[level-membership-for-cardiovascular-category]18
Macromolecular Complexes and Regulation of the Sodium Channel Nav1.5
Nav1.5 and Interacting Proteins
Among the many ionic currents known to be involved in the genesis of the cardiac action potential (AP), the Na+ current (INa) has been studied for more than 50 years1 and remains a major focus of research. The basic structure and function of the cardiac Na+ channel Nav1.5 and its central role in cardiac pathologies are covered in Chapters 1, 9, and 50 of this book. This chapter summarizes recently published data on the proteins interacting with Nav1.5 that form distinct macromolecular and multiprotein complexes. The roles of the four β-subunits are not reviewed because that topic is covered in other chapters of this book. Over the past several years, Nav1.5 has been shown to interact with a growing list of regulatory proteins (Figure 18-1, A and B; Table 18-1). The genes coding for several of these interacting proteins have been found in patients with inherited arrhythmias, such as congenital long QT syndrome (LQTS)2 and Brugada syndrome (BrS).3 The proteins interacting with Nav1.5 have been classified as (1) anchoring/adaptor proteins involved in trafficking, targeting, and anchoring the channel protein to specific membrane compartments; (2) enzymes interacting with and modifying the channel structure via posttranslational modifications such as protein kinases or ubiquitin ligases; and (3) proteins modulating the biophysical properties of Nav1.5 upon binding (see Table 18-1). These classifications are not mutually exclusive.
Table 18-1
The 16 Proteins (or Families of Proteins) Reported to Interact and Regulate Nav1.5
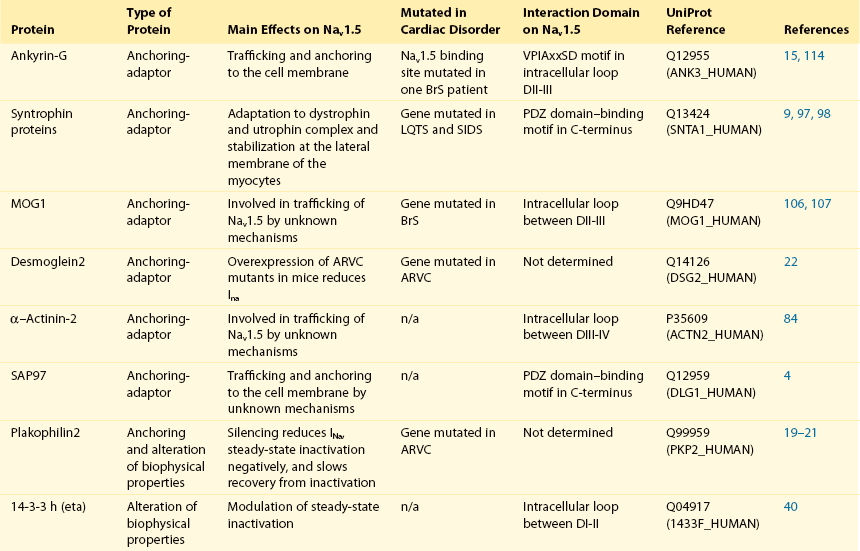
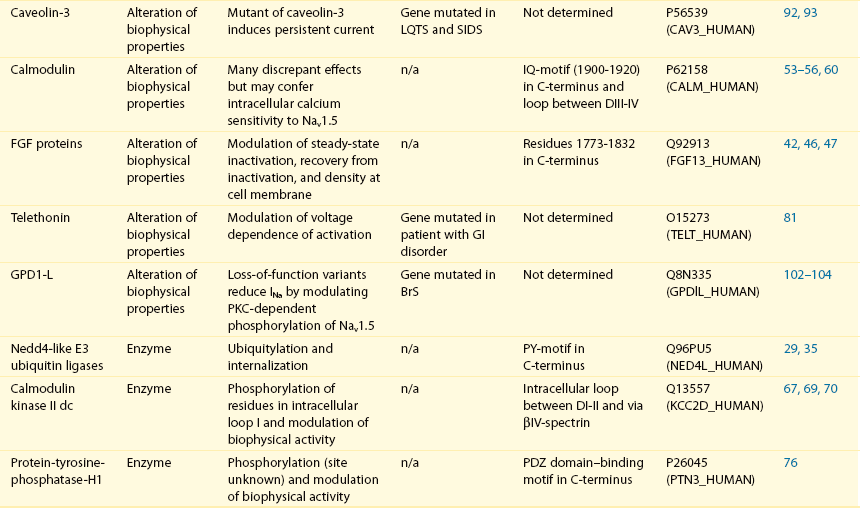
This table does not take into account the four β sodium channel subunits.
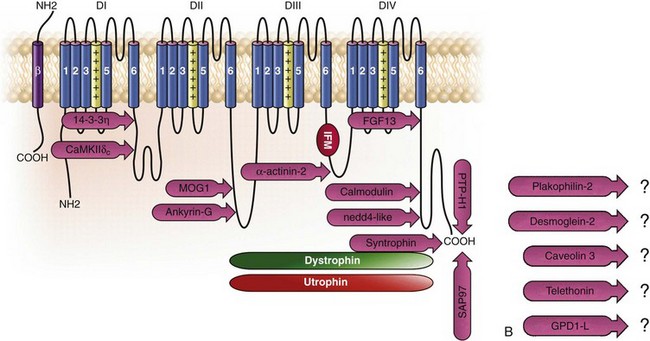
Figure 18-1 Schematic representation of the α- and β-subunits of the Nav1.5 and interacting proteins. A, The predicted membrane topology of the α-subunit of Nav1.5 is illustrated. DI-DIV indicates the four homologous domains of the α-subunit; segments S5 and S6 are the pore-lining segments and the S4 helices (yellow) serve as voltage sensors. In the connecting loop between DIII and DIV, the IFM red symbol presents the three residues (isoleucine, phenylalanine, and methionine) known to play a key role in the fast inactivation process. The two intracellular loops display many sites for phosphorylation, whereas the C-terminal domain bears many protein-protein interaction motifs. Only one β-subunit of the four known to be able to interact with sodium channels is shown (purple = transmembrane domain protein on the left side).The eleven proteins, in addition to the β-subunits, are reported to interact with Nav1.5, for which a binding site to one of the intracellular domains is represented schematically. B, For five proteins, the interaction has been shown only by coimmunoprecipitation experiments, but the binding sites are still unknown (question marks). These interactions may be indirect (i.e., requiring adaptor proteins).
Localization of Nav1.5 in Cardiac Cells: Evidence for Distinct Pools
Immunofluorescence staining experiments have shown that Nav1.5 is expressed in distinct membrane compartments (i.e., the intercalated discs and the “lateral membrane” of cardiac cells) (Figure 18-2).4–6 This notion has been under debate because earlier studies described a quasi-exclusive localization of Nav1.5 at the intercalated discs.7,8 Our group proposed a model of Nav1.5 localization at two distinct pools based on strong molecular and in vivo evidence showing that Nav1.5 belongs to the dystrophin multiprotein complex.9 Dystrophin and syntrophin proteins are not expressed at the intercalated discs of human,10 rat,11 or mouse cardiomyocytes.4 Recent studies have presented evidence supporting this multiple-pool model of Nav1.5. Roden’s group investigated a knock-in mouse model harboring the p.D1275N mutation in the gene SCN5A, which codes for Nav1.5 and is found in patients with dilated cardiomyopathy.12 They observed a marked reduction in the expression of Nav1.5 exclusively at the lateral membrane.12 More recently, Lin et al.13 obtained functional evidence for two pools of cardiac Na+ channels with different biophysical properties by performing macropatch experiments at different locations in cardiac cells. It is very possible that this two-pool expression model of Nav1.5 is an oversimplification because there is both functional14 and morphologic evidence6,15 of a T-tubular population of Nav1.5 (Figure 18-3). In a recent paper, Nav1.5 was found to be colocalized with SAP97 at the T-tubules.16 Our recent findings suggest that this third pool does not depend on the syntrophin/dystrophin complex because it is well distinguished in dystrophin-deficient (mdx) myocytes (see Figure 18-3).
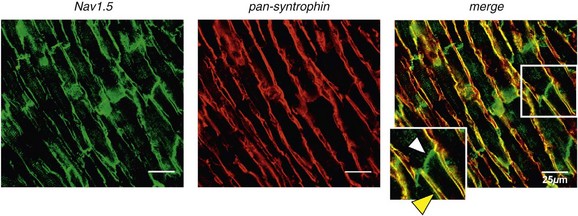
Figure 18-2 Stainings of cardiac sections leading to the concept of multiple pools of Nav1.5 channels in cardiac cells. Sections of mouse ventricular myocardium with anti-Nav1.5 staining (left panel), anti-pan-syntrophin staining (middle panel), and overlay (right panel, merge). From these stainings, it is clear that syntrophin proteins are not expressed at the intercalated discs (white arrowhead) where Nav1.5 is present. This defines the intercalated disc pool of Nav1.5 that has been proposed to interact with SAP97 (see Figure 1, A, and Figure 4). The yellow arrowhead shows the lateral membrane pool of Nav1.5 colocalized with syntrophin proteins; white bar = 25 µm.
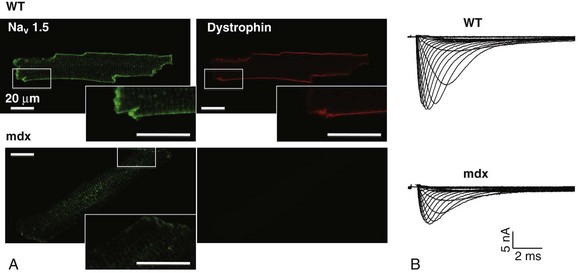
Figure 18-3 Reduction of Nav1.5 and sodium current in dystrophin-deficient mouse cardiomyocytes. A, Isolated mouse cardiomyocytes from wild type and mdx (dystrophin-deficient) mice with Nav1.5 staining (green) and dystrophin staining (red). Whereas Nav1.5 channels are found at the intercalated discs and at the lateral membrane compartment, it is apparent that dystrophin is excluded from the intercalated discs. The Nav1.5 pool at the discs has been shown to colocalize with SAP97. B, Sodium current recordings from whole-cell patch-clamp experiments of freshly isolated wild type and mdx mouse cardiomyocytes. It can be inferred that the reduction in current is caused by the specific loss of Nav1.5 channels from the lateral membrane pool. (A, From Petitprez S, Zmoos AF, Ogrodnik J, et al: SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108:294–304, 2011; with permission from Wolters Kluwer Health. B, Modified from Gavillet B, Rougier JS, Domenighetti AA, et al: Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res 18:407–414, 2006; with permission from Wolters Kluwer Health.)
The relationship between the expression of Nav1.5 at the intercalated discs and other disc proteins has recently been studied. It was observed that the intercalated disc pool of Nav1.5 is dependent on the expression of key proteins known to be well expressed at the discs, such as connexin43,17,18 plakophilin2,19–21 and desmoglein2.22 Thus, Nav1.5 not only interacts with many different partners, but its localization in different cellular and membrane compartments is also very diverse. One of the obvious conclusions of these observations is that there is not one cardiac Na+ channel Nav1.5, but rather a multiplicity of them with variable functions and regulatory mechanisms.
Proteins Interacting with Nav1.5 Without Demonstrated Roles in Arrhythmias
This section lists the proteins that interact with Nav1.5 that currently have no demonstrable pathologic roles. These interacting proteins were discovered by either performing protein-protein interaction screens, such as yeast two-hybrid assays, or by using proteomic-based protein identification assays. The sites of interaction, often protein-protein interaction domains, were mapped on the sequence of Nav1.5 as described in Figure 18-1, A. This list does not follow any specific logic related to importance but is dictated by the chronologic order of the published studies.
Ubiquitin-Protein Ligases of the Nedd4/Nedd4-like Family
Ubiquitylation of target proteins tags them for proteasome- or lysosome-dependent degradation23 but also serves multiple nondegradative functions, in particular the trafficking of membrane proteins.24 Ubiquitin is a small protein of 76 amino acids found in all animal cells.25 It binds covalently to lysine residues of target proteins. This protein ubiquitylation is performed by E3 ubiquitin-protein ligases.25 Ubiquitylated membrane proteins are subsequently internalized and can be targeted for lysosomal or proteasomal degradation. Alternatively, they can also be deubiquitylated by specific proteases and recycled back to the membrane.26,27 Many different ion channels have been reported recently to be regulated by the Nedd4-like family of E3 ubiquitin-protein ligases. Nedd4-like enzymes bind specifically to target proteins that have consensus domains known as PY motifs with the sequence [L/P]PxY.28 Such PY motifs are found in the C-termini of all voltage-gated Na+ channels (with the exception of Nav1.4, Nav1.9, and Nax29,30), as well as in other cardiac ion channels.27,31,32 Nedd4/Nedd4-like enzymes harbor several WW domains33 that can interact with these PY motifs. When expressed in Xenopus oocytes, Nav1.5-mediated INa is decreased by Nedd4-2–mediated ubiquitylation.34 Our group investigated the molecular determinants of this regulation35 and found that the ubiquitin-protein ligase Nedd4-2 binds directly to the PY motif of Nav1.5 and ubiquitylates the channel in mammalian cells. Because there was no reduction in the total level of Nav1.5 protein upon coexpression with Nedd4-2, we concluded that this was most likely caused by an increased internalization rate rather than by Nav1.5 degradation.29 Inhibition of the proteasome has been linked recently to an increase in INa and Nav1.5 expression in neonatal rat cardiomyocytes.36 Ubiquitylated Nav1.5 was found to be present in mouse cardiac tissue,35 further suggesting that membrane turnover or the stability of Nav channels can be regulated in vivo via ubiquitylation. Nine of such Nedd4-like E3 enzymes are present in the human genome,37 at least eight of which are expressed at the RNA level in human cardiac tissue.31 It remains to be determined which of these Nedd4/Nedd4-like proteins regulates Nav1.5 in a physiologic context and whether the PY motif plays a role in cardiac disease. Altogether these results suggest that the ubiquitin-proteasome system is involved in several aspects of Nav1.5 regulation, the intricacies of which remain to be elucidated.
14-3-3η (eta) Protein
The 14-3-3 protein family is composed of dimeric cytosolic adaptor ubiquitous proteins.38 The members of this family are involved in many cellular functions such as the binding and regulation of trafficking of various membrane proteins.39 Allouis et al. performed yeast two-hybrid and coimmunoprecipitation experiments40 showing that the isoform 14-3-3η interacts with the N-terminal part of the intracellular loop linking domains I to II (see Figure 18-1, A and Table 18-1). Furthermore, it was observed that 14-3-3 and Nav1.5 are colocalized at the intercalated discs of myocytes. No influence on the Nav1.5-mediated peak current was observed when Nav1.5 and 14-3-3η were coexpressed in COS cells, suggesting that this protein does not influence Nav1.5 trafficking. In this expression system, 14-3-3 shifted the inactivation curve toward negative potentials and delayed recovery from inactivation, illustrating that 14-3-3 proteins are able to modify the biophysical properties of ion channels. Because different isoforms of 14-3-3 proteins are expressed in cardiac cells,40 their exact roles in normal cardiac function and their implications in disease states require further investigation.
Fibroblast Growth Factor Homologous Factors
Although fibroblast growth factor homologous factor (FHF) family members are highly homologous in sequence and structure to fibroblast growth factors (FGF), their functional roles are quite different. FHFs are found in the cytosol because their N-termini lack signal sequences that are required for secretion.41 The FGF homologous factors FHF3, FHF1, FHF2, and FHF4 are also called FGF11, FGF12, FGF13, and FGF14, respectively. One of the best characterized functions of the FHFs is their ability to bind to and modulate voltage-gated Na+ channels. Liu et al.42 were the first to demonstrate that FGF12 interacts with the proximal portion of the C-terminus of Nav1.5 (see Figure 18-1, A). This finding was then confirmed by Goetz et al.,43 who also showed that FGF11 and FGF14 interact with the same domain of Nav1.5. In HEK293 cells, coexpression of FGF12 with Nav1.5 shifted the steady-state inactivation relationship toward hyperpolarized values without affecting the other parameters studied.42 Interestingly, several LQTS type 3 and BrS mutations are located in the domain that interacts with FHF1B (see Table 18-1). The SCN5A mutation D1790G44 disrupted the binding of this protein with Nav1.5 and abolished the FHF 12-induced shift of steady-state inactivation. Another member of the FGF family, FGF14, has been shown to downregulate the activity of Nav1.5.45 Because this regulatory protein is expressed in the brain, and not in the heart,45 the significance of this interaction is likely restricted to the central nervous system. A recent study46 demonstrated that FGF13 is expressed in mouse cardiac myocytes. FGF13 was shown to directly bind to and colocalize with Nav1.5 in mouse cardiac cells. Reducing the expression of FGF13 decreased the Na+ current, shifted the availability curve toward more negative potentials, and slowed recovery from inactivation. Furthermore, decreased membrane expression of Nav1.5 was observed in cells with reduced expression of FGF13. These results underscore the specific role of FGF13 in modulating Nav1.5 function and suggest that this gene may be a susceptibility gene for cardiac arrhythmias. A crystal structure of the Nav1.5 C-terminal domain complexed with FGF13 and calmodulin was recently published.47 The functional relationship between FGFs and calmodulin remains to be studied in more detail.
Calmodulin
Intracellular Ca2+ has been shown to modulate the function of many ion channels, including the voltage-gated Na+ channels.48,49 Many cardiac ion channels use calmodulin (CaM), a ubiquitous intracellular Ca2+-binding protein involved in many different cellular processes,50 as a Ca2+-sensing partner. The Nav1.5 C-terminal domain has an IQ motif with a consensus sequence of IQxxxRxxxxR (see Table 18-1), which is very similar to that found in voltage-gated calcium channels.51 The IQ motif is also found in all isoforms of the Nav family.52 Several studies47,53–55 have shown a direct interaction between CaM and the IQ motif of Nav1.5. Recent work by Wang et al. described a crystal structure with an IQ motif ternary complex of Nav1.5, FGF13, and CaM. The functional consequences of this CaM-Nav1.5 interaction are controversial. Several studies52,56,57 have shown inconsistent results that have been difficult to reconcile. A few groups53,55–57 reported that the voltage dependence and stability of the inactivated state were dependent on CaM and the IQ motif. In addition, it has been proposed that CaM may not be the only sensor for the Ca2+-dependent regulation of Nav1.5, because the proximal part of the Nav1.5 C-terminal domain is similar to an EF-hand motif, which is known to bind Ca2+.58,59 Whether this domain binds intracellular calcium is still under debate.47,60–62 The role of intracellular Ca2+ in Nav1.5 regulation was recently investigated by Casini et al.63 They demonstrated that Nav1.5 single-channel conductance is decreased (most likely directly) with an increase in the intracellular Ca2+ concentration. Intracellular Ca2+ appears to be an important regulator of Nav1.5 function, but more studies are needed to clarify its role.
Ca2+/Calmodulin-dependent Protein Kinase II
Ca2+/calmodulin-dependent protein kinases II (CaMKII) are serine/threonine protein kinases expressed in many cell types, where they transduce intracellular Ca2+ increases into the phosphorylation of target proteins, including cardiac ion channels.64 CaMKIIδc is the predominant cardiac isoform upregulated in human and animal heart failure models.65,66 Nav1.5 has been found to colocalize and coimmunoprecipitate with CaMKIIδc.67–69 Ashpole et al.69 have shown that CaMKIIδc interacts with the first intracellular loop of Nav1.5 (see Figure 18-1) and that the residues Ser-516 and Thr-594 may be phosphorylated by this kinase. Hund et al.70 found that Ser-571 was also a target of CaMKII within the same intracellular loop of Nav1.5, and that this regulation was dependent on the correct expression of βIV-spectrin, which interacts with ankyrin-G at the intercalated discs. Overexpression of the CaMKIIδc enzyme in rabbit myocytes and transgenic mice induced a Ca2+-dependent hyperpolarizing shift of the steady-state inactivation curve, slowed the recovery from inactivation, and increased the persistent INa. These biophysical property alterations of INa are similar to some congenital LQTS type 3 mutation-dependent modifications.71 In mice, transgenic overexpression of CaMKIIδc leads to chronic heart failure and episodes of ventricular tachycardia. In dog ventricular myocytes, it has also been shown that CaMKII activity increases the persistent/late INa,72 but it is unclear whether these arrhythmias are the direct consequence of the Nav1.5 biophysical alterations or are related to other heart failure mechanisms. Nevertheless, CaMKII is an important component in the pathogenesis of arrhythmias and potentially a new drug target.64
Proteins Tyrosine Phosphatase PTPH1
Ion channels are also regulated by the phosphorylation of tyrosine residues, a process that depends on both the phosphorylation activity of tyrosine protein kinases and the dephosphorylation activity of phosphatases.73 It was demonstrated that the overexpression of the protein tyrosine kinase Fyn in HEK293 cells altered several of the biophysical properties of Nav1.5 via the phosphorylation of Tyr-1495.74 This residue is close to the Ile-Phe-Met cluster (IFM) in the intracellular loop linking domains III-IV (see Figure 18-1, A), which is known to mediate the rapid inactivation process of voltage-gated Na+ channels.75 Fyn coexpression in HEK293 cells shifted the steady-state inactivation curve toward depolarized potentials and accelerated the recovery from Nav1.5 inactivation.74 The site of interaction of Fyn with Nav1.5 remains to be investigated. We reported76 that the protein tyrosine phosphatase, PTPH1, interacts with the PDZ-domain binding motif of Nav1.5, which is also known to interact with two other PDZ-bearing proteins, syntrophin9 and SAP97.4 PTPH1 coexpression in HEK293 cells shifted the availability curve of wild-type Nav1.5 toward hyperpolarized potentials. This effect was abolished when the PDZ domain–binding motif of the Nav1.5 C-terminus was removed by an early stop mutation. These results suggest that tyrosine phosphorylation of Nav1.5 modulates the stability of the inactivated state. The fact that several proteins (e.g., PTPH1, syntrophin proteins and SAP97) interact with the same binding domain supports the coexistence of different multiprotein complexes with Nav1.5 in cardiac cells.
Telethonin
Telethonin is a small 19-kDa protein expressed in striated muscle cells, including cardiac cells. Mutations in the gene coding for telethonin, TCAP, lead to hypertrophic and dilated cardiomyopathy and limb-girdle muscular dystrophy.77,78 This protein has been shown to interact with the sarcomeric protein titin,79 as well as the β-subunit of the KCNQ1 channel (KCNE1), which mediates the cardiac IKs current.80 Telethonin can be coprecipitated with Nav1.5 from mouse cardiac tissue, and the two proteins colocalize in cardiomyocytes.81 The site of interaction on Nav1.5 has not yet been mapped. When the endogenous expression of telethonin in HEK293 cells was reduced, the voltage-dependent activation of the Nav1.5-mediated current shifted toward positive potential values. In addition, coexpression experiments of telethonin and Nav1.5 in HEK293 cells altered several of the kinetic properties of the channel.81 Furthermore, a point mutation in TCAP was found in a patient with abnormal gut motility,81 consistent with the fact that Nav1.5 and telethonin are expressed in the neurons of the intestinal mucosa.82 The precise role of telethonin in the regulation Nav1.5 in the heart (and other tissues) remains to be clarified.
α–Actinin-2
α–Actinin-2 is a protein of the F actin–cross-linking protein family (similar to spectrin and dystrophin) that interacts with and regulates the trafficking of several potassium channels,83 in particular Kv1.5. A recent study used84 pull-down experiments to show that α–actinin-2 interacts with the intracellular III-IV loop of Nav1.5. Coexpression of α–actinin-2 in tsA201 cells increased the measured INa densities as well as the Nav1.5 protein at the cell surface. Colocalization of Nav1.5 and α–actinin-2 was found mainly at the lateral membrane at the level of the Z-lines. α–actinin-2 seems to be an alternative to the dystrophin/syntrophin complex and ankyrin-g for the connection of Nav1.5 to the cytoskeletal network. Whether these anchoring proteins have overlapping or clearly distinct functions remains to be investigated.
Synapse-associated Protein 97
Proteins of the membrane-associated guanylate kinase (MAGUK) family are expressed mainly at the cell-cell junctions and are characterized by numerous protein-protein interaction domains, including PDZ domains.85 The MAGUK proteins regulate the function and localization of many membrane proteins (including ion channels) in neurons, epithelial cells, and cardiomyocytes. Synapse-associated protein 97 (SAP97) and zonula occludens 1 are the predominant MAGUK proteins expressed in cardiac cells.86 SAP97 regulates the targeting, localization and function of cardiac K+ channels, such as Kir2.x,87 Kv1.5,88 and Kv4.x89 via their PDZ domain–binding motifs located in the C-termini. We recently obtained evidence for the coexistence of at least two pools of Nav1.5 channels in cardiomyocytes: one located at the lateral membrane with the dystrophin multiprotein complex, and the other with SAP97 at the intercalated discs (Figure 18-4).4 We demonstrated that the interaction of Nav1.5 and SAP97 was dependent on the PDZ domain–binding motif that most likely interacts with one of the PDZ domains of SAP97. Reduction of the expression of SAP97 in both HEK293 and atrial cardiac cells led to a decreased INa without modification of any biophysical properties, suggesting that SAP97 may play a role in controlling the density of Nav1.5 at the cell surface of cardiac cells. The detailed mechanisms underlying this regulation have not yet been investigated. SAP97 expression has been found at the intercalated discs4,88 and the T-tubule compartment of cardiac cells.16 Milstein et al.16 recently demonstrated that Nav1.5 expression is coregulated with the potassium channel Kir2.1, and both interact with SAP97 (see Chapter 21 of this book).
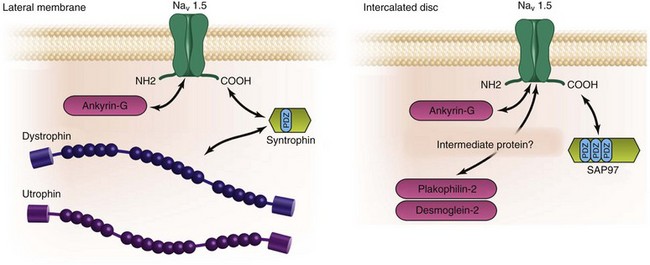
Figure 18-4 Schematic presentation of the proteins defining the two proposed pools of Nav1.5. The findings of our group have led to the concept of two distinct macromolecular complexes with Nav1.5: (left) at the lateral membrane with the dystrophin/syntrophin complex and (right) at the intercalated discs with the MAGUK protein SAP97. Note that when dystrophin is absent (as in mdx mouse myocytes), utrophin may also interact with Nav1.5 (see Figure 18-1, A).
Proteins Interacting With Nav1.5 That Are Linked to Cardiac Arrhythmias
Caveolin-3
Caveolae are plasma membrane invaginations with an enrichment of signaling molecules and ion channels.90 Caveolin proteins are important constituents of these caveolae. Caveolin-3, encoded by the gene CAV3, is the predominant isoform expressed in striated muscle cells, including cardiomyocytes. Mutations in CAV3 have been linked to limb-girdle muscular dystrophy, rippling muscle disease, and familial hypertrophic cardiomyopathy.91 CAV3 has also recently been shown to be mutated in patients with congenital LQTS type 992 and sudden infant death syndrome (SIDS).93 Caveolin-3 was coimmunoprecipitated with Nav1.5 in rat cardiac tissue94 and HEK293 cells.92 The site of interaction between caveolin-3 and Nav1.5 has not yet been determined (see Figure 18-1, B). Immunohistostaining of cardiac cells demonstrated that these two proteins are mainly colocalized at the lateral membrane.92,94,95 Dystrophin is also a component of caveolae,96 suggesting that the interaction between caveolin-3 and Nav1.5 could be indirect via proteins of the dystrophin multiprotein complex.9 The precise role of the Nav1.5/caveolin-3 interaction in normal physiology remains to be clarified. The coexpression of Nav1.5 and the mutants of caveolin-3 found in patients with LQTS and SIDS in HEK293 cells was shown to increase the inward Na+-persistent current.92,93 In an earlier study, it was reported that β-adrenergic stimulation by isoproterenol led to a rapid increase of peak INa in rat cardiac myocytes.94 This phenomenon is most likely independent of protein kinase A, because a protein kinase A inhibitor did not reduce this effect. The increase was, however, completely abolished by antibodies against caveolin-3. The precise molecular and cellular mechanisms underlying these observations require further investigation.
α-1 Syntrophin and the Dystrophin/Utrophin Complex
Earlier work has shown that Nav1.5 is part of the dystrophin multiprotein complex.97 Our group demonstrated that Nav1.5 interacts with dystrophin via adaptor syntrophin proteins.9 Similar to the binding with SAP97 and PTPH1, this interaction is dependent on the C-terminal PDZ domain–binding motif of Nav1.5 (see Figure 18-1, A), which is composed of the last three residues of the protein. In the hearts of dystrophin-deficient mice (mdx), the best-studied animal model of Duchenne muscular dystrophy, the protein level of Nav1.5 was decreased.9 The decreased expression of the Na+ channel resulted in reduced cellular INa and conduction defects, which were reflected on the electrocardiogram by a prolongation of the QRS complex duration, as well as conduction slowing in optical mapping experiments.4 The reduced expression of the Nav1.5 protein could not be explained by a decrease in the SCN5A mRNA level,9 suggesting that a defect in the translational process or a lack of dystrophin may reduce the stability of the Nav1.5 protein. Albesa et al.98 showed that utrophin, a homologue protein of dystrophin, was upregulated in mdx hearts and interacted with Nav1.5 similarly to dystrophin. Cardiac cells deficient in both dystrophin and utrophin displayed a larger decrease in Nav1.5 expression and INa.98 Two missense mutations in SNTA1, which encodes α1-syntrophin, have been described in patients with congenital LQTS,99,100 further supporting the important role of this multiprotein complex in the regulation of Nav1.5. The SNTA1 mutation, p.A390V,99 was reported to disrupt a presently undescribed macromolecular complex comprising neuronal nitric oxide synthase, plasma membrane Ca-ATPase type 4b with syntrophin, and Nav1.5. The overexpression of the mutant syntrophin protein in cardiac myocytes increased the persistent Na+ current, a finding consistent with the LQTS phenotype in affected individuals. The authors also described increased nitrosylation of Nav1.5 when mutant syntrophin was coexpressed in HEK293 cells, suggesting that under basal conditions with wild-type syntrophin, Nav1.5 nitrosylation is low because of the inhibition of neuronal nitric oxide synthase activity by Ca-ATPase. The syntrophin mutation may disrupt this complex and lead to increased nitrosylation of the channel, thus increasing the Nav1.5-persistent current. Because syntrophin seems to be excluded from the intercalated discs,4 syntrophin-dependent regulation of the persistent current is most likely exclusively related to the lateral pool of Nav1.5, suggesting that Nav1.5-dependent late currents at the lateral membrane may be different from those at the intercalated discs.
Glycerol-3-Phosphate Dehydrogenase-like Protein
Mutations in the gene coding for Nav1.5, SCN5A, are found in approximately 20% of patients with BrS. Other possible causative genes are still being investigated (see Chapter 92 ). London et al. described a locus on chromosome 3101 in a large family with BrS but simultaneously excluded SCN5A. A missense mutation of the gene coding for the glycerol-3-phosphate dehydrogenase-like protein (GPDlL) was later found.102 GPDlL is expressed in cardiac tissue and coexpression experiments using HEK293 cells showed that mutant GPDlL reduced the INa. Three more mutations of the GPDlL gene have been found in infants who died of SIDS.103 Expression of these variants in neonatal mouse cardiomyocytes decreased INa, suggesting that a proportion of SIDS patients may have decreased INa, as is observed in BrS patients. Valdivia et al.104 observed an interaction between Nav1.5 and GPDlL by performing pull-down experiments. The site of interaction has not yet been investigated. The mechanisms by which the genetic variants of GPDlL reduce the Na+ current have been studied in expression systems.104 A pathway has been proposed whereby Ser-1503 can be phosphorylated by protein kinase C (PKC) and leads to reduced INa. It has been shown that the activity of PKC depends on GPDlL function and that the mutant GPDlL variants lead to a further decrease of the INa. These observations linking the redox state of the cells with the activity of Nav1.5 are very interesting and should be investigated using native cardiac cells and tissues.
Multicopy Suppressor of gsp1 (MOG1)
MOG1 is a small 29-kDa protein that is encoded by the RANGRF gene. MOG1 binds to the GTP-binding nuclear protein Ran and is involved in regulating nuclear protein trafficking.105 MOG1 is found in the nucleus and the cytosol of cardiac cells, and its mRNA is well expressed in cardiac tissue.105 Wu et al.106 demonstrated that MOG1 interacts with the intracellular loop between domains II and III of Nav1.5 (see Figure 18-1, A). This interaction was first described by performing a yeast two-hybrid screen, followed by pull-down and coimmunoprecipitation experiments.106 The two proteins were also shown to colocalize in mouse ventricular cells, mostly at the intercalated discs. MOG1 coexpression in HEK293 cells increased the Nav1.5-mediated current without altering its biophysical properties, suggesting that MOG1 is a cofactor for optimal channel expression at the cell membrane. The role of MOG1 in regulating the surface expression of Nav1.5 was confirmed in a more recent study.107 This study described two genetic variants of MOG1 found in patients with BrS that led to reduced expression of Nav1.5 at the cell membrane of rat atrial cardiomyocytes and a decreased current. This work puts RANGRF on the increasing long list of susceptibility genes for BrS and illustrates the heterogeneity of mechanisms found in this syndrome.
Plakophilin2
Plakophilin2 is a desmosomal protein found in the intercalated discs of cardiac cells. The human gene (PKP2), which encodes plakophilin2, is mutated in patients with arrhythmogenic right ventricular cardiomyopathy.108 By performing pull-down and coimmunoprecipitation experiments, Delmar’s group19,20 demonstrated that Nav1.5 interacts not only with plakophilin2 but also with ankyrin-G and connexin43.20 Whether the interactions between these different proteins of the intercalated discs are direct or indirect, as well as the site of interaction with Nav1.5, remains to be determined. It was, however, convincingly shown that these proteins are colocalized at the intercalated discs, and that silencing of plakophilin2 in cardiomyocytes reduces the INa and alters some of its biophysical properties. The influence of plakophilin2 integrity on Nav1.5 was demonstrated in vivo by studying a mouse model expressing only one allele of plakophilin2.21 Reduced expression of plakophilin2 decreased INa without changing the localization or total expression of Nav1.5 in the mouse heart. These findings clearly support the notion of the coregulation of desmosomal proteins and the Nav1.5 complex at the intercalated discs. The molecular and cellular mechanisms underlying these observations are still unclear.
Desmoglein2
The most recently described protein interacting with Nav1.5 is desmoglein2, another protein of the desmosomal complex of cardiac cells found to be mutated in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC).109 Desmoglein2 was shown22 to coimmunoprecipitate with Nav1.5 in mouse cardiac tissue. The study of a transgenic mouse model overexpressing one mutant of desmoglein2 (p.N271S) revealed a reduction of the INa and electrical impulse propagation. There was, however, no reduction in Nav1.5 total expression. These results are very similar to those seen with the plakophilin2 mutants (discussed earlier) and further support a cross-talk mechanism between the desmosomal proteins and Nav1.5, whose molecular details remain to be investigated.
Ankyrin-G
The ankyrin proteins organize, transport, and anchor membrane proteins to the actin and spectrin cytoskeleton of cells.110 In the human genome, ANK1-3 are the three genes encoding ankyrins. The expression of ankyrin-B (ANK2) and ankyrin-G (ANK3) has been demonstrated in the heart.110 Mutations of ANK2 have been linked to congenital LQTS type 4,111 sinoatrial node dysfunction, atrial fibrillation, conduction slowing, and sudden cardiac death, the collection of which define “ankyrin-B cardiac syndrome.”112 There is currently no evidence that Nav1.5 is directly regulated by ankyrin-B, even though in ankyrin-B–deficient mouse cardiac cells, Na+ channels display late openings similar to those seen in Nav1.5 congenital LQTS type 3 mutant channels.113 On the other hand, ankyrin-G was shown to directly interact with the ankyrin-binding motif of the linker loop between domains II and III of Nav1.5114 (see Figure 18-1, A, and Table 18-1). Ankyrin-G is predominantly located at the intercalated discs, where it interacts with not only Nav1.5 but also the actin-associated protein βIV-spectrin and CaMKII.70 A clear T-tubular localization of ankyrin-G has also been observed.70 A SCN5A mutation, p.E1053K, in this motif was found in a BrS patient and was shown to disrupt the interaction between Nav1.5 and ankyrin-G.15 Expression of wild type and mutant Nav1.5 channels in adult rat myocytes15 was achieved using viral vectors, and although wild type Nav1.5 channels were correctly transported to the intercalated discs and lateral membranes, the E1053K channels remained in the cytoplasm of the transduced cells.15 These findings were recently confirmed115 by silencing the expression of ankyrin-G in neonatal cardiomyocytes, which resulted in the reduction of Nav1.5 expression and incorrect trafficking of most of the channels to the cell membrane.
Acknowledgments
The author thanks Dr. A. Felley for her useful comments on this chapter and Dr. L. Gillet for his help in collecting information. We also thank J. Gramling for the preparation of preliminary versions of Figure 18-3 and 18-4.
References
1. Fozzard, HA. Cardiac sodium and calcium channels: A history of excitatory currents. Cardiovasc Res. 2002; 55(1):1–8.
2. Cerrone, M, Priori, SG. Genetics of sudden death: focus on inherited channelopathies. Eur Heart J. 2011; 32(17):2109–2118.
3. Mizusawa, Y, Wilde, AAM. Brugada syndrome. Circ Arrhythmia Electrophysiol. 2012; 5(3):606–616.
4. Petitprez, S, Zmoos, AF, Ogrodnik, J, et al. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1. 5 in cardiomyocytes. Circ Res. 2011; 108:294–304.
5. Haufe, V, Camacho, JA, Dumaine, R, et al. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol. 2005; 564(3):683–696.
6. Leoni, AL, Gavillet, B, Rougier, JS, et al. Variable Na(v)1. 5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a mouse model. PLoS ONE. 2010; 5(2):e9298.
7. Maier, SKG, Westenbroek, RE, McCormick, KA, et al. Distinct subcellular localization of different sodium channel α and β subunits in single ventricular myocytes from mouse heart. Circulation. 2004; 109(11):1421–1427.
8. Kucera, JP, Rohr, S, Rudy, Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002; 91(12):1176–1182.
9. Gavillet, B, Rougier, JS, Domenighetti, AA, et al. Cardiac sodium channel Nav1. 5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006; 18(99):407–414.
10. Kaprielian, RR, Stevenson, S, Rothery, SM, et al. Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation. 2000; 101(22):2586–2594.
11. Stevenson, SA, Cullen, MJ, Rothery, S, et al. High-resolution en-face visualization of the cardiomyocyte plasma membrane reveals distinctive distributions of spectrin and dystrophin. Eur J Cell Biol. 2005; 84(12):961–971.
12. Watanabe, H, Yang, T, Stroud, DM, et al. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011; 124(9):1001–1011.
13. Lin, X, Liu, N, Lu, J, et al. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011; 8(12):1923–1930.
14. Brette, F, Orchard, CH. Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem Biophys Res Commun. 2006; 348(3):1163–1166.
15. Mohler, PJ, Rivolta, I, Napolitano, C, et al. Nav1. 5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1. 5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004; 101(50):17533–17538.
16. Milstein, ML, Musa, H, Balbuena, DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012; 109(31):E2134–E2143.
17. Jansen, JA, Noorman, M, Musa, H, et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1. 5 expression and reduced sodium current which accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2011; 9(4):600–607.
18. Desplantez, T, McCain, M, Beauchamp, P, et al. Connexin 43 ablation in fetal atrial myocytes decreases electrical coupling, partner connexins and sodium current. Cardiovasc Res. 2012; 94(1):58–65.
19. Sato, PY, Musa, H, Coombs, W, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009; 105(6):523–526.
20. Sato, PY, Coombs, W, Lin, X, et al. Interactions between ankyrin-G, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res. 2011; 109(2):193–201.
21. Cerrone, M, Noorman, M, Lin, X, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012; 95(4):460–468.
22. Rizzo, S, Lodder, EM, Verkerk, AO, et al. Intercalated disc abnormalities, reduced Na+ current density and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012; 95(4):409–418.
23. Glickman, MH, Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2):373–428.
24. Staub, O, Rotin, D. Role of ubiquitylation in cellular membrane transport. Physiol Rev. 2006; 86(2):669–707.
25. Hershko, A, Ciechanover, A. The ubiquitin system. Annu Rev Biochem. 1998; 67:425–479.
26. Hicke, L, Dunn, R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003; 19:141–172.
27. Abriel, H, Staub, O. Ubiquitylation of ion channels. Physiology. 2005; 20(6):398–407.
28. Rotin, D, Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009; 10(6):398–409.
29. Rougier, J-S, van Bemmelen, MX, Bruce, MC, et al. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am J Physiol Cell Physiol. 2005; 288(3):C692–C701.
30. Fotia, AB, Ekberg, J, Adams, DJ, et al. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J Biol Chem. 2004; 279(28):28930–28935.
31. Jespersen, T, Membrez, M, Nicolas, CS, et al. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4like family. Cardiovasc Res. 2007; 74:64–74.
32. Albesa, M, Grilo, LS, Gavillet, B, et al. Nedd4-2-dependent ubiquitylation and regulation of the cardiac potassium channel hERG1. J Mol Cell Cardiol. 2011; 51(1):90–98.
33. Staub, O, Rotin, D. WW domains. Structure. 1996; 4(5):495–499.
34. Abriel, H, Kamynina, E, Horisberger, JD, et al. Regulation of the cardiac voltage-gated Na+ channel (H1) by the ubiquitin-protein ligase Nedd4. FEBS Lett. 2000; 466(2-3):377–380.
35. van Bemmelen, MX, Rougier, J-S, Gavillet, B, et al. Cardiac voltage-gated sodium channel Nav1. 5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res. 2004; 95(3):284–291.
36. Kang, L, Zheng, M, Morishima, M, et al. Bepridil up-regulates cardiac Na(+) channels as a long-term effect by blunting proteasome signals through inhibition of calmodulin activity. Br J Pharmacol. 2009; 157(3):404–414.
37. Ingham, RJ, Gish, G, Pawson, T. The Nedd4 family of E3 ubiquitin ligases: Functional diversity within a common modular architecture. Oncogene. 2004; 23(11):1972–1984.
38. Morrison, DK. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009; 19(1):16–23.
39. Mrowiec, T, Schwappach, B. 14-3-3 proteins in membrane protein transport. Biol Chem. 2006; 387(9):1227–1236.
40. Allouis, M, Le Bouffant, F, Wilders, R, et al. 14-3-3 is a regulator of the cardiac voltage-gated sodium channel Nav1. 5. Circ Res. 2006; 98(12):1538–1546.
41. Olsen, SK, Garbi, M, Zampieri, N, et al. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003; 278(36):34226–34236.
42. Liu, CJ, Dib-Hajj, SD, Renganathan, M, et al. Modulation of the cardiac sodium channel Nav1. 5 by fibroblast growth factor homologous factor 1B. J Biol Chem. 2003; 278(2):1029–1036.
43. Goetz, R, Dover, K, Laezza, F, et al. Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFS for binding and modulation of voltage-gated sodium channels. J Biol Chem. 2009; 284(26):17883–17896.
44. Wehrens, XHT, Abriel, H, Cabo, C, et al. Arrhythmogenic mechanism of an LQT-3 mutation of the human heart Na(+) channel alpha-subunit: A computational analysis. Circulation. 2000; 102(5):584–590.
45. Lou, JY, Laezza, F, Gerber, BR, et al. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol. 2005; 569(Pt 1):179–193.
46. Wang, C, Hennessey, JA, Kirkton, RD, et al. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res. 2011; 109(7):775–782.
47. Wang, C, Chung, BC, Yan, H, et al. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012; 20(7):1167–1176.
48. Saimi, Y, Kung, C. Calmodulin as an ion channel subunit. Annu Rev Physiol. 2002; 64:289–311.
49. Pitt, GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007; 73(4):641–647.
50. Chin, D, Means, AR. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000; 10(8):322–328.
51. Pitt, GS, Zuhlke, RD, Hudmon, A, et al. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001; 276(33):30794–30802.
52. Herzog, RI, Liu, C, Waxman, SG, et al. Calmodulin binds to the C terminus of sodium channels Nav1. 4 and Nav1. 6 and differentially modulates their functional properties. J Neurosci. 2003; 23(23):8261–8270.
53. Tan, HL, Kupershmidt, S, Zhang, R, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002; 415(6870):442–447.
54. Deschenes, I, Neyroud, N, DiSilvestre, D, et al. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ Res. 2002; 90(4):E49–E57.
55. Kim, J, Ghosh, S, Liu, H, et al. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004; 279(43):45004–45012.
56. Young, KA, Caldwell, JH. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J Physiol. 2005; 565(2):349–370.
57. Motoike, HK, Liu, H, Glaaser, IW, et al. The Na+ channel inactivation gate is a molecular complex: A novel role of the COOH-terminal domain. J Gen Physiol. 2004; 123(2):155–165.
58. Shah, VN, Wingo, TL, Weiss, KL, et al. Calcium-dependent regulation of the voltage-gated sodium channel hH1: Intrinsic and extrinsic sensors use a common molecular switch. PNAS. 2006; 103(10):3592–3597.
59. Wingo, TL, Shah, VN, Anderson, ME, et al. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004; 11(3):219–225.
60. Potet, F, Chagot, B, Anghelescu, M, et al. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009; 284(13):8846–8854.
61. Chagot, B, Potet, F, Balser, JR, et al. Solution NMR structure of the C-terminal EF-hand domain of human cardiac sodium channel NaV1. 5. J Biol Chem. 2008; 284(10):6436–6445.
62. Miloushev, VZ, Levine, JA, Arbing, MA, et al. Solution structure of the NaV1. 2 C-terminal EF-hand domain. J Biol Chem. 2009; 284(10):6446–6454.
63. Casini, S, Verkerk, AO, van Borren, MM, et al. Intracellular calcium modulation of voltage-gated sodium channels in ventricular myocytes. Cardiovasc Res. 2009; 81(1):72–81.
64. Couchonnal, LF, Anderson, ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology. 2008; 23(3):151–159.
65. Maier, LS. Role of CaMKII for signaling and regulation in the heart. Front Biosci. 2009; 14:486–496.
66. Maier, LS, Bers, DM. Calcium, calmodulin, and calcium-calmodulin kinase II: Heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002; 34(8):919–939.
67. Wagner, S, Dybkova, N, Rasenack, EC, et al. Ca/calmodulin-dependent protein kinase II regulates cardiac Na channels. J Clin Invest. 2006; 116(12):3127–3128.
68. Yoon, JY, Ho, WK, Kim, ST, et al. Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J Mol Cell Cardiol. 2009; 47(4):475–484.
69. Ashpole, NM, Herren, AW, Ginsburg, KS, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1. 5 gating by multiple phosphorylation sites. J Biol Chem. 2012; 287(24):19856–19869.
70. Hund, TJ, Koval, OM, Li, J, et al. A bIV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010; 120(10):3508–3519.
71. Kass, RS. The channelopathies: Novel insights into molecular and genetic mechanisms of human disease. J Clin Invest. 2005; 115(8):1986–1989.
72. Maltsev, VA, Reznikov, V, Undrovinas, NA, et al. Modulation of the late sodium current by Ca2+, calmodulin, and CaMKI I in normal and failing dog cardiomyocytes: Similarities and differences. Am J Physiol Heart Circ Physiol. 2008; 294(4):1597–1608.
73. Davis, MJ, Wu, X, Nurkiewicz, TR, et al. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol. 2001; 281(5):H1835–H1862.
74. Ahern, CA, Zhang, JF, Wookalis, MJ, et al. Modulation of the cardiac sodium channel NaV1. 5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005; 96(9):991–998.
75. Hartmann, HA, Tiedeman, AA, Chen, SF, et al. Effects of III-IV linker mutations on human heart Na+ channel inactivation gating. Circ Res. 1994; 75(1):114–122.
76. Jespersen, T, Gavillet, B, van Bemmelen, MX, et al. Cardiac sodium channel Nav1. 5 interacts with and is regulated by the protein tyrosine phosphatase PTPH1. Biochem Biophys Res Commun. 2006; 348:1456–1463.
77. Faulkner, G, Lanfranchi, G, Valle, G. Telethonin and other new proteins of the Z-disc of skeletal muscle. IUBMB Life. 2001; 51(5):275–282.
78. Hayashi, T, Arimura, T, Itoh-Satoh, M, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004; 44(11):2192–2201.
79. Mues, A, van der Ven, PFM, Young, P, et al. Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Lett. 1998; 428(1-2):111–114.
80. Furukawa, T, Ono, Y, Tsuchiya, H, et al. Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol. 2001; 313(4):775–784.
81. Mazzone, A, Strege, PR, Tester, DJ, et al. A mutation in telethonin alters Nav1. 5 function. J Biol Chem. 2008; 283(24):16537–16544.
82. Ou, Y, Gibbons, SJ, Miller, SM, et al. SCN5A is expressed in human jejunal circular smooth muscle cells. Neurogastroenterol Motil. 2002; 14(5):477–486.
83. Steele, DF, Eldstrom, J, Fedida, D. Mechanisms of cardiac potassium channel trafficking. J Physiol (Lond). 2007; 582(Pt 1):17–26.
84. Ziane, R, Huang, H, Moghadaszadeh, B, et al. Cell membrane expression of cardiac sodium channel Na(v)1. 5 is modulated by alpha-actinin-2 interaction. Biochemistry. 2010; 49(1):166–178.
85. Funke, L, Dakoji, S, Bredt, DS. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu Rev Biochem. 2000; 74:219–245.
86. Godreau, D, Vranckx, R, Maguy, A, et al. Expression, regulation and role of the MAGUK protein SAP-97 in human atrial myocardium. Cardiovasc Res. 2002; 56(3):433–442.
87. Leonoudakis, D, Conti, LR, Anderson, S, et al. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2. x) associated proteins. J Biol Chem. 2004; 279(21):22331–22346.
88. Godreau, D, Vranckx, R, Maguy, A, et al. Different isoforms of synapse-associated protein, SAP97, are expressed in the heart and have distinct effects on the voltage-gated K+ channel Kv1. 5. J Biol Chem. 2003; 278(47):47046–47052.
89. El-Haou, S, Balse, E, Neyroud, N, et al. Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res. 2009; 104(6):758–769.
90. Williams, TM, Lisanti, MP. The Caveolin genes: From cell biology to medicine. Ann Med. 2004; 36(8):584–595.
91. Cohen, AW, Hnasko, R, Schubert, W, et al. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004; 84(4):1341–1379.
92. Vatta, M, Ackerman, MJ, Ye, B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006; 114(20):2104–2112.
93. Cronk, LB, Ye, B, Kaku, T, et al. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007; 4(2):161–166.
94. Yarbrough, TL, Lu, T, Lee, HC, et al. Localization of cardiac sodium channels in caveolin-rich membrane domains: Regulation of sodium current amplitude. Circ Res. 2002; 90(4):443–449.
95. Shibata, EF, Brown, TL, Washburn, ZW, et al. Autonomic regulation of voltage-gated cardiac ion channels. J Cardiovasc Electrophysiol. 2006; 17(Suppl 1):S34–S42.
96. Doyle, DD, Goings, G, Upshaw-Earley, J, et al. Dystrophin associates with caveolae of rat cardiac myocytes: Relationship to dystroglycan. Circ Res. 2000; 87(6):480–488.
97. Gee, SH, Madhavan, R, Levinson, SR, et al. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998; 18(1):128–137.
98. Albesa, M, Ogrodnik, J, Rougier, J-S, et al. Regulation of the cardiac sodium channel Nav1. 5 by utrophin in dystrophin-deficient mice. Cardiovasc Res. 2011; 89(2):320–328.
99. Ueda, K, Valdivia, C, Medeiros-Domingo, A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. PNAS. 2008; 105(27):9355–9360.
100. Wu, G, Ai, T, Kim, JJ, et al. Alpha-1-syntrophin mutation and the long QT syndrome: A disease of sodium channel disruption. Circ Arrhythm Electrophysiol. 2008; 1:193–201.
101. Weiss, R, Barmada, MM, Nguyen, T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: A novel gene locus on chromosome 3. Circulation. 2002; 105(6):707–713.
102. London, B, Michalec, M, Mehdi, H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPDl-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007; 116:2260–2268.
103. Van Norstrand, DW, Valdivia, CR, Tester, DJ, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPDl-L) mutations in sudden infant death syndrome. Circulation. 2007; 116:2253–2259.
104. Valdivia, CR, Ueda, K, Ackerman, MJ, et al. GPDlL links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol. 2009; 297(4):H1446–H1452.
105. Marfatia, KA, Harreman, MT, Fanara, P, et al. Identification and characterization of the human MOG1 gene. Gene. 2001; 266(1-2):45–56.
106. Wu, L, Yong, SL, Fan, C, et al. Identification of a new co-factor, MOG1, required for the function of cardiac sodium channel Nav1. 5. J Biol Chem. 2008; 283(11):6968–6978.
107. Kattygnarath, D, Maugenre, S, Neyroud, N, et al. MOG1: A new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet. 2011; 4(3):261–268.
108. Gerull, B, Heuser, A, Wichter, T, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004; 36(11):1162–1164.
109. Pilichou, K, Nava, A, Basso, C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006; 113(9):1171–1179.
110. Cunha, SR, Mohler, PJ. Cardiac ankyrins: Essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc Res. 2006; 71(1):22–29.
111. Mohler, PJ, Schott, JJ, Gramolini, AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003; 421(6923):634–639.
112. Mohler, PJ, Le Scouarnec, S, Denjoy, I, et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants. Human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007; 115(4):432–441.
113. Chauhan, VS, Tuvia, S, Buhusi, M, et al. Abnormal cardiac Na(+) channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ Res. 2000; 86(4):441–447.
114. Lemaillet, G, Walker, B, Lambert, S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003; 278(30):27333–27339.
115. Lowe, JS, Palygin, O, Bhasin, N, et al. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008; 180(1):173–186.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]18
Macromolecular Complexes and Regulation of the Sodium Channel Nav1.5
Nav1.5 and Interacting Proteins
Among the many ionic currents known to be involved in the genesis of the cardiac action potential (AP), the Na+ current (INa) has been studied for more than 50 years1 and remains a major focus of research. The basic structure and function of the cardiac Na+ channel Nav1.5 and its central role in cardiac pathologies are covered in Chapters 1, 9, and 50 of this book. This chapter summarizes recently published data on the proteins interacting with Nav1.5 that form distinct macromolecular and multiprotein complexes. The roles of the four β-subunits are not reviewed because that topic is covered in other chapters of this book. Over the past several years, Nav1.5 has been shown to interact with a growing list of regulatory proteins (Figure 18-1, A and B; Table 18-1). The genes coding for several of these interacting proteins have been found in patients with inherited arrhythmias, such as congenital long QT syndrome (LQTS)2 and Brugada syndrome (BrS).3 The proteins interacting with Nav1.5 have been classified as (1) anchoring/adaptor proteins involved in trafficking, targeting, and anchoring the channel protein to specific membrane compartments; (2) enzymes interacting with and modifying the channel structure via posttranslational modifications such as protein kinases or ubiquitin ligases; and (3) proteins modulating the biophysical properties of Nav1.5 upon binding (see Table 18-1). These classifications are not mutually exclusive.
Table 18-1
The 16 Proteins (or Families of Proteins) Reported to Interact and Regulate Nav1.5
This table does not take into account the four β sodium channel subunits.
Figure 18-1 Schematic representation of the α- and β-subunits of the Nav1.5 and interacting proteins. A, The predicted membrane topology of the α-subunit of Nav1.5 is illustrated. DI-DIV indicates the four homologous domains of the α-subunit; segments S5 and S6 are the pore-lining segments and the S4 helices (yellow) serve as voltage sensors. In the connecting loop between DIII and DIV, the IFM red symbol presents the three residues (isoleucine, phenylalanine, and methionine) known to play a key role in the fast inactivation process. The two intracellular loops display many sites for phosphorylation, whereas the C-terminal domain bears many protein-protein interaction motifs. Only one β-subunit of the four known to be able to interact with sodium channels is shown (purple = transmembrane domain protein on the left side).The eleven proteins, in addition to the β-subunits, are reported to interact with Nav1.5, for which a binding site to one of the intracellular domains is represented schematically. B, For five proteins, the interaction has been shown only by coimmunoprecipitation experiments, but the binding sites are still unknown (question marks). These interactions may be indirect (i.e., requiring adaptor proteins).
Localization of Nav1.5 in Cardiac Cells: Evidence for Distinct Pools
Immunofluorescence staining experiments have shown that Nav1.5 is expressed in distinct membrane compartments (i.e., the intercalated discs and the “lateral membrane” of cardiac cells) (Figure 18-2).4–6 This notion has been under debate because earlier studies described a quasi-exclusive localization of Nav1.5 at the intercalated discs.7,8 Our group proposed a model of Nav1.5 localization at two distinct pools based on strong molecular and in vivo evidence showing that Nav1.5 belongs to the dystrophin multiprotein complex.9 Dystrophin and syntrophin proteins are not expressed at the intercalated discs of human,10 rat,11 or mouse cardiomyocytes.4 Recent studies have presented evidence supporting this multiple-pool model of Nav1.5. Roden’s group investigated a knock-in mouse model harboring the p.D1275N mutation in the gene SCN5A, which codes for Nav1.5 and is found in patients with dilated cardiomyopathy.12 They observed a marked reduction in the expression of Nav1.5 exclusively at the lateral membrane.12 More recently, Lin et al.13 obtained functional evidence for two pools of cardiac Na+ channels with different biophysical properties by performing macropatch experiments at different locations in cardiac cells. It is very possible that this two-pool expression model of Nav1.5 is an oversimplification because there is both functional14 and morphologic evidence6,15 of a T-tubular population of Nav1.5 (Figure 18-3). In a recent paper, Nav1.5 was found to be colocalized with SAP97 at the T-tubules.16 Our recent findings suggest that this third pool does not depend on the syntrophin/dystrophin complex because it is well distinguished in dystrophin-deficient (mdx) myocytes (see Figure 18-3).
Figure 18-2 Stainings of cardiac sections leading to the concept of multiple pools of Nav1.5 channels in cardiac cells. Sections of mouse ventricular myocardium with anti-Nav1.5 staining (left panel), anti-pan-syntrophin staining (middle panel), and overlay (right panel, merge). From these stainings, it is clear that syntrophin proteins are not expressed at the intercalated discs (white arrowhead) where Nav1.5 is present. This defines the intercalated disc pool of Nav1.5 that has been proposed to interact with SAP97 (see Figure 1, A, and Figure 4). The yellow arrowhead shows the lateral membrane pool of Nav1.5 colocalized with syntrophin proteins; white bar = 25 µm.
Figure 18-3 Reduction of Nav1.5 and sodium current in dystrophin-deficient mouse cardiomyocytes. A, Isolated mouse cardiomyocytes from wild type and mdx (dystrophin-deficient) mice with Nav1.5 staining (green) and dystrophin staining (red). Whereas Nav1.5 channels are found at the intercalated discs and at the lateral membrane compartment, it is apparent that dystrophin is excluded from the intercalated discs. The Nav1.5 pool at the discs has been shown to colocalize with SAP97. B, Sodium current recordings from whole-cell patch-clamp experiments of freshly isolated wild type and mdx mouse cardiomyocytes. It can be inferred that the reduction in current is caused by the specific loss of Nav1.5 channels from the lateral membrane pool. (A, From Petitprez S, Zmoos AF, Ogrodnik J, et al: SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108:294–304, 2011; with permission from Wolters Kluwer Health. B, Modified from Gavillet B, Rougier JS, Domenighetti AA, et al: Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res 18:407–414, 2006; with permission from Wolters Kluwer Health.)
The relationship between the expression of Nav1.5 at the intercalated discs and other disc proteins has recently been studied. It was observed that the intercalated disc pool of Nav1.5 is dependent on the expression of key proteins known to be well expressed at the discs, such as connexin43,17,18 plakophilin2,19–21 and desmoglein2.22 Thus, Nav1.5 not only interacts with many different partners, but its localization in different cellular and membrane compartments is also very diverse. One of the obvious conclusions of these observations is that there is not one cardiac Na+ channel Nav1.5, but rather a multiplicity of them with variable functions and regulatory mechanisms.
Proteins Interacting with Nav1.5 Without Demonstrated Roles in Arrhythmias
This section lists the proteins that interact with Nav1.5 that currently have no demonstrable pathologic roles. These interacting proteins were discovered by either performing protein-protein interaction screens, such as yeast two-hybrid assays, or by using proteomic-based protein identification assays. The sites of interaction, often protein-protein interaction domains, were mapped on the sequence of Nav1.5 as described in Figure 18-1, A. This list does not follow any specific logic related to importance but is dictated by the chronologic order of the published studies.
Ubiquitin-Protein Ligases of the Nedd4/Nedd4-like Family
Ubiquitylation of target proteins tags them for proteasome- or lysosome-dependent degradation23 but also serves multiple nondegradative functions, in particular the trafficking of membrane proteins.24 Ubiquitin is a small protein of 76 amino acids found in all animal cells.25 It binds covalently to lysine residues of target proteins. This protein ubiquitylation is performed by E3 ubiquitin-protein ligases.25 Ubiquitylated membrane proteins are subsequently internalized and can be targeted for lysosomal or proteasomal degradation. Alternatively, they can also be deubiquitylated by specific proteases and recycled back to the membrane.26,27 Many different ion channels have been reported recently to be regulated by the Nedd4-like family of E3 ubiquitin-protein ligases. Nedd4-like enzymes bind specifically to target proteins that have consensus domains known as PY motifs with the sequence [L/P]PxY.28 Such PY motifs are found in the C-termini of all voltage-gated Na+ channels (with the exception of Nav1.4, Nav1.9, and Nax29,30), as well as in other cardiac ion channels.27,31,32 Nedd4/Nedd4-like enzymes harbor several WW domains33 that can interact with these PY motifs. When expressed in Xenopus oocytes, Nav1.5-mediated INa is decreased by Nedd4-2–mediated ubiquitylation.34 Our group investigated the molecular determinants of this regulation35 and found that the ubiquitin-protein ligase Nedd4-2 binds directly to the PY motif of Nav1.5 and ubiquitylates the channel in mammalian cells. Because there was no reduction in the total level of Nav1.5 protein upon coexpression with Nedd4-2, we concluded that this was most likely caused by an increased internalization rate rather than by Nav1.5 degradation.29 Inhibition of the proteasome has been linked recently to an increase in INa and Nav1.5 expression in neonatal rat cardiomyocytes.36 Ubiquitylated Nav1.5 was found to be present in mouse cardiac tissue,35 further suggesting that membrane turnover or the stability of Nav channels can be regulated in vivo via ubiquitylation. Nine of such Nedd4-like E3 enzymes are present in the human genome,37 at least eight of which are expressed at the RNA level in human cardiac tissue.31 It remains to be determined which of these Nedd4/Nedd4-like proteins regulates Nav1.5 in a physiologic context and whether the PY motif plays a role in cardiac disease. Altogether these results suggest that the ubiquitin-proteasome system is involved in several aspects of Nav1.5 regulation, the intricacies of which remain to be elucidated.
14-3-3η (eta) Protein
The 14-3-3 protein family is composed of dimeric cytosolic adaptor ubiquitous proteins.38 The members of this family are involved in many cellular functions such as the binding and regulation of trafficking of various membrane proteins.39 Allouis et al. performed yeast two-hybrid and coimmunoprecipitation experiments40 showing that the isoform 14-3-3η interacts with the N-terminal part of the intracellular loop linking domains I to II (see Figure 18-1, A and Table 18-1). Furthermore, it was observed that 14-3-3 and Nav1.5 are colocalized at the intercalated discs of myocytes. No influence on the Nav1.5-mediated peak current was observed when Nav1.5 and 14-3-3η were coexpressed in COS cells, suggesting that this protein does not influence Nav1.5 trafficking. In this expression system, 14-3-3 shifted the inactivation curve toward negative potentials and delayed recovery from inactivation, illustrating that 14-3-3 proteins are able to modify the biophysical properties of ion channels. Because different isoforms of 14-3-3 proteins are expressed in cardiac cells,40 their exact roles in normal cardiac function and their implications in disease states require further investigation.
Fibroblast Growth Factor Homologous Factors
Although fibroblast growth factor homologous factor (FHF) family members are highly homologous in sequence and structure to fibroblast growth factors (FGF), their functional roles are quite different. FHFs are found in the cytosol because their N-termini lack signal sequences that are required for secretion.41 The FGF homologous factors FHF3, FHF1, FHF2, and FHF4 are also called FGF11, FGF12, FGF13, and FGF14, respectively. One of the best characterized functions of the FHFs is their ability to bind to and modulate voltage-gated Na+ channels. Liu et al.42 were the first to demonstrate that FGF12 interacts with the proximal portion of the C-terminus of Nav1.5 (see Figure 18-1, A). This finding was then confirmed by Goetz et al.,43 who also showed that FGF11 and FGF14 interact with the same domain of Nav1.5. In HEK293 cells, coexpression of FGF12 with Nav1.5 shifted the steady-state inactivation relationship toward hyperpolarized values without affecting the other parameters studied.42 Interestingly, several LQTS type 3 and BrS mutations are located in the domain that interacts with FHF1B (see Table 18-1). The SCN5A mutation D1790G44 disrupted the binding of this protein with Nav1.5 and abolished the FHF 12-induced shift of steady-state inactivation. Another member of the FGF family, FGF14, has been shown to downregulate the activity of Nav1.5.45 Because this regulatory protein is expressed in the brain, and not in the heart,45 the significance of this interaction is likely restricted to the central nervous system. A recent study46 demonstrated that FGF13 is expressed in mouse cardiac myocytes. FGF13 was shown to directly bind to and colocalize with Nav1.5 in mouse cardiac cells. Reducing the expression of FGF13 decreased the Na+ current, shifted the availability curve toward more negative potentials, and slowed recovery from inactivation. Furthermore, decreased membrane expression of Nav1.5 was observed in cells with reduced expression of FGF13. These results underscore the specific role of FGF13 in modulating Nav1.5 function and suggest that this gene may be a susceptibility gene for cardiac arrhythmias. A crystal structure of the Nav1.5 C-terminal domain complexed with FGF13 and calmodulin was recently published.47 The functional relationship between FGFs and calmodulin remains to be studied in more detail.
Calmodulin
Intracellular Ca2+ has been shown to modulate the function of many ion channels, including the voltage-gated Na+ channels.48,49 Many cardiac ion channels use calmodulin (CaM), a ubiquitous intracellular Ca2+-binding protein involved in many different cellular processes,50 as a Ca2+-sensing partner. The Nav1.5 C-terminal domain has an IQ motif with a consensus sequence of IQxxxRxxxxR (see Table 18-1), which is very similar to that found in voltage-gated calcium channels.51 The IQ motif is also found in all isoforms of the Nav family.52 Several studies47,53–55 have shown a direct interaction between CaM and the IQ motif of Nav1.5. Recent work by Wang et al. described a crystal structure with an IQ motif ternary complex of Nav1.5, FGF13, and CaM. The functional consequences of this CaM-Nav1.5 interaction are controversial. Several studies52,56,57 have shown inconsistent results that have been difficult to reconcile. A few groups53,55–57 reported that the voltage dependence and stability of the inactivated state were dependent on CaM and the IQ motif. In addition, it has been proposed that CaM may not be the only sensor for the Ca2+-dependent regulation of Nav1.5, because the proximal part of the Nav1.5 C-terminal domain is similar to an EF-hand motif, which is known to bind Ca2+.58,59 Whether this domain binds intracellular calcium is still under debate.47,60–62 The role of intracellular Ca2+ in Nav1.5 regulation was recently investigated by Casini et al.63 They demonstrated that Nav1.5 single-channel conductance is decreased (most likely directly) with an increase in the intracellular Ca2+ concentration. Intracellular Ca2+ appears to be an important regulator of Nav1.5 function, but more studies are needed to clarify its role.
Ca2+/Calmodulin-dependent Protein Kinase II
Ca2+/calmodulin-dependent protein kinases II (CaMKII) are serine/threonine protein kinases expressed in many cell types, where they transduce intracellular Ca2+ increases into the phosphorylation of target proteins, including cardiac ion channels.64 CaMKIIδc is the predominant cardiac isoform upregulated in human and animal heart failure models.65,66 Nav1.5 has been found to colocalize and coimmunoprecipitate with CaMKIIδc.67–69 Ashpole et al.69 have shown that CaMKIIδc interacts with the first intracellular loop of Nav1.5 (see Figure 18-1) and that the residues Ser-516 and Thr-594 may be phosphorylated by this kinase. Hund et al.70 found that Ser-571 was also a target of CaMKII within the same intracellular loop of Nav1.5, and that this regulation was dependent on the correct expression of βIV-spectrin, which interacts with ankyrin-G at the intercalated discs. Overexpression of the CaMKIIδc enzyme in rabbit myocytes and transgenic mice induced a Ca2+-dependent hyperpolarizing shift of the steady-state inactivation curve, slowed the recovery from inactivation, and increased the persistent INa. These biophysical property alterations of INa are similar to some congenital LQTS type 3 mutation-dependent modifications.71 In mice, transgenic overexpression of CaMKIIδc leads to chronic heart failure and episodes of ventricular tachycardia. In dog ventricular myocytes, it has also been shown that CaMKII activity increases the persistent/late INa,72 but it is unclear whether these arrhythmias are the direct consequence of the Nav1.5 biophysical alterations or are related to other heart failure mechanisms. Nevertheless, CaMKII is an important component in the pathogenesis of arrhythmias and potentially a new drug target.64
Proteins Tyrosine Phosphatase PTPH1
Ion channels are also regulated by the phosphorylation of tyrosine residues, a process that depends on both the phosphorylation activity of tyrosine protein kinases and the dephosphorylation activity of phosphatases.73 It was demonstrated that the overexpression of the protein tyrosine kinase Fyn in HEK293 cells altered several of the biophysical properties of Nav1.5 via the phosphorylation of Tyr-1495.74
[/not-level-membership-for-cardiovascular-category]
