CHAPTER 73 Liver Chemistry and Function Tests
BILIRUBIN (see Chapter 20)
BILIRUBIN METABOLISM
Bilirubin is a breakdown product of heme (ferroprotoporphyrin IX). About 4 mg/kg body weight of bilirubin is produced each day, nearly 80% from the breakdown of hemoglobin in senescent red blood cells and prematurely destroyed erythroid cells in the bone marrow and the remainder from the turnover of hemoproteins such as myoglobin and cytochromes distributed throughout the body.1 The initial steps of bilirubin metabolism occur in reticuloendothelial cells, predominantly in the spleen. Heme is converted to biliverdin by the microsomal enzyme heme oxygenase. Biliverdin is then converted to bilirubin by the cytosolic enzyme biliverdin reductase.
After entering the hepatocyte, unconjugated bilirubin is bound in the cytosol to a number of proteins, including proteins in the glutathione S-transferase superfamily.2 These proteins serve to reduce efflux of bilirubin back into the serum and present the bilirubin for conjugation. The enzyme uridine-5′-diphosphate (UDP) glucuronyl transferase found in the endoplasmic reticulum solubilizes bilirubin by conjugating it to glucuronic acid to produce bilirubin monoglucuronide and diglucuronide.3 The now hydrophilic bilirubin diffuses to the canalicular membrane for excretion into the bile canaliculi. Conjugated bilirubin is transported across the canalicular membrane by the multiple drug resistance-associated protein 2 (MRP2) via an adenosine triphosphate (ATP)-dependent process.4 This is the only energy-dependent step in bilirubin metabolism and explains why even patients with fulminant hepatic failure have a predominantly conjugated hyperbilirubinemia. Once in the bile, conjugated bilirubin passes undisturbed until it reaches the distal ileum and colon, where bacteria containing β-glucuronidases hydrolyze conjugated bilirubin to unconjugated bilirubin, which is further reduced by bacteria to colorless urobilinogen.5 The urobilinogen is either excreted unchanged, oxidized and excreted as urobilin, which has an orange color, or absorbed passively by the intestine into the portal system as urobilinogen. The majority of the absorbed urobilinogen is re-excreted by the liver. A small percentage filters across the renal glomerulus and is excreted in urine. Unconjugated bilirubin is never found in urine because in the serum it is bound to albumin and not filtered by the glomerulus. The presence of bilirubin in urine indicates a conjugated hyperbilirubinemia and hepatobiliary disease.
MEASUREMENT OF SERUM BILIRUBIN
The terms direct and indirect bilirubin, which correspond roughly to conjugated and unconjugated bilirubin, respectively, derive from the original van den Bergh reaction.6 Serum bilirubin is still measured in clinical laboratories by some modification of this diazo reaction.7 In this assay, bilirubin is exposed to diazotized sulfanilic acid. The conjugated fraction of bilirubin reacts promptly, or “directly,” with the diazo reagent without the need for an accelerant and thereby allows measurement of the conjugated bilirubin fraction by photometric analysis within 30 to 60 seconds. The total bilirubin is measured 30 to 60 minutes after the addition of an accelerant such as alcohol or caffeine. The unconjugated, or indirect, fraction is then determined by subtracting the direct component from the total bilirubin.
Using the diazo method, normal values of total serum bilirubin are between 1.0 and 1.5 mg/dL, with 95% of a normal population falling between 0.2 and 0.9 mg/dL.8 Normal values for the indirect component are between 0.8 and 1.2 mg/dL. The diazo method, however, tends to overestimate the amount of conjugated bilirubin, particularly within the normal range. As a result, “normal” ranges for conjugated bilirubin have crept upward over time. In general, if the direct acting fraction is less than 15% of the total, the bilirubin can be considered to be entirely indirect. The most frequently reported upper limit of normal for conjugated bilirubin is 0.3 mg/dL. The presence of even a mild increase in conjugated bilirubin in the serum should raise the possibility of liver injury. The measurement and fractionation of serum bilirubin in patients with jaundice does not allow differentiation between parenchymal (hepatocellular) and obstructive (cholestatic) jaundice.
The magnitude and duration of hyperbilirubinemia have not been critically assessed as prognostic markers. In general, the higher the serum bilirubin level in patients with viral hepatitis, the greater the hepatocellular damage and the longer the course of disease. Patients may die, however, of acute liver failure with only a modest elevation of serum bilirubin. Total serum bilirubin correlates with poor outcomes in alcoholic hepatitis and is a critical component of the model for end-stage liver disease (MELD) score, which is used to estimate survival of patients with end-stage liver disease (see later and Chapter 95).
APPROACH TO THE PATIENT WITH AN ELEVATED BILIRUBIN LEVEL
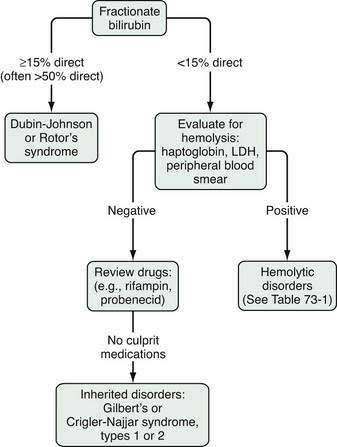
The evaluation of the patient with an isolated elevation of the serum bilirubin level is quite different from that of the patient with an elevated bilirubin associated with elevated liver enzyme levels; the latter suggests either a hepatocellular or cholestatic process, as discussed later. The first step in the evaluation of a patient with an isolated elevation of the serum bilirubin level is to fractionate the bilirubin to determine if it is conjugated or unconjugated bilirubin (Fig. 73-1). If less than 15% of the total is conjugated, one can be assured that virtually all the serum bilirubin is unconjugated. An overproduction of bilirubin as a result of excessive breakdown of hemoglobin can occur with any of a number of inherited or acquired disorders (Table 73-1). The patient’s medication history should be reviewed for drugs that can cause impaired hepatocellular uptake of bilirubin. If no cause is identified, a genetic enzyme deficiency that results in impaired conjugation of bilirubin, the most common of which is Gilbert’s syndrome, is likely.
| CAUSE | MECHANISM |
|---|---|
| Indirect Hyperbilirubinemia | |
| Hemolytic Disorders | Overproduction of bilirubin |
| Inherited | |
| Red cell enzyme defects (e.g., glucose-6-phosphate dehydrogenase deficiency) | |
| Sickle cell disease | |
| Spherocytosis and elliptocytosis | |
| Acquired | |
| Drugs and toxins | |
| Hypersplenism | |
| Immune mediated | |
| Paroxysmal nocturnal hemoglobinuria | |
| Traumatic: macro- or microvascular injury | |
| Ineffective Erythropoiesis | Overproduction of bilirubin |
| Cobalamin deficiency | |
| Folate deficiency | |
| Profound iron deficiency | |
| Thalassemia | |
| Drugs: Rifampin, Probenecid | Impaired hepatocellular uptake |
| Inherited Conditions Crigler-Najjar syndrome types I and II | Impaired conjugation of bilirubin |
| Gilbert’s syndrome | |
| Other | |
| Hematoma | Overproduction of bilirubin |
| Direct Hyperbilirubinemia | |
| Inherited Conditions | |
| Dubin-Johnson syndrome Rotor’s syndrome | Impaired excretion of conjugated bilirubin |
As discussed in Chapter 20, Gilbert’s syndrome is common, with a reported incidence of 6% to 12% (see Table 20-2). A mutation in the TATAA element in the 5′ promoter region of the UDP glucuronyl transferase gene results in a reduction in enzyme activity to approximately one third of normal. The mildly elevated indirect serum hyperbilirubinemia seen in Gilbert’s syndrome is of no clinical consequence. This benign clinical course contrasts with those of much rarer conditions, Crigler-Najjar syndrome, types I and II (see Table 20-2). The mutations in these conditions result in significantly reduced UDP glucuronyl transferase activity: <10% in Crigler-Najjar type II and complete absence of enzyme activity in Crigler-Najjar type I, resulting in much greater elevations of unconjugated serum bilirubin to levels that carry an increased risk of kernicterus.
When an isolated hyperbilirubinemia is associated with a conjugated fraction of >15%, and typically >50%, the diagnosis is either the uncommon Dubin-Johnson syndrome or the even rarer Rotor’s syndrome (see Fig. 73-1, Table 20-2, and Table 64-4). The defect in Dubin-Johnson syndrome is in the MRP2 gene. The defect in Rotor’s syndrome has yet to be defined, but in both syndromes excretion of conjugated bilirubin across the bile canalicular membrane is reduced, resulting in an increased conjugated serum bilirubin level. Neither syndrome is associated with adverse clinical outcomes. Additional genetic disorders of bile acid transport that may be associated with hyperbilirubinemia are discussed in Chapters 64 and 76.
AMINOTRANSFERASES
The serum aminotransferases (also called transaminases), the most sensitive markers of acute hepatocellular injury, have been used to identify liver disease since the 1950s.9 ALT (formerly serum glutamic pyruvic transaminase, or SGPT) and AST (formerly serum glutamic oxaloacetic transaminase, or SGOT) catalyze the transfer of the α-amino groups of alanine and l-aspartic acid, respectively, to the α-keto group of ketoglutaric acid. AST, found in cytosol and mitochondria, is widely distributed throughout the body; it is found, in order of decreasing concentration, in liver, cardiac muscle, skeletal muscle, kidney, brain, pancreas, lung, leukocytes, and erythrocytes. ALT, a cytosolic enzyme also found in many organs, is present in greatest concentration by far in the liver and is, therefore, a more specific indicator of liver injury. Increases in serum values of the aminotransferases reflect either damage to tissues rich in these enzymes or changes in cell membrane permeability that allow ALT and AST to leak into serum; hepatocyte necrosis is not required for the release of aminotransferases, and the degree of elevation of the aminotransferases does not correlate with the extent of liver injury.10
Aminotransferases have no function in serum and act like other serum proteins. They are distributed in plasma and interstitial fluid and have half-lives measured in days. The activity of ALT and AST at any moment reflects the relative rate at which they enter and leave the circulation. They are probably cleared by cells in the reticuloendothelial system, with AST cleared more rapidly than ALT. Normal values for aminotransferases in serum vary widely among laboratories, but values gaining general acceptance are <30 U/L for men and <19 U/L for women. The inter-laboratory variation in the normal range is the result of technical issues; no reference standards exist to establish the upper limits of normal for serum ALT and AST levels. Therefore, each reference laboratory is responsible for identifying a locally defined reference population or for using a normal range first established in the 1950s.9 The normal range is defined as the mean of the reference population plus 2 standard deviations; approximately 95% of a uniformly distributed population will fall within this “normal” range. Some investigators have recommended revisions of normal values for the aminotransferases with adjustments for sex and body mass index, but others have raised concern about the potential costs and unclear benefits of implementing such a change.11–15
APPROACH TO THE PATIENT WITH AN ELEVATED AMINOTRANSFERASE LEVEL
Serum aminotransferase levels are typically elevated in all forms of liver injury; levels up to 300 U/L are nonspecific. In certain circumstances the degree and pattern of elevation of the aminotransferases, evaluated in the context of a patient’s characteristics, symptoms, and physical examination findings, can suggest particular diagnoses and direct the subsequent evaluation (Table 73-2). The differential diagnosis of marked elevations of aminotransferase levels (>1000 U/L) includes viral hepatitis (A to E), toxin or drug-induced liver injury, ischemic hepatitis, and less commonly, autoimmune hepatitis, acute Budd-Chiari syndrome, fulminant Wilson disease, and acute obstruction of the biliary tract.
Table 73-2 Causes of Elevated Serum Aminotransferase Levels*
| Chronic, Mild Elevations, ALT > AST (<150 U/L or 5 × normal) |
| Hepatic Causes |
ALT, alanine aminotransferase; AST, aspartate aminotransferase.
* Virtually any liver disease can cause moderate aminotransferase elevations (5-15 × normal)
The ratio of AST to ALT in serum is helpful in a few specific circumstances—perhaps most importantly in the recognition of alcoholic liver disease. If the AST level is less than 300 U/L, a ratio of AST to ALT of more than 2 suggests alcoholic liver disease, and a ratio of more than 3 is highly suggestive of alcoholic liver disease.16 The ratio results from a deficiency of pyridoxal 5′-phosphate in patients with alcoholic liver disease; ALT synthesis in the liver requires pyridoxal phosphate more than does AST synthesis.17 When a patient with chronic alcoholic liver disease sustains a superimposed liver injury, particularly acetaminophen hepatotoxicity, the aminotransferase level can be strikingly elevated, yet the AST/ALT ratio is maintained.
An increased ratio of AST to ALT may also be seen in muscle disorders. The degree of elevation is typically less than 300 U/L, but in rare cases, such as rhabdomyolysis, levels observed in patients with acute hepatocellular disease can be reached. In cases of acute muscle injury, the AST/ALT ratio may initially be greater than 3 : 1, but the ratio quickly declines toward 1 : 1 because of the shorter serum half-life of AST.18 The ratio typically is close to 1 : 1 in patients with chronic muscle diseases.
Although the AST to ALT ratio is typically less than 1 in patients with chronic viral hepatitis and nonalcoholic fatty liver disease (NAFLD), a number of investigators have observed that, as cirrhosis develops, the ratio rises and may become greater than 1. Studies have shown that an AST/ALT ratio of greater than 1 as an indicator of cirrhosis in patients with chronic hepatitis C has a high specificity (94% to 100%) but a relatively low sensitivity (44% to 75%).19 The increase in AST/ALT ratio with the development of cirrhosis is believed to result from impaired functional hepatic blood flow, with a consequent decrease in hepatic sinusoidal uptake of AST.20
The majority of patients evaluated for elevated serum aminotransferase levels are asymptomatic and have mild elevations (≤5-fold) identified during routine screening. The first step in the evaluation of mildly elevated serum aminotransferase levels is to repeat the test to confirm persistence of the elevated value. If the aminotransferase level remains elevated, the recommended evaluation is illustrated in Figure 73-2. The initial step is to take a careful history focused on identifying all of the patient’s medications, including over-the-counter (OTC) medications, complementary and alternative medications (CAM), and substances of abuse. Correlating the use of medications temporally with the laboratory abnormalities will sometimes reveal a specific culprit. Almost any medication, including OTC medications, CAM, and substances of abuse, has the potential to elevate serum aminotransferase levels. Relatively common offending agents include nonsteroidal anti-inflammatory drugs, antibiotics, hydroxymethylglutaryl-coenzyme A reductase inhibitors, antiepileptics, and antituberculous medications (see Chapter 86). The association between use of a medication and liver enzyme elevations is readily established by stopping the medication and observing return of the enzyme levels to normal. Re-challenge with the suspect medication followed by a rise in serum aminotransferase levels is confirmatory but often not undertaken. Muscle disease should also be excluded by obtaining serum creatine kinase and aldolase levels.
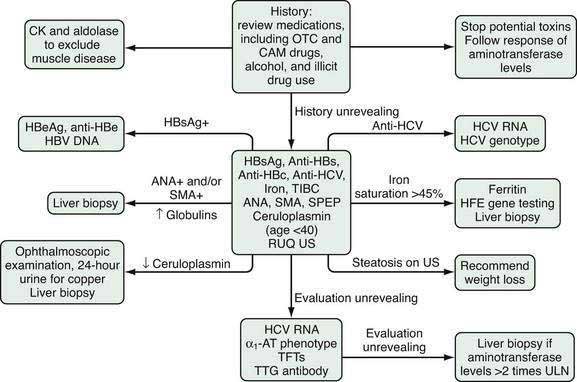
The next step in the evaluation is to assess the patient for the more common and treatable causes of liver disease, including chronic hepatitis B and C, hemochromatosis, autoimmune hepatitis, Wilson disease, and NAFLD. Although autoimmune hepatitis is commonly considered a disease of young to middle-aged women, it also is seen in men and has been reported in all ethnic groups (see Chapter 88). The clinical onset of Wilson disease is usually between the ages of 5 and 25 years; the diagnosis should be considered initially in all patients age 40 or younger and those older than age 40 with aminotransferase elevations that remain unexplained after other causes are excluded (see Chapter 75). NAFLD is the most common cause of elevated serum aminotransferase levels in the United States (see Chapter 85).
ALKALINE PHOSPHATASE
The term alkaline phosphatase applies generally to a group of isoenzymes distributed widely throughout the body.21 The isoenzymes of greatest clinical importance in adults are in the liver and bone because these organs are the major sources of serum alkaline phosphatase. Other isoenzymes originate from the placenta, small intestine, and kidneys. In the liver, alkaline phosphatase is found on the canalicular membrane of hepatocytes; its precise function is undefined. Alkaline phosphatase has a serum half-life of approximately seven days, and although the sites of degradation are unknown, clearance of alkaline phosphatase from serum is independent of either patency of the biliary tract or functional capacity of the liver. Hepatobiliary disease leads to increased serum alkaline phosphatase levels through induced synthesis of the enzyme and leakage into the serum, a process mediated by bile acids.22
A number of individual physiologic variations in serum alkaline phosphatase levels have been identified. Patients with blood groups O and B have elevations in serum alkaline phosphatase levels caused by release of intestinal alkaline phosphatase after a fatty meal.23 This observation is the basis for the recommendation by some authorities that the serum alkaline phosphatase level be checked in the fasting state. An increased serum alkaline phosphatase level of intestinal origin is seen in a benign familial elevation of serum alkaline phosphatase. Serum alkaline phosphatase values vary with age. Male and female adolescents have serum alkaline phosphatase levels twice the level seen in adults; the level correlates with bone growth, and the increase in serum is in bone alkaline phosphatase. Although the level of serum alkaline phosphatase increases after age 30 years in both men and women, the increase is more pronounced in women than in men; a healthy 65-year-old woman has a serum alkaline phosphatase level 50% higher than that of a healthy 30-year-old woman.24 The reason for this difference is not known. In a person with isolated elevation of the serum alkaline phosphatase level, the serum GGTP or 5′NT are used to distinguish a liver origin from bone origin of the alkaline phosphatase elevation.
A low serum alkaline phosphatase level may occur in patients with Wilson disease, especially those presenting with fulminant hepatitis and hemolysis, possibly because of reduced activity of the enzyme owing to displacement of the co-factor zinc by copper (see Chapter 75).
Gamma Glutamyl Transpeptidase
Gamma glutamyl transpeptidase (GGTP) is found in the cell membranes of a wide distribution of tissues including liver (both hepatocytes and cholangiocytes), kidney, pancreas, spleen, heart, brain, and seminal vesicles. It is present in the serum of healthy persons. Serum levels are not different between men and women and do not rise in pregnancy. Although an elevated serum GGTP level has high sensitivity for hepatobiliary disease, its lack of specificity limits its clinical utility. The primary use of serum GGTP levels is to identify the source of an isolated elevation in the serum alkaline phosphatase level; GGTP is not elevated in bone disease (Fig. 73-3).25 GGTP is elevated in patients taking phenytoin, barbiturates, and some drugs used in highly active antiretroviral therapy, including non-nucleoside reverse transcriptase inhibitors and the protease inhibitor abacavir.26,27
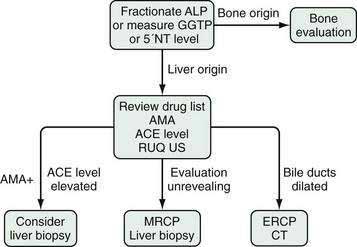
Serum GGTP levels are also elevated in patients who drink alcohol, and some experts have advocated use of the GGTP level for identifying unreported alcohol use (see Chapter 84). The sensitivity of an elevated serum GGTP level for alcohol use ranges from 52% to 94%, but a low specificity limits its usefulness for this purpose.28 One study has suggested an association between high serum GGTP levels and the risk of hepatocellular carcinoma.29 The GGTP level had a negative predictive value of 97.9%, higher than that for alkaline phosphatase, total bilirubin, ALT, and AST, for detecting bile duct stones in patients undergoing laparoscopic cholecystectomy.30
5′-Nucleotidase
5′-Nucleotidase (5′NT) is associated with the canalicular and sinusoidal plasma membranes. Its function is undefined. 5′NT is also found in the intestine, brain, heart, blood vessels, and endocrine pancreas. Serum levels of 5′NT are unaffected by sex or race, but age affects the level; values are lowest in children and increase gradually, reaching a plateau at approximately age 50 years. As with GGTP, the primary role of the serum 5′NT level is to identify the organ source of an isolated serum alkaline phosphatase elevation (see Fig. 73-3). The 5′NT level is not increased in bone disease and is primarily increased in hepatobiliary disease.
APPROACH TO THE PATIENT WITH AN ELEVATED ALKALINE PHOSPHATASE LEVEL
The first step in the evaluation of a patient with an isolated and asymptomatic elevation of the serum alkaline phosphatase is to identify the tissue source (see Fig. 73-3). The most precise way of doing this is via fractionation through electrophoresis; each isoenzyme of alkaline phosphatase has different electrophoretic mobilities.31 Tests used in the past involving heat and urea denaturation of alkaline phosphatase are neither sensitive nor specific. An acceptable alternative method is to check either the serum GGTP or 5′NT level; elevation of either verifies that the elevated alkaline phosphatase is the result of hepatobiliary disease. A normal 5′NT level, however, does not rule out the possibility of hepatobiliary disease because the 5′NT and alkaline phosphatase do not necessarily increase in parallel in early or mild hepatic injury, thus making GGTP the preferred test.
The primary value of an elevated serum level of alkaline phosphatase of liver origin is to allow the recognition of cholestatic disorders (i.e., disorders associated with impaired bile flow, often with jaundice). In general, a serum alkaline phosphatase elevation out of proportion to the level of the aminotransferases suggests a cholestatic disorder. A four-fold elevation of the serum alkaline phosphatase is seen in approximately 75% of patients with chronic cholestasis, both intrahepatic or extrahepatic, whereas lesser elevations are nonspecific and can occur in a wide range of conditions. Figures 73-3 and 73-4 illustrate the recommended evaluation of cholestatic liver enzymes—either an isolated alkaline phosphatase elevation (see Fig. 73-3) or a disproportionate elevation of the alkaline phosphatase compared with the aminotransferases (see Fig. 73-4).
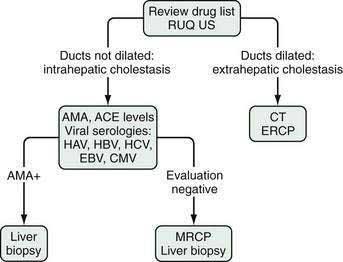
Central to the evaluation of an elevated alkaline phosphatase level is imaging of the biliary tract. An absence of dilated intrahepatic bile ducts focuses the search on intrahepatic causes of cholestasis (Table 73-3), whereas dilated ducts should lead to an evaluation of extrahepatic causes of cholestasis (Table 73-4). As with elevated aminotransferase levels, the evaluation of intrahepatic causes of cholestatic liver enzymes begins with a careful history of medication use, including OTC medications, CAM, and drugs of abuse, and temporal correlation of their use with elevation of the liver enzymes. A liver biopsy generally is not required for the diagnosis; withdrawal of the offending agent and resolution of the liver enzymes is sufficient to confirm the diagnosis. The rate of improvement can be slow, and if bile duct destruction has developed (“vanishing bile duct syndrome”), the changes may be irreversible.
Table 73-3 Intrahepatic Causes of Cholestatic Liver Enzyme Elevations in Adults
| Drugs* |
* Categorized by histologic pattern. Drug lists are not meant to be comprehensive.
Table 73-4 Extrahepatic Causes of Cholestatic Liver Enzymes in Adults
| Intrinsic |
| Choledocholithiasis |
| Immune-Mediated Duct Injury |
| Malignancy |
| Infections |
| Extrinsic |
| Malignancy |
| Mirizzi’s syndrome* |
| Pancreatitis |
| Pancreatic pseudocyst |
AIDS, acquired immunodeficiency syndrome.
* Compression of the common hepatic duct by a stone in the neck of the gallbladder.
Primary biliary cirrhosis (PBC) is a classic autoimmune disease. The immunologic injury is characterized by a T cell–mediated destruction of the intrahepatic bile ducts. Although predominantly a disease of middle-aged women with a median age at diagnosis of approximately 50 years, 5% to 10% of affected patients are men. The reported age range is 22 to 93 years. Antimitochondrial antibodies (AMA) are found in serum in 95% of patients and are diagnostic; a liver biopsy that demonstrates characteristic histologic findings is confirmatory (see Chapter 89).
Primary sclerosing cholangitis (PSC) is a disease of altered immunity marked by inflammation and fibrosis of the intra- or extrahepatic bile ducts, or both. The disorder is strongly associated with inflammatory bowel disease and is found most commonly in younger men. The diagnosis is confirmed by cholangiography, either endoscopic retrograde cholangiopancreatography (ERCP) or magnetic resonance cholangiopancreatography (MRCP) (see Chapter 68).
Granulomatous liver disease can be caused by a number of disorders (see Table 73-3). Infectious etiologies must be excluded because treatment for many of the other causes of granulomatous liver disease is immunosuppressive therapy. Sarcoidosis is the most common etiology. The diagnosis is based on an elevated angiotensin-converting enzyme (ACE) level and typical extrahepatic manifestations. Hepatic involvement, however, is uncommonly the impetus for initiating therapy for sarcoidosis (see Chapter 35).
Viral hepatitis, particularly cases cased by Epstein-Barr virus (EBV) and cytomegalovirus (CMV), can manifest with a prominent cholestatic liver enzyme pattern (see Chapter 81). A number of familial conditions produce intrahepatic cholestasis (see Table 64-5). Progressive forms of these disorders manifest in childhood, whereas the benign forms—benign recurrent intrahepatic cholestasis types 1 and 2—can manifest for the first time in adulthood. Other intrahepatic causes of cholestatic liver enzymes are listed in Table 73-3.
If imaging shows intrahepatic ductal dilatation, the evaluation focuses on the extrahepatic biliary tract to identify an intrinsic or extrinsic process causing biliary obstruction (see Table 73-4). The evaluation often includes an ERCP for tissue acquisition and placement of a biliary stent if obstruction is present (see Chapter 70). Computed tomography (CT) provides assessment for an extrinsic process, and tissue acquisition can be performed with CT or endoscopic ultrasound guidance.
TESTS OF HEPATIC SYNTHETIC FUNCTION
ALBUMIN
Quantitatively, the most important plasma protein, albumin, accounts for 75% of the plasma colloid oncotic pressure and is synthesized exclusively by hepatocytes. The average adult produces approximately 15 g/day and has 300 to 500 g of albumin distributed in body fluids. The liver has the ability to double the rate of synthesis in the setting of rapid albumin loss or a dilutional decrease in the serum albumin concentration.32 The half-life of albumin is 14 to 21 days; the site of degradation is not known. Albumin synthesis is regulated by changes in nutritional status, osmotic pressure, systemic inflammation, and hormone levels.33 Thus, the differential diagnosis of serum hypoalbuminemia, in addition to hepatocellular dysfunction, includes malnutrition, excessive loss from protein-losing enteropathy or nephrotic syndrome, chronic systemic inflammatory conditions, and hormonal imbalances.
The long serum half-life of albumin in serum accounts for its unreliability as a marker of hepatic synthetic function in acute liver injury. Serum albumin levels less than 3 g/dL in a patient with newly diagnosed hepatitis should raise suspicion of a chronic process. Serum albumin is an excellent marker of hepatic synthetic function in patients with chronic liver disease and cirrhosis, with the exception of patients with cirrhosis and ascites, who may have normal or increased albumin production but an increased volume of distribution that results in a low serum albumin level. Albumin has no utility as a screening test in patients for whom there is low suspicion of liver disease; a study in which the serum albumin level was measured in 449 consecutive patients yielded 56 abnormal results, of which only 2 (0.4%) were of clinical importance.34
PROTHROMBIN TIME
Clotting is the end result of a complex series of enzymatic reactions involving clotting factors, all of which are produced in the liver except factor VIII, which is produced by vascular endothelial cells. The prothrombin time is a measure of the rate at which prothrombin is converted to thrombin, reflecting the extrinsic pathway of coagulation. Factors involved in the synthesis of prothrombin include II, V, VII, and X. The international normalized ratio (INR) is used to express the degree of anticoagulation on warfarin therapy. The INR standardizes prothrombin time measurement according to the characteristics of the thromboplastin reagent used in a particular laboratory; the initial measurement is expressed as an international sensitivity index (ISI), which is then used in calculating the INR. Because the ISI is validated only for patients taking a vitamin K antagonist, concern has been raised about the validity of using the ISI (and INR) in patients with chronic liver disease.35 Two studies have demonstrated, in fact, that the ISI, as currently determined, is not accurate for calculating the INR in patients with cirrhosis, and the investigators have proposed that specific ISI and INR determinations using control patients with liver disease be used to eliminate inter-laboratory variability in calculating the INR in patients with cirrhosis.36,37
Measurement of the prothrombin time in patients with liver disease is most useful in cases of acute liver disease. Unlike the serum albumin, the prothrombin time allows an assessment of current hepatic synthetic function; factor VII has the shortest serum half-life (six hours) of all the clotting factors. The prothrombin time has prognostic value in patients with acute acetaminophen- and non-acetaminophen-related liver failure (see Chapter 93), as well as alcoholic hepatitis (see Chapter 84). The INR, along with total serum bilirubin and creatinine levels, are components of the MELD score, which is used to allocate donor organs for liver transplantation. The MELD score accurately predicts survival in patients with decompensated cirrhosis (see later).
TESTS TO DETECT HEPATIC FIBROSIS
Hyaluronan is a glucosaminoglycan produced in mesenchymal cells and widely distributed in the extracellular space. Typically degraded by hepatic sinusoidal cells, serum levels of hyaluronan are elevated in patients with cirrhosis as a result of sinusoidal capillarization (see Chapter 90). A fasting hyaluronan level greater than 100 mg/L had a sensitivity of 83% and specificity of 78% for the detection of cirrhosis in patients with a variety of chronic liver diseases.38 Hyaluronan has been shown to be useful for identifying advanced fibrosis in patients with chronic hepatitis C, chronic hepatitis B, alcoholic liver disease, and nonalcoholic steatohepatitis.39 Preoperative serum hyaluronan levels also have been shown to correlate with the development of hepatic dysfunction after hepatectomy.40
FibroTest (marketed as FibroSure in the United States) is the best evaluated of the multiparameter blood tests. The test incorporates haptoglobin, bilirubin, GGTP, apolipoprotein A-I, and α2-macroglobulin and has been found to have high positive and negative predictive values for diagnosing advanced fibrosis in patients with chronic hepatitis C. One study showed that use of a more sensitive index cut-off had a sensitivity of 90%, specificity of 36%, positive predictive value of 88%, and negative predictive value of 40% for the diagnosis of bridging fibrosis in patients with chronic hepatitis C.41 The test has similar performance characteristics in patients with chronic hepatitis B and alcoholic liver disease and has been shown to predict advanced fibrosis in patients taking methotrexate for psoriasis.42 The newer FIBROSpect II assay incorporates hyaluronate, tissue inhibitor of metalloproteinase 1, and α2-macroglobulin. In a group of patients with chronic hepatitis C, FIBROSpect II had a sensitivity of 72% and a specificity of 74% for identifying advanced fibrosis.43
Transient elastography (TE), marketed as FibroScan, uses ultrasound waves to measure hepatic stiffness noninvasively. Central to this technique’s development was the principle that fibrosis leads to increased stiffness of the hepatic tissue and that a shear wave would propagate faster through stiff material than through elastic material.44 The ultrasound transducer emits a low-frequency (50 Hz) shear wave, and the amount of time required for the wave to go through a set “window” of tissue is measured.45 The window of tissue is 1 cm by 4 cm, 100 times the area of an average liver biopsy. A meta-analysis showed that TE performed best at differentiating cirrhosis from absence of cirrhosis but was less accurate for the estimation of lesser degrees of fibrosis.46 TE has been shown to be accurate for identifying advanced fibrosis in patients with chronic hepatitis C, PBC, hemochromatosis, NAFLD, and recurrent chronic hepatitis after liver transplantation.47–50
Magnetic resonance elastography (MRE) is another noninvasive technique under study. The shear elasticity of the liver is measured after low-frequency (65 Hz) waves are transmitted into the right lobe of the liver. In one study,51 MRE was found to be superior to TE for staging liver fibrosis in patients with a variety of chronic liver diseases.
QUANTITATIVE LIVER FUNCTION TESTS
CAFFEINE CLEARANCE
Caffeine clearance tests quantify functional hepatic capacity by assessing the activity of cytochrome P450 1A2, N-acetyltransferase, and xanthine oxidase. Caffeine is given orally (200 to 366 mg), and levels are measured in blood, urine, saliva, breath, or scalp hair. The alternative methods correlate well with the plasma clearance method. Tobacco use increases caffeine clearance, and drug interactions can affect results. Increasing age correlates with decreased caffeine clearance. Overnight salivary caffeine clearance has been shown to correlate with ICG measurements and galactose clearance as well as with results of the aminopyrine breath test (see later).52
LIDOCAINE METABOLITE FORMATION
Lidocaine is metabolized to its major metabolite monoethylglycinexylidide (MEGX) by the hepatic cytochrome P450 system.53 Serum samples are taken 15, 30, and 60 minutes after an intravenous administration of lidocaine (1 mg/kg). Neither MEGX nor galactose elimination were found to be superior to the Child-Turcotte-Pugh (CTP) (see Chapter 90) or MELD score in predicting prognosis in patients with cirrhosis secondary to viral hepatitis.54 Other studies have suggested that a decline in MEGX concentration correlates well with histologic worsening in patients with chronic liver disease.55
AMINOPYRINE BREATH TEST
The 14C and 13C aminopyrine breath tests measure hepatic mixed-function oxidase mass. The radioactive methyl groups of aminopyrine undergo demethylation and eventual conversion to labeled CO2, which is then exhaled and can be measured. After an overnight fast, a known dose of 14C aminopyrine (1 to 2 µCi) is administered orally, and breath samples are taken every 30 minutes for 4 hours; some investigators check a single sample at either 1 or 2 hours. Healthy subjects excrete 6.6% ± 1.3% of the administered dose in the breath in two hours; patients with hepatocellular injury excrete considerably less. The degree of decrease in excretion of aminopyrine overlaps considerably in patients with all types of severe liver disease, including cirrhosis, chronic hepatitis, alcoholic liver disease, and hepatocellular carcinoma.56 Data are conflicting regarding the ability of this test to predict survival in patients with chronic liver disease.
BILE ACIDS
Bile acids are synthesized from cholesterol in hepatocytes, conjugated to glycine or taurine, and secreted into bile (see Chapter 64). After passage into the small intestine, most bile acids are actively reabsorbed. The liver efficiently extracts bile acids from the portal blood. In healthy persons, all bile acids in serum emanate from the reabsorption of bile acids in the small intestine. Maintenance of normal serum bile acid concentrations depends on hepatic blood flow, hepatic uptake, secretion of bile acids, and intestinal transit. Serum bile acids are sensitive but nonspecific indicators of hepatic dysfunction and allow some quantification of functional hepatic reserve. Serum bile acid levels correlate moderately well with the results of aminopyrine breath tests in patients with chronic hepatitis.57 Unfortunately, the correlation between serum bile acid levels and the histologic severity of chronic hepatitis and alcoholic liver disease is poor.58 Serum bile acids are elevated in patients with cholestatic liver diseases but normal in patients with Gilbert’s syndrome and Dubin-Johnson syndrome and can be used to make the distinction. Although decreased serum bile acid levels are highly specific indicators of liver dysfunction, they are not as sensitive as initially hoped.
SPECIFIC APPLICATIONS OF LIVER BIOCHEMICAL TESTING
DRUG-INDUCED LIVER INJURY
Most drugs that are hepatotoxic cause idiosyncratic liver injury, defined as injury that is unpredictable, occurs at therapeutic drug levels, and is infrequent. The estimated frequency of idiosyncratic drug-induced liver injury for any particular medication ranges from 1 in 1000 to 1 in 100,000. These reactions are marked by a variable latency period ranging from 5 to 90 days, or even longer.59 Other drugs produce dose-dependent toxicity. These injuries are predictable, have a high incidence, and generally have a well understood mechanism. Acetaminophen is the classic example of a drug that causes dose-dependent liver injury. The dose of acetaminophen exceeds 15 g, almost four times the recommended daily dose, in 80% of cases. Acetaminophen doses within the therapeutic range (<4 g/day) can be sufficient to cause liver injury in susceptible persons, such as those who use ethanol chronically. The King’s College criteria identify patients with a poor prognosis from acetaminophen-induced liver injury: those with an arterial pH <7.3 or those with an INR >6.5, serum creatinine level >3.4 g/dL, and stage 3 to 4 hepatic encephalopathy (see Chapter 86).60
Most occurrences of drug-induced liver injury are mild and respond promptly to drug withdrawal with complete resolution. Isolated elevation of the serum aminotransferase levels, even to values greater than three times the upper limit of normal, is associated with a positive outcome. When aminotransferase elevations are associated with clinical jaundice (so-called Hy’s Law, after the late Dr. Hyman Zimmerman), the risk of mortality is increased to as high as 10%.61
SURGICAL CANDIDACY AND ORGAN ALLOCATION
Patients with acute and chronic liver disease are potentially at increased risk of morbidity and mortality if they undergo surgery. The risk depends on the etiology of the liver disease, severity of the liver disease, and planned operation. Although routine preoperative liver biochemical testing is not recommended in otherwise healthy people, the identification of unexpected elevated liver enzyme levels should prompt a postponement of surgery until the cause of the abnormalities has been identified. A retrospective analysis found that patients with acute viral hepatitis who undergo laparotomy had an operative mortality rate of approximately 9.5%.62 Elective surgery should be postponed in patients with acute hepatitis. The surgical risk in patients with chronic hepatitis correlates with the severity of histologic inflammation in the liver. Those with only portal inflammation and interface hepatitis have low operative risk, whereas those with panlobular hepatitis have an increased risk. The etiology of chronic hepatitis does not influence outcome.
An estimated 10% of patients with advanced liver disease undergo surgery in the last two years of their lives. Cirrhosis is associated with increased operative risk, particularly with certain types of surgery, including cardiothoracic surgery, hepatic resection, and other abdominal operations. The data evaluating the surgical risk in these patients were derived retrospectively but point consistently toward the usefulness of the CTP scoring system for predicting perioperative mortality. Two studies performed more than 10 years apart examined mortality after abdominal surgery in cirrhotic patients and reported nearly identical rates of mortality for patients with Child’s class A, B, and C cirrhosis: 10%, 30% to 31%, and 76% to 82%, respectively.63,64 In general, surgery may be undertaken in patients with Child’s class A cirrhosis, whereas the medical condition of patients with Child’s class B cirrhosis should be optimized prior to planned surgery. The mortality rate in patients with Child’s class C cirrhosis is prohibitive, and surgery should be avoided.
The MELD score was created originally to predict survival in patients with cirrhosis and portal hypertension undergoing placement of a transjugular intrahepatic portosystemic shunt (TIPS).65 The score has subsequently been validated as an accurate predictor of survival in patients with advanced liver disease. The MELD score incorporates three objective variables into a mathematical formula: 9.57 × loge(creatinine) + 3.78 × loge(total bilirubin) + 11.2 × loge(INR) + 6.43. The working range is 6 to 40, and the score has been shown to correlate with mortality in patients undergoing surgery other than liver transplantation, including hepatic resection, other abdominal procedures, and cardiac surgery.66–68
MELD is used most often for prioritizing the allocation of donor organs for liver transplantation. Before 2002, organs were allocated by the United Network for Organ Sharing (UNOS) on the basis of the CTP score and time on the wait list. Questions of fairness with this system were raised in light of subjective variables in the CTP score (specifically, the amount of ascites and grade of encephalopathy). Wait time was also believed to be an unfair component that placed patients referred at a late stage of disease at a disadvantage. The MELD score addressed these concerns by using only three objective variables and eliminating wait time as a criterion for organ allocation.69 Since implementation of the MELD score for prioritizing organ allocation, the number of deaths among patients on the wait list has decreased, suggesting that use of the MELD score is achieving its primary goal—allocation of organs to the sickest patients first (see Chapter 95).
Addario L, Scaglione G, Tritto G, et al. Prognostic value of quantitative liver function tests in viral cirrhosis: A prospective study. Eur J Gastroenterol Hepatol. 2006;18:713-20. (Ref 54.)
Bellest L, Eschwege V, Poupon R, et al. A modified international normalized ratio as an effective way of prothrombin time standardization in hepatology. Hepatology. 2007;46:528-34. (Ref 36.)
Friedrich-Rust M, Martens S, Sarrazin C, et al. Performance of transient elastography for the staging of liver fibrosis: A meta-analysis. Gastroenterology. 2008;134:960-74. (Ref 46.)
Kamath PS, Kim WR. The Model for End-Stage Liver Disease (MELD). Hepatology. 2007;45:797-805. (Ref 65.)
Kaneda H, Hashimoto E, Yatsuji S, et al. Hyaluronic acid levels can predict severe fibrosis and platelet counts can predict cirrhosis in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2006;21:1459-65. (Ref 39.)
Kaplan M. Alkaline phosphatase. Gastroenterology. 1972;62:452-68. (Ref 21.)
Kim HC, Nam CM, Jee SH, et al. Normal serum aminotransferase concentration and risk of mortality from liver diseases: Prospective cohort study. BMJ. 2004;328:983. (Ref 13.)
Kunde SS, Lazenby AJ, Clements RH, et al. Spectrum of NAFLD and diagnostic implications of the proposed new normal range for serum ALT in obese women. Hepatology. 2005;42:650-6. (Ref 15.)
Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349:474-85. (Ref 59.)
Mansour A, Watson W, Shayani V, et al. Abdominal operation in patients with cirrhosis: Still a major surgical challenge. Surgery. 1997;122:730-6. (Ref 64.)
Nathwani RA, Pais S, Reynolds TB, et al. Serum alanine aminotransferase in skeletal muscle diseases. Hepatology. 2005;41:380-2. (Ref 18.)
Poynard T, McHutchison J, Manns M, et al. Biochemical surrogate markers of liver fibrosis and activity in a randomized trial of peginterferon alpha-2b and ribavirin. Hepatology. 2003;38:481-92. (Ref 41.)
Prati D, Taioli E, Zanella A, et al. Updated definitions of healthy ranges for serum alanine aminotranferase levels. Ann Intern Med. 2002;137:1-10. (Ref 12.)
Tripodi A, Chantarangkul V, Prirnignani M, et al. The international normalized ratio calibrated for cirrhosis normalizes prothrombin time results for model for end-stage liver disease calculation. Hepatology. 2007;46:520-7. (Ref 37.)
Wiesner R, Edwards E, Freeman R, et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology. 2003;124:91-6. (Ref 69.)
1. Lester R, Schmid R. Bilirubin metabolism. N Engl J Med. 1964;270:779-86.
2. Levi AJ, Gatmaitan Z, Arias IM. Two hepatic cytoplasmic protein fractions, Y and Z, and their possible role in the hepatic uptake of bilirubin sulfobromophthalein, and other anions. J Clin Invest. 1969;48:2156-67.
3. Bosma PJ, Seppen J, Goldhoorn B, et al. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem. 1994;269:17960-4.
4. Gatmaitan ZC, Arias IM. ATP-dependent transport systems in the canalicular membrane of the hepatocyte. Physiol Rev. 1995;75:261-75.
5. Poland RL, Odell GB. Physiologic jaundice: The enterohepatic circulation of bilirubin. N Engl J Med. 1971;284:1-6.
6. van den Bergh AA, Muller P. Uber eine direkte und eine indirekte Diazoreaktion auf Bilirubin. Biochem Z. 1916;77:90.
7. Zieve L, Hill E, Hanson M, et al. Normal and abnormal variations and clinical significance of the one-minute and total serum bilirubin determinations. J Lab Clin Med. 1951;38:446-69.
8. Bloomer JR, Berk PD, Howe RB, et al. Interpretation of plasma bilirubin levels based on studies with radioactive bilirubin. JAMA. 1971;218:216-20.
9. Karmen A, Wroblewski F, Ladue JS. Transaminase activity in human blood. JCI. 1955;34:126-31.
10. Kallai L, Hahn A, Roeder V, et al. Correlation between histological findings and transaminase values in chronic diseases of the liver. Acta Medica Scandinavica. 1964;175:49-56.
11. Piton A, Poynard T, Imbert-Bismut F, et al. Factors associated with serum alanine transaminase activity in healthy subjects: Consequences for the definition of normal values, for selection of blood donors, and for patients with chronic hepatitis C. Hepatology. 1998;27:1213-19.
12. Prati D, Taioli E, Zanella A, et al. Updated definitions of healthy ranges for serum alanine aminotranferase levels. Ann Intern Med. 2002;137:1-10.
13. Kim HC, Nam CM, Jee SH, et al. Normal serum aminotransferase concentration and risk of mortality from liver diseases: Prospective cohort study. BMJ. 2004;328:983.
14. Kaplan MM. Alanine aminotransferase levels: What’s normal? Ann Intern Med. 2002;137:49-51.
15. Kunde SS, Lazenby AJ, Clements RH, et al. Spectrum of NAFLD and diagnostic implications of the proposed new normal range for serum ALT in obese women. Hepatology. 2005;42:650-6.
16. Cohen JA, Kaplan MM. The SGOT/SGPT ratio: An indicator of alcoholic liver disease. Dig Dis Sci. 1979;24:835-8.
17. Diehl AM, Boitnott J, Van Duyn MA, et al. Relationship between pyridoxal 5′-phosphate deficiency and aminotransferase levels in alcoholic hepatitis. Gastroenterology. 1984;86:632-6.
18. Nathwani RA, Pais S, Reynolds TB, et al. Serum alanine aminotransferase in skeletal muscle diseases. Hepatology. 2005;41:380-2.
19. Sheth SG, Flamm SL, Gordon FD, et al. AST/ALT ratio predicts cirrhosis in patients with chronic hepatitis C virus infection. Am J Gastroenterol. 1998;93:44-8.
20. Giannini E, Botta F, Fasoli A, et al. Progressive liver functional impairment is associated with an increase in AST/ALT ratio. Dig Dis Sci. 1999;44:1249-53.
21. Kaplan M. Alkaline phosphatase. Gastroenterology. 1972;62:452-68.
22. Kaplan MM. Serum alkaline phosphatase: Another piece is added to the puzzle. Hepatology. 1986;6:526.
23. Bamford KF, Harris H, Luffman J, et al. Serum-alkaline phosphatase and the ABO blood groups. Lancet. 1965;1:530-1.
24. Wolf PL. Clinical significance of an increased or decreased serum alkaline phosphatase level. Arch Pathol Lab Med. 1978;102:497-501.
25. Rutenberg AM, Goldbarg JA, Pineda GP, et al. Serum γ-glutamyl transpeptidase activity in hepatobiliary pancreatic disease. Gastroenterology. 1963;45:43.
26. Rosalki SB, Tarlow D, Rau D. Plasma gamma-glutamyl transpeptidase elevation in patients receiving enzyme-inducing drugs. Lancet. 1971;2:376-7.
27. Bani-Sadr F, Miailhes P, Rosenthal E, et al. Risk factors for grade 3 or 4 gamma-glutamyl transferase elevation in HIV/hepatitis C virus-coinfected patients. AIDS. 2008;22:1234-6.
28. Moussavian SN, Becker RC, Piepmeyer JL, et al. Serum gamma-glutamyl transpeptidase and chronic alcoholism: Influence of alcohol ingestion and liver disease. Dig Dis Sci. 1985;30:211-14.
29. Hu G, Tuomilehto J, Pukkala E, et al. Joint effects of coffee consumption and serum gamma-glutamyltransferase on the risk of liver cancer. Hepatology. 2008;48:7-9.
30. Yang MH, Chen TH, Wang SE, et al. Biochemical predictors for absence of common bile duct stones in patients undergoing laparoscopic cholecystectomy. Surg Endosc. 2008;22:1620-4.
31. Kaplan MM, Rogers L. Separation of serum alkaline phosphatase isoenzymes by polyacrylamide gel electrophoresis. Lancet. 1968;2:1029-31.
32. Rothschild MA, Oratz M, Zimmon D, et al. Albumin synthesis in cirrhotic subjects studied with carbonate 14C. J Clin Invest. 1969;48:344-50.
33. Rothschild MA, Oratz M, Schreiber SS. Serum albumin. Hepatology. 1988;8:385-401.
34. van Zantsen SJO, Depla ACTM, Dekker PC, et al. The clinical importance of routine measurement of liver enzymes, total protein, and albumin in a general medicine outpatient clinic: A prospective study. N Engl J Med. 1992;40:53-61.
35. Trotter JF, Brimhall B, Arjal R. Specific laboratory methodologies achieve higher model for end-stage liver disease (MELD) scores for patients listed liver transplantation. Liver Transpl. 2004;10:995-1000.
36. Bellest L, Eschwege V, Poupon R, et al. A modified international normalized ratio as an effective way of prothrombin time standardization in hepatology. Hepatology. 2007;46:528-34.
37. Tripodi A, Chantarangkul V, Prirnignani M, et al. The international normalized ratio calibrated for cirrhosis normalizes prothrombin time results for model for end-stage liver disease calculation. Hepatology. 2007;46:520-7.
38. Plevris JN, Kaydon GH, Simpson KJ, et al. Serum hyaluronan: A non-invasive test for diagnosing liver cirrhosis. Eur J Gastroenterol Hepatol. 2000;12:1121-7.
39. Kaneda H, Hashimoto E, Yatsuji S, et al. Hyaluronic acid levels can predict severe fibrosis and platelet counts can predict cirrhosis in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2006;21:1459-65.
40. Mizguchi T, Katsuramaki T, Nobuoka T, et al. Serum hyaluronate level for predicting subclinical liver dysfunction after hepatectomy. World J Surg. 2004;28:971-6.
41. Poynard T, McHutchison J, Manns M, et al. Biochemical surrogate markers of liver fibrosis and activity in a randomized trial of peginterferon alpha-2b and ribavirin. Hepatology. 2003;38:481-92.
42. Berends MA, Snoek J, de Jong EM, et al. Biochemical and biophysical assessment of MTX-induced liver fibrosis in psoriasis patients. Liver Int. 2007;27:639-45.
43. Zaman A, Rosen HR, Ingram K, et al. Assessment of FIBROSpect II to detect hepatic fibrosis in chronic hepatitis C patients. Am J Med. 2007;280:e9-14.
44. Sandrin L, Fourquet B, Hasquenoph JM, et al. Transient elastography: A new noninvasive method for assessment of hepatic fibrosis. Ultrasound Med Biol. 2003;29:1705-13.
45. Nguyen-Khac E, Capron D. Noninvasive diagnosis of liver fibrosis by ultrasonic transient elastography (Fibroscan). Eur J Gastroenterol Hepatol. 2006;18:1321-5.
46. Friedrich-Rust M, Martens S, Sarrazin C, et al. Performance of transient elastography for the staging of liver fibrosis: A meta-analysis. Gastroenterology. 2008;134:960-74.
47. Yoneda M, Mawatari H, Fujita K, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with nonalcoholic fatty liver disease (NAFLD). Dig Liver Dis. 2008;40:371-8.
48. Adhoute X, Foucher J, Laharie D, et al. Diagnosis of liver fibrosis using FibroScan and other noninvasive methods in patients with hemochromatosis: A prospective study. Gastroenterol Clin Biol. 2008;32:180-7.
49. Rigamonti C, Donato MF, Fraquelli M, et al. Transient elastography predicts fibrosis progression in patients with recurrent hepatitis C after liver transplantation. Gut. 2008;576:821-7.
50. Gomez-Dominguez E, Mendoza J, Garcia-Buey L, et al. Transient elastography to assess hepatic fibrosis in primary biliary cirrhosis. Aliment Pharmacol Ther. 2008;27:441-7.
51. Huwart L, Sempoux C, Vicaut E, et al. Magnetic resonance elastography for the noninvasive staging of liver fibrosis. Gastroenterology. 2008;135:32-40.
52. Jost G, Wahllander A, von Mandach U, et al. Overnight salivary caffeine clearance: A liver function test suitable for routine use. Hepatology. 1987;7:338-44.
53. Oellerich M, Armstrong VW. The MEGX test: A tool for the real-time assessment of hepatic function. Ther Drug Monit. 2001;23:81-92.
54. Addario L, Scaglione G, Tritto G, et al. Prognostic value of quantitative liver function tests in viral cirrhosis: A prospective study. Eur J Gastroenterol Hepatol. 2006;18:713-20.
55. Shiffman M, Luketic VA, Sanyal AJ, et al. Hepatic lidocaine metabolism and liver histology in patients with chronic hepatitis and cirrhosis. Hepatology. 1994;19:933-40.
56. Carlisle R, Galambos JT, Warren WD. The relationship between conventional liver tests, quantitative function tests, and histopathology in cirrhosis. Dig Dis Sci. 1979;24:358-62.
57. Monroe PS, Baker AL, Schneider JF, et al. The aminopyrine breath test and serum bile acids reflect histologic severity in chronic hepatitis. Hepatology. 1982;2:317-22.
58. Einarsson K, Angelin B, Bjorkhem I, et al. The diagnostic value of fasting individual serum bile acids in anicteric alcoholic liver disease: Relation to liver morphology. Hepatology. 1985;5:108-11.
59. Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349:474-85.
60. O’Grady J, Alexander G, Hayllar K, et al. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97:439-45.
61. Maddrey WC. Drug-induced hepatotoxicity. J Clin Gastroenterol. 2005;39:S83-9.
62. Powell-Jackson P, Greenway B, William R. Adverse effects of laparotomy in patients with unsuspected liver disease. Br J Surg. 1982;69:449-51.
63. Garrison RN, Cryer HM, Howard DA, et al. Clarification of risk factors for abdominal operations in patients with hepatic cirrhosis. Ann Surg. 1984;199:648-55.
64. Mansour A, Watson W, Shayani V, et al. Abdominal operation in patients with cirrhosis: Still a major surgical challenge. Surgery. 1997;122:730-6.
65. Kamath PS, Kim WR. The Model for End-Stage Liver Disease (MELD). Hepatology. 2007;45:797-805.
66. Teh SH, Christein J, Donohue J, et al. Hepatic resection of hepatocellular carcinoma in patients with cirrhosis: Model of end-stage liver disease (MELD) score predicts perioperative mortality. J Gastrointest Surg. 2005;9:1207-15.
67. Seaman A, Barnes DS, Zein NN, et al. Predicting outcome after cardiac surgery in patients with cirrhosis: A comparison of Child-Pugh and MELD score. Clin Gastroenterol Hepatol. 2004;2:719-23.
68. Northup PG, Wanamaker RC, Lee VD, et al. Model for end-stage liver disease (MELD) predicts non-transplant surgical mortality in patients with cirrhosis. Ann Surg. 2005;242:244-51.
69. Wiesner R, Edwards E, Freeman R, et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology. 2003;124:91-6.