Chapter 6 Local regulation of vascular function
Even in the absence of unusual structural design features like those discussed at the end of the last chapter, there is not always a straightforward relationship between regional microcirculatory blood flow and the applied pressure gradient. In many tissues, changes in perfusion pressure do not cause corresponding changes in flow because local adjustments of microcirculatory resistance tend to maintain blood flow constant (autoregulation). In addition, in some tissues with relatively low metabolic rates the blood flow is distributed unevenly across the capillary bed, with intermittent perfusion of capillaries in different areas (vasomotion). Finally, local vascular conductance is proportional to local tissue metabolism, with increased metabolic rate causing vasodilation and increased blood flow (functional hyperaemia). These three phenomena are due to a mixture of factors, the most universally important of which are the local vasodilator actions of chemicals released from the endothelium, local metabolites and the response of vascular smooth muscle cells to altered transmural pressure.
We will first examine the mechanisms involved in each of these three categories of local control.
ENDOTHELIAL CONTROL OF LOCAL CONDUCTANCE
Identity of endothelium-derived vasoactive factors
Endothelial cells release several factors that affect vascular smooth muscle cell activity. The most studied of these is the vasodilator nitric oxide (NO), endothelial synthesis of which is dependent on free intracellular Ca2+. NO acts by diffusing into the muscle cells and activating guanylate cyclase. There is good evidence that continual release of NO from the endothelium leads to a tonic dilator effect on the vascular smooth muscle, the magnitude of which depends on the balance between rate of release and rate of dispersal into the bloodstream. In addition, increased shear stress on the endothelium increases NO release. This probably constitutes an important factor in regulation of vessel calibre in large conduit arteries (see Chapter 7, p. 81) but, because of the large total cross-sectional area and concomitant low flow velocity in microvascular beds, the effect of shear stress on release of endothelial mediators in arterioles is likely to be much less substantial.
A number of additional biologically active factors of endothelial origin also exist, including EDRFs, that causes muscle cell hyperpolarization (endothelium-derived hyperpolarizing factor, EDHF) and for which there are several candidate molecules, together with the endothelins that cause vasoconstriction. None of these factors have been studied to the same extent as NO and their physiological significance is not yet clear, but it seems probable that at least collectively they may play a significant role in regulation of peripheral resistance (Quyyumi & Ozkor 2006).
Assessment of endothelial control of local conductance
Reactive hyperaemia
If the arterial inflow to a tissue is occluded for more than a few seconds, the conduit and microcirculatory arterial vessels dilate downstream of the occlusion. As a result, release of the occlusion is accompanied by a temporary elevation of local blood flow above the pre-occlusion level, termed reactive hyperaemia. The reactive hyperaemic responses of both conduit and microcirculatory vessels are attenuated after administration of substances that prevent NO production, suggesting that both involve EDRFs and might, therefore, be utilized as indices of endothelial dilator function. It is uncertain whether EDRFs other than NO are implicated.
Reactive hyperaemia is usually elicited in the forearm with occlusion of the brachial artery above the elbow for a period of between 2 and 3 min. In these circumstances, the peak flow immediately after release of occlusion is typically two to four times the pre-occlusion value and returns to this resting level over 30–50 s. Conduit vessel dilation is assessed using ultrasound imaging of brachial artery diameter, and the reactive hyperaemic response is referred to as flow-mediated dilation. Microcirculatory dilation is usually assessed either by measurement of blood flow through forearm or finger using venous occlusion plethysmography (Fig. 6.1) or by laser-Doppler measurement of forearm skin flow.
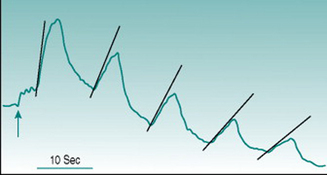
Comparative studies show poor correlation between the magnitudes of large and small vessel responses in different subject populations (Hansell et al 2004), which is perhaps not surprising if we consider the mechanisms likely to underlie dilation in the two vessel types. Assuming that all endothelial cells release EDRFs continuously, local accumulation of these factors during the period of arterial occlusion should have similar dilator consequences in large and small vessels once the normal pressure gradient is re-established. In conduit vessels, however, the increased volume flow will be accompanied by a high rate of shear stress on the endothelium that will increase local EDRF release further. As flow velocity is much lower in the microcirculation, shear stress is not likely to generate factor release at that site. A second complicating factor arises from the fact that cessation of blood flow through a tissue results in local accumulation of dilator metabolites (see Chapter 6, p. 67). Depending on the metabolic rate of the tissue and the duration of arterial occlusion, metabolic dilation in the microcirculation may, therefore, act additively with EDRFs.
Endothelial turnover
The endothelium is turning over continually, with dying cells being shed into the circulation and being replaced by new cells that originate from circulating bone marrow-derived progenitors. These processes may provide additional indices by which the functional status of the endothelium can be assessed at a whole-body level. On the one hand, large numbers of dislodged endothelial cells in the blood stream could indicate reduced viability, consistent with reduced EDRF function. A number of studies have reported elevated numbers of circulating endothelial cells (CECs) in various disease states associated with other evidence of reduced endothelial dilator capacity, including hypertension and diabetes. Conversely, it could be argued that rapid cell turnover would lower the average age of the endothelial population and so reduce the proportion of old, metabolically sub-optimal cells. This possibility is consistent with observations that CECs rise in parallel with endothelial dilator capacity in healthy subjects during aerobic training (see Chapter 11, p. 134).
CEC numbers alone, therefore, provide only ambiguous information on endothelial status and additional markers of endothelial viability must be used concomitantly in order to decide whether high CEC values denote healthy or unhealthy endothelium. One marker that may be useful is Von Willebrand factor, which is released from endothelial cells in response to inflammatory stimuli. Von Willebrand factor levels appear to be elevated in at least most pathological situations where CEC numbers are increased. By contrast, the one study that has compared these parameters with fitness levels in healthy subjects (O’Sullivan 2003) found that the rise in CECs associated with training was not linked to any change in plasma Von Willebrand levels.
Assessment of nitric-oxide involvement in endothelial control of local conductance
Release of nitric oxide
It can be anticipated that NO released from endothelium will diffuse into the bloodstream as well as into the vascular wall, so that plasma levels of NO could provide an index of endothelial dilator function. Detection of NO itself is not practicable, since the NO molecule is metabolized too rapidly to allow blood sampling and assay. However, the main breakdown product of NO, nitrite ion, is stable and recent studies have demonstrated good correlation between serum nitrite levels and other markers of NO-dependent dilation (Rassaf et al 2006).
METABOLIC CONTROL OF LOCAL CONDUCTANCE
All known cellular metabolites, including adenosine nucleotide degradation products, CO2, phosphates, lactate and protons, have dilator actions on vascular smooth muscle. This provides a mechanism by which local vascular conductance can, in theory, be matched automatically to tissue metabolic demands. At least some of the activity of certain metabolites (for example adenine molecules) is produced via their stimulating release of NO and other endothelial dilator factors; other metabolites, such as protons, appear to act directly on the vascular smooth muscle. Since the balance between different factors depends on the precise metabolic pathways utilized in a tissue, the dilator contributions of each metabolite vary between different organ systems. Identifying the relative importance of different factors in particular situations may be important since variations in those factors that are most potent, or in highest concentration, may be the most reliable index of hyperaemia. Because of this, much debate has taken place in the research literature as regards the roles of specific factors in, for example skeletal muscle (Clifford & Hellsten 2004).
In terms of functional significance, on the other hand, the evidence to date suggests that no single identified factor is overwhelmingly important in any tissue (see Functional hyperaemia during exercise, p. 72, for further discussion). This does not necessarily imply that another unknown factor is the main contributor. The non-specificity of functional dilatation can be ascribed partly to the additive dilator effects of all metabolites present. It also reflects the fact that absolute interstitial osmolality is a powerful vasodilator influence regardless of the identity of the osmotically active particles. In addition, it is possible that certain metabolites may have far greater biological efficacy when present at the outer surface of the vascular media than when presented in the bloodstream as is common in experimental studies.
The effect of metabolites is amplified in some tissues by several other vasodilator influences that are proportional in intensity to tissue activity. The most widespread of these is mediated by a population of ion channels on vascular smooth muscle cells that open in response to locally reduced dissolved oxygen tension; that is, that function as hypoxia receptors (Quayle et al 2006). Opening of these channels results in cell hyperpolarization and relaxation, so falls in interstitial oxygenation following increased tissue oxygen consumption have a similar effect to that of increased metabolite accumulation. Interestingly, the hypoxia receptors in pulmonary arterioles produce local vasoconstriction rather than dilatation, a feature that is very important in regulation of gas exchange in the lung (see Chapter 8, p. 96).
Additional dilator factors are liberated during metabolic activity from particular tissue cell types. These include potassium ions, NO and PGs in the case of striated muscle, short-chain peptides termed kinins in the case of exocrine glands and secretomotor peptide hormones in the case of the gastrointestinal tract. Further information on these factors and how they may interact during the circulatory adjustments to exercise will be found in the section Functional hyperaemia during exercise, p. 72.
MYOGENIC CONTROL OF LOCAL CONDUCTANCE
Stretching a vascular smooth muscle cell causes graded depolarization of the cell membrane and this opens voltage-dependent calcium channels. As a result, the level of free intracellular Ca2+ rises and the cell contracts; a phenomenon known as the myogenic response. Experimental studies using isolated arterial segments suggest that the amount of membrane stretch in arterioles due to normal levels of intravascular pressure depolarizes the cells by 20–30 mV, which is sufficient to cause 40–50% constriction relative to their passive diameter (Dora 2005). Thus, a substantial proportion of ongoing total peripheral resistance related to blood pressure maintenance is due to the myogenic response, independent of vasoconstrictor influences by sympathetic nerves or circulating hormones.
As well as contributing to resting peripheral resistance, the myogenic response is elicited whenever intravascular pressure changes. An increase in arterial perfusion pressure will result in depolarization and arteriolar constriction and, conversely, decreased perfusion pressure will cause hyperpolarization and dilation. The molecular details of the process that monitors membrane distortion are still unclear. The end-result, however, is that the contractile state of the cell does not alter to maintain a stable vessel diameter in the face of altered perfusion pressure. Rather, increased pressure results in a net decrease in diameter and decreased pressure in a net increase. This implies that what is being monitored is not membrane distortion itself, but wall stress.
AUTOREGULATION
From the pressure/flow relationships discussed in Chapter 5, you would expect changes in perfusion pressure gradient through a regional vascular bed to produce approximately proportionate changes in blood flow. In skeletal and cardiac muscles, brain, gastrointestinal tract and kidney, however, this occurs only with extremely low or high pressures. Over the range typical of most normal circumstances (from somewhere around 60–70 mmHg up to around 150 mmHg) flow changes transiently when pressure is altered but returns over 20–60 s to its previous value, so that flow is effectively independent of pressure over this so-called autoregulatory pressure range (Fig. 6.2).
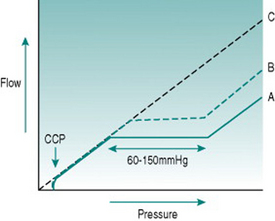
In cardiac and skeletal muscles, brain and gastrointestinal tract, changes in local metabolite concentration appear to be the dominant mechanism. At any time, absolute microvascular resistance reflects the ambient metabolite concentration. This is a balance between metabolite production and the rate at which these molecules are being washed out of the tissue by the blood flow. If perfusion pressure is altered, the rate of washout will change, resulting in accumulation of metabolites and reduced vascular resistance if perfusion pressure falls or depletion of metabolites and vasoconstriction if pressure rises.
VASOMOTION
Although autoregulation can maintain stable total organ blood flow despite altered perfusion pressure, provided that metabolic rate remains constant, this does not necessarily mean that the organ is perfused evenly. When the tissue cells are inactive, as in resting skeletal muscles, blood flow may be so low that intravascular pressure within the microcirculation is not sufficient to prevent some arteriolar vessels collapsing under the constraints of Laplace’s law (see Chapter 5, p. 52). Under these circumstances there is intermittent perfusion of neighbouring portions of the capillary bed, a process known as vasomotion.
The phenomenon of vasomotion relies on the fact that absolute critical closing pressure (CCP) is inversely related to the local concentration of interstitial metabolites. The sequence of events involved is most easily visualized by simplifying the vasculature to one arteriole giving rise to two sequential metarterioles and capillaries (Fig. 6.3) (in real life, three or more sequential metarterioles would be present). Imagine that, initially, the interstitial metabolite concentration is homogenous so that both metarterioles have similar CCPs. That which is more distally located along the parent arteriole will be most likely to close, since perfusion pressure must be lower in that region. A situation will, therefore, be created where the upstream capillary is perfused while the more distal capillary has no flow (Fig 6.3A). This will progressively wash metabolites out of the interstitium around the perfused capillary and allow metabolite accumulation around the non-perfused capillary (Fig. 6.3B). In parallel, the relaxant effect of the metabolites will change so that CCP of the perfused metarteriole rises and that of the non-perfused vessel falls. Eventually, CCP in the non-perfused vessel falls below the perfusion pressure so that flow is re-established, while CCP in the perfused vessel will rise above its perfusion pressure and so that metarteriole will shut (Fig. 6.3C–D).
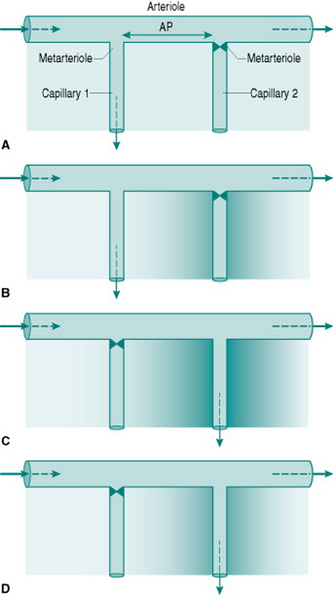
It is worth remembering that when vascular smooth muscle is maximally relaxed, CCP approaches zero. Thus, during exercise, the metarterioles in active skeletal muscles will all be open despite the presence of sympathetic vasoconstriction in the arteriolar bed further upstream. As is discussed in Chapter 8 (p. 94), this allows a substantial increase in nutritional muscle perfusion without the need for abolition of local sympathetic tone.
FUNCTIONAL HYPERAEMIA DURING EXERCISE
Skeletal muscle circulation
During exercise, nutritional blood flow through the active skeletal muscles rises by 10–20-fold from its resting value of 2–5 ml/min/100 g. This functional hyperaemia reaches a plateau within 2 s of muscle contraction beginning and is maintained throughout the exercise. Although a number of dilator metabolites are known to be produced within contracting muscles, including protons, adenine breakdown products and lactate, none of these substances alone appears to be the primary mediator of the functional hyperaemia, since in no case does the timecourse and concentration profile of metabolite appearance in the interstitium parallel exactly the timecourse and magnitude of hyperaemia (Clifford & Hellsten 2004).
Other sets of factors also help bring about hyperaemia in exercising muscle, although it is not possible to estimate how much each contribute to blood flow in different patterns of exercise. One is circulating adrenaline (epinephrine), which may activate dilator β-adrenoceptors on the arteriolar muscle, although the effect of this is limited by the presence also of α-adrenoceptors (see Chapter 7, p. 85). As well, contraction leads to release from muscle cells of NO and PGs, more prominently in oxidative than in glycolytic fibres. Thirdly, there is a direct relaxant effect on vascular smooth muscle of the increased tissue temperature associated with increased metabolism (McMeeken & Bell 1990).
These effects on arteriolar tone are supplemented by dilation of the vessels immediately upstream, including the most distal conducting arteries. Despite the arterioles contributing most to regional vascular resistance, arterial dilation reduces regional resistance somewhat and so enhances absolute tissue perfusion. The dilation of conducting arteries appears to involve retrograde propagation of a relaxing signal from the arteriolar level by direct coupling between cells in the vessel wall. The mechanisms underlying this conducted (or propagated) dilation are not well-defined, but may involve endothelial NO and ATP-sensitive potassium channel activation (Takano et al 2005). Finally, the increased blood flow due to all these factors will increase endothelial shear stress in the conduit arteries supplying the active muscles, producing local release of NO and PGs, and reducing overall regional vascular resistance further.
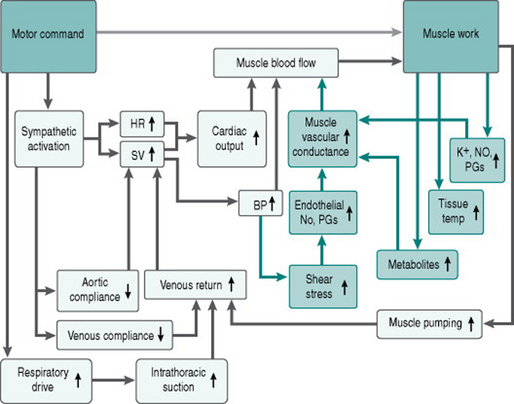
Here is our flow chart (Fig. 6.4) of the matrix of responses to acute exercise, updated to include the effects of these local vasodilator processes.
Coronary circulation
Local processes are involved also in the hyperaemic effects of exercise on perfusion of the coronary and cutaneous circulations. The coronary circulation is of course in a state of functional hyperaemia even under non-exercising conditions, reflecting the fact that cardiac muscle is continually active. This means that there is no spare capillary capacity to be drawn on when cardiac workload is increased as evidenced by the fact that oxygen extraction in the coronary capillary bed is always around 60%. By contrast, extraction of oxygen in tissues such as skeletal muscle while at rest, where only a fraction of the capillary bed is perfused at any moment (see Vasomotion, above), is around 30–40%. Under these circumstances, coronary blood flow cannot rise during exercise to the same extent as that in skeletal muscle, the absolute change being proportional to the rise in cardiac output; that is, reaching a ceiling of four to fivefold the resting value in an untrained subject.
β-adrenoceptor activation contributes around 25% of this hyperaemia, the effect being more profound than in skeletal muscle because coronary smooth muscle has a higher ratio of β- to α-adrenoceptors. The magnitude of this sympathetic effect alone is probably sufficient to account for the initial rapid rise in coronary perfusion that is needed to maintain aerobic metabolism over the first few seconds of exercise. The remaining, dominant fraction of the sustained flow increase is presumably mediated primarily by myocardial metabolites and endothelial factors, as in skeletal muscle. However, again as with skeletal muscle, it is clear that no one identified factor is responsible (Tune et al 2004). Assuming that the full response involves cooperative effects of a range of mediators is simpler than postulating that a potent, as-yet undiscovered dilator process exists.
Skin circulation
Onset of heat-loss processes during sustained exercise involves withdrawal of sympathetic vasomotor drive to the resistance vessels of the skin and a substantial rise in cutaneous blood flow. This passive neural mechanism, however, accounts only for around 50% of the level to which skin blood flow rises during intensive exercise. The remainder of the cutaneous hyperaemic response is secondary to activation of eccrine sweat glands. In these and in other exocrine glands, the secretory process results in liberation of short-chain peptides of the kinin group (bradykinin and kallidin) into the gland interstitium. Kinins are potent vasodilators that act at least partly by stimulating release of endothelial dilator factors (Quyumi & Ozkor 2006). As well as being vasodilators, kinins increase capillary permeability and this facilitates movement of water and electrolytes out of the plasma into the secretory cells.
Splanchnic circulation
Barry MA. Effects of smoking and ageing on cardiovascular function. University of Dublin, 2003. PhD thesis
Clifford PS, Hellsten Y. Vasodilatory mechanisms in contracting skeletal muscle. Journal of Applied Physiology. 2004;97:393-403.
Dora KA. Does arterial myogenic tone determine blood flow in vivo? American Journal of Physiology. 2005;289:H1323-H1325.
Hansell J, Hemareh L, Agewall S, Norman M. Non-invasive assessment of endothelial function relation between vasodilatory responses in skin microcirculation and brachial artery. Clinical Physiology and Functional Imaging. 2004;24:317-322.
McMeeken JM, Bell C. Microwave irradiation of the human forearm and hand. Physiotherapy, Theory Practice. 1990;6:171-176.
O’Sullivan SE. The effects of exercise training on markers of endothelial function in young healthy men. International Journal of Sports Medicine. 2003;24:404-409.
Quayle JM, Turner MR, Burrell HE, Kamishima T. Effects of hypoxia, anoxia, and metabolic inhibitors on KATP channels in rat femoral artery myocytes. American Journal of the Heart, Circulation and Physiology. 2006;291:H71-H80.
Quyyumi AA, Ozkor M. Vasodilation by hyperpolarization: beyond NO. Hypertension. 2006;48:1023-1025.
Rassaf T, Heiss C, Hendgen-Cotta U, et al. Plasma nitrite reserve and endothelial function in the human forearm circulation. Free Radical Biology and Medicine. 2006;41:295-301.
Takano H, Dora KA, Garland CJ. Spreading vasodilatation in resistance arteries. Journal of Smooth Muscle Research. 2005;41:303-311.
Tune JD, Gorman MW, Feigl EO. Matching coronary blood flow to myocardial oxygen consumption. Journal of Applied Physiology. 2004;97:404-415.