Table 9.13, along with those in which radiotherapy has curative potential when used in addition to surgery (adjuvant radiotherapy). Palliative treatments are frequently used to provide relief of symptoms to improve quality if not duration of survival (Box 9.5). Palliative treatment is usually given in as few fractions as possible over as short a time as possible. Radiotherapy planning, by the use of CT scanning guidance, has been complemented by the introduction of 3-dimensional planning and intensity modulated radiotherapy (IMRT) which can deliver curved dose distributions to enable an improved therapeutic ratio. This allows a greater differential in dose between the tumour and critical normal structures, in turn allowing dose escalation or a reduced risk of toxicity. 4D radiotherapy planning is also becoming widely used varying radiation dose over time, e.g. the respiratory cycle during lung cancer treatment. Stereotactic focused irradiation using the γ-knife or Cyberknife can concentrate gamma radiation from multiple sources onto a small volume to generate an ablative dose for treating tumours of the CNS and isolated metastases.
| Primary modality | Adjuvant to primary surgery |
|---|---|
|
Retina |
Lung |
|
CNS |
Breast |
|
Skin |
Uterus |
|
Pharynx and larynx |
Bladder |
|
Cervix and vagina |
Rectum |
|
Prostate |
Testis-seminoma |
|
Lymphoma |
Sarcoma |
 Box 9.5
Box 9.5
Palliative benefits of radiotherapy
 Pain relief, e.g. bone metastases
Pain relief, e.g. bone metastases
 Reduction of headache and vomiting in raised intracranial pressure from CNS metastases
Reduction of headache and vomiting in raised intracranial pressure from CNS metastases
 Relief of obstruction of bronchus, oesophagus, ureter and lymphatics
Relief of obstruction of bronchus, oesophagus, ureter and lymphatics
 Preservation of skeletal integrity from metastases in weight-bearing bones
Preservation of skeletal integrity from metastases in weight-bearing bones
 Reversal of neurological impairment from spinal cord or optic nerve compression by metastases
Reversal of neurological impairment from spinal cord or optic nerve compression by metastases
Side-effects of radiotherapy
Early radiotherapy side-effects may occur within days to weeks of treatment when they are usually self-limiting but associated with general systemic disturbance (Table 9.14). The side-effects will depend upon tissue sensitivity, fraction size and treatment volume and are managed with supportive measures until normal tissue repair occurs. The toxicity may also be enhanced by exposure to other radiation-sensitizing agents, especially some cytotoxics, e.g. bleomycin, actinomycin, anthracyclines, cisplatin and 5-fluorouracil.
|
Acute temporary side-effects/dependent on region being treated |
|
|
Anorexia, nausea, malaise |
|
|
Mucositis, oesophagitis, diarrhoea |
|
|
Alopecia |
|
|
Myelosuppression |
|
|
Late side-effects |
|
|
Skin |
Ischaemia, ulceration |
|
Bone |
Necrosis, fracture, sarcoma |
|
Mouth |
Xerostomia, ulceration |
|
Bowel |
Stenosis, fistula, diarrhoea |
|
Bladder |
Fibrosis |
|
Vagina |
Dyspareunia, stenosis |
|
Lung |
Fibrosis |
|
Heart |
Pericardial fibrosis, cardiomyopathy, vasculopathy |
|
CNS |
Myelopathy |
|
Gonads |
Infertility, menopause |
|
Second malignancies |
e.g. leukaemia, cancer, e.g. thyroid |
|
Other |
Carotid artery stenosis |
Acute oncology
The acute care of all patients admitted to hospital with a cancer diagnosis has become known as acute oncology, in order to ensure a coordinated patient care, with access to all facets of the multidisciplinary team and thus the most efficient use of high cost inpatient facilities. In addition, there are a number of common oncological emergencies for which urgent treatment is critical for success (Table 9.15).
|
Fever |
Neutropenic sepsis |
|
Breathlessness |
Pulmonary embolus, pleural effusion |
|
Neutropenic sepsis |
|
|
Bronchial obstruction and lobar collapse |
|
|
Tense ascites |
|
|
Hypotension |
Neutropenic sepsis |
|
Embolus, pericardial tamponade |
|
|
Swollen facies |
Superior vena caval obstruction |
|
Leg weakness |
Spinal cord compression |
|
Mental deterioration |
Hypercalcaemia |
|
Raised intracranial pressure |
|
|
Renal failure |
Obstructive uropathy, sepsis |
|
Drugs; NSAIDs, methotrexate, cisplatin |
|
|
Metabolic; calcium, uric acid, myeloma protein, tumour lysis |
|
|
Haemorrhage |
Tumour erosion, thrombocytopenia, DIC |
|
Bone pain |
Pathological fracture |
|
Acute abdomen |
Intestinal obstruction and perforation |
|
Jaundice |
Obstructing mass, parenchymal destruction by tumour or drugs |
Neutropenic sepsis
This is the most common cause of attendance in the emergency department for any cancer patient and must be always considered in any patient who is unwell within a month of chemotherapy. Neutropenic patients are at high risk of bacterial and fungal infection, most often from enteric bowel flora. Patients must be warned of the possibility of neutropenic fever occurring. Nonspecific symptoms are also common, e.g. nausea, diarrhoea, drowsiness, breathlessness. A fever >37.5°C may not always be present. The critical test is the full blood count and patients with neutrophils <1.0 × 109/L are managed by the immediate introduction of broad-spectrum antibiotics and fluid resuscitation. Treatment can be risk stratified and low-risk patients managed with oral antibiotics such as co-amoxiclav. However, signs of systemic illness such as tachycardia, hypotension, oliguria mandate urgent admission and resuscitation with intravenous treatment (Box 9.6). Initial empirical therapy should be reviewed following microbiological results. All such patients need to be discussed with the appropriate specialist oncology team and hospitals should have clear protocols for the rapid institution of antibiotics in such patients within an hour of arrival in the A&E department. In units practised in the assessment of febrile neutropenia it is possible to follow a more risk-stratified antibiotic policy and avoid or curtail admission with oral co-amoxiclav plus ciprofloxacin when low-risk features are present: i.e. absence of tachycardia, hypotension, hypoxia and mucositis and an expected short duration of myelosuppression.
 Box 9.6
Box 9.6
Treatment of febrile neutropenia*
 One-off reading ≥38.5°C or a reading ≥38°C sustained for 1 hour
One-off reading ≥38.5°C or a reading ≥38°C sustained for 1 hour
 Patients on antipyretics or steroids and elderly patients may not mount a febrile response
Patients on antipyretics or steroids and elderly patients may not mount a febrile response

Immediate intervention is essential
 Resuscitation with intravenous fluids to restore circulatory function, monitor urine output, GCS and central venous pressure
Resuscitation with intravenous fluids to restore circulatory function, monitor urine output, GCS and central venous pressure
 Cultures of blood, urine, sputum and stool
Cultures of blood, urine, sputum and stool
 Empirical antibiotics as per local policy and sensitivities
Empirical antibiotics as per local policy and sensitivities
 Intensive care review and consideration for inotropic support at an early stage
Intensive care review and consideration for inotropic support at an early stage


Pulmonary embolus
This is a common complication of the coagulopathy of cancer and as a side-effect of chemotherapy (Fig. 9.11). It often presents with unexplained breathlessness and episodic exacerbations from multiple small emboli, rather than chest pain. A high level of suspicion should be kept in any cancer patient with hypoxia or chest pain. CT pulmonary angiogram is the investigation of choice. Prophylactic anticoagulation is given to all immobilized patients (see p. 429). Warfarin is ineffective in reversing the coagulopathy of cancer and heparin is required as long as treatment continues or the cancer is active.
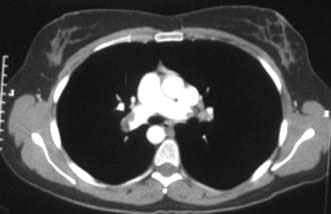
Superior vena caval obstruction
Superior vena caval obstruction (Fig. 9.12) can arise from any upper mediastinal mass but is most commonly associated with lung cancer and lymphoma. The patient presents with difficulty breathing and/or swallowing, with stridor, swollen, oedematous facies and arms with venous congestion in the neck and dilated veins in the upper chest and arms. Treatment is with immediate steroids, vascular stents, anticoagulation and mediastinal radiotherapy or chemotherapy. Some tumours, e.g. lymphomas, small-cell lung cancers and germ cell tumours, are so sensitive to chemotherapy that this is preferred to radiotherapy, as the masses are likely to be both large and associated with more disseminated disease elsewhere. An early decision is necessary on the patient’s likely prognosis, as ventilatory support may be required until treatment has had time to relieve the obstruction.
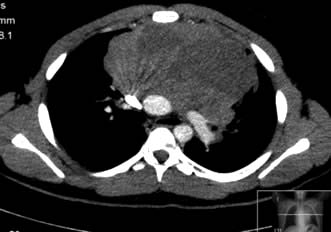
Spinal cord compression
Spinal cord compression (see p. 1135) needs to be rapidly diagnosed and urgent treatment arranged within 24 hours of onset of paresis to salvage as much functional capacity as possible. Early neurological clinical features may be incomplete, more subjective than objective and gradual in onset. MR scanning is the investigation of choice. Treatment should begin with high-dose steroids and a joint neurosurgical and oncological consultation. Good prognosis patients with limited disease require surgical decompression and radiotherapy to the affected vertebrae to achieve the best disease control and palliation.
Acute hypercalcaemia
This presents with vomiting, confusion, constipation and oliguria. Treatment is by resuscitation with intravenous fluids to establish a saline diuresis and i.v. bisphosphonate, e.g. pamidronate or the more potent zoledronic acid (see Emergency Box 19.2). Treating the cause is crucial. Denosumab and calcitonin can be used in intractable cases.
Malignant bile duct obstruction
FURTHER READING
Hoeijmakers JH. DNA damage, aging and cancer. N Engl J Med 2009; 361:1475–1485.
McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011; 364:340–350.
Smith TJ, Khatcheressian J, Lyman GH et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 2006; 24:3187–3205.
Haematological malignancies
The leukaemias, the lymphomas and multiple myeloma are an interrelated spectrum of malignancies of the myeloid and lymphoid systems. They are uncommon but not rare, the lymphomas alone being the fifth commonest cancer in the UK. The aetiology of these diseases for the most part is unknown, although viruses, irradiation, cytotoxic poisons and immune suppression have been implicated in a small proportion of cases (see p. 434). The pathogenesis involves at least one or usually more molecular abnormalities and non-random chromosomal abnormalities have been detected in several leukaemias and lymphomas. Classification has become increasingly complex, with the universally applied WHO scheme demanding morphological, cytogenetic and sometimes molecular criteria to be fulfilled. Transformation from low-grade to high-grade pathologic subtype may occur. Treatment options are multiple. Patients need to be supported through treatment involving prolonged myelosuppression and immunosuppression. These are potentially life-threatening but can also be curative. This has given rise to the need for highly skilled staff and specialist facilities; patients should be referred to these centres for treatment.
Haematological malignancies can be divided on the basis of the speed of evolution of the disease, according to the cell of origin (myeloid or lymphoid) and according to whether the presentation is primarily a marrow-based leukaemic presentation or a nodal or extranodal lymphomatous presentation where soft tissue masses predominate. This is summarized in Table 9.16. This is an arbitrary division and there is movement across the divisions, e.g. CML can transform to either AML or ALL, myeloproliferative neoplasms (MPN) and MDS can transform to AML, CLL and low-grade NHL can transform to a high-grade form ‘Richter’s transformation’.
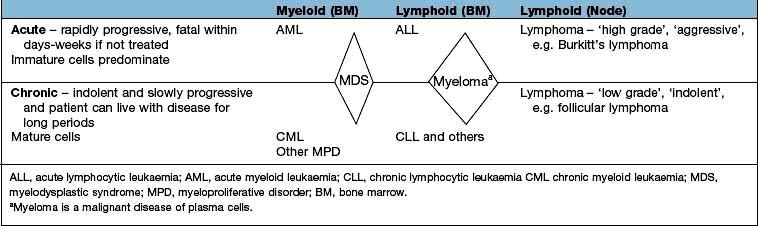 |
The myelodysplastic syndromes are considered pre-leukaemic and are discussed on page 405. Similarly the myeloproliferative disorders may also transform to acute leukaemia and are discussed on page 402.
The leukaemias
There are four main subtypes, as discussed above:
1. Acute myeloid leukaemia (AML)
2. Acute lymphoblastic leukaemia (ALL)
Aetiology
In the majority of patients, this is unknown but several factors have been associated:
 Radiation. This can induce genetic damage to haemopoietic precursors and ALL, AML and CML have been seen in increased incidences in survivors of Hiroshima and Nagasaki and in patients treated with ionizing radiation.
Radiation. This can induce genetic damage to haemopoietic precursors and ALL, AML and CML have been seen in increased incidences in survivors of Hiroshima and Nagasaki and in patients treated with ionizing radiation.



Genetic abnormalities in leukaemia
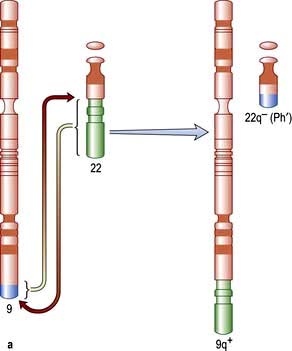
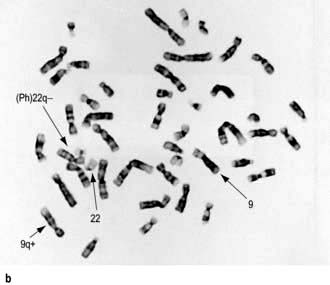
The first non-random chromosomal abnormality to be described was the Philadelphia (Ph) chromosome, which is associated with CML in 97% of cases (see Fig. 9.16a). The Ph chromosome is also found in ALL, the incidence in the latter illness increasing with age. The translocation is shown schematically in Figure 9.16a. The Ph chromosome is an abnormal chromosome 22, resulting from a reciprocal translocation between part of the long arm of chromosome 22 and chromosome 9. The resulting karyotype is described as t(9; 22)(q34; q11). The molecular consequences of the translocation are that part of the Abelson proto-oncogene (c-ABL) normally present on chromosome 9 is translocated to chromosome 22, where it comes into juxtaposition with a region of chromosome 22 named the ‘breakpoint cluster region’ (BCR). The new ‘fusion’ gene BCR-ABL is capable of being expressed as a chimeric messenger RNA, which has been identified in cells from patients with CML. When translated, this produces a fusion protein that has tyrosine kinase activity and enhanced phosphorylating activity compared with the normal protein, resulting in altered cell growth, stromal attachment and apoptosis. The breakpoint differs in CML and Ph-positive ALL, leading to the production of two different tyrosine kinase proteins with molecular weights of 210 kDa and 190 kDa, respectively. It is unclear whether the presence of BCR-ABL is sufficient for the development of the disease. It has been shown that normal subjects can carry low levels of the BCR-ABL fusion gene in their blood without developing leukaemia. Other genetic and cytogenetic abnormalities are often seen in leukaemic cells (Table 9.17).
|
|
a The entities included in this group are defined almost identically to the corresponding entity in the French–American–British (FAB) classification.
Modified from: Jaffe ES, Harris NL, Stein H et al. (eds) World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008, with permission from the World Health Organization.
Acute leukaemias
The acute leukaemias increase in incidence with advancing age. Acute myeloid (myeloblastic, myelogenous) leukaemia (AML) has a median age at presentation of 65 years and may arise de novo or against a background of myelodysplasia or prior cytotoxic chemotherapy (‘secondary’). Acute lymphoid (lymphoblastic) leukaemia (ALL) has a substantially lower median age at presentation and in addition is the commonest malignancy in childhood. The WHO classification is shown in Table 9.17.
Clinical features
The majority of patients with acute leukaemia, regardless of subtype, present with symptoms reflecting inadequate haematopoiesis secondary to infiltration of the bone marrow by leukaemic cells, symptoms due to tissue infiltration by leukaemic cells, the consequences of a high WBC or substance release from the tumour cells (Table 9.18).
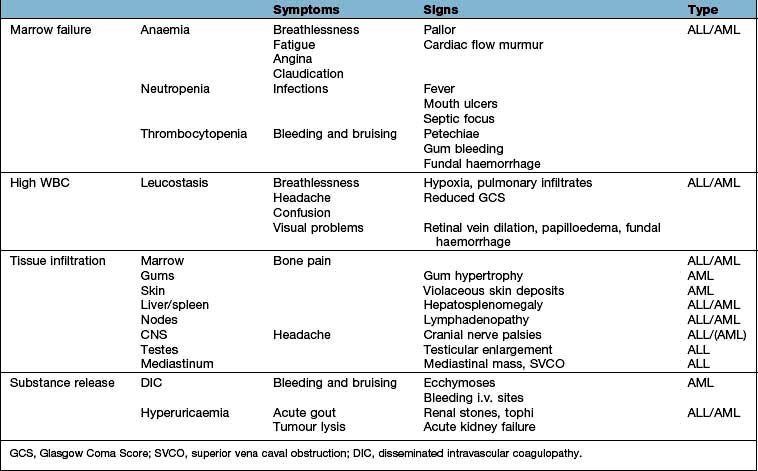
Investigations
Confirmation of diagnosis (Fig. 9.13)
 Blood count. Hb low, WBC raised usually (sometimes low), platelets low.
Blood count. Hb low, WBC raised usually (sometimes low), platelets low.
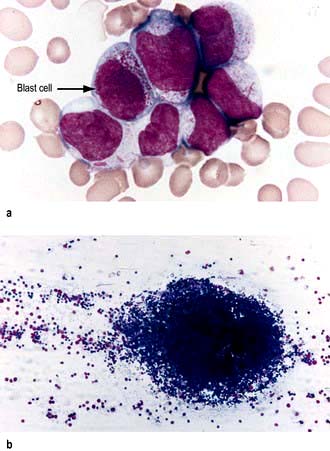
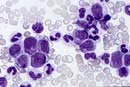
 Blood film. Blast cells almost invariably seen (Fig. 9.13a), lineage may be identified morphologically, e.g. presence of Auer rods is consistent with a diagnosis of AML (Box 9.7).
Blood film. Blast cells almost invariably seen (Fig. 9.13a), lineage may be identified morphologically, e.g. presence of Auer rods is consistent with a diagnosis of AML (Box 9.7).
 Bone marrow aspirate. Increased cellularity, reduced erythropoiesis, reduced megakaryocytes. Replacement by blast cells >20% (often approaching 100%) (Fig. 9.13b). Lineage confirmation by immunophenotyping, e.g. AML – CD33 or CD13, B lineage ALL – CD10 and CD19 and T lineage – CD3. Cytogenetic FISH analysis in real-time PCR and molecular genetics for prognostication.
Bone marrow aspirate. Increased cellularity, reduced erythropoiesis, reduced megakaryocytes. Replacement by blast cells >20% (often approaching 100%) (Fig. 9.13b). Lineage confirmation by immunophenotyping, e.g. AML – CD33 or CD13, B lineage ALL – CD10 and CD19 and T lineage – CD3. Cytogenetic FISH analysis in real-time PCR and molecular genetics for prognostication.
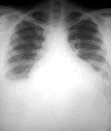
 Chest X-ray. Mediastinal widening often present in T lymphoblastic leukaemia.
Chest X-ray. Mediastinal widening often present in T lymphoblastic leukaemia.


 Box 9.7
Box 9.7







Active therapy
Supportive care
This forms the basis of treatment whether for cure or palliation:
 Avoidance of symptoms of anaemia (haemoglobin >80–100 g/L) – repeated transfusion of packed red cells (sometimes irradiation of cells is required)
Avoidance of symptoms of anaemia (haemoglobin >80–100 g/L) – repeated transfusion of packed red cells (sometimes irradiation of cells is required)

 Treatment of infection (see Box 9.6):
Treatment of infection (see Box 9.6):
 Control of hyperuricaemia with hydration, prophylactic allopurinol and occasionally rasburicase (see p. 479).
Control of hyperuricaemia with hydration, prophylactic allopurinol and occasionally rasburicase (see p. 479).
Specific treatment
Successful remission induction is always followed by further treatment (consolidation). The details of this are determined by the type of leukaemia, the patient’s risk factors and the patient’s tolerance of treatment. Recurrence is almost invariable if ‘consolidation’ therapy is not given. This reflects the lack of sensitivity of the definition of ‘complete remission’, which has been solely morphological. Cytogenetics and molecular genetic techniques can however identify residual leukaemic cells not detected morphologically and they are highly predictive of recurrence. Recommendations have recently been made to modify the definition of remission to reflect this. Failure to achieve morphological CR with two cycles of therapy (‘refractory’) carries almost as bad a prognosis as the untreated leukaemia. If CR can be achieved, e.g. by new experimental approaches, cure may still be possible with stem cell transplantation (see p. 444). A small proportion of patients with refractory disease may also be cured by a myeloablative allograft.
Acute myeloid leukaemia (AML)
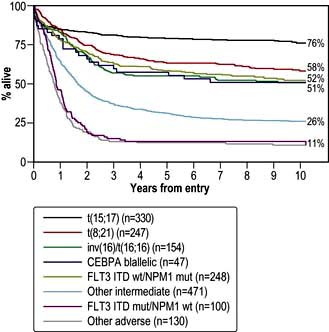
In AML, the prognosis is dependent on a range of key variables, the two main ones being age and cytogenetics (Fig. 9.14; Table 9.19).
| Good risk | Poor risk |
|---|---|
Acute promyelocytic leukaemia (APML)
This is a variant of AML, occurring in 10–15% of cases, that is characterized by the translocation t(15; 17) and with particular morphological features. There is an almost invariable coagulopathy, which was a major cause of early death. The empirical discovery that all-trans-retinoic acid (ATRA) causes differentiation of promyelocytes and rapid reversal of the bleeding tendency was a major breakthrough. APML is treated with ATRA combined with several courses of chemotherapy. Complete remission and molecular remission occur in at least 90% of younger adults with APML and at least 70% will expect to be cured. Transplantation may be necessary either if the leukaemia is not eliminated at the molecular level, or following recurrence and reinduction therapy. Arsenic trioxide, which induces apoptosis via activation of the caspase cascade (see p. 33), is used with resistant or relapsed disease and is under investigation as first-line therapy.
Acute lymphoblastic leukaemia (ALL)
Prognostic factors
A number of clinical and laboratory features are determinants of treatment response and survival in ALL (Table 9.20). Increasingly, therapeutic strategies based upon prognostic risk are being used in the management of the disease.
| Risk factor | Good | Poor |
|---|---|---|
|
Age |
Younger age |
Older age |
|
WBC |
<50 × 109/L for B-lineage <100 × 109/L for T-lineage |
>50 × 109/L for B-lineage >100 × 109/L for T-lineage |
|
Immunophenotype |
CD10 + common ALL |
Pro-B ALL |
|
Cytogenetic aberrations |
t(12;21) hyperdiploidy |
t(9;22) or t(4;11) hypodiploidy |
|
Time to response |
Early clearance of blasts |
Failure to achieve a CR within 3–4 weeks |
|
Minimal residual disease |
MRD negative |
MRD positive |
|
Extramedullary disease |
CSF clear |
CSF positive |
Prognosis
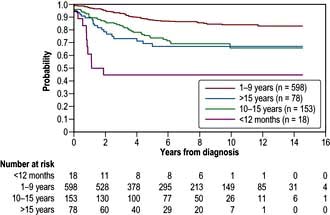
The prognosis (Fig. 9.15) of ALL in childhood is now excellent: complete remission is achieved in almost all, with up to 80% being alive without recurrence at 5 years. Failure occurs most frequently in those with high presentation blast counts or a t(9;22) translocation. Current treatment strategies are to lessen therapy for ‘good risk’ children in order to avoid some of the long-term consequences of therapy such as avascular necrosis of bone, infertility, neurotoxicity and cardiotoxicity.
Chronic leukaemias
Chronic myeloid leukaemia (CML)
Chronic myeloid leukaemia (CML), which accounts for about 14% of all leukaemias, is one of the family of myeloproliferative neoplasms (MPNs) and is almost exclusively a disease of adults with the peak of presentation being between 40 and 60 years. It is defined by the presence of the Philadelphia chromosome (Fig. 9.16), either demonstrated cytogenetically (95%) or molecularly (5%). Unlike the acute leukaemias which are either rapidly reversed or rapidly fatal, CML has a more slowly progressive course, which if not initially cured, will be followed eventually by ‘blast crisis’ transformation to acute leukaemia (75% myeloid, 25% lymphoid) or myelofibrosis with death in a median of 3–4 years.
Clinical features
CML usually presents in the chronic phase and some patients have no symptoms. Symptoms will include:











Investigations




Chronic lymphocytic leukaemia (CLL)
This is the most common leukaemia, occurring predominantly in later life and increasing in frequency with advancing years (median age of presentation between 65 and 67 years). It results from the clonal expansion of small lymphocytes and is almost invariably (95%) B cell in origin. The majority of patients are asymptomatic, identified as a chance finding on a blood count performed for another indication. Other patients, however, present with the features of marrow failure or immunosuppression. The median survival is about 10 years and prognosis correlates with clinical stage at presentation (Table 9.21). A number of cytogenetic and molecular abnormalities are now recognized as being of prognostic significance (see below). This condition may present in leukaemic phase with significant marrow involvement (CLL) or may present as localized disease (small lymphocytic lymphoma, SLL). The tumour cells in each condition are indistinguishable and a similar therapeutic approach is therefore used. A pre-malignant condition, monoclonal B cell lymphocytosis (MBL) exists where there are less than the 5 × 109/L B-cells required for a diagnosis of CLL. Some of these have CLL phenotype and may progress to CLL.
Clinical features
The majority of patients are asymptomatic at presentation. Common symptoms will be:
 Recurrent infection because of (functional) leucopenia and immune failure (reduced immunoglobulins)
Recurrent infection because of (functional) leucopenia and immune failure (reduced immunoglobulins)







Investigations
 Blood count. Hb normal or low; WBC raised and may be very high; with lymphocytosis (criteria for diagnosis >5 × 109/L), platelets normal or low.
Blood count. Hb normal or low; WBC raised and may be very high; with lymphocytosis (criteria for diagnosis >5 × 109/L), platelets normal or low.

 Bone marrow. Reflects peripheral blood, often very heavily infiltrated with lymphocytes.
Bone marrow. Reflects peripheral blood, often very heavily infiltrated with lymphocytes.

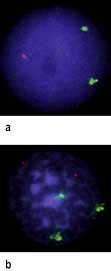
 Cytogenetics/FISH analyses are not essential for diagnosis but help in the assessment of prognosis.
Cytogenetics/FISH analyses are not essential for diagnosis but help in the assessment of prognosis.
 Direct Coombs’ test. May be positive if there is haemolysis.
Direct Coombs’ test. May be positive if there is haemolysis.

Management
In CLL, the major consideration is when to treat, indeed 30% of patients will never require intervention. Treatment depends on the ‘stage’ (Table 9.21) of the disease. Choice of therapy will depend upon patient-related factors such as age and co-morbidity, adverse prognostic features and anticipated response and toxicities to therapy. Intervention, when indicated, usually causes improvement in symptoms and in the blood count. The effect on survival is unclear.
 Marrow failure manifest by worsening anaemia and/or thrombocytopenia
Marrow failure manifest by worsening anaemia and/or thrombocytopenia

 Massive or progressive splenomegaly or lymphadenopathy
Massive or progressive splenomegaly or lymphadenopathy
 Progressive disease manifest by doubling of the lymphocyte count in 6 months
Progressive disease manifest by doubling of the lymphocyte count in 6 months


General/supportive treatment
Anaemia due to haemolysis is treated with steroids. Anaemia and thrombocytopenia due to marrow infiltration is treated with chemotherapy and, when necessary, transfusion. Erythropoietin (see p. 374) may avoid the need for transfusions, particularly in patients receiving chemotherapy.