[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 86 Liver Disease Caused by Drugs
DEFINITIONS AND IMPORTANCE
The term drug-induced liver disease should be confined to cases in which the nature of liver injury has been characterized histologically. With the exception of acetaminophen, anticancer drugs, and some botanical or industrial hepatotoxins (see Chapter 87), most cases of drug-induced liver disease represent adverse drug reactions or hepatic drug reactions. These effects are noxious and unintentional and occur at doses recommended for prophylaxis or therapy. The latent period is longer (typically one week to three or six months) than that for direct hepatotoxins (hours to a few days), and extrahepatic features of drug hypersensitivity may be present.
Although drug-induced liver disease is a relatively uncommon cause of jaundice or acute hepatitis in the community, it is an important cause of more severe types of acute liver disease, particularly among older people (see Epidemiology). The overall mortality rate among patients hospitalized for drug-induced liver injury is approximately 10%1 but varies greatly for individual drugs.2,3 The reported frequencies of individual hepatic drug reactions are often underestimated because of the inadequacy of spontaneous reporting by physicians and pharmacists.2,3 With more reliable prospective and epidemiologic techniques, the frequency (or risk) of most types of drug-induced liver disease is between one per 10,000 and one per 100,000 persons exposed.4 Because these responses to drug exposure are clearly rare and unpredictable, they are often termed idiosyncratic drug reactions. Their rarity blunts diagnostic acumen because most clinicians will see few, if any, cases and therefore do not have an appropriate level of clinical suspicion. This concern applies especially to complementary and alternative medicine (CAM), as discussed in Chapter 87. Failure to withdraw the causative agent after the onset of symptoms of drug hepatitis or inadvertent reexposure to such a drug is a common and avoidable factor in acute liver failure attributable to drug-induced liver injury.5–8 Another challenge is that hepatic drug reactions produce an array of clinical syndromes and pathologic findings that mimic known hepatobiliary diseases. Furthermore, although individual agents (and some drug classes) typically produce a characteristic “signature syndrome,” they can also be associated with other and sometimes multiple clinicopathological syndromes.
Drug-induced liver injury is the most common reason for withdrawal of an approved drug from the market. The subject therefore has medicoeconomic, legal, and regulatory ramifications. Because of the low frequency of most types of idiosyncratic drug reactions that involve the liver, serious hepatotoxicity is not usually detected until post-marketing surveillance is conducted. Historically, drugs that have developed a reputation for potential hepatotoxicity usually have been replaced by more acceptable alternatives. Examples include troglitazone, the prototypic thiazolidinedione and bromfenac, a nonsteroidal anti-inflammatory drug (NSAID), both of which were withdrawn from the market because of several cases of fatal acute liver failure.5–9
The burgeoning number of available conventional medications and CAM preparations now includes many hundreds that can be cited as rare causes of drug-induced liver disease. This increasing number of potentially causative agents poses several challenges to clinicians,5–10 including concern about what constitutes an adequate level of patient information at the time a drug is prescribed and the reliability of evidence linking an individual agent to a particular type of liver injury.5,11–13 Another development is the appreciation that drug toxicity, in the context of complex medical situations, can interact with other causes of liver injury. Noteworthy examples of such situations are bone marrow transplantation; cancer chemotherapy; highly active antiretroviral therapy (HAART) for human immunodeficiency virus (HIV) infection and the acquired immunodeficiency syndrome (AIDS); use of antituberculosis drugs in patients with chronic viral hepatitis; rifampin hepatitis in patients with primary biliary cirrhosis (Chapter 89); nonalcoholic fatty liver disease (NAFLD)—particularly nonalcoholic steatohepatitis (NASH)—precipitated by tamoxifen; and possibly other drugs in overweight persons with type 2 diabetes mellitus and the metabolic syndrome.
EPIDEMIOLOGY
Frequency or risk—the number of adverse reactions for a given number of persons exposed—is the best term for expressing how common a drug reaction is. Time-dependent terms such as incidence and prevalence are not appropriate for drug reactions because the frequency is not linearly related to the duration of exposure. For most reactions, the onset occurs within a relatively short exposure time, or latent period, although some rarer types of chronic liver disease occur after many months or years. The frequency of drug-induced liver disease is usually based on the reported rate of drug reactions; such reports are usually a voluntary part of post-marketing surveillance and are submitted to pharmaceutical companies or adverse drug reaction monitoring bodies. In the United States, following approval by the U.S. Food and Drug Administration (FDA), pharmaceutical companies are required to report serious adverse events (any incident resulting in death, a threat to life, hospitalization, or permanent disability [Code of Federal Regulations]). Surveillance becomes a more passive process, however, when a drug is approved for marketing, and physicians and pharmacists are encouraged to file voluntary written reports through the MediWatch program. Similar systems operate in most industrialized countries. Nevertheless, MediWatch receives reports for fewer than 10% of adverse drug reactions,2 and in France fewer than 6% of hepatic adverse drug reactions are reported.3 The situation may be somewhat better in Sweden, but the annual reported incidence of adverse drug reactions of 2.2 per 100,000 in the population over the age of 15 is still much lower than the predicted incidence of 14 per 100,000.3 A prospective surveillance study in Spain measured the annual incidence of drug-related acute serious liver disease as 7.4 per million inhabitants.4
CASE DEFINITION: WHICH AGENT?
At least 300 agents have been implicated in drug-induced liver injury.10 The evidence for most drugs, however, is confined to individual or small numbers of case reports, especially in letters to scientific journals or to regulatory authorities, or small observational series. Therefore, for most agents, the evidence that they could cause liver injury is circumstantial and incomplete. Reports often lack pathologic definition, full exclusion of other disorders (for older reports), and logistic imputation of causality, especially with respect to temporal associations (see Diagnosis).5,9,10 Overall, probably fewer than 50 agents have been implicated reliably as causes of drug-induced liver disease. In general, agents used most commonly in clinical practice and in the community, including antimicrobials, antineoplastic agents, and NSAIDs, are those that have been implicated most often in drug-induced liver injury in larger series. The challenge of identifying the culprit drug among multiple candidates is discussed later.1,4,5,6,11
FREQUENCIES OF HEPATIC DRUG REACTIONS
Because of incomplete reporting, frequencies of hepatic drug reactions may often be underestimated. These estimated frequencies are also crude indicators of risk because of the inherent inaccuracies of case definitions (see Diagnosis)5,9,10 and because case recognition and reporting depend on the skill and motivation of observers. The increased interest of prescribers when initial cases of drug-induced liver disease have been described, together with inappropriate prescribing (e.g., prolonged use of bromfenac, which was approved only for seven days of use, and overprescribing of flucloxacillin and amoxicillin-clavulanic acid in some countries) can give rise to apparent “mini-epidemics.” More appropriate epidemiologic methods applied to hepatotoxicity have included prescription event monitoring, record linkage, and case-control studies. Prescription event monitoring and record linkage have been used to estimate the frequency of liver injury with some antimicrobials (erythromycins, sulfonamides, tetracyclines, flucloxacillin, amoxicillin-clavulanate) and NSAIDs.11
Epidemiologic studies confirm the rarity of drug-induced liver disease with currently used agents. For NSAIDs, the risk of liver injury is between 1 and 10 per 100,000 individuals exposed1,4,5,6; amoxicillin-clavulanic acid has been associated with cholestatic hepatitis in 1 to 2 per 100,000 exposed persons1,4,5,6,8; and low-dose tetracyclines have caused hepatotoxicity in less than one case per million persons exposed.1,4–6 The frequency of liver injury may be higher for agents that exert a metabolic type of hepatotoxicity. For example, isoniazid causes liver injury in up to 2% of persons exposed; the risk depends on the patient’s age and gender, concomitant exposure to other agents, and presence of hepatitis B virus (HBV) and possibly hepatitis C virus (HCV) infections.12 For some drugs in which other host factors play an etiopathogenic role, case-control studies have been used to define attributable risk. Examples include the implication of aspirin in Reye’s syndrome and oral contraceptives in liver tumors and hepatic vein thrombosis.
A relationship may exist between the frequency and severity of serum ALT elevations that indicate liver injury and the risk of severe hepatotoxicity. This relationship was proposed in the 1970s by the late Hyman Zimmerman.6 According to “Hy’s rule,” elevations of serum ALT levels to eight-fold or more above the upper limit of normal or associated increases in the serum bilirubin concentration indicate a potential for the drug to cause acute liver failure at a rate of about 10% of the number of cases of jaundice. Therefore, if two cases of jaundice associated with drug-induced liver injury are observed in a total phase 3 clinical trial experience of 2500 patients, approximately one case of acute liver failure would be expected for every 12,500 subjects who were prescribed the drug during the marketing phase.
IMPORTANCE OF DRUGS AS A CAUSE OF LIVER DISEASE
Hepatotoxicity accounts for less than 5% of cases of jaundice or acute hepatitis in the community and for even fewer cases of chronic liver disease5,6; however, drugs are an important cause of more severe types of liver disease and for liver disease in older people. They account for 10% of cases of severe hepatitis admitted to the hospital in France6 and for 43% of cases of hepatitis among patients 50 years of age or older.7 Drugs account for more than half of the cases of acute liver failure referred to special units in the United States7 and between 20% and 75% of cases of acute liver failure in other industrialized countries.4,7 The pattern of agents incriminated varies among countries; for example, herbal medicines are a relatively more common cause in Asian countries than in other countries (Chapter 87).
RISK FACTORS
For dose-dependent hepatotoxins such as acetaminophen and methotrexate and for some idiosyncratic reactions that are partly dependent on dose (e.g., bromfenac, tetracyclines, dantrolene, tacrine, oxypenicillins), the factors that influence the risk of drug-induced liver disease include the dose of the drug, blood level of the drug, and duration of intake. For idiosyncratic reactions, however, host determinants appear to be central to liver injury. The most critical determinant is likely to be genetic predisposition, but other “constitutional” and environmental factors can influence the risk of liver injury, as summarized in Table 86-1. The most important factors are age,4 gender, exposure to other substances, a history or family history of previous drug reactions, other risk factors for liver disease, and concomitant medical disorders.
Table 86-1 Factors Influencing the Risk of Liver Diseases Caused by Drugs
| FACTOR | EXAMPLES OF DRUGS AFFECTED | INFLUENCE |
|---|---|---|
| Age | Isoniazid, nitrofurantoin, halothane, troglitazone | Age >60 years: increased frequency, increased severity |
| Valproic acid, salicylates | More common in children | |
| Gender | Halothane, minocycline, nitrofurantoin | More common in women, especially with chronic hepatitis |
| Amoxicillin-clavulanic acid, azathioprine | More common in men | |
| Dose | Acetaminophen, aspirin; some herbal medicines | Blood levels are directly related to the risk of hepatotoxicity |
| Tetracycline, tacrine, oxypenicillins | Idiosyncratic reactions, but partial relationship to dose | |
| Methotrexate, vitamin A | Total dose, dosing frequency, duration of exposure is related to the risk of hepatic fibrosis | |
| Genetic factors | Halothane, phenytoin, sulfonamides | Multiple cases in families |
| Amoxicillin-clavulanic acid | Strong HLA association | |
| Valproic acid | Familial cases, association with mitochondrial enzyme deficiencies | |
| History of other drug reactions | Isoflurane, halothane, enflurane | Instances of cross-sensitivity have been reported among members of each drug class but are rare |
| Erythromycins | ||
| Diclofenac, ibuprofen, tiaprofenic acid | ||
| Sulfonamides, COX-2 inhibitors | ||
| Other drugs | Acetaminophen | Isoniazid, zidovudine, and phenytoin lower dose threshold and increase severity of hepatotoxicity |
| Valproic acid | Other antiepileptics increase risk of hepatotoxicity | |
| Anticancer drugs | Interactive vascular toxicity | |
| Excessive alcohol use | Acetaminophen hepatotoxicity | Lowered dose threshold, poorer outcome |
| Isoniazid, methotrexate | Increased risk of liver injury, hepatic fibrosis | |
| Nutritional status: | ||
| Obesity | Halothane, troglitazone, tamoxifen, methotrexate | Increased risk of liver injury; hepatic fibrosis |
| Fasting | Acetaminophen | Increased risk of hepatotoxicity |
| Preexisting liver disease | Hycanthone, pemoline | Increased risk of liver injury |
| Antituberculosis drugs, ibuprofen | Increased risk of liver injury with chronic hepatitis B and C | |
| Other diseases/conditions: | ||
| Diabetes mellitus | Methotrexate | Increased risk of hepatic fibrosis |
| HIV infection/AIDS | Sulfonamides | Increased risk of hypersensitivity |
| Renal failure | Tetracycline, methotrexate | Increased risk of liver injury, hepatic fibrosis |
| Organ transplantation | Azathioprine, thioguanine, busulfan | Increased risk of vascular toxicity |
AIDS, acquired immunodeficiency syndrome; COX-2, cyclooxygenase-2; HIV, human immunodeficiency virus; HLA, human leukocyte antigen.
Genetic Factors
Genetic determinants predispose to drug-induced liver disease,13 as they do for other types of drug reaction, such as penicillin allergy. Atopic patients have been thought to have an increased risk of some types of drug hepatitis, but this increase in risk has not been proved. Genetic factors determine the activity of drug-activating and antioxidant pathways, encode pathways of canalicular bile secretion, and modulate the immune response, tissue stress responses, and cell death pathways (see Chapter 72). Documented examples of drugs associated with a familial predisposition to adverse hepatic drug reactions are few and include valproic acid and phenytoin.5,8,13 Inherited mitochondrial diseases are a risk factor for valproic acid–induced hepatotoxicity.14 Some forms of drug-induced liver disease, particularly drug-induced hepatitis and granulomatous reactions, can be associated with the reactive metabolite syndrome (see later).
Weak associations have been reported between specific human leukocyte antigen (HLA) haplotypes and some types of drug-induced liver disease. Andrade and colleagues13 found positive associations between the class II HLA haplotype and cholestatic or mixed liver damage for some drugs. They suggested that no specific HLA allele predisposed to the overall risk of drug-induced liver disease but that the pattern of liver injury could be influenced by these genetic determinants. Other investigators have found stronger associations between the HLA haplotype and cholestatic reactions to amoxicillin-clavulanic acid and ticlopidine (see later).13,15
Age
Most hepatic drug reactions are more common in adults than in children. Exceptions include valproic acid hepatotoxicity, which is most common in infants younger than three years of age and rare in adults, and Reye’s syndrome, in which salicylates play a key role.16,17 As discussed later, both may be examples of mitochondrial toxicity.14 In adults, the risk of isoniazid-associated hepatotoxicity is greater in persons older than 40 years of age. Similar observations have been made for nitrofurantoin, halothane, etretinate, diclofenac, and troglitazone.1,4,5,10 The increased frequency of adverse drug reactions in older subjects is largely the result of increased exposure, the use of multiple agents, and altered drug disposition.1,4,5,10 In addition, clinical severity of hepatotoxicity increases strikingly with age, as exemplified by fatal reactions to isoniazid and halothane.
Gender
Women are particularly predisposed to drug-induced hepatitis, a difference that cannot be attributed simply to increased exposure. Examples include toxicity caused by halothane, nitrofurantoin, sulfonamides, flucloxacillin, minocycline, and troglitazone.5,6 Drug-induced chronic hepatitis caused by nitrofurantoin, diclofenac, or minocycline has an even more pronounced female preponderance.6 Conversely, equal sex frequency or even male preponderance is common for some drug reactions characterized by cholestasis, as for amoxicillin-clavulanic acid. Azathioprine-induced liver disease is more likely to develop in male renal transplant recipients than in female recipients.18
Concomitant Exposure to Other Agents
Patients who are taking multiple drugs are more likely to experience an adverse reaction than those who are taking one agent.5,9,10 The mechanisms include enhanced cytochrome P450 (CYP)-mediated metabolism of the second drug to a toxic intermediate (see later). Examples discussed later include toxicity caused by acetaminophen, isoniazid, valproic acid, other anticonvulsants, and anticancer drugs. Alternatively, drugs may alter the disposition of other agents by reducing bile flow or competing with canalicular pathways for biliary excretion (phase 3 drug elimination) (see later). This mechanism may account for apparent interactions between oral contraceptive steroids and other drugs to produce cholestasis. Drugs or their metabolites may also interact in mechanisms of cellular toxicity and cell death that involve mitochondrial injury, intracellular signaling pathways, activation of transcription factors, and regulation of hepatic genes involved in controlling the response to stress and injury that triggers pro-inflammatory and cell death processes.19,20
Previous Drug Reactions
A history of an adverse drug reaction generally increases the risk of reactions to the same drug and also to some other agents (see later). Nevertheless, instances of cross-sensitivity to related agents in cases of drug-induced liver disease are surprisingly uncommon. Examples of cross-sensitivity between drugs (or drug classes) include the haloalkane anesthetics (see Chapter 87), erythromycins, phenothiazines and tricyclic antidepressants, isoniazid and pyrazinamide, sulfonamides and other sulfur-containing compounds (e.g., some clyclooxygenase-2 [COX-2] inhibitors), and some NSAIDs. A crucial point is that a previous reaction to the same drug is a major risk factor for an increase in the severity of drug-induced liver injury.6
Nutritional Status
Obesity is strongly associated with the risk of halothane hepatitis (see Chapter 87) and appears to be an independent risk factor for NASH and hepatic fibrosis in persons taking methotrexate or tamoxifen. Fasting also predisposes to acetaminophen hepatotoxicity,21 and a role for undernutrition has been proposed in isoniazid hepatotoxicity.22
Preexisting Liver Disease
In general, liver diseases such as alcoholic cirrhosis and cholestasis do not predispose to adverse hepatic reactions. Exceptions include toxicity to some anticancer drugs, niacin, pemoline, and hycanthone. Preexisting liver disease is a critical determinant of methotrexate-induced hepatic fibrosis (discussed later). Patients with chronic HBV infection12 and possibly those with chronic HCV infection or HIV/AIDS appear to be at heightened risk of liver injury during anti-tuberculosis or HAART therapy,23 after exposure to ibuprofen and possibly other NSAIDs, after myeloablative therapy in preparation for bone marrow transplantation (resulting in sinusoidal obstruction syndrome [see later]),24 and possibly after taking antiandrogens, such as flutamide and cyproterone acetate.25 A particularly strong association has been observed between HCV infection (present in 33% of patients with HIV/AIDS) and the risk of liver injury during HAART; the risk may be increased 2- to 10-fold.26–30
Other Diseases
Rheumatoid arthritis appears to increase the risk of salicylate hepatotoxicity, and a curious, unexplained observation is that hepatitis associated with sulfasalazine appears to be more common in patients with rheumatoid arthritis than in those with inflammatory bowel disease.8–10,31,32 Diabetes mellitus, obesity, and chronic kidney disease predispose to methotrexate-induced hepatic fibrosis, whereas HIV/AIDS confers a heightened risk of sulfonamide hypersensitivity.31–33 A retrospective cohort study of five health maintenance organizations found that the age- and sex-standardized incidence of drug-induced acute liver failure in patients with diabetes mellitus was 0.08 to 0.15 per 1000 person-years, irrespective of the therapeutic agent used (the number using troglitazone was small); the incidence was highest (approximately 0.3 per 1000) during the first six months of exposure.32 Renal transplantation is a risk factor for azathioprine-associated vascular injury, whereas kidney disease predisposes to tetracycline-induced fatty liver.6 Finally, sinusoidal obstruction syndrome induced by anticancer drugs is more common after bone marrow transplantation24 and in persons with HCV infection.5,6,8–10,26
PATHOPHYSIOLOGY
PATHWAYS OF DRUG METABOLISM
As reviewed elsewhere,5,34 phase 1 pathways of drug metabolism include oxidation, reduction, and hydrolytic reactions. The products can be readily conjugated or excreted without further modification.
Cytochrome P450
Most type 1 reactions are catalyzed by microsomal drug oxidases, the key component of which is a hemoprotein of the CYP gene superfamily. The apparent promiscuity of drug oxidases toward drugs, environmental toxins, steroid hormones, lipids, and bile acids results from the existence of multiple closely related CYP proteins. More than 20 CYP enzymes are present in the human liver.34
The reaction cycle involves binding of molecular oxygen to the iron in the heme prosthetic group, with subsequent reduction of oxygen by acceptance of an electron from nicotinamide-adenine dinucleotide phosphate (NADPH) cytochrome P450 reductase, a flavoprotein reductase. The resulting “activated oxygen” is incorporated into the drug or another lipophilic compound. Reduction of oxygen and insertion into a drug substrate (“mixed function oxidation”) can result in formation of chemically reactive intermediates, including free radicals, electrophilic “oxy-intermediates” (e.g., unstable epoxides, quinone imines), and reduced (and therefore reactive) oxygen species (ROS). The quintessential example is the CYP2E1-catalyzed metabolite of acetaminophen, N-acetyl-p-benzoquinone imine (NAPQI), an oxidizing and arylating metabolite that is responsible for liver injury associated with acetaminophen hepatotoxicity. Other quinone compounds are potential reactive metabolites of troglitazone, quinine, and methyldopa. Epoxide metabolites of diterpenoids may be hepatotoxic products of the hepatic metabolism of some plant toxins (see Chapter 87).35 ROS have broad significance in the production of tissue injury, particularly by contributing to the production of oxidative stress and triggering tissue stress responses and cell death pathways, as discussed later.
The hepatic content of CYP proteins is higher in acinar zone 3 (see Chapter 71). Localization of CYP2E1 is usually confined to a narrow rim of hepatocytes 1 to 2 cells thick around the terminal hepatic venule. This finding explains in part the zonality of hepatic lesions produced by drugs and toxins, such as acetaminophen and carbon tetrachloride, which are converted to reactive metabolites.
Genetic and Environmental Determinants of Cytochrome P450 Enzymes
Pharmacogenetics and Polymorphisms of Cytochrome P450 Expression
The hepatic expression of each CYP enzyme is genetically determined. This finding largely explains the four-fold or greater differences in rates of drug metabolism among healthy subjects. Some CYPs, particularly minor forms, are also subject to polymorphic inheritance; therefore, occasional persons completely lack the encoded protein.34 One example is CYP2D6, the enzyme responsible for the metabolism of debrisoquine and perhexiline. Poor metabolizers lack CYP2D6 and accumulate perhexiline when given usual doses; lack of CYP2D6 is the critical determinant in serious adverse effects of perhexiline, including chronic hepatitis and cirrhosis.36 Other examples include CYPs 2C9 and 2C19, which affect the metabolism of S-warfarin, omeprazole, tolbutamide, and phenytoin and of S-mephenytoin, respectively34; 3% of white populations and 15% of Asians are poor metabolizers of S-mephenytoin.
Nutrition and Disease-Related Changes
A person’s nutritional status influences the expression of certain CYPs, both in health and with liver disease.5,10,20,34 Expression of CYP2E1 is increased by obesity, high fat intake, and fasting.20,34 Diseases that alter the expression of hepatic CYPs include diabetes mellitus (increased CYP2E1), hypothyroidism (decreased CYP1A), and hypopituitarism (decreased CYP3A4).34 Cirrhosis is associated with decreased levels of total cytochrome P450 and also with reduced hepatic perfusion; the result is a decrease in the clearance of drugs such as propranolol that are metabolized rapidly by the liver.34 The effects of cirrhosis vary, however, among individual CYP families (e.g., CYP1A levels are lowered, but CYP2C and CYP2D6 levels often are preserved) and with the type of liver disease (e.g., CYP3A4 levels are preserved with cholestatic liver disease but lowered with hepatocellular liver disease).
Adaptive Response and Enzyme Induction
Exposure to lipophilic substances results in an adaptive response that usually involves synthesis of new enzyme protein, a process termed enzyme induction. The molecular basis for genetic regulation of constitutive and inducible expression of the major human hepatic cytochrome P450, CYP3A4, has been determined.37 Agents such as rifampin interact with the pregnane X-receptor (PXR), a member of the orphan nuclear receptor family of transcriptional regulators.37 Activated PXR and the analogous constitutive androstane receptor (CAR) in turn bind to cognate nucleotide sequences upstream to the CYP3A4 structural gene within a “xenobiotic-regulatory enhancer module” (XREM). This interaction regulates the CYP3A4 promoter downstream and ultimately the transcription of CYP3A4 protein. Similar control mechanisms apply to several other CYP pathways,37,38 particularly those involved with bile acid synthesis in which the nuclear receptors implicated include the farnesoid X-receptor (FXR), which down-regulates bile acid synthesis and up-regulates bile salt excretory pathways, and liver X receptor, a positive regulator of bile acid synthesis via CYP7A (see also Chapter 64).37
Common examples of the induction of microsomal enzymes by environmental compounds include the effect of smoking cigarettes and cannabis on CYP1A238 and of alcohol on CYP2E1 and possibly CYP3A4.39 Several drugs are potent inducers of CYP enzymes. Isoniazid induces CYP2E1, whereas phenobarbital and phenytoin increase the expression of multiple CYPs.34 Rifampin is a potent inducer of CYP3A4, as is hypericum,40 the active ingredient of St John’s wort, a commonly used herbal medicine, thereby causing interactions between conventional medicines and a CAM preparation. Further descriptions of the regulation of hepatic drug metabolizing enzymes have been published elsewhere.34,38
Phase 3 Pathways
Several transporters secrete drugs, drug metabolites, or their conjugates into bile, and this mechanism is often referred to as phase 3 of hepatic drug elimination. These pathways involve ATP-binding cassette (ABC) proteins, which derive the energy for their transport functions from hydrolysis of ATP. ABC transport proteins are widely distributed in nature and include the cystic fibrosis transmembrane conductance regulator (CFTR) (see Chapter 76) and the canalicular and intestinal copper transporters (see Chapter 75). The role of ABC transport proteins in secretion of bile has been reviewed (see Chapter 64).37,38,41,42
Multidrug resistance protein 1 (MDR1) is highly expressed on the apical (canalicular) plasma membrane of hepatocytes, where it transports cationic drugs, particularly anticancer agents, into bile. Another family of ABC transporters, the multidrug resistance-associated proteins (MRPs), is also expressed in the liver. At least two members of this family excrete drug (and other) conjugates from hepatocytes: MRP-3 on the basolateral surface facilitates passage of drug conjugate into the sinusoidal circulation, and MRP-2, expressed on the canalicular membrane, pumps endogenous compounds (e.g., bilirubin diglucuronide, leukotriene-glutathionyl conjugates, glutathione) and drug conjugates into bile. The bile salt export pump (BSEP) and MDR3 (in humans) and Mdr2 (in mice) are other canalicular transporters concerned, respectively, with bile acid and phospholipid secretion into bile. Genetic polymorphisms of these genes are associated with human cholestatic liver diseases (see Chapters 64 and 76). BSEP interacts with several drugs.42
Regulation of the membrane expression and activity of these drug elimination pathways is complex. The possibility that their altered expression or impaired activity (by competition between agents, changes in membrane lipid composition, or damage from reactive metabolites or covalent binding) could lead to drug accumulation, impairment of bile flow, or cholestatic liver injury has been demonstrated for estrogens,43,44 troglitazone,45 terbinafine,46 and flucloxacillin47 and may have wider mechanistic importance for drug-induced cholestasis and other forms of liver injury.42
TOXIC MECHANISMS OF LIVER INJURY
Oxidative Stress and the Glutathione System
The liver is exposed to oxidative stress by the propensity of hepatocytes to reduce oxygen, particularly in mitochondria and also in microsomal electron transport systems (such as CYP2E1), and by NADPH-oxidase-catalyzed formation of ROS and nitroradicals in Kupffer cells, endothelial cells, and stimulated polymorphs and macrophages. To combat oxidative stress, the liver is well-endowed with antioxidant mechanisms, including micronutrients, such as vitamin E and vitamin C, thiol-rich proteins (e.g., metallothionein, ubiquinone), metal-sequestering proteins (e.g., ferritin), and enzymes that metabolize reactive metabolites (e.g., epoxide hydrolases), ROS (e.g., catalase, superoxide dismutase), and lipid peroxides (e.g., glutathione peroxidases). Glutathione (l-gamma-glutamyl-l-cyteine-glycine) is the most important antioxidant in the mammalian liver.19
Hepatocytes are the exclusive site of glutathione synthesis. Hepatic levels of glutathione are high (5 to 10 mmol/L) and can be increased by enhancing the supply of cysteine for glutathione synthesis; this mechanism is the cornerstone of thiol antidote therapy for acetaminophen poisoning. Hepatocyte glutathione synthesis increases in response to pro-oxidants, as occurs when CYP2E1 is overexpressed as a result of signaling via the redox-sensitive transcription factor Nrf.19,20,48,49 Glutathione is a critical cofactor for several antioxidant pathways, including thiol-disulfide exchange reactions and glutathione peroxidase. Glutathione peroxidase has a higher affinity for hydrogen peroxide than does catalase, and it disposes of lipid peroxides, free radicals, and electrophilic drug metabolites. Reduced glutathione is a cofactor for conjugation reactions catalyzed by the glutathione S-transferases. Other reactions proceed nonenzymatically. In turn, the products include glutathione-protein mixed disulfides and oxidized glutathione. The latter can be converted back to glutathione by proton donation catalyzed by glutathione reductase.
Normally, most glutathione within the hepatocyte is in the reduced state, indicating the importance of this pathway for maintenance of the redox capacity of the cell. The reduced form of NADPH is an essential cofactor for glutathione reductase; NADPH formation requires ATP, thereby illustrating a critical link between the energy-generating capacity of the liver and its ability to withstand oxidative stress. Glutathione is also compartmentalized within the hepatocyte, with the highest concentrations found in the cytosol. Adequate levels of glutathione are essential in mitochondria, where ROS are constantly being formed as a minor by-product of oxidative respiration and in response to some drugs or metabolites that interfere with the mitochondrial respiratory chain. Mitochondrial glutathione is maintained by active uptake from the cytosol, a transport system that is altered by chronic ethanol exposure, and is, therefore, another potential target of drug toxicity.19
Biochemical Mechanisms of Cellular Injury
Mechanisms once thought to be central to hepatotoxicity, such as covalent binding to cellular enzymes and peroxidation of membrane lipids, are no longer regarded as exclusive pathways of cellular damage. Rather, oxidation of proteins, phospholipid fatty acyl side chains (lipid peroxidation), and nucleosides appear to be components of the biochemical stress that characterizes toxic liver injury. Secondary reactions also may play a role; these reactions include post-translational modification of proteins via adenosine diphosphate (ADP) ribosylation or protease activation, cleavage of DNA by activation of endogenous endonucleases, and disruption of lipid membranes by activated phospholipases.20 Some of these catabolic reactions could be initiated by a rise in the cytosolic ionic calcium concentration [Ca2+]i, as a result of increased Ca2+ entry or release from internal stores in the endoplasmic reticulum and mitochondria.19,20
Types of Cell Death
Apoptosis
Apoptosis is an energy-dependent, genetically programmed form of cell death that typically results in controlled deletion of individual cells. In addition to its major roles in developmental biology, tissue regulation, and carcinogenesis, apoptosis is important in toxic, viral, and immune-mediated liver injury.50–53 The ultrastructural features of apoptosis are cell and nuclear shrinkage, condensation and margination of nuclear chromatin, plasma membrane blebbing, and ultimately fragmentation of the cell into membrane-bound bodies that contain intact mitochondria and other organelles. Engulfment of these apoptotic bodies by surrounding epithelial and mesenchymal cells conserves cell fragments that contain nucleic acid and intact mitochondria. These fragments are then digested by lysosomes and recycled without release of bioactive substances. As a consequence, apoptosis in it purest form (usually found only in vitro) does not incite an inflammatory tissue reaction. The cellular processes that occur in apoptosis are often mediated by caspases, a family of proteolytic enzymes that contain a cysteine at their active site and cleave polypeptides at aspartate residues; non–caspase-mediated programmed cell death has also been described in experimental hepatotoxicity (see also Chapter 72).
Apoptosis rarely, if ever, is the sole form of cell death in common forms of liver injury, such as ischemia-reperfusion injury, cholestasis, and toxic liver injury, all of which are typically associated with a hepatic inflammatory response. Whether or not activation of pro-death signals causes cell death depends on several factors, including pro-survival signals, the rapidity of the process, the availability of glutathione and ATP, and the role of other cell types. Some of these issues are discussed briefly here and are reviewed in more detail elsewhere.20,50–53
Mitochondria play a pivotal role in pathways that provoke or oppose apoptosis.50,51,53 In the external pathway, activation of the death domain of pro-apoptotic receptors recruits adapter molecules—Fas-associated death domain (FADD) and TNF receptor-associated death domain (TRADD)—which bind and activate procaspase 8 to form the death-inducing signaling complex (DISC). In turn, caspase 8 cleaves Bid, a pro-apoptotic member of the B cell lymphoma/leukemia (Bcl-2) family, to tBid. tBid causes translocation of Bax to the mitochondria, where it aggregates with Bak to promote permeability of the mitochondria.50 Release of cytochrome c and other pro-death molecules, including Smac (which binds caspase inhibitor proteins, such as inhibitor of apoptosis proteins [IAPs]) and apoptosis-inducing factor (AIF, also known as Apaf)51 allows formation of the “aptosome,” which activates caspase 9 and eventually caspase 3 to execute cell death (Fig. 86-1). Intracellular stresses in various sites release other mitochondrial permeabilizing proteins (e.g., Bmf from the cytoskeleton and Bim from the endoplasmic reticulum), whereas members of the Bcl-2 family, Bcl-2 and Bcl-XL, antagonize apoptosis and serve as survival factors by regulating the integrity of mitochondria; the protective mechanism is poorly understood. Stress-activated protein kinases, particularly c-jun N-terminal kinase (JNK) may also be pro-apoptotic52 by phosphorylating and inactivating the mitochondrial protective protein Bcl-XL.
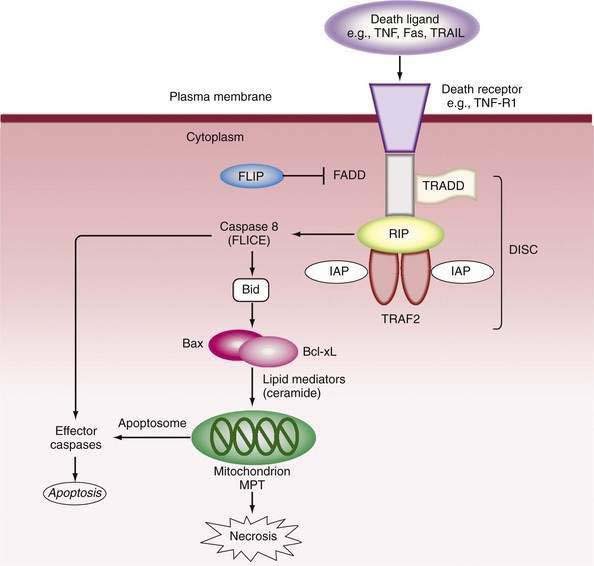
Execution of cell death by apoptosis usually occurs via activation of caspase 3, but more than one caspase-independent pathway of programmed cell death has been described.53 Stresses to the endoplasmic reticulum can bypass mitochondrial events by activation of caspase 12, which in turn activates caspase 9 independently of the apoptosome. The final steps of programmed cell death are energy dependent. Therefore, depletion of ATP abrogates the controlled attempt at “cell suicide,” resulting instead in necrosis (see later) or an overlapping pattern that has been designated as “apoptotic necrosis” or “necraptosis.”54,55 Furthermore, when apoptosis is massive, the capacity for rapid phagocytosis can be exceeded, and “secondary” necrosis can occur.55
Inhibition of caspases is an important protective mechanism against cell death. Such anti-apoptotic pathways include chemical blockade of the cysteine thiol group by nitric oxide (NO) or ROS and cellular depletion of glutathione.20 Protein inhibitors include IAP family members, heat shock proteins (HSPs), and FLICE (caspase-8)-inhibitory proteins (FLIP).50–52 FLIP inhibit caspase-8 activation as a decoy for FADD binding. Bcl-2 and Bcl-XL inhibit mitochondrial permeability, whereas phosphatidylinositol 3-kinase/Akt phosphorylates caspase 9 and activates NF-κB.
Necrosis
In contrast to apoptosis, necrosis has been conceptualized as a relatively uncontrolled process that can result from extensive damage to the plasma membrane with disturbance of ion transport, dissolution of membrane potential, cell swelling, and eventually rupture of the cell. Drug-induced injury to the mitochondrion can impair energy generation, whereas MPT can release stored Ca2+ into the cytosol and perturb other ionic gradients. Mitochondrial enzymes appear to be a particular target of NAPQI, the reactive metabolite of acetaminophen. Reye’s syndrome–like disorders (e.g., toxicity caused by valproic acid; some nucleoside analogs, such as fialuridine, didanosine, zidovudine, zalcitabine; and possibly “ecstasy” [see Chapter 87]) may also result from mitochondrial injury. Mitochondrial injury can result in cell death by either apoptosis or necrosis54,55; the type of cell death pathway may depend primarily on the energy state of the cell, as well as the rapidity and severity of the injury process. In the presence of ATP, cell death can proceed by apoptosis, but when mitochondria are de-energized, the mechanism of cell death is necrosis. This apparent dichotomy between cell death processes is probably artificial, and apoptosis and necrosis more likely represent the morphologic and mechanistic ends of a spectrum of overlapping cell death processes.19,55
Role of Hepatic Nonparenchymal Cells and the Innate Immune Response
In addition to migratory cells, activation of nonparenchymal liver cell types is likely to play an important role in drug and toxin-induced liver injury. Kupffer cells function as resident macrophages and antigen-presenting cells. Some of the toxic effects of activated Kupffer cells, as well as of recruited leukocytes, may be mediated by release of cytokines, such as TNF and Fas-L, which under some circumstances can induce cell death in hepatocytes by apoptosis or necrosis.55 In addition, activated Kupffer cells release ROS, nitroradicals, leukotrienes, and proteases.
Endothelial cells of the hepatic sinusoids or terminal hepatic veins are vulnerable to injury by some hepatotoxins because of their low glutathione content. Such hepatotoxins include the pyrrolizidine alkaloids, which are an important cause of the sinusoidal obstruction syndrome (hepatic veno-occlusive disease).56 Other types of drug-induced vascular injury may be caused primarily by involvement of the sinusoidal endothelial cells (see Chapter 83).
IMMUNOLOGIC MECHANISMS
In addition to the activation of innate inflammatory processes in the liver by toxic mechanisms, (extrinsic) immunologic mechanisms could account for certain aspects of idiosyncratic drug-induced liver disease. Immune attack involves liganding of death receptors, as discussed earlier, or porin-mediated introduction of granzyme.19 The most convincing evidence for drug allergy includes (1) delayed onset after initial exposure and accelerated onset after rechallenge, (2) hepatic inflammatory infiltrates with neutrophils and eosinophils, and (3) fever, rash, lymphadenopathy, peripheral eosinophilia, and involvement of other organs. In some types of drug hepatitis, the liver is clearly implicated as part of a systemic hypersensitivity reaction, as described later for the reactive metabolite syndrome (RMS); sulfonamides, phenytoin, nitrofurantoin, minocycline, nevirapine, and some Chinese herbal medicines are causative agents. Why the liver is the predominant site of injury in some persons whereas other organs are involved in other persons is unclear; genetic factors relevant to tissue-specific gene expression could be involved.
One possible immunopathogenic mechanism for drug-induced liver disease is the altered antigen concept, in which an initial interaction between drug metabolites and cellular proteins results in the formation of neoantigens (haptens) or drug-protein adducts. An example is the formation of trifluoroacetylated (TFA) adducts after exposure to halothane or other haloalkane anesthetics (see Chapter 87). For these adducts to initiate tissue-damaging immune responses (1) processing should be presented in an immunogenic form (e.g., by Kupffer cells, in association with major histocompatibility complex [MHC] molecules); (2) appropriately responsive CD4+ T cells must be present to provide help to induce an immune response; and (3) the drug-derived antigen, together with a class II MHC molecule, must be expressed on the target cells in order to attract CD8+ (cytotoxic) T cells. That bile duct epithelial cells are more likely to express class II MHC antigens may explain why they are possible targets in drug-induced cholestatic hepatitis.
Although antibodies directed against TFA-protein adducts circulate in the majority of patients following recovery from halothane-induced liver injury,57 the specificity and pathogenicity of these antibodies remain in doubt. Another way in which circulating drug-induced antibodies could result in immune-mediated lysis of hepatocytes is through molecular mimicry of host enzymes.58 Experimental evidence suggests that for diclofenac antibody-dependent cell-mediated immunity could operate as a mechanism for drug-induced liver disease.59
CLINICOPATHOLOGIC FEATURES OF DRUG-INDUCED LIVER DISEASE
CLASSIFICATION
Drugs are often divided into dose-dependent, or predictable, hepatotoxins and dose-independent, or unpredictable (idiosyncratic), hepatotoxins. Dose-dependent hepatotoxins generally require metabolic activation to toxic metabolites or interfere with subcellular organelles and biochemical processes at key sites, such as mitochondria or canalicular bile secretion.43 Liver injury produced by dose-dependent hepatotoxins usually occurs after a short latent period (hours), is characterized by zonal necrosis or microvesicular steatosis, and can be reproduced in other species. By contrast, idiosyncratic hepatotoxins cause a wide range of histologic changes and do not reliably cause injury in other species; in addition, the latent period before the onset of injury is variable in duration. The distinction between dose-dependent and idiosyncratic hepatotoxins is blurred with agents such as dantrolene, tacrine, perhexiline, flucloxacillin, cyclophosphamide, nucleoside analogs, bromfenac, anticancer drugs, and cyclosporine. Liver injury caused by each of these drugs is partly dose dependent, but reactions occur in only a small proportion of exposed persons.
Two general types of mechanisms may account for idiosyncratic hepatotoxicity: metabolic idiosyncrasy and immunoallergy. Metabolic idiosyncrasy refers to the susceptibility of rare persons to hepatotoxicity from a drug that, in conventional doses, is usually safe. Such susceptibility may result from genetic or acquired differences in drug metabolism or canalicular secretion, mitochondrial defects, or cell death receptor signaling. Immunoallergy indicates operation of the immune system in mediating the response to a drug. These two mechanisms may be interrelated (see later). Other pathogenic mechanisms may include indirect mediation of liver injury, as in vascular and possibly hyperthermic changes produced by cocaine, ecstasy, intraarterial fluroxuridine, and possibly anesthetics (see Chapter 87).
The most practical classification of drug hepatotoxicity is based on clinical and laboratory features and liver histology, as summarized in Table 86-2. This classification provides a framework for discussing drug-induced hepatic disease in comparison with other hepatobiliary disorders but is imperfect because the clinical and pathologic features are not always congruent. Moreover, much overlap between categories exists, particularly in the spectrum from severe necrosis (which may result from dose-dependent or idiosyncratic hepatotoxicity) to focal necrosis with lobular inflammation (hepatitis) to cholestasis. Many drugs produce a spectrum of syndromes from hepatitis to cholestasis, and some authorities include a further category of mixed cholestatic-hepatocellular reactions. Granulomatous hepatitis is associated with liver biochemical test abnormalities that are usually indistinguishable from those typical of hepatitis, cholestasis, or mixed reactions.
Table 86-2 Clinicopathologic Classification of Drug-Induced Liver Disease
| CATEGORY | DESCRIPTION | IMPLICATED DRUGS: EXAMPLES |
|---|---|---|
| Hepatic adaptation | No symptoms; raised serum GGTP and AP levels (occasionally raised ALT) | Phenytoin, warfarin |
| Hyperbilirubinemia | Rifampin, flavaspidic acid | |
| Dose-dependent hepatotoxicity | Symptoms of hepatitis; zonal, bridging, and massive necrosis; serum ALT level >5-fold increased, often >2000 U/L | Acetaminophen, nicotinic acid, amodiaquine, hycanthone |
| Other cytopathic toxicity, acute steatosis | Microvesicular steatosis, diffuse or zonal; partially dose dependent, severe liver injury, features of mitochondrial toxicity (lactic acidosis) | Valproic acid, didanosine, HAART agents, fialuridine, l-asparaginase, some herbal medicines |
| Acute hepatitis | Symptoms of hepatitis; focal, bridging, and massive necrosis; serum ALT level >5-fold increased; extrahepatic features of drug allergy in some cases | Isoniazid, dantrolene, nitrofurantoin, halothane, sulfonamides, phenytoin, disulfiram, acebutolol, etretinate, ketoconazole, terbinafine, troglitazone |
| Chronic hepatitis | Duration >3 months; interface hepatitis, bridging necrosis, fibrosis, cirrhosis; clinical and laboratory features of chronic liver disease; autoantibodies with some types of reaction (see Table 86-6) | Nitrofurantoin, etretinate, diclofenac, minocycline, nefazodone (see also Table 86-6) |
| Granulomatous hepatitis | Hepatic granulomas with varying hepatitis and cholestasis; raised serum ALT, AP, GGTP levels | Allopurinol, carbamazepine, hydralazine, quinidine, quinine (see also Table 86-5) |
| Cholestasis without hepatitis | Cholestasis, no inflammation; serum AP levels >twice-normal | Oral contraceptives, androgens |
| Cholestatic hepatitis | Cholestasis with inflammation; symptoms of hepatitis; raised serum ALT and AP levels | Chlorpromazine, tricyclic antidepressants, erythromycins, amoxicillin-clavulanic acid |
| Cholestasis with bile duct injury | Bile duct lesions and cholestatic hepatitis; clinical features of cholangitis | Chlorpromazine, flucloxacillin, dextropropoxyphene |
| Chronic cholestasis: | Cholestasis present >3 months | |
| Vanishing bile duct syndrome | Paucity of small bile ducts; resembles primary biliary cirrhosis, but AMA negative | Chlorpromazine, flucloxacillin, trimethoprim-sulfamethoxazole |
| Sclerosing cholangitis | Strictures of large bile ducts | Intra-arterial floxuridine, intralesional scolicidals |
| Steatohepatitis | Steatosis, focal necrosis, Mallory’s hyaline, pericellular fibrosis, cirrhosis; chronic liver disease, portal hypertension | Perhexiline, amiodarone, others (see Chapter 85) |
| Vascular disorders | Sinusoidal obstruction syndrome, nodular regenerative hyperplasia, others | Many (see Table 86-8) |
| Tumors | Hepatocellular carcinoma, adenoma, angiosarcoma, others | Many (see Chapter 94) |
ALT, alanine aminotransferase; AMA, antimitochondrial antibodies; AP, alkaline phosphatase; AST, aspartate aminotransferase; GGTP, gamma glutamyl transpeptidase; HAART, highly active antiretroviral therapy.
Drugs can alter liver test results without causing significant liver injury. Such adaptive responses include hyperbilirubinemia associated with rifampin, cyclosporine, and indinavir and raised serum GGTP and alkaline phosphatase levels associated with phenytoin and warfarin.5,6 The latter effect is probably attributable to microsomal enzyme induction. For agents such as isoniazid, however, the distinction between adaptation and minor injury is blurred; adaptation in such cases may be a response to oxidative injury. Conversely, liver tumors or hepatic fibrosis may develop insidiously without significant abnormalities of liver biochemical tests—the former in association with sex steroids or vinyl chloride monomer and the latter with methotrexate, arsenic, or hypervitaminosis A.
The duration of the disorder is another consideration in classifying drug-induced liver diseases. In general, chronic liver disease is much less commonly attributable to drugs and toxins than are acute reactions,8 but not to consider drugs as a possible eitology of chronic liver disease can lead to a missed diagnosis, with serious clinical consequences.8,9 In contrast to most other types of hepatic pathobiology, drugs and toxins constitute the most important cause of vascular disorders of the liver (see later). Drugs also have been associated with chronic cholestasis, chronic hepatitis, steatohepatitis, hepatic fibrosis, cirrhosis, and benign and malignant liver tumors.
HISTOPATHOLOGIC FEATURES
Although no pathognomonic hallmarks of drug-induced liver disease have been identified, certain histologic patterns suggest drug-induced liver injury. These patterns include zonal necrosis or microvesicular steatosis (which accompanies mitochondrial injury) and mixed histologic features of hepatocellular necrosis and cholestasis. Necrotic lesions that are disproportionately severe compared with the clinical picture also indicate a possible drug cause, whereas destructive bile duct lesions, prominent neutrophils, and eosinophils (at least 25% of inflammatory cells) are suggestive of drug-induced cholestatic hepatitis. Hepatic granuloma formation is another common type of hepatic drug reaction. In cases of steatohepatitis, hepatic fibrosis, or liver tumors, no specific clues to a drug cause have been recognized, although sex steroids increase the vascularity of hepatic tumors and are frequently associated with sinusoidal dilatation or peliosis hepatis. Drug-induced steatohepatitis caused by amiodarone and perhexilene tends to be associated with severe lesions that more closely resemble alcoholic hepatitis than NASH.60 Other drugs (e.g., tamoxifen, methotrexate) cause lesions that are indistinguishable from NASH associated with diabetes mellitus and the metabolic syndrome.6,8,61
CLINICAL FEATURES
The history and physical examination can provide important clues to the diagnosis of hepatic drug reactions. Most important is the temporal pattern of disease evolution in relation to exposure to drugs or toxins. The identification of specific risk factors for hepatotoxicity (e.g., chronic excessive alcohol intake in a person taking acetaminophen) and the presence of systemic features of drug hypersensitivity may indicate the correct diagnosis. Systemic features include fever, rash, mucositis, eosinophilia, lymphadenopathy, a mononucleosis-like syndrome, bone marrow suppression, vasculitis, renal failure, pneumonitis, and pancreatitis. These features may be part of a characteristic syndrome thought to have a genetic basis and likely mediated by formation of drug metabolites that act as haptens to initiate an immunodestructive tissue reaction termed the reactive metabolite syndrome (RMS).62
Reactive Metabolite Syndrome
Drugs implicated as a cause of RMS include sulfonamides, aminopenicillins, fluoroquinolones, clozapine, anticonvulsants (phenytoin, lamotrigine, phenobarbital, carbamazepine), minocycline, protease inhibitors (nevirapine, abacavir), some NSAIDs, and Chinese herbal medicines.62 Risk factors for RMS include a family history of an affected first-degree relative (increases the risk to 1 in 4). Use of other drugs, such as glucocorticoids or valproic acid, at the time the new agent is started increases the risk 4- to 10-fold. The presence of a disorder associated with immune dysregulation (e.g., systemic lupus erythematosus) increases the risk 10-fold, whereas HIV/AIDS increases the risk 100-fold.
A PRACTICAL APPROACH TO DIAGNOSIS
In the absence of specific diagnostic tests, diagnosis of drug-induced liver disease requires clinical suspicion, a thorough drug history, careful consideration of the temporal relationships between drug ingestion and liver disease, and exclusion of other disorders. The objective weighing of evidence for and against an individual agent—causality assessment—is a probabilistic form of diagnosis.63,64 Several clinical scales that incorporate and weigh various features of hepatic adverse drug reactions have been described.9,65–67 A liver biopsy may be indicated in some cases to exclude other diseases and to provide further clues to a drug etiology.68 In the future, in vitro tests may provide confirmatory evidence for particular drugs,57–65 but rechallenge is currently the standard test for drug-induced liver disease.66
PHYSICIAN AWARENESS
Physician awareness is critical for the diagnosis of drug-induced liver disease. The sources of potential hepatotoxins include not only prescribed medications, but also over-the-counter drugs (e.g., ibuprofen), CAM preparations (see Chapters 87 and 127), substances taken for recreational use (e.g., cocaine, ecstasy) or self-poisoning, and environmental contaminants in food and water supplies, the home, the workplace, and the community. Unfortunately, patients and physicians do not always heed early nonspecific symptoms associated with reactions to hepatotoxic drugs. For example, preventable deaths from liver failure still occur more than 40 years after the recognition that isoniziad can cause drug hepatitis.69 Although continuing education and availability of information about potentially hepatotoxic drugs are important issues, physicians have a professional and legal obligation to inform patients about possible adverse drug reactions. A study from Switzerland found that the frequency of new cases of drug-induced liver injury among over 4000 hospital admissions was 1.4% (57 cases). Nevertheless, the drug reaction was not mentioned as a diagnosis in the physicians’ discharge note in 52% to 67% of cases.66
Drug toxicity should be considered a possibility in cases of obscure or poorly explained liver disease, particularly in cases in which mixed or atypical patterns of cholestasis and hepatitis, cholestasis in which common causes have been excluded, especially in the elderly, and histologic features suggestive of a drug etiology are observed. In such cases, the drug history must be addressed as a special investigation, with attention paid to additional sources of information (household members, primary care providers), household drugs, non-prescribed medications, and environmental toxins (see Chapter 87).
EXCLUSION OF OTHER DISORDERS
Other diseases must be excluded before hepatobiliary disease can be ascribed to a drug. For acute and chronic hepatocellular reactions, viral and autoimmune causes of hepatitis and vascular and metabolic disorders must be considered. Some types of drug-induced chronic hepatitis are associated with autoantibodies and superficially resemble autoimmune hepatitis. An approach to the correct diagnosis is described later (see Nitrofurantoin). Drug-induced cholestasis should be considered only when dilatation of the bile duct has been excluded by imaging. In older patients, and particularly when drug exposure does not include agents known to cause cholestasis, cholangiography (e.g., magnetic resonance imaging [MRCP], endoscopic retrograde cholangiography [ERCP]) is obligatory, as is liver biopsy. Drugs and metabolic factors may interact to cause steatohepatitis, as discussed later.
EXTRAHEPATIC FEATURES
The constellation of rash, eosinophilia, and other organ involvement is relatively specific for an adverse drug reaction as a cause of liver disease (see earlier). These findings, however, are present in only a minority of cases, so their absence is not helpful. In particular, drugs that cause idiosyncratic liver injury by nonimmunologic mechanisms are not usually associated with extrahepatic features. Specific diagnostic tests for individual drug-induced liver diseases have been described57 but are not generally accepted or available. In the case of dose-dependent hepatotoxins, blood levels may be helpful (see later).
CHRONOLOGIC RELATIONSHIPS
For most drugs, the chronologic relationship among drug ingestion, onset, and resolution of liver injury remains the most important consideration in diagnosis. The criteria for temporal eligibility include the relationship of drug ingestion to onset, course of the reaction after discontinuation of the drug, and response to readministration of the drug.5,6–9 Deliberate rechallenge can be hazardous and is rarely indicated for logistic and ethical reasons, but inadvertent rechallenge may have occurred already. The rechallenge is regarded as positive if the serum ALT or alkaline phosphatase level increases at least two-fold.1,6,9 Deliberate rechallenge may be considered to ascertain whether a drug that is important for an individual patient is responsible for hepatotoxicity (e.g., amiodarone needed for refractory ventricular tachycardia). In other cases, documenting the propensity of newer agents, hitherto unrecognized as hepatotoxins, to cause liver injury may be desirable. Written informed consent is required for a deliberate rechallenge.
CONSIDERATIONS IN PATIENTS WITH VIRAL HEPATITIS
Patients with chronic hepatitis B or C may be at higher risk of liver injury from antituberculosis chemotherapy, ibuprofen and possibly other NSAIDs, anti-cancer drugs, and HAART compared with persons without viral hepatitis.26–30 A more common clinical problem is the finding of a serum ALT level greater than 300 U/L at a routine office visit in a patient with previous levels less than 150 U/L. In patients with hepatitis C, the rise in serum ALT is more likely the result of drug toxicity than a spontaneous change in the activity of the hepatitis C, particularly when the ALT level is greater than 1000 U/L.68 The most commonly implicated agents are acetaminophen taken in moderate doses under conditions of increased risk (e.g., fasting, alcohol excess, use of other medication) and CAM preparations, typically Chinese herbal remedies (see Chapter 87). Clinical suspicion is essential for recognizing the drug cause of liver injury so that appropriate advice can be given. Determination of blood levels of acetaminophen also may be useful in difficult cases, but levels (particularly undetectable levels) can be difficult to interpret in the context of regular ingestion, as opposed to a single episode of self-poisoning.
PREVENTION AND MANAGEMENT
For dose-dependent hepatotoxins, prevention depends on adherence to dosage guidelines or use of blood levels. This approach has virtually abolished some forms of drug-induced liver injury, such as tetracycline-induced fatty liver, aspirin hepatitis, and methotrexate-induced hepatic fibrosis. In cases with specific risk factors, strategies to prevent toxicity are essential (e.g., avoid use of valproic acid with other drugs in the very young; do not prescribe methotrexate to persons who consume alcohol in excess). Moderate doses of acetaminophen are contraindicated in heavy drinkers and after fasting,21 and administration of halothane should not be repeated within 28 days or in persons suspected of previous sensitivity to a haloalkane anesthetic.
A more difficult issue is whether regular (protocol) screening with liver biochemical tests should be performed when a drug is prescribed. Although such screening often is recommended by authors and drug manufacturers, the efficiency and cost-effectiveness of this approach are unknown. The onset of liver injury is often rapid, rendering once-a-month or every-second-week screening futile. Furthermore, 7.5% of persons who receive placebo in clinical trials have persistently raised serum ALT levels.70 If liver biochemical test levels are monitored, the level of abnormality at which a drug should be discontinued is uncertain, as illustrated by isoniazid, which causes some liver biochemical test abnormality in 30% of exposed subjects. Generally, the recommendation is that isoniazid be stopped if serum ALT levels exceed 250 U/L or more than five times the upper limit of normal, but elevation of the serum bilirubin or albumin concentration or prolongation of the prothrombin time provides a clearer indication to stop the drug. Conversely, a rise in the serum GGTP level or a minor elevation of serum alkaline phosphastase level usually indicates hepatic adaptation rather than liver injury. We do not routinely recommend protocol screening, but this approach could be useful for agents such as valproic acid, isoniazid, pyrazinamide, ketoconazole, dantrolene, tacrine, thiazolidinediones, and synthetic retinoids, either because the onset of liver injury may be delayed and gradual in some cases or because such screening can emphasize the hepatotoxic potential of these drugs to patients and physicians. Liver biopsy has a role in the assessment of hepatic fibrosis in patients who take methotrexate (see later).
Highly toxic solvents should be avoided in the workplace, and such agents have been abandoned. Adequate ventilation and use of masks and protective clothing are vital to prevent occupational exposure to hepatotoxic chemicals. In some cases, liver biochemical tests are performed routinely in exposed persons, but abnormalities are more likely to reflect diseases such as chronic hepatitis C, alcoholism, and NAFLD than toxic liver injury. In the case of vinyl chloride exposure, periodic physical examination (for hepatomegaly) and hepatic imaging with ultrasonography may be useful (see Chapter 87).
Active management of drug-induced liver injury includes removal of the drug and administration of antidotes and anti-inflammatory and cytoprotective agents. In practice, treatment usually is confined to discontinuation of the hepatotoxic drug. Failure to discontinue a drug that is the cause of liver injury is the single most important factor leading to poor outcomes, such as acute liver failure and chronic liver disease.8,9 For ingested toxins such as metals, poisonous mushrooms, and acetaminophen, removal of the unabsorbed drug through the aspiration of stomach contents may be appropriate. Methods to remove absorbed toxins, such as hemodialysis through a charcoal column and forced diuresis, are not effective for hepatotoxins. For chlordecone, an organochlorine insecticide that is lipid-soluble and excreted in bile, oral administration of cholestyramine enhances removal of the agent from the body by interrupting the enterohepatic cycle.71 Thiol replacement therapy, usually with N-acetylcysteine (NAC), is indicated as an antidote for acetaminophen poisoning. Whether NAC or other antioxidants have a role in other types of acute hepatotoxicity is unclear, but the flavonoid, silybin (silymarin), is traditionally used for Amanita phalloides toxicity72 and tocopherol analogs show promise in experimental hepatotoxicity (see Chapter 87).
Beyond discontinuation of the offending agent, the management of drug hepatitis and cholestasis is symptomatic and supportive. In cases of acute liver failure, hepatic transplantation should be considered (see Chapters 93 and 95).7 Ursodeoxycholic acid has shown some promise in the management of chronic cholestasis and pruritus caused by drug hepatotoxicity. Glucocorticoids have little role in the management of drug-induced cholestasis or hepatitis and are ineffective in chlorpromazine-, methyldopa-, and isoniazid-induced hepatitis and in drug-induced fulminant hepatic failure. Case reports attest to the occasional effectiveness of glucocorticoids in protracted cases of hepatitis caused by etretinate, allopurinol, diclofenac, or ketoconazole.5 Glucocorticoids should be reserved for atypical and refractory cases, particularly those associated with vasculitis. Clinical evidence of the effectiveness of putative hepatoprotective agents, such as prostaglandin analogs, is lacking.
DOSE-DEPENDENT HEPATOTOXICITY
Few dose-dependent hepatotoxins are clinically important today. Examples include acetaminophen, some herbal medicines (CAM preparations), plant and fungal toxins, amodiaquine, hycanthone, vitamin A, methotrexate, cyclophosphamide, anti-cancer drugs, carbon tetrachloride, phosphorus, and metals (especially iron, copper, and mercury). Acetaminophen is by far the most important of these; hepatotoxicity caused by CAM preparations is discussed in Chapter 87.
ACETAMINOPHEN
General Nature, Frequency, and Predisposing Factors
Acetaminophen (paracetamol) is a widely used analgesic available without prescription. It is safe when taken in the recommended therapeutic dose of 1 to 4 g daily, but hepatotoxicity produced by self-poisoning with acetaminophen has been recognized since the 1960s. Despite the effectiveness of thiol-based antidotes, acetaminophen remains the most common cause of drug-induced liver injury in most countries and an important cause of acute liver failure.7,73 Parasuicide and suicide are the usual reasons for overdose.73,74 Although controversial,75,76 hepatologists and pediatricians see cases of acetaminophen poisoning that have arisen through what Zimmerman and Maddrey termed therapeutic misadventure.77 This occurrence is especially common in persons who habitually drink alcohol to excess and has also been recognized after daily ingestion of moderate therapeutic doses (10 to 20 g over three days) of acetaminophen in adults and children who are fasting or malnourished21 or who are taking drugs that interact with the metabolism of acetaminophen.77
Single doses of acetaminophen that exceed 7 to 10 g (140 mg/kg body weight in children) may cause liver injury, but this outcome is not inevitable. Severe liver injury (serum ALT level greater than 1000 U/L) or fatal cases usually involve doses of at least 15 to 25 g, but because of interindividual variability, survival is possible even after ingestion of a massive single dose of acetaminophen (greater than 50 g).78 Among persons with an untreated acetaminophen overdose, severe liver injury occurred in only 20%, and among those with severe liver injury, the mortality rate was 20%.78 Conversely, among heavy drinkers, daily acetaminophen doses of 2 to 6 g have been associated with fatal hepatotoxicity.75–78
Risk factors for acetaminophen-induced hepatotoxicity are summarized in Table 86-3. Children are relatively resistant to acetaminophen-induced hepatotoxicity,79 possibly because of their tendency to ingest smaller doses, greater likelihood of vomiting, or biological resistance. Therapeutic misadventure after multiple doses, especially during fasting and when weight-based recommendations have been exceeded, has a high mortality rate. By contrast, the presence of underlying liver disease does not predispose to acetaminophen hepatotoxicity.
Table 86-3 Risk Factors for Acetaminophen-Induced Hepatotoxicity
| FACTOR | RELEVANCE |
|---|---|
| Age | Children may be more resistant than adults |
| Dose | Minimal hepatotoxic dose: 7.5g (≈100 mg/kg) in adults, 150 mg/kg in children |
| Severe toxicity possible with dose >15 g | |
| Blood level | Influenced by dose, time after ingestion, gastric emptying |
| Best indicator of risk of hepatotoxicity (see text and Fig. 86-2) | |
| Chronic excessive alcohol ingestion | Toxic dose threshold lowered; worsens prognosis (also related to late presentation); nephrotoxicity common |
| Fasting | Toxic dose threshold lowered—therapeutic misadventure (see text) |
| Concomitant medication | Toxic dose threshold lowered—therapeutic misadventure; worsens prognosis (e.g., isoniazid, phenytoin, zidovudine) |
| Time of presentation | Late presentation or delayed treatment (>16 hr) predicts worse outcome |
Self-poisoning with acetaminophen is most common in young women, but fatalities are most frequent in men, possibly because of alcoholism and late presentation.73–75 The time of presentation is critical because thiol therapy given within 12 hours of acetaminophen poisoning virtually abolishes significant liver injury (see later). Therapeutic misadventure is also associated with a worse outcome.76 Concomitant use of agents such as phenobarbital, phenytoin, isoniazid, and zidovudine is another risk factor for acetaminophen hepatotoxicity. These drugs may promote the oxidative metabolism of acetaminophen to NAPQI by inducing CYP2E1 (for isoniazid) or CYP3A4 (for phenytoin) or by competing with glucuronidation pathways (for zidovudine). Alcohol and fasting have dual effects by enhancing expression of CYP2E1 and by depleting hepatic glutathione. Fasting also may impair acetaminophen conjugation by depleting cofactors for the glucuronidation and sulfation pathways.21
Clinical Course, Outcomes, and Prognostic Indicators
In the first two days after acetaminophen self-poisoning, features of liver injury are not present. Nausea, vomiting, and drowsiness are often caused by concomitant ingestion of alcohol and other drugs. After 48 to 72 hours, serum ALT levels may be elevated, and symptoms such as anorexia, nausea and vomiting, fatigue, and malaise may occur. Hepatic pain may be pronounced. In severe cases, the course is characterized by repeated vomiting, jaundice, hypoglycemia, and other features of acute liver failure, particularly coagulopathy and hepatic encephalopathy. The liver may shrink as a result of severe necrosis. Serum levels of ALT are often between 2000 and 10,000 U/L. These high levels, together with high levels of other intracellular proteins (ferritin, glutathione S-transferases), may provide a clue to the diagnosis in complex settings, as may occur with alcoholic patients and those with viral hepatitis.77
Indicators of a poor outcome73–76 include grade IV hepatic coma, acidosis, severe and sustained impairment of coagulation factor synthesis, renal failure, and a pattern of falling serum ALT levels in conjunction with a worsening prothrombin time (see also Chapter 93). Renal failure reflects acute tubular necrosis or the hepatorenal syndrome. Myocardial injury also has been attributable to acetaminophen toxicity.78 Death occurs between 4 and 18 days after the overdose and generally results from cerebral edema and sepsis complicating hepatic and multiorgan failure. A majority of patients recover completely. Cases of apparent chronic hepatotoxicity rarely have been attributed to continued ingestion of acetaminophen (2 to 6 g/day), usually in a susceptible host, such as a heavy drinker or a person with preexisting, unrecognized liver disease.5,6 Rare cases of acetaminophen hypersensitivity, typically involving skin or lung, have been reported in association with liver injury.80,81
Management
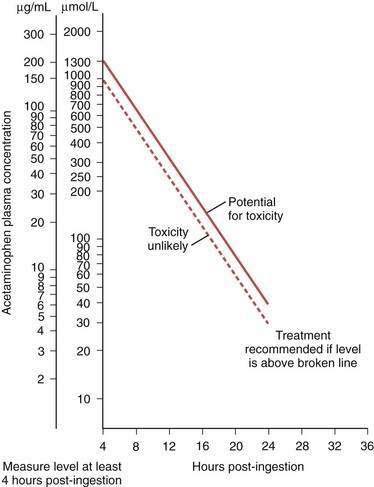
Blood levels of acetaminophen should be measured at the time of presentation. Because of delayed gastric emptying, however, blood levels within four hours of ingestion may underestimate the extent of exposure. After four hours, acetaminophen blood levels give a reliable indicator of the risk of liver injury in patients with an acute overdose (not in those with a therapeutic misadventure). The risk of liver injury is then estimated by reference to the Prescott nomogram (Fig. 86-2).78 Indications for antidote therapy include a reliable history of major poisoning (more than 10 g) or blood acetaminophen levels in the moderate or high-risk bands on the monogram, or both.74,78 At-risk patients should be hospitalized for monitoring.
Hepatic necrosis occurs only when glutathione concentrations fall below a critical level, thereby allowing NAPQI to produce liver injury. Administration of cysteine donors stimulates hepatic synthesis of glutathione. Many cysteine precursors or thiol donors could be used, but NAC has become the agent of choice. Oral administration is preferred in the United States,73,78 with a loading dose of 140 mg/kg followed by administration of 70 mg/kg every 4 hours for 72 hours. This regimen is highly effective, despite the theoretical disadvantage that delayed gastric emptying and vomiting may reduce intestinal absorption of NAC. In Europe and Australia, NAC is administered by slow bolus intravenous injection followed by infusion (150 mg/kg over 15 minutes in 200 mL of 5% dextrose, with a second dose of 50 mg/kg 4 hours later, if the blood acetaminophen levels indicate a high risk of hepatoxicity, and a total dose over 24 hours of 300 mg/kg).78 The intravenous route may be associated with a higher rate of hypersensitivity reactions because of the higher systemic blood levels achieved.5 Adverse reactions to NAC may be severe, with rash, angioedema, and shock, which occasionally is fatal.5 Therefore, NAC must be administered under close supervision and only for appropriate indications. In patients known to be sensitized to NAC, methionine is probably just as effective but is not available in a commercial preparation; it must be made up fresh and often causes vomiting.78
Cases of acetaminophen-induced severe liver injury are virtually abolished if NAC is administered within 12 hours and possibly within 16 hours of acetaminophen ingestion.73,74,78 After 16 hours, thiol donation is unlikely to affect the development of liver injury because oxidation of acetaminophen to NAPQI with consequent oxidation of thiol groups is complete and mitochondrial injury and activation of cell death pathways are likely to be established. Nevertheless, NAC has been reported to decrease the mortality associated with acetaminophen-induced hepatotoxicity when administered 16 to 36 hours after self-poisoning,73,74,78 possibly because NAC stabilizes vascular reactivity in patients with liver failure. Therefore, administration of NAC is recommended for patients with a late presentation after acetaminophen overdose. Other strategies to protect the liver against acetaminophen poisoning, such as inhibition of CYP-dependent metabolism through the use of cimetidine or administration of prostaglandin analogs, which are efficacious in rats, have not been established as clinically useful. The constitutive androstane receptor (CAR) has been identified as a regulator of acetaminophen metabolism and hepatotoxicity in mice.82 Inhibition of CAR activity by administration of androstanol one hour after acetaminophen dosing blocks liver injury.83
Liver transplantation has been advocated as a therapeutic option for select patients in whom liver failure develops after acetaminophen poisoning.73,74 The selection of cases is based on the prognostic indicators discussed earlier and is strongly influenced by the prospects for successful psychological rehabilitation (see Chapter 95).74 In several series, about 60% of listed patients have been transplanted, and survival rates have exceeded 70%.74
Prevention
Safe use of acetaminophen involves adherence to the recommended maximum dose for healthy adults and children and education about the risk factors that lower the toxic dose threshold. Acetaminophen doses of more than 2 g a day are contraindicated in heavy drinkers, in those taking other medications (particularly phenytoin, zidovudine, and isoniazid), and during fasting. Prolonged use of acetaminophen requires caution in patients with severe cardiorespiratory disease or advanced cirrhosis. Use of acetaminophen for self-poisoning continues despite attempts at public education about the risks involved. The chances of harm from a suicidal gesture may be reduced by the sale of acetaminophen in smaller package sizes and in blister packs, which hamper ready access to the tablets or capsules.84,85
OTHER TYPES OF CYTOPATHIC LIVER INJURY
Some hepatotoxins are not as clearly dose dependent as acetaminophen but cause cytopathic or cytotoxic changes, such as extensive hydropic change, diffuse or zonal microvesicular steatosis, and zonal necrosis.5,6 Injury likely represents metabolic idiosyncrasy, in which the drug or one of its metabolites accumulates and interferes with protein synthesis or intermediary metabolism, or both. The mitochondrion often appears to be the main subcellular target, and other metabolically active tissues can be involved. Pancreatitis and renal tubular injury may accompany severe liver injury caused by valproic acid, tetracycline, and HAART, and metabolic acidosis with a shock-like state is common. The first agent recognized to cause this clinicopathologic syndrome was tetracycline administered in high doses (greater than 2 g/day for more than four days, usually intravenously) to pregnant women, to men taking estrogens, or to patients with renal failure.6 With appropriate dose limitations, this reaction is entirely preventable.
Niacin (Nicotinic Acid)
Hepatotoxicity associated with use of niacin, or nicotinic acid (3-pyridinecarboxylic acid), has been noted since the 1960s. When used to treat hypercholesterolemia, niacin has been an important cause of liver injury.86 It is a dose-dependent hepatotoxin; liver injury usually occurs at doses that exceed 2 g/day, but in rare instances, low-dose (500 mg/day) sustained-release niacin has been implicated in fulminant hepatic failure.87 Patients who are taking sulfonylurea drugs and those with preexisting liver disease, particularly alcoholic hepatitis, are at increased risk. No association with age, diet, or insulin-managed diabetes mellitus has been recognized. The symptoms of niacin hepatotoxicity begin as early as one week to as long as four years after the drug is started. The clinicopathologic spectrum encompasses mild and transient increases in serum ALT levels, jaundice, acute hepatitis, and cholestasis. Liver injury resolves completely when the drug is stopped. Liver biopsy specimens show hepatic necrosis and centrilobular cholestasis. Well-documented cases of fulminant hepatitis, some necessitating liver transplantation, also have been attributed to niacin. Substitution of one niacin preparation for another without a dose adjustment should be avoided; switching from immediate- to sustained-release preparations requires a 50% to 70% reduction in the dose of niacin.
Valproic Acid (Sodium Valproate)
Valproic acid-associated hepatic injury occurs almost exclusively in children, particularly those younger than three years of age. Also at risk are persons with a family history of a mitochondrial enzyme deficiency (particularly involving the urea cycle or long-chain fatty acid metabolism), Friedreich’s ataxia, or Reye’s syndrome or with a sibling affected by valproic acid hepatotoxicity. Another risk factor is multiple drug therapy. Cases in adults have been described rarely. The overall risk of liver injury among persons taking valproic acid varies from 1 per 500 persons exposed among high-risk groups (children under age 3, polypharmacy, genetic defects of mitochondrial enzymes) to less than 1 in 37,000 in low-risk groups.88
No relationship exists between valproic acid toxicity and dose, but blood levels of valproic acid tend to be high in one half of affected persons. The metabolite 4-en-valproic acid, produced by CYP-catalyzed metabolism of valproic acid, is a dose-dependent hepatotoxin in animals and in vitro. The concept has emerged that valproic acid is an occult dose-dependent toxin in which accumulation of a hepatotoxic metabolite (favored by coexposure to CYP-inducing antiepileptic agents) produces mitochondrial injury in a susceptible host (e.g., young children, especially those with partial deficiencies of mitochondrial enzymes).89
Symptoms begin 4 to 12 weeks after treatment with valproic acid is started and are often nonspecific, including lethargy, malaise, poor feeding, somnolence, worsening seizures, muscle weakness, and facial swelling. In typical cases, features of hepatotoxicity follow, including anorexia, nausea, vomiting, abdominal discomfort over the liver, and weight loss.88,89 When jaundice ensues, hypoglycemia, ascites, coagulation disorders, and encephalopathy indicate liver failure with imminent coma and death. In some cases, a neurologic syndrome characterized by ataxia, mental confusion, and coma predominates, with little evidence of hepatic involvement. In other cases, fever and tender hepatomegaly suggestive of Reye’s syndrome may be present (see later); such cases tend to have a better prognosis. Additional extrahepatic features may include alopecia, hypofibrinogenemia, thrombocytopenia, and pancreatitis. The terminal phase is often indicated by renal failure, hypoglycemia, metabolic acidosis, and severe bacterial infection.
Laboratory features include modest elevations of serum bilirubin and aminotransferase levels; the aspartate aminotransferase (AST) level is usually higher than the ALT level. A profound decrease in clotting factor levels, hypoalbuminemia, and raised serum ammonia levels are common. A small liver with increased echogenicity suggestive of steatosis or extensive necrosis is seen on hepatic imaging. Histologic examination of the liver shows submassive or massive hepatic necrosis in two thirds of cases with either zonal or generalized microvesicular steatosis.89 Ultrastructural studies indicate conspicuous abnormalities of the mitochondria.
Antiretroviral Agents
Elevated liver biochemical test levels and clinical evidence of liver disease are common in patients with HIV/AIDS. Reasons for this finding include HBV or HCV infection, other hepatobiliary infections, lymphoma and other tumors, and possibly effects of HIV infection itself. The frequency of hepatic injury with HAART (which often includes three or four agents) is at least 10%.23,27,28,90 The agents used can be broadly categorized as nucleoside (or nucleotide) reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, and protease inhibitors. Because HIV co-infection with HBV or HCV increases the risk of toxicity, all patients should be screened for viral hepatitis before starting HAART.28
Nucleoside (or Nucleotide) Reverse Transcriptase Inhibitors
Nucleosides and nucleotides that block HIV reverse transcriptase are also weak inhibitors of mitochondrial DNA polymerase gamma in vitro; the order of their potency is: zalcitabine > didanosine > stavudine > lamivudine > zidovudine > abacavir.91 The mechanisms of hepatotoxicity may also involve oxidative stress, resulting in further deletion of mitochondrial DNA, and the consequences of impaired oxidative phosphorylation, fatty acyl β-oxidation, and insulin resistance.
In clinical studies, zidovudine, didanosine, and stavudine are the agents implicated most often in liver injury.90–93 Risk factors for mitochondrial drug toxicity among persons with HIV infection include obesity, female gender, and absence of an AIDS-defining illness.90–94 Hallmarks of mitochondrial hepatotoxicity include extensive microvesicular or macrovesicular steatosis (or both), lactic acidosis, and liver biochemical test abnormalities with progression to acute liver failure. Asymptomatic hyperlactatemia is common (especially with stavudine) among persons treated with HAART,94 but life-threatening lactic acidosis with hepatic steatosis is rare, with an estimated risk of 1.3 per 1000 person-years of antiretroviral use. The onset is a median of 6 months (with a range of 3 to 17 months) after treatment is started. Patients present with symptoms that are nonspecific and include nausea, vomiting, diarrhea, dyspnea, lethargy, and abdominal pain. Extrahepatic manifestations, such as myopathy or peripheral neuropathy, and in severe cases pancreatitis and renal failure, may follow the onset of the lactic acidosis and liver injury. Discontinuation of the drug is mandatory but does not prevent fatalities. Nevertheless, the overall mortality rate is low. One suggested approach to prevention is to monitor therapy with nucleos(t)ide reverse transcriptase inhibitors by coupling serum ALT and AST testing with serial measurements of the HIV load and CD4 count. Any new aminotransferase elevation should be followed immediately by measurement of serum lactate, muscle, and pancreatic enzyme levels.92
Non-nucleoside Reverse Transcriptase Inhibitors
Non-nucleoside reverse transcriptase inhibitors occasionally may cause hepatitis as part of a hypersensitivity reaction within the first six weeks of use.90,95,96 Reactions are usually accompanied by peripheral and tissue eosinophilia, skin rash, and lymphadenopathy. Resolution occurs within four weeks of discontinuing the drug.95 Nevirapine also has been implicated in several instances of severe hepatotoxicity,97,98 including cases among healthcare workers in whom nevirapine was used for post-exposure prophylaxis against HIV infection.97 The FDA received 12 reports of such hepatotoxic reactions between 1997 and 2000; liver failure requiring hepatic transplantation developed in one person, seven had clinical features of hepatitis (jaundice, fever, nausea, vomiting, abdominal pain, and hepatomegaly), and four others had elevated serum aminotransferase levels without symptomatic illness. The recommended two-week dose escalation regimen was not adhered to in some of the cases.99 Sequential toxicity with nevirapine followed by efavirenz has been reported in an HIV-HCV coinfected person.100
Protease Inhibitors
Elevation of liver enzymes occurs commonly with protease inhibitors, but clinical hepatitis is infrequent. The agents most often implicated in liver injury are ritonavir and indinavir. The latter also may be associated with unconjugated hyperbilirubinemia in 7% of treated persons, a finding that is of no clinical consequence.9 Severe acute hepatitis may occur rarely. The association with peripheral or tissue (in liver biopsy specimens) eosinophilia in some cases suggests an immunoallergic basis for liver injury.101,102 Acute hepatitis also has been reported in 2.9% to 30% of persons who take ritonavir.103 The course of the illness is generally mild, and the liver injury responds favorably to drug withdrawal. Rarely, acute liver failure may develop; in these cases, liver histologic examination has shown severe microvesicular steatosis, cholestasis, and extensive fibrosis.
Several studies have addressed the potential influence of underlying chronic viral hepatitis on the toxicity of protease inhibitors. Although hepatotoxicity appeared to be more common, liver injury was rapidly reversible in most cases; this observation suggests that the overall effect of protease inhibitors in coinfected persons is not detrimental.104 Many protease induce or inhibit CYP3A4, thereby causing important drug-drug interactions.105 Furthermore, the immune reconstitution that can follow successful HAART may cause a flare-up of previously quiescent chronic hepatitis B (see Chapter 33).
Aspirin
Aspirin occasionally has been associated with major increases in serum ALT levels suggestive of drug hepatitis, but hepatotoxicity occurs only when blood salicylate concentrations exceed 25 mg/100 mL.106 In addition, individual susceptibility factors include hypoalbuminemia, active juvenile rheumatoid arthritis, and systemic lupus erythematosus. Most cases of aspirin-induced hepatotoxicity have been identified by biochemical testing, rather than clinical features. If present, symptoms usually begin within the first few days or weeks of high-dose aspirin therapy. Acute liver failure and fatalities have been rare. Resolution occurs rapidly after drug withdrawal, and salicylates can be reintroduced at a lower dose. All salicylates appear to carry hepatotoxic potential so there is no advantage to replacing aspirin with another salicylate. Liver biopsy specimens reveal a nonspecific focal hepatitis with hepatocellular degeneration and hydropic changes. Steatosis is not usually present, and the absence of steatosis distinguishes aspirin hepatotoxicity from Reye’s syndrome.
Reye’s syndrome has been linked with use of aspirin in febrile children. Although Reye’s syndrome is not simply a form of drug-induced liver disease, aspirin plays an important role in its multifactorial pathogenesis. Reye’s syndrome usually occurs between three and four days after an apparently minor viral infection. It is characterized by acute encephalopathy and hepatic injury, the latter documented by a three-fold or greater rise of serum aminotransferase or ammonia levels and by characteristic histologic findings. Because of effective public health campaigns against the use of aspirin in young febrile children, the incidence of Reye’s syndrome has declined markedly.107
The incidence of Reye’s syndrome in the United States, Great Britain, and elsewhere has fallen dramatically since aspirin use has been avoided in children with a viral illness,16,17 although misdiagnosis in the past of cases that subsequently were shown to be caused by inborn errors of metabolism that mimic Reye’s syndrome may account in part for the apparent decline in the incidence of Reye’s syndrome.107
Other Drugs
l–asparaginase is an antileukemic drug that often causes hepatotoxicity, which usually is reversible but can result in liver failure associated with diffuse microvesicular steatosis.108
Amodiaquine, a 4-aminoquinolone antimalarial agent, has been associated with fatal hepatotoxicity, as well as with agranulocytosis.109 Toxicity may be related to the total dose of the drug. Amodiaquine should be reserved for active treatment of chloroquine-resistant falciparum malaria, and dose recommendations should be strictly observed.
Hycanthone is an antischistosomal agent that causes dose-dependent hepatoxicity. Risk factors for hepatotoxicity include concomitant administration of phenothiazines or estrogens, preexisting liver injury, and bacterial infection, but the most important risk factor is dose.110
The hepatotoxic effects of environmental toxins, illicit drug abuse substances, and metals are discussed in Chapter 87.
DRUG-INDUCED ACUTE HEPATITIS
The term acute hepatitis is used to describe lesions characterized by the presence of hepatic inflammation with conspicuous hepatocyte cell death or degeneration. More severe lesions include zonal and bridging necrosis or massive (panlobular) hepatic necrosis; these lesions may be associated with acute (fulminant or subfulminant) hepatic failure.5,6 Acute hepatitis accounts for nearly 50% of reported adverse drug reactions involving the liver,1–4 and potential causative agents are numerous.5,6,8,110,111
Two broad types of drug hepatitis are those cases with clinical and laboratory features consistent with drug allergy (immunoallergic reactions) and those without such features. The latter type could be the result of metabolic idiosyncrasy; partial dose dependence, a relationship between hepatitis and metabolism of the drug, and histologic or ultrastructural features consistent with chemical toxicity are often found. The clinical and laboratory features that suggest one or the other type of drug hepatitis are summarized in Table 86-4. Nitrofurantoin is discussed as an example of immunoallergy, and isoniazid is used to illustrate metabolic idiosyncrasy. Other relatively frequent examples of drug hepatitis are described briefly, including those associated with granulomatous reactions and chronic hepatitis.
Table 86-4 Types of Drug-Induced Acute Hepatitis: Immunoallergic Reaction versus Metabolic Idiosyncrasy
| CHARACTERISTIC | IMMUNOALLERGIC REACTION | METABOLIC IDIOSYNCRASY |
|---|---|---|
| Frequency | <1 case per 10,000 persons exposed | 1-50 cases per 10,000 persons exposed |
| Gender predilection | Women, often ≥2:1 | Variable, slightly more common in women |
| Latent period to onset | Fairly constant, 2-10 weeks | More variable, 2-24 weeks, occasionally longer than 1 year |
| Relationship to dose | None | Usually none (occasional exceptions) |
| Interactions with other agents | None | Alcohol; occasionally other drugs (e.g., isoniazid with rifampin) |
| Course after stopping drug | Prompt improvement (rare exceptions, e.g., minocycline) | Variable; occasionally slow improvement or deterioration (e.g., troglitazone) |
| Positive rechallenge | Always; often fever within 3 days | Usual (in two thirds of cases), abnormal liver biochemical test levels in 2-21 days |
| Fever | Usual; often initial symptom, part of prodrome | Infrequent, less prominent |
| Extrahepatic features (rash, lymphadenopathy) | Common | Rare |
| Eosinophilia | ||
| Blood | 33%-67% of cases | <10% of cases |
| Tissue | Usual, pronounced | Common but mild |
| Autoantibodies | Often present | Rarely present |
| Examples | Nitrofurantoin, phenytoin, methyldopa, sulfonamides, etretinate, minocycline | Isoniazid, pyrazinamide, ketoconazole, dantrolene, troglitazone |
IMMUNOALLERGIC REACTIONS
Nitrofurantoin
Nitrofurantoin, a synthetic furan-based compound, is a urinary antiseptic agent that continues to lead to cases of hepatic injury.112 The frequency of nitrofurantoin hepatic injury ranges from 0.3 to 3 cases per 100,000 exposed persons.113,114 The risk increases with age, particularly after the age of 64. Two thirds of acute cases occur in women, and the female-to-male ratio is 8 : 1 for chronic hepatitis.113,114 The range of liver diseases associated with nitrofurantoin includes acute hepatitis, occasionally with features of cholestasis, hepatic granulomas, chronic hepatitis with autoimmune phenomena, acute liver failure, and cirrhosis.113,114 Causality has been proved by rechallenge, and no relationship to dose has been observed; cases have even been described after ingestion of milk from a nitrofurantoin-treated cow.115
The relative frequencies of hepatocellular and cholestatic or mixed reactions and of acute and chronic hepatitis caused by nitrofurantoin have been the subject of debate. The nature of the adverse reactions covers a spectrum of biochemical and histologic features that have no apparent relevance to the patient’s clinical outcome. Chronicity depends mostly on the duration of drug ingestion, which has been less than six weeks in acute cases but more than six months in 90% of chronic cases.113,114 Patients with chronic hepatitis often have continued to take nitrofurantoin despite symptoms attributable to an adverse drug effect or have been exposed to another course of the drug after previous toxicity. The mortality rate for chronic nitrofurantoin hepatitis is 20%, compared with 5% to 10% for acute hepatitis.113
Liver biochemical testing may show pronounced elevation of serum ALT levels, but more often the pattern is mixed, with an increase in the serum alkaline phosphatase level as well. In other cases, the results suggest cholestasis. Serum bilirubin levels tend to be increased in proportion to the severity of the reaction. In contrast to most types of acute drug hepatitis, serum albumin concentrations often are low. Hypergammaglobulinemia is more likely in patients with chronic hepatitis than in those with acute hepatitis.113 Eosinophilia occurs in 33% of cases. Autoantibodies (antinuclear antibodies and smooth muscle antibodies) are present in some patients with acute hepatitis and in 80% of those with chronic hepatitis. The presence of autoantibodies can make differentiation of nitrofurantoin-induced fulminant hepatitis from autoimmune hepatitis challenging.116 In contrast to idiopathic autoimmune hepatitis, the frequency of the human leukocyte antigens HLA-B8 and -DRw3 is not increased.113,114
Other Drugs
Methyldopa was one of the first drugs reported to cause immunoallergic drug hepatitis. Cases are now rare because better antihypertensive agents are available, except for pregnancy-related cases.117 Hepatic reactions to methyldopa vary from abnormal liver biochemical test levels, severe acute hepatitis, granuloma formation, and cholestasis to chronic hepatitis with bridging necrosis and cirrhosis. The female predilection, clinical and laboratory changes, course, and extrahepatic features of drug allergy are similar to those for nitrofurantoin.
Phenytoin causes severe acute drug hepatitis in less than one per 10,000 persons exposed.118 Incidence rates are equal in men and women, and cases can occur in childhood. Blacks may be more often affected than whites. Rash, fever, eosinophilia, lymphadenopathy, a pseudomononucleosis syndrome, and other allergic features are common. The clinical features are suggestive of immunoallergy as part of RMS. An individual or familial enzymatic defect that leads to reduced disposal of phenytoin arene oxide has been detected among patients with phenytoin reactions.118 This finding implicates a possible metabolic factor in the pathogenesis of phenytoin toxicity.
Barbiturates, including phenobarbital, rarely are associated with acute hepatitis. Described cases have been similar to phenytoin reactions; fever and rash are usual, and the rate of mortality as a result of liver failure is high.119 Among newer antiepileptic drugs, felbamate120 and topiramate121 have been associated with acute liver failure.
Sulfonamides are a cause of drug hepatitis that is relatively common with combination drugs such as co-trimoxazole (sulfamethoxazole and trimethoprim).122 Trimethoprim alone has been associated with some cases of cholestatic hepatitis; the estimated risk is 1.4 cases per 100,000 exposed persons.122 Reactions to co-trimoxazole resemble those associated with trimethoprim more closely than they resemble sulfonamide reactions; cholestasis or cholestatic hepatitis is more common than is hepatitis. Patients with HIV/AIDS are predisposed to sulfonamide hypersensitivity. Some other drugs have a sulfa moiety that differs from that of sulfonamides but that may increase the risk of cross-sensitivity reactions; for example, celecoxib, a COX-2 inhibitor, has been observed to cause severe hepatitis in two women with a history of sulfonamide sensitivity.123 Likewise, sulfonylureas, such as gliclazide, rarely have been associated with drug hepatitis with features of immunoallergy.124
Sulfasalazine (salicylazosulfapyrine, salazopyrine) has been associated with rare cases of often severe acute hepatitis.125 Although the sulfonamide moiety has been associated to be responsible, this assumption has been challenged by the report of one patient in whom hepatitis recurred after exposure to mesalamine (mesalazine, 5-aminosalicylate).126 This finding implicates the salicylate moiety, and like salicylate hepatitis (discussed earlier) sulfasalazine hepatotoxicity appears to be more common in patients with rheumatoid arthritis than in those with inflammatory bowel disease. A case of mesalamine-induced chronic hepatitis with autoimmune features was diagnosed after 21 months of treatment with the drug.127 Granulomatous hepatitis has been reported with mesalamine.128
Minocycline and other tetracyclines used in conventional low doses are a rare but important cause of drug hepatitis,129,130 including cases that have resulted in acute liver failure requiring liver transplantation.131 Minocycline is one of the few agents in current use that can lead to drug-induced autoimmune hepatitis, as discussed later.
Disulfiram (Antabuse) rarely has been associated with acute hepatitis, occasionally leading to liver failure.132 Disulfiram hepatitis usually is easy to distinguish from alcoholic hepatitis by the ten-fold or greater elevation of serum ALT activity.
β-adrenergic blocking agents rarely have been incriminated in hepatotoxicity. Acebutolol,133 carvedilol,134 labetalol,135 and metoprolol136 each have been associated with cases of acute hepatitis; some cases were proved by rechallenge. Reactions were hepatocellular and severe. Data are insufficient to determine whether or not immunoallergy is likely.
The calcium channel blockers, nifedipine,137 verapamil,138 diltiazem,139 and amlodipine140 have good safety records, but rare cases of acute hepatitis with a short incubation period (five days to six weeks) and other features of immunoallergy have been reported.
Of the angiotensin II receptor blockers, irbesartan has been linked to two reports of cholestasis.141,142 In both patients, jaundice developed within one month of the start of therapy. Liver biochemical testing showed predominant cholestasis, and findings on ultrasonography were normal. Histologic examination revealed marked cholestatic features in both patients, with an inflammatory infiltrate and eosinophils in one patient. Clinical resolution occurred when the medication was stopped; however, liver biochemical abnormalities persisted for more than one year in one patient. Biliary ductopenia is a rare complication. Other angiotensin II receptor blockers implicated in cases of acute hepatitis or cholestatic hepatitis include losartan, valsartan, and candesartan.143–145
Angiotensin-converting enzyme inhibitor-induced liver disease is a rare but important complication of this widely prescribed class of drugs. The incidence has been estimated to be 9 per 100,000 patients treated.146 Reactions to captopril (the oldest and possibly most hepatotoxic representative) and enalapril usually manifest as cholestatic hepatitis, but hepatocellular or mixed hepatocellular reactions can occur.147–149 Features of hypersensitivity, such as fever, skin rashes, and eosinophilia have been observed in patients with captopril hepatotoxicity.147 Histologic examination reveals marked centrilobular cholestasis with eosinophilic portal infiltrates.147 Liver biochemical abnormalities usually resolve after withdrawal of the drug, but resolution may take up to six months in some cases. Fulminant hepatic failure has been attributed to lisinopril,150 whereas fosinopril has been associated with bland cholestasis.151 Ramipril has been implicated in three cases of cholestatic liver injury, one of which progressed to biliary cirrhosis.152 Biliary ductopenia also has been observed with enalapril.153
Hydroxymethylglutaryl-coenzyme A reductase inhibitors (“statins”), as a class of drugs, are not strongly associated with important hepatic injury, although literature reports and data contributed to drug safety surveillance authorities appear to be discordant. Use of statins has increased greatly as new guidelines have lowered the target level for low-density lipoprotein (LDL) cholesterol, thereby resulting in use of higher doses of statins. A dose-related rise in serum aminotransferase levels develops in 1% to 3% of people who take statins.154 A minor (less than two-fold normal) rise in the serum ALT and AST levels (without symptoms) is the most common manifestation of liver injury with these compounds. These elevations usually reverse rapidly with discontinuation of the statin and also reverse if therapy is not interrupted. Lovastatin,155 pravastatin,156 atorvastatin,157 simvastatin,158 and rosuvastatin159 have been implicated in a few reports of acute hepatitis or cholestatic hepatitis. Use of some members of this group of lipid-lowering agents in combination with gemfibrozil is associated with an increased rate of myositis but apparently not with an increased rate of drug-induced liver injury.
Prescribing guidelines for statins invariably warn about the risk of liver injury when these agents are prescribed to persons with preexisting liver abnormalities. Nevertheless, evidence that fatty liver (or steatohepatitis), hepatitis C, or other common liver disorders predispose to drug-induced liver disease is lacking. Moreover, a controlled trial of high-dose pravastatin in patients with preexisting liver disease has demonstrated the safety of statins in this setting.160 Similar conclusions were reached in the Dallas Heart Study, in which statin users were no more likely to exhibit serum ALT elevations than non-users.161 Likewise, statin hepatotoxicity has been unrelated to the serum ALT level at baseline in other retrospective studies162,163 and has even been shown to reduce overall progression of hepatic fibrosis.163 Monitoring serum amionotransferase levels is recommended, but this approach is unlikely to predict toxicity164 or to be cost-effective.165 In the Air Force Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TEXCAPS) increases of more than three times the upper limit of normal were observed in only 18 of 100,000 aminotransferase determinations in users of lovastatin, and in no case did hepatitis develop.165
Etretinate, a synthetic retinoid, is useful for treating several skin diseases. Unlike vitamin A (see Chapter 87), synthetic retinoids are not predictable hepatotoxins, but etretinate has been associated with elevated liver biochemical test levels in 10% to 25% of treated patients.166 Levels may normalize with a reduction in drug dose, thereby suggesting partial dose dependency. Approximately 10 cases of severe hepatitis have been attributed to etretinate, and some have been proved by rechallenge.166 Most patients were women older than age 50; two cases were associated with chronicity, and one patient appeared to respond to glucocorticoids. Because etretinate has a half-life of 100 days, monitoring serum ALT levels is recommended in users of the drug. A progressive increase in the serum ALT level to values above twice the upper limit of normal are an indication to stop the etretinate or to perform a liver biopsy.166 Acitretin is another synthetic retinoid that has been associated with a few instances of acute hepatitis, including cases associated with bile duct injury and progressive hepatic fibrosis.167,168
Gastric acid suppression drugs have an excellent safety record, although rare adverse hepatic reactions have been reported. The histamine H2 receptor antagonist oxmetidine was removed from clinical trials because of hepatotoxicity, and subsequently, ebrotidine was withdrawn because of many cases of liver injury.169 Cimetidine,170 ranitidine,171 and famotidine172 have been associated with cases of acute hepatitis, mostly mild and often with cholestatic features. Some cases have been proved by rechallenge. Features of immunoallergy were present in some of the cimetidine reactions. Cases of hepatotoxicity have been attributed to the proton pump inhibitors omeprazole, lansoprazole, and pantoprazole; these reports have been isolated and causality was not established unequivocally in some of the cases.173,174
Zafirlukast, a leukotriene receptor antagonist effective against asthma, has been reported to cause severe liver injury, with several instances of acute liver failure.175,176 Montelukast has been implicated in three cases of acute hepatitis or cholestatic hepatitis.177
Ticlopidine, an antiplatelet agent, has been associated with more than 30 reports of hepatotoxicity. Examination of liver histology has shown bland cholestasis in most cases and occasionally microvesicular steatosis; cholestatic hepatitis with bile duct injury also has been reported.178 An association with a specific HLA haplotype (A*3303) was observed in Japanese patients with ticlopidine hepatotoxicity.179 Currently, clopidogrel is preferred to ticlopidine, but clopidogrel also can cause hepatocellular or mixed hepatocellular-cholestatic liver injury.180
METABOLIC IDIOSYNCRASY
Isoniazid
Isoniazid-induced liver injury has been characterized since the 1970s, but deaths still occur.181–184 Hepatitis develops in about 21 per 1000 persons exposed to isoniazid; 5% to 10% of cases are fatal. The risk and severity of isoniazid hepatitis increase with age; the risk is 0.3% in the third decade of life and increases to 2% or higher after age 50.181,182 Overall frequency rates are the same in men and women, but 70% of fatal cases are in women; black and Hispanic women may be at particular risk.181,182 The risk of toxicity is not related to the dose or blood level of isoniazid. The role of genetic factors has been controversial. Associations have been described with specific genes that code for enzymes involved in aspects of drug metabolism or detoxification (CYP2E1, N-acetyltransferase, glutathione-S-transferase), but data are conflicting.185–187 Chronic excessive alcohol intake increases the frequency and severity of isoniazid hepatotoxicity,181,182 as may rifampin and pyrazinamide.188 Concomitant use of pyrazinamide or acetaminophen have been associated with several cases that were fatal or led to liver transplantation.189 Some studies have found that the risk of liver injury from isoniazid and other anti-tuberculosis drugs is increased among persons with chronic HBV infection, but reports are conflicting.189 Malnutrition may play a role in isoniazid hepatotoxicity in some countries. Likewise, in patients with HCV or HIV infection (or both), the risk of significant serum ALT elevations during anti-tuberculosis treatment has been reported to be increased several-fold; successful antiviral treatment of hepatitis C allowed the safe reintroduction of anti-tuberculosis drugs in four patients.
Serum ALT levels increase in 10% to 36% of persons taking isoniazid in the first 10 weeks. The elevations typically are minor and resolve spontaneously. In persons in whom hepatitis develops, the latent period from exposure to disease ranges from 1 week to more than 6 months, with a median of approximately 8 weeks and, in severe cases, 12 weeks.181,182 Re-exposure to isoniazid may be associated with an accelerated onset, although the experience in India is that gradual reintroduction of isoniazid and rifampin therapy can be achieved in a majority of cases after drug hepatitis has resolved. Prodromal symptoms occur in one third of patients and include malaise, fatigue, and early symptoms of hepatitis, such as anorexia, nausea, and vomiting. Jaundice appears several days later and is the only feature in approximately 10% of cases. Fever, rash, arthralgias, and eosinophilia are uncommon.
Liver biochemical testing indicates hepatocellular injury; serum AST levels exceed serum ALT levels in one half of patients. Serum bilirubin levels usually are elevated; values that are increased more than ten-fold indicate a poor prognosis. In one study,181 one third of patients had a prolonged prothrombin time, and 60% of these cases were fatal. Liver biopsy samples generally show hepatocellular injury, which is focal in approximately 50% of cases, often with marked hydropic change in residual hepatocytes. In the remaining cases, hepatocellular necrosis is zonal, submassive, or massive, with inflammation confined to the portal tracts. Cholestasis and lobular regeneration suggestive of early cirrhosis is a rare feature.
Cases with a fatal outcome have been associated with a longer duration of treatment with isoniazid or continued ingestion of isoniazid after the onset of symptoms.181,182 Therefore, most deaths from isoniazid hepatitis could be prevented if patients report symptoms early in the course and isoniazid is discontinued. In the United States, isoniazid hepatotoxicity is second only to acetaminophen as an indication for liver transplantation for drug-induced liver injury.189 Children are less susceptible than adults, but serious hepatotoxicity can occur in children; over a 10-year period (1987-1997), eight children required liver transplantation for isoniazid hepatotoxicity in the United States (0.2% of pediatric liver transplants).190
Recovery is rapid if isoniazid is discontinued before severe liver injury is established. Management of liver failure is supportive (see Chapter 93); liver transplantation is indicated in the most severe cases. Prevention is the most appropriate way to prevent isoniazid hepatotoxicity, and determining whether the risks of isoniazid preventive therapy outweigh those of latent tuberculosis is critical. The optimal approach to monitoring a patient for isoniazid toxicity is uncertain; every-other-week or monthly monitoring of serum ALT levels will not always prevent the rapid onset of severe hepatotoxicity. Effective prevention depends on awareness of early symptoms, no matter how nonspecific.
Other Drugs
Other Antituberculosis Drugs
Most cases in which rifampin has been implicated in liver injury have occurred in patients who also are taking isoniazid,191 but a few cases have been observed when rifampin was given alone to patients with underlying liver disease.192
Pyrazinamide (and the related ethionamide) has long been known to be a dose-dependent hepatotoxin. The drug is now used in lower doses (1.5 to 2 g/day) because of the emergence of resistant strains of mycobacteria. Hepatotoxicity in patients who are taking combinations that include isoniazid and pyrazinamide may be particularly severe.183 Monitoring of serum ALT levels during therapy is recommended. Cross-sensitivity among isoniazid, pyrazinamide, and ethionamide may occur. Treatment of latent tuberculosis with the combination of pyrazinamide and rifampin, levofloxacin, or ethambutol has been associated with an increased risk of hepatic injury.193–195
Antifungal Agents
Ketoconazole is associated with raised serum levels of aminotransferases in 5% to 17% of treated patients.196,197 Symptomatic hepatitis occurs in 7 to 20 of 100,000 exposed persons. Women (with a female-to-male ratio of 2 : 1) and persons older than 40 years of age are particularly susceptible to ketoconazole-induced liver injury.196–198 Concurrent use of drugs (e.g., lovastatin) that share similar metabolic pathways of elimination (CYP3A4) with ketoconazole can lead to hepatotoxicity.199 Reactions are usually mild but can be severe, with rare cases of acute liver failure.200 The mortality rate is 3% to 7%.196,197 The onset of toxicity is 6 to 12 weeks after ketoconazole is started, and rarely after the drug is stopped. Toxicity is unrelated to the dose of the drug. Continued ingestion of ketoconazole after the onset of symptoms leads to an adverse outcome. Jaundice occurs in 50% of patients in whom acute hepatitis develops, and up to one third may present with nonspecific symptoms, such as nausea, anorexia, and vomiting. Fever, rash, eosinophilia, and other immunoallergic characteristics are rare. Liver biochemical test levels are primarily hepatocellular or mixed, but cholestatic hepatitis or bland cholestasis may occur.196 Jaundice usually resolves within 12 weeks, but resolution may take months.196,197 Cirrhosis is a rare complication following acute hepatic injury.201 The role of glucocorticoid treatment in cases that are slow to resolve is unclear. Fulminant hepatic failure requiring liver transplantation has been reported.202 Fatal liver failure also has followed the use of ketoconazole to reduce hypercortisolism in patients with Cushing’s syndrome.203
Terbinafine is an allylamine antifungal agent that is effective in the treatment of onychomycosis. Several cases of cholestatic hepatitis attributed to terbinafine have been reported.204 The frequency of hepatotoxicity associated with this drug has been estimated to be two to three cases per 100,000 persons exposed.204 The onset is usually within four to six weeks after the drug is started. Liver biopsy specimens show hepatocyte degeneration and canalicular cholestasis with variable portal tract inflammation. Recovery is usual with discontinuation of the drug, although prolonged cholestasis with ductopenia has been reported.205 Ursodeoxycholic acid has been prescribed to affected patients to hasten recovery when cholestasis is protracted.205,206 Fulminant hepatic failure also has been described,207 and a case of sinusoidal obstruction syndrome has been associated with use of terbinafine in a liver transplant recipient.208 The FDA has received at least 16 reports of fulminant liver failure possibly linked to terbinafine209; the frequency of this outcome has been estimated to be 1 per million persons exposed.210
Fluconazole and itraconazole appear to be less hepatotoxic than ketoconazole and terbinafine211; elevations of liver biochemical test levels occur in fewer than 5% of patients. Rare cases of severe hepatic necrosis have been ascribed to fluconazole, but other causes were not excluded. Instances of acute liver failure associated with itraconazole have been reported.212–214 Among more than 69,000 patients who received an oral antifungal agent, ketoconazole and itraconazole were most often associated with liver injury; the relative risks were 228 and 17.7, respectively, in comparison with non-users.215
Antidiabetic Drugs
Thiazolidinediones
Troglitazone was the first peroxisome proliferator-activated receptor-γ (PPARγ) agonist used in patients with type 2 diabetes mellitus for improving glycemic control and lowering serum lipid levels by reducing insulin resistance. Elevations of serum aminotransferase levels were noted in 0.5% to 1.9% of recipients in early trials, which failed to reveal serious hepatotoxicity.216 Reports of acute liver failure emerged in the post-marketing phase,217 in which troglitazone was associated with more than 75 instances of fatal hepatotoxicity or liver failure requiring hepatic transplantation.218
Reported cases of troglitazone hepatotoxicity generally were in older women and obese persons—the common phenotype of persons with type 2 diabetes mellitus. Detailed epidemiologic studies have not been performed to define the risk factors clearly. Evidence that preexisting liver disease or other drugs predispose to troglitazone hepatotoxicity is lacking, although a progressive course in one patient was attributed to concurrent use of simvastatin and troglitazone.219 Mitochondrial injury is favored as the mechanism of hepatic injury, but other mechanisms (e.g., reactive metabolites, inhibition of BSEP) have been proposed.220–222
The onset of troglitazone hepatotoxicity was often as late as 9 to 12 months after treatment was started223,224; rare cases had a much earlier onset (8 days).225 Presenting symptoms included nausea, fatigue, jaundice, vomiting, and symptoms of liver failure. Progression to acute liver failure was often rapid, and in some cases, deterioration continued despite discontinuation of troglitazone.226 Histologic examination of liver biopsy specimens, explanted livers, or autopsy material showed submassive or massive hepatic necrosis, with post-collapse scarring, bile duct proliferation, and some eosinophils.227 Severe cholestasis also was reported,228 as is sometimes observed with other causes of fulminant hepatic failure (e.g., valproic acid) and does not necessarily imply a pathogenic mechanism different from that in cases not associated with cholestasis. Troglitazone was withdrawn from the market in 1999.
Serious liver injury appears to be rare with the second-generation thiazolidinediones rosiglitazone and pioglitazone. In clinical trials, a raised serum ALT level (to greater than three times the upper limit of normal) was reported in 0.25% of patients treated with rosiglitazone and 0.26% treated with pioglitazone.218 Six reports of hepatotoxicity associated with rosiglitazone have been published.229 In some cases, the onset was earlier (e.g., 8 days, 21 days) than in most reported cases of troglitazone hepatotoxicity. On the other hand, the presentation of hepatic injury can be delayed beyond one year.230 An alternative diagnosis of ischemic hepatitis (or ischemia-reperfusion injury) was proposed for one of the cases, and confounding factors were present in two other cases.230,231 Examination of liver histologic findings showed cholestatic hepatitis or granulomatous hepatitis.232 One death has been reported, but all other patients have recovered. Pioglitazone has been implicated in five reports of acute hepatocellular injury.229 Cholestatic hepatitis with bile duct injury was observed in two cases.233 Most patients have recovered after discontinuing pioglitazone. Acute liver failure is a rare complication.234 An isolated reversible increase in the serum alkaline phosphatase level has been described in a patient taking pioglitazone.235
Before treatment with drugs of this class is begun, the FDA recommends that liver biochemical test levels be measured; the pretreatment serum ALT level should be less than 2.5 times the upper limit of normal. Monitoring the serum ALT level every two months during the first year of therapy and periodically thereafter is advised. If ALT levels remain persistently elevated (greater than three times the upper limit of normal), the thiazolidinedione should be discontinued. Symptoms suggestive of hepatitis should be assessed immediately. Persons in whom jaundice developed with troglitazone should not take other thiazolidinediones.236
Other Oral Hypoglycemic Drugs
Hepatocellular liver injury was common with older sulfonylureas, such as carbutamide, metahexamide, and chlorpropamide.237 Tolbutamide, tolazamide, glimepiride, and glibenclamide, which are currently used agents, rarely have been associated with cholestasis or cholestatic hepatitis.238–242 Hypersensitivity phenomena (fever, skin rash, eosinophilia) were present in some cases, as would be expected in view of the structural relationship between sulfonylureas and sulfonamides. Most cases resolved after withdrawal of the drug; however, chronic cholestasis progressing to vanishing bile duct syndrome (see Chapter 20) has been described with tolbutamide and tolazamide.241,242 Fatal liver failure has been reported in at least two patients, one of whom had underlying cirrhosis.243 Gliclazide15,244 and glibenclamide also have been associated with hepatocellular liver injury and, with the latter drug, hepatic granulomas.239 Metformin, acarbose, repaglinide, and human insulin rarely have been associated with liver injury.245–248
Drugs Used for Neurologic Disorders
Several neuroleptic agents have been associated with drug hepatitis. Some reactions appear to be immunoallergic, whereas others conform to the pattern of apparent metabolic idiosyncrasy, depending on the structure of the drug. Such reactions have been reported for commonly used antidepressants, such as fluoxetine,249,250 aroxetine,251 venlafaxine,252 trazodone,253 tolcapone,254 and nefazodone255; the last two drugs also have been implicated in several cases of acute liver failure.
Antidepressants
Monoamine Oxidase Inhibitors
Iproniazid was one of the first drugs associated with acute hepatitis. Reactions occurred in 1% of recipients and were often severe, with instances of fatal fulminant liver failure. The hydrazine substituent (which iproniazid shares in part with isoniazid, ethionamide, pyrazinamide, and niacin) was determined to be hepatotoxic moiety.256 Phenelzine and isocarboxazid also have been associated with occasional instances of hepatocellular injury, but monoamine oxidase inhibitors are now prescribed infrequently.257
Tricyclic Antidepressants
Tricyclic antidepressants bear a structural resemblance to the phenothiazines and are occasional causes of cholestatic or, less commonly, hepatocellular injury. Recovery following cessation of the drug is usual, but prolonged cholestasis has been observed with amitriptyline258 and imipramine.259
Selective Serotonin Reuptake Inhibitors and Other Modern Antidepressants
Selective serotonin reuptake inhibitors (SSRIs) have a better overall safety profile than tricyclic antidepressants. Liver enzyme elevations have been observed in asymptomatic persons taking fluoxetine and paroxetine.249 A few reports of acute and chronic hepatitis have been attributed to the use of SSRIs.249,250 Nefazodone has been associated with cases of subacute liver failure.255,260 Centrilobular, massive, or submassive hepatic necrosis was observed on histologic examination of the livers of affected persons. Nefazodone has been withdrawn from the market. Trazodone has been implicated in cases of acute and chronic hepatocellular injury.253,261 The onset can be delayed as long as 18 months or can occur within 5 days of the start of the drug.262 Occasional reports have noted the occurrence of severe hepatotoxicity with combinations of antidepressants or with antidepressants used in combination with other neuroleptic agents.263 Drug regulatory authorities have been alerted about hepatic adverse events with atomoxetine, a norepinephrine reuptake inhibitor used in treating attention deficit hyperactivity disorder. Only three of the reported cases have been linked conclusively with the drug; all showed a pattern of acute hepatocellular injury.264
Other Neurologic Drugs
Tolcapone, a catechol-o-methyl transferase inhibitor used in the treatment of Parkinson’s disease, has been associated with at least four cases of acute liver failure.265,266 All reported cases occurred in women older than 70 years of age, who presented with jaundice and high serum ALT levels. Centrilobular necrosis was observed on liver histologic examination at autopsy in one case.265 Serious liver injury has not been reported in users of the drug who adhered to monitoring guidelines.266 Postmarketing surveillance has identified three additional patients with acute hepatocellular injury caused by tolcapone. The general consensus, however, is that tolcapone is safe if users are monitored appropriately.267 Current FDA guidelines recommend serum ALT testing every two to four weeks for the first six months. Thereafter, the frequency of testing is left to the discretion of the treating doctor. Patients in whom signs of hepatic impairment or a rise in the serum ALT level (at least one to two times the upper limit of normal) develops should be monitored closely; persistent serum ALT elevations (more than two times the upper limit of normal) are an indication to discontinue the drug.268
Alpidem,269 zolpidem,270 and bentazepam271 are sedative hypnotics that have been implicated in hepatotoxicity. In three reported cases of bentazepam hepatotoxicity, the clinicopathologic pattern resembled chronic hepatitis, but without autoantibodies or other immunologic features.271
Tacrine is a reversible choline esterase inhibitor that improves cognition in patients with Alzheimer’s disease. In a survey of tacrine-related adverse effects in 2446 patients with Alzheimer’s disease, serum ALT levels more than three times the upper limit of normal occurred in 25% of patients and more often in women than in men; levels were elevated more than 20-fold in 2% of patients.272 No dose effect was observed. Serum ALT levels rose abruptly, rather than gradually, and the elevations resolved after discontinuation of tacrine. Symptoms were rare; only nausea and vomiting correlated with major serum ALT elevations. In liver biopsy specimens from three patients, steatosis and mild lobular hepatitis were observed. According to this study,272 minor degrees of hepatocellular injury occur in one half of users of tacrine, but tolerance to this minor form of liver injury eventually develops. Isolated reports of patients in whom jaundice developed indicate a rare potential for tacrine to cause more severe hepatotoxicity. Weekly monitoring of serum ALT levels during the first three months of therapy with tacrine and discontinuation of the drug if values reach three times the upper limit of normal should prevent important hepatotoxicity.272 Although the mechanism of tacrine-induced hepatotoxicity is unclear, a metabolic idiosyncrasy seems likely. Genetic factors that may underlie individual susceptibility to tacrine toxicity have been suggested (e.g., glutathione S-transferase polymorphisms),273 but examination of a panel of 19 candidate genes failed to reveal relevant mutations.274 Mitochondrial injury has been implicated in an animal model of tacrine hepatotoxicity.275
Dantrolene, a skeletal muscle relaxant used to treat spasticity, causes hepatitis in about 1% of exposed persons, with a case-fatality rate of approximately 28%.276 Most affected patients have been older than 30 years of age. One third of patients are asymptomatic, and the remainder present with jaundice and symptoms of hepatitis. Hepatocellular necrosis, often submassive or massive, has been noted on liver biopsy specimens.276 When therapy with dantrolene is initiated, liver biochemical tests should be monitored every two weeks. Liver enzyme elevations are an indication to stop dantrolene.
Other neurotropic drugs and muscle relaxants implicated as idiosyncratic hepatotoxins include tizanidine (a centrally acting muscle relaxant),277 alverine (a smooth muscle relaxant),278 and riluzole.279 Patients with cirrhosis who take tizanidine are at risk of developing hypotension; levels of this CYP1A2-metabolized drug are increased in persons with cirrhosis as a consequence of diminished cytochrome activity.280 Riluzole is a glutamate antagonist approved for the treatment of amyotrophic lateral sclerosis. Increased serum ALT levels were reported in 1.3% to 10% of users of the drug in clinical trials. Two cases of acute hepatitis with microvesicular steatosis have since been reported, with onset at four and eight weeks, respectively, after the start of treatment.279,281 Rarely, hepatocellular injury may be delayed for as long as six months. Liver biochemical test abnormalities resolve rapidly after riluzole is discontinued.
Nonsteroidal Anti-Inflammatory Drugs
Bromfenac, a phenylacetic acid derivative, was withdrawn from the U.S. market in 1998 because of several cases of severe hepatotoxic reactions that resulted in acute liver failure leading to liver transplantation or death.282 Most affected patients had received therapeutic doses of bromfenac for more than 90 days before experiencing a prodrome of malaise and fatigue, followed by symptoms of severe hepatitis and progressive liver failure for 5 to 37 days; use of the drug had been recommended for only up to 7 days. No features suggestive of immunoallergy were evident. Histologic examination of the liver showed zonal or confluent necrosis with a predominantly lymphocytic infiltrate.
Most COX-2 inhibitors appear to be relatively free of hepatoxicity, although a small number of cases have been reported in association with nimesulide and celecoxib.283 An exception is lumiracoxib, which was associated with severe hepatotoxicity and was withdrawn from use. Celecoxib has a low potential for liver injury. In a review of 14 controlled trials, the frequency of hepatic dysfunction (0.8%) was not significantly different from that in placebo-treated patients (0.9%) and appeared to be lower than that observed with other NSAIDs.284 Elevations in serum aminotransferase levels often occurred in persons taking diclofenac concurrently. When serious hepatocellular injury was attributed to celecoxib, female gender was a predisposing factor.285 The onset of symptoms was between four days and four weeks after the drug was started. Liver biochemical abnormalities were consistent with a pattern of hepatocellular or mixed liver injury. Eosinophilia and skin rash suggestive of RMS occurred in some patients. All patients recovered within one to four months of discontinuation of the drug. Acute liver failure has been a rare complication of celecoxib toxicity. The manufacturer currently recommends that celecoxib not be administered to persons with a documented sulfonamide allergy because of published reports of cross-reactivity and toxicity.
Nimesulide is an NSAID that has COX-2 selectivity. It has been linked to several cases of acute hepatitis and fatal hepatic failure, especially in women,286 although the risk of liver injury is very small.287 The time to the onset of symptoms has ranged from 1 to 15 weeks, although a delay of up to 8 months is possible.288,289 Hypersensitivity features with peripheral eosinophilia may occur. Centrilobular or bridging necrosis and occasionally bland cholestasis have been described on liver histologic examination. Resolution usually occurs 2 to 17 months after nimesulide is discontinued.289
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 86 Liver Disease Caused by Drugs
DEFINITIONS AND IMPORTANCE
The term drug-induced liver disease should be confined to cases in which the nature of liver injury has been characterized histologically. With the exception of acetaminophen, anticancer drugs, and some botanical or industrial hepatotoxins (see Chapter 87), most cases of drug-induced liver disease represent adverse drug reactions or hepatic drug reactions. These effects are noxious and unintentional and occur at doses recommended for prophylaxis or therapy. The latent period is longer (typically one week to three or six months) than that for direct hepatotoxins (hours to a few days), and extrahepatic features of drug hypersensitivity may be present.
Although drug-induced liver disease is a relatively uncommon cause of jaundice or acute hepatitis in the community, it is an important cause of more severe types of acute liver disease, particularly among older people (see Epidemiology). The overall mortality rate among patients hospitalized for drug-induced liver injury is approximately 10%1 but varies greatly for individual drugs.2,3 The reported frequencies of individual hepatic drug reactions are often underestimated because of the inadequacy of spontaneous reporting by physicians and pharmacists.2,3 With more reliable prospective and epidemiologic techniques, the frequency (or risk) of most types of drug-induced liver disease is between one per 10,000 and one per 100,000 persons exposed.4 Because these responses to drug exposure are clearly rare and unpredictable, they are often termed idiosyncratic drug reactions. Their rarity blunts diagnostic acumen because most clinicians will see few, if any, cases and therefore do not have an appropriate level of clinical suspicion. This concern applies especially to complementary and alternative medicine (CAM), as discussed in Chapter 87. Failure to withdraw the causative agent after the onset of symptoms of drug hepatitis or inadvertent reexposure to such a drug is a common and avoidable factor in acute liver failure attributable to drug-induced liver injury.5–8 Another challenge is that hepatic drug reactions produce an array of clinical syndromes and pathologic findings that mimic known hepatobiliary diseases. Furthermore, although individual agents (and some drug classes) typically produce a characteristic “signature syndrome,” they can also be associated with other and sometimes multiple clinicopathological syndromes.
Drug-induced liver injury is the most common reason for withdrawal of an approved drug from the market. The subject therefore has medicoeconomic, legal, and regulatory ramifications. Because of the low frequency of most types of idiosyncratic drug reactions that involve the liver, serious hepatotoxicity is not usually detected until post-marketing surveillance is conducted. Historically, drugs that have developed a reputation for potential hepatotoxicity usually have been replaced by more acceptable alternatives. Examples include troglitazone, the prototypic thiazolidinedione and bromfenac, a nonsteroidal anti-inflammatory drug (NSAID), both of which were withdrawn from the market because of several cases of fatal acute liver failure.5–9
The burgeoning number of available conventional medications and CAM preparations now includes many hundreds that can be cited as rare causes of drug-induced liver disease. This increasing number of potentially causative agents poses several challenges to clinicians,5–10 including concern about what constitutes an adequate level of patient information at the time a drug is prescribed and the reliability of evidence linking an individual agent to a particular type of liver injury.5,11–13 Another development is the appreciation that drug toxicity, in the context of complex medical situations, can interact with other causes of liver injury. Noteworthy examples of such situations are bone marrow transplantation; cancer chemotherapy; highly active antiretroviral therapy (HAART) for human immunodeficiency virus (HIV) infection and the acquired immunodeficiency syndrome (AIDS); use of antituberculosis drugs in patients with chronic viral hepatitis; rifampin hepatitis in patients with primary biliary cirrhosis (Chapter 89); nonalcoholic fatty liver disease (NAFLD)—particularly nonalcoholic steatohepatitis (NASH)—precipitated by tamoxifen; and possibly other drugs in overweight persons with type 2 diabetes mellitus and the metabolic syndrome.
EPIDEMIOLOGY
Frequency or risk—the number of adverse reactions for a given number of persons exposed—is the best term for expressing how common a drug reaction is. Time-dependent terms such as incidence and prevalence are not appropriate for drug reactions because the frequency is not linearly related to the duration of exposure. For most reactions, the onset occurs within a relatively short exposure time, or latent period, although some rarer types of chronic liver disease occur after many months or years. The frequency of drug-induced liver disease is usually based on the reported rate of drug reactions; such reports are usually a voluntary part of post-marketing surveillance and are submitted to pharmaceutical companies or adverse drug reaction monitoring bodies. In the United States, following approval by the U.S. Food and Drug Administration (FDA), pharmaceutical companies are required to report serious adverse events (any incident resulting in death, a threat to life, hospitalization, or permanent disability [Code of Federal Regulations]). Surveillance becomes a more passive process, however, when a drug is approved for marketing, and physicians and pharmacists are encouraged to file voluntary written reports through the MediWatch program. Similar systems operate in most industrialized countries. Nevertheless, MediWatch receives reports for fewer than 10% of adverse drug reactions,2 and in France fewer than 6% of hepatic adverse drug reactions are reported.3 The situation may be somewhat better in Sweden, but the annual reported incidence of adverse drug reactions of 2.2 per 100,000 in the population over the age of 15 is still much lower than the predicted incidence of 14 per 100,000.3 A prospective surveillance study in Spain measured the annual incidence of drug-related acute serious liver disease as 7.4 per million inhabitants.4
CASE DEFINITION: WHICH AGENT?
At least 300 agents have been implicated in drug-induced liver injury.10 The evidence for most drugs, however, is confined to individual or small numbers of case reports, especially in letters to scientific journals or to regulatory authorities, or small observational series. Therefore, for most agents, the evidence that they could cause liver injury is circumstantial and incomplete. Reports often lack pathologic definition, full exclusion of other disorders (for older reports), and logistic imputation of causality, especially with respect to temporal associations (see Diagnosis).5,9,10 Overall, probably fewer than 50 agents have been implicated reliably as causes of drug-induced liver disease. In general, agents used most commonly in clinical practice and in the community, including antimicrobials, antineoplastic agents, and NSAIDs, are those that have been implicated most often in drug-induced liver injury in larger series. The challenge of identifying the culprit drug among multiple candidates is discussed later.1,4,5,6,11
FREQUENCIES OF HEPATIC DRUG REACTIONS
Because of incomplete reporting, frequencies of hepatic drug reactions may often be underestimated. These estimated frequencies are also crude indicators of risk because of the inherent inaccuracies of case definitions (see Diagnosis)5,9,10 and because case recognition and reporting depend on the skill and motivation of observers. The increased interest of prescribers when initial cases of drug-induced liver disease have been described, together with inappropriate prescribing (e.g., prolonged use of bromfenac, which was approved only for seven days of use, and overprescribing of flucloxacillin and amoxicillin-clavulanic acid in some countries) can give rise to apparent “mini-epidemics.” More appropriate epidemiologic methods applied to hepatotoxicity have included prescription event monitoring, record linkage, and case-control studies. Prescription event monitoring and record linkage have been used to estimate the frequency of liver injury with some antimicrobials (erythromycins, sulfonamides, tetracyclines, flucloxacillin, amoxicillin-clavulanate) and NSAIDs.11
Epidemiologic studies confirm the rarity of drug-induced liver disease with currently used agents. For NSAIDs, the risk of liver injury is between 1 and 10 per 100,000 individuals exposed1,4,5,6; amoxicillin-clavulanic acid has been associated with cholestatic hepatitis in 1 to 2 per 100,000 exposed persons1,4,5,6,8; and low-dose tetracyclines have caused hepatotoxicity in less than one case per million persons exposed.1,4–6 The frequency of liver injury may be higher for agents that exert a metabolic type of hepatotoxicity. For example, isoniazid causes liver injury in up to 2% of persons exposed; the risk depends on the patient’s age and gender, concomitant exposure to other agents, and presence of hepatitis B virus (HBV) and possibly hepatitis C virus (HCV) infections.12 For some drugs in which other host factors play an etiopathogenic role, case-control studies have been used to define attributable risk. Examples include the implication of aspirin in Reye’s syndrome and oral contraceptives in liver tumors and hepatic vein thrombosis.
A relationship may exist between the frequency and severity of serum ALT elevations that indicate liver injury and the risk of severe hepatotoxicity. This relationship was proposed in the 1970s by the late Hyman Zimmerman.6 According to “Hy’s rule,” elevations of serum ALT levels to eight-fold or more above the upper limit of normal or associated increases in the serum bilirubin concentration indicate a potential for the drug to cause acute liver failure at a rate of about 10% of the number of cases of jaundice. Therefore, if two cases of jaundice associated with drug-induced liver injury are observed in a total phase 3 clinical trial experience of 2500 patients, approximately one case of acute liver failure would be expected for every 12,500 subjects who were prescribed the drug during the marketing phase.
IMPORTANCE OF DRUGS AS A CAUSE OF LIVER DISEASE
Hepatotoxicity accounts for less than 5% of cases of jaundice or acute hepatitis in the community and for even fewer cases of chronic liver disease5,6; however, drugs are an important cause of more severe types of liver disease and for liver disease in older people. They account for 10% of cases of severe hepatitis admitted to the hospital in France6 and for 43% of cases of hepatitis among patients 50 years of age or older.7 Drugs account for more than half of the cases of acute liver failure referred to special units in the United States7 and between 20% and 75% of cases of acute liver failure in other industrialized countries.4,7 The pattern of agents incriminated varies among countries; for example, herbal medicines are a relatively more common cause in Asian countries than in other countries (Chapter 87).
RISK FACTORS
For dose-dependent hepatotoxins such as acetaminophen and methotrexate and for some idiosyncratic reactions that are partly dependent on dose (e.g., bromfenac, tetracyclines, dantrolene, tacrine, oxypenicillins), the factors that influence the risk of drug-induced liver disease include the dose of the drug, blood level of the drug, and duration of intake. For idiosyncratic reactions, however, host determinants appear to be central to liver injury. The most critical determinant is likely to be genetic predisposition, but other “constitutional” and environmental factors can influence the risk of liver injury, as summarized in Table 86-1. The most important factors are age,4 gender, exposure to other substances, a history or family history of previous drug reactions, other risk factors for liver disease, and concomitant medical disorders.
Table 86-1 Factors Influencing the Risk of Liver Diseases Caused by Drugs
| FACTOR | EXAMPLES OF DRUGS AFFECTED | INFLUENCE |
|---|---|---|
| Age | Isoniazid, nitrofurantoin, halothane, troglitazone | Age >60 years: increased frequency, increased severity |
| Valproic acid, salicylates | More common in children | |
| Gender | Halothane, minocycline, nitrofurantoin | More common in women, especially with chronic hepatitis |
| Amoxicillin-clavulanic acid, azathioprine | More common in men | |
| Dose | Acetaminophen, aspirin; some herbal medicines | Blood levels are directly related to the risk of hepatotoxicity |
| Tetracycline, tacrine, oxypenicillins | Idiosyncratic reactions, but partial relationship to dose | |
| Methotrexate, vitamin A | Total dose, dosing frequency, duration of exposure is related to the risk of hepatic fibrosis | |
| Genetic factors | Halothane, phenytoin, sulfonamides | Multiple cases in families |
| Amoxicillin-clavulanic acid | Strong HLA association | |
| Valproic acid | Familial cases, association with mitochondrial enzyme deficiencies | |
| History of other drug reactions | Isoflurane, halothane, enflurane | Instances of cross-sensitivity have been reported among members of each drug class but are rare |
| Erythromycins | ||
| Diclofenac, ibuprofen, tiaprofenic acid | ||
| Sulfonamides, COX-2 inhibitors | ||
| Other drugs | Acetaminophen | Isoniazid, zidovudine, and phenytoin lower dose threshold and increase severity of hepatotoxicity |
| Valproic acid | Other antiepileptics increase risk of hepatotoxicity | |
| Anticancer drugs | Interactive vascular toxicity | |
| Excessive alcohol use | Acetaminophen hepatotoxicity | Lowered dose threshold, poorer outcome |
| Isoniazid, methotrexate | Increased risk of liver injury, hepatic fibrosis | |
| Nutritional status: | ||
| Obesity | Halothane, troglitazone, tamoxifen, methotrexate | Increased risk of liver injury; hepatic fibrosis |
| Fasting | Acetaminophen | Increased risk of hepatotoxicity |
| Preexisting liver disease | Hycanthone, pemoline | Increased risk of liver injury |
| Antituberculosis drugs, ibuprofen | Increased risk of liver injury with chronic hepatitis B and C | |
| Other diseases/conditions: | ||
| Diabetes mellitus | Methotrexate | Increased risk of hepatic fibrosis |
| HIV infection/AIDS | Sulfonamides | Increased risk of hypersensitivity |
| Renal failure | Tetracycline, methotrexate | Increased risk of liver injury, hepatic fibrosis |
| Organ transplantation | Azathioprine, thioguanine, busulfan | Increased risk of vascular toxicity |
AIDS, acquired immunodeficiency syndrome; COX-2, cyclooxygenase-2; HIV, human immunodeficiency virus; HLA, human leukocyte antigen.
Genetic Factors
Genetic determinants predispose to drug-induced liver disease,13 as they do for other types of drug reaction, such as penicillin allergy. Atopic patients have been thought to have an increased risk of some types of drug hepatitis, but this increase in risk has not been proved. Genetic factors determine the activity of drug-activating and antioxidant pathways, encode pathways of canalicular bile secretion, and modulate the immune response, tissue stress responses, and cell death pathways (see Chapter 72). Documented examples of drugs associated with a familial predisposition to adverse hepatic drug reactions are few and include valproic acid and phenytoin.5,8,13 Inherited mitochondrial diseases are a risk factor for valproic acid–induced hepatotoxicity.14 Some forms of drug-induced liver disease, particularly drug-induced hepatitis and granulomatous reactions, can be associated with the reactive metabolite syndrome (see later).
Weak associations have been reported between specific human leukocyte antigen (HLA) haplotypes and some types of drug-induced liver disease. Andrade and colleagues13 found positive associations between the class II HLA haplotype and cholestatic or mixed liver damage for some drugs. They suggested that no specific HLA allele predisposed to the overall risk of drug-induced liver disease but that the pattern of liver injury could be influenced by these genetic determinants. Other investigators have found stronger associations between the HLA haplotype and cholestatic reactions to amoxicillin-clavulanic acid and ticlopidine (see later).13,15
Age
Most hepatic drug reactions are more common in adults than in children. Exceptions include valproic acid hepatotoxicity, which is most common in infants younger than three years of age and rare in adults, and Reye’s syndrome, in which salicylates play a key role.16,17 As discussed later, both may be examples of mitochondrial toxicity.14 In adults, the risk of isoniazid-associated hepatotoxicity is greater in persons older than 40 years of age. Similar observations have been made for nitrofurantoin, halothane, etretinate, diclofenac, and troglitazone.1,4,5,10 The increased frequency of adverse drug reactions in older subjects is largely the result of increased exposure, the use of multiple agents, and altered drug disposition.1,4,5,10 In addition, clinical severity of hepatotoxicity increases strikingly with age, as exemplified by fatal reactions to isoniazid and halothane.
Gender
Women are particularly predisposed to drug-induced hepatitis, a difference that cannot be attributed simply to increased exposure. Examples include toxicity caused by halothane, nitrofurantoin, sulfonamides, flucloxacillin, minocycline, and troglitazone.5,6 Drug-induced chronic hepatitis caused by nitrofurantoin, diclofenac, or minocycline has an even more pronounced female preponderance.6 Conversely, equal sex frequency or even male preponderance is common for some drug reactions characterized by cholestasis, as for amoxicillin-clavulanic acid. Azathioprine-induced liver disease is more likely to develop in male renal transplant recipients than in female recipients.18
Concomitant Exposure to Other Agents
Patients who are taking multiple drugs are more likely to experience an adverse reaction than those who are taking one agent.5,9,10 The mechanisms include enhanced cytochrome P450 (CYP)-mediated metabolism of the second drug to a toxic intermediate (see later). Examples discussed later include toxicity caused by acetaminophen, isoniazid, valproic acid, other anticonvulsants, and anticancer drugs. Alternatively, drugs may alter the disposition of other agents by reducing bile flow or competing with canalicular pathways for biliary excretion (phase 3 drug elimination) (see later). This mechanism may account for apparent interactions between oral contraceptive steroids and other drugs to produce cholestasis. Drugs or their metabolites may also interact in mechanisms of cellular toxicity and cell death that involve mitochondrial injury, intracellular signaling pathways, activation of transcription factors, and regulation of hepatic genes involved in controlling the response to stress and injury that triggers pro-inflammatory and cell death processes.19,20
Previous Drug Reactions
A history of an adverse drug reaction generally increases the risk of reactions to the same drug and also to some other agents (see later). Nevertheless, instances of cross-sensitivity to related agents in cases of drug-induced liver disease are surprisingly uncommon. Examples of cross-sensitivity between drugs (or drug classes) include the haloalkane anesthetics (see Chapter 87), erythromycins, phenothiazines and tricyclic antidepressants, isoniazid and pyrazinamide, sulfonamides and other sulfur-containing compounds (e.g., some clyclooxygenase-2 [COX-2] inhibitors), and some NSAIDs. A crucial point is that a previous reaction to the same drug is a major risk factor for an increase in the severity of drug-induced liver injury.6
Nutritional Status
Obesity is strongly associated with the risk of halothane hepatitis (see Chapter 87) and appears to be an independent risk factor for NASH and hepatic fibrosis in persons taking methotrexate or tamoxifen. Fasting also predisposes to acetaminophen hepatotoxicity,21 and a role for undernutrition has been proposed in isoniazid hepatotoxicity.22
Preexisting Liver Disease
In general, liver diseases such as alcoholic cirrhosis and cholestasis do not predispose to adverse hepatic reactions. Exceptions include toxicity to some anticancer drugs, niacin, pemoline, and hycanthone. Preexisting liver disease is a critical determinant of methotrexate-induced hepatic fibrosis (discussed later). Patients with chronic HBV infection12 and possibly those with chronic HCV infection or HIV/AIDS appear to be at heightened risk of liver injury during anti-tuberculosis or HAART therapy,23 after exposure to ibuprofen and possibly other NSAIDs, after myeloablative therapy in preparation for bone marrow transplantation (resulting in sinusoidal obstruction syndrome [see later]),24 and possibly after taking antiandrogens, such as flutamide and cyproterone acetate.25 A particularly strong association has been observed between HCV infection (present in 33% of patients with HIV/AIDS) and the risk of liver injury during HAART; the risk may be increased 2- to 10-fold.26–30
Other Diseases
Rheumatoid arthritis appears to increase the risk of salicylate hepatotoxicity, and a curious, unexplained observation is that hepatitis associated with sulfasalazine appears to be more common in patients with rheumatoid arthritis than in those with inflammatory bowel disease.8–10,31,32 Diabetes mellitus, obesity, and chronic kidney disease predispose to methotrexate-induced hepatic fibrosis, whereas HIV/AIDS confers a heightened risk of sulfonamide hypersensitivity.31–33 A retrospective cohort study of five health maintenance organizations found that the age- and sex-standardized incidence of drug-induced acute liver failure in patients with diabetes mellitus was 0.08 to 0.15 per 1000 person-years, irrespective of the therapeutic agent used (the number using troglitazone was small); the incidence was highest (approximately 0.3 per 1000) during the first six months of exposure.32 Renal transplantation is a risk factor for azathioprine-associated vascular injury, whereas kidney disease predisposes to tetracycline-induced fatty liver.6 Finally, sinusoidal obstruction syndrome induced by anticancer drugs is more common after bone marrow transplantation24 and in persons with HCV infection.5,6,8–10,26
PATHOPHYSIOLOGY
PATHWAYS OF DRUG METABOLISM
As reviewed elsewhere,5,34 phase 1 pathways of drug metabolism include oxidation, reduction, and hydrolytic reactions. The products can be readily conjugated or excreted without further modification.
Cytochrome P450
Most type 1 reactions are catalyzed by microsomal drug oxidases, the key component of which is a hemoprotein of the CYP gene superfamily. The apparent promiscuity of drug oxidases toward drugs, environmental toxins, steroid hormones, lipids, and bile acids results from the existence of multiple closely related CYP proteins. More than 20 CYP enzymes are present in the human liver.34
The reaction cycle involves binding of molecular oxygen to the iron in the heme prosthetic group, with subsequent reduction of oxygen by acceptance of an electron from nicotinamide-adenine dinucleotide phosphate (NADPH) cytochrome P450 reductase, a flavoprotein reductase. The resulting “activated oxygen” is incorporated into the drug or another lipophilic compound. Reduction of oxygen and insertion into a drug substrate (“mixed function oxidation”) can result in formation of chemically reactive intermediates, including free radicals, electrophilic “oxy-intermediates” (e.g., unstable epoxides, quinone imines), and reduced (and therefore reactive) oxygen species (ROS). The quintessential example is the CYP2E1-catalyzed metabolite of acetaminophen, N-acetyl-p-benzoquinone imine (NAPQI), an oxidizing and arylating metabolite that is responsible for liver injury associated with acetaminophen hepatotoxicity. Other quinone compounds are potential reactive metabolites of troglitazone, quinine, and methyldopa. Epoxide metabolites of diterpenoids may be hepatotoxic products of the hepatic metabolism of some plant toxins (see Chapter 87).35 ROS have broad significance in the production of tissue injury, particularly by contributing to the production of oxidative stress and triggering tissue stress responses and cell death pathways, as discussed later.
The hepatic content of CYP proteins is higher in acinar zone 3 (see Chapter 71). Localization of CYP2E1 is usually confined to a narrow rim of hepatocytes 1 to 2 cells thick around the terminal hepatic venule. This finding explains in part the zonality of hepatic lesions produced by drugs and toxins, such as acetaminophen and carbon tetrachloride, which are converted to reactive metabolites.
Genetic and Environmental Determinants of Cytochrome P450 Enzymes
Pharmacogenetics and Polymorphisms of Cytochrome P450 Expression
The hepatic expression of each CYP enzyme is genetically determined. This finding largely explains the four-fold or greater differences in rates of drug metabolism among healthy subjects. Some CYPs, particularly minor forms, are also subject to polymorphic inheritance; therefore, occasional persons completely lack the encoded protein.34 One example is CYP2D6, the enzyme responsible for the metabolism of debrisoquine and perhexiline. Poor metabolizers lack CYP2D6 and accumulate perhexiline when given usual doses; lack of CYP2D6 is the critical determinant in serious adverse effects of perhexiline, including chronic hepatitis and cirrhosis.36 Other examples include CYPs 2C9 and 2C19, which affect the metabolism of S-warfarin, omeprazole, tolbutamide, and phenytoin and of S-mephenytoin, respectively34; 3% of white populations and 15% of Asians are poor metabolizers of S-mephenytoin.
Nutrition and Disease-Related Changes
A person’s nutritional status influences the expression of certain CYPs, both in health and with liver disease.5,10,20,34 Expression of CYP2E1 is increased by obesity, high fat intake, and fasting.20,34 Diseases that alter the expression of hepatic CYPs include diabetes mellitus (increased CYP2E1), hypothyroidism (decreased CYP1A), and hypopituitarism (decreased CYP3A4).34 Cirrhosis is associated with decreased levels of total cytochrome P450 and also with reduced hepatic perfusion; the result is a decrease in the clearance of drugs such as propranolol that are metabolized rapidly by the liver.34 The effects of cirrhosis vary, however, among individual CYP families (e.g., CYP1A levels are lowered, but CYP2C and CYP2D6 levels often are preserved) and with the type of liver disease (e.g., CYP3A4 levels are preserved with cholestatic liver disease but lowered with hepatocellular liver disease).
Adaptive Response and Enzyme Induction
Exposure to lipophilic substances results in an adaptive response that usually involves synthesis of new enzyme protein, a process termed enzyme induction. The molecular basis for genetic regulation of constitutive and inducible expression of the major human hepatic cytochrome P450, CYP3A4, has been determined.37 Agents such as rifampin interact with the pregnane X-receptor (PXR), a member of the orphan nuclear receptor family of transcriptional regulators.37 Activated PXR and the analogous constitutive androstane receptor (CAR) in turn bind to cognate nucleotide sequences upstream to the CYP3A4 structural gene within a “xenobiotic-regulatory enhancer module” (XREM). This interaction regulates the CYP3A4 promoter downstream and ultimately the transcription of CYP3A4 protein. Similar control mechanisms apply to several other CYP pathways,37,38 particularly those involved with bile acid synthesis in which the nuclear receptors implicated include the farnesoid X-receptor (FXR), which down-regulates bile acid synthesis and up-regulates bile salt excretory pathways, and liver X receptor, a positive regulator of bile acid synthesis via CYP7A (see also Chapter 64).37
Common examples of the induction of microsomal enzymes by environmental compounds include the effect of smoking cigarettes and cannabis on CYP1A238 and of alcohol on CYP2E1 and possibly CYP3A4.39 Several drugs are potent inducers of CYP enzymes. Isoniazid induces CYP2E1, whereas phenobarbital and phenytoin increase the expression of multiple CYPs.34 Rifampin is a potent inducer of CYP3A4, as is hypericum,40 the active ingredient of St John’s wort, a commonly used herbal medicine, thereby causing interactions between conventional medicines and a CAM preparation. Further descriptions of the regulation of hepatic drug metabolizing enzymes have been published elsewhere.34,38
Phase 3 Pathways
Several transporters secrete drugs, drug metabolites, or their conjugates into bile, and this mechanism is often referred to as phase 3 of hepatic drug elimination. These pathways involve ATP-binding cassette (ABC) proteins, which derive the energy for their transport functions from hydrolysis of ATP. ABC transport proteins are widely distributed in nature and include the cystic fibrosis transmembrane conductance regulator (CFTR) (see Chapter 76) and the canalicular and intestinal copper transporters (see Chapter 75). The role of ABC transport proteins in secretion of bile has been reviewed (see Chapter 64).37,38,41,42
Multidrug resistance protein 1 (MDR1) is highly expressed on the apical (canalicular) plasma membrane of hepatocytes, where it transports cationic drugs, particularly anticancer agents, into bile. Another family of ABC transporters, the multidrug resistance-associated proteins (MRPs), is also expressed in the liver. At least two members of this family excrete drug (and other) conjugates from hepatocytes: MRP-3 on the basolateral surface facilitates passage of drug conjugate into the sinusoidal circulation, and MRP-2, expressed on the canalicular membrane, pumps endogenous compounds (e.g., bilirubin diglucuronide, leukotriene-glutathionyl conjugates, glutathione) and drug conjugates into bile. The bile salt export pump (BSEP) and MDR3 (in humans) and Mdr2 (in mice) are other canalicular transporters concerned, respectively, with bile acid and phospholipid secretion into bile. Genetic polymorphisms of these genes are associated with human cholestatic liver diseases (see Chapters 64 and 76). BSEP interacts with several drugs.42
Regulation of the membrane expression and activity of these drug elimination pathways is complex. The possibility that their altered expression or impaired activity (by competition between agents, changes in membrane lipid composition, or damage from reactive metabolites or covalent binding) could lead to drug accumulation, impairment of bile flow, or cholestatic liver injury has been demonstrated for estrogens,43,44 troglitazone,45 terbinafine,46 and flucloxacillin47 and may have wider mechanistic importance for drug-induced cholestasis and other forms of liver injury.42
TOXIC MECHANISMS OF LIVER INJURY
Oxidative Stress and the Glutathione System
The liver is exposed to oxidative stress by the propensity of hepatocytes to reduce oxygen, particularly in mitochondria and also in microsomal electron transport systems (such as CYP2E1), and by NADPH-oxidase-catalyzed formation of ROS and nitroradicals in Kupffer cells, endothelial cells, and stimulated polymorphs and macrophages. To combat oxidative stress, the liver is well-endowed with antioxidant mechanisms, including micronutrients, such as vitamin E and vitamin C, thiol-rich proteins (e.g., metallothionein, ubiquinone), metal-sequestering proteins (e.g., ferritin), and enzymes that metabolize reactive metabolites (e.g., epoxide hydrolases), ROS (e.g., catalase, superoxide dismutase), and lipid peroxides (e.g., glutathione peroxidases). Glutathione (l-gamma-glutamyl-l-cyteine-glycine) is the most important antioxidant in the mammalian liver.19
Hepatocytes are the exclusive site of glutathione synthesis. Hepatic levels of glutathione are high (5 to 10 mmol/L) and can be increased by enhancing the supply of cysteine for glutathione synthesis; this mechanism is the cornerstone of thiol antidote therapy for acetaminophen poisoning. Hepatocyte glutathione synthesis increases in response to pro-oxidants, as occurs when CYP2E1 is overexpressed as a result of signaling via the redox-sensitive transcription factor Nrf.19,20,48,49 Glutathione is a critical cofactor for several antioxidant pathways, including thiol-disulfide exchange reactions and glutathione peroxidase. Glutathione peroxidase has a higher affinity for hydrogen peroxide than does catalase, and it disposes of lipid peroxides, free radicals, and electrophilic drug metabolites. Reduced glutathione is a cofactor for conjugation reactions catalyzed by the glutathione S-transferases. Other reactions proceed nonenzymatically. In turn, the products include glutathione-protein mixed disulfides and oxidized glutathione. The latter can be converted back to glutathione by proton donation catalyzed by glutathione reductase.
Normally, most glutathione within the hepatocyte is in the reduced state, indicating the importance of this pathway for maintenance of the redox capacity of the cell. The reduced form of NADPH is an essential cofactor for glutathione reductase; NADPH formation requires ATP, thereby illustrating a critical link between the energy-generating capacity of the liver and its ability to withstand oxidative stress. Glutathione is also compartmentalized within the hepatocyte, with the highest concentrations found in the cytosol. Adequate levels of glutathione are essential in mitochondria, where ROS are constantly being formed as a minor by-product of oxidative respiration and in response to some drugs or metabolites that interfere with the mitochondrial respiratory chain. Mitochondrial glutathione is maintained by active uptake from the cytosol, a transport system that is altered by chronic ethanol exposure, and is, therefore, another potential target of drug toxicity.19
Biochemical Mechanisms of Cellular Injury
Mechanisms once thought to be central to hepatotoxicity, such as covalent binding to cellular enzymes and peroxidation of membrane lipids, are no longer regarded as exclusive pathways of cellular damage. Rather, oxidation of proteins, phospholipid fatty acyl side chains (lipid peroxidation), and nucleosides appear to be components of the biochemical stress that characterizes toxic liver injury. Secondary reactions also may play a role; these reactions include post-translational modification of proteins via adenosine diphosphate (ADP) ribosylation or protease activation, cleavage of DNA by activation of endogenous endonucleases, and disruption of lipid membranes by activated phospholipases.20 Some of these catabolic reactions could be initiated by a rise in the cytosolic ionic calcium concentration [Ca2+]i, as a result of increased Ca2+ entry or release from internal stores in the endoplasmic reticulum and mitochondria.19,20
Types of Cell Death
Apoptosis
Apoptosis is an energy-dependent, genetically programmed form of cell death that typically results in controlled deletion of individual cells. In addition to its major roles in developmental biology, tissue regulation, and carcinogenesis, apoptosis is important in toxic, viral, and immune-mediated liver injury.50–53 The ultrastructural features of apoptosis are cell and nuclear shrinkage, condensation and margination of nuclear chromatin, plasma membrane blebbing, and ultimately fragmentation of the cell into membrane-bound bodies that contain intact mitochondria and other organelles. Engulfment of these apoptotic bodies by surrounding epithelial and mesenchymal cells conserves cell fragments that contain nucleic acid and intact mitochondria. These fragments are then digested by lysosomes and recycled without release of bioactive substances. As a consequence, apoptosis in it purest form (usually found only in vitro) does not incite an inflammatory tissue reaction. The cellular processes that occur in apoptosis are often mediated by caspases, a family of proteolytic enzymes that contain a cysteine at their active site and cleave polypeptides at aspartate residues; non–caspase-mediated programmed cell death has also been described in experimental hepatotoxicity (see also Chapter 72).
Apoptosis rarely, if ever, is the sole form of cell death in common forms of liver injury, such as ischemia-reperfusion injury, cholestasis, and toxic liver injury, all of which are typically associated with a hepatic inflammatory response. Whether or not activation of pro-death signals causes cell death depends on several factors, including pro-survival signals, the rapidity of the process, the availability of glutathione and ATP, and the role of other cell types. Some of these issues are discussed briefly here and are reviewed in more detail elsewhere.20,50–53
Mitochondria play a pivotal role in pathways that provoke or oppose apoptosis.50,51,53 In the external pathway, activation of the death domain of pro-apoptotic receptors recruits adapter molecules—Fas-associated death domain (FADD) and TNF receptor-associated death domain (TRADD)—which bind and activate procaspase 8 to form the death-inducing signaling complex (DISC). In turn, caspase 8 cleaves Bid, a pro-apoptotic member of the B cell lymphoma/leukemia (Bcl-2) family, to tBid. tBid causes translocation of Bax to the mitochondria, where it aggregates with Bak to promote permeability of the mitochondria.50 Release of cytochrome c and other pro-death molecules, including Smac (which binds caspase inhibitor proteins, such as inhibitor of apoptosis proteins [IAPs]) and apoptosis-inducing factor (AIF, also known as Apaf)51 allows formation of the “aptosome,” which activates caspase 9 and eventually caspase 3 to execute cell death (Fig. 86-1). Intracellular stresses in various sites release other mitochondrial permeabilizing proteins (e.g., Bmf from the cytoskeleton and Bim from the endoplasmic reticulum), whereas members of the Bcl-2 family, Bcl-2 and Bcl-XL, antagonize apoptosis and serve as survival factors by regulating the integrity of mitochondria; the protective mechanism is poorly understood. Stress-activated protein kinases, particularly c-jun N-terminal kinase (JNK) may also be pro-apoptotic52 by phosphorylating and inactivating the mitochondrial protective protein Bcl-XL.
Execution of cell death by apoptosis usually occurs via activation of caspase 3, but more than one caspase-independent pathway of programmed cell death has been described.53 Stresses to the endoplasmic reticulum can bypass mitochondrial events by activation of caspase 12, which in turn activates caspase 9 independently of the apoptosome. The final steps of programmed cell death are energy dependent. Therefore, depletion of ATP abrogates the controlled attempt at “cell suicide,” resulting instead in necrosis (see later) or an overlapping pattern that has been designated as “apoptotic necrosis” or “necraptosis.”54,55 Furthermore, when apoptosis is massive, the capacity for rapid phagocytosis can be exceeded, and “secondary” necrosis can occur.55
[/not-level-membership-for-gastroenterology-and-hepatology-category]
