[level-membership-for-pediatrics-category]
56 Lymphomas
Hodgkin’s Lymphoma
ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; CMV, cytomegalovirus; EBV, Epstein-Barr virus.
Evaluation and Staging
Staging of pediatric HL is based on the Ann Arbor Classification and is outlined in Figure 56-1.
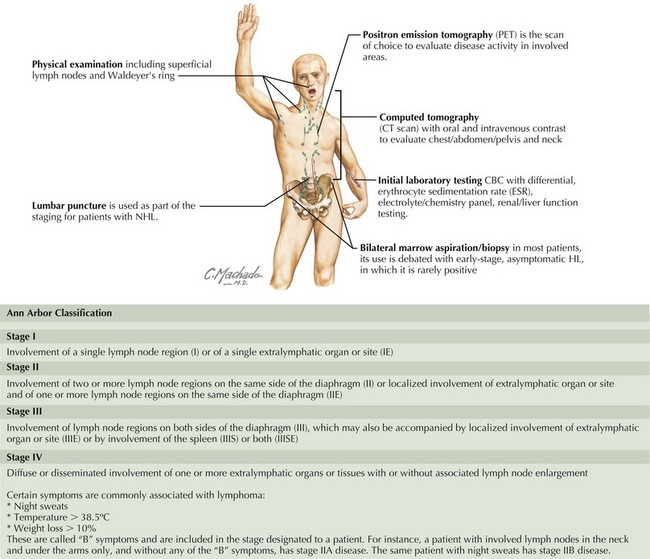
Management
Current treatment for pediatric HL is based on risk-adapted therapy and includes chemotherapy, radiation, or both. Two principles were critical for the development of chemotherapy treatment regimens for HL. First, multiple chemotherapeutic agents with different mechanisms of action are used to circumvent potential drug resistance. In addition, the treatment is structured to avoid combinations of medications with overlapping toxicities. Common agents used to treat HL include bleomycin, vinblastine/vincristine, etoposide, procarbazine, prednisone, methotrexate, doxorubicin, and cyclophosphamide. The combination and doses of medications depend on risk stratification. Table 56-2 depicts the current risk stratification system for The Children’s Oncology Group; however, there may be small differences in this scheme for patients treated in other countries or cooperative groups.
Table 56-2 Risk Stratification of Patients with Hodgkin’s Lymphoma
| Risk Category | Stages |
|---|---|
| Low risk | IA, IIA; no bulk disease* |
| Intermediate risk | IA and IIA with bulk disease* All IB and IIB All IIIA and IVA |
| High risk | All IIIB and IVB |
* Bulk disease is defined as a mediastinal mass greater than 33% of the chest width or any nodal aggregate that is larger than 6 cm.
Non-Hodgkin’s Lymphoma
Evaluation and Staging
In B-cell lymphomas, the primary presentation is not confined to a specific organ, nor is there a specific pattern of spread. Therefore, staging systems for NHL reflect tumor volume more than the degree of spread from the primary site. Several staging systems have been established for NHL. However, the Murphy staging system is the most widely accepted (Table 56-3).
| Stage | Definition |
|---|---|
| I | A single tumor (extranodal) or single anatomic area (nodal), excluding mediastinum or abdomen |
| II | A single tumor (extranodal) with regional node involvement; or a primary GI tract tumor with or without associated mesenteric node involvement, grossly completely resected; or, on the same side of the diaphragm, two or more nodal areas or two single (extranodal) tumors with or without regional node involvement |
| III | All primary intrathoracic tumors (mediastinal, pleural, thymic); or all extensive primary intraabdominal disease that is unresectable; or all primary paraspinal or epidural tumors regardless of other sites; or on both sides of the diaphragm, two single tumors (extranodal) or two or more nodal areas |
| IV | Any of the above with initial CNS or bone marrow involvement (<25%) |
CNS, central nervous system; GI, gastrointestinal.
Gross TG, Termuhlen AM. Pediatric non-Hodgkin’s lymphoma. Curr Oncol Rep. 2007;9(6):459-465.
Olson MR, Donaldson SS. Treatment of pediatric Hodgkin lymphoma. Curr Treat Options Oncol. 2008;9:81-94.
Smith MA, Ries LA, Gurney JG, et al. Leukemia. In: Ries LAG, Smith MA, Gurney JG, et al, editors. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975-1995, NIH Pub. No. 99-4649. Bethesda, MD: National Cancer Institute, SEER Program, 1999.
Thomas RK, Re D, Zander T, Wolf J, Diehl V. Epidemiology and etiology of Hodgkin’s lymphoma. Ann Oncol. 2002;13(suppl 4):147-152.
Weinstein HJ, Hudson MM, Link MP. Pediatric Lymphomas. Heidelberg, Germany: Springer; 2007.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
56 Lymphomas
Hodgkin’s Lymphoma
ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; CMV, cytomegalovirus; EBV, Epstein-Barr virus.
