Lymphomas
Lymphomas are a result of chromosomal alterations resulting in the uncontrolled growth of cells of lymphoid origin. Among all ages, lymphomas constitute just 5% of all cancers diagnosed annually in the USA. In children, however, this percentage increases to 8%.1 Combined, Hodgkin and non-Hodgkin lymphoma are the second most common childhood solid tumors (behind brain tumors and ahead of soft tissue sarcomas and neuroblastoma).
Lymphomas have classically been divided into two distinct groups: Hodgkin disease (HD) and non-Hodgkin lymphoma (NHL). In 2001, HD was designated Hodgkin lymphoma (HL) by the World Health Organization (WHO) lymphoma classification system.2 HL and NHL have a relatively similar prevalence among children and young adults, but NHL becomes significantly more common after 40 years of age (Fig. 69-1). Typically, patients with both HL and NHL are initially seen with enlarged lymph nodes and may have systemic symptoms of fever, fatigue and/or extralymphatic spread. However, these two types of lymphoma also have clear differences. HL usually is seen as an indolent process, whereas NHL is most often seen in children with a rapid onset of symptoms. Due to this propensity for rapid growth, children with NHL often have associated anatomic and metabolic co-morbidities to such a degree that their recognition and need for treatment constitutes a medical emergency. With HL, treatment is based primarily on staging and less on histologic subtype. In contrast, the current treatment of NHL depends on the histologic and immunophenotypic subtypes in addition to stage.
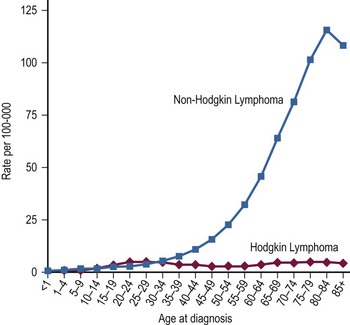
FIGURE 69-1 Age-specific incidence rates per 100,000 population for Hodgkin lymphoma and non-Hodgkin lymphoma from 2000–2009. (Howlader N, Noone AM, Krapcho M, et al, editors. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). Bethesda, MD: National Cancer Institute. http://seer.cancer.gov/csr/1975_2009_pops09/, based on November 2011 SEER data submission, posted to the SEER web site, 2012)
These two lymphomas are truly a study of contrasts. This is no more evident than in the evolution of their therapy. For years, HL has been one of the most curable cancers. Now, with markedly improved treatment protocols, NHL has a nearly equivalent cure rate.3 Children under 15 years of age had 5-year relative survival rates of 96% for HL and 86% for NHL from 2001–2007, up from 81% and 43%, respectively, from 1975–1977.1 Owing to the historic high survival rate with HL, its therapy has focused on a reduction in intensity. In contrast, because of its previously poor prognosis, NHL therapy has focused on intensification of therapy. The use of higher doses of chemotherapy over a short period (as compared with prior methods) has resulted in the dramatic improvement in cure and response of NHL.
Hodgkin Lymphoma
Thomas Hodgkin, in his classic thesis in 1832, described the gross necropsy examinations of seven patients.4 He noted the association of generalized lymphadenopathy and splenomegaly in six patients without evidence of infection or inflammation. Histologic descriptions of the Reed–Sternberg (RS) cell, the pathognomonic multinucleated giant cell, did not occur until after the turn of the century.5,6 Even though the etiology was unclear, therapeutic interventions began soon after the discovery of X-rays. More successful application of radiation therapy awaited the description of the disease’s propensity for contiguous spread. With this knowledge, application of radiation to the involved and adjacent nodal areas (extended-field technique) resulted in improvements in survival in the late 1930s.7 In the early 1960s, due to limitations in the radiologic techniques of that era, the practice of systematic laparotomy, splenectomy, and celiac node and liver biopsy at the time of initial presentation was developed for the purpose of staging and for targeted therapy.8 This has properly been described as the model for the careful staging of cancer as a required prerequisite to the design of therapy, which is a hallmark of oncologic practice today.9,10
During this same time, combination chemotherapy entered into the treatment armamentarium, and remission and cure rates markedly improved. These improvements have made HL one of the most curable cancers today, with a five-year survival of 96% for pediatric patients diagnosed between 2002 and 2008.11 With this high expectation for cure, attention over the past decade has focused on reducing the long-term sequelae of treatment. To this end, chemotherapy has evolved from an adjunctive role to a primary one, with the hope of eliminating the need for irradiation (and its attendant sequelae) altogether. When irradiation is needed, if used in combination with chemotherapy, the focus has been to reduce the size of the fields (from extended to involved) and the doses used. The two classic chemotherapy combinations (MOPP: nitrogen mustard, vincristine [Oncovin], procarbazine, prednisone; and ABVD: doxorubicin [Adriamycin], bleomycin, vinblastine, dacarbazine) have evolved. Hybrids of these combinations are now being utilized to reduce the toxicity to the patient.
Incidence and Epidemiology
It is estimated that 9,060 individuals will be diagnosed with HL in the USA each year, accounting for just 0.6% of all cancers and only 11% of all lymphomas.1 However, in children, it is the sixth most common type of cancer, with approximately 400 children diagnosed annually.1,12 This constitutes 4% of all childhood cases of cancer and approximately half of all childhood cases of lymphoma.1 HL has an incidence of 3.2 cases/100,000 teens aged 15–19 years.13 A bimodal distribution exists when considering all ages, but in children alone, a gradual trend is seen of increasing incidence with increasing age. HL is exceedingly rare in children younger than age 2 years and peaks in the adolescent years.14 Beyond age 11 years, it is the most common of the two types of lymphoma and accounts for about 15% of all cancer in young adults ages 15 to 24 years.13 A slight male predominance (1.3 : 1)11 is noted, but in the youngest children, the male-to-female ratio is much larger (12–19 : 1).15
Monozygotic twins of HL patients have been found to be at greater risk of developing HL than are dizygotic twins, strongly implicating genetics as a principal risk factor.16 In young adults, an increased risk of HL is found with higher socioeconomic status.17 Young adults with HL come from smaller families, have fewer infectious exposures as young children, and/or have later exposure to infections than do control populations.17,18 This correlates closely with socioeconomic status and implicates delayed exposure to infections as a principal risk factor.
Most likely, a combination of genetic risk and infectious exposure predisposes a young adult to HL. Immunodeficiency may be the link between these two risk factors, at least in a subgroup of HL patients. HL is more prevalent in human immunodeficiency virus (HIV)-infected patients.19–21 Also, patients with HL have a higher incidence of cellular immunodeficiency at the time of diagnosis.22 Etiologic theories encompass these two risk factors and focus primarily on the Epstein–Barr virus (EBV). Genomic material from EBV has been found in the RS cells in up to 79% of HL cases.23–25 A higher risk of HL has been noted in individuals with a history of infectious mononucleosis26–28 and with previously high titers to EBV.29 One hypothesis that incorporates these factors suggests the following sequence: (1) a genetic, iatrogenic, or viral immunosuppression; (2) subsequently or coincidentally, an EBV infection or oncogenetic rearrangement in a lymphoid precursor cell; (3) further genetic alterations; followed by (4) clonal expansion of lymphoid cells with morphologic features of RS cells; finally resulting in (5) the clinical syndrome known as HL, diagnosed by the presence of RS cells.30
Classification and Histologic Subtyping
The diagnosis of classical HL requires the dual finding of the diagnostic Hodgkin and RS cells (HRS cells) plus a reactive cellular background.31 The RS cell is a large cell (15–45 mm) with an ‘owl’s eye’ appearance (Fig. 69-2). It has a multilobed nucleus (or is multinucleated), each with a prominent eosinophilic nucleolus surrounded by a clear zone (halo) and an intensely stained nuclear membrane. The ‘owl’s eye’ appearance is the result of a bilobed nucleus. The RS cell often makes up no more than 2% of the involved cells. Hodgkin cells are the mononuclear variant of RS cells. The cellular background is a reactive, pleomorphic mixture of inflammatory cells including reactive lymphocytes, histiocytes, plasma cells, eosinophils, neutrophils, and fibroblasts, with varying degrees of fibrosis and sclerosis. The HRS cell is a clonal, neoplastic cell seen in classical HL and is thought to induce the reactive background through the abundant release of various cytokines.32 HRS cells typically are CD15 and CD30 positive and negative for CD45 and B-lineage antigens.33 In contrast, the nodular lymphocyte predominant (LP) HL cells (popcorn cells) are usually positive for B-lineage antigens, CD15 and CD30 expressions are lacking, and the immunoglobulin genes are expressed.33
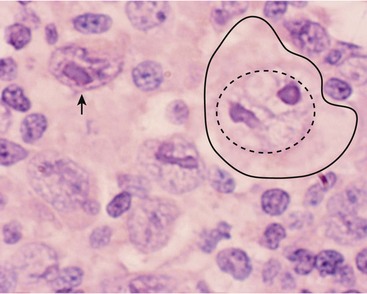
FIGURE 69-2 This photograph depicts a RS cell, which is pathognomonic for HD. On the right side of the slide, the large nucleolus is outlined by the dotted circle and the entire cell is outlined by the solid line. Note the relatively pale nuclear chromatin. The nucleolus has the appearance of an ‘owl’s eye’ from which it receives its name. The arrow points to a mononuclear variant of the RS cell, which has reticulated nuclear chromatin surrounding an almost rectangular macronucleus.
For histologic typing, the Rye classification was commonly used for three decades but has been supplanted by the WHO classification. The 2008 WHO classification lists two main types of HL: classical HL and nodular LP. Classical HL is further divided into four subtypes by morphology. These subtypes include nodular sclerosis (NS, the most common), mixed cellularity (MC), lymphocyte-rich (LRHL), and lymphocyte-depleted (LDHL).34 The NS subtype is seen in 40% of younger patients and 70% of adolescents.35 It is characterized by tumor nodules surrounded by broad sclerotic bands arising from a thickened fibrotic capsule.31 This subtype has a strong predilection for involving the lower cervical, supraclavicular, and mediastinal lymph nodes. The MC subtype is found in 30% of cases and has an increased incidence in younger children.15 HRS cells are typically increased in number. The lymph node architecture is often completely effaced by the HRS cells and their surrounding reactive cells. This subtype often is first seen with advanced, widely disseminated disease in extranodal sites. In addition to its relatively common incidence among all HL patients, it is the most common histologic type seen in HIV-infected patients.21
From 2001 to 2009, the National Cancer Institute (NCI)-sponsored SEER data revealed the following 5-year survival rates for patients age 0–19 years: LRHL 100%; NS 95.7%; MC 92%; and nodular LP 100% (Table 69-1). In patients of all ages, the 5-year survival rate for LDHL was 58%.11 Reports have shown that LPHL has a better prognosis and needs markedly reduced therapy to achieve cure.36,37 This differentiation of therapeutic response between LPHL and the other classic HL histologic types appears to validate the distinction observed in the immunophenotyped RS cells.38 The worse outcome of the MC and LDHL types may reflect their typically higher stage at diagnosis.
TABLE 69-1
Hodgkin Lymphoma: Sites of Involvement at the Time of Initial Diagnosis
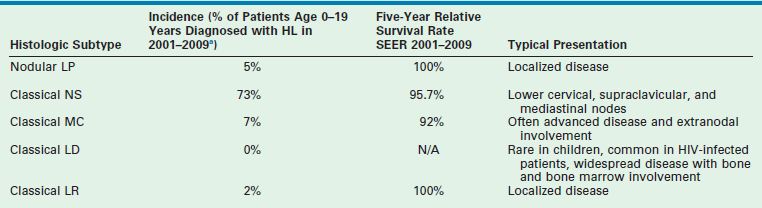
aEleven per cent of patients were classified as NOS (not otherwise specified) and are not included in the histologic subtypes listed above.
Data from Howlader N, Noone AM, Krapcho M, et al, editors. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). Bethesda, MD: National Cancer Institute. http://seer.cancer.gov/csr/1975_2009_pops09/, based on November 2011 SEER data submission, posted to the SEER web site, 2012.
Clinical Presentation
Classically, children with HL present with painless enlarged lymph nodes, typically in the cervical or supraclavicular nodal groups (see Table 69-1). Nodes are often described as rubbery and fixed. They may be either single or matted with other nodes. Occasionally, because of rapid growth, tenderness may develop. Tumor lysis syndrome, a result of rapid and extensive tumor growth and a common complication in children with NHL, is rarely seen in children with HL.
HL tends to spread in a contiguous manner. Therefore, at presentation, one must examine carefully the nodal groups adjacent to the initially identified nodes. More than 90% of patients have involvement of either the cervical or mediastinal nodal groups (or both).39 Interestingly, HL tends to spread from the cervical nodes of one side of the neck to the mediastinum before it spreads to the contralateral cervical nodes. When laparotomy was included in the staging process (which is no longer routinely performed), the spleen was noted to be involved in 27% of patients.39 When evaluating the histologic subtypes and patterns of initial involvement, the MC and LD subtypes of HL have more widespread involvement than do the NS or LP HL subtypes (see Table 69-1).
Mediastinal disease, in addition to a predilection for certain histologic subtypes, is most common in girls older than 12 years, and in those with constitutional symptoms (also known as B symptoms).40 Mediastinal disease may appear with significant respiratory compromise due to compression of the trachea, carina, (or both), including the major bronchi.41 These patients may have dyspnea on exertion or at rest, persistent cough, or stridor. They may have recently been treated for presumed asthma or bronchiolitis, without radiographic imaging. Patients with this presentation may have a history of orthopnea and are most comfortable in an upright forward-leaning position to relieve the pressure on the airway (from the anterior mediastinal mass). The physician must be vigilant for mediastinal disease because it can be silent until a patient is sedated for a radiologic or surgical procedure. These patients may prove impossible to ventilate, even with intubation, because of distal tracheal or bronchial obstruction. It is imperative that all patients with suspected lymphoma (HL or NHL) have a chest radiograph or chest CT scan before any sedation or procedure. Signs of superior vena caval obstruction, including edema and cyanosis of the face and jugular venous distention, may also be present. Extralymphatic involvement can include the liver (the most common extralymphatic organ involved), lungs, bone, bone marrow, and skin, among other sites. Whereas bone marrow involvement is present in only 4–14% of patients overall, among those patients with stage IV disease it occurs one-third of the time.42
Most patients have no systemic symptoms at the time of initial diagnosis. About one-quarter of patients will have one or more B symptoms, defined as weight loss of more than 10% in the previous 6 months, unexplained recurrent fevers greater than 38°C, or drenching night sweats.39 Pruritus, fatigue, and anorexia are other nonspecific symptoms seen in HL patients. Laboratory findings at diagnosis are nonspecific and typically are indicative of an inflammatory process. The erythrocyte sedimentation rate (ESR), serum copper, and ferritin levels are frequently elevated and may be useful later as monitors for relapse. A high ferritin (>142 ng/mL) level or increased ESR (>50) has been associated with a worse prognosis.43,44 The lactate dehydrogenase (LDH) may be elevated as well. Although not common, leukopenia may be indicative of bone marrow involvement.42
Diagnosis
The diagnostic evaluation should include a physical examination and laboratory and radiologic studies (Box 69-1). The physical examination should be directed to the obviously involved nodal groups and also to adjacent groups, keeping in mind the natural history of HL and its propensity for contiguous spread. More than four involved nodal groups in stage II patients is associated with a worse prognosis.45 Bulky disease (nodes or nodal aggregates >10 cm and/or mediastinal tumor width more than one-third of intrathoracic width on a posteroanterior chest radiograph or CT) is associated with a worse outcome in low-stage patients, and necessitates additional therapy to achieve equivalent outcomes.46–48 Auscultation of the airway, palpation of the abdomen, and examination of distant nodal groups are all critical as well.
Staging
Further evaluation of a patient with HL is required to determine the extent of disease at diagnosis and thus the stage of disease (Table 69-2). The common staging system for HL was adopted in 1971.49 This system is based on the observation of contiguous nodal spread in HL. Patients are further divided into asymptomatic (A) and symptomatic (B) subcategories. This subclassification for symptomatic patients is based on the findings of a worse prognosis for B patients and the need for a systemic therapy approach (i.e., chemotherapy in addition to radiation). This likely reflects the finding that patients with B symptoms are more likely to have distant, widespread disease when histologically staged.50
TABLE 69-2
Ann Arbor Staging Classification for Hodgkin Lymphoma
| Stage | Definition |
| I | Involvement of a single lymph node region (I) or of a single extralymphatic organ or site (IE) |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or localized involvement of an extralymphatic organ or site and its regional lymph node(s) with involvement of one or more lymph node regions on the same side of the diaphragm (IIE) |
| III | Involvement of lymph node regions on both sides of the diaphragm (III), which may be accompanied by involvement of the spleen (IIIS) or by localized involvement of an extralymphatic organ or site (IIIE) or both (IIISE) |
| IV | Disseminated (multifocal) involvement of one or more extralymphatic organs or tissues with or without associated lymph node involvement or isolated extralymphatic organ involvement with distant (nonregional) nodal involvement |
For HL, the decision for the type and intensity of therapy rests on the staging results. Traditionally, two methods of staging were used in HL patients: clinical and histologic. In the past, all patients underwent both methods. Clinical staging includes physical, laboratory, and radiologic evaluations. Histologic staging requires a staging laparotomy with splenectomy, nodal sampling, and wedge biopsies of both hepatic lobes. Radiologic evaluations continue to evolve. Lymphangiograms, once a critical component of staging in HL, have been supplanted by more modern and less invasive imaging modalities. CT examination is used most frequently.51 For those who will be treated by irradiation alone, accurate assessment of abdominal disease is critical. Staging laparotomy with splenectomy, nodal sampling, and wedge biopsies of both hepatic lobes has been shown to increase the stage of disease in up to 35% of patients initially evaluated with CT (i.e., the difference between clinical and histologic staging).52,53 This would seem to indicate that abdominal exploration is important. However, with the use of systemic chemotherapy and the de-emphasis on irradiation, this discrepancy between clinical and histologic staging no longer appears to have a significant impact on treatment or outcome.54,55
For the majority of children with HL, staging is based on clinical criteria. Laparotomy (or laparoscopy) is not encouraged or recommended. However, abdominal staging should continue to be used in patients destined to be treated with irradiation alone (although this is now rare in children) because abdominal disease would have a significant impact on planned therapy.56 Staging laparotomy (or laparoscopy) with splenectomy is not without its risks. There are the typical postoperative complications of abdominal surgery. Moreover, with splenectomy, there is a lifelong risk of overwhelming sepsis with encapsulated organisms and these patients require lifelong antibiotic prophylaxis.57 An increased risk of secondary leukemia also exists in those HL patients treated with chemotherapy who have undergone splenectomy (5.9%) compared with those who have not (0.7%) as part of their staging procedure.58–60
Nuclear medicine scans are another modality used for staging HL patients. 18flurodeoxyglocose (FDG) imaging has gradually supplanted gallium scans. Fluorodeoxyglucose-labeled positron emission tomography (FDG-PET) has been found to be more sensitive and specific than either gallium or CT.61–64 Similar to gallium scanning, it leads to a higher staging in a significant percentage of patients. FDG-PET during and after therapy has been highly predictive of patient outcome65,66 and helps to differentiate residual scar tissue from residual lymphoma,67 although false-positive findings with inflammatory conditions have been reported.66 In children, it is important also to recognize the phenomena of thymic rebound after therapy. This may result in both an enlarging mediastinal mass on CT and a positive nuclear medicine scan. An experienced radiologist will recognize this phenomenon by its timing (within the first 6 months after therapy has been completed) and by the normal (although enlarged) homogeneous appearance of the thymic tissue. However, false-negative interpretations can occur. Thus, close follow-up of these patients is critical. Finally, the bone marrow examination continues to be important, regardless of planned methods of therapy, because its involvement would upgrade the patient’s disease to stage IV status and necessitate more intensive chemotherapy.
Treatment
Principles of Radiation Therapy in the Treatment of Hodgkin Lymphoma
Despite the goal of eliminating radiation from the therapeutic regimens for children with early stage HL, it must be recognized that HL is a very radiosensitive neoplasm. A long record of efficacy exists, using radiation either alone or in combination with chemotherapy for this neoplasm. Radiation therapy has traditionally been given to the sites of disease and contiguous, clinically uninvolved, areas. This is known as extended-field irradiation. More recently, involved-field irradiation has become more widely used. This is a more attractive option when combined with chemotherapy. In children, involved-field irradiation has been shown to provide excellent local control (97%).68 A study from Germany found that not only were the remission rate and disease-free survival (DFS) no different between involved-field and extended-field irradiation, but the side effects (leukopenia, thrombocytopenia, nausea, gastrointestinal toxicity, and pharyngeal toxicity) were significantly reduced when using only involved-field irradiation.69
The use of radiation therapy alone remains an option for therapy in adults with low-stage (I to III) HL because it allows them the opportunity to avoid the toxicity associated with chemotherapy.70–73 Even if relapse occurs in those treated with radiation only, the ability to salvage a long-term cure does not appear to be compromised by delaying the use of chemotherapy until the first relapse.
Currently, however, combined-modality therapy remains the standard of care for children and adolescents with HL.74
Principles of Chemotherapy
Chemotherapy is the therapeutic backbone for children with both early and advanced-stage HL. A large number of chemotherapy combinations have been used for HL. Historically, two regimens have been the most widely and effectively used for patients with early stage HL. MOPP or ABVD was administered over a 12-month period and resulted in excellent outcomes.75,76 However, these combinations have significant long-term sequelae when administered in full doses for a year. The recognition that successful treatment with chemotherapy for children with HL would have significant impact on their quality of life and ultimate survival has led to newer combinations of chemotherapy. In general, these regimens have been variations of MOPP and ABVD. These hybrids have either replaced those agents having the worst sequelae (e.g., cyclophosphamide for nitrogen mustard) or have involved the originals being given at significantly lower doses, or both.
Newer regimens in low-stage patients have been examined with lower-dose alkylating agents, which are the causes of the majority of the long-term sequelae seen in these patients.77 In addition, the number of cycles or overall duration has been significantly decreased as well.48,78 Typically, a complete therapeutic protocol currently is given over 3 to 6 months. Radiation therapy sometimes remains a part of these regimens, although it is given at lower doses and encompasses smaller fields. In some studies, the chemotherapy regimens that have been given without irradiation have produced equivalent results to regimens with irradiation in patients with low-stage disease.79–81 For those with high-risk HL, therapeutic regimens that are response-based and intensifying in both dose and timing are showing improved outcomes over the traditional regimens, with DFS now in excess of 90%.82
Stage, Histology, and Response-Based Therapy
Until recently, therapy for HL was primarily dictated by the stage at which the child was first seen. Now histology and response to therapy are added to the equation.36,83 Those with LPHL histology and low-stage disease may be considered for further reductions in chemotherapy. If the disease is completely resected via an excisional biopsy, no further treatment may be needed. In a European study of 58 children with low-stage LPHL treated with surgery alone, outcomes were good with 67% progression free survival (PFS) for those who achieved a complete response (CR) with surgery only, and with an overall survival (OS) rate of 100%.84 A COG clinical trial is attempting to confirm the results of smaller studies showing favorable outcomes with resection alone in stage I patients with LPHL, a single involved lymph node, and a complete resection. Many regimens now incorporate this concept into their design, with fewer cycles of chemotherapy or elimination of irradiation for those with early CRs.
Currently, blood or marrow stem cell transplantation is reserved for those patients whose disease is refractory to systemic chemotherapy or who have experienced relapse. Recent trials have shown that regardless of the duration of initial remission, those who are treated with high-dose chemotherapy and stem cell rescue have less treatment failure than do those treated with conventional chemotherapy.85,86 Autologous stem cell transplant (ASCT) has become the standard of care for relapsed HL.87
Promising new therapies are being investigated, including brentuximab (SGN-35), a monoclonal antibody to CD-30 that has been linked to an antitubulin agent. In a study of 42 relapsed HL patients, 15 patients had objective responses with nine CRs.88 Other novel, targeted therapies are being studied, including rituximab, an anti-CD 20 antibody; Bortezomib, a reversible proteosome inhibitor; and histone deacetylase inhibitors.87
Results
Most patients treated with combinations of chemotherapy and radiation enter into CR (>90%).89,90 Many patients, especially those with the NS subtype, may have persistent adenopathy or mediastinal enlargement for months or years after therapy. Although most prove to be cured, close follow-up is necessary. For those who do not enter remission with today’s front-line chemotherapy/irradiation combinations, the prognosis is poor. Therapeutic intensification with subsequent stem cell transplant will likely be needed.86,91
For low-risk patients, combined-modality (chemotherapy and radiation) therapy typically results in greater than 90% five-year DFS and OS rates. For intermediate risk patients, greater than 80% 5-year EFS rates are found. For high-risk patients, significant advances have been made, with EFS and OS greater that 90% in relatively recent trials.82,92,93
Long-Term Sequelae
The concern over long-term sequelae guides much of modern therapy for HL, both in adults and particularly in children. These sequelae result from both radiation therapy and chemotherapy.94,95 The long-term sequelae of irradiation in growing children are the overriding reason for the efforts to reduce or eliminate it from therapeutic regimens. Bone irradiation may result in shortening of the clavicles in those patients receiving mantle radiation or a shortened height in those receiving radiation to the spine.96 Radiation to the neck often results in permanent hypothyroidism97 and increases the risk of thyroid cancer.98,99 If radiation is to be given to the pelvis of a female patient, consideration should be given to positioning the ovaries away from the field of irradiation.100
Second malignancies are a major concern after therapy for HL.101–103 The most frequent cause of death in long-term survivors of HL is a second malignancy.104 The relative risk of a second malignancy in HL patients has been estimated to be five- to 11-fold that of the general population.102,105 This represents a 15- to 25-year actuarial risk of 7% to 23%.102,105–107 Second malignancies are more prevalent in those with HL treated before age 21 years than in the older age groups for all tumors except lung cancer.102 These second cancers include leukemia and solid tumors. The risk of leukemia seems primarily related to the type of chemotherapy used,105,108 with a cumulative incidence of 3.3%, with a plateau after about ten years. However, one study found a decrease in secondary leukemia among those treated with the newer hybrid regimens.107 This likely is a result of the reduction in nitrogen mustard and procarbazine (in MOPP) doses, the principal culprits in the development of secondary leukemia.109,110 Patients treated with ABVD do not have an increased risk of leukemia. The reduction in the incidence of leukemia may be a result of the decreasing use of splenectomy for staging as this operation has been shown to increase the risk of leukemia in HL patients treated with chemotherapy.109,111
Solid tumors, including those of lung, stomach, melanoma, bone, and soft tissue, have accounted for most of the second malignancies, with a cumulative incidence of 13–22% at 15 to 25 years. No plateau has been appreciated.102,105,106 This risk in HL survivors has not decreased when cohorts treated in the 1960s are compared with those in the 1980s.102 This increased risk of solid tumors is related primarily to irradiation,112,113 with some added risk when subsequent chemotherapy is used in relapse patients.114 It has been recognized that radiation exposure to the breast tissue in adult women has resulted in a fourfold increase in rates of subsequent breast cancer,115–118 whereas the risk of subsequent breast cancer is increased by 39-fold if the breasts are irradiated during adolescence.119 For an adolescent, this increases the probability of developing breast cancer between the ages of 20 and 30 years from 0.04% to 1.6%120 and may be as high as 35% by age 40 years. Other long-term sequelae include cardiac complications secondary to mantle irradiation and/or the use of doxorubicin (Adriamycin in ABVD regimens) that affect up to 13% of patients.121,122
Non-Hodgkin Lymphoma
In contrast to the similarities between adult and pediatric HL, the types of NHL that occur in adults and children, their presentation, their treatment, and their outcome are dramatically different. Most adults with NHL have low- or intermediate-grade lymphomas. In contrast, children with these types of lymphomas are exceedingly rare. Instead, virtually all children with NHL have one of four high-grade, diffuse types: Burkitt lymphoma (BL, formerly small, noncleaved cell lymphoma [SNCCL]), lymphoblastic lymphoma (formerly precursor T-cell lymphoblastic lymphoma [T-LL]), diffuse large B-cell lymphoma (DLBCL), or anaplastic large cell lymphoma (ALCL). Most patients present with advanced or disseminated disease (stages III and IV).
These lymphomas typically appear as a rapidly expanding mass with a short symptomatic history. This propensity for rapid growth makes the diagnostic evaluation in a child with suspected NHL a medical urgency, if not emergency. Of all the childhood tumors, NHL has the greatest chance of presenting with complications. Anatomic impingement of adjacent structures (mediastinal tumors on the trachea and bronchi, nasopharyngeal tumors on the orbits, bowel obstruction with or without intussusception) and metabolic derangements due to tumor lysis (before and after therapy is initiated) are not infrequent results of its very rapid growth. Better management of the initial anatomic and metabolic complications, improved methods of determining the subtypes of NHL (perhaps the most important reason for improved survival), and better chemotherapy combinations (more intensive, yet shorter) have brought the most dramatic improvements in DFS and OS for children with NHL over the past several decades.123 Five-year relative survival (adjusted for normal life expectancy) was 86% for patients diagnosed with NHL from 2001 to 2007.1 Between 1975 and 2006, a 75% decline in mortality was seen in children with NHL.124 In addition to more intense therapy of shorter duration, the other major change in therapy for children with NHL is the virtual elimination of radiation from treatment regimens. This should reduce the long-term sequelae that would have otherwise resulted. For the surgeon seeing the child with suspected lymphoma, rapid evaluation and proper handling of biopsy material will have dramatic beneficial effects on the outcomes.
Incidence and Epidemiology
NHL accounts for 4% of all cancers in adult and pediatric patients. It is estimated that 70,130 men and women will be diagnosed with NHL in the USA in 2012.11 In children (ages 0 to 14 years), NHL patients accounted for 4% of childhood cancers1 and approximately 50% of all lymphomas.1 The annual incidence is 1.3 cases/100,000 children ages 10–14 and 1.8 cases/100,000 children ages 15–19 years.11 Before age 11 years, it is the most common of the two types of lymphoma. A high male-to-female ratio of 3.0 is found,11 making it the most disproportionately occurring tumor between the two genders during childhood. This large difference is found in all ages of childhood. The age distribution demonstrates two small peaks in incidence from 6 to 7 years and between 12 and 14 years (see Fig. 69-1). The 6- and 7-year-olds overwhelmingly have BL, and the teenagers typically have T-LL.
NHL of B-cell origin, either BL or DLBCL, occurs more often in patients with prior EBV exposure, in individuals with a history of immune suppression, and in equatorial Africa.125–127 It is known that in patients with an iatrogenic (e.g., post-transplant, immunosuppressive therapy) or acquired immunodeficiency, congenital-EBV infection has an etiologic role in either the development or the predisposition to B-cell NHL.128,129 Correlations have been made between the viral load and the risk of post-transplant lymphoproliferative disorders (PTLDs).130–132 Patients at greatest risk for PTLDs are those in whom their primary infection with EBV occurs within the first 3 to 4 months after transplantation. For T-LL or ALCL, no such etiologic correlations have been found.
Classification
Over the years, several classification schemes have been used.133 The Revised European American Lymphoma Classification is the basis for the WHO classification that was updated in 2008.34 Childhood NHL primarily consists of four major subtypes in the WHO system: (1) BL/leukemia and Burkitt-like (mature B-cell neoplasms accounting for 39% of NHL patients3); (2) lymphoblastic lymphoma (28%3); (3) DLBCL; and (4) ALCL. DLBCL and ALCL were previously lumped together as large cell lymphomas and comprised about one-third of the NHL of childhood. With the separation of these two entities, the cases previously identified as large cell lymphoma are now divided almost equally between DLBCL and ALCL.134 The first two classifications are part of the ‘small, round, blue cell tumors,’ which presents the pathologist with the challenge of proper identification. To differentiate these from the other three classic small round blue cell tumors (neuroblastoma, rhabdomyosarcoma, and Ewing sarcoma) requires the presence of the immunocytochemical marker leukocyte common antigen CD45 (LCA), which is absent in the other tumor cell types.
BL has classically been divided into Burkitt and non-Burkitt (Burkitt-like in the REAL classification) subtypes. These are of a mature B-cell origin, with flow cytometric immunophenotyping revealing the presence of surface immunoglobulin IgM, CD10, CD19, CD20, CD22, CD79a, and human leukocyte antigen (HLA)-DR antigens. The histologic appearance of these two types differs in the degree of pleomorphism, with Burkitt being more uniform appearing than non-Burkitt. Although a distinction has been made for years between Burkitt and non-Burkitt subtypes of diffuse SNCCL lymphomas, no clinical differences are found between these two subtypes.135 Histologically, the cells of BL are medium sized with round nuclei containing two to five nucleoli, abundant basophilic cytoplasm, and cytoplasmic lipid vacuoles. Owing to its extreme rates of proliferation and spontaneous cell death, a number of macrophages are seen within this monomorphic field, consuming the dying cells and giving rise to the classic ‘starry sky’ appearance of BL.136
Lymphoblastic lymphomas (LL) are distinguished by round or convoluted nuclei, finely dispersed chromatin, inconspicuous nucleoli, and scant cytoplasm. In the vast majority of these tumors, flow cytometry reveals the presence of the T-cell markers CD3 and CD7, with variable positivity for CD2 and CD5. These cells are typically Tdt positive, whereas BLs are Tdt negative. This subtype is classified as a precursor T-cell LL in the WHO classification. A small number of LL cases are B-cell precursor and express pre–B-cell antigen profiles.134
Large cell lymphomas are a heterogeneous group of neoplasms. Histologically, approximately half are immunoblastic, 40% are large noncleaved cell, and fewer than 5% are large cleaved cell.137 Flow cytometry shows relatively equal frequencies of B- or T-cell origin (36% and 33%, respectively) with 30% indeterminate.138,139 A unique subset, identified by the immunophenotype CD30+ (the antigen identified by the Ki-1 monoclonal antibody),140 is recognized morphologically by its anaplastic characteristics, including very large cells with abundant cytoplasm, atypical lobulated nuclei, and prominent nucleoli. These cells exhibit a cohesive pattern with lymph node sinusoidal invasion. In the WHO classification, this is referred to as ALCL. In the past, this subtype has also been referred to as malignant histiocytosis. The majority (60%) of these children have a T-cell immunophenotype.138,141 Although it was originally thought to be uncommon in children, this subtype accounts for 40–50% of the large cell lymphoma cases.138,139,142,143
Clinical Presentation
By Initial Site of Disease
Overall, unlike those with HL and adult NHL, children with NHL are often initially seen with extranodal disease and typically have disease that spreads by routes other than contiguous nodal pathways. In children, the abdomen is the originating site of disease in 31%, the mediastinum in 27%, and the head and neck in 29%.3 Other sites include peripheral nodes, bone, and skin. Most abdominal disease primary lesions are due to BL, whereas most mediastinal/intrathoracic primary lesions are due to LL. Disease that occurs primarily in the peripheral nodes and bones is often due to large cell lymphomas, and skin involvement is primarily associated with the Ki-1+ large cell lymphoma subtype (ALCL).139,144,145 Correlating with this distribution and the known age peaks of the two types of small cell lymphomas, abdominal primary lesions occur more often in children younger than 10 years, whereas mediastinal primary lesions are more likely to occur in adolescents. Children with abdominal primary lesions may present with nausea, vomiting, abdominal pain, and changes in bowel habits. On physical examination, they may be found to have an abdominal mass in any of the quadrants. Also, they may present with an acute abdomen due to either intussusception (typically due to infiltration of Peyer’s patches) (Fig. 69-3) or small bowel obstruction, perforation of an involved bowel wall, or an ileocecal mass mimicking acute appendicitis.146

FIGURE 69-3 This 12-year-old child presented with abdominal pain, nausea, and vomiting. An abdominal mass was palpable in the right abdomen. (A) The CT scan shows a large ileocolic intussuspection in the right mid-abdomen. The mesenteric vessels within the intussuceptum are marked with the arrow. The lead point for this intussusception was a non-Hodgkin lymphoma. (B) The resected specimen shows a lymphoma in the small bowel which led to the intussusception.
Children with mediastinal primary lesions may have minimal symptoms, such as a mild cough or audible wheeze, or can have impending airway obstruction. These latter patients can also have significant engorgement of the vasculature in the head, face, and upper thorax because of superior vena cava compression. Thrombosis may be present in these vessels as well. Often these patients will assume a forward-leaning position and cannot tolerate being placed in the supine position because of the anterior mediastinal mass. The patient’s history may reveal orthopnea as well as shortness of breath and dyspnea on exertion. The recent onset of asthma symptoms is not uncommon. Shortness of breath also may be due to pleural effusions. A chest radiograph or chest CT scan is an essential component of the patient’s initial evaluation before sedation or any procedure (Fig. 69-4). Chest radiography and chest CT will reveal the widened mediastinum with often dramatic narrowing of the trachea and bronchi. Pericardial effusions are often present and may be seen on CT, magnetic resonance imaging (MRI), or echocardiography.147
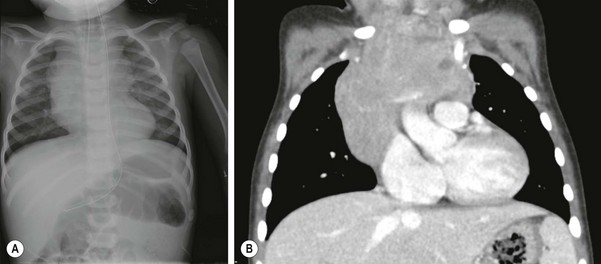
FIGURE 69-4 This 17-month-old boy had a 1-month history of stridor and several episodes of perioral cyanosis when he was fussy. The patient was seen in an emergency room where the (A) chest radiograph showed a large anterior mediastinal mass. (B) A contrasted CT scan showed the large heterogeneous, noncalcified mediastinal mass which extends across the anterior mediastinum. The mass encases and abuts, but does not occlude, the left subclavian and innominate veins. The mass measures 4.6 cm × 6.8 cm × 6.5 cm. On another view, the distal trachea was seen to narrow to 2 mm in AP diameter just proximal to the carina. Bone marrow examination and lumbar puncture were normal. CT-guided biopsy of the mediastinal mass yielded NHL. This infant responded nicely to chemotherapy and has completely recovered.
Patients with head and neck lymphomas may have a history of rapidly progressive adenopathy, recent onset of snoring at night, mouth breathing, halitosis, epistaxis, proptosis or periorbital edema, diplopia, extraocular muscle paralysis due to entrapment, cranial nerve paralysis, sudden blindness, or a combination of these symptoms. Physical examination of the nares, oral cavity, and extraocular movements is important and may reveal signs not appreciated as abnormal by the child. The presence of asymmetric and painless tonsillar hypertrophy should also alert the clinician to the possibility of NHL.148
Evaluation with CT often reveals a homogeneous mass that may show destruction of the adjacent bony structures. Bone NHL primary disease is usually seen as lytic lesions found on radiographs obtained for various reasons, including localized tenderness.149–151 Skin lesions are typically ulcerative and fail to heal, but also may be completely subcutaneous.144 Patients with central nervous system (CNS) involvement may be asymptomatic, have seizures, or have signs and symptoms related to tumor infiltration in the brain.
Laboratory findings at the time of diagnosis are dependent on the amount of tumor present (regardless of the histologic subtype). Generally, patients will have an elevated ESR or C-reactive protein (CRP) level. Those with large tumor burdens typically have high LDH levels as an indicator of tumor lysis risk,152,153 disease regression, and disease progression. The degree of LDH elevation has been used as an adverse prognostic factor.3,154,155 For those with a high tumor burden at presentation, laboratory signs of tumor lysis will also include elevated uric acid, phosphorus, and potassium levels along with a low calcium level. Some patients may already be in renal failure at the time of presentation or have an elevated creatinine.152,153 Hematologic values are nonspecific, and the presence of cytopenias should raise the suspicion of marrow involvement. Cerebrospinal fluid (CSF) pleocytosis may or may not be present in those with CNS involvement.
More than 60% of patients have advanced or disseminated (stages III and IV) disease at diagnosis.3,156 Bone marrow metastasis is defined as greater than 5% but less than 25% involvement. Patients with more than 25% disease in the bone marrow are classified as having leukemia. Fourteen per cent of patients initially have some bone marrow involvement, and 3% have CNS involvement.3
By Histologic Subtype
BL was first described by the surgeon Denis Burkitt in Uganda, where he identified the common finding of enormous involvement of the nodes around the jaw.157 Later, it was determined that, although this was a common presentation of those patients with endemic Burkitt (African) lymphoma, those with sporadic Burkitt (American) lymphoma more typically had presentation of disease either in the abdomen or the nasopharynx.158 Patients with endemic BL have accompanying abdominal disease in roughly half the cases, and patients with sporadic BL have jaw involvement 15–20% of the time.159 Patients with sporadic BL have a higher incidence of bone marrow involvement (21% vs 7%) but lower CNS dissemination (11% vs 17%). Approximately two thirds of BL patients will have disseminated or advanced disease (defined as stages III and IV) at diagnosis.160
T-LL patients most often are adolescents with supradiaphragmatic disease, affecting either the intrathoracic region or the head and neck. Disseminated disease is present in nearly 90% of T-LL patients at diagnosis.160 In T-LL patients, involvement of the bone marrow has been found in approximately one fourth of children, and CNS disease is present in fewer than 10%.160
ALCL patients may present with disease in all sites but there is a higher prevalence than the other two subtypes for skin, bone, and peripheral nodes.144,151,161 ALCL is found in two distinct clinical forms: primary cutaneous ALCL and primary systemic ALCL (as fevers and weight loss, and in advanced stage).134 Disseminated disease in ALCL patients is present at diagnosis in up to 65% of patients.160 Involvement of the bone marrow or CNS in ALCL patients is rare. DLBCL may present as a mediastinal primary lesion or as nodal or extranodal disease, most commonly in the abdomen or head and neck.134
In Immunodeficient Patients
For patients with congenital or acquired immunodeficiency, NHL presentation will vary from polyclonal plasmacytic hyperplasia, most often localized in nasopharyngeal nodes or tonsils, to a clonal polymorphic lymphoma slowly arising in the lymph nodes or extranodal sites, to widely disseminated, rapidly progressive immunoblastic lymphoma.162,163 Symptoms may be nonspecific, with fever and malaise. Hepatosplenomegaly and lymphadenopathy may be presenting signs. Gastrointestinal symptoms of longer than 14 days duration with anorexia, weight loss, and diarrhea should raise suspicion for this condition.132 NHL has become more common with the use of very potent antirejection drugs after solid organ or bone marrow transplantation. Involvement of the transplanted organ is not unusual.132,164
Diagnosis
Children initially suspected of having NHL should be evaluated immediately because of the high risk of either metabolic or anatomic complications before therapy begins. The rapid growth of these tumors may create a life-threatening complication overnight in a child who seemed relatively healthy the previous day (Box 69-2).
Similar to HL, for patients critically ill at diagnosis, such as those with severe airway obstruction, diagnosis by alternative methods may be required. NHL is a systemic disease that requires chemotherapy so debulking operations or attempts at local control are not necessary. The one exception in which initial total resection may be considered are those patients with an abdominal mass in whom bowel resection is already required because of perforation or obstruction. In this case, total resection of the tumor should be considered. In this setting, resection reduces the stage of the patient’s disease, improves survival, and reduces the amount of therapy required.165 For all other patients, resection of the mass provides no improvement in staging or long-term cure, and delays the time to initiation of chemotherapy. It should be remembered that most patients have disseminated disease at presentation. Also, it is important to note that with chemotherapy alone, more than 90% will achieve a complete remission.
Once NHL is suspected, a concerted and well-conceived plan of evaluation is important to achieve a diagnosis as quickly as possible. This should include laboratory examination to evaluate tumor burden and presence or risk of tumor lysis syndrome. The radiographic evaluation in these patients is extremely important.51 No procedures or sedation should be attempted until a mediastinal mass has been excluded. To identify the extent of disease, CTs of the neck, chest, abdomen, and pelvis are required. An examination of the head, either CT or MRI, should be obtained in those patients with CNS symptoms, with CSF pleocytosis, or in whom the primary lesions are parameningeal based. Bone scans should typically be obtained. 18FDG-PET or gallium scans are currently recommended. 18FDG-PET scans have gradually replaced gallium scans and are now considered an essential tool in the initial evaluation of patients with NHL. Advantages of PET over gallium include same-day imaging, improved resolution, and a higher target-to-background ratio.166 Diagnostic PET scans are reliable (greater than 90% positive) in patients with DLBCL. Studies of patients with HL and NHL have found PET to be superior to gallium for diagnosing disease sites.66 Also, PET has been shown to be more sensitive than CT or gallium scans in staging NHL.167 PET may be fused with CT for improved imaging. PET is also an important component in evaluating response to treatment.
Histologic evaluation of the biopsy material should include general histochemical techniques to confirm the lymphoma and its subtype. Additional tests include flow cytometric analysis of cell-surface markers to determine the immunophenotype of the lymphoma, cytogenetic evaluation for diagnostic translocations, fluorescent in situ hybridization (FISH), and DNA analysis using either Southern blotting or the polymerase chain reaction (PCR) for detection of the pathognomonic oncogenes (gene rearrangements), even in the absence of identifiable cytogenetic translocations.168 Examination of markers in tumor cells for EBV is important in the evaluation of PTLDs. Therapy differences between the subtypes of lymphoma are such that assignment to the wrong subtype due to a lack of adequate diagnostic material will adversely affect the chance of cure. These evaluations may be performed with biopsy material from any involved site, including the primary mass, enlarged lymph nodes, effusions, and bone marrow. Gene expression profiling, which uses DNA microarrays, has been shown to categorize patients further into specific histologic and genetic subsets of lymphoma, with much greater predictability of the clinical outcome.169 This technique has revolutionized diagnostic and prognostic characterization for NHL.
Staging
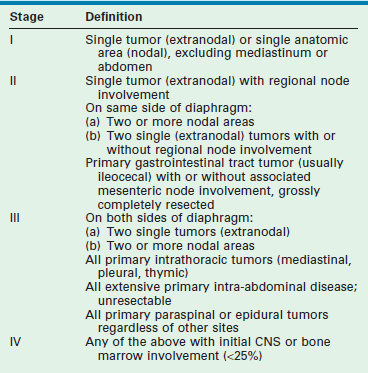
Staging is important in the determination of therapeutic planning. The most widely used staging schema today is the St. Jude’s or Murphy system (Table 69-3).170 This is an adaptation of the Ann Arbor scheme and is applicable to all types of childhood NHL. It divides patients into localized (stage I or II) and disseminated or advanced (stage III or IV) disease. Involvement of the CNS or bone marrow immediately places the patient in the stage IV category. Patients with more than 25% bone marrow involvement are, by definition, diagnosed with leukemia rather than with lymphoma. These include B-cell or Burkitt leukemia (L3 leukemia morphologic classification) and T-cell leukemia. The former patients are treated on B-cell NHL protocols with much better results than previously obtained on acute lymphoblastic leukemia (ALL) regimens. Many of the B-cell NHL protocol results reported in the literature include these patients in their stage IV populations. The T-cell leukemia patients remain on ALL protocols, but many similarities exist between these protocols and those used in T-LL therapy.
Prognostic Risk Factors
When all patients are treated similarly, the stage of the lymphoma at diagnosis is a strong predictor of outcome.3 Prediction of a patient’s eventual outcome stratifies patients at high risk for relapse to more intensive or novel therapies and patients at low risk to shorter, more moderate therapies. Many prognostic factors have been evaluated over the years. All prognostic factors are dependent on the therapy subsequently given.3 It has been definitively shown that histology-based therapy is of critical importance in the successful outcome of these patients.156
CNS involvement in both SNCCL and LL patients has predictably worse outcomes.156 In patients with BL, the adverse effect of CNS disease on outcome has, in some studies, been more attributable to tumor burden at diagnosis than to the presence of CNS disease alone (i.e., those with greater tumor burden are more likely to have CNS disease).171 Patients with BL older than 15 years of age have a worse prognosis than patients younger than 15 years old. An LDH greater than 500 IU/L also predicts a worse outcome for patients with BL.172
Treatment
Therapy for childhood NHL has evolved over the past several decades, and is based on the knowledge that this tumor is extremely chemosensitive. For BL and large cell lymphoma, the duration of therapy has become shorter as it became apparent that most, if not all, patients were experiencing relapse within the first 6 to 8 months of therapy.160,173 Despite reducing therapy to 6 months or less, no increase in relapse has been seen. Relapses for the most part have occurred within the first 6 to 8 months after diagnosis and virtually all have occurred within the first 2 years.154–156,173,174 Therapy for BL has shown a clear improvement as methotrexate and cytarabine doses have been increased. These two agents, in addition to cyclophosphamide, vincristine, doxorubicin (Adriamycin), and prednisone (and etoposide for the stage IV patients), now play a critical role in the successful outcome of these children.160,165,174,175 The addition of radiation has not shown improvements in DFS or OS.142,144,176 The addition of rituximab (an anti-CD20 monoclonal antibody) is currently being studied.
NHL in immunodeficient individuals is most often a B-cell lymphoma, either small or large cell. Therapy for these patients has typically been directed toward these histologic types. For patients with ongoing iatrogenic immune suppression, a reduction in the immunosuppressive agent, with or without acyclovir, may be adequate to induce a remission in up to 75% of cases.132,177 In the past few years, for resistant disease, new methods using monoclonal antibody therapy, primarily rituximab, have been used with promising results alone, but these antibodies are most efficacious when combined with chemotherapy.178
Results
When reviewing the outcomes of children treated for NHL, it is quickly apparent that considerable improvement in DFS and OS has occurred over the past several decades.3 Today, typically 90–100% of patients will achieve complete remission.142,173,179 Five-year relative survival rates for children ages birth to 19 years, and diagnosed between 1996 and 2004, were 83.3%.13
Patients with localized disease have an overall excellent prognosis, regardless of histologic subtype, with DFS typically exceeding 90–95%. BL patients with advanced disease have experienced DFS exceeding 80% in the recent trials. Patients with LL with disseminated disease are not faring as well, but DFS for these patients is exceeding 65–70% in most trials. Overall, when they occur, treatment failures typically happen within the first two years after diagnosis. Patients with BL who experience relapse primarily do so within the first 6 to 8 months. LL patients will have an occasional late failure after two years, although even in this group of patients the vast majority of failures will occur early.179 The prognosis for patients with DLBCL is excellent with three-year EFS of ≥90% in a recent trial.180 Patients with advanced-stage ALCL have 2-year EFS of approximately 70%.181
Early Complications
Depending on the tumor burden at diagnosis, patients may initially have a constellation of significant metabolic derangements known as tumor lysis syndrome.152,153 These problems include hyperuricemia, hyperphosphatemia, hyperkalemia, and hypocalcemia. Recognition of this syndrome is critical to prevent life-threatening complications, including acute renal failure. Without treatment, the incidence of acute renal failure may be as high as 30%.182 Tumor lysis syndrome is the result of the rapid turnover of cells within the tumor. The fraction of tumor cells in S phase at any given time can approach 27% in some patients.183 These tumors have a high degree of spontaneous lysis at the time of diagnosis because they rapidly outgrow their blood supply. Any manipulation, including transfusion or operation, may induce a sudden worsening of this syndrome.
Therapy is primarily based on preventing hyperuricemia. For those at high risk, determined by the presence of an elevated LDH, creatinine, or uric acid value, intervention is important. For most patients with little or no elevation in these values, adequate hydration (>3000 mL/m2/day) and pH monitoring (maintain between 7.0 and 7.5) is adequate, along with the initiation of allopurinol to reduce the production of uric acid through inhibition of xanthine oxidase.184 Rasburicase cleaves uric acid into allantoin, a soluble by-product. This agent, administered daily for one to five days, dramatically reduces measurable uric acid levels to immeasurable levels, thus allowing the clinician to focus on prevention or treatment of hyperphosphatemia, which requires maintaining acidic urine.185,186 Despite these measures, it may be necessary to place patients on dialysis either to treat oliguria/anuria or to prevent it in the presence of rapidly increasing uric acid, phosphorus (typically >10 mg/dL), or potassium (>7.5 mEq/L) levels.160,187 In an effort to avoid this complication, an initial low-dose therapy (usually 1 week) to more slowly reduce the tumor burden is used in some treatment regimens.
As a result of the much more myelosuppressive protocols required in NHL therapy, infection is a much greater risk for NHL patients as compared with HL patients.156 In one recent study, 63% of the deaths were due to infection. Most patients require transfusion support during treatment because of the myelosuppression. The chemotherapy itself may cause acute complications, including severe chemical burns due to extravasation of certain vesicant agents (vincristine, anthracyclines). As most children require the placement of right atrial catheters to facilitate their therapy, thrombosis of this area and the surrounding vasculature has become more frequent.188 Mucositis is seen in a significant number of patients during therapy for Burkitt lymphoma as well.
Long-Term Sequelae
As long-term survival has improved, the concern over lifelong sequelae has increased in these patients. With current therapy, these complications include cardiac toxicity,122 infertility as a result of the alkylating agents used,189 and secondary leukemias due to epipodophyllotoxins (etoposide, tenoposide) and the alkylating agents used in the NHL regimens.190 The risk for developing cardiac toxicity is related to several factors, including the irradiation dose, cumulative anthracycline dose, and age at exposure. Patients are at an increased risk for anthracycline-related cardiomyopathy if they are female, have received doses greater than 200–300 mg/m2, and were younger when given anthracyclines.191
References
1. Siegel, R, Naishadham, D, Jemal, A. Cancer Statistics, 2012. CA Cancer J Clin. 2012; 62:10–29.
2. InJaffe ES, Harris NL, Stein H, et al, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2001.
3. Murphy, SB, Fairclough, DL, Hutchison, RE, et al. Non-Hodgkin’s lymphomas of childhood: An analysis of the histology, staging, and response to treatment of 338 cases at a single institution. J Clin Oncol. 1989; 7:186–193.
4. Hodgkin, T. On some morbid appearances of the absorbent glands and spleen. Med Chirurg Trans. 1832; 17:68–114.
5. Sternberg, C. Uber eine eigenartige unter dem Bilde der Pseudoleukemaemie verlaufende Tuberculose des lymphatischen Apparetes. Z Heilkd. 1898; 19:21–92.
6. Reed, D. On the pathological changes in Hodgkin’s disease: With especial reference to its relation to tuberculosis. Johns Hopkins Hosp Rep. 1902; 10:133–196.
7. Gilbert, R. Radiotherapy in Hodgkin’s disease. AJR Am J Roentgenol. 1939; 41:198–240.
8. Kaplan, HS, Rosenberg, SA. The management of Hodgkin’s disease. Cancer. 1975; 36:796–803.
9. Hellman, S. Thomas Hodgkin and Hodgkin’s disease: Two paradigms appropriate to medicine today. JAMA. 1991; 265:1007–1010.
10. Zantinga, AR, Coppes, MJ. Thomas Hodgkin (1798–1866): Pathologist, social scientist, and philanthropist. Med Pediatr Oncol. 1996; 27:122–127.
11. Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). National Cancer Institute: Bethesda, MD, 201. http://seer.cancer.gov/csr/1975_2009_pops09/
12. American Cancer Society. Cancer Facts and Figures 2007. Atlanta: American Cancer Society; 2007.
13. Ries LAG, Melbert D, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975–2005. National Cancer Institute: Bethesda, MD, 200. http//seer.cancer.gov/csr/1975_2005/
14. Gurney, JG, Severson, RK, Davis, S, et al. Incidence of cancer in children in the United States. Cancer. 1995; 75:2186–2195.
15. Kung, FH. Hodgkin’s disease in children 4 years of age or younger. Cancer. 1991; 67:1428–1430.
16. Mack, TM, Cozen, W, Shibata, DK, et al. Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med. 1995; 332:413–418.
17. Grufferman, S, Delzell, E. Epidemiology of Hodgkin’s disease. Epidemiol Rev. 1984; 6:76–106.
18. Gutensohn, N, Cole, P. Childhood social environment and Hodgkin’s disease. N Engl J Med. 1981; 304:135–140.
19. Reynolds, P, Sunders, LD, Layefsky, ME, et al. The spectrum of acquired immunodeficiency syndrome (AIDS)—associated malignancies in San Francisco, 1980–1987. Am J Epidemiol. 1993; 137:19–30.
20. Hessol, NA, Katz, MH, Liu, JY, et al. Increased incidence of Hodgkin disease in homosexual men with HIV infection. Ann Intern Med. 1992; 117:309–311.
21. Volberding, P, Baker, K, Levine, A. Human immunodeficiency virus hematology. Hematology Am Soc Hematol Educ Program. 2003; 294–313.
22. Riggs, S, Hagemeister, FB. Immunodeficiency states: A predisposition to lymphoma. In: Fuller LM, Hagemeister FB, Sullivan M, eds. Hodgkin’s Disease and Non-Hodgkin’s Lymphomas in Adults and Children. New York: Raven; 1988:451.
23. Ambinder, RF, Browning, PJ, Lorenzana, I, et al. Epstein-Barr virus and childhood Hodgkin’s disease in Honduras and the United States. Blood. 1993; 81:462–467.
24. Herbst, H, Steinbrecher, E, Niedobitek, G, et al. Distribution and phenotype of Epstein-Barr virus–harboring cells in Hodgkin’s disease. Blood. 1992; 80:484–491.
25. Knecht, H, Odermatt, B, Bachmann, E, et al. Frequent detection of Epstein-Barr virus DNA by the polymerase chain reaction in lymph node biopsies from patients with Hodgkin’s disease without genomic evidence of B- or T-cell clonality. Blood. 1991; 78:760–767.
26. Rosdahl, N, Larsen, SO, Clemmesen, J. Hodgkin’s disease in patients with previous infectious mononucleosis: 30 years’ experience. BMJ. 1974; 2:253–256.
27. Hjalgrim, H, Askling, J, Sorenson, P, et al. Risk of Hodgkin’s disease and other cancers after infectious mononucleosis. J Natl Cancer Inst. 2000; 92:1522–1528.
28. Hjalgrim, H, Askling, J, Rostgaard, K, et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N Engl J Med. 2003; 349:1324–1332.
29. Evans, AS, Gutensohn, NM. A population-based case-control study of EBV and other viral antibodies among persons with Hodgkin’s disease and their siblings. Int J Cancer. 1984; 34:149–157.
30. Haluska, FG, Brufsky, AM, Canellos, GP. The cellular biology of the Reed-Sternberg cell. Blood. 1994; 84:1005–1019.
31. Lukes, RJ, Butler, JJ. The pathology and nomenclature of Hodgkin’s disease. Cancer Res. 1966; 26:1063–1083.
32. Gruss, HJ, Pinto, A, Duyster, J, et al. Hodgkin’s disease: A tumor with disturbed immunological pathways. Immunol Today. 1997; 18:156–163.
33. Hudson, MM, Onciu, M, Donaldson, SS. Hodgkin lymphoma. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:695–721.
34. Swerdlow, SH, Campo, E, Harris, NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. Lyon, France: IARC Press; 2008.
35. Donaldson, SS, Link, MP. Childhood lymphomas: Hodgkin’s disease and non-Hodgkin’s lymphoma. In: Moosa AR, Robson MC, Schimpff SC, eds. Comprehensive Textbook of Oncology. Baltimore: Williams & Wilkins; 1986:1161.
36. Murphy, S, Morgan, E, Katzenstein, H, et al. Results of little or no treatment for lymphocyte-predominant Hodgkin’s disease in children and adolescents. Am J Pediatr Hematol Oncol. 2003; 25:684–687.
37. Pellegrino, B, Terrier-Lacombe, M, Oberlin, O, et al. Lymphocyte-predominant Hodgkin’s lymphoma in children: Therapeutic abstention after initial lymph node resection: A study of the French Society of Pediatric Oncology. J Clin Oncol. 2003; 21:2948–2952.
38. Stein, H, Diehl, V, Marafioti, T, et al. The nature of Reed-Sternberg cells, lymphocytic and histiocytic cells and their molecular biology in Hodgkin’s disease. In: Mauch PM, Armitage JO, Diehl V, et al, eds. Hodgkin’s disease. Philadelphia: Lippincott Williams & Wilkins; 1999:121.
39. Mauch, PM, Kalish, LA, Kadin, M, et al. Patterns of Hodgkin disease: Implications for etiology and pathogenesis. Cancer. 1993; 71:2062–2071.
40. Maity, A, Goldwein, JW, Lange, B, et al. Mediastinal masses in children with Hodgkin’s disease. Cancer. 1992; 69:2755–2760.
41. Jeffery, GM, Mead, GM, Whitehouse, JM. Life-threatening airway obstruction at the presentation of Hodgkin’s disease. Cancer. 1991; 67:506–510.
42. Munker, R, Hasenclaver, D, Brosteanu, O, et al. Bone marrow involvement in Hodgkin’s disease: An analysis of 135 consecutive cases. J Clin Oncol. 1995; 13:403–409.
43. Hann, HL, Lange, B, Stahlhut, MW, et al. Prognostic importance of serum transferrin and ferritin in childhood Hodgkin’s disease. Cancer. 1990; 66:313–316.
44. Tubiana, M, Henry-Arnar, M, Burgers, MV, et al. Prognostic significance of erythrocyte sedimentation rate in clinical stages I-II of Hodgkin’s disease. J Clin Oncol. 1984; 2:194–200.
45. Cosset, J, Henry Amar, M, Meerwadt, J, et al. The EORTC trials for limited stage Hodgkin’s disease. Eur J Cancer. 1992; 28A:1847–1850.
46. Longo, D, Russo, A, Duffey, P, et al. Treatment of advanced stage massive mediastinal Hodgkin’s disease: The case for combined modality therapy. J Clin Oncol. 1991; 9:227–235.
47. Maity, A, Goldwein, J, Lange, B, et al. Mediastinal mass in children with Hodgkin’s disease. Cancer. 1992; 69:2755–2760.
48. Vecchi, V, Pileri, S, Burnelli, R, et al. Treatment of pediatric Hodgkin’s disease tailored to stage, mediastinal mass, and age. Cancer. 1993; 72:2049–2057.
49. Carbone, PP, Kaplan, HS, Husshoff, K, et al. Report of the committee on Hodgkin’s disease staging classification. Cancer Res. 1971; 31:1860–1861.
50. Mauch, P, Larson, D, Osteen, R, et al. Prognostic factors for positive surgical staging in patients with Hodgkin’s disease. J Clin Oncol. 1990; 8:257–265.
51. Castellino, RA. Diagnostic imaging evaluation of Hodgkin’s disease and non-Hodgkin’s lymphoma. Cancer. 1991; 67:1177–1180.
52. Muraji, T, Hays, DM, Siegel, SE, et al. Evaluation of the surgical aspects of staging laparotomy for Hodgkin’s disease in children. J Pediatr Surg. 1982; 17:843–848.
53. Mendenhall, NP, Cantor, AB, Williams, JL, et al. With modern imaging techniques, is staging laparotomy necessary in pediatric Hodgkin’s disease? A Pediatric Oncology Group Study. J Clin Oncol. 1993; 11:2218–2225.
54. Gomez, GA, Reese, PA, Nava, H, et al. Staging laparotomy and splenectomy in early Hodgkin’s disease: No therapeutic benefit. Am J Med. 1984; 77:205–210.
55. Jenkin, D, Chan, H, Freedman, M, et al. Hodgkin’s disease in children: Treatment results with MOPP and low-dose, extended field irradiation. Ca Treatment Rep. 1982; 66:949–959.
56. Russel, KJ, Donaldson, SS, Cox, RS, et al. Childhood Hodgkin’s disease: Patterns of relapse. J Clin Oncol. 1984; 2:80–87.
57. American Academy of Pediatrics, Committee on Infectious Diseases. Visual Red Book on CD-ROM, 2001 Update. Elk Grove Village, IL: American Academy of Pediatrics; 2001.
58. Kaldor, JM, Day, NE, Clarke, EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med. 1990; 322:7–13.
59. Tura, S, Fiacchini, M, Zinzani, PL, et al. Splenectomy and the increasing risk of secondary acute leukemia in Hodgkin’s disease. J Clin Oncol. 1993; 11:925–930.
60. Dietrich, PY, Henry-Amar, M, Cosset, JM, et al. Second primary cancers in patients continuously disease-free from Hodgkin’s disease: A protective role for the spleen? Blood. 1994; 84:1209–1215.
61. Kostakoglu, L, Leonard, J, Kuji, I, et al. Comparison of fluorine-18 fluorodeoxyglucose positron emission tomography and Ga-67 scintigraphy in evaluation of lymphoma. Cancer. 2002; 94:879–888.
62. Wirth, A, Seymour, J, Hicks, R, et al. Fluorine-18 fluorodeoxyglucose positron emission tomography, gallium-67 scintigraphy, and conventional staging for Hodgkin’s disease and non-Hodgkin’s lymphoma. Am J Med. 2002; 112:262–268.
63. Shen, Y, Kao, A, Yen, R. Comparison of 18F-fluoro-2-deoxyglucose positron emission tomography and gallium-67 citrate scintigraphy for detecting malignant lymphoma. Oncol Rep. 2002; 9:321–325.
64. Buchmann, I, Reinhardt, M, Elsner, K. 2-(Fluorine-18)fluoro-2-deoxy-d-glucose positron emission tomography in the detection and staging of malignant lymphoma: A bicenter trial. Cancer. 2001; 91:889–899.
65. Kostakoglu, L, Coleman, M, Leonard, J, et al. PET predicts prognosis after 1 cycle of chemotherapy in aggressive lymphoma and Hodgkin’s disease. J Nucl Med. 2002; 43:1018–1027.
66. Friedberg, J, Chengazi, V. PET scans in the staging of lymphoma: Current status. Oncologist. 2003; 8:438–447.
67. Weihrauch, M, Re, D, Scheidhauer, K, et al. Thoracic positron emission tomography using 18F-fluorodeoxyglucose for the evaluation of residual mediastinal Hodgkin disease. Blood. 2001; 98:2930–2934.
68. Donaldson, SS, Link, MP. Combined modality treatment with low-dose radiation and MOPP chemotherapy for children with Hodgkin’s disease. J Clin Oncol. 1987; 5:742–749.
69. Engert, A, Schiller, P, Josting, A, et al. Involved-field radiotherapy is equally effective and less toxic compared with extended-field radiotherapy after four cycles of chemotherapy in patients with early-stage unfavorable Hodgkin’s lymphoma: Results of the HD8 trial of the German Hodgkin’s Lymphoma Study Group. J Clin Oncol. 2003; 21:3601–3608.
70. Sears, JD, Greven, KM, Ferree, CR, et al. Definitive irradiation in the treatment of Hodgkin’s disease. Cancer. 1997; 79:145–151.
71. Mauch, PM, Canellos, GP, Shulman, LN, et al. Mantle irradiation alone for selected patients with laparotomy-staged IA to IIA Hodgkin’s disease: Preliminary results of a prospective trial. J Clin Oncol. 1995; 13:947–952.
72. Wasserman, TH, Trenkner, DA, Fineberg, B, et al. Cure of early-stage Hodgkin’s disease with subtotal nodal irradiation. Cancer. 1991; 68:1208–1215.
73. Donaldson, SS, Whitaker, SJ, Plowman, PN, et al. Stage I-II pediatric Hodgkin’s disease: Long-term follow-up demonstrates equivalent survival rates following different management schemes. J Clin Oncol. 1990; 8:1128–1137.
74. Nachman, J, Sposto, R, Herzog, P, et al. Randomized comparison of low-dose involved-field radiotherapy and no radiotherapy for children with Hodgkin’s disease who achieve a complete response to chemotherapy. J Clin Oncol. 2002; 20:3765–3771.
75. Devita, VT, Serpick, A, Carbone, PP. Combination chemotherapy in the treatment of advanced Hodgkin’s disease. Ann Intern Med. 1970; 73:881–895.
76. Bonnadonna, G, Zucali, R, Monfardini, S, et al. Combination chemotherapy of Hodgkin’s disease with Adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer. 1975; 36:252–259.
77. Donaldson, S, Link, M, Weinstein, H, et al. Final results of a prospective clinical trial with VAMP and low-dose involved-field radiation for children with low risk Hodgkin’s disease. J Clin Oncol. 2007; 25:332–337.
78. Hutchinson, R, Fryer, C, Krailo, M, et al. Comparison of MOPP/ABVD with ABVD/XRT for treatment of advanced Hodgkin’s disease in children (CCG-521). Proc ASCO. 1992; 11:340.
79. Ekert, H, Waters, K, Smith, P, et al. Treatment with MOPP or CHLVPP chemotherapy only for all stages of childhood Hodgkin’s disease. J Clin Oncol. 1988; 6:1845–1850.
80. Ekert, H, Fox, L, Dalla-Pozzo, K, et al. A pilot study of EVAP/ABV chemotherapy in 25 newly diagnosed children with Hodgkin’s disease. Br J Cancer. 1993; 67:159–162.
81. Behrendt, H, Brinkhuis, M, Van Leeuwen, EF. Treatment of childhood Hodgkin’s disease with ABVD without radiotherapy. Med Pediatr Oncol. 1996; 26:244–248.
82. Kelly, KM, Sposto, R, Hutchinson, R, et al. BEACOPP chemotherapy is a highly effective regimen in children and adolescents with high risk Hodgkin lymphoma: A report from the Children’s Oncology Group CCG-59704 clinical trial. Blood. 2011; 117:2596–2603.
83. Weiner, MA, Leventhal, B, Brecher, ML, et al. Randomized study of intensive MOPP-ABVD with or without low-dose total-nodal radiation therapy in the treatment of stages IIB, IIIA2, IIIB, and IV Hodgkin’s disease in pediatric patients: A Pediatric Oncology Group study [see comments]. J Clin Oncol. 1997; 15:2769–2779.
84. Mauz-Korholz, C, Gorde-Grosjean, S, Hasenclever, D, et al. Resection alone in 58 children with limited stage, lymphocyte-predominant Hodgkin lymphoma-experience from the European network group on pediatric Hodgkin lymphoma. Cancer. 2007; 110(1):179–185.
85. Friedman DI, Wolden S, Constine L, et al. AHOD0031: A Phase III study of dose-intensive therapy for intermediate risk Hodgkin Lymphoma: A report from the Children’s Oncology Group. 53rd American Society of Hematology Annual Meeting 2010; Abstract #766.
86. Schmitz, N, Pfistner, B, Sextro, M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haematopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: A randomized trial. Lancet. 2002; 359:2065–2071.
87. Freed, J, Kelly, KM. Current approaches to the management of pediatric Hodgkin Lymphoma. Pediatr Drugs. 2010; 12(2):85–98.
88. Younes, A, Bartlett, NL, Leonard, JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010; 363:1812–1821.
89. Oberlin, O, Leverger, G, Pacquement, H, et al. Low-dose radiation therapy and reduced chemotherapy in childhood Hodgkin’s disease: The experience of the French Society of Pediatric Oncology. J Clin Oncol. 1992; 10:1602–1608.
90. Hunger, SP, Link, MP, Donaldson, SS. ABVD/MOPP and low-dose involved-field radiotherapy in pediatric Hodgkin’s disease: The Stanford experience. J Clin Oncol. 1994; 12:2160–2166.
91. Bonfante, V, Santoro, A, Viviani, S, et al. Outcome of patients with Hodgkin’s disease failing after primary MOPP-ABVD. J Clin Oncol. 1997; 15:528–534.
92. Diehl, V, Franklin, J, Pfreundschuh, M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin’s disease. N Engl J Med. 2003; 348:2386–2395.
93. Sieber, M, Bredenfeld, H, Josting, A, et al. 14-day variant of the bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone regimen in advanced-stage Hodgkin’s lymphoma: Results of a pilot study of the German Hodgkin’s Lymphoma Study Group. J Clin Oncol. 2003; 21:1734–1739.
94. Bookman, MA, Longo, DL, Young, RC. Late complications of curative treatment in Hodgkin’s disease. JAMA. 1988; 260:680–683.
95. Aleman, B, van den Belt-Dusebout, AW, Klokman, WJ, et al. Long-term cause-specific mortality of patients treated for Hodgkin’s disease. J Clin Oncol. 2003; 21:3431–3439.
96. Willman, KY, Cox, RS, Donaldson, SS. Radiation induced height impairment in pediatric Hodgkin’s disease. Int J Radiat Oncol Biol Phys. 1994; 28:85–92.
97. Constine, LS, Donaldson, SS, McDougall, JR, et al. Thyroid dysfunction after radiotherapy in children with Hodgkin’s disease. Cancer. 1984; 53:878–883.
98. McHenry, C, Jarosz, H, Calandra, D, et al. Thyroid neoplasia following radiation therapy for Hodgkin’s lymphoma. Arch Surg. 1987; 122:684–686.
99. Sankila, R, Garwicz, S, Olsen, JH, et al. Risk of subsequent malignant neoplasms among 1641 Hodgkin’s disease patients diagnosed in childhood and adolescence: A population-based cohort study in the five Nordic countries. J Clin Oncol. 1996; 14:1442–1446.
100. Thibaud, E, Ramirez, M, Brauner, R, et al. Preservation of ovarian function by ovarian transposition performed before pelvic irradiation during childhood. J Pediatr. 1992; 121:880–884.
101. Bhatia, S, Robison, LL, Oberlin, O, et al. Breast cancer and other second neoplasms after childhood Hodgkin’s disease. N Engl J Med. 1996; 334:745–751.
102. Dores, G, Metayer, C, Curtis, R, et al. Second malignant neoplasms among long-term survivors of Hodgkin’s disease: A population-based evaluation over 25 years. J Clin Oncol. 2002; 20:3484–3494.
103. Longo, D. Radiation therapy in the treatment of Hodgkin’s disease: Do you see what I see? J Natl Cancer Inst. 2003; 95:928–929.
104. Hoppe, RT. Hodgkin’s disease: Complications of therapy and excess mortality. Ann Oncol. 1997; 8(Suppl. 1):S115–S118.
105. Tucker, MA, Coleman, CN, Cox, RS, et al. Risk of second cancers after treatment for Hodgkin’s disease. N Engl J Med. 1988; 318:76–81.
106. Cimino, G, Papa, G, Tura, S, et al. Second primary cancer following Hodgkin’s disease: Updated results of an Italian multicentric study. J Clin Oncol. 1991; 9:432–437.
107. Van Leeuwen, FE, Klokman, WJ, Hagenbeek, A, et al. Second cancer risk following Hodgkin’s disease: A 20-year follow-up study. J Clin Oncol. 1994; 12:312–325.
108. Kaldor, JM, Day, NE, Clarke, EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med. 1990; 322:7–13.
109. Tura, S, Fiacchini, M, Zinzani, PL, et al. Splenectomy and the increasing risk of secondary acute leukemia in Hodgkin’s disease. J Clin Oncol. 1993; 11:925–930.
110. Van Leeuwen, FE, Chorus, AM, van den Belt-Dusebout, AW, et al. Leukemia risk following Hodgkin’s disease: Relation to cumulative dose of alkylating agents, treatment with teniposide combinations, number of episodes of chemotherapy, and bone marrow damage. J Clin Oncol. 1994; 12:1063–1073.
111. Dietrich, PY, Henry-Amar, M, Cosset, JM, et al. Second primary cancers in patients continuously disease-free from Hodgkin’s disease: A protective role for the spleen? Blood. 1994; 84:1209–1215.
112. Biovin, JF, O’Brien, K. Solid cancer risk after treatment of Hodgkin’s disease. Cancer. 1988; 61:2541–2546.
113. Salloum, E, Doria, R, Shubert, W, et al. Second solid tumors in patients with Hodgkin’s disease cured after radiation or chemotherapy plus adjuvant low-dose radiation. J Clin Oncol. 1996; 14:2435–2443.
114. Doria, R, Holford, T, Farber, LR, et al. Second solid malignancies after combined modality therapy for Hodgkin’s disease. J Clin Oncol. 1995; 13:2016–2022.
115. Curtis, RE, Boice, JD, Jr. Second cancers after radiotherapy for Hodgkin’s disease. N Engl J Med. 1988; 319:244–245.
116. Prior, P, Pope, DJ. Hodgkin’s disease: Subsequent primary cancers in relation to treatment. Br J Cancer. 1988; 58:512–517.
117. Kaldor, JM, Day, NE, Band, P, et al. Second malignancies following testicular cancer, ovarian cancer, and Hodgkin’s disease: An international collaborative study among cancer registries. Int J Cancer. 1987; 39:571–585.
118. Wahner-Roedler, D, Nelson, D, Croghan, I, et al. Risk of breast cancer and breast cancer characteristics in woman treated with supradiaphragmatic radiation for Hodgkin lymphoma: Mayo Clinic experience. Mayo Clin Proc. 2003; 78:708–715.
119. Hancock, SL, Horning, SJ, Hoppe, RT. Breast cancer after the treatment of Hodgkin’s disease [abstract]. Int J Radiat Oncol Biol Phys. 1991; 21:157.
120. Shapiro, CL, Mauch, PM. Radiation-associated breast cancer after Hodgkin’s disease: Risks and screening in perspective. J Clin Oncol. 1992; 10:1662–1665.
121. Hancock, SL, Tucker, MA, Hoppe, RT. Factors affecting late mortality from heart disease after treatment of Hodgkin’s disease. JAMA. 1993; 270:1949–1955.
122. Steinherz, LJ, Steinherz, PG, Tan, CT, et al. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA. 1991; 266:1672–1677.
123. Novakovic, B. USA childhood cancer survival, 1973–1987. Med Pediatr Oncol. 1994; 23:480–486.
124. Smith, MA, Seibel, NL, Altekruse, SF, et al. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J Clin Oncol. 2010; 28:2625–2634.
125. Magrath, IT. The pathogenesis of Burkitt’s lymphoma. Recent Adv Cancer Res. 1990; 55:133–270.
126. Hanto, DW, Frizzera, G, Gajl-Peczalska, KJ. Epstein-Barr virus, immunodeficiency, and B cell lymphoproliferation. Transplantation. 1985; 39:461–472.
127. Cohen, JI. Epstein-Barr virus lymphoproliferative disease associated with acquired immune deficiency. Medicine. 1991; 70:137–160.
128. Shibata, D, Weiss, LM, Nathwani, BN, et al. Epstein-Barr virus in benign lymph node biopsies from individuals infected with the human immunodeficiency virus is associated with concurrent or subsequent development of non-Hodgkin’s lymphoma. Blood. 1991; 77:1527–1533.
129. Neri, A, Barriga, F, Inghirami, G, et al. Epstein-Barr virus infection precedes clonal expansion in Burkitt’s and acquired immunodeficiency syndrome-associated lymphomas. Blood. 1991; 77:1092–1095.
130. Savoie, A, Perpete, C, Carpentier, L, et al. Direct correlation between the load of Epstein-Barr virus–infected lymphocytes in the peripheral blood of pediatric transplant patients and risk of lymphoproliferative disease. Blood. 1994; 83:2715–2722.
131. Rooney, CM, Loftin, SK, Holladay, HS, et al. Early identification of Epstein-Barr virus–associated post-transplantation lymphoproliferative disease. Br J Haematol. 1995; 89:98–103.
132. Holmes, R, Sokol, R. Epstein-Barr virus and post-transplant lymphoproliferative disease. Pediatr Transplant. 2002; 6:456–464.
133. Sreenan, JJ, Tubbs, RR. The influence of immunology and genetics on lymphoma classification: A historical perspective. Ca Invest. 1996; 14:572–588.
134. Link, MP, Weinstein, HJ. Malignant Non-Hodgkin lymphomas in children. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:722–747.
135. Hutchison, RE, Murphy, SB, Fairclough, DL, et al. Diffuse small noncleaved cell lymphoma in children: Burkitt’s versus non-Burkitt’s types. Cancer. 1989; 64:23–28.
136. Harris, NL, Jaffe, ES, Stein, H, et al. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood. 1994; 84:1361–1392.
137. Nathwani, BN, Griffith, RC, Kelly, DR, et al. A morphologic study of childhood lymphoma of the diffuse ‘histiocytic’ type: The pediatric oncology experience. Cancer. 1987; 59:1138–1142.
138. Hutchison, RE, Berard, CW, Shuster, JJ, et al. B-cell lineage confers favorable outcome among children and adolescents with large-cell lymphoma: A Pediatric Oncology Group study. J Clin Oncol. 1995; 13:2023–2032.
139. Sandland, JT, Pui, CH, Santana, VM, et al. Clinical features and treatment outcome for children with CD30+ large-cell non-Hodgkin’s lymphoma. J Clin Oncol. 1994; 12:895–898.
140. Falini, B, Pileri, S, Pizzolo, G, et al. CD30 (Ki-1) molecule: A new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995; 85:1–14.
141. Fillipa, DA, Ladanyi, M, Wollner, N, et al. CD30 (Ki-1)-positive malignant lymphomas: Clinical, immunophenotypic, histologic, and genetic characteristics and differences with Hodgkin’s disease. Blood. 1996; 87:2905–2917.
142. Reiter, A, Schrappe, M, Tiemann, M, et al. Successful treatment strategy for Ki-1 anaplastic large-cell lymphoma of childhood: A prospective analysis of 62 patients enrolled in three consecutive Berlin-Frankfurt-Munster Group studies. J Clin Oncol. 1994; 12:899–908.
143. Kaden, ME. Ki-1/CD30+ (anaplastic) large-cell lymphoma: Maturation of a clinicopathologic entity with prospects of effective therapy. J Clin Oncol. 1994; 12:884–887.
144. Kadin, ME, Sako, D, Berliner, N, et al. Childhood Ki-1 lymphoma presenting with skin lesions and peripheral lymphadenopathy. Blood. 1986; 68:1042–1049.
145. Howat, AJ, Thomas, H, Waters, KD, et al. Malignant lymphoma of bone in children. Cancer. 1987; 59:335–339.
146. Meyers, PA, Potter, VP, Wolner, N, et al. Bowel perforation during initial treatment of childhood non-Hodgkin’s lymphoma. Cancer. 1985; 56:259–261.
147. Tesoro-Tess, JD, Biasi, S, Balzarini, L, et al. Heart involvement in lymphomas. Cancer. 1993; 72:2484–2490.
148. Ridgway, D, Wolff, LJ, Neerhout, RC, et al. Unsuspected non-Hodgkin’s lymphoma of the tonsils and adenoids in children. Pediatrics. 1987; 79:399–402.
149. Ghelman, B. Radiology of bone tumors. Orthop Clin North Am. 1989; 20:287–312.
150. Clayton, F, Butler, JJ, Ayala, AG, et al. Non-Hodgkin’s lymphoma in bone. Cancer. 1987; 60:2494–2501.
151. Wollner, N, Lane, JM, Marcove, RC, et al. Primary skeletal non-Hodgkin’s lymphoma in the pediatric age group. Med Pediatr Oncol. 1992; 20:506–513.
152. Tsokos, GC, Balow, JE, Spiegel, RJ, et al. Renal and metabolic complications of undifferentiated and lymphoblastic lymphomas. Medicine. 1981; 60:218–229.
153. Hande, KR, Garrow, GC. Acute tumor lysis syndrome in patients with high-grade non-Hodgkin’s lymphoma. Am J Med. 1993; 94:133–139.
154. Finlay, JL, Anderson, JR, Cecalupo, AJ, et al. Disseminated nonlymphoblastic lymphoma of childhood: A Children’s Cancer Group study, CCG-552. Med Pediatr Oncol. 1994; 23:453–463.
155. Schwenn, MR, Blattner, SR, Lynch, SR, et al. hic-COM: A 2-month intensive chemotherapy regimen for children with stage III and IV Burkitt’s lymphoma and B-cell acute lymphoblastic leukemia. J Clin Oncol. 1991; 9:133–138.
156. Anderson, JR, Jenkin, RDT, Wilson, JF, et al. Long-term follow-up of patients treated with COMP or LSA2L2 therapy for childhood non-Hodgkin’s lymphoma: A report of CCG-551 from the Children’s Cancer Group. J Clin Oncol. 1993; 11:1024–1032.
157. Burkitt, D. A sarcoma involving the jaws in African children. Br J Surg. 1958; 46:218–223.
158. Shad, A, Magrath, I. Malignant non-Hodgkin’s lymphomas in children. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 3rd ed. Philadelphia: Lippincott-Raven; 1997:545–587.
159. Sariban, E, Donahue, A, Magrath, IT. Jaw involvement in American Burkitt’s lymphoma. Cancer. 1984; 53:141–146.
160. Reiter, A, Schrappe, M, Parwaresch, R, et al. Non-Hodgkin’s lymphomas of childhood and adolescence: Results of a treatment stratified for biologic subtypes and stage: A report of the Berlin-Frankfurt-Munster Group. J Clin Oncol. 1995; 13:359–372.
161. Stein, H, Foss, H, Durkop, H, et al. CD30+ anaplastic large cell lymphoma: A review of its histopathologic, genetic, and clinical features. Blood. 2000; 96:3681–3695.
162. Levine, AM. Acquired immunodeficiency syndrome-related lymphoma. Blood. 1992; 80:8–20.
163. Knowles, DM, Cesarman, E, Chadburn, A, et al. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of post-transplantation lymphoproliferative disorders. Blood. 1995; 85:552–565.
164. Ho, M. Risk factors and pathogenesis of post-transplant lymphoproliferative disorders. Transplant Proc. 1995; 27:38–40.
165. Reiter, A, Zimmerman, W, Zimmerman, M, et al. The role of initial laparotomy and second look surgery in the treatment of abdominal B-cell non-Hodgkin’s lymphoma of childhood: A report of the BFM group. Eur J Pediatr Surg. 1994; 4:74–81.
166. Palestro, CJ, Rini, JN, Tomas, MB. Lymphoma. In: Charron M, ed. Practical Pediatric PET Imaging. New York: Springer; 2006:220–242.
167. Hernandez-Pampaloni, M, Takalkar, A, Yu, JQ, et al. F-18 FDG-PET imaging and correlation with CT in staging and follow-up of pediatric lymphomas. Pediatr Radiol. 2006; 36:524–531.
168. Downing, JR, Shurtleff, SA, Zielenska, M, et al. Molecular detection of the (2;5) translocation of non-Hodgkin’s lymphoma by reverse transcriptase-polymerase chain reaction. Blood. 1995; 85:3416–3422.
169. Staudt, L. Molecular diagnosis of the hematologic cancers. N Engl J Med. 2003; 348:1777–1785.
170. Murphy, SB. Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas: Dissimilarities from lymphomas in adults. Semin Oncol. 1980; 7:332–339.
171. Haddy, TB, Adde, MA, Magrath, IT. CNS involvement in small noncleaved cell lymphoma: Is CNS disease per se a poor prognostic sign? J Clin Oncol. 1991; 9:1973–1982.
172. Cairo, MS, Sposto, R, Perkins, SL, et al. Burkitt’s and Burkitt-like lymphoma in children and adolescents: A review of the Children’s Cancer Group experience. Br J Haematol. 2003; 120:660–670.
173. Meadows, AT, Sposto, R, Jenkin, RD, et al. Similar efficacy of 6 and 18 months of therapy with four drugs (COMP) for localized non-Hodgkin’s lymphoma of children: A report from the Children’s Cancer Study Group. J Clin Oncol. 1989; 7:92–99.
174. Patte, C, Philip, T, Rodary, C, et al. High survival rate in advanced-stage B-cell lymphomas and leukemias without CNS involvement with a short intensive polychemotherapy: Results from the French Pediatric Oncology Society of a randomized trial of 216 children. J Clin Oncol. 1991; 9:123–132.
175. Bowman, WP, Shuster, JJ, Cook, B, et al. Improved survival for children with B-cell acute lymphoblastic leukemia and stage IV small noncleaved-cell lymphoma: A Pediatric Oncology Group study. J Clin Oncol. 1996; 14:1252–1261.
176. Sullivan, MP, Ramirez, I. Curability of Burkitt’s lymphoma with high-dose cyclophosphamide, high-dose methotrexate therapy and intrathecal chemoprophylaxis. J Clin Oncol. 1985; 3:627–636.
177. Swinnen, LJ, Mullen, GM, Carr, TJ, et al. Aggressive treatment for postcardiac transplant lymphoproliferation. Blood. 1995; 86:3333–3340.
178. Orjuela, M, Gross, T, Cheung, Y, et al. A pilot study of chemoimmunotherapy (cyclophosphamide, prednisone, and rituximab) in patients with post-transplant lymphoproliferative disorder following solid organ transplantation. Clin Cancer Res. 2003; 9:3945S–3952S.
179. Link, MP, Donaldson, SS, Berard, CW, et al. Results of treatment of childhood localized non-Hodgkin’s lymphoma with combination chemotherapy with or without radiotherapy. N Engl J Med. 1990; 322:1169–1174.
180. Oschlies, I, Klapper, W, Zimmermann, M, et al. Diffuse large B-cell lymphoma in pediatric patients belongs predominantly to the germinal-center type B-cell lymphomas: Clinicopathologic analysis of cases included in the German BFM (Berlin-Frankfurt-Münster) Multicenter Trial. Blood. 2006; 107:4047–4052.
181. Le Deley, MC, Rosolen, A, Reiter, A, et al. The impact of the association of vinblastine during induction chemotherapy and as maintenance treatment in children and adolescents with high-risk anaplastic large cell lymphoma: Results of a randomized trial of the EICNHL Group. Blood. 2008; 112:577.
182. Cohen, LF, Balow, JE, Magrath, IT, et al. Acute tumor lysis syndrome: A review of 37 patients with Burkitt’s lymphoma. Am J Med. 1980; 68:486–491.
183. Murphy, SB, Melvin, SL, Mauer, AM, et al. Correlation of tumor cell kinetic studies with surface marker results in childhood non-Hodgkin’s lymphoma. Cancer Res. 1979; 39:1534–1538.
184. Smalley, R, Guaspari, A, Haase-Statz, S, et al. Allopurinol: Intravenous use for prevention and treatment of hyperuricemia. J Clin Oncol. 2000; 18:1758–1763.
185. Pui, C, Mahmoud, H, Wiley, J, et al. Recombinant urate oxidase for the prophylaxis or treatment of hyperuricemia in patients with leukemia or lymphoma. J Clin Oncol. 2001; 19:697–704.
186. Goldman, S, Holcenberg, J, Finklestein, J, et al. A randomized comparison between rasburicase and allopurinol in children with lymphoma or leukemia at high risk for tumor lysis. Blood. 2001; 97:2998–3003.
187. Allegretta, GJ, Weisman, SJ, Altman, AJ. Oncologic emergencies I: Metabolic and space-occupying consequences of cancer and cancer treatment. Pediatr Clin North Am. 1985; 32:601–611.
188. Korones, DN, Buzzard, CJ, Asselin, BL. Right atrial thrombi in children with cancer and indwelling catheters. J Pediatr. 1996; 128:841–846.
189. Pryzant, RM, Meistrich, ML, Wilson, G, et al. Long-term reduction in sperm count after chemotherapy with and without radiation therapy for non-Hodgkin’s lymphomas. J Clin Oncol. 1993; 11:239–247.
190. Sandoval, C, Pui, CH, Bowman, LC, et al. Secondary acute myeloid leukemia in children previously treated with alkylating agents, intercalating topoisomerase II inhibitors, and irradiation. J Clin Oncol. 1993; 11:1039–1045.
191. Friedman, DL, Meadows, AT. Late effects following lymphoma treatment. In: Weinstein HJ, Hudson MM, Link MP, eds. Pediatric Lymphomas. Berlin: Springer; 2007:259–280.