CHAPTER 87 Liver Disease Caused by Anesthetics, Toxins, and Herbal Preparations
Although halothane hepatitis is now largely of historical interest, it holds an important place in the annals of causality assessment in drug-induced liver disease.1 In contrast with the largely unpredictable hepatotoxicity seen with more modern anesthetics and most other medicinal agents (as discussed in Chapter 86), liver damage caused by occupationally and environmentally encountered chemical compounds and other toxins often is more predictable, dose related, and predominantly cytotoxic in nature.1–4 Industrial exposure to hepatotoxic chemicals is a less frequent occupational hazard today than in the past, but reports of toxicity from chemical agents, as well as metals, adulterated cooking oils, and botanical toxins, have not disappeared.3,4 Additionally, the use of complementary and alternative medicine (CAM) preparations continues to increase, and reports of liver injury from potentially hepatotoxic herbal and weight loss products continue to appear (see Chapter 127).5,6 Anesthetics, herbal products, mushrooms, and other toxins continue to account for a substantial percentage of emergency liver transplants for acute liver failure.7
ANESTHETIC AGENTS
The volatile inhalational anesthetics in current use are derivatives of some of the most potent chemical hepatotoxins developed for medicinal purposes. Chloroform, the original haloalkane anesthetic, has long been abandoned but remains an important experimental hepatotoxin, as does carbon tetrachloride (another chlorinated aliphatic hydrocarbon), which found use as an early vermifuge and is still employed as a household reagent in some parts of the world.1,8 Halothane (fluothane), introduced in the 1950s as a safer, nonexplosive alternative to ether, is a haloalkane compound that produced a well-described but rare syndrome of acute hepatotoxicity, usually after repeat exposure.9 The anesthetics that followed-methoxyflurane, enflurane, isoflurane—all have been implicated as a cause of similar injury, albeit less commonly for enflurane and isoflurane than for halothane; even fewer instances have been reported for the newest agents, sevoflurane and desflurane,10,11 because of their proportionally lower degree of metabolism.12 Halothane is no longer being produced in the United States but continues to be employed in other countries13 and is a case study in the elucidation of immunologic-mediated liver injury.14
HALOTHANE
The retrospective National Halothane Study, cited in the past as the basis for exonerating halothane as a cause of hepatotoxicity,15 is now considered flawed.1 Nearly 1000 cases of halothane hepatotoxicity were reported worldwide during the 1960s and 1970s.1,9,16 A fairly uniform clinical picture of postoperative fever, eosinophilia, jaundice, and hepatic necrosis occurred a few days to weeks after administration of anesthesia, usually after repeat exposure to halothane, and the case-fatality rate was high (Table 87-1). Rare cases of halothane-induced liver injury were reported after workplace exposure among anesthesiologists, surgeons, nurses, and laboratory staff and after halothane sniffing for “recreational” use; in affected persons, antibodies to trifluoroacetylated (TFA) proteins were demonstrated, indicating previous exposure.17
Table 87-1 Clinicopathologic Features of Halothane Hepatitis
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ULN, upper limit of normal.
Two types of postoperative liver injury have been associated with halothane. A minor form is seen in 10% to 30% of patients, in whom mild asymptomatic elevations in serum alanine aminotransferase (ALT) levels develop between the first and tenth postoperative days; the risk of hepatotoxicity is higher after two or more exposures to halothane than with subsequent use of alternative agents such as enflurane, isoflurane, and desflurane. Evidence of immune activation is lacking in these patients,18 in whom the ALT elevations generally are rapidly reversible. The major form of halothane-induced hepatotoxicity is a rare, dose-independent, severe hepatic drug reaction with elements of immunoallergy and metabolic idiosyncrasy (see Table 87-1). After an initial exposure to halothane, the frequency of this form of toxicity is only approximately 1 per 10,000,19 but the rate increases to approximately 1 per 1000 after two or more exposures, especially when the anesthetic agent is readministered within a few weeks.1 Typically, zone 3 (centrilobular) hepatic necrosis is seen histologically.20 The case-fatality rate ranged from 14% to 71% in the pre-liver transplantation era.1
Risk Factors for Halothane Hepatitis
Host-related risk factors for halothane hepatitis are listed in Table 87-2. The reaction is rare in childhood11; patients younger than 10 years of age represent only about 3% of the total, and cases in persons younger than 30 years account for less than 10%.11,16 The disease tended to be more severe in persons older than 40 years of age. Two thirds of cases have been in women, and repeat exposure to halothane (especially within a few weeks or months) is documented in as many as 90% of cases.1 The time between exposures can be as long as 28 years.21 After repeat exposure, hepatitis is earlier in onset and more severe. Obesity is another risk factor, possibly because of storage of halothane in body fat. The induction of cytochrome P450 (CYP) enzymes (especially CYP2E1) that metabolize halothane to its toxic intermediate has been produced experimentally with phenobarbital, alcohol, and isoniazid; valproate inhibits and phenytoin has no specific effect on halothane hepatotoxicity.1 Inhibition of CYP2E1 by administration of a single dose of disulfiram has been suggested as a means of preventing halothane hepatitis—by inhibiting the production of the metabolite responsible for neoantigen formation.22
Table 87-2 Risk Factors for Halothane Hepatitis
CYP2E1, cytochrome P450 2E1.
Familial predisposition to halothane-induced liver injury has been reported in closely related family members.23 Serum antibodies to volatile anesthetics have been found in pediatric anesthesiologists,17 who, like patients with halothane hepatitis, had higher levels of serum autoantibodies to CYP2E1 and to endoplasmic reticulum protein (ERp58) than those found in general anesthesiologists and control subjects who had never been exposed to inhalational anesthetics. The autoantibodies are not thought to have a role in pathogenesis.1
Pathology
In a study of 77 cases of halothane hepatitis reviewed by the Armed Forces Institute of Pathology,20 various degrees of liver injury were seen, depending on the severity of the reaction. Massive or submassive necrosis involving zone 3 was present in all autopsy specimens, whereas biopsy material revealed a broader range of injury—from spotty necrosis in about one third of cases to zone 3 necrosis in two thirds. The zone 3 injury is sharply demarcated, and the inflammatory response is less severe than in acute viral hepatitis.
Pathogenesis
Halothane injury occurs by one or more of three potential mechanisms: hypersensitivity, production of hepatotoxic metabolites, and hypoxia, in decreasing order of importance.1 Evidence for the role of hypersensitivity is found in the increased susceptibility and shortened latency after repeat exposure, the hallmark symptoms and signs of drug allergy (fever, rash, eosinophilia, and granuloma formation), and the detection of neoantigens and antibodies. Halothane oxidation yields trifluoroacetylchloride, which acts on hepatocyte proteins to produce neoantigens that are responsible for the major form of injury. By contrast, reductive pathways produce free radicals that can act as reactive metabolites that may have a role in causing minor injury. A unifying hypothesis set forth by Zimmerman1 suggests that halothane injury most likely is the result of immunologic enhancement of zone 3 necrosis produced by the reductive metabolite(s). Accordingly, the hepatotoxic potential of halothane depends on the susceptibility of the patient and on factors that promote production of hepatotoxic or immunogenic metabolites.1 A protective role for zinc pretreatment has been proposed based on studies in rats.24
Course and Outcome
Mortality rates for halothane hepatitis were high in early series; since then, successful treatment has been achieved with liver transplantation in many patients.13 When spontaneous recovery occurs, symptoms usually resolve within 5 to 14 days, and recovery is complete within several weeks.1 Immunosuppressive agents have only rarely been reported to improve the outcome.11 It is doubtful that halothane causes chronic hepatitis.1 Adverse prognostic factors include age older than 40 years, obesity, severe coagulopathy, serum bilirubin level greater than 20 mg/dL, and a shorter interval to onset of jaundice.1,16,19
OTHER ANESTHETIC AGENTS
The likelihood that individual haloalkane anesthetics will cause liver injury appears to be related to the extent to which they are metabolized by hepatic CYP enzymes: 20% to 30% for halothane, greater than 30% for methoxyflurane, 2% for enflurane, 1% for sevoflurane, and 0.2% or less for isoflurane and desflurane.12 Accordingly, the estimated frequency of hepatitis from the newer agents is much less than that for halothane (Table 87-3).
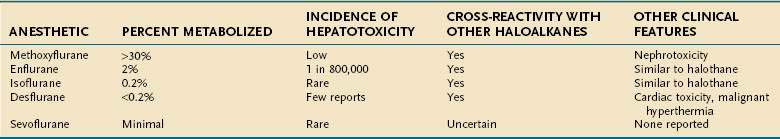
Methoxyflurane caused hepatotoxicity and a high frequency of nephrotoxicity that led to its withdrawal.25 Enflurane caused a clinical syndrome similar to that for halothane, with the onset of fever within 3 days and jaundice in 3 to 19 days after anesthesia26,27; the estimated incidence of enflurane-induced liver injury was about 1 in 800,000 exposed patients.10
Despite its low rate of metabolism,12 numerous instances of isoflurane-associated liver injury have been reported.28–30 In one case, cross-sensitivity was suspected 22 years after an initial exposure to enflurane.29 TFA liver proteins have been detected in patients with suspected isoflurane liver toxicity.30
The newest haloalkane anesthetics, desflurane and sevoflurane, appear to be nearly free of adverse hepatic effects. Desflurane undergoes minimal biotransformation and was not associated with the development of TFA antibodies in exposed rats.12 Only isolated reports of liver injury in patients receiving desflurane anesthesia have been published.31 The biotransformation of sevoflurane also is minimal, and only rare reports have implicated this agent in postoperative hepatic dysfunction.32 Ether, nitrous oxide, and cyclopropane apparently are devoid of significant hepatotoxic potential because of their lack of halogen moieties.1
JAUNDICE IN THE POSTOPERATIVE PERIOD
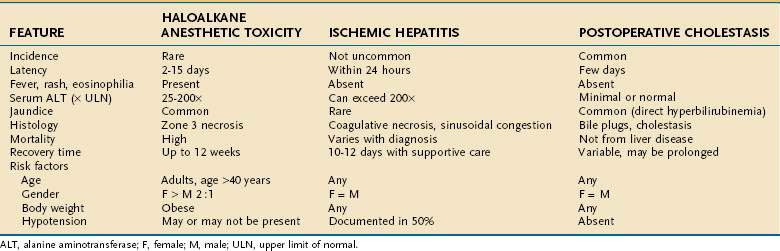
From 25% to 75% of patients undergoing surgery experience postoperative hepatic dysfunction, ranging from mild elevations in liver biochemical tests to hepatic failure; jaundice has been reported in nearly 50% of patients with underlying cirrhosis in the postoperative period (see Chapter 20).33 Patients undergoing upper abdominal surgical procedures are at highest risk of postoperative liver dysfunction, as well as pancreatitis, cholecystitis, and bile duct injury because of impaired blood flow to the liver.33 Table 87-4 lists many causes of postoperative jaundice and hepatic dysfunction, broadly divided into hepatocellular injury, cholestasis, and indirect hyperbilirubinemia. Drugs that may cause hepatoxicity in this setting include antibiotics (e.g., erythromycin, telithromycin, amoxicillin-clavulanate, trimethoprim-sulfamethoxazole) and the halogenated anesthetics discussed earlier; most produce injury by hypersensitivity mechanisms within one to two weeks of administration.1,2 Table 87-5 contrasts the features of halogenated anesthetic-induced hepatitis, ischemic hepatitis (shock liver) (see Chapter 83), and cholestatic injury (see Chapter 86) in the early postoperative period.
Table 87-4 Causes of Postoperative Hepatic Dysfunction
ALT, alanine aminotransferase; G6PD, glucose-6-phosphate dehydrogenase; NASH, nonalcoholic steatohepatitis.
CHEMICALS
COMMERCIAL AND INDUSTRIAL CHEMICALS
Among tens of thousands of chemical compounds in commercial and industrial use, several hundred are listed as causing liver injury by the National Institute for Occupational Safety and Health (NIOSH), as published in their Pocket Guide to Chemical Hazards.34 The National Library of Medicine also maintains a database of chemical toxins in its Toxicology and Environmental Health Information Program (TEHIP).35
Toxic exposure to chemical agents occurs most often from inhalation or absorption by the skin and less often from absorption by the gastrointestinal tract after oral ingestion or through a parenteral route. Because most chemical toxins are lipid soluble, when absorbed they can easily cross biological membranes to reach their target organ(s), including the liver.3,4 Hepatotoxic chemical exposure (as with carbon tetrachloride and phosphorus) usually results in an acute cytotoxic injury that typically consists of three distinct phases, similar to those observed after an acetaminophen overdose or ingestion of toxic mushrooms (Table 87-6).1,3 Less commonly, acute cholestatic injury may occur.36 Many chemicals (e.g., vinyl chloride) also are carcinogenic, and hepatic malignancies have been part of the clinicopathologic spectrum of chemical injury (Table 87-7).37 Although liver injury is the dominant toxicity for some agents, hepatic damage may be only one facet of more generalized toxicity for other agents.3
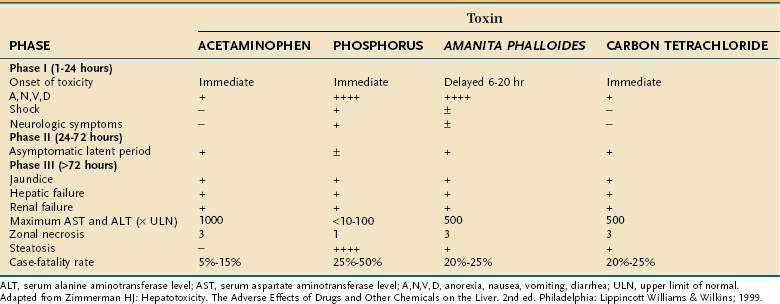
Table 87-7 Clinicopathologic Spectrum of Chemical Hepatotoxins
| Acute Injury |
| Necrosis |
Data from references 1, 3, 4, 36, and 37.
Carbon Tetrachloride and Other Chlorinated Aliphatic Hydrocarbons
Carbon tetrachloride (CCl4) is a classic example of a zone 3 hepatotoxin that causes necrosis leading to hepatic failure (see Table 87-6). Injury is mediated by its metabolism to a toxic trichloromethyl radical catalyzed by CYP2E1.7,38 Alcohol potentiates the injury through induction of this cytochrome.1 Most cases have been the result of industrial or domestic accidents, such as inhalation of CCl4-containing dry cleaning fluids that are used as household reagents or ingestion of these compounds by alcoholics who mistake them for potable beverages.1 At the cellular level, direct damage to cellular membranes results in leakage of intracellular enzymes and electrolytes, leading in turn to calcium shifts and lipid peroxidation.8 Hepatic steatosis develops as a result of triglyceride accumulation caused by haloalkylation-dependent inhibition of lipoprotein micelle transport out of the hepatocyte.38 CCl4 is more toxic than other haloalkanes and haloalkenes because toxicity correlates inversely with the level of bond dissociation energy, number of halogen atoms, and chain length (Table 87-8).1,38 In older series, complete clinical and histologic recovery from CCl4-induced liver damage was the rule with modest exposures, but supervening acute tubular necrosis and gastrointestinal hemorrhage were associated with a case-fatality rate of 10% to 25%.1,3 Activation of endonucleases, causing chromosomal damage and mutations, may result in carcinogenesis.38
| COMPOUND | RELATIVE TOXICITY |
|---|---|
| Carbon tetrachloride | ++++ |
| Tetrachlorethane | ++++ |
| Chloroform | ++ |
| Trichloroethylene | + to ++ |
| 1,1,2-Trichloroethane | + to ++ |
| Tetrachloroethylene | + |
| 1,1,1-Trichloroethane | + |
| Dichloromethane | ± |
| Dibromomethane | ± |
| Methylchloride | − |
Scale from ++++, maximal injury to −, trivial or no injury.
Chloroform remains an important experimental hepatotoxin, although its use as an anesthetic has long been abandoned (see later).1,3 Hepatic injury, including chronic hepatitis, has been reported with 1,1,1-trichloroethane.39
Hydrochlorofluorocarbons (HCFCs) have been associated with liver injury in several industrial workers exposed to dichlorotrifluoroethane (HCFC 123) and 1-chlorotetraflu-oroethane (HCFC 124), both of which are metabolized to reactive trifluoroacetyl halide intermediates similar to those implicated in halothane toxicity.40 Zone 3 necrosis is present on liver biopsy specimens, and autoantibodies against CYP2E1 or P58 are detected in the serum of many affected persons. As with halothane, liver toxicity may be potentiated by ethanol.41
Vinyl Chloride and Other Chlorinated Ethylenes
In the past, exposure to vinyl chloride monomer (VCM), or monochloroethylene, occurred in polymerization plants where vinyl chloride was heated to form polyvinyl chloride (PVC) in the manufacture of plastics; the toxic gas containing VCM was inhaled in this process.1 Vinyl chloride is ubiquitous in the environment and has been estimated by the Environmental Protection Agency to exist in at least 10% of toxic waste sites.4 Although PVC appears to be nontoxic, long-term exposure to VCM has led to chronic liver injury, including nodular subcapsular fibrosis, sinusoidal dilatation, peliosis hepatis, and periportal fibrosis associated with portal hypertension.1,3 Nonalcoholic fatty liver disease, including lipogranulomas, has been described in more than 50% of nonobese chemical workers with high exposure levels to VCM; some of these workers continued to have nonalcoholic steatohepatitis up to six years later.42
Vinyl chloride is carcinogenic. Angiosarcoma develops after a mean latency of 25 years after exposure; the risk is related to the duration and extent of contact.43 Alcohol appears to enhance the hepatocarcinogenicity of vinyl chloride, in rodents and possibly in humans, by inducing CYP2E1, which converts vinyl chloride to a toxic or carcinogenic metabolite (e.g., 2-chloroethylene oxide).1 A history of vinyl chloride exposure was found in 15% to 25% of all cases of hepatic angiosarcoma reported in the late 1970s,3 and strict hygienic measures instituted in 1974 have resulted in a marked decrease in the frequency of angiosarcoma since then; however, persons with the highest exposure still have a four-fold increased risk of developing periportal hepatic fibrosis, which may be a precursor of angiosarcoma.44 Persons previously exposed to vinyl chloride should undergo regular clinical examination for early detection of liver tumors, and those with known chronic liver disease or high levels of exposure should undergo regular hepatic imaging. Persons who work in PVC plants should undergo regular monitoring of liver biochemical test levels, and those with persistent abnormalities should be removed from workplace exposure.44 High serum levels of hyaluronic acid were correlated with the development of angiocarcinoma in 26 of 82 workers occupationally exposed to PVC in Kentucky.45
Nonhalogenated Organic Compounds
Benzene has been associated with minor hepatic injury in animals. Toluene led to steatosis and necrosis in a “glue sniffer”46 and has been associated with acute fatty liver of pregnancy; it caused elevations in serum gamma glutamyl transpeptidase levels after industrial exposure. Xylene can cause mild hepatic steatosis, and styrene (vinyl benzene) has led to elevated serum aminotransferase levels after prolonged exposure.
Trinitrotoluene and Other Nitroaromatic Compounds
Trinitrotoluene (TNT), or nitroglycerin, was first observed to be hepatotoxic during World War I, when severe acute and subacute hepatic necrosis developed in munitions workers in England, Germany, and the United States; the case-fatality rate was more than 25%.1,3 The frequency of hepatotoxicity during World War II was lower, with approximately 1 in 500 workers affected, but the estimated frequencies of methemoglobinemia and aplastic anemia were 50 times higher.3 Subacute hepatic necrosis followed two to four months of regular exposure to TNT. Percutaneous absorption was the major source of exposure. In some patients, rapidly progressive liver failure and death occurred within days to months, with massive hepatic necrosis at autopsy. In others, the subacute injury progressed over several months to micronodular cirrhosis and portal hypertension. The relatively low incidence of injury suggests that formation of a toxic metabolite was involved.1 Nitrobenzene and dinitrobenzene also were observed to be hepatotoxic during World War I. As with TNT, excessive exposure led to methemoglobinemia.
Nitroaliphatic Compounds
Nitromethane, nitroethane, and nitropropane cause variable degrees of hepatic injury. 2-Nitropropane (2-NP) has caused fatal massive hepatic necrosis after occupational exposure as a solvent, fuel additive, varnish remover, and rocket propellant. Toxic hepatitis associated with the chronic inhalation of propane and butane also has been reported.47
Polychlorinated Biphenyls and Other Halogenated Aromatic Compounds
Polychlorinated biphenyls (PCBs) are mixtures of trichloro-, tetrachloro-, pentachloro-, and hexachloro-derivatives of biphenyls, naphthalenes, and triphenyls that are used in the manufacture of electrical transformers, condensers, capacitors, insulating materials for electrical cables, and industrial fluids. Acute and chronic hepatotoxicity from PCB exposure seen during World War II resembled that caused by TNT.3,4 Inhalation of toxic fumes released by the melting of PCBs and chloronaphthalene mixtures during soldering of electrical materials was the most common means of exposure.1 The severity of liver injury correlated with the number of chlorine molecules.3 Liver damage appeared as early as seven weeks after ongoing exposure and was accompanied by anorexia, nausea, and edema of the face and hands. Acne-like skin lesions (chloracne) usually preceded hepatic injury. Once jaundice appeared, death occurred within two weeks in fulminant cases, which were characterized by massive necrosis (so-called acute yellow atrophy), or after one to three months in the subacute form. Cirrhosis developed in some persons who survived the acute injury.1
Polybrominated biphenyls (PBBs) appear to be even more toxic than PCBs. Consumption of milk and meat from livestock given feed mistakenly contaminated by a PBB has led to hepatomegaly and minor elevations in liver enzyme levels in exposed persons.3
Miscellaneous Chemical Compounds
Dimethylformamide is a solvent used in the synthetic resin and leather industries that causes dose-related massive hepatic necrosis in animals48 and is capable of producing focal hepatic necrosis and microvesicular steatosis in humans.3 Most persons exposed for more than one year have symptomatic disease that slowly resolves when they are removed from the workplace. Disulfiram-like symptoms can occur.49 Alcohol use, hepatitis B virus infection, and a high body mass index are risk factors.50
Hydrazine and its derivatives used in jet and rocket fuel cells are also experimental hepatotoxins and carcinogens and have been reported to cause hepatic steatosis in animals1 and reversible injury in humans after inhalation.51 Bromoalkanes and iodoalkanes, used in insecticides and aircraft fuels, have rarely caused hepatic injury.3 Ethylene dibromide (dibromoethane) has led to zone 3 hepatic necrosis after ingestion in attempted suicide or as fatal hepatotoxicity associated with nephrotoxicity and cardiotoxicity following occupational exposure or inadvertent poisoning.52
PESTICIDES
Although exposure to insecticides, herbicides, and other pesticides is common, acute liver injury resulting from these compounds, many of which are chlorinated hydrocarbons, is rare.1,3 Evidence that dichlorodiphenyl-trichloroethane (DDT) and other organochlorines (aldrin, amitrole, chlordane, dieldrin, lindane, mirex) lead to liver damage or carcinogenicity is limited.1 Agent Orange (2.4-dichlorophenoxyacetic acid), the defoliant widely used in Vietnam, has been reported to cause acute hepatitis after chronic exposure; however, contaminating dioxins have been suggested to be responsible for the toxic effects.53,54 Moreover, chronic liver injury among Vietnam veterans is more likely to have been related to viral infections or alcohol than to Agent Orange,55 and hepatocarcinogenesis is more likely to have been related to chronic hepatitis B infection.56
Ingestion of or dermal exposure to dichloride dimethyldipyridilium (paraquat) has been implicated in several instances of hepatotoxicity as a result of attempted suicide and homicide.57 Patients may present with severe vomiting and profuse diarrhea leading to hypokalemia and often have evidence of oral, pharyngeal, and esophageal caustic injury after ingestion. Death results from a combination of renal, respiratory, cardiac, and hepatic failure; mortality rates are as high as 70%, and death often occurs within the first 48 hours. Treatment with charcoal hemoperfusion in conjunction with cyclophosphamide, dexamethasone, furosemide, and vitamins B and C—the so-called Caribbean scheme—has been attempted, but persons who ingest more than 45 mL are likely to die with or without this treatment.57 Histopathologic changes include zone 3 necrosis followed by injury to small- and medium-sized interlobular bile ducts.58
Chlordecone (Kepone) has been shown to impair biliary excretion and lipid transport and storage,59 but neurologic toxicity appears to dominate the clinical injury. Occupational exposure has led to hepatic steatosis and elevated serum aminotransferase levels. Trivial hepatic enzyme abnormalities have been seen in persons heavily exposed to chloretone.3 Hexachlorobenzene in contaminated grain has been associated with an epidemic of porphyria cutanea tarda and liver injury.3
Inorganic arsenic has long been used as a homicidal or suicidal agent, and toxic exposure in the past also followed ingestion of Fowler’s solution (arsenic trioxide) used as a treatment for psoriasis and asthma.1,3 Other sources of exposure are contaminated ground and well water and homemade alcohol. Doses greater than 3 g can cause death in one to three days; hepatic injury generally is overshadowed by gastrointestinal, neurologic, and vascular effects,1 leading ultimately to central nervous system depression and vascular collapse. A lesion resembling hepatic sinusoidal obstruction syndrome (hepatic veno-occlusive disease) can develop,3 and noncirrhotic portal hypertension developed in more than 90% of 248 patients who consumed contaminated drinking water for up to 15 years.60
Occupational exposure to arsenic is still observed among vineyard workers, farmers, and gold miners,61 although its use as an insecticide has been curtailed since the 1940s. Lumber treated with chromated copper arsenate as a preservative may be an additional source of exposure.62 The clinical syndrome associated with arsenicosis includes skin lesions (blackfoot disease), anemia, diabetes mellitus, hearing loss, neurobehavioral disorders, and cardiovascular diseases, in addition to benign and malignant liver disease.63 Chronic hepatic injury, including cirrhosis and noncirrhotic portal hypertension, may be a precursor to hepatic neoplasms, such as angiosarcomas, hemangioendotheliomas, and hepatocellular carcinomas, after exposure of more than 10 years.64 Treatment with thiol chelators has had variable success in cases of prolonged exposure, and coadministration of antioxidants, such as vitamins C and E, may be of added benefit.65
METALS
IRON
Most of the 5000 cases of accidental iron poisoning in the United States each year occur in young children who mistake iron supplements for candy.1 The severity of injury correlates with the dose ingested3; ingestion of less than 20 mg/kg of elemental iron is unlikely to produce serious toxicity, whereas doses of more than 200 mg/kg can be fatal.1 Severe injury has been seen only with serum iron concentrations above 700 −g/dl measured within the first 12 hours after ingestion.66 Iron, per se, is not hepatotoxic, but ferric and ferrous ions can act through free radicals and lipid peroxidation to cause membrane disruption and necrosis.67 Clinically evident liver injury is uncommon, but zone 1 necrosis occurs in the most severe cases.1 Clinical illness is characterized by sequential phases of gastrointestinal injury, subsidence of symptoms, and overt hepatotoxicity accompanied by renal failure. Deferiprone, an oral iron chelator, was implicated in causing worsening hepatic fibrosis in a long-term study of patients with thalassemia68; however, these findings were not confirmed by subsequent histopathologic analysis,69 and injury from this agent appears unlikely.
PHOSPHORUS
Poisoning by white phosphorus has been rare since its use in firecrackers and matches was outlawed in the mid-twentieth century.3 Cases reported since then usually have been the result of ingestion of rat or roach poison.1 Shortly after ingestion, vomiting, gastrointestinal bleeding, convulsions, shock, and death occur within 24 hours. Phosphorescence of the vomitus and stools and a typical garlic-like odor on the breath are characteristic, when present. The predominant hepatic lesion is steatosis and necrosis, most prominent in the periportal region. Serum aminotransferase levels generally are no higher than 10 times the upper limit of normal.1
COPPER SALTS
Acute poisoning by copper leads to a syndrome resembling iron toxicity. Ingestion of toxic amounts (1 to 10 mg) usually is seen with suicidal intent, especially on the Indian subcontinent.3,67 Vomiting, diarrhea, and abdominal pain accompanied by a metallic taste are seen during the first few hours after ingestion. Gastrointestinal tract erosions, renal tubular necrosis, and rhabdomyolysis often accompany zone 3 hepatic necrosis by the second or third day. Jaundice results from both hepatic injury and acute hemolysis caused by high blood copper levels.3 The mortality rate is 15%, with early deaths resulting from shock and circulatory collapse and late deaths resulting from hepatic and renal failure.1
THORIUM DIOXIDE
Thorium dioxide (Thorotrast) was used as an intravenous contrast medium for radiographic procedures in the first half of the twentieth century; more than 50,000 persons may have been exposed.1 Thorotrast was subsequently found to cause hepatic angiosarcomas after latency periods of 20 to 40 years. As with arsenic, reports of hepatic sinusoidal obstruction syndrome and a Budd-Chiari-like syndrome of portal hypertension also have appeared. Given the extraordinarily long half-life (hundreds of years) of the compound, which is a radioactive alpha emitter, exposed persons remain at risk for the development of leukemia, in addition to hepatocellular cancer.70 Histologically, thorium dioxide is found in Kupffer cells and macrophages as dark brown refractile granules, the identity of which can be confirmed by spectrographic analysis.38
OTHER METALS
Although cadmium produces hepatic necrosis and cirrhosis in laboratory animals,71 evidence is lacking that exposure to cadmium causes important human injury.1 Several metals are associated with apoptosis, which might explain their potential for hepatotoxicity.72 Beryllium has led to midzonal liver necrosis as a result of phagocytosis of insoluble beryllium phosphate by Kupffer cells.1 Chronic industrial exposure (usually by inhalation of high concentrations of oxide or phosphorus mixtures) is associated with the formation of hepatic (and pulmonary) granulomas.3 Therapy with chelating agents and antioxidants has been used in animal models of beryllium toxicity.73 Lead hepatotoxicity may be seen as part of the larger symptom complex of abdominal pain, constipation, and encephalopathy that occurs with chronic ingestion or environmental exposure.74
ADULTERATED COOKING OILS AND CONTAMINATED FOODS
The Spanish toxic oil syndrome occurred in 1981, after exposure of up to 100,000 Spaniards to rapeseed cooking oil that was contaminated by anilines and acetanilides. Nearly 20,000 persons became ill, many with hepatic injury and jaundice. Approximately 2500 died.75 Among 332 patients followed for up to eight years, hepatic injury developed in 43%, usually at the onset of a multisystem disease. A mixed cholestatic-hepatocellular injury pattern was seen, with jaundice or hepatomegaly in fewer than 20%. After an eight-year follow-up, liver disease persisted in only four patients.75
Epping jaundice refers to an epidemic of toxic liver injury that occurred in Epping, England, in 1965.3,36,76 The outbreak involved 84 persons who had eaten bread contaminated with methylenedianiline that had spilled onto the floor of a van carrying flour. The clinical syndrome consisted of abdominal pain, fever, and chills, followed by cholestatic jaundice resembling that seen with biliary obstruction; eosinophilia was seen in about one half of patients. Liver biopsy specimens revealed Kupffer cell hyperplasia with portal inflammation but little or no necrosis.3 Most persons recovered in four to six weeks, with jaundice lasting up to four months in a few. The mechanism of injury was thought to be a chemically induced cholangitis, possibly as a result of a hypersensitivity reaction. Cholangiocarcinoma later developed in one patient.76
Yusho oil disease in western Japan, and a related epidemic referred to as yu-cheng in Taiwan, involved nearly 2000 persons who had eaten rice prepared in oil contaminated by PCBs, dioxins, and polychlorinated dibenzofurans in 1968. The disease was characterized by chloracne, skin hyperpigmentation, eyelid edema, and neuropathy, with jaundice reported in approximately 10% of patients.77 Exposed persons still harbored high levels of these agents nearly three decades after the outbreak.77
Hexachlorobenzene contamination of wheat in the 1950s led to an epidemic of toxic porphyria cutanea tarda and severe liver disease involving more than 3000 Turkish Kurds, with a mortality rate that exceeded 10%. This fungicide had been added to seed grain that was used for food during a famine.3,36
DRUGS OF ABUSE
COCAINE
Cocaine is a dose-dependent hepatotoxin.3 Acute cocaine intoxication affects the liver in 60% of patients,73 and many affected persons have markedly elevated serum ALT levels (greater than 1000 U/L). Associated features include rhabdomyolysis, hypotension, hyperpyrexia, disseminated intravascular coagulation, and renal failure. Hepatic injury probably is the result of toxic metabolites (e.g., norcocaine nitroxide) formed by the CYP system, specifically CYP2E1 and CYP2A,79 and enhanced hepatotoxicity is seen in persons who regularly consume alcohol.3 In animals, pretreatment with N-acetylcysteine decreases the risk of cocaine hepatotoxicity,80 although the usefulness of N-acetylcysteine for treating human cocaine-induced hepatic injury has not been determined.
OTHERS
“Ecstasy” (3,4-methylenedioxymethamphetamine) is a euphorigenic and psychedelic amphetamine derivative that can lead to hepatic necrosis as part of a heat stroke–like syndrome resulting from exhaustive dancing in hot nightclubs (“raves”).81 The injury can be fatal and has necessitated liver transplantation in some instances.82,83 The role of CYP enzymes in the toxicity of this and other so-called “designer drugs” may relate to specific genetic polymorphisms of CYP2D6 or other cytochromes.84
Phencyclidine (“angel dust”) is another stimulant that can lead to hepatic injury as part of a syndrome of malignant hyperthermia that produces zone 3 hepatic necrosis, congestion, and collapse, with high serum aspartate aminotransferase (AST) and ALT levels reminiscent of ischemic hepatitis.85
BOTANICAL AND ENVIRONMENTAL HEPATOTOXINS
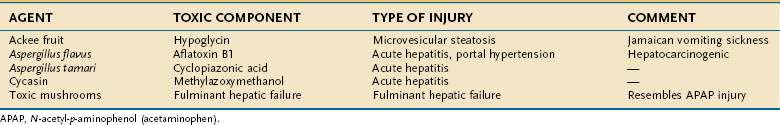
Examples of hepatotoxic mushrooms, fruits, and other foodstuffs, including grains and nuts contaminated by fungal mycotoxins or other potentially injurious compounds, are listed in Table 87-9.
MUSHROOMS
Poisonous varieties of mushrooms number approximately 100 among the more than 5000 species, and more than 8000 mushroom poisonings were reported in the United States in 2001.86 Greater than 90% of cases of fatal poisoning are caused by Amanita phylloides (death cap) or Amanita verna (destroying angel), found in the Pacific Northwest and eastern United States.87 A fatal outcome can follow ingestion of a single 50-g (2-oz) mushroom; the toxin is one of the most potent and lethal in nature.88 Alpha-amatoxin is thermostable, can resist drying for years, and is not inactivated by cooking. Rapidly absorbed through the gastrointestinal tract, the amatoxin reaches hepatocytes through the enterohepatic circulation and inhibits production of messenger RNA and protein synthesis, leading in turn to cell necrosis. A second toxin, phalloidin, is responsible for the severe gastroenteritis that precedes hepatic and central nervous system injury.89 Phalloidin disrupts cell membranes by interfering with polymerization of actin. A latent period of 6 to 20 hours after ingestion of a mushroom precedes the first symptoms of intense abdominal pain, vomiting, and diarrhea. Hepatocellular jaundice and renal failure occur over the next 24 to 48 hours and are followed by confusion, delirium, convulsions, and eventually coma by 72 hours.1,89 The characteristic hepatic lesion is steatosis and zone 3 hepatic necrosis, with nucleolar inclusions seen on electron microscopy.3
In a case series of eight patients,90 the mean serum AST level was 5488 U/L (range, 1486 to 12,340), ALT 7618 (range, 3065 to 15,210), and bilirubin 10.5 mg/dL (range, 1.8 to 52), with peak levels on days four and five. Acute kidney injury requiring dialysis developed in one patient, and three exhibited encephalopathy. Mortality rates traditionally have been high, especially when the serum ALT level exceeds 1000 U/L, and emergency liver transplantation often is required90; however, some patients survive with conservative management, which includes nasogastric lavage with activated charcoal, intravenous penicillin G, N-acetylcysteine (using a standard acetaminophen [N-acetyl-p-aminophenol, or APAP] protocol of a loading dose of 140 mg/kg orally followed by 15 additional oral doses of 70 mg/kg or one of two intravenous protocols: for presentation within 10 hours of exposure, a loading dose of 150 mg/kg infused over 1 hour, followed by 50 mg/kg over 4 hours, followed by 100 mg/kg over 16 hours; for presentation more than 10 hours after exposure, a loading dose of 140 mg/kg over 1 hour, followed by 70 mg/kg every 4 hours for at least 12 doses), and milk thistle (Silybum marianum).89 The use of these therapeutic modalities is not always effective, and in a large review of 2108 cases over a 20-year period in the United States and Europe,91 penicillin G, either alone or in combination with other therapy, demonstrated limited benefit. Similarly, no role for glucocorticoids was found. The utility of plasmapheresis or hemoperfusion is unproved.
OTHER FOODSTUFFS
The unripe fruit of the ackee tree (Blighia sapida), native to Jamaica, contains a hepatotoxin, hypoglycin A, that produces a clinical syndrome of gastrointestinal distress and microvesicular steatosis known as Jamaican vomiting sickness, which resembles Reye’s syndrome (see Chapter 86).3,36,92 Cholestatic jaundice has been described after chronic ingestion.93
Cycasin is a potent hepatotoxin and hepatocarcinogen found in the fruit of the cycad tree (Cycas circinalis, Cycas revoluta). A small epidemic of acute hepatic injury attributable to the ingestion of cycad nuts was reported from Japan. The purported toxin is methylazoxymethanol, which normally is eliminated or rendered inactive in preparing the nuts before ingestion.3
Aflatoxins are a family of mycotoxins found in Aspergillus flavus and related fungi that are ubiquitous in tropical and subtropical regions. They contaminate peanuts, cashews, soybeans, and grains stored under warm, moist conditions and are well-known hepatotoxins and hepatocarcinogens.1,3 Aflatoxin B1, a potent inhibitor of RNA synthesis, is the most hepatotoxic member of the family. Reactive metabolites are formed by the CYP system, and malnutrition is a possible potentiating factor (perhaps because of the depletion of glutathione). When consumed in large quantities, aflatoxin B1 is responsible for a clinical syndrome characterized by fever, malaise, anorexia, and vomiting followed by jaundice. Portal hypertension with splenomegaly and ascites may develop over the next few weeks. In large epidemics, mortality rates have approached 25% and correlate with the dose ingested.3 Zone 3 hepatic necrosis without inflammation is the characteristic lesion. Other histologic findings include cholestasis, microvesicular steatosis, and bile duct proliferation.3
The risk of hepatocellular carcinoma (HCC) correlates with the amount of aflatoxin consumed, especially in sub-Saharan Africa and eastern China, where wheat often exceeds rice as a staple in the diet (see Chapter 94).3 Alcohol and possibly exposure to DDT may play an enhancing role in hepatocarcinogenesis.94 An even more important cofactor may be the hepatitis B virus.95 The frequency of a mutation in the TP53 tumor suppressor gene correlates with the development of HCC in these regions, but this mutation is rare in HCC from Western countries (see Chapter 94).95
VITAMINS AND HERBAL PREPARATIONS
The use of vitamins, dietary supplements, and herbal and nonproprietary remedies is an important aspect of CAM. This field continues to grow in the United States and around the world (see Chapter 127).96 In the United States, alternative medicines were used by 34% of the population in 1990 and 42% in 1997; nearly 20% of the population took complementary medicines at the same time as conventional prescriptions.97 The use of herbal products is even more popular among patients with chronic liver disease,98,99 despite the absence of controlled clinical trials to assess safety and efficacy in this setting.100 Many so-called health foods, dietary and weight loss supplements, and herbal products are potent hepatotoxins that have led to acute liver failure and the need for emergency liver transplantation.5,6,8,101,102 Dietary supplements containing anabolic androgenic steroids may cause severe cholestatic liver injury.103,104
HYPERVITAMINOSIS A
Vitamin A (retinol) is a dose- and duration-dependent hepatotoxin capable of causing injury ranging from asymptomatic elevations in serum aminotransferase levels with minor hepatic histologic changes to perisinusoidal fibrosis leading to noncirrhotic portal hypertension and, in some cases, cirrhosis.105 Approximately one third of the U.S. population is estimated to take vitamin supplements containing vitamin A, with as many as 3% of products providing a daily dose of at least 25,000 IU. Hypervitaminosis A usually is the result of self-ingestion, rather than intentional overdose, and all age groups are represented.106 The average daily dose of vitamin A in reported cases of liver disease has been nearly 100,000 IU over an average duration of 7.2 years, for a mean cumulative dose of 229 million IU, but liver injury has been described with daily doses of 10,000 to 45,000 IU,107 and cirrhosis has occurred after a daily intake of 25,000 IU for at least 6 years.105,107 Long-term use of low-dose vitamin A supplements (250 to 5000 retinol equivalents per day) does not appear to be toxic.108
Because of the long half-life of vitamin A in the liver (50 days to 1 year),107,109 the fibrotic process may continue due to the slow release of hepatic vitamin A stores despite discontinuation of oral intake of the vitamin. Genetic factors may play a role, and apparent familial hypervitaminosis A occurred in four siblings who ingested large doses as treatment for congenital ichthyosis.110 Vitamin A toxicity has been reported in native Alaskans who ingest large amounts of fresh polar bear liver.105 Water-soluble, emulsified, and solid formulations of vitamin A are up to 10 times as toxic as oil-based preparations.111
Hepatotoxicity from vitamin A has been attributed to activation of hepatic stellate cells, the body’s principal storage site of the vitamin. Resulting hyperplasia and hypertrophy produce sinusoidal obstruction and increased collagen synthesis, leading in turn to portal hypertension.112 Rare cases of peliosis hepatis also have been attributed to hypervitaminosis A. Beta carotene, a precursor of vitamin A, is involved in the neoplastic transformation of squamous cell lung and tracheal tissues, especially in smokers who consume alcohol.113
Liver biopsy specimens show increased storage of vitamin A, seen as characteristic greenish autofluorescence on irradiation with ultraviolet light.105 The excess vitamin A is stored initially in stellate cells that lie in the space of Disse and become hyperplastic and hypertrophic. The enlarged clear stellate cells compress the hepatic sinusoids, giving rise to a “Swiss cheese,” or honeycombed, appearance.105 Hepatocellular injury usually is minor, with microvesicular steatosis and focal degeneration and without significant necrosis or inflammation. Hepatic fibrosis in a perisinusoidal distribution can arise from activated stellate cells that transform into myofibroblasts. In one series,105 cirrhosis was present in 59%, chronic hepatitis in 34%, microvesicular steatosis in 21%, perisinusoidal fibrosis in 14%, and peliosis in 3% of cases.
Hypervitaminosis A also can involve the skin and central nervous system.1 Hepatomegaly is common, and in severe cases, splenomegaly, ascites, and esophageal variceal bleeding may be features.1,5 Liver biochemical test abnormalities, present in two thirds of cases, are nonspecific, with only modest elevations in serum aminotransferase and alkaline phosphatase levels.
The diagnosis of vitamin A toxicity rests on a dietary and medication history and clinical suspicion. Plasma vitamin A levels may be normal, and the diagnosis is supported by the demonstration of increased hepatic stores of vitamin A and characteristic histologic findings.114 The diagnosis may be delayed for several years if hepatotoxicity is not recognized or is misdiagnosed.105,107
Symptoms resolve and liver enzymes normalize gradually after discontinuation of vitamin A ingestion in less severe cases, but deterioration may continue in cases of severe intoxication, particularly when cirrhosis is already present.107 Features of liver failure and cirrhosis at the time of diagnosis indicate a poor prognosis, and liver transplantation may be required.1 Alcohol can potentiate hepatotoxicity and should be avoided. Vitamin A supplements generally should be avoided in other types of liver disease because of possible accentuation of hepatic injury and fibrosis.113 Severe liver injury rarely has been reported with the use of acitretin, a vitamin A metabolite.115
HERBAL REMEDIES AND NUTRITIONAL SUPPLEMENTS
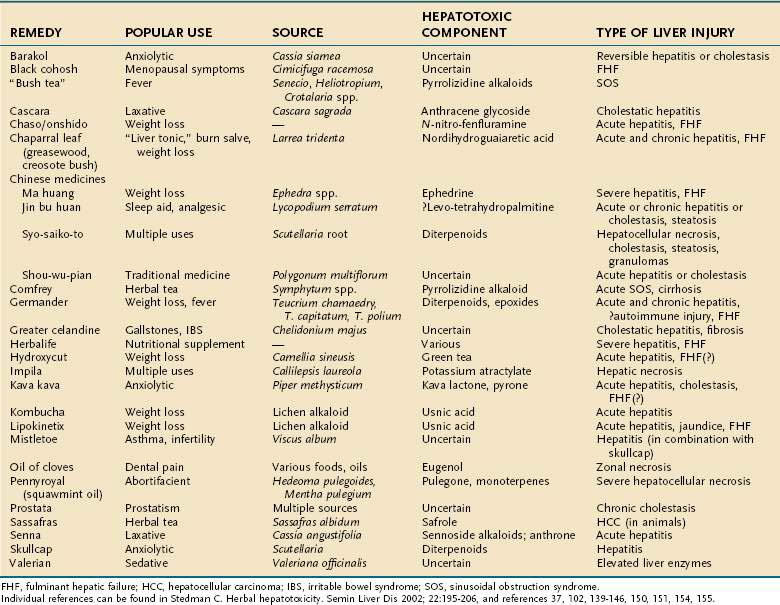
The increasing use of CAM is well described in patients with liver disease.96–99 Silymarin (Silybum marianum, milk thistle) is the most commonly used herbal preparation among these patients, and while it appears to be quite safe, there is an increasing number of reports of hepatotoxicity from several classes of herbal and weight reduction agents that has paralleled the rise in CAM therapies.5,6,37,102 Warnings have been issued for several agents, and in a few instances, the U.S. Food and Drug Administration (FDA) and other health authorities have requested their removal from the marketplace (e.g., kava kava, ephedra [ma huang], Lipokinetix, and Hydroxycut in the United States [see FDA.gov/Food/ResourcesforYou/Consumers/ucm08542.htm] and germander in France). Any patient with liver disease should be questioned about the ingestion of herbal remedies. Estes and colleagues101 documented the use of several commonly promoted herbal agents (including Lipokinetix, skullcap, ma huang, chaparral, and kava kava) in 50% of their patients with acute liver failure over a two-year period. The agents were used by an equal number of men and women, for several months to 15 years. Six of 10 patients underwent emergency liver transplantation (with 2 deaths), 3 died before transplantation, and only 1 patient recovered spontaneously. Table 87-10 lists various herbal remedies according to their toxic constituent and the nature of the associated liver disease.
Pyrrolizidine Alkaloids
Pyrrolizidine alkaloids are found in approximately 3% of all flowering plant species throughout the world, and ingestion of such plants, often as medicinal teas or in other formulations, can produce acute and chronic liver disease, including sinusoidal obstruction syndrome (SOS), in humans and livestock.116 SOS was first reported in the 1950s as a disease of Jamaican children, manifesting with acute abdominal distention, marked hepatomegaly, and ascites, a triad that resembled Budd-Chiari syndrome (see Chapter 83).3 The disease was linked to consumption of “bush tea,” made largely from plants of Senecio, Heliotropium, and Crotalaria species and taken as a folk remedy for acute childhood illnesses that are characterized histologically by centrilobular hepatic congestion with occlusion of the hepatic venules leading to congestive cirrhosis. Comfrey (Symphytum officinale) remains commercially available even though it is a dose-dependent hepatotoxin.6,116 In Afghanistan, ingestion of pyrrolizidine alkaloid-contaminated grains and bread led to a large epidemic of SOS, affecting 8000 persons and innumerable sheep.3
Hepatotoxic pyrrolizidine alkaloids are cyclic diesters, and some forms (e.g., fulvine, monocrotaline) cause both liver and lung injury.116 The mechanism of injury is postulated to be impairment of nucleic acid synthesis by reactive metabolites of pyrrolizidine alkaloids generated by hepatic microsomes, leading, in turn, to progressive loss of sinusoid cells and sinusoidal hemorrhage, as well as injury to the endothelium of the terminal hepatic venule, with deposition of fibrin.116,117
SOS causes acute, subacute, and chronic injury. The acute form is characterized by zone 3 necrosis and sinusoidal dilatation, leading to a Budd-Chiari-like syndrome with abdominal pain and the rapid onset of ascites within three to six weeks of ingestion. In Jamaica, the course was rapidly fatal in 15% to 20% of affected persons. Approximately one half of the patients with the acute form recovered spontaneously; transition to a more chronic form of injury occurred in the remainder.1,3 In the subacute and chronic forms, central fibrosis and bridging between central veins led to a form of cirrhosis similar to that seen with chronic passive hepatic congestion (so-called cardiac cirrhosis). At one time, this form of injury accounted for one third of the cases of cirrhosis seen in Jamaica, with death often resulting from complications of portal hypertension in as few as one to three years.3 Certain pyrrolizidine alkaloids, such as comfrey extracts, are hepatocarcinogenic and, like aflatoxins, induce mutations of the TP53 gene.116
Germander
The blossoms of plants from the Labiatae family (Teucrium chamaedrys) were used for years in herbal teas and in the mid-1980s as capsules for weight reduction in France, until several dozen cases of liver injury, including fatal hepatic failure,118,119 forced its withdrawal from the French marketplace in 1992.119 Most patients were middle-aged women who had ingested germander for 3 to 18 weeks, with consequent development of acute hepatocellular injury, often with jaundice.119 The injury usually resolved within 1.5 to 6 months after the germander was discontinued, with prompt recurrence after rechallenge in many persons. The cause of germander hepatotoxicity is an interplay between toxic metabolites and immunoallergic mechanisms. Germander is composed of several compounds, including glycosides, flavonoids, and furan-containing diterpenoids, all of which are converted by the CYP system (especially CYP3A) to reactive metabolites. Covalent binding to cellular proteins, depletion of hepatic glutathione, apoptosis, and cytoskeleton membrane injury (bleb formation) cause cell disruption in animal models.6,8 Epoxide hydrolase on plasma membranes is a target of germander antibodies, which have been found in the sera of patients who have consumed germander teas over long periods of time.120
Reports of liver injury also have appeared with other species of Teucrium, including Teucrium capitatum121 and Teucrium polium.122
Chaparral
The dried leaf of the desert shrub chaparral (Larrea tridentata), also known as greasewood or creosote bush, is ground into a tea or used in capsules or tablets for various ailments. Multiple reports of hepatitis have appeared; most cases have occurred within 1 to 12 months of use and resolved within a few weeks to months of discontinuation.123 Among 13 cases reported to the FDA,123 acute hepatocellular or cholestatic injury was observed, with 2 cases of fulminant hepatitis requiring liver transplantation and 4 cases of progression to cirrhosis. Renal toxicity and skin rash can accompany liver injury. The active ingredient, nordihydroguaiaretic acid, an inhibitor of cyclooxygenase and lipoxygenase pathways, is the likely cause of hepatic injury, although the mechanism also may involve phytoestrogen-induced effects on the liver.101 A case of recurrence on rechallenge suggests a possible role for immunoallergy.6
Pennyroyal
The leaves of pennyroyal (the common name for two related plant species, Hedeoma pulegoides and Mentha pulegium) are used to make oils (squawmint oil), tablets, and home-brewed mint teas. The plant contains pulegone and smaller amounts of other monoterpene ketones. Oxidative metabolites of pulegone (e.g., menthofuran) bind to cellular proteins and deplete hepatic glutathione, thereby leading to liver injury.124 Cases of hepatocellular injury, including fatal necrosis, were associated with gastrointestinal and central nervous system toxicity within a few hours of ingestion. In animals, inhibition of pulegone metabolism by the CYP system with disulfiram and cimetidine has limited pennyroyal hepatotoxicity.125 The use of N-acetylcysteine may protect against pennyroyal toxicity in human cases.124
Chinese Herbal Medications
Jin bu huan (Lycopodium serratum) is a traditional herbal remedy that has been used as a sedative and analgesic for more than 1000 years.6 Numerous cases of hepatic injury have appeared,126,127 with a mean latency of 20 weeks (range, 7 to 52 weeks) after the start of jin bu huan in recommended doses. Associated symptoms and signs included fever, fatigue, nausea, pruritus, abdominal pain, hepatomegaly, and jaundice. Liver biopsy specimens from a small number of patients showed a range of histopathologic changes, including lobular hepatitis with prominent eosinophils, mild hepatitis with microvesicular steatosis, and fibrotic expansion of the portal tracts. The injury resolved within a mean of 8 weeks (range, 2 to 30 weeks) but could recur on rechallenge.126 The only predisposing factor was female gender. Serum ALT levels were increased 20- to 50-fold, with minor increases in the alkaline phosphatase level, except in one patient with cholestasis. Hyperbilirubinemia was prominent in the more severe cases. A case of chronic hepatitis has been described.6 The mechanism of injury may involve levo-tetrahydropalmatine, a neuroactive metabolite with structural similarity to pyrrolizidine alkaloids. At present, the FDA has banned the importation of jin bu huan anodyne tablets into the United States.6
Syo-saiko-to (xiao-chai-hu-tang, dai-saiko-to) contains Scutellaria root (skullcap), which is a postulated hepatotoxin.128 The spectrum of liver injury has included hepatocellular necrosis, microvesicular steatosis, cholestasis, granuloma formation, and a flare of autoimmune hepatitis.129 Reversible acute hepatitis or cholestasis has followed the consumption of shou-wu-pian, a product derived from Polygonum multiflorum.130
Ma huang, derived from plants of Ephedra species, has been reported to cause acute, sometimes severe, hepatitis, including acute liver failure.101,131,132 The active constituent, ephedrine, also has been linked to severe adverse cardiovascular and central nervous system effects, including fatalities, when used as a stimulant and weight loss aid.133 The FDA has issued a ruling that ephedra-containing products present an unreasonable risk and should be avoided.134
Weight Loss Products
Chaso and onshido are Chinese herbal dietary weight loss supplements that were reported to cause severe liver injury, with a mean serum ALT level of 1978 U/L (range, 283 to 4074 U/L), in 12 patients.135 Fulminant hepatic failure developed in two persons: one died, and the other survived after receiving a liver transplant. The suspected hepatotoxic ingredient was N-nitroso-fenfluramine, a derivative of the appetite suppressant fenfluramine, which was withdrawn from the U.S. market in 1997.136
Another dietary supplement used for weight loss, Lipokinetix (composed of norephedrine, sodium usniate [usnic acid], diiodothyronine, yohimbine, and caffeine), has been associated with acute hepatitis, including fulminant hepatic failure requiring liver transplantation.101,137 In a case series of seven previously healthy patients (four women, three men; mean age, 27 years), acute hepatitis developed after a latent period of less than 4 weeks in five patients and 8 to 12 weeks in the other two. Mean serum ALT levels were 4501 U/L (range, 438 to 14,150 U/L), and mean serum bilirubin levels were 6.5 mg/dL (range, 2.2 to 14.6 mg/dL). No evidence of immunoallergy was evident. All of the patients recovered spontaneously, with normalization of serum ALT and bilirubin levels within four months.
Fulminant hepatic failure necessitating emergency liver transplantation was reported in a previously healthy 28-year-old nonobese woman who had taken an over-the-counter preparation of usnic acid for weight loss,138 suggesting that this agent may be the hepatotoxic component of Lipokinetix. Usnic acid also is a component of Kombucha tea, which has been associated with hepatic injury.6 Usnic acid is a potent inhibitor of CYP2C19 and CYP2C9 and may interact with other medications or supplements to produce hepatotoxic drug-drug interactions.139 Herbalife is the latest nutritional supplement to be reported to cause severe liver injury, including the need for liver transplantation.140,141 A mixed hepatic infiltrate with eosinophils and lymphocytes was seen on liver biopsy specimens, along with other changes, including necrosis, cholestasis, and sinusoidal obstruction. A positive rechallenge reaction was observed in a number of patients after their liver biochemical abnormalities had normalized.140
Hydroxycut was withdrawn from the U.S. market in 2009 after an FDA review of reports found that the slimming aid was associated with possible hepatotoxicity.142 One of its active ingredients, green tea (Camellia sinensis), despite being used widely for millennia,143 has been implicated in liver injury.144 A safety review of green tea by the U.S. Pharmacopeia145 found that in each reported case a relation with liver injury was possible, although the study used the Naranjo causality scale, which is not specific for assessing hepatotoxicity.146
Kava Kava
Kava kava is a natural sedative and antianxiety agent derived from the root of the pepper plant (Piper methysticum). This herbal product has been the subject of an FDA consumer alert6 after it was banned in the European Union and Canada147 because of severe hepatotoxicity, including fatal liver failure.101,148,149 A review of 78 cases of hepatic injury reported to the FDA included 11 cases of liver failure requiring liver transplantation and 4 deaths.149 Other investigators, however, have questioned the validity of the causality assessment, and only rare instances of hepatotoxicity are found when a more accurate causality scale is used.150,151 Although kavalactone has been shown to inhibit cytochrome P450 enzymes, deplete hepatic glutathione, and possibly inhibit cyclooxygenase,149 the hepatotoxic component may be the major kava alkaloid pipermethystine.152 Induction of apoptosis and mitochondrial toxicity are the suspected hepatotoxic mechanisms.153
Chitturi S, Farrell GC. Hepatotoxic slimming agents and other herbal hepatotoxins. J Gastroenterol Hepatol. 2008;23:366-73. (Ref 5.)
Chojkier M. Hepatic sinusoidal-obstruction syndrome: Toxicity of pyrrolizidine alkaloids. J Hepatol. 2003;39:437-46. (Ref 116.)
Elinav E, Pinsker G, Safadi R, et al. Association between consumption of Herbalife nutritional supplements and acute hepatotoxicity. J Hepatol. 2007;47:514-20. (Ref 140.)
Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis. 2004;8:151-66. (Ref 33.)
Favreau JT, Ryu ML, Braunstein G, et al. Severe hepatotoxicity associated with the dietary supplement LipoKinetix. Ann Intern Med. 2002;136:590-5. (Ref 137.)
Foti RS, Dickmann LJ, Davis JA, et al. Metabolism and related human risk factors for hepatic damage by usnic acid containing nutritional supplements. Xenobiotica. 2008;38:264-80. (Ref 139.)
Kenna JG. Mechanism, pathology, and clinical presentation of hepatotoxicity of anesthetic agents. In: Kaplowitz N, DeLeve L, editors. Drug-Induced Liver Disease. New York: Marcel Dekker; 2004:405-24. (Ref 11.)
Lewis JH, Kleiner D. Hepatic injury due to drugs, chemicals and toxins. In: Burt AD, Portmann BC, Ferrell LD, editors. MacSween’s Pathology of the Liver. 5th ed. Philadelphia: Churchill Livingstone Elsevier; 2007:649-759. (Ref 37.)
Mandibusan MK, Odin M, Eatmond DA. Postulated carbon tetrachloride mode of action: A review. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2007;25:185-209. (Ref 38.)
Mohi-ud-din R, Lewis JH. Drug- and chemical-induced cholestasis. Clin Liver Dis. 2004;8:95-132. (Ref 93.)
Rengstorff DS, Osorio RW, Bonacini M. Recovery from severe hepatitis caused by mushroom poisoning without liver transplantation. Clin Gastroenterol Hepatol. 2003;1:392-6. (Ref 89.)
Schoepfer AM, Engel A, Fattinger K, et al. Herbal does not mean innocuous: Ten cases of severe hepatotoxicity associated with dietary supplements from Herbalife products. J Hepatol. 2007;47:521-6. (Ref 141.)
Seeff LB, Lindsay KL, Bacon BR, et al. Complementary and alternative medicine in chronic liver disease. Hepatology. 2001;34:595-603. (Ref 98.)
Verma S, Thuluvath PJ. Complementary and alternative medicine in hepatology: Review of evidence of efficacy. Clin Gastroenterol Hepatol. 2007;5:408-16. (Ref 99.)
Zimmerman HJ. Hepatotoxicity. The Adverse Effects of Drugs and Other Chemicals on the Liver, 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 1999. (Ref 1.)
Zimmerman HJ, Lewis JH. Chemical- and toxin-induced hepatotoxicity. Gastroenterol Clin North Am. 1995;24:1027-45. (Ref 3.)
1. Zimmerman HJ. Hepatotoxicity. The Adverse Effects of Drugs and Other Chemicals on the Liver, 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 1999.
2. Lewis JH. Drug-induced liver disease. Med Clin North Am. 2000;84:1275-311.
3. Zimmerman HJ, Lewis JH. Chemical- and toxin-induced hepatotoxicity. Gastroenterol Clin North Am. 1995;24:1027-45.
4. Tolman KG, Sirrine RW. Occupational hepatotoxicity. Clin Liver Dis. 1998;2:563-89.
5. Chitturi S, Farrell GC. Hepatotoxic slimming agents and other herbal hepatotoxins. J Gastroenterol Hepatol. 2008;23:366-73.
6. Schiano TD. Hepatotoxicity and complementary and alternative medicines. Clin Liver Dis. 2003;7:453-73.
7. Mindikoglu AL, Magder LS, Regev A. Outcome of liver transplantation for drug-induced acute liver failure in the United States: Analysis of the United Network for Organ Sharing database. Liver Transplant. 2009;15:719-29.
8. Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105-36.
9. Inman WH, Mushin WW. Jaundice after repeated exposure to halothane: A further analysis of reports to the Committee on Safety of Medicines. BMJ. 1978;2:1455-6.
10. Holt C, Csete M, Martin P. Hepatotoxicity of anesthetics and other central nervous system drugs. Gastroenterol Clin North Am. 1995;24:853-74.
11. Kenna JG. Mechanism, pathology, and clinical presentation of hepatotoxicity of anesthetic agents. In: Kaplowitz N, DeLeve L, editors. Drug-Induced Liver Disease. New York: Marcel Dekker; 2004:405-24.
12. Njoku D, Laster MJ, Gong DH, et al. Biotransformation of halothane, enflurane, isoflurane, and desflurane to trifluoroacetylated liver proteins: Association between protein acylation and hepatic injury. Anesth Analg. 1997;84:173-8.
13. Lo SK, Wendon J, Mieli-Vergani G, et al. Halothane-induced acute liver failure: Continuing occurrence and use of liver transplantation. Eur J Gastroenterol Hepatol. 1998;10:635-9.
14. Vergani D, Mieli-Vergani G, Alberti A, et al. Antibodies to the surface of halothane-altered rabbit hepatocytes in patients with severe halothane-associated hepatitis. N Engl J Med. 1980;303:66-71.
15. Summary of the National Halothane Study. Possible association between halothane anesthesia and postoperative hepatic necrosis. JAMA. 1966;197:775-88.
16. Neuberger J. Halothane and hepatitis. Incidence, predisposing factors and exposure guidelines. Drug Saf. 1990;5:28-38.
17. Njoku DB, Greenberg RS, Bourdi M, et al. Autoantibodies associated with volatile anesthetic hepatitis found in the sera of a large cohort of pediatric anesthesiologists. Anesth Analg. 2002;94:243-9.
18. Sakaguchi Y, Inaba S, Irita K, et al. Absence of antitrifluoroacetate antibody after halothane anaesthesia in patients exhibiting no or mild liver damage. Can J Anaesth. 1994;41:398-403.
19. Cousins MJ, Plummer JL, Hall PD. Risk factors for halothane hepatitis. Aust N Z J Surg. 1989;59:5-14.
20. Benjamin SB, Goodman ZD, Ishak KG, et al. The morphologic spectrum of halothane-induced hepatic injury: Analysis of 77 cases. Hepatology. 1985;5:1163-71.
21. Martin JL, Dubbink DA, Plevak DJ, et al. Halothane hepatitis 28 years after primary exposure. Anesth Analg. 1992;74:605-8.
22. Spracklin DK, Emery ME, Thummel KE, et al. Concordance between trifluoroacetic acid and hepatic protein trifluoroacetylation after disulfiram inhibition of halothane metabolism in rats. Acta Anaesthesiol Scand. 2003;47:765-70.
23. Farrell GC, Prendergast D, Murray M. Halothane hepatitis. Detection of a constitutional susceptibility factor. N Engl J Med. 1985;313:1310-14.
24. Unsal C, Celik JB, Toy H, et al. Protective role of zinc pretreatment in hepatotoxicity induced by halothane. Eur J Anaesthesiol. 2008; June 5:1-6. [Epub ahead of print]
25. Joshi PH, Conn HO. The syndrome of methoxyflurane-associated hepatitis. Ann Intern Med. 1974;80:395-401.
26. Lewis JH, Zimmerman HJ, Ishak KG, et al. Enflurane hepatotoxicity. A clinicopathologic study of 24 cases. Ann Intern Med. 1983;98:984-92.
27. Eger EIII, Smuckler EA, Ferrell LD, et al. Is enflurane hepatotoxic? Anesth Analg. 1986;65:21-30.
28. Turner GB, O’Rourke D, Scott GO, et al. Fatal hepatotoxicity after re-exposure to isoflurane: A case report and review of the literature. Eur J Gastroenterol Hepatol. 2000;12:955-9.
29. Martin JL, Keegan MT, Vasdev GMS, et al. Fatal hepatitis associated with isoflurane exposure and CYP2A6 autoantibodies. Anesthesiology. 2001;95:551-3.
30. Njoku DB, Shrestha S, Soloway R, et al. Subcellular localization of trifluoroacetylated liver proteins in association with hepatitis following isoflurane. Anesthesiology. 2002;96:757-61.
31. Singhal S, Gray T, Grace G, et al. Sevoflurane hepatotoxicity: A case report of sevoflurane hepatic necrosis and review of the literature. Am J Ther. 2009. [Epub ahead of print]
32. Turillazzi E, D’Errico S, Neri M, et al. A fatal case of fulminant hepatic necrosis following sevoflurane anesthesia. Toxicol Pathol. 2007;35:840-5.
33. Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis. 2004;8:151-66.
34. . National Institute for Occupational Safety and Health (NIOSH) Pocket Guide to Chemical Hazards September 2007. DHHS Publication No. 2005-149. Government Printing Office, P.O. Box 371954, Pittsburgh, PA 15250-7954 www.cdc.gov/niosh/npg/npg.html
35. Wexler P. The U.S. National Library of Medicine’s Toxicology and Environmental Health Information Program. Toxicology. 2004;198:161-8.
36. Lewis JH, Zimmerman HJ. Drug- and chemical-induced cholestasis. Clin Liver Dis. 1999;3:433-64.
37. Lewis JH, Kleiner D. Hepatic injury due to drugs, chemicals and toxins. In: Burt AD, Portmann BC, Ferrell LD, editors. MacSween’s Pathology of the Liver. 5th ed. Philadelphia: Churchill Livingstone Elsevier; 2007:649-759.
38. Mandibusan MK, Odin M, Eatmond DA. Postulated carbon tetrachloride mode of action: A review. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2007;25:185-209.
39. Croquet V, Fort J, Oberti F, et al. 1,1,1-trichloroethane-induced chronic active hepatitis. Gastroenterol Clin Biol. 2003;27:120-2.
40. Boucher R, Hanna C, Rusch GM, et al. Hepatotoxicity associated with overexposure to 1,1-dichloro-2,2,2-trifluoroethane (HCFC-123). AIHA J (Fairfax, Va). 2003;64:68-79.
41. Hoet P, Buchet JP, Sempoux C, et al. Potentiation of 2,2-dichloro-1,1,1-trifluoroethane (HCFC-123)-induced liver toxicity by ethanol in guinea pigs. Arch Toxicol. 2002;76:707-14.
42. Cave MC, Mahalingashetty A, Khan R, et al. Non-alcoholic fatty liver disease in non-obese American chemical workers. Hepatology. 2007;46(4 suppl 1):746A.
43. Du CL, Wang JD. Increased morbidity odds ratio of primary liver cancer and cirrhosis of the liver among vinyl chloride monomer workers. Occup Environ Med. 1998;55:528-32.
44. Maroni M, Mocci F, Visentin S, et al. Periportal fibrosis and the liver ultrasonography findings in vinyl chloride workers. Occup Environ Med. 2003;60:60-5.
45. Cave MC, Groce R, Mahalingashetty A, et al. Elevated serum hyaluronic acid may identify vinyl chloride workers at high risk for the subsequent development of hepatic angiosarcoma. Hepatology. 2007;46(4 suppl 1):427A.
46. Meadows R, Verghese A. Medical complications of glue sniffing. South Med J. 1996;89:455-62.
47. Aydin Y, Ozcakar L. Occupational hepatitis due to chronic inhalation of propane and butane gases. Int J Clin Pract. 2003;57:546.
48. Senoh H, Katagiri T, Arito H, et al. Toxicity due to 2- and 13-wk inhalation exposures of rats and mice to N,N-dimethylformamide. J Occup Health. 2003;45:365-75.
49. Fiorito A, Larese F, Molinari S, et al. Liver function alterations in synthetic leather workers exposed to dimethylformamide. Am J Ind Med. 1997;32:255-60.
50. Luo JC, Kuo HW, Cheng TJ, et al. Abnormal liver function associated with occupational exposure to dimethylformamide and hepatitis B virus. J Occup Environ Med. 2001;43:474-82.
51. Kao YH, Chong CH, Ng WT, et al. Hydrazine inhalation hepatotoxicity. Occup Med (Lond). 2007;57:535-7.
52. Singh N, Jatav OP, Gupta RK, et al. Outcome of sixty four cases of ethylene bromide ingestion treated in a tertiary care hospital. J Assoc Physicians India. 2007;55:842-5.
53. Niittynen M, Tuomisto JT, Auriola S, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced accumulation of biliverdin and hepatic peliosis in rats. Toxicol Sci. 2003;71:112-13.
54. Michalek JE, Ketchum NS, Longnecker MP. Serum dioxin and hepatic abnormalities in veterans of Operation Ranch Hand. Ann Epidemiol. 2001;11:304-11.
55. Tamburro CH. Chronic liver injury in phenoxy herbicide-exposed Vietnam veterans. Environ Res. 1992;59:175-88.
56. Cordier S, Le TB, Verger P, et al. Viral infections and chemical exposures as risk factors for hepatocellular carcinoma in Vietnam. Int J Cancer. 1993;55:196-201.
57. Botella de Maglia J, Belenguer Tarin JE. Paraquat poisoning. A study of 29 cases and evaluation of the effectiveness of the “Caribbean scheme.”. Med Clin (Barc). 2000;115:530-3.
58. Mullick FG, Ishak KG, Mahabir R, et al. Hepatic injury associated with paraquat toxicity in humans. Liver. 1981;1:209-21.
59. Carpenter HM, Hedstrom OR, Siddens LK, et al. Ultrastructural, protein, and lipid changes in liver associated with chlordecone treatment of mice. Fundam Appl Toxicol. 1996;34:157-64.
60. Santra A, Das Gupta J, De BK, et al. Hepatic manifestations in chronic arsenic toxicity. Indian J Gastroenterol. 1999;18:152-5.
61. Eisler R. Arsenic hazards to humans, plants, and animals from gold mining. Rev Environ Contam Toxicol. 2004;180:133-65.
62. Rice KC, Conko KM, Hornberger GM. Anthropogenic sources of arsenic and copper to sediments in a suburban lake, Northern Virginia. Environ Sci Technol. 2002;36:4962-7.
63. Guha Mazumder DN. Chronic arsenic toxicity: Clinical features, epidemiology, and treatment: Experience in West Bengal. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2003;38:141-63.
64. Chen Y, Ahsan H. Cancer burden from arsenic in drinking water in Bangladesh. Am J Public Health. 2004;94:741-4.
65. Kannan GM, Flora SJ. Chronic arsenic poisoning in the rat: Treatment with combined administration of succimers and an antioxidant. Ecotoxicol Environ Saf. 2004;58:37-43.
66. Robertson A, Tenenbein M. Hepatotoxicity in acute iron poisoning. Hum Exp Toxicol. 2005;24:559-62.
67. Britton RS. Metal-induced hepatotoxicity. Semin Liver Dis. 1996;16:3-12.
68. Olivieri NF, Brittenham GM, McLaren CE, et al. Long-term safety and effectiveness of iron-chelation therapy with deferiprone for thalassemia major. N Engl J Med. 1998;339:417-23.
69. Wanless IR, Sweeney G, Dhillon AP, et al. Lack of progressive hepatic fibrosis during long-term therapy with deferiprone in subjects with transfusion-dependent beta-thalassemia. Blood. 2002;100:1566-9.
70. Ito Y, Kojiro N, Nakashima T, et al. Pathomorphologic characteristics of 102 cases of Thorotrast-related hepatocellular carcinoma, cholangiocarcinoma and hepatic angiosarcoma. Cancer. 1988;62:1153-62.
71. Newairy AA, El-Sharaky AS, Badreideen MM, et al. The hepatoprotective effects of selenium against cadmium toxicity in rats. Toxicology. 2007;242:23-30.
72. Rana SV. Metals and apoptosis: Recent developments. J Trace Elem Med Biol. 2008;22:262-84.
73. Johri S, Shukla S, Sharma P. Role of chelating agents and antioxidants in beryllium induced toxicity. Indian J Exp Biol. 2002;40:575-82.
74. Mudipalli A. Lead hepatotoxicity and potential health effects. Indian J Med Res. 2007;126:518-27.
75. Sanchez-Porro Valades P, Posada de la Paz M, de Andres Copa P, et al. Toxic oil syndrome: Survival in the whole cohort between 1981 and 1995. J Clin Epidemiol. 2003;56:701-8.
76. Hall AJ, Harrington JM, Waterhouse JA. The Epping jaundice outbreak: A 24 year follow up. J Epidemiol Community Health. 1992;46:327-8.
77. Yoshimura T. Yusho in Japan. Ind Health. 2003;41:139-48.
78. Silva MO, Roth D, Reddy KR, et al. Hepatic dysfunction accompanying acute cocaine intoxication. J Hepatol. 1991;12:312-15.
79. Aoki K, Takimoto M, Ota H, et al. Participation of CYP2A in cocaine-induced hepatotoxicity in female mice. Pharmacol Toxicol. 2000;87:26-32.
80. Labib R, Abdel-Rahman MS, Turkall R. N-acetylcysteine pretreatment decreases cocaine- and endotoxin-induced hepatotoxicity. J Toxicol Environ Health A. 2003;66:223-39.
81. Henry JA, Jeffreys KJ, Dawling S. Toxicity and deaths from 3,4-methylenedioxymethamphetamine (“ecstasy”). Lancet. 1992;340:384-7.
82. Garbino J, Henry JA, Mentha G, et al. Ecstasy ingestion and fulminant hepatic failure: Liver transplantation to be considered as a last therapeutic option. Vet Human Toxicol. 2001;43:99-102.
83. Lange-Brock N, Berg T, Muller AR, et al. Acute liver failure following the use of ecstasy (MDMA). Z Gastroenterol. 2002;40:581-6.
84. Maurer HH, Kraemer T, Springer D, et al. Chemistry, pharmacology, toxicology, and hepatic metabolism of designer drugs of the amphetamine (ecstasy), piperazine, and pyrrolidinophenone types: A synopsis. Ther Drug Monit. 2004;26:127-31.
85. Armen R, Kanel G, Reynolds T. Phencyclidine-induced malignant hyperthermia causing submassive liver necrosis. Am J Med. 1984;77:167-72.
86. Litovitz L, Klein-Schwartz W, Rodgers GC, et al. 2001 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2002;20:391-452.
87. Nordt SP, Manoguerra A, Clark RF. 5-year analysis of mushroom exposures in California. West J Med. 2000;173:314-17.
88. Vetter J. Toxins of Amanita phalloides. Toxicon. 1998;36:13-24.
89. Rengstorff DS, Osorio RW, Bonacini M. Recovery from severe hepatitis caused by mushroom poisoning without liver transplantation. Clin Gastroenterol Hepatol. 2003;1:392-6.
90. Broussard CN, Aggarwal A, Lacey SR, et al. Mushroom poisoning-from diarrhea to liver transplantation. Am J Gastroenterol. 2001;96:3195-8.
91. Enjalbert F, Rapior S, Nouguier-Soule J, et al. Treatment of amatoxin poisoning: 20-Year retrospective analysis. J Toxicol Clin Toxicol. 2002;40:715-57.
92. Toxic hypoglycemic syndrome-Jamaica, 1989-1991. MMWR Morb Mortal Wkly Rep. 1992;41:53-5.
93. Mohi-ud-din R, Lewis JH. Drug- and chemical-induced cholestasis. Clin Liver Dis. 2004;8:95-132.
94. Angsubhakorn S, Pradermwong A, Phanwichien K, et al. Promotion of aflatoxin B1-induced hepatocarcinogenesis by dichlorodiphenyl trichloroethane (DDT). Southeast Asian J Trop Med Public Health. 2002;33:613-23.
95. Kew MC. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003;23:405-9.
96. Rhee SM, Garg VK, Hershey CO. Use of complementary and alternative medicines by ambulatory patients. Arch Intern Med. 2004;164:1004-9.
97. Eisenberg DM, Kessler RC, Van Rompay MI. Perceptions about complementary therapies relative to conventional therapies among adults who use both. Results from a national survey. Ann Intern Med. 2001;135:344-51.
98. Seeff LB, Lindsay KL, Bacon BR, et al. Complementary and alternative medicine in chronic liver disease. Hepatology. 2001;34:595-603.
99. Verma S, Thuluvath PJ. Complementary and alternative medicine in hepatology: Review of evidence of efficacy. Clin Gastroenterol Hepatol. 2007;5:408-16.
100. Angell M, Kassirer JP. Alternative medicine—the risks of untested and unregulated remedies. N Engl J Med. 1998;339:839-41.
101. Estes JD, Stolpman D, Olyaei A, et al. High prevalence of potentially hepatotoxic herbal supplement use in patients with fulminant hepatic failure. Arch Surg. 2003;138:852-8.
102. Stickel F, Egerer G, Seitz HK. Hepatotoxicity of botanicals. Public Health Nutr. 2000;3:113-24.
103. Kafrouni MI, Anders RA, Verma S. Hepatotoxicity associated with dietary supplements containing anabolic steroids. Clin Gastroenterol Hepatol. 2007;5:809-12.
104. Shah NL, Zacharias I, Khettry U, et al. Methasteron-associated cholestatic liver injury: Clinicopathologic findings in 5 cases. Clin Gastroenterol Hepatol. 2008;6:255-8.
105. Geubel AP, De Galocsy C, Alves N, et al. Liver damage caused by therapeutic vitamin A administration: Estimation of dose-related toxicity in 41 cases. Gastroenterology. 1991;100:1701-9.
106. Leo MA, Lieber CS. Hypervitaminosis A. A liver lover’s lament. Hepatology. 1988;8:412-7.
107. Kowalski TE, Falestiny M, Furth E, et al. Vitamin A hepatotoxicity: A cautionary note regarding 25,000 IU supplements. Am J Med. 1994;97:523-8.
108. Johnson EJ, Krall EA, Dawson-Hughes B, et al. Lack of an effect of multivitamins containing vitamin A on serum retinyl esters and liver function tests in healthy women. J Am Coll Nutr. 1992;11:682-6.
109. Jorens PG, Michielsen PP, Pelckmans PA, et al. Vitamin A abuse: Development of cirrhosis despite cessation of vitamin A. A six-year clinical and histopathologic follow-up. Liver. 1992;12:381-6.
110. Sarles J, Scheiner C, Sarran M, et al. Hepatic hypervitaminosis A: A familial observation. J Pediatr Gastroenterol Nutr. 1990;10:71-6.
111. Myhre AM, Carlsen MH, Bohn SK, et al. Water-miscible, emulsified, and solid forms of retinol supplements are more toxic than oil-based preparations. Am J Clin Nutr. 2003;78:1152-9.
112. Hautekeete ML, Geerts A. The hepatic stellate (Ito) cell: Its role in human liver disease. Virchows Arch. 1997;430:195-207.
113. Leo MA, Lieber CS. Alcohol, vitamin A, and β-carotene: Adverse interactions, including hepatotoxicity and carcinogenicity. Am J Clin Nutr. 1999;69:1071-85.
114. Ukleja A, Scolapio JS, McConnell JP, et al. Nutritional assessment of serum and hepatic vitamin A levels in patients with cirrhosis. JPEN J Parenter Enteral Nutr. 2002;26:184-8.
115. Leithead JA, Simpson KJ, MacGilchrist AJ. Fulminant hepatic failure following overdose of the vitamin A metabolite acitretin. Eur J Gastroenterol Hepatol. 2009;21:230-2.
116. Chojkier M. Hepatic sinusoidal-obstruction syndrome: Toxicity of pyrrolizidine alkaloids. J Hepatol. 2003;39:437-46.
117. Copple BL, Ganey PE, Roth RA. Liver inflammation during monocrotaline hepatotoxicity. Toxicology. 2003;190:155-69.
118. Perez Alvarez J, Saez-Royuela F, Gento Pena E, et al. Acute hepatitis due to ingestion of Teucrium chamaedrys infusions. Gastroenterol Hepatol. 2001;24:240-3.
119. Larrey D, Vial T, Pauwels A, et al. Hepatitis after germander (Teucrium chamaedrys) administration: Another instance of herbal medicine hepatotoxicity. Ann Intern Med. 1992;117:129-32.
120. De Berardinis V, Moulis C, Maurice M, et al. Human microsomal epoxide hydrolase is the target of germander induced autoantibodies on the surface of human hepatocytes. Mol Pharmacol. 2000;58:542-51.
121. Dourakis S, Papanikolaou IS, Tzemanakis EN, et al. Acute hepatitis associated with herb (Teucrium capatatum L.) administration. Eur J Gastroenterol Hepatol. 2002;14:693-5.
122. Polymeros D, Kamberoglou D, Tzias V. Acute cholestatic hepatitis caused by Teucrium polium (golden germander) with transient appearance of antimitochondrial antibody. J Clin Gastroenterol. 2002;34:100-1.
123. Sheikh NM, Philen RM, Love LA. Chaparral-associated hepatotoxicity. Arch Intern Med. 1997;157:913-19.
124. Anderson IB, Mullen WH, Meeker JE, et al. Pennyroyal toxicity: Measurement of toxic metabolite levels in two cases and review of the literature. Ann Intern Med. 1996;124:726-34.
125. Sztajnkrycer MD, Otten EJ, Bond GR, et al. Mitigation of pennyroyal oil hepatotoxicity in the mouse. Acad Emerg Med. 2003;10:1024-8.
126. Woolf GM, Petrovic LM, Rojter SE, et al. Acute hepatitis associated with the Chinese herbal product Jin Bu Huan. Ann Intern Med. 1994;121:729-35.
127. Horowitz RS, Feldhaus K, Dart RC, et al. The clinical spectrum of jin bu huan toxicity. Arch Intern Med. 1996;156:899-903.
128. Itoh S, Marutani K, Nishijima T, et al. Liver injuries induced by herbal medicine, syo-saiko-to (xiao-chai-hu-tang). Dig Dis Sci. 1995;40:1845-8.
129. Kamiyama T, Nouchi T, Kojima S, et al. Autoimmune hepatitis triggered by administration of an herbal medicine. Am J Gastroenterol. 1997;92:703-4.
130. Mazzanti G, Battinelli L, Daniele C, et al. New case of acute hepatitis following the consumption of Shou Wu Pian, a Chinese herbal product derived from Polygonum multiflorum. Ann Intern Med. 2004;140:W30.
131. Nadir A, Agarwal S, King PD, et al. Acute hepatitis associated with the use of a Chinese herbal product, ma-huang. Am J Gastroenterol. 1996;91:1436-8.
132. Bajaj J, Know JF, Komorowski R, et al. The irony of herbal hepatitis: Ma-huang-induced hepatotoxicity associated with compound heterozygosity for hereditary hemochromatosis. Dig Dis Sci. 2003;48:1925-8.
133. Shekelle PG, Hardy ML, Morton SC, et al. Efficacy and safety of ephedra and ephedrine for weight loss and athletic performance: A meta-analysis. JAMA. 2003;289:1537-45.
134. Rados C. Ephedra ban: No shortage of reasons. FDA Consumer. 2004;38:6.
135. Adachi M, Saito H, Kobayashi H, et al. Hepatic injury in 12 patients taking the herbal weight loss aids Chaso or Onshido. Ann Intern Med. 2003;139:488-92.
136. Kawaguchi T, Harada M, Arimatsu H, et al. Severe hepatotoxicity associated with a N-nitrosofenfluramine-containing weight-loss supplement: Report of three cases. J Gastroenterol Hepatol. 2004;19:349-50.
137. Favreau JT, Ryu ML, Braunstein G, et al. Severe hepatotoxicity associated with the dietary supplement LipoKinetix. Ann Intern Med. 2002;136:590-5.
138. Durazo FA, Lassman C, Han SHB, et al. Fulminant liver failure due to usnic acid for weight loss. Am J Gastroenterol. 2004;99:950-2.
139. Foti RS, Dickmann LJ, Davis JA, et al. Metabolism and related human risk factors for hepatic damage by usnic acid containing nutritional supplements. Xenobiotica. 2008;38:264-80.
140. Elinav E, Pinsker G, Safadi R, et al. Association between consumption of Herbalife nutritional supplements and acute hepatotoxicity. J Hepatol. 2007;47:514-20.
141. Schoepfer AM, Engel A, Fattinger K, et al. Herbal does not mean innocuous: Ten cases of severe hepatotoxicity associated with dietary supplements from Herbalife products. J Hepatol. 2007;47:521-6.
142. Dara L, Hewett J, Lim JK. Hydroxycut hepatotoxicity: A case series and review of liver toxicity from herbal weight loss supplements. World J Gastroenterol. 2008;14:6999-7004.
143. Schneider C, Segre T. Green tea: Potential health benefits. Am Fam Physician. 2009;79:591-4.
144. Mazzanti G, Menniti-Ippolito F, Moro PA, et al. Hepatotoxicity from green tea: A review of the literature and two unpublished cases. Eur J Clin Pharmacol. 2009;65:331-41.
145. Sarma DN, Barrett ML, Chavez ML, et al. Safety of green tea extracts: A systematic review by the US Pharmacopeia. Drug Saf. 2008;31:469-84.
146. Liss G, Lewis JH. Drug-induced liver injury: What was new in 2008? Expert Opin Drug Metab Toxicol. 2009;5:1-18.
147. Schulze J, Raasch W, Siegers CP. Toxicity of kava pyrones, drug safety and precautions—a case study. Phytomedicine. 2003;10(suppl 4):68-73.
148. Stickel F, Baumuller HM, Seitz K, et al. Hepatitis induced by kava (Piper methysticum rhizome). J Hepatol. 2003;39:62-7.
149. Clouatre DL. Kava kava: Examining new reports of toxicity. Toxicol Lett. 2004;150:85-96.
150. Teschke R, Schwarzenboeck A, Hennermann KH. Kava hepatotoxicity: A clinical survey and critical analysis of 26 suspected cases. Eur J Gastroenterol Hepatol. 2008;20:1182-93.
151. Teschke R, Schwartzenboeck A, Akinci A. Kava hepatotoxicity: A European view. N Z Med J. 2008;121:90-8.
152. Nerurkar PV, Dragull K, Tang CS. In vitro toxicity of kava alkaloid, pipermethystine, in HepG2 cells compared to kavalactones. Toxicol Sci. 2004;79:106-11.
153. Lude S, Torok M, Dieterle S, et al. Hepatocellular toxicity of kava leaf and root extracts. Phytomedicine. 2008;15:120-31.
154. Mahady GB, Low Dog T, Barrett ML, et al. United States Pharmacopeia review of the black cohosh case reports of hepatotoxicity. Menaopause. 2008;15(4 pt 1):628-38.
155. Teschke R, Bahre R, Genthner A, et al. Suspected black cohosh hepatotoxity—challenges and pitfalls of causality assessment. Maturitas. 2009;63:302-14.