Chapter 7 Liver, biliary tract and pancreatic disease
The liver
Structure of the liver and biliary system
The liver
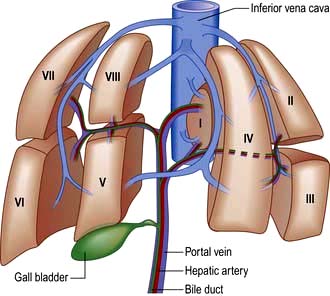
The liver is the body’s largest internal organ (1.2–1.5 kg) and is situated in the right hypochondrium. A functional division into the larger right lobe (containing caudate and quadrate lobes) and the left lobe is made by the middle hepatic vein. The liver is further subdivided into eight segments (Fig. 7.1) by divisions of the right, middle and left hepatic veins. Each segment has its own portal pedicle, permitting individual segment resection at surgery.
 The hepatic artery, a branch of the coeliac axis, supplies 25% of the hepatic blood flow. The hepatic artery autoregulates flow ensuring a constant total blood flow.
The hepatic artery, a branch of the coeliac axis, supplies 25% of the hepatic blood flow. The hepatic artery autoregulates flow ensuring a constant total blood flow.

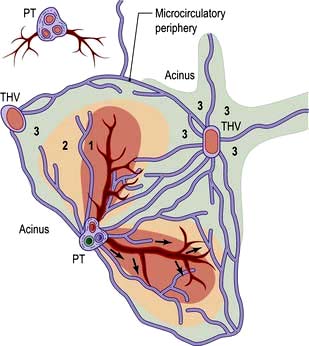
The acinus is the functional hepatic unit. This consists of parenchyma supplied by the smallest portal tracts containing portal vein radicles, hepatic arterioles and bile ductules (Fig. 7.2). The hepatocytes near this triad (zone 1) are well supplied with oxygenated blood and are more resistant to damage than the cells nearer the terminal hepatic (central) veins (zone 3).
The sinusoids lack a basement membrane and are loosely surrounded by specialist fenestrated endothelial cells and Kupffer cells (phagocytic cells). Sinusoids are separated by plates of liver cells (hepatocytes). The subendothelial space between the sinusoids and hepatocytes is the space of Disse, which contains a matrix of basement membrane constituents and stellate cells (see Fig. 7.23).
Stellate cells store retinoids in their resting state and contain the intermediate filament, desmin. When activated (to myofibroblasts) they are contractile and probably regulate sinusoidal blood flow. Endothelin and nitric oxide play a major role in modulating stellate cell contractility. Activated stellate cells produce signal proteins for synthesis or inhibition of degradation of extracellular matrix components, including collagen, as well as cytokines and chemotactic signals (see p. 328).
Functions of the liver
Protein metabolism (see also p. 197)
Synthesis and storage
The liver also synthesizes all factors involved in coagulation (apart from one-third of factor VIII) – that is, fibrinogen, prothrombin, factors V, VII, IX, X and XIII, proteins C and S and antithrombin (see Ch. 8) as well as components of the complement system. The liver stores large amounts of vitamins, particularly A, D and B12, and lesser amounts of others (vitamin K and folate), and also minerals – iron in ferritin and haemosiderin and copper.
Carbohydrate metabolism
Glucose homeostasis and the maintenance of the blood sugar is a major function of the liver. It stores approximately 80 g of glycogen. In the immediate fasting state, blood glucose is maintained either by glucose release from breaking down glycogen (glycogenolysis) or by synthesizing new glucose (gluconeogenesis). Sources for gluconeogenesis are lactate, pyruvate, amino acids from muscles (mainly alanine and glutamine) and glycerol from lipolysis of fat stores. In prolonged starvation, ketone bodies and fatty acids are used as alternative sources of fuel as the body tissues adapt to a lower glucose requirement (see Ch. 5).
Lipid metabolism
Fats are insoluble in water and are transported in plasma as protein-lipid complexes (lipoproteins). These are discussed in detail on page 1005.
The liver has a major role in metabolizing of lipoproteins. It synthesizes very-low-density lipoproteins (VLDLs) and high-density lipoproteins (HDLs). HDLs are the substrate for lecithin-cholesterol acyltransferase (LCAT), which catalyses the conversion of free cholesterol to cholesterol ester (see below). Hepatic lipase removes triglyceride from intermediate-density lipoproteins (IDLs) to produce low-density lipoproteins (LDLs) which are degraded by the liver after uptake by specific cell-surface receptors (see Fig. 20.19).
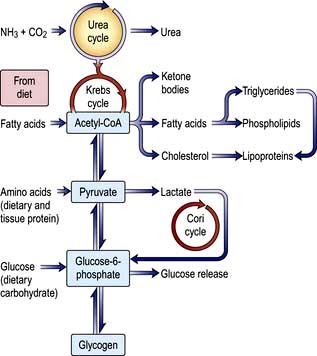
Cholesterol may be of dietary origin but most is synthesized from acetyl-CoA mainly in the liver, intestine, adrenal cortex and skin. It occurs either as free cholesterol or is esterified with fatty acids; this reaction is catalysed by LCAT. This enzyme is reduced in severe liver disease, increasing the ratio of free cholesterol to ester, which alters membrane structures. One result of this is the red cell abnormalities (e.g. target cells) seen in chronic liver disease. Phospholipids (e.g. lecithin) are synthesized in the liver. The complex interrelationships between protein, carbohydrate and fat metabolism are shown in Figure 7.3.
Formation of bile
Bile secretion and bile acid metabolism
Bile formation requires uptake of bile acids and other organic and inorganic ions across the basolateral (sinusoidal) membranes by multiple transport proteins (sodium taurocholate co-transporting polypeptide (NTCP) and sodium independent organic anion transporting polypeptide 2 (OATP2), Fig. 7.4). This process is driven by Na+/K+-ATPase in the basolateral membranes. Intracellular transport across hepatocytes is partly through microtubules and partly by cytosol transport proteins.
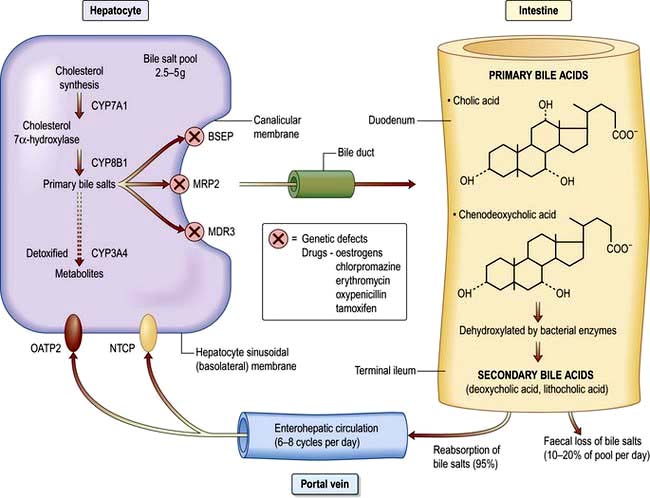
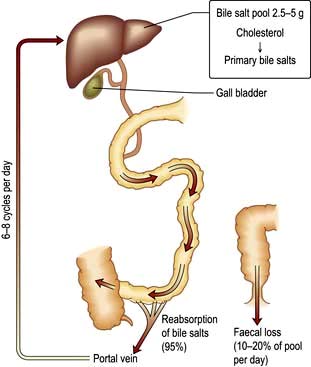
The bile acids are excreted into bile and then pass via the common bile duct into the duodenum. The two primary bile acids – cholic acid and chenodeoxycholic acid (Fig. 7.4) – are conjugated with glycine or taurine, which increases their solubility. Intestinal bacteria convert these acids into secondary bile acids, deoxycholic and lithocholic acid. Figure 7.5 shows the enterohepatic circulation of bile acids.
Bilirubin metabolism
Normally, 250–300 mg (425–510 mmol) of bilirubin are produced daily. The iron and globin are removed from haem and are reused. Biliverdin is formed from haem and then reduced to form bilirubin. The bilirubin produced is unconjugated and water-insoluble, due to internal hydrogen bonding, and is transported to the liver attached to albumin. Bilirubin dissociates from albumin and is taken up by hepatic cell membranes and transported to the endoplasmic reticulum by cytoplasmic proteins, where it is conjugated with glucuronic acid and excreted into bile. The microsomal enzyme, uridine diphosphoglucuronosyl transferase, catalyses the formation of bilirubin monoglucuronide and then diglucuronide. This conjugated bilirubin is water-soluble and is actively secreted into biliary canaliculi and excreted into the intestine within bile (Fig. 8.5). It is not absorbed from the small intestine because of its large molecular size. In the terminal ileum, bacterial enzymes hydrolyse the molecule, releasing free bilirubin which is then reduced to urobilinogen, some of which is excreted in the stools as stercobilinogen. The remainder is absorbed by the terminal ileum, passes to the liver via the enterohepatic circulation, and is re-excreted into bile. Urobilinogen bound to albumin enters the circulation and is excreted in urine via the kidneys. When hepatic excretion of conjugated bilirubin is impaired, a small amount is strongly bound to serum albumin and is not excreted by the kidneys; it accounts for the persistent hyperbilirubinaemia for a time after cholestasis has resolved.
Hormone and drug inactivation
The liver catabolizes hormones such as insulin, glucagon, oestrogens, growth hormone, glucocorticoids and parathyroid hormone. It is also the prime target organ for many hormones (e.g. insulin). It is the major site for the metabolism of drugs (see p. 348) and alcohol (see p. 225). Fat-soluble drugs are converted to water-soluble substances that facilitate their excretion in the bile or urine. Cholecalciferol is converted to 25-hydroxycholecalciferol.
Investigations
Investigative tests can be divided into:

 Urine tests – for bilirubin and urobilinogen
Urine tests – for bilirubin and urobilinogen


Blood tests
Useful blood tests for certain liver diseases are shown in Table 7.1.
Table 7.1 Useful blood and urine tests for certain liver diseases
| Test | Disease |
|---|---|
|
Anti-mitochondrial antibody |
Primary biliary cirrhosis |
|
Anti-nuclear, smooth muscle (actin), liver/kidney microsomal antibody |
Autoimmune hepatitis |
|
Raised serum immunoglobulins: |
|
|
IgG |
Autoimmune hepatitis |
|
IgM |
Primary biliary cirrhosis |
|
Viral markers |
Hepatitis A, B, C, D, E and others |
|
α-Fetoprotein |
Hepatocellular carcinoma |
|
Serum iron, transferrin saturation, serum ferritin |
Hereditary haemochromatosis |
|
Serum and urinary copper, serum caeruloplasmin |
Wilson’s disease |
|
α1-Antitrypsin |
Cirrhosis (± emphysema) |
|
Anti-nuclear cytoplasmic antibodies |
Primary sclerosing cholangitis |
|
Markers of liver fibrosis |
Non-alcoholic fatty liver disease |
|
|
Hepatitis C |
|
Genetic analyses |
e.g. HFE gene (hereditary haemochromatosis) |
Liver biochemistry


This is a microsomal enzyme present in liver, but also in many tissues. Its activity can be induced by drugs such as phenytoin and by alcohol. If the ALP is normal, a raised serum γ-GT can be a useful guide to alcohol intake (see p. 1182). However, mild elevations of γ-GT are common, even with a small alcohol consumption and is also raised with fatty liver disease. It does not necessarily indicate liver disease if the other liver biochemical tests are normal. In cholestasis the γ-GT rises in parallel with the ALP as it has a similar pathway of excretion. This is also true of 5-nucleotidase, another microsomal enzyme that can be measured in blood.
Additional blood investigations
Haematological
 Bleeding produces a hypochromic, microcytic picture.
Bleeding produces a hypochromic, microcytic picture.
 Alcohol causes macrocytosis, sometimes with leucopenia and thrombocytopenia.
Alcohol causes macrocytosis, sometimes with leucopenia and thrombocytopenia.
 Hypersplenism results in pancytopenia.
Hypersplenism results in pancytopenia.
 Cholestasis can often produce abnormal-shaped cells, and also deficiency of vitamin K.
Cholestasis can often produce abnormal-shaped cells, and also deficiency of vitamin K.
 Haemolysis may accompany acute liver failure and jaundice.
Haemolysis may accompany acute liver failure and jaundice.
 Aplastic anaemia occurs in up to 2% of patients with acute viral hepatitis.
Aplastic anaemia occurs in up to 2% of patients with acute viral hepatitis.
 A raised serum ferritin with transferrin saturation (>60%) is seen in hereditary haemochromatosis.
A raised serum ferritin with transferrin saturation (>60%) is seen in hereditary haemochromatosis.
Biochemical



Immunological tests
 Anti-mitochondrial antibody (AMA) in serum is found in over 95% of patients with primary biliary cirrhosis (PBC) (p. 338). Several different AMA subtypes are described, depending on their antigen specificity, and are also found in autoimmune hepatitis and other autoimmune diseases. AMA is demonstrated by an immunofluorescent technique and is neither organ- nor species-specific. M2 subtype is specific for PBC.
Anti-mitochondrial antibody (AMA) in serum is found in over 95% of patients with primary biliary cirrhosis (PBC) (p. 338). Several different AMA subtypes are described, depending on their antigen specificity, and are also found in autoimmune hepatitis and other autoimmune diseases. AMA is demonstrated by an immunofluorescent technique and is neither organ- nor species-specific. M2 subtype is specific for PBC.


Genetic analysis
These tests are performed routinely for haemochromatosis (HFE gene) and for α1-antitrypsin deficiency. Markers are also available for the most frequent abnormal genes in Wilson’s disease (see p. 341).
Imaging techniques
Ultrasound examination


 the detection of gallstones (Fig. 7.6)
the detection of gallstones (Fig. 7.6)
 focal liver disease – lesions >1 cm
focal liver disease – lesions >1 cm
 general parenchymal liver disease
general parenchymal liver disease


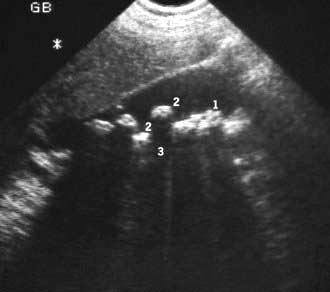
Other abdominal masses can be delineated and biopsies obtained under ultrasonic guidance.
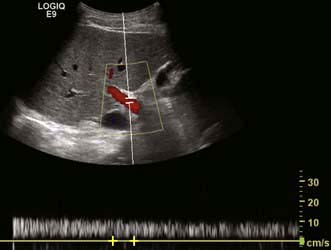
Colour Doppler ultrasound will demonstrate vascularity within a lesion and the direction of portal and hepatic vein blood flow (Fig. 7.7).
Computed tomography (CT) examination
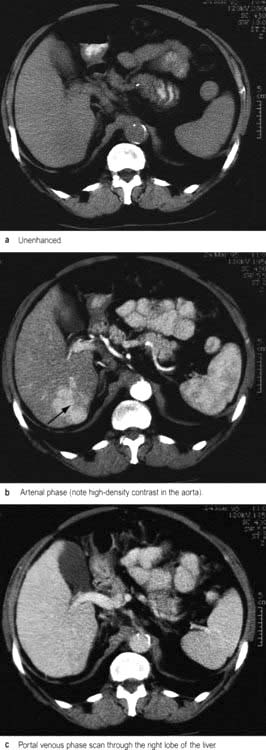
CT during or immediately after i.v. contrast shows both arterial and portal venous phases of enhancement, enabling more precise characterization of a lesion and its vascular supply (Fig. 7.8). Retrospective analysis of data allows multiple overlapping slices to be obtained with no increase in the radiation dose, providing excellent visualization of the size, shape and density of the liver, pancreas, spleen, lymph nodes and lesions in the porta hepatis. Multi-planar and three-dimensional reconstruction in the arterial phase can create a CT angiogram, often making formal invasive angiography unnecessary. CT also provides guidance for biopsy. It has advantages over US in detecting calcification and is useful in obese subjects, although US is usually the imaging modality used first to investigate liver disease.






Endoscopic retrograde cholangiopancreatography (ERCP)
 Common bile duct stones can be removed after performing a diathermy cut of the sphincter to facilitate their withdrawal. Sphincterotomy has a morbidity rate of 3–5%: acute pancreatitis is the commonest, severe haemorrhage is rare. There is an overall mortality of 0.4%.
Common bile duct stones can be removed after performing a diathermy cut of the sphincter to facilitate their withdrawal. Sphincterotomy has a morbidity rate of 3–5%: acute pancreatitis is the commonest, severe haemorrhage is rare. There is an overall mortality of 0.4%.

 Brachytherapy can be administered after placement at ERCP for therapy of cholangiocarcinoma.
Brachytherapy can be administered after placement at ERCP for therapy of cholangiocarcinoma.
Liver biopsy (Practical Box 7.1)
Histological examination of the liver is valuable in the differential diagnosis of diffuse or localized parenchymal disease. Liver biopsy can be performed on a day-case basis. The indications and contraindications are shown in Table 7.2. The mortality rate is less than 0.02% when performed by experienced operators.
 Practical Box 7.1
Practical Box 7.1
Needle biopsy of the liver









Table 7.2 Indications and contraindications for liver biopsy
|
|
Symptoms of liver disease
Chronic liver disease
 Right hypochondrial pain due to liver distension
Right hypochondrial pain due to liver distension
 Abdominal distension due to ascites
Abdominal distension due to ascites
 Ankle swelling due to fluid retention
Ankle swelling due to fluid retention
 Haematemesis and melaena from gastrointestinal haemorrhage
Haematemesis and melaena from gastrointestinal haemorrhage
 Pruritus due to cholestasis – this is often an early symptom of primary biliary cirrhosis
Pruritus due to cholestasis – this is often an early symptom of primary biliary cirrhosis
 Breast swelling (gynaecomastia), loss of libido and amenorrhoea due to endocrine dysfunction
Breast swelling (gynaecomastia), loss of libido and amenorrhoea due to endocrine dysfunction
 Confusion and drowsiness due to neuropsychiatric complications (portosystemic encephalopathy).
Confusion and drowsiness due to neuropsychiatric complications (portosystemic encephalopathy).
Signs of liver disease
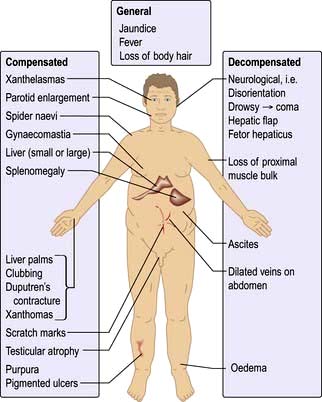
Jaundice
 Haemolytic jaundice – increased bilirubin load for the liver cells
Haemolytic jaundice – increased bilirubin load for the liver cells


Haemolytic jaundice
The increased breakdown of red cells (see p. 375) leads to an increase in production of bilirubin. The resulting jaundice is usually mild (serum bilirubin of 68–102 µmol/L or 4–6 mg/dL) as normal liver function can easily handle the increased bilirubin derived from excess haemolysis. Unconjugated bilirubin is not water-soluble and therefore does not pass into urine; hence the term ‘acholuric jaundice’. Urinary urobilinogen is increased.
The causes of haemolytic jaundice are those of haemolytic anaemia (p. 375). The clinical features depend on the cause; anaemia, jaundice, splenomegaly, gallstones and leg ulcers may be seen.
Investigations show features of haemolysis (p. 375). The level of unconjugated bilirubin is raised but the serum ALP, transferases and albumin are normal. Serum haptoglobulins are low. The differential diagnosis is from other forms of jaundice.
Congenital hyperbilirubinaemias (non-haemolytic)
Conjugated
Dubin–Johnson (autosomal recessive) and Rotor syndromes are due to defects in hepatic bilirubin handling. The prognosis is good in both. In the Dubin–Johnson syndrome there are mutations in both MRP2 (p. 305) transporter genes; the liver is black owing to melanin deposition.
Benign recurrent intrahepatic cholestasis
This is rare and presents in early adulthood. Recurrent attacks of acute cholestasis occur without progression to chronic liver disease. Jaundice, severe pruritus, steatorrhoea and weight loss develop. Serum γ-GT is normal. The gene has been mapped to the FIC1 locus, but the precise relation to cholestasis is unclear. It may be associated with intrahepatic cholestasis of pregnancy (p. 346).
Progressive familial intrahepatic cholestasis (PFIC) syndromes
This is a heterogeneous group of conditions defined by defective secretion of bile acids (see Figs 7.4 and 7.29). They are autosomal recessive. In type 1 (PFIC1), with cholestasis in the first weeks of life, the γ-GT is normal. The gene is on the familial intrahepatic cholestasis-1 gene (FIC1) locus, and has been mapped to a region encoding P type ATPases (ATP8BI) on chromosome 18q21. Type 2 (PFIC2) has been mapped to the bile salt export pump gene (BSEP, also called ABCB1). The protein is located in the canalicular domain of the hepatocyte plasma membrane. The phenotypic expression frequently is a nonspecific giant cell hepatitis progressing to cholestasis – in both types, the γ-GT is normal. Type 3 is due to a multidrug resistance protein 3-P-glycoprotein PGY3 (MDR-3) gene mutation (also called ABCB4 gene) leading to deficient canalicular phosphatidylcholine transport and thus toxic bile acids causing liver damage, which can lead to cirrhosis. Liver transplantation is the only cure for these syndromes.
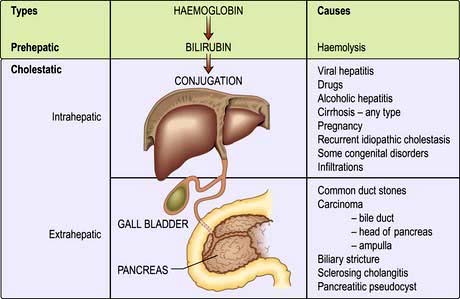
Cholestatic jaundice (acquired)
This can be divided into extrahepatic and intrahepatic cholestasis. The causes are shown in Figure 7.10.
 Extrahepatic cholestasis is due to large duct obstruction of bile flow at any point in the biliary tract distal to the bile canaliculi.
Extrahepatic cholestasis is due to large duct obstruction of bile flow at any point in the biliary tract distal to the bile canaliculi.

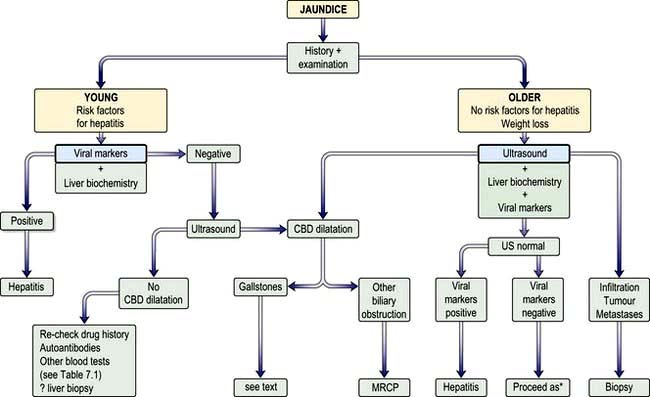
Differential diagnosis of jaundice
 Country of origin. The incidence of hepatitis B virus (HBV) infection is increased in many parts of the world (p. 318).
Country of origin. The incidence of hepatitis B virus (HBV) infection is increased in many parts of the world (p. 318).

 Recent outbreak of jaundice. An outbreak in the community suggests hepatitis A virus (HAV).
Recent outbreak of jaundice. An outbreak in the community suggests hepatitis A virus (HAV).
 Recent consumption of shellfish. This suggests HAV infection.
Recent consumption of shellfish. This suggests HAV infection.

 Men having sex with men. This increases the chance of HBV infection.
Men having sex with men. This increases the chance of HBV infection.
 Female sex workers. This increases the chance of HBV infection.
Female sex workers. This increases the chance of HBV infection.


 Drugs taken (particularly in the previous 2–3 months). Many drugs cause jaundice (see p. 348).
Drugs taken (particularly in the previous 2–3 months). Many drugs cause jaundice (see p. 348).



 Recent surgery on the biliary tract or for carcinoma.
Recent surgery on the biliary tract or for carcinoma.

 Fevers or rigors. These are suggestive of cholangitis or possibly a liver abscess.
Fevers or rigors. These are suggestive of cholangitis or possibly a liver abscess.
Clinical features
The signs of acute and chronic liver disease should be looked for (p. 312). Certain additional signs may be helpful:
 Hepatomegaly. A smooth tender liver is seen in hepatitis and with extrahepatic obstruction, but a knobbly irregular liver suggests metastases. Causes of hepatomegaly are shown in Table 7.3.
Hepatomegaly. A smooth tender liver is seen in hepatitis and with extrahepatic obstruction, but a knobbly irregular liver suggests metastases. Causes of hepatomegaly are shown in Table 7.3.

 Ascites. This is found in cirrhosis but can also be due to carcinoma (particularly ovarian) and many other causes (see Table 7.14).
Ascites. This is found in cirrhosis but can also be due to carcinoma (particularly ovarian) and many other causes (see Table 7.14).



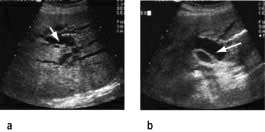
Hepatitis
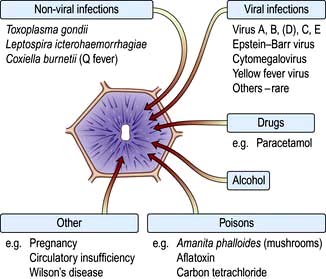
Acute parenchymal liver damage can be caused by many agents (Fig. 7.13).
Chronic hepatitis is defined as any hepatitis lasting for 6 months or longer and is classified according to the aetiology (Table 7.4). Chronic viral hepatitis is the principal cause of chronic liver disease, cirrhosis and hepatocellular carcinoma worldwide.
Chronic hepatitis




Viral hepatitis
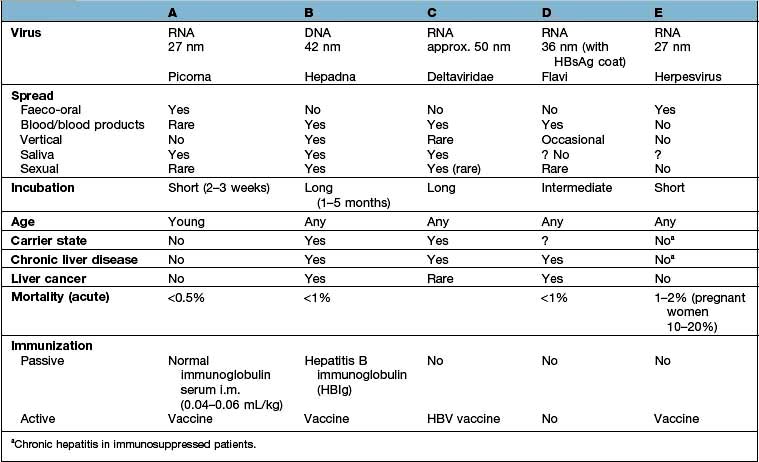
The differing features of the common forms of viral hepatitis are summarized in Table 7.5.
Hepatitis A
Epidemiology
Hepatitis A virus (HAV)
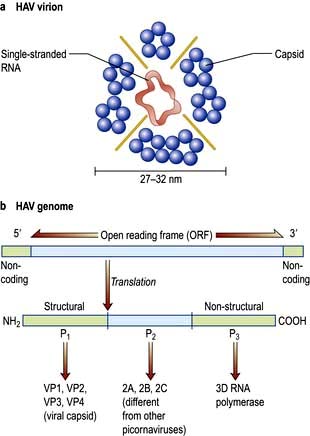
HAV is a picornavirus, having the structure shown in Figure 7.14. It has a single serotype as only one epitope is immuno-dominant. It replicates in the liver, is excreted in bile and then excreted in the faeces for about 2 weeks before the onset of clinical illness and for up to 7 days after. The disease is maximally infectious just before the onset of jaundice. HAV particles can be demonstrated in the faeces by electron microscopy.
Clinical features
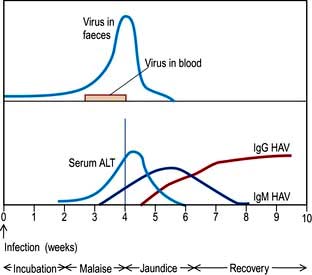
After 1 or 2 weeks, some patients become jaundiced and symptoms often improve. Persistence of nausea, vomiting or any mental confusion warrants assessment in hospital. As the jaundice deepens, the urine becomes dark and the stools pale owing to intrahepatic cholestasis. The liver is moderately enlarged and the spleen is palpable in about 10% of patients. Occasionally, tender lymphadenopathy is seen, with a transient rash in some cases. Thereafter the jaundice lessens and in the majority of cases the illness is over within 3–6 weeks. Extrahepatic complications are rare but include arthritis, vasculitis, myocarditis and acute kidney injury. A biphasic illness occasionally occurs, with the return of jaundice. Rarely the disease may be very severe with fulminant hepatitis, liver coma and death. The typical sequence of events after HAV exposure is shown in Figure 7.15.
Hepatitis B
Epidemiology
FURTHER READING
Amon JJ. Hepatitis in drug users: time for attention, time for action. Lancet 2011; 378:543–544.
Dienstag JL. Hepatitis B viral infection. N Engl J Med 2008; 359:1486–1500.
Nelson PK, Mathers BM, Cowie B et al. Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet 2011; 378:578–583.
Hepatitis B virus (HBV)
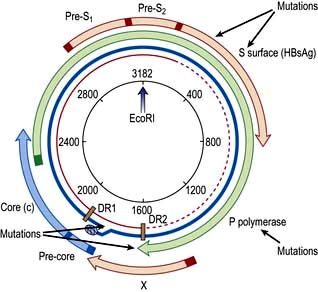
The core or nucleocapsid is formed of core protein (HBcAg) containing incompletely double-stranded circular DNA and DNA polymerase/reverse transcriptase. One strand is almost a complete circle and contains overlapping genes that encode both structural proteins (pre-S, surface (S), core (C)) and replicative proteins (polymerase and X). The other strand is variable in length. DR1 and DR2 are direct repeats necessary for HBV synthesis during viral replication (Fig. 7.16).
Pathogenesis
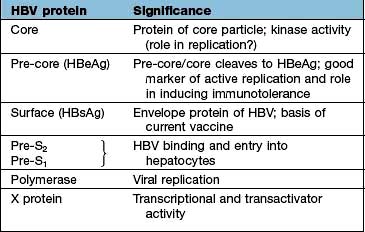
Translation into HBV proteins (Table 7.6) as well as replication of the genome takes place in the endoplasmic reticulum; they are then packaged together and exported from the cell. There is an excess production of non-infective HbsAg particles which are extruded into the circulation.
 |
Chronic HBV infection progresses through a replicative and an integrated phase. In the replicative phase there is active viral replication with hepatic inflammation and the patient is highly infectious with HBeAg and HBV DNA positivity. At some stage the viral genome becomes integrated into the host DNA and the viral genes are then transcribed along with those of the host. At this stage, the level of HBV DNA in the serum is low and the patient is HBeAg negative and HBe antibody positive. The aminotransferases are now normal or only slightly elevated and liver histology shows little inflammation, often with cirrhosis. Hepatocellular carcinoma (HCC) develops in patients with this late-stage disease, but the mechanism is still unclear. The REVEAL Study (Risk Evaluation of Viral load Elevation and Associated Liver disease) showed that the risk of HCC was related to levels of HBV DNA rather than a raised aminotransferase (ALT). Integration of the viral DNA with the host-cell chromosomal DNA does appear to have a major role in carcinogenesis. There is evidence to implicate inactivation of p53-induced apoptosis by protein X (Table 7.6), allowing accumulation of abnormal cells and, eventually, carcinogenesis.
Clinical features of acute hepatitis
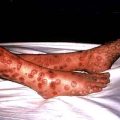
The sequence of events following acute HBV infection is shown in Figure 7.17. However, in many, the infection is subclinical. When HBV infection is acquired perinatally, an acute hepatitis usually does not occur as there is a high level of immunological tolerance and the virus persists in over 90%. If there is an acute clinical episode the virus is cleared in approximately 99% of patients as there is a good immune reaction. The clinical picture is the same as that found in HAV infection, although the illness may be more severe. In addition, a serum sickness-like immunological syndrome may be seen. This consists of rashes (e.g. urticaria or a maculopapular rash) and polyarthritis affecting small joints occurring in up to 25% of cases in the prodromal period. Fever is usual. Extrahepatic immune complex-mediated conditions such as an arteritis or glomerulonephritis are occasionally seen.
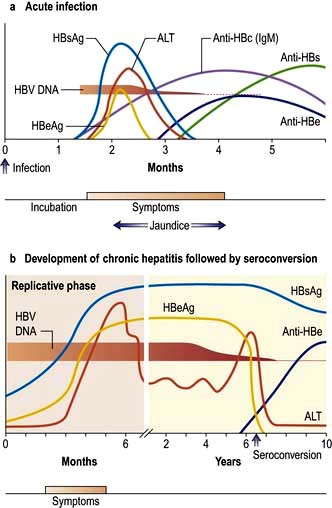
Investigations
These are generally the same as for hepatitis A.
Specific tests. The markers for HBV are shown in Table 7.7. HBsAg is looked for initially; if it is found, a full viral profile is then performed. In acute infection, as HBsAg may be cleared rapidly, anti-HBc IgM is diagnostic. HBV DNA is the most sensitive index of viral replication. HBV DNA has been shown to persist (using polymerase chain reaction (PCR) techniques) even when the e antibody has developed.
|
Antigens |
|
|
HBsAg |
Acute or chronic infection |
|
HBeAg |
Acute hepatitis B |
|
Persistence implies: |
|
|
Continued infectious state development of chronicity |
|
|
Increased severity of disease |
|
|
HBV DNA |
Implies viral replication |
|
Found in serum and liver |
|
|
Levels indicate response to treatment |
|
|
Antibodies |
|
|
Anti-HBs |
Immunity to HBV; previous exposure; vaccination |
|
Anti-HBe |
Seroconversion |
|
Anti-HBc |
|
|
IgM |
Acute hepatitis B (high titre) |
|
Chronic hepatitis B (low titre) |
|
|
IgG |
Past exposure to hepatitis B (HBsAg-negative) |
Course
The majority of patients recover completely, fulminant hepatitis occurring in up to 1%. Some patients go on to develop chronic hepatitis (p. 321), cirrhosis (p. 328) and hepatocellular carcinoma (p. 347) or have inactive chronic HBV infection. The outcome depends upon several factors, including the virulence of the virus and the immuno-competence and age of the patient. Some genetic factors, e.g. the presence of MHC class II genotype, may alter host defence to HBV.
Chronic HBV infection
Clinical features and investigations of active chronic hepatitis B
Investigations show a moderate rise in aminotransferases and a slightly raised ALP. The serum bilirubin is often normal. HBsAg and HBV DNA are found in the serum, usually with HBe antigen, unless a mutant virus is involved (see p. 319).
Treatment for chronic hepatitis B
 Patients with moderate to severe active necroinflammation and/or fibrosis in the liver biopsy with HBV DNA above 2000 iu/mL (approximately 10 000 copies/mL) and/or ALT above the upper limit of normal. Age and co-morbidities also affect the decision to treat and with which agents.
Patients with moderate to severe active necroinflammation and/or fibrosis in the liver biopsy with HBV DNA above 2000 iu/mL (approximately 10 000 copies/mL) and/or ALT above the upper limit of normal. Age and co-morbidities also affect the decision to treat and with which agents.


Interferon, entecavir and tenofovir are the most commonly used drugs (p. 93). Response to therapy is judged by a reduction in the HBV DNA level, and if HBeAg is present, by sero-conversion to anti-HBe.
Overall, the response rate (i.e. disappearance of HbeAg) is 25–40%. The success rate depends on factors shown in Table 7.8.
Table 7.8 Factors predictive of a sustained response to treatment in patients with chronic hepatitis B
|
Duration of disease |
Short |
|
Liver biochemistry |
High serum aminotransferases |
|
Histology |
Active liver disease (mild to moderate) |
|
Viral levels |
Low HBV DNA levels |
|
Other |
Absence of immunosuppression |
|
Female gender |
|
|
Adult acquired |
|
|
Delta virus negative |
|
|
Rapidity of response to oral therapy |
Oral therapy
 Entecavir is very effective and reduces HBV DNA quickly, and there is little viral resistance. Serum HBV DNA becomes negative in 67% (HBeAg-positive patients) and 90% (HBeAg-negative patients) by 48 weeks. It should not be used in patients with lamivudine-induced mutants, but can be used if adefovir mutations have occurred.
Entecavir is very effective and reduces HBV DNA quickly, and there is little viral resistance. Serum HBV DNA becomes negative in 67% (HBeAg-positive patients) and 90% (HBeAg-negative patients) by 48 weeks. It should not be used in patients with lamivudine-induced mutants, but can be used if adefovir mutations have occurred.


The duration of all treatment is probably lifelong and is still being assessed.
Hepatitis D
 Co-infection of HDV and HBV which is clinically indistinguishable from an acute icteric HBV infection, but a biphasic rise of serum aminotransferases may be seen. Diagnosis is confirmed by finding serum IgM anti-HDV in the presence of IgM anti-HBc. IgM anti-delta appears at 1 week and disappears by 5–6 weeks (occasionally 12 weeks) when serum IgG antidelta is seen. The HDV RNA is an early marker of infection. The infection may be transient but the clinical course is variable.
Co-infection of HDV and HBV which is clinically indistinguishable from an acute icteric HBV infection, but a biphasic rise of serum aminotransferases may be seen. Diagnosis is confirmed by finding serum IgM anti-HDV in the presence of IgM anti-HBc. IgM anti-delta appears at 1 week and disappears by 5–6 weeks (occasionally 12 weeks) when serum IgG antidelta is seen. The HDV RNA is an early marker of infection. The infection may be transient but the clinical course is variable.

Hepatitis C
Epidemiology
Hepatitis C virus (HCV)
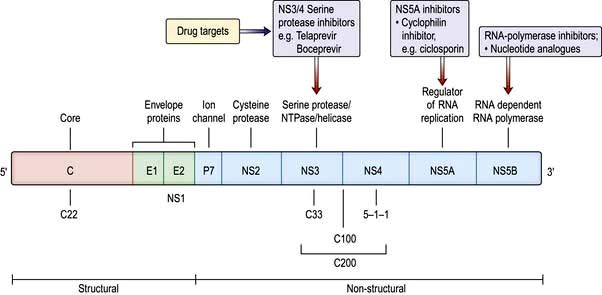
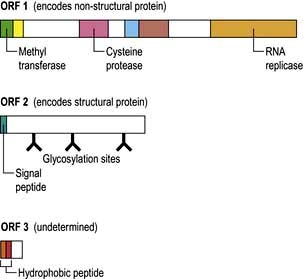
HCV is a single-stranded RNA virus of the Flaviviridae family. The RNA genome is approximately 10 kb in length, encoding a polyprotein product consisting of structural (capsid and envelope) and non-structural viral proteins (Fig. 7.18). Comparisons of subgenomic regions, such as E1, NS4 or NS5, have allowed variants to be classified into six genotypes. Variability is distributed throughout the genome with the non-structural gene of different genotypes showing 30–50% nucleotide sequence disparity. Genotypes 1a and 1b account for 70% of cases in the USA and 50% in Europe. There is a rapid change in envelope proteins, making it difficult to develop a vaccine. Antigens from the nucleocapsid regions have been used to develop enzyme-linked immunosorbent assays (ELISA). The current assay, ELISA-3, incorporates antigens NS3, NS4 and NS5 regions.
Course
Some 85–90% of asymptomatic patients develop chronic liver disease. A higher percentage of symptomatic patients ‘clear’ the virus with only 48–75% going on to chronic liver disease (p. 312). Cirrhosis develops in about 20–30% within 10–30 years and of these patients between 7% and 15% will develop hepatocellular carcinoma. The course is adversely affected by co-infection with HBV and/or HIV, and by alcohol consumption, which should be discouraged.
Chronic hepatitis C infection
Clinical features
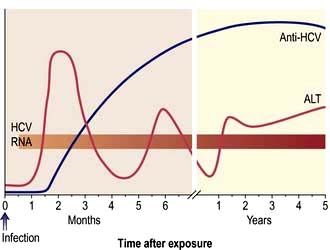
Patients with chronic hepatitis C infection are usually asymptomatic, the disease being discovered only following a routine biochemical test when mild elevations in the aminotransferases (usually ALT) are noticed (50%). The elevation in ALT may be minimal and fluctuating (Fig. 7.19) and some patients have a persistently normal ALT (25%), the disease being detected by checking HCV antibodies (e.g. in blood donors).
Diagnosis
The HCV genotype should be characterized in patients who are to be given treatment (see below).
Liver biopsy is indicated if treatment is being considered, especially for genotypes 1 and 4. Non-invasive methods for the diagnosis of fibrosis such as serum markers and elastography (p. 309) can replace the need for biopsy in many cases and are useful in follow-up. The changes on liver biopsy are highly variable. Sometimes only minimal inflammation is detected, but in most cases the features of chronic hepatitis are present, as described (p. 316). Lymphoid follicles are often present in the portal tracts, and fatty change is frequently seen. Histological scoring systems such as METAVIR and Ishak evaluate the inflammation and fibrosis and are used to guide therapy.
Treatment
Treatment (Fig. 7.20) is appropriate for patients with chronic hepatitis on liver histology and/or who have HCV RNA in their serum whether or not serum aminotransferases are raised. The presence of cirrhosis is not a contraindication, but therapeutic responses are less likely. Patients with decompensated cirrhosis should be considered for transplantation. The aim of treatment is to eliminate the HCV RNA from the serum in order to:


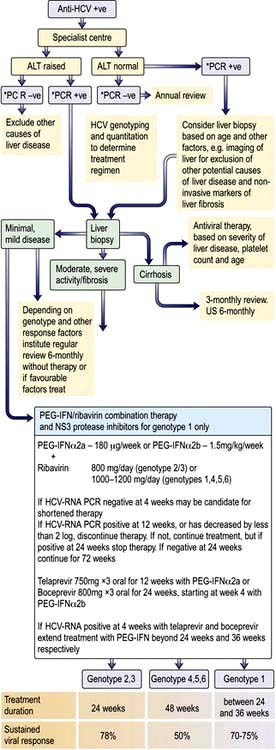
Side-effects of interferon are described on page 321. Ribavirin is usually well tolerated but side-effects include a dose-related haemolysis, pruritus and nasal congestion. Telaprevir causes a rash and anaemia, and boceprevir causes dysgeusia and anaemia. Pregnancy must be avoided with antiviral therapy.
Hepatitis E
Hepatitis E virus (HEV) is an RNA virus (herpesvirus) (Fig. 7.21) causing a hepatitis clinically very similar to hepatitis A. It is enterally transmitted, usually by contaminated water, with 30% of dogs, pigs and rodents carrying the virus. Epidemics have been seen in many developing countries and sporadically in developed countries, in patients who have had contacts with farm animals or travel abroad. It has a mortality from fulminant hepatic failure of 1–2%, which rises to 20% in pregnant women. There is no carrier state and it does not progress to chronic liver disease except in some immunosuppressed patients. An ELISA for IgG and IgM anti-HEV is available for diagnosis. HEV RNA can be detected in the serum or stools by PCR. Prevention and control depend on good sanitation and hygiene; a vaccine has been developed and used successfully in China.
Acute hepatitis due to other infectious agents
Infectious mononucleosis (see also p. 99). This is due to the Epstein–Barr (EB) virus. Mild jaundice associated with minor abnormalities of liver biochemistry is extremely common, but ‘clinical’ hepatitis is rare. Hepatic histological changes occur within 5 days of onset; the sinusoids and portal tracts are infiltrated with large mononuclear cells but the liver architecture is preserved. A Paul–Bunnell or Monospot test is usually positive, and atypical lymphocytes are present in the peripheral blood. Treatment is symptomatic.
Cytomegalovirus (CMV) (see also p. 99). This can cause acute hepatitis, usually a glandular fever type syndrome in healthy individuals, but is more severe in those with an impaired immune response. Only the latter need treatment with valganciclovir or ganciclovir.
CMV DNA is positive in blood; CMV IgM is also positive, but there are false-positive reactions.
The liver biopsy shows intranuclear inclusions and giant cells.
Herpes simplex (see also p. 97). Very occasionally the herpes simplex virus causes a generalized acute infection, particularly in the immunosuppressed patient, and occasionally in pregnancy. Aminotransferases are usually massively elevated. Liver biopsy shows extensive necrosis. Aciclovir is used for treatment.
Toxoplasmosis (see also p. 149). The clinical picture is similar to infectious mononucleosis, with abnormal liver biochemistry, but the Paul–Bunnell test is negative.
Yellow fever (see also p. 329). This viral infection is carried by the mosquito Aedes aegypti and can cause acute hepatic necrosis. There is no specific treatment.
Fulminant hepatic failure (FHF)
This is defined as severe hepatic failure in which encephalopathy develops in under 2 weeks in a patient with a previously normal liver (occasionally in some patients with previous liver damage; e.g. D virus superinfection in a previous carrier of HBsAg, Budd–Chiari syndrome or Wilson’s disease). Cases that evolve at a slower pace (2–12 weeks) are called subacute or subfulminant hepatic failure. FHF is a rare but often life-threatening syndrome that is due to acute hepatitis from many causes (Table 7.9). The causes vary throughout the world; most cases are due to viral hepatitis, but paracetamol overdose is common in the UK (50% of cases). HCV does not usually cause FHF although exceptional cases have been reported from Japan and India.
|
|
Histologically, there is multiacinar necrosis involving a substantial part of the liver. Severe fatty change is seen in pregnancy (p. 346), Reye’s syndrome (p. 348) or following tetracycline administration intravenously.
Treatment
There is no specific treatment, but patients should be managed in a specialized unit. Transfer criteria to such units are shown in Box 7.1. Supportive therapy as for hepatic encephalopathy is necessary (see p. 337). When signs of raised intracranial pressure (which is sometimes measured directly) are present, 20% mannitol (1 g/kg bodyweight) should be infused intravenously; this dose may need to be repeated. Dexamethasone is of no value. Hypoglycaemia, hypokalaemia, hypomagnesaemia, hypophosphataemia and hypocalcaemia should be anticipated and corrected with 10% dextrose infusion (checked by 2-hourly dipstick testing), potassium, calcium, phosphate and magnesium supplements. Hyponatraemia should be corrected with hypertonic saline. Coagulopathy is managed with intravenous vitamin K, platelets, blood or fresh frozen plasma. Haemorrhage may be a problem and patients are given a proton pump inhibitor (PPI) to prevent gastrointestinal bleeding. Prophylaxis against bacterial and fungal infection is routine, as infection is a frequent cause of death and may preclude liver transplantation. Suspected infection should be treated immediately with suitable antibiotics. Renal and respiratory failure should be treated as necessary. Liver transplantation has been a major advance for patients with FHF. It is difficult to judge the timing or the necessity for transplantation, but there are guidelines based on validated prognostic indices of survival (see below).
 Box 7.1
Box 7.1




Course and prognosis
In mild cases (grades I and II encephalopathy with drowsiness and confusion), two-thirds of the patients will survive. The outcome of severe cases (grades III and IV encephalopathy with stupor or deep coma) is related to the aetiology. In special units, 70% of patients with paracetamol overdosage and grade IV coma survive, as do 30–40% patients with HAV or HBV hepatitis. Poor prognostic variables indicating a need to transplant the liver are shown in Box 7.2.
 Box 7.2
Box 7.2








Autoimmune hepatitis
Pathogenesis
FURTHER READING
Czaja AJ. Genetic factors affecting the occurrence, clinical phenotype, and outcome of autoimmune hepatitis. Clin Gastroenterol Hepatol 2008; 6(4):379–388.
Manns MP, Woynarowski M, Kreisel W et al. Budesonide induces remission more effectively than prednisolone in a controlled trial of patients with autoimmune hepatitis. Gastroenterology 2010; 139:1198–1206.