[level-membership-for-pediatrics-category]
Lesions of The Stomach
Hypertrophic Pyloric Stenosis
Hypertrophic pyloric stenosis (HPS) is one of the most common surgical conditions of the newborn.1–9 It occurs at a rate of 1 to 4 per 1,000 live births in Caucasian infants, but is seen less often in non-Caucasian children.1–4 Males are affected more often with a 4 : 1 male-to-female ratio. Risk factors for HPS include family history, gender, younger maternal age, being a first-born infant, and maternal feeding patterns.4,9,10 Premature infants are diagnosed with HPS later than term or post-term infants.4
Etiology
The cause of HPS is unknown, but genetic and environmental factors appear to play a large role in the pathophysiology. Circumstantial evidence for a genetic predisposition includes race discrepancies, the increased frequency in males, and the birth order (first-born infants with a positive family history). Environmental factors associated with HPS include the method of feeding (breast vs formula), seasonal variability, exposure to erythromycin, and transpyloric feeding in premature infants.5–7 Additionally, there has been interest in several gastrointestinal peptides or growth factors that may facilitate pyloric hypertrophy. Some of these include excessive substance P, decreased neurotrophins, deficient nitric oxide synthase, and gastrin hypersecretion.8,9 Thus, the etiology of HPS is likely multifactorial with environmental influences.
Diagnosis
The classic presentation of HPS is nonbilious, projectile vomiting in a full-term neonate who is between 2 and 8 weeks old. Initially, the emesis is infrequent and may appear to be gastroesophageal reflux disease. However, over a short period of time, the emesis occurs with every feeding and becomes forceful (i.e., projectile). The contents of the emesis are usually the recent feedings, but signs of gastritis are not uncommon (‘coffee-ground’ emesis). On physical examination, the neonate usually appears well if the diagnosis is made early. However, depending on the duration of symptoms and degree of dehydration, the neonate may be gaunt and somnolent. Visible peristaltic waves may be present in the mid to left upper abdomen. The pylorus may be palpable in 72–89% of patients.11,12 To palpate the hypertrophied pylorus, the baby must be relaxed. Techniques for relaxing the infant include bending the knees and flexing the hips, and using a pacifier with sugar water. These techniques should be attempted after the stomach has been decompressed with a 10 French to 12 French orogastric tube. After palpating the liver edge, the examiner’s fingertips should slide underneath the liver in the midline. Slowly, the fingers are pulled back and down, trying to trap the ‘olive.’ Palpating the hypertrophied pylorus requires patience and an optimal examination setting. If palpated, no further studies are needed. If the pylorus cannot be palpated, ultrasound (US) should be performed.
Ultrasound has become the standard technique for diagnosing HPS and has supplanted the physical examination at most institutions. The diagnostic criteria for pyloric stenosis is a muscle thickness greater than or equal to 4 mm and a pyloric channel length greater than or equal to 16 mm (Fig. 29-1).12 A thickness of more than 3 mm is considered positive if the neonate is younger than 30 days of age.13 The study is dependent on the expertise of the ultrasound technician and radiologist.
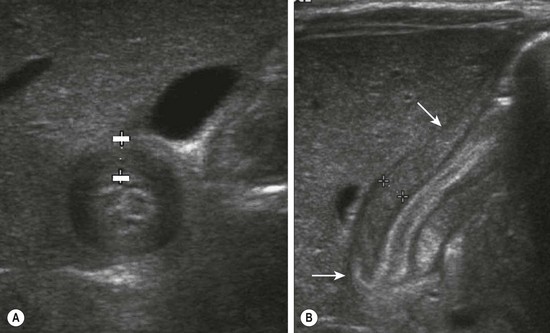
FIGURE 29-1 Ultrasonography has become the standard imaging study for diagnosing pyloric stenosis and has supplanted physical examination at most institutions. The (A) transverse and (B) longitudinal views of hypertrophic pyloric stenosis are seen here. Muscle thickness greater than or equal to 4 mm on the transverse view or a length greater than or equal to 16 mm on the longitudinal view is diagnostic of pyloric stenosis. On this study, the pyloric wall thickness was 5 mm and the length (arrows) was 20 mm.
There are reports of non-radiologists performing ultrasound for HPS, which would obviously reduce the need for the ultrasound technician.14,15 If the ultrasound findings are equivocal, then an upper gastrointestinal series can be helpful in confirming the diagnosis (Fig. 29-2).
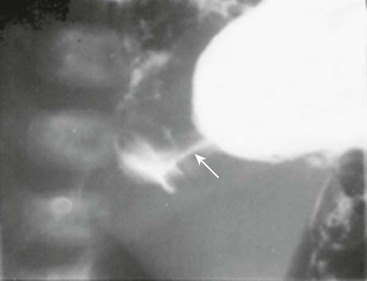
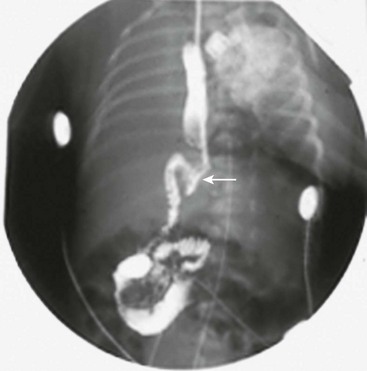
FIGURE 29-2 At some hospitals outside of urban centers, ultrasound technicians and radiologists proficient in performing an ultrasound study for pyloric stenosis are not available. Also, in some instances, an ultrasound study can be equivocal. An upper gastrointestinal series can be helpful in making the diagnosis of pyloric stenosis or confirming an equivocal ultrasound study. In this upper gastrointestinal study, note the ‘string sign’ indicating a markedly diminished pyloric channel (arrow) and subsequent gastric outlet obstruction. It is important to evacuate the contrast material after this study to reduce the risk of aspiration and pulmonary complications.
Treatment
The mainstay of therapy is typically resuscitation followed by pyloromyotomy. There are reports of medical treatment with atropine and pyloric dilation, but these treatments require long periods of therapy and are often not effective.16–20
The Open Approach
Several incisions have been described for the open approach. The typical right upper quadrant transverse incision seems to be used most commonly (Fig. 29-3). An alternate more cosmetically pleasing incision involves an omega-shaped incision around the superior portion of the umbilicus followed by incising the linea alba cephalad. With either incision, the pylorus is exteriorized through the incision. A longitudinal serosal incision is made in the pylorus approximately 2 mm proximal to the junction of the duodenum and is carried onto the anterior gastric wall for approximately 5 mm. Blunt dissection is used to initially divide the firm pyloric fibers. This can be performed using the handle of a scalpel. Once a good edge of fibers has been developed, a pyloric spreader or hemostat can be used to spread the fibers until the pyloric submucosal layer is seen. The pyloromyotomy is then completed by ensuring that all fibers are divided throughout the entire length of the pyloromyotomy. This is confirmed by visualizing the circular muscle of the stomach proximally as well as a slight protrusion of the submucosa. The most common point of mucosal entry is at the distal part of the incision at the duodenal–pyloric junction. Therefore, care must be exercised when dividing the fibers in this region. The pyloromyotomy can be evaluated for completeness by rocking the superior and inferior edges of the myotomy back and forth to ensure independent movement. The mucosal integrity can be checked by instilling air through the previously placed suction catheter. If there are no leaks, the air should be suctioned. Minor bleeding is common and should be ignored because it will cease after the venous congestion is reduced when the pylorus is returned to the abdominal cavity. The abdominal incision is then closed in layers.
The Laparoscopic Operation
Neonatal laparoscopy has grown in popularity with the refinement in technique and smaller instruments. The first reported laparoscopic pyloromyotomy in the English language was in 1991 (the authors had reported the first case in the French literature in 1990).21 Since then, this procedure has been accepted by most pediatric surgeons. Critics of this approach argue that laparoscopic pyloromyotomy exposes the patient to undue risks compared with the open technique. However, recent randomized prospective trials have not shown any difference in complication rates.22,23 Operative times can vary depending on the experience of the surgeon. The minimally invasive approach for pyloromyotomy is similar to laparoscopic appendectomy in terms of acceptance and has become the standard technique for pyloromyotomy in many centers.
Local anesthesia is instilled at all incisions. An atraumatic bowel grasper is inserted through the patient’s right incision, and a pylorotome or spatula cautery tip is introduced through the patient’s left incision (Fig. 29-4). The duodenum is grasped firmly just distal to the pylorus, and the pylorus is maneuvered into view. Occasionally, a transabdominal stay suture wrapping around the falciform ligament is helpful to elevate the liver away from the pylorus. A longitudinal pyloromyotomy is then made with the knife or cautery in a similar manner as the open technique (Fig. 29-5). Initially, a retractable arthrotomy knife was used; however, it is no longer available in the USA. Thus, most USA pediatric surgeons now use an unguarded arthrotomy knife or the cautery. Once the seromuscular layer is incised, a laparoscopic pyloric spreader or a box-type grasper is inserted to perform the myotomy. Completeness of the myotomy and mucosal integrity are checked in a similar manner as the open technique. Omentum can be placed over the myotomy to help with hemostasis. The pneumoperitoneum is evacuated after the instruments are removed. The umbilicus is closed with absorbable suture, and the stab incisions are closed with skin adhesive (see Fig. 29-4B).
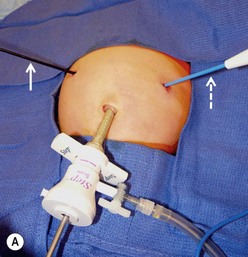
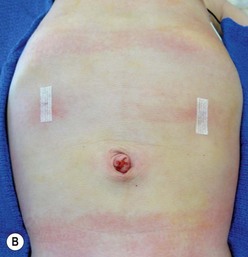
FIGURE 29-4 Laparoscopic pyloromyotomy has become a common approach for pyloric stenosis in infants. In the USA, the sheathed arthrotomy knife is no longer available. Therefore, other techniques are now utilized. (A) The atraumatic grasper that is holding the duodenum is seen on the patient’s right (solid arrow). In the patient’s left upper abdomen, a spatula tipped cautery (dotted arrow) has been introduced to incise the serosa of the stomach. The 5 mm cannula has been placed in the umbilicus through which an angled telescope is introduced for visualization. (B) The stab incisions have been closed with steri-strips.
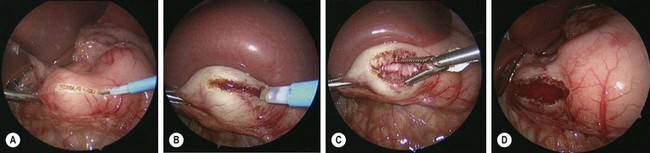
FIGURE 29-5 These intraoperative photographs depict a laparoscopic pyloromyotomy. (A) The spatula tipped cautery is being used to incise the serosa and outer muscular layer of the hypertrophied pylorus. (B) The tip of the cautery is introduced into the hypertrophied muscle and twisted to break up the muscle fibers and create a space for insertion of the pyloric spreader. (C) The pyloric spreader is introduced into the muscle and gently opened to split the hypertrophied muscle fibers. The submucosa is visualized through the myotomy. (D) Air is introduced into the stomach to assess the integrity of the mucosa.
Postoperative Care
Postoperative care is similar for both operative approaches, assuming the mucosal integrity of the stomach is intact. Complicated feeding regimens have been advocated in the past. However, more recent studies support the use of ad libitum feedings in the early postoperative period. This results in a faster time to full feeding and earlier discharge.24,25 In many centers, if postoperative emesis is encountered, it is suggested to ‘feed through it.’ At our institution, we limit the feedings to a maximum of 3 oz every three hours. There are data to suggest that the degree and duration of preoperative metabolic derangement affects the postoperative feeding schedule. Babies who required more complicated resuscitation tend to take longer to reach full feeding and discharge.26
A survey about postoperative feeding regimens in pediatric surgery residency training centers in North America was recently performed. Thirty-two of the 47 institutions responded to the survey. The average time from operation to initiation of feedings was 4.3 hours. There was a wide variability in responses, but 26 of the 32 responding programs employ a protocol-based feeding regimen.27
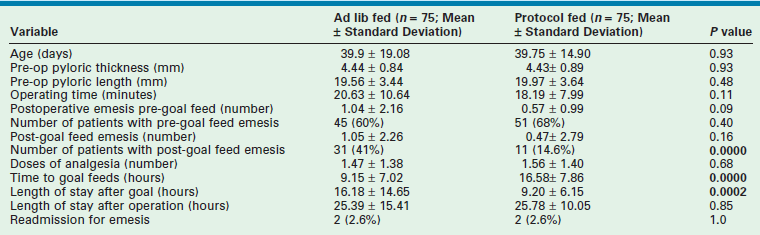
A prospective randomized trial recently compared a protocol-based feeding regimen to ad libitum feeding after laparoscopic pyloromyotomy.28 Feeding was begun two hours after laparoscopic pyloromyotomy in both groups. The ad libitum group was allowed formula or breast milk two hours after the operation and was considered ready for discharge after tolerating three consecutive feeds without emesis. The babies who underwent feeding via protocol were given Pedialyte two hours after the operation followed by another round of Pedialyte, which was followed by two rounds of half strength formula or breast milk, followed by two rounds of full strength formula or breast milk, followed by the home feeding regimen. The baby was discharged on the home feeding regimen if doing well. With a power of 0.9 and an alpha of 0.05, a sample size of 150 patients was calculated. There were no differences in patient characteristics at presentation. The ad libitum group reached goal feeding sooner than the group who were fed via protocol (Table 29-1). However, this did not translate into a difference in length of postoperative hospitalization. There were more patients with emesis in the ad libitum group after reaching goal feedings. There was no difference in readmission rates, as two patients in each group were readmitted after discharge.28
Complications
The major complications of pyloromyotomy include mucosal perforation, wound infection, incisional hernia, prolonged postoperative emesis, incomplete myotomy, and duodenal injury. There are prospective randomized trials that do not show any difference in complication rates between the laparoscopic and open techniques.22,23
Mucosal perforation occurs in 1–2% of cases.22,23 If the disruption occurs at the duodenopyloric junction, a simple interrupted absorbable suture can be used to close the defect and a patch of omentum can be positioned over the closure. This can be accomplished laparoscopically depending on the surgeon’s experience. Otherwise, the laparoscopic procedure should be converted to open. If the perforation is large or in the middle of the myotomy, then the myotomy should be closed with absorbable suture. A new myotomy can then be made 90–180° from the original incision. Feedings should be held for 24 hours and then restarted. A water-soluble contrast study can be performed if desired.
Duodenal injuries also can occur with either the laparoscopic or open approach. In a 25-year retrospective review of 901 open pyloromyotomies performed between 1969 and 1994, there were 39 duodenal perforations that were recognized intraoperatively and repaired. There were no unrecognized duodenal perforations that developed after the operation.29
Wound infections also occur in 1–2% of cases.22,23 There are no data to support the use of prophylactic perioperative antibiotics because a pyloromyotomy is considered a clean procedure. Local wound care is usually sufficient to treat these infections.
Incisional hernias and wound dehiscence occurs in approximately 1% of cases.22 Most hernias require repair at some point. Laparoscopically, port site hernias usually involve omentum protruding through the incision. This can sometimes be managed at the bedside by cleansing the area with povidone-iodine, ligating and trimming the extracorporeal omentum, elevating the abdominal wall to get the omentum back into the peritoneal cavity, and using fine absorbable suture to close the skin.
Postoperative emesis is common, occurring in most infants to some degree. Prolonged emesis is less common and ranges in incidence from 2–26%. Most commonly, this is due to gastroesophageal reflux but can occur secondary to an incomplete myotomy. It has been suggested that the laparoscopic approach may be a risk factor for inadequate myotomy, but this is likely related to the surgeon’s experience with this technique.24
Pyloric Atresia
Pyloric atresia is a rare disease (1 : 100,000 live births) and presents with symptoms of gastric outlet obstruction. The disease is difficult to characterize because it is so rare. However, several generalizations can be made from looking at relatively large series. Other congenital anomalies are found in up to 40% of patients with pyloric atresia.30–32 The most common associated condition is epidermolysis bullosa.33–36 There is a suggestion of autosomal recessive transmission in patients with familial disease.30
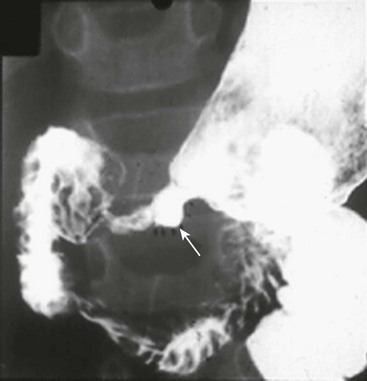
Three anatomic variants have been described. With type I, there is a mucosal membrane or web. In type II, the pyloric channel is a solid cord (Fig. 29-6). In type III, there is a gap between the stomach and the duodenum.31 These babies present with nonbilious emesis and have similar electrolyte abnormalities to infants with HPS. A ‘single bubble’ is usually found on an abdominal radiograph (see Figure 29-6). If necessary, the diagnosis can be confirmed with a contrast study. The differential diagnosis includes malrotation with volvulus, proximal duodenal atresia, gastric volvulus, pyloric duplication, retrograde duodenal gastric intussusception, and aberrant pancreatic tissue plugging the pylorus.37,38
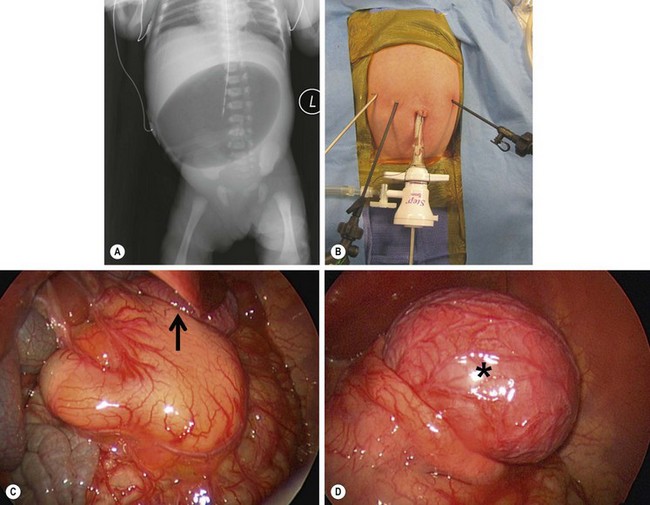
FIGURE 29-6 This baby developed nonbilious emesis shortly after birth. (A) An abdominal film shows a single bubble. This is classic for pyloric atresia. (B) The baby underwent laparoscopic repair and the position of the instruments is shown. (C) At operation, the pylorus was a solid core (type II). On close inspection, note a gastric duplication in the superior aspect of the greater curve of the stomach (arrow). This gastric duplication is better seen in (D) and was resected. The baby has recovered nicely and has not developed any problems two years later.
The operative management depends on the type of atresia. Excision of the web or membrane with Heinecke–Mikulicz pyloroplasty is usually possible for a type I atresia. However, for types II or III, often a Billroth I gastroduodenostomy is needed if the solid core or gap is long (see Fig 29-6). There is also a report of successful treatment with gastroduodenal mucosal advancement for this condition.39 Gastrojejunostomy is not recommended as there is a reported 60% failure rate and mortality rate of 50%.40 Laparoscopic repair for a type II atresia was recently described.41
Generally, long-term outcomes are very good. Morbidity and mortality are usually related to the associated anomalies. Patients with pyloric atresia and epidermolysis bullosa often die due to septicemia, electrolyte imbalance, protein loss, or failure to thrive due to the exudative skin lesions. However, there are reports of long-term survival in neonates with pyloric atresia and epidermolysis bullosa so aggressive surgical intervention for these patients should not be withheld.42
Gastric Perforation
Neonatal gastric perforations most commonly occur in premature infants. About half of neonatal perforations are spontaneous, and the other half are iatrogenic from instrumentation.43 Prematurity is associated with an increased mortality.44 The perforations are usually managed with laparotomy or laparoscopy. The perforation can usually be closed primarily with or without an omental patch.
Gastric perforation due to peptic ulcer disease in infants and children is very rare. Typically, perforation occurs at the site of a prepyloric ulcer. Again, this may be repaired primarily via laparotomy or laparoscopy with or without an omental patch.45
Peptic Ulcer Disease
Peptic ulcer disease and its complications are rarely seen in children. However, there have been reports of neonatal and childhood perforated ulcers as well as gastric outlet obstruction in children due to peptic ulcer disease.45–47 Peptic ulcer disease appears to be associated with Helicobacter pylori in the majority of pediatric cases. Treatment is primarily directed at acid reduction and eradication of H. pylori. Triple therapy with a proton pump inhibitor, amoxicillin, and clarithromycin is typically used initially. For strains that are resistant to clarithromycin, metronidazole is substituted.48 Operative treatment is usually reserved for complications of peptic ulcer disease, such as perforation or gastric outlet obstruction (Fig. 29-7). If ulcer perforation is suspected, it is reasonable to start with exploratory laparoscopy because there are reports of successful laparoscopic treatment in children.46 A gastric resection operation is not usually needed due to effective proton pump inhibitors.
Gastric Duplications
Gastric duplications are rare anomalies that generally occur along the greater curvature (see Fig. 29-6D). If the lesion is near the pylorus, the presentation may be very similar to HPS. The diagnosis can be differentiated from pyloric stenosis by ultrasound. The lesion rarely communicates with the lumen. If it does, the patient may present with hematemesis or melena. Gastric duplications represent approximately 4% of all gastrointestinal duplications. Approximately half are discovered in the neonatal period and are seen when the neonate presents with vomiting, poor feeding, and an epigastric mass.49 Ectopic gastric mucosa is common in other duplications throughout the gastrointestinal tract. These are not considered gastric duplications.
Treatment of gastric duplications is complete resection. Gastric duplications have been found associated with pancreatic ductal abnormalities.50 In this instance, care must be taken during the dissection not to injure normal pancreas, although it may be necessary to resect an accessory pancreas.
Microgastria
Congenital microgastria is a rare disorder that usually occurs in conjunction with other congenital anomalies or, more rarely, alone (Fig. 29-8). Associated anomalies include the VACTERL association (vertebral anomalies, anorectal atresia, cardiac anomalies, tracheoesophageal fistula and esophageal atresia, renal and limb anomalies), tracheoesophageal cleft, malrotation, and asplenia. There are currently only three reported cases of isolated microgastria.51,52 Microgastria is usually temporized with jejunal feedings. Operative intervention consists of jejunal feeding tubes and Hunt–Lawrence gastric augmentation. There are a few patients who have been reported with successful follow-up after a Hunt–Lawrence pouch.51,53,54
Antral Web
The first modern description of an antral web was in 1969.55 Early case reports in children described incomplete gastric outlet obstruction due to an antral web.56–59 The etiology is unknown and is generally thought to be congenital or the result of an inflammatory process. In adults there is a case report that strongly suggested peptic ulcer disease led to an antral web.60 These patients typically present with gastric outlet obstruction. In the infant, an antral web may be confused with HPS. The baby with an antral web may have a normal abdominal sonogram. The abdominal examination may be normal. However, an upper gastrointestinal series will show the lesion.
Treatment of an antral web consists of resuscitation (see earlier section on HPS) and operative correction which can be performed via laparotomy or laparoscopy (Fig. 29-9). This is a diagnosis that may be amenable to endoluminal treatment in the future.
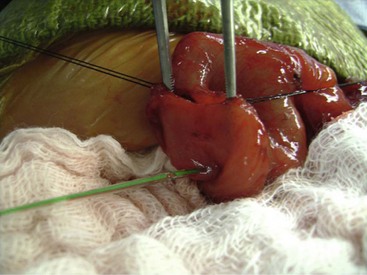
FIGURE 29-9 This neonate developed nonbilious emesis shortly after birth and was thought to have an antral web. At laparotomy, the antral web is visualized. The forceps are proximal to the web and the feeding tube (arrow) has been placed through the web. After resection of the web, the patient recovered uneventfully and has not developed any further problems.
Gastric Volvulus
Gastric volvulus can occur from primary or secondary causes. Primary gastric volvulus is thought to be due to laxity of the gastric ligaments. Secondary disease may occur due to a paraesophageal hernia or other diaphragmatic hernia. The presenting symptoms can be intermittent or complete gastric obstruction, ischemia, pain, and/or bleeding. The most common signs and symptoms of gastric volvulus in children include acute abdominal pain, intractable retching, and the inability to pass a nasogastric tube into the stomach lumen.61,62
The average age at presentation is 2.5 years. Equal numbers of males and females are affected.61 Gastric volvulus is classified based on the axis of gastric rotation. Mesenteroaxial gastric volvulus is rotation about the gastric short axis, transecting the greater and lesser curvatures. Organoaxial gastric volvulus is rotation around the long axis of the stomach (Figs 29-10 and 29-11).
FIGURE 29-10 These drawings depict the development of a gastric volvulus. (A) Normal anatomy. (B) The axis of rotation for an organoaxial volvulus is seen on the left and a mesoaxial volvulus is on the right. (C) Demonstration of an organoaxial volvulus. (D) Demonstration of a mesoaxial volvulus. (E) Combined mesoaxial and organoaxial volvulus. (Adapted from Cribbs RK, Gow KW, Wulkan ML. Gastric volvulus in infants and children. Pediatrics 2008;122:e752–62.)
FIGURE 29-11 These two contrast studies depict a gastric volvulus. (A) This contrast study depicts an organoaxial volvulus in which the stomach has rotated on its long axis. Note the relatively normal position of the pylorus (arrow). (B) This contrast study shows a mesoaxial volvulus. In this study, the pylorus is in the left upper abdomen due to rotation around the short axis of the stomach. In both studies, the gastroesophageal junction is in a relatively normal position.
The principles of treatment are patient resuscitation, nasogastric decompression, and operative correction. Diaphragmatic defects, if found, are repaired in secondary gastric volvulus. A gastropexy is then performed. This has traditionally been accomplished with a gastrostomy tube or button. However, there are recent reports of successful laparoscopic gastropexy in which the anterior stomach, along the greater curvature, is sutured to the abdominal wall.63
Foreign Bodies and Bezoar
Gastric bezoars are relatively uncommon causes of gastric outlet obstruction and chronic abdominal pain in children. Phytobezoars are made up of vegetable matter. Trichobezoars are made up of hair that is swallowed. This is referred to as the Rapunzel syndrome.64 The hair usually fills the stomach and extends into the duodenum. However, it can extend to the ileum. Attempts at gastroscopic removal are usually futile, except in cases of small bezoars. The bezoar has been typically removed through a gastrotomy at laparotomy. However, there are recent reports of laparoscopic removal.65,66 At our institution, we recently removed a large gastric trichobezoar laparoscopically with the aid of an endoscopic bag (Fig. 29-12).
References
1. Pedersen, RN, Garne, E, Loane, M, et al. Infantile hypertrophic pyloric stenosis: A comparative study of incidence and other epidemiological characteristics in seven European regions. J Matern Fetal Neonatal Med. 2008; 21:599–604.
2. Sommerfield, T, Chalmers, J, Youngson, G, et al. The changing epidemiology of infantile hypertrophic pyloric stenosis in Scotland. Arch Dis Child. 2008; 93:1007–1011.
3. Persson, S, Ekbom, A, Granath, F, et al. Parallel incidences of sudden infant death syndrome and infantile hypertrophic pyloric stenosis: A common cause? Pediatrics. 2001; 108:E70.
4. Schechter, R, Torfs, CP, Bateson, TF. The epidemiology of infantile hypertrophic pyloric stenosis. Paediatr Perinat Epidemiol. 1997; 11:407–427.
5. Honein, MA, Paulozzi, LJ, Himelright, IM, et al. Infantile hypertrophic pyloric stenosis after pertussis prophylaxis with erythromycin: A case review and cohort study. Lancet. 1999; 354:2101–2105.
6. Mitchell, LE, Risch, N. The genetics of infantile hypertrophic pyloric stenosis: A re-analysis. Am J Dis Child. 1993; 147:1203–1211.
7. Rasmussen, L, Green, A, Hansen, LP. The epidemiology of infantile hypertrophic pyloric stenosis in a Danish population, 1950-84. Int J Epidemiol. 1989; 18:413–417.
8. Spitz, L, Zail, SS. Serum gastrin levels in congenital hypertrophic pyloric stenosis. J Pediatr Surg. 1976; 11:33–35.
9. Vanderwinden, JM, Mailleux, P, Schiffmann, SN, et al. Nitric oxide synthase activity in infantile hypertrophic pyloric stenosis. N Engl J Med. 1992; 327:511–515.
10. White, MC, Langer, JC, Don, S, et al. Sensitivity and cost minimization analysis of radiology versus olive palpation for the diagnosis of hypertrophic pyloric stenosis. J Pediatr Surg. 1998; 33:913–917.
11. Breaux, CW, Jr., Georgeson, KE, Royal, SA, et al. Changing patterns in the diagnosis of hypertrophic pyloric stenosis. Pediatrics. 1988; 81:213–217.
12. Keller, H, Waldmann, D, Greiner, P. Comparison of preoperative sonography with intraoperative findings in congenital hypertrophic pyloric stenosis. J Pediatr Surg. 1987; 22:950–952.
13. Lamki, N, Athey, PA, Round, ME, et al. Hypertrophic pyloric stenosis in the neonate—diagnostic criteria revisited. Can Assoc Radiol J. 1993; 44:21–24.
14. Malcom, GE, 3rd., Raio, CC, Del Rios, M, et al. Feasibility of emergency physician diagnosis of hypertrophic pyloric stenosis using point-of-care ultrasound: A multi-center case series. J Emerg Med. 2009; 37:283–286.
15. Boneti, C, McVay, MR, Kokoska, ER, et al. Ultrasound as a diagnostic tool used by surgeons in pyloric stenosis. J Pediatr Surg. 2008; 43:87–91.
16. Meissner, PE, Engelmann, G, Troeger, J, et al. Conservative treatment of infantile hypertrophic pyloric stenosis with intravenous atropine sulfate does not replace pyloromyotomy. Pediatr Surg Int. 2006; 12:1021–1024.
17. Kawahara, H, Takama, Y, Yoshida, H, et al. Medical treatment of infantile hypertrophic pyloric stenosis: Should we always slice the “olive”? J Pediatr Surg. 2005; 40:1848–1851.
18. Kawahara, H, Imura, K, Nishikawa, M, et al. Intravenous atropine treatment in infantile hypertrophic pyloric stenosis. Arch Dis Child. 2002; 87:71–74.
19. Ogawa, Y, Higashimoto, Y, Nishijima, E, et al. Successful endoscopic balloon dilatation for hypertrophic pyloric stenosis. J Pediatr Surg. 1996; 31:1712–1714.
20. Yusuf, TE, Brugge, WR. Endoscopic therapy of benign pyloric stenosis and gastric outlet obstruction. Curr Opin Gastroenterol. 2006; 22:570–573.
21. Alain, JL, Grousseau, D, Terrier, G. Extramucosal pylorotomy by laparoscopy. J Pediatr Surg. 1991; 26:1191–1192.
22. St. Peter, SD, Holcomb, GW, Calkins, CM, et al. Open versus laparoscopic pyloromyotomy for pyloric stenosis: A prospective, randomized trial. Ann Surg. 2006; 244:363–370.
23. Leclair, MD, Plattner, V, Mirallie, E, et al. Laparoscopic pyloromyotomy for hypertrophic pyloric stenosis: A prospective, randomized controlled trial. J Pediatr Surg. 2007; 42:692–698.
24. Adibe, OO, Nichol, PF, Lim, FY, et al. Ad libitum feeds after laparoscopic pyloromyotomy: A retrospective comparison with a standardized feeding regimen in 227 infants. J Laparoendosc Adv Surg Tech A. 2007; 17:235–237.
25. Georgeson, KE, Corbin, TJ, Griffen, JW, et al. An analysis of feeding regimens after pyloromyotomy for hypertrophic pyloric stenosis. J Pediatr Surg. 1993; 28:1478–1480.
26. St. Peter, SD, Tsao, K, Sharp, SW, et al. Predictors of emesis and time to goal intake after pyloromyotomy: Analysis from a prospective trial. J Pediatr Surg. 2008; 43:2038–2041.
27. Juang, D, Adibe, OO, Laituri, CA, et al. Distribution of feeding styles after pyloromyotomy among pediatric surgical training programs in North America. Eur J Pediatr Surg. 2012; 22:409–411.
28. Adibe OO, Iqbal CS, Sharp SW, et al. Protocol versus ad lib feeds after laparoscopic pyloromyotomy: A prospective randomized trial. 2012 (accepted for publication).
29. Hulka, F, Harrison, MW, Campbell, TJ, et al. Complications of pyloromyotomy for infantile hypertrophic pyloric stenosis. Am J Surg. 1997; 173:450–452.
30. Ilce, Z, Erdogan, E, Kara, C, et al. Pyloric atresia: 15-year review from a single institution. J Pediatr Surg. 2003; 38:1581–1584.
31. Okoye, BO, Parikh, DH, Buick, RG, et al. Pyloric atresia: Five new cases, a new association, and a review of the literature with guidelines. J Pediatr Surg. 2000; 35:1242–1245.
32. Al-Salem, AH. Congenital pyloric stenosis and associated anomalies. Pediatr Surg Int. 2007; 23:559–563.
33. Almaani, N, Liu, L, Dopping-Hepenstal, PJ, et al. Autosomal dominant junctional epidermolysis bullosa. Br J Dermatol. 2009; 160:1094–1097.
34. Birnhaum, RY, Landau, D, Elbedour, K, et al. Deletion of the first pair of fibronectin type III repeats of the integrin beta-4 gene is associated with epidermolysis bullosa, pyloric atresia and aplasia cutis congenita in the original Carmi syndrome patients. Am J Med Genet A. 2008; 146:1063–1066.
35. Nakamura, H, Sawamura, D, Goto, M, et al. Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1). J Mol Diagn. 2005; 7:28–35.
36. Samad, L, Siddiqui, EF, Arain, MA, et al. Pyloric atresia associated with epidermolysis bullosa: Three cases presenting in three months. J Pediatr Surg. 2004; 39:1267–1269.
37. Moore, CC. Congenital gastric outlet obstruction. J Pediatr Surg. 1989; 24:1241–1246.
38. Bronsther, B, Nadeau, MR, Abrams, MW. Congenital pyloric atresia: A report of three cases and a review of the literature. Surgery. 1971; 69:130–136.
39. Dessanti, A, Iannuccelli, M, Dore, A, et al. Pyloric atresia: An attempt at anatomic pyloric sphincter reconstruction. J Pediatr Surg. 2000; 35:1372–1374.
40. Kourolinka, CW, Steward, JR. Pyloric atresia. Am J Dis Child. 1978; 132:903–905.
41. Juang D, Holcomb GW III. Laparoscopic repair of pyloric atresia. Video presentation, 2012 American College of Surgeons meeting.
42. Hayashi, AH, Galliani, CA, Gilis, DA. Congenital pyloric atresia and junctional epidermolysis bullosa: A report of long-term survival and a review of the literature. J Pediatr Surg. 1991; 26:1341–1345.
43. Abadir, J, Emil, S, Nguyen, N. Abdominal foregut perforations in children: A 10-year experience. J Pediatr Surg. 2005; 40:1903–1907.
44. Lin, CM, Lee, HC, Kao, HA, et al. Neonatal gastric perforation: Report of 15 cases and review of the literature. Pediatr Neonatol. 2008; 49:65–70.
45. Edwards, MJ, Kollenberg, SJ, Brandt, ML, et al. Surgery for peptic ulcer disease in children in the post-histamine2-blocker era. J Pediatr Surg. 2005; 40:850–854.
46. Wong, BP, Chao, NS, Leung, MW, et al. Complications of peptic ulcer disease in children and adolescents: Minimally invasive treatments offer feasible surgical options. J Pediatr Surg. 2006; 41:2073–2075.
47. Hua, MC, Kong, MS, Lai, MW, et al. Perforated peptic ulcer in children: A 20-year experience. J Pediatr Gastroenterol Nutr. 2007; 45:71–74.
48. Kato, S, Konno, M, Maisawa, S, et al. Results of triple eradication therapy in Japanese children: A retrospective multicenter study. J Gastroenterol. 2004; 39:838–843.
49. Cooper, S, Abrams, RS, Carbaugh, RA. Pyloric duplications: Review and case study. Am Surg. 1995; 61:1092–1094.
50. Muraoka, A, Tsuruno, M, Katsuno, G, et al. A gastric duplication cyst with an aberrant pancreatic ductal system: Report of a case. Surg Today. 2002; 32:531–535.
51. Jones, VS, Cohen, RC. An eighteen-year follow-up after surgery for congenital microgastria—case report and review of literature. J Pediatr Surg. 2007; 42:1957–1960.
52. Kroes, EJ, Festen, C. Congenital microgastria: A case report and review of literature. Pediatr Surg Int. 1998; 13:416–418.
53. Velasco, AL, Holcomb, GW, Templeton, JM, et al. Management of congenital microgastria. J Pediatr Surg. 1990; 25:192–197.
54. Neifeld, JP, Berman, WF, Lawrence, W, et al. Management of congenital microgastria with a jejunal reservoir pouch. J Pediatr Surg. 1980; 15:882–885.
55. Banks, PA, Waye, JD. The gastroscopic appearance of antral web. Gastrointest Endosc. 1969; 15:228–229.
56. Hait, G, Esselstyn, CB, Jr., Rankin, GB. Prepyloric mucosal diaphragm (antral web): Report of a case and review of the literature. Arch Surg. 1972; 105:486–490.
57. Campbell, DP, Vanhoutte, JJ, Smith, EI. Partially obstructing antral web—a distinct clinical entity. J Pediatr Surg. 1973; 8:723–728.
58. Patnaik, DN, Sun, S, Groff, DB. Newborn gastric outlet obstruction caused by an antral web. J Med Soc N J. 1976; 73:736–737.
59. Bell, MJ, Ternberg, JL, McAlister, W, et al. Antral diaphragm—a cause of gastric outlet obstruction in infants and children. J Pediatr. 1977; 90:196–202.
60. Huggins, MJ, Friedman, AC, Lichtenstein, JE, et al. Adult acquired antral web. Dig Dis Sci. 1982; 27:80–83.
61. Miller, DL, Pasquale, MD, Seneca, RP, et al. Gastric volvulus in the pediatric population. Arch Surg. 1991; 126:1146–1149.
62. Heldrich, FJ, Kumarasena, D, Hakim, J, et al. Acute gastric volvulus in children: A rare disorder. Pediatr Emerg Care. 1993; 9:221–223.
63. Cribbs, RK, Gow, KW, Wulkan, ML. Gastric volvulus in infants and children. Pediatrics. 2008; 122:e752–e762.
64. Naik, S, Gupta, V, Naik, S, et al. Rapunzel syndrome reviewed and redefined. Dig Surg. 2007; 24:157–161.
65. Shami, SB, Jararaa, AA, Hamade, A, et al. Laparoscopic removal of a huge gastric trichobezoar in a patient with trichotillomania. Surg Laparosc Endosc Percutan Tech. 2007; 17:197–200.
66. Nirasawa, Y, Mori, T, Ito, Y, et al. Laparoscopic removal of a large gastric trichobezoar. J Pediatr Surg. 1998; 33:663–665.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Lesions of The Stomach
Hypertrophic Pyloric Stenosis
Hypertrophic pyloric stenosis (HPS) is one of the most common surgical conditions of the newborn.1–9 It occurs at a rate of 1 to 4 per 1,000 live births in Caucasian infants, but is seen less often in non-Caucasian children.1–4 Males are affected more often with a 4 : 1 male-to-female ratio. Risk factors for HPS include family history, gender, younger maternal age, being a first-born infant, and maternal feeding patterns.4,9,10 Premature infants are diagnosed with HPS later than term or post-term infants.4
Etiology
The cause of HPS is unknown, but genetic and environmental factors appear to play a large role in the pathophysiology. Circumstantial evidence for a genetic predisposition includes race discrepancies, the increased frequency in males, and the birth order (first-born infants with a positive family history). Environmental factors associated with HPS include the method of feeding (breast vs formula), seasonal variability, exposure to erythromycin, and transpyloric feeding in premature infants.5–7 Additionally, there has been interest in several gastrointestinal peptides or growth factors that may facilitate pyloric hypertrophy. Some of these include excessive substance P, decreased neurotrophins, deficient nitric oxide synthase, and gastrin hypersecretion.8,9 Thus, the etiology of HPS is likely multifactorial with environmental influences.
Diagnosis
The classic presentation of HPS is nonbilious, projectile vomiting in a full-term neonate who is between 2 and 8 weeks old. Initially, the emesis is infrequent and may appear to be gastroesophageal reflux disease. However, over a short period of time, the emesis occurs with every feeding and becomes forceful (i.e., projectile). The contents of the emesis are usually the recent feedings, but signs of gastritis are not uncommon (‘coffee-ground’ emesis). On physical examination, the neonate usually appears well if the diagnosis is made early. However, depending on the duration of symptoms and degree of dehydration, the neonate may be gaunt and somnolent. Visible peristaltic waves may be present in the mid to left upper abdomen. The pylorus may be palpable in 72–89% of patients.11,12 To palpate the hypertrophied pylorus, the baby must be relaxed. Techniques for relaxing the infant include bending the knees and flexing the hips, and using a pacifier with sugar water. These techniques should be attempted after the stomach has been decompressed with a 10 French to 12 French orogastric tube. After palpating the liver edge, the examiner’s fingertips should slide underneath the liver in the midline. Slowly, the fingers are pulled back and down, trying to trap the ‘olive.’ Palpating the hypertrophied pylorus requires patience and an optimal examination setting. If palpated, no further studies are needed. If the pylorus cannot be palpated, ultrasound (US) should be performed.
Ultrasound has become the standard technique for diagnosing HPS and has supplanted the physical examination at most institutions. The diagnostic criteria for pyloric stenosis is a muscle thickness greater than or equal to 4 mm and a pyloric channel length greater than or equal to 16 mm (Fig. 29-1).12 A thickness of more than 3 mm is considered positive if the neonate is younger than 30 days of age.13 The study is dependent on the expertise of the ultrasound technician and radiologist.
FIGURE 29-1 Ultrasonography has become the standard imaging study for diagnosing pyloric stenosis and has supplanted physical examination at most institutions. The (A) transverse and (B) longitudinal views of hypertrophic pyloric stenosis are seen here. Muscle thickness greater than or equal to 4 mm on the transverse view or a length greater than or equal to 16 mm on the longitudinal view is diagnostic of pyloric stenosis. On this study, the pyloric wall thickness was 5 mm and the length (arrows) was 20 mm.
There are reports of non-radiologists performing ultrasound for HPS, which would obviously reduce the need for the ultrasound technician.14,15 If the ultrasound findings are equivocal, then an upper gastrointestinal series can be helpful in confirming the diagnosis (Fig. 29-2).
FIGURE 29-2 At some hospitals outside of urban centers, ultrasound technicians and radiologists proficient in performing an ultrasound study for pyloric stenosis are not available. Also, in some instances, an ultrasound study can be equivocal. An upper gastrointestinal series can be helpful in making the diagnosis of pyloric stenosis or confirming an equivocal ultrasound study. In this upper gastrointestinal study, note the ‘string sign’ indicating a markedly diminished pyloric channel (arrow) and subsequent gastric outlet obstruction. It is important to evacuate the contrast material after this study to reduce the risk of aspiration and pulmonary complications.
Treatment
The mainstay of therapy is typically resuscitation followed by pyloromyotomy. There are reports of medical treatment with atropine and pyloric dilation, but these treatments require long periods of therapy and are often not effective.16–20
The Open Approach
Several incisions have been described for the open approach. The typical right upper quadrant transverse incision seems to be used most commonly (Fig. 29-3). An alternate more cosmetically pleasing incision involves an omega-shaped incision around the superior portion of the umbilicus followed by incising the linea alba cephalad. With either incision, the pylorus is exteriorized through the incision. A longitudinal serosal incision is made in the pylorus approximately 2 mm proximal to the junction of the duodenum and is carried onto the anterior gastric wall for approximately 5 mm. Blunt dissection is used to initially divide the firm pyloric fibers. This can be performed using the handle of a scalpel. Once a good edge of fibers has been developed, a pyloric spreader or hemostat can be used to spread the fibers until the pyloric submucosal layer is seen. The pyloromyotomy is then completed by ensuring that all fibers are divided throughout the entire length of the pyloromyotomy. This is confirmed by visualizing the circular muscle of the stomach proximally as well as a slight protrusion of the submucosa. The most common point of mucosal entry is at the distal part of the incision at the duodenal–pyloric junction. Therefore, care must be exercised when dividing the fibers in this region. The pyloromyotomy can be evaluated for completeness by rocking the superior and inferior edges of the myotomy back and forth to ensure independent movement. The mucosal integrity can be checked by instilling air through the previously placed suction catheter. If there are no leaks, the air should be suctioned. Minor bleeding is common and should be ignored because it will cease after the venous congestion is reduced when the pylorus is returned to the abdominal cavity. The abdominal incision is then closed in layers.
The Laparoscopic Operation
Neonatal laparoscopy has grown in popularity with the refinement in technique and smaller instruments. The first reported laparoscopic pyloromyotomy in the English language was in 1991 (the authors had reported the first case in the French literature in 1990).21 Since then, this procedure has been accepted by most pediatric surgeons. Critics of this approach argue that laparoscopic pyloromyotomy exposes the patient to undue risks compared with the open technique. However, recent randomized prospective trials have not shown any difference in complication rates.22,23 Operative times can vary depending on the experience of the surgeon. The minimally invasive approach for pyloromyotomy is similar to laparoscopic appendectomy in terms of acceptance and has become the standard technique for pyloromyotomy in many centers.
Local anesthesia is instilled at all incisions. An atraumatic bowel grasper is inserted through the patient’s right incision, and a pylorotome or spatula cautery tip is introduced through the patient’s left incision (Fig. 29-4). The duodenum is grasped firmly just distal to the pylorus, and the pylorus is maneuvered into view. Occasionally, a transabdominal stay suture wrapping around the falciform ligament is helpful to elevate the liver away from the pylorus. A longitudinal pyloromyotomy is then made with the knife or cautery in a similar manner as the open technique (Fig. 29-5). Initially, a retractable arthrotomy knife was used; however, it is no longer available in the USA. Thus, most USA pediatric surgeons now use an unguarded arthrotomy knife or the cautery. Once the seromuscular layer is incised, a laparoscopic pyloric spreader or a box-type grasper is inserted to perform the myotomy. Completeness of the myotomy and mucosal integrity are checked in a similar manner as the open technique. Omentum can be placed over the myotomy to help with hemostasis. The pneumoperitoneum is evacuated after the instruments are removed. The umbilicus is closed with absorbable suture, and the stab incisions are closed with skin adhesive (see Fig. 29-4B).
FIGURE 29-4 Laparoscopic pyloromyotomy has become a common approach for pyloric stenosis in infants. In the USA, the sheathed arthrotomy knife is no longer available. Therefore, other techniques are now utilized. (A) The atraumatic grasper that is holding the duodenum is seen on the patient’s right (solid arrow). In the patient’s left upper abdomen, a spatula tipped cautery (dotted arrow) has been introduced to incise the serosa of the stomach. The 5 mm cannula has been placed in the umbilicus through which an angled telescope is introduced for visualization. (B) The stab incisions have been closed with steri-strips.
FIGURE 29-5 These intraoperative photographs depict a laparoscopic pyloromyotomy. (A) The spatula tipped cautery is being used to incise the serosa and outer muscular layer of the hypertrophied pylorus. (B) The tip of the cautery is introduced into the hypertrophied muscle and twisted to break up the muscle fibers and create a space for insertion of the pyloric spreader. (C) The pyloric spreader is introduced into the muscle and gently opened to split the hypertrophied muscle fibers. The submucosa is visualized through the myotomy. (D) Air is introduced into the stomach to assess the integrity of the mucosa.
Postoperative Care
Postoperative care is similar for both operative approaches, assuming the mucosal integrity of the stomach is intact. Complicated feeding regimens have been advocated in the past. However, more recent studies support the use of ad libitum feedings in the early postoperative period. This results in a faster time to full feeding and earlier discharge.24,25
[/not-level-membership-for-pediatrics-category]
