SECTION IV LOCALISED NEUROLOGICAL DISEASE AND ITS MANAGEMENT C. PERIPHERAL NERVE AND MUSCLE
THE POLYNEUROPATHIES – FUNCTIONAL ANATOMY
STRUCTURE OF THE NERVE CELL AND AXON
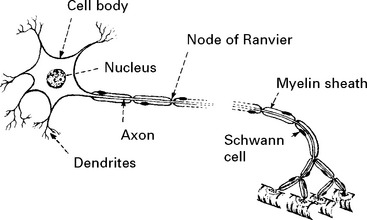
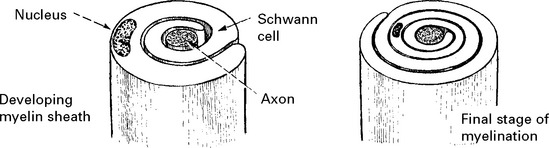
All axons have a cellular sheath – Schwann cell – but not all axons are myelinated.
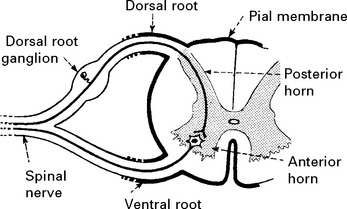
SPINAL PERIPHERAL NERVOUS SYSTEM
Sensation can be divided into:
These different forms of sensation are carried from the periphery by axons with specific characteristics. The central connections and pathways vary also (see page 200).

The thoracic nerves supply skeletal muscles and subserve sensation of the thorax and abdomen.
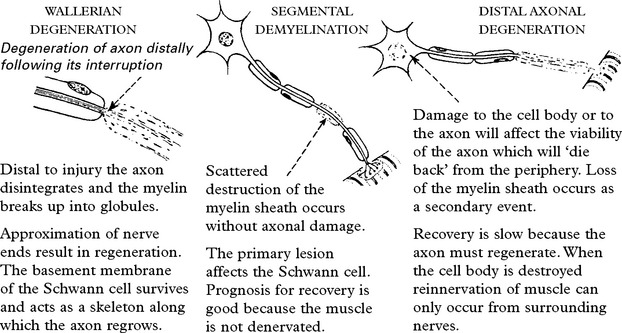
THE POLYNEUROPATHIES – SYMPTOMS
Sensory
Negative phenomena – loss of sensation.
Disease of large myelinated fibres produces loss of touch and joint position perception.
Disease of small unmyelinated fibres produces painful positive phenomena:
International Association for the Study of Pain (IASP) definitions has clarified the following.
THE POLYNEUROPATHIES – SIGNS
SENSORY EXAMINATION


Examination of gait is important; with joint position impairment, sensory ataxia is evident. Romberg’s test is positive (see page 28). Neuropathic burns/ulcers or joints may be present.
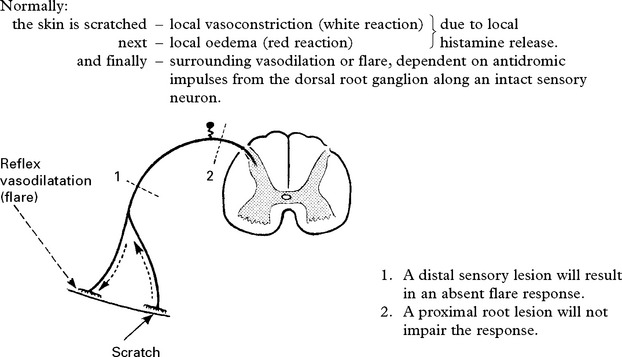
The AXON REFLEX can be used to ‘place’ lesions in the sensory pathway.
THE POLYNEUROPATHIES – CLASSIFICATION
There are several approaches to classification:
The following table based primarily on mode of onset is for reference. Certain neuropathies will be dealt with separately (see pages 439–444).
| CAUSE | FUNCTIONAL DISTURBANCE | PATHOLOGY |
|---|---|---|
| ACUTE: days to 4 weeks | ||
| Inflammatory (Guillain Barré syndrome) | Predominantly motor | Demyelinative with perivascular lymphocytic infiltration |
| Distal or proximal | ||
| Autonomic disturbance | ||
| Diphtheria—— | Cranial nerve onset | Demyelinative. No inflammatory infiltration. |
| Mixed motor/sensory | ||
| Porphyria—— | Motor (may begin in arm). | Axonal |
| Autonomic disturbance | ||
| Minimal sensory loss. | ||
| SUBACUTE – occasionally CHRONIC: months and years | ||
| ASYMETRICAL and MULTIFOCAL | ||
| Infections | ||
| Leprosy | Sensory neuropathy, often multifocal; associated depigmentation | Spectrum from paucibacillary (few organisms with intense inflammation) to multibacillary (many organisms with little inflammation) |
| HIV | Range of associated neuropathies | |
| Vasculitic disorders | ||
| Polyarteritis nodosa; | Usually presents with mononeuritis multiplex or an asymmetrical sensorimotor neuropathy. | Vasculitis with Wallerian degeneration in distal nerves |
| Wegner’s granulomatosis; | ||
| Churg-Strauss syndrome | ||
| Often painful | ||
| Non-systemic vasculitis | As above without systemic features | |
| SUBACUTE and CHRONIC: months and years | ||
| SYMMETRICAL | ||
| Metabolic and endocrine disorders | ||
| Diabetes | Most commonly distal sensorimotor | |
| But wide range of other forms (see later) | ||
| Ureamia | Distal sensorimotor | Axonal degeneration |
| Hypothyroidism | Distal sensorimotor | |
| Acromegaly | Distal sensorimotor | |
| CAUSE | FUNCTIONAL DISTURBANCE | PATHOLOGY |
|---|---|---|
| Nutritional deficiencies | ||
| Vitamin B1 (thiamine) | Predominantly sensory, with burning feet. | Axonal degeneration with segmental demyelination |
| Includes alcoholic neuropathy | Weakness may develop. | |
| Autonomic involvement common but mild | ||
| B12 deficiency | Predominantly sensory; may be associated spinal cord involvement | |
| Malignant disease | ||
| Paraneoplastic | Sensory or sensorimotor | Axonal; may be associated antibodies (anti-Hu) |
| Infiltrative | Multifocal, often a polyradiculopathy | More common with lymphoma |
| Paraprotein associated | ||
| Monoclonal gammopathy (IgG, IgA, IgM) | Sensorimotor neuropathy | Axonal with segmental demyelination |
| Chronic inflammatory demyelinating polyneuropathy (CIDP) (see later) | ||
| Amyloid | ||
| Primary, secondary or familial | Sensorimotor neuropathy often with autonomic involvement | Thickened nerves with amyloid deposition and small fibre neuropathy |
| May present as multiple mononeuropathies | ||
| Inherited neuropathies | ||
| Charcot-Marie-Tooth disease (see below) | ||
| Refsum’s disease | Phytanic acid storage disorder. Sensorimotor neuropathy with ichthyosis, retinitis pigmentosa and deafness | |
| Drug induced | ||
| Wide range of drugs induce neuropathies including: | ||
| Antibiotics | Metronidazole; ethambutol; isoniazid; nitrofurantoin; dapsone | |
| Oncology drugs | Adriamycin; cisplatin; taxanes; vincristine | |
| HIV drugs | Didanosine; stavudine; zalcitabine | |
| Others | Amiodarone; gold; phenytoin | |
| Toxin induced | ||
| Solvents | ||
| Heavy metals | Lead; arsenic; thallium | |
INVESTIGATION OF NEUROPATHY
For a patient with a distal symmetrical sensorimotor neuropathy:
Further investigations (depending on clinical history):
THE POLYNEUROPATHIES – SPECIFIC TYPES
GUILLAIN BARRÉ SYNDROME (ACUTE INFLAMMATORY DEMYELINATING POLYNEUROPATHY)
Investigations
Treatment
Treatment is generally given to those who can no longer walk and is deferred in milder cases.
CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY (CIDP)
Prevalence – 3% of all neuropathies
Age of onset: mean 35 yrs (fluctuating course – younger, progressive – older)
| Diagnosis: | Electrophysiology |
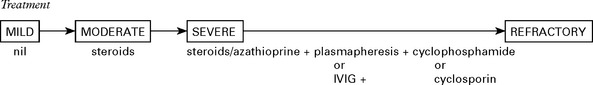
Outcome with treatment – 30% symptom free – 45% mild disability – 25% severe disability
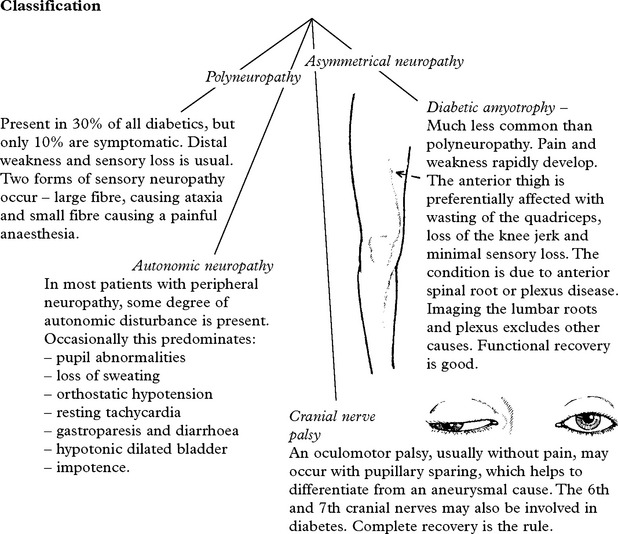
DIABETIC NEUROPATHY
This condition is uncommon in childhood and increases with age.
CARCINOMATOUS POLYNEUROPATHY
THE POLYNEUROPATHIES – SPECIFIC TYPES – INHERITED NEUROPATHIES

PLEXUS SYNDROMES AND MONONEUROPATHIES
Cranial nerve mononeuropathies have been dealt with separately.
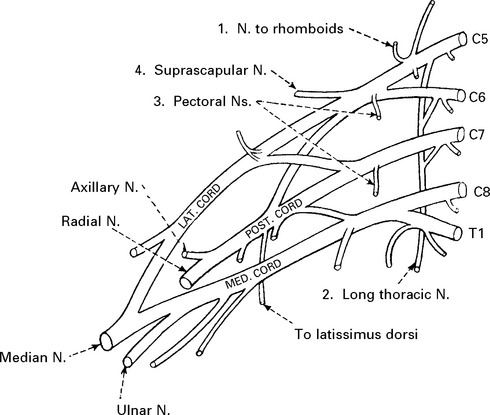
BRACHIAL PLEXUS SYNDROMES
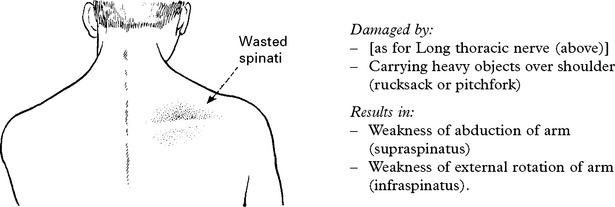
UPPER PLEXUS LESION (C5C6)

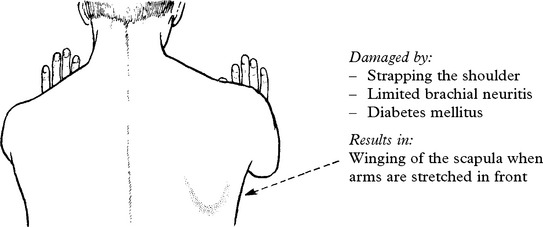
When damage to C5C6 is more proximal, nerve to rhomboids and long thoracic nerve may be affected.

LOWER PLEXUS LESION (C8T1)
Forced abduction of the arm at birth (Klumpke’s paralysis) or trauma may produce damage to the lower plexus. This results in paralysis of the intrinsic hand muscles producing a claw hand, C8T1 sensory loss and a Horner’s syndrome (page 145) if the T1 root is involved.
TOTAL BRACHIAL LESION
This results in complete flaccid paralysis and anaesthesia of the arm.
The presence of a Horner’s syndrome indicates proximal T1 nerve root involvement.
THORACIC OUTLET SYNDROME
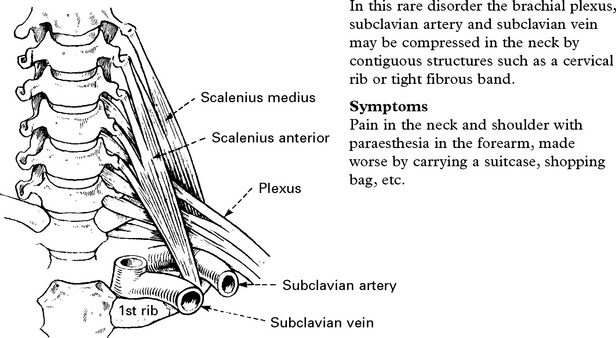
Signs
Sensory loss in a T1 distribution.
Wasting and weakness of thenar and occasionally interosseous muscles.
BRACHIAL NEURITIS (Neuralgic amyotrophy)
Brachial neuritis is a relatively common disorder sometimes associated with:
In most cases it develops without any evident precipitating cause.
Treatment
Narcotic analgesics may be required if pain is extreme. Corticosteriods are normally given though the value of immunotherapy is uncertain. By 2 yrs – 75% have fully recovered. Brachial neuritis may be familial. Recurrent attacks occur in Hereditary Neuropathy with liability to Pressure Palsies – HNPP (page 444). Autosomal dominant forms of Hereditary Neuralgic Amyotrophy (HNA), both acute and chronic are described, some linked to chromosome 17q.
UPPER LIMB MONONEUROPATHIES

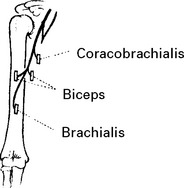
MUSCULOCUTANEOUS NERVE (Lateral cord) (C5C6)

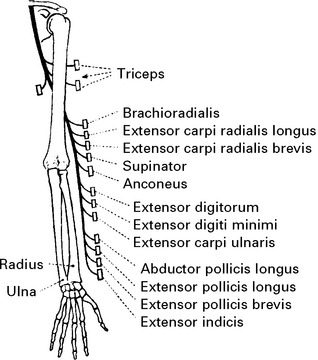
RADIAL NERVE (Posterior cord) (C6C7C8)
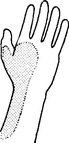

MEDIAN NERVE (Lateral and medial cords) (C7C8)
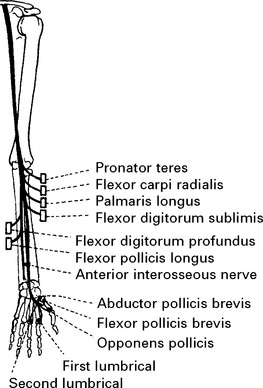
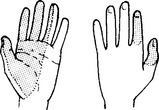
Palmar surfaces of the radial border of the hand.
Carpal tunnel syndrome
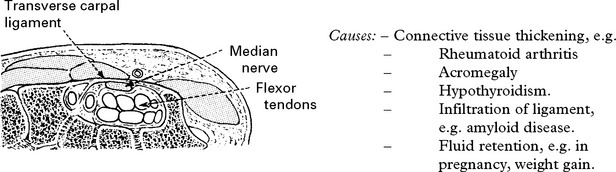
| Causes: | – Connective tissue thickening, e.g. | |
| – | Rheumatoid arthritis | |
| – | Acromegaly | |
| – | Hypothyroidism. | |
| – | Infiltration of ligament, e.g. amyloid disease. | |
| – | Fluid retention, e.g. in pregnancy, weight gain. | |
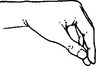
ULNAR NERVE (Medial cord) (C7C8)
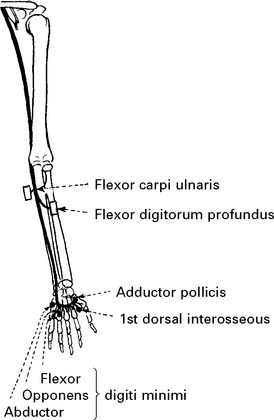
Both palmar and dorsal surfaces of the ulnar border of the hand.
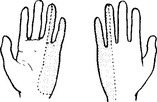
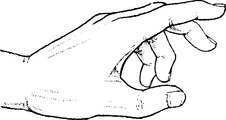
In the hand, close to the hamate bone, it divides into deep and superficial branches.
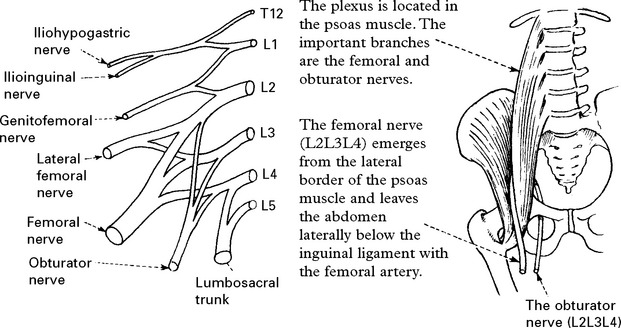
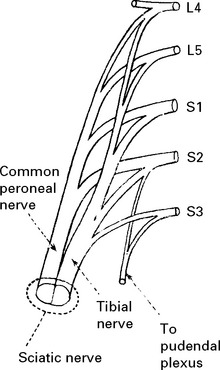
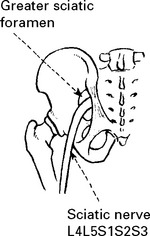
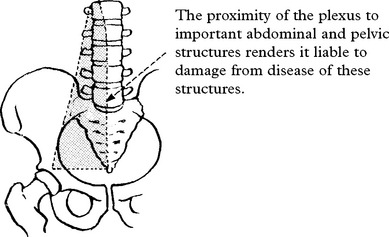
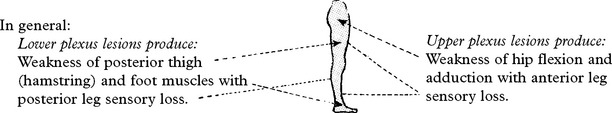
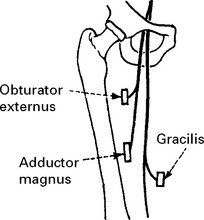
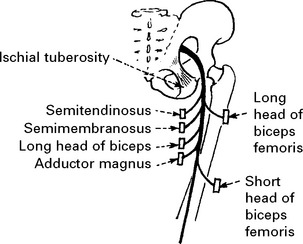
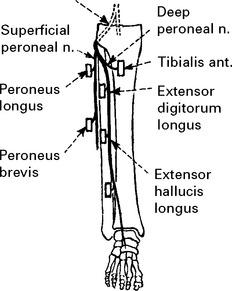
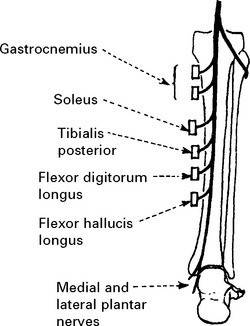
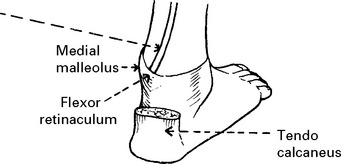
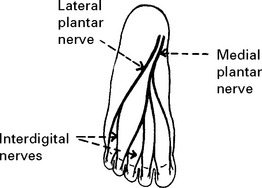
LOWER LIMB MONONEUROPATHIES
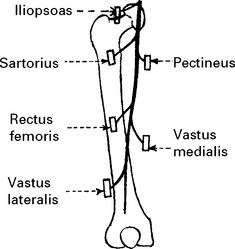
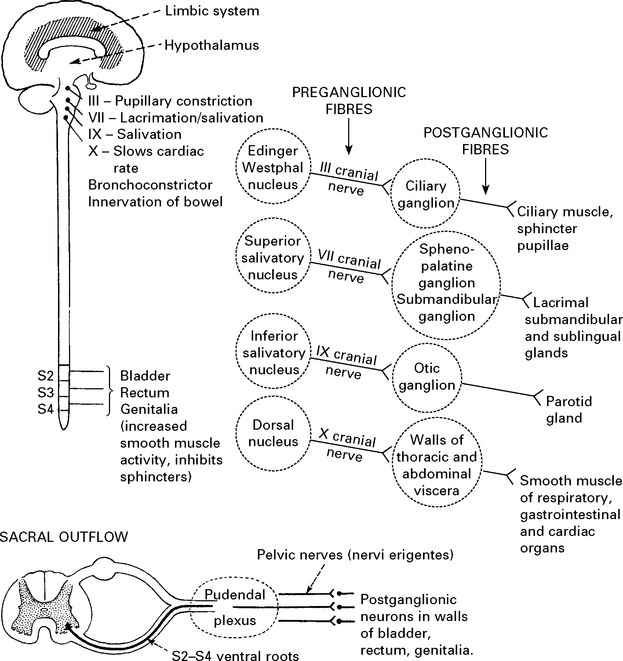
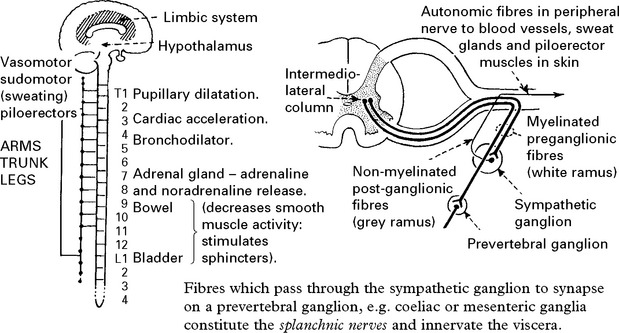
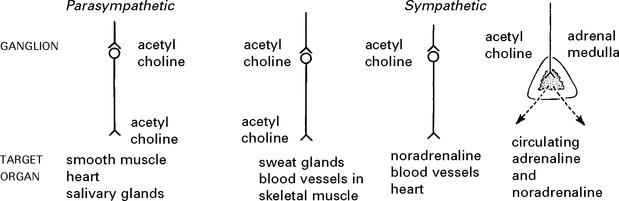
AFFERENT AUTONOMIC NERVOUS SYSTEM
Sympathetic
Terminate in spinal cord in intermediate zone of grey matter – in relation to preganglionic neurons.
TESTS OF AUTONOMIC FUNCTION
BLOOD PRESSURE CONTROL
HEART RATE
AUTONOMIC NERVOUS SYSTEM – SPECIFIC DISEASES
The following are less common disorders which primarily may affect the autonomic nervous system –
IDIOPATHIC ORTHOSTATIC HYPOTENSION
Two types of this condition are recognised:
In the latter disorder, features of extrapyramidal system involvement are also found.
DIABETIC AUTONOMIC NEUROPATHY
Symptoms of autonomic dysfunction are common in long-standing insulin-dependent diabetics:
ADIE’S SYNDROME
A tonic pupil (page 144) associated with areflexia and occasionally widespread autonomic dysfunction, e.g. segmental hypohidrosis (absent sweating) and diarrhoea.
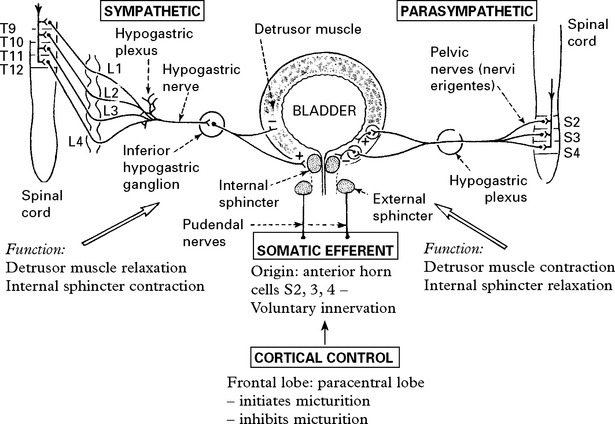
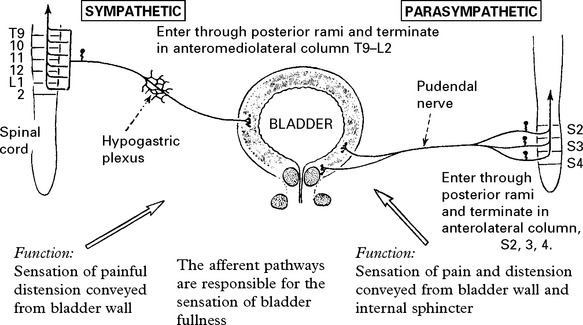
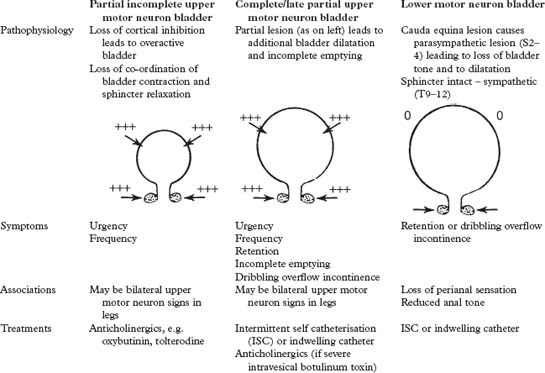
MICTURITION
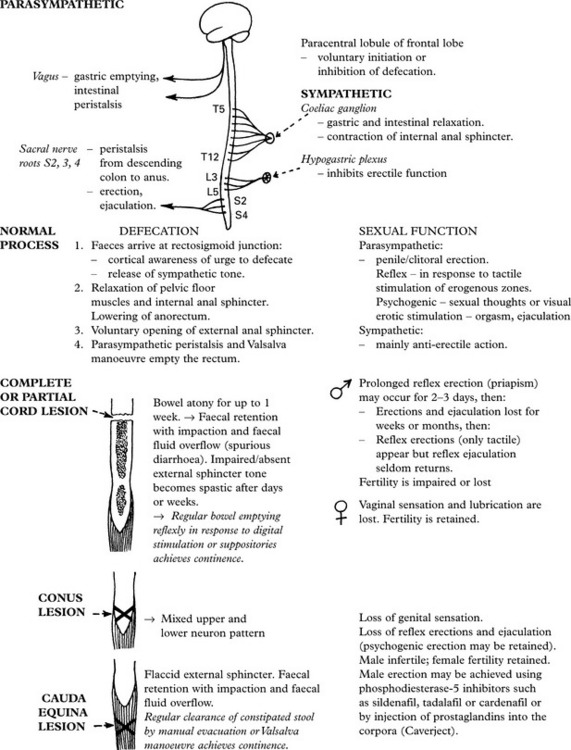
DISEASES OF SKELETAL (VOLUNTARY) MUSCLE
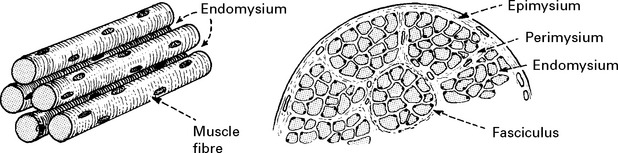
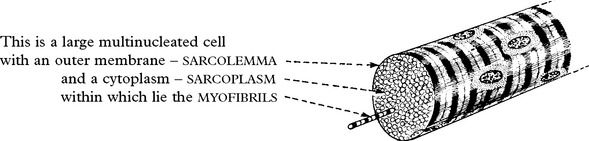

MUSCLE MORPHOLOGY AND FUNCTION
Fibre type
Type I: Slow twitch, fatigue resistant.
| Characteristics: | Type I | Type II |
|---|---|---|
| ATPase stain: Light | ATPase stain: Dark | |
| Oxidative metabolism | Glycolytic metabolism | |
| Abundant mitochondria | High glucogen content |
Neuromuscular junction
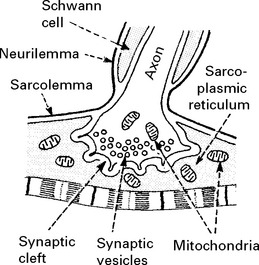
Physiology
Muscle contraction results from the following:
Biochemistry
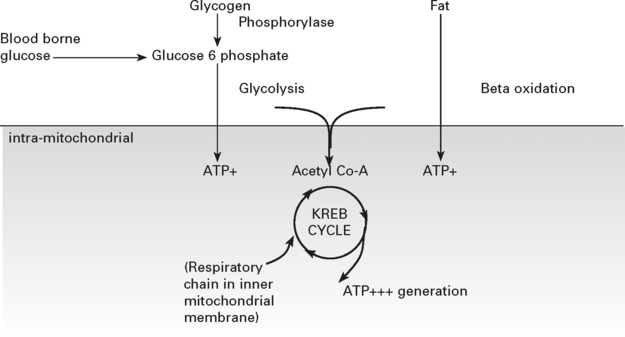
The biochemical pathways yielding energy from the Krebs cycle reactions are dependent on proteins coded for by both the nuclear and mitochondrial genome. Mitochondrial DNA (mtDNA) is present in many copies per mitochondrion, with many mitochondria per cell. The usual state is that all an individual’s mtDNA has the same sequence – homoplasmy – but in the mitochondrial disorders mutations are frequently present in only a proportion of the mtDNA – heteroplasmy. The distribution of these populations is not homogeneous across tissues and these features make the diagnosis of disorders associated with abnormalities of mtDNA difficult when the mutation may not be detected in blood but may be present in varying amounts in muscle or other affected tissues (see page 481).
MUSCLE DISEASE – HISTORY, EXAMINATION AND INVESTIGATIONS
History taking
Examination
Investigations
INHERITED MUSCLE DISORDERS
DUCHENNE DYSTROPHY
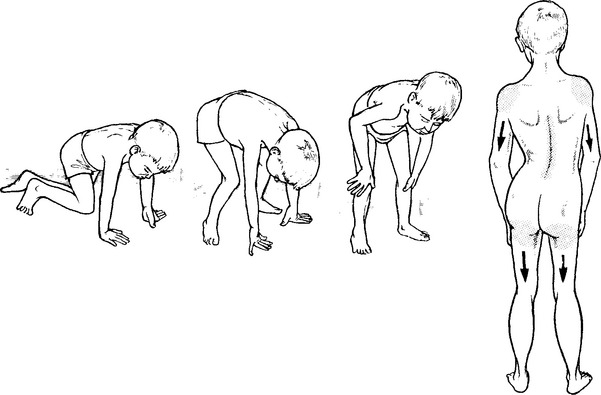
MUSCULAR DYSTROPHIES
DYSTROPHIES WITH PARTICULAR PATTERNS OF WEAKNESS
Facioscapulohumeral (FSH)
Limb girdle syndromes and limb girdle muscular dystrophy (LGMD)
| CATEGORIES | EXAMPLES | SUGGESTIVE FEATURES |
|---|---|---|
| Non-dystrophic genetic myopathies | Desmin myopathy, congenital structural myopathies (nemaline etc.) | Early onset, presence of contractures, often very thin muscles yet only mild weakness |
| Metabolic myopathies | Acid Maltase deficiency, McArdles disease, mitochondrial disorders | Pain, variability, exercise intolerance |
| Endocrine | Hypo-and hyperthyroidism, osteomalacic myopathy, Cushing’s syndrome | Diffuse pattern of weakness, endocrine features may not be prominent |
| Toxic/metabolic | Steroid therapy, alcohol, statins | Should be apparent from history |
| Inflammatory | Polymyositis | See discussion below |
| Limb Girdle Muscular Dystrophy (LGMD) | At least 3 dominant and 9 recessive forms. Precise diagnosis requires specialised investigation | Symmetry, focal involvement of individual muscles, cardiac conduction defects, contractures from early stages |

Clinical features

DYSTROPHIES: GENERAL PRINCIPLES
INFLAMMATORY MYOPATHY
Inflammatory myopathy associated with malignant disease
Sarcoid myopathy – some with this multi-system disease have granulomas in skeletal muscle.
POLYMYOSITIS/DERMATOMYOSITIS
An autoimmune basis for these disorders is supported by:
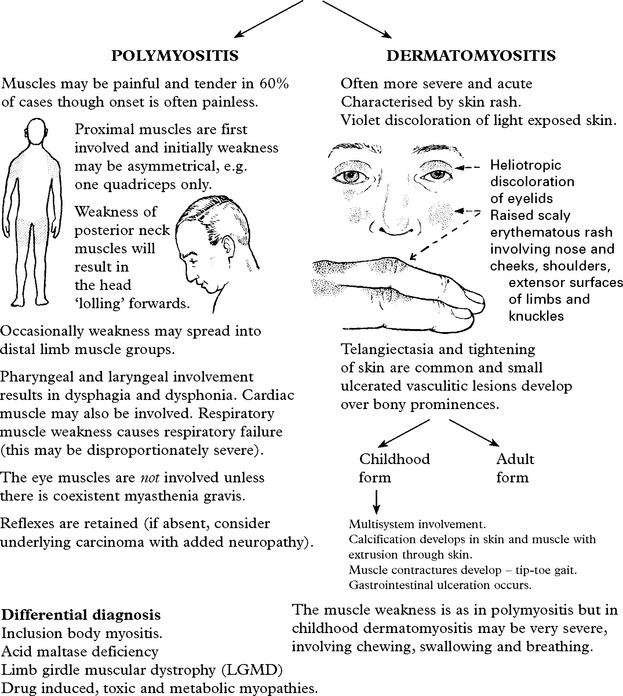
INCLUSION BODY MYOSITIS (IBM)
Investigations
| Muscle enzymes | Circulating antibodies |
|---|---|
| Creatine kinase (CK) is elevated. | e.g. rheumatoid factor, antinuclear factor. Present in 40%. |
| Released from necrotic muscle, it is an indicator of disease activity and severity | |
| Electromyography | Erythrocyte sedimentation rate (ESR) |
| Shows a typical myopathic pattern. | Elevated in most patients. |
ENDOCRINE MYOPATHIES
CHANNELOPATHIES: PERIODIC PARALYSES AND MYOTONIA
Importantly episodes of weakness, with alterations in serum potassium, are most commonly secondary to drugs (e.g. diuretics and corticosteroids) or disorders such as alcoholism, renal and endocrine disease. (See page 478.)
| Hypokalaemic periodic paralysis | Hyperkalaemic periodic paralysis | Paramyotonia congenita |
|---|---|---|
|
The gene has been mapped on chromosome 1, mutations resulting in upset of the dihydropyridine receptor, a voltage-gated calcium channel.
|
| Normokalaemic periodic paralysis | Thyrotoxic periodic paralysis | Congenital myotonia |
|---|---|---|
| There are patients with episodic weakness in whom no alteration in serum potassium can be found. Many are sensitive to the administration of oral potassium salts. Treatment is the same as for the hyperkalaemic form but there is no response to acetazolamide. Muscle biopsy in these patients showed occasional vacuoles and prominent tubular aggregates. | Attacks of paralysis are associated with hypokalaemia and are clinically similar to those of the hypokalaemic form. Mainly occurs in Asians and rarely in non-Asians. The majority of patients experience their first attack in their 30s. There is a marked (20 to 1) male to female predominance. | Dominant form (Thomsen’s disease) and recessive (Becker’s) are both caused by mutation in the chloride channel gene. Myotonia can be triggered by cold, improving with exercise. May have muscle hypertrophy. Treatment with quinine, phenytoin or mexilitene reduces myotonia. |
METABOLIC AND TOXIC MYOPATHIES
Metabolic myopathies
The following disorders are representative but not comprehensive.
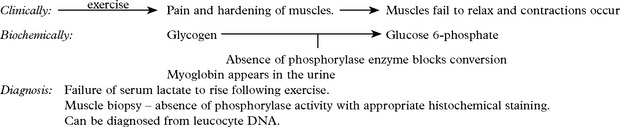
| Diagnosis: |
Treatment with oral fructose may help.
Systemic carnitine deficiency – childhood onset weakness with hypoglycaemic encephalopathy, precipitated by fasting and resembling Reye’s syndrome (page 508). Serum and muscle carnitine levels are low. Biopsy shows an excessive number of lipid droplets in type 1 fibres. The liver, kidney, and heart contain excessive lipid. Cardiomyopathy is fatal.
MITOCHONDRIAL DISORDERS
Certain specific syndromes are recognised though overlap and diversity of phenotype is common.
| CPEO (Chronic progressive external ophthalmoplegia) | MERRF (Myoclonic epilepsy with ragged red fibres) |
MYASTHENIA GRAVIS
Myasthenia gravis is a disorder of neuromuscular transmission characterised by:
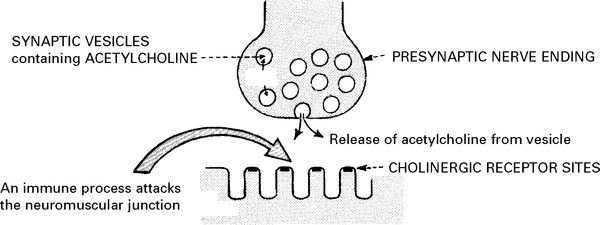
MYASTHENIA GRAVIS – PATHOLOGY
Changes are found in the thymus gland and in muscle.
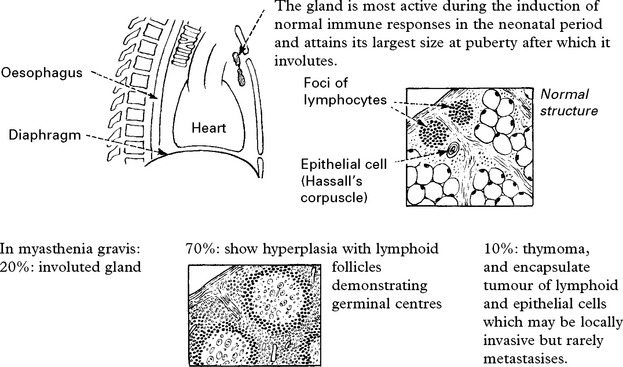
Muscle biopsy may show abnormalities:
CLINICAL FEATURES
Several clinical subdivisions are recognised:

Respiratory muscle involvement accompanies severe illness.
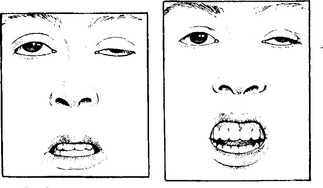

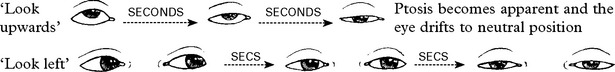
Limb and trunk signs and symptoms
Limb reflexes are often hyperactive and fatigue on repeated testing.
| Natural history: (Before treatment became available) | 10% of patients entered a period of remission of long duration. |
| 20% experienced short periods of remission (1 to several months). | |
| 30% progressed to death. | |
| The remainder showed varying degrees of disability accentuated by exercise. |
MYASTHENIA GRAVIS – DIFFERENTIAL DIAGNOSIS

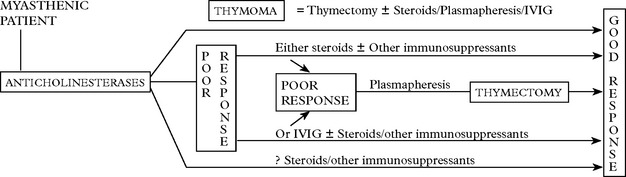
MYASTHENIA GRAVIS – TREATMENT
Anticholinesterase drugs
This is the longest established form of treatment (1930s).
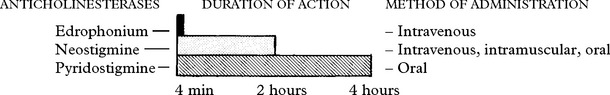
Atropine may mask early warning symptoms of this potential life-threatening state.
NEONATAL form of myasthenia gravis: this develops in a number of infants of myasthenic mothers.