[level-membership-for-pediatrics-category]
Lesions of The Pancreas
The pancreas originates during week 4 of gestation as dual evaginations from the foregut endoderm. The dorsal pancreatic bud gives rise to the body and tail of the pancreas, its minor duct (Santorini) and papilla, and the continuation of the major duct (Wirsung) into the body and tail. The ventral pancreatic bud arises from the biliary diverticulum and swings around the dorsal aspect of the duodenal anlage during gut rotation to give rise to the head of the pancreas as well as the proximal portion of Wirsung’s duct (Fig. 46-1).1
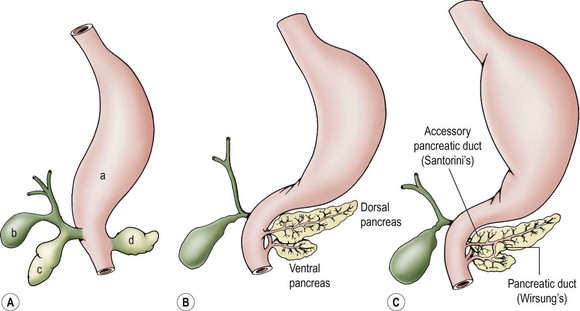
FIGURE 46-1 Pancreatic embryology. (A) Stomach (a), gallbladder (b), and ventral (c) and dorsal (d) pancreatic buds develop separately at embryologic week 4. The pancreas develops as an evagination of the developing foregut. The dorsal bud evaginates directly off of the duodenal anlage. (B) The ventral bud evaginates from the biliary bud and then swings around to the left, with gut rotation occurring simultaneously. (C) The main pancreatic duct of Wirsung and the minor accessory duct of Santorini are shown.
The two pancreatic buds fuse to form one pancreas at approximately 7 weeks’ gestation, although it appears that complete fusion of the two ducts to form the main pancreatic duct is delayed until the perinatal period.2 The endocrine component of the pancreas, the islets of Langerhans, starts to differentiate before evagination of the pancreatic buds from the wall of the foregut. The islets comprise 10% of the pancreas during early embryonic and fetal life, but this contribution decreases to less than 1% in adulthood. Pancreatic acini begin to form at 12 weeks gestation and begin to accumulate organelles and zymogen granules at this stage, but do not secrete appreciable amounts of enzyme until birth.1
The pancreas is convex and its midportion is reflected over the anterior surface of the upper lumbar vertebrae and aorta. Its lateral aspects falls posteriorly toward each kidney (Fig. 46-2). The arterial supply of the pancreas is from the celiac and SMA, which form the pancreaticoduodenal arcade. The pancreas also has anastomoses from the splenic artery.3
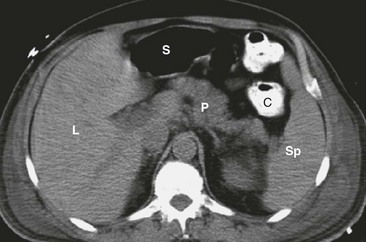
FIGURE 46-2 CT scan showing the cross-sectional anatomy of the pancreas. The pancreas (P) lies convexly across the lumbar spine with the tail of the pancreas next to the spleen (Sp) and the hilum of the left kidney. The head of the pancreas lies to the right of the spine near the hilum of the right kidney. L, Liver; S, stomach; C, colon. (From Maher MM, Hahn PF, Gervais DA, et al. Portable abdominal CT: Analysis of quality and clinical impact in more than 100 consecutive cases. AJR Am J Roentgenol 2004;183:663–70.)
Congenital Anomalies
Ectopic pancreatic rests are frequently encountered along foregut derivatives such as stomach, duodenum, jejunum, and colon, but are also infrequently encountered in the thorax and other sites.4–6 These lesions are found in approximately 2% of autopsy series and represent the most common anomaly of the gastric antrum. Moreover, they may cause gastric outlet obstruction.7 Their origin is unknown, but may be the result of aberrant epithelial–mesenchymal interactions leading to the transdifferentiation of embryonic epithelium into pancreatic epithelium. Several studies have implicated defects in hedgehog signaling and Notch signaling as the cause of ectopic pancreatic rests.8 Ectopic rests are typically asymptomatic and are encountered incidentally at laparotomy or during endoscopy. They can be identified as pancreatic tissue visually, because the surface has the same granular acinar appearance as the normal pancreas. These ectopic pancreatic rests usually do not become inflamed, possibly because they contain numerous small drainage ducts that usually do not obstruct; however, they can occasionally cause intestinal obstruction or bleeding. When encountered at laparotomy, ectopic rests should probably be excised, unless the excision would entail significant risk.
An annular pancreas is thought to result from faulty rotation of the ventral pancreatic bud in its course around the posterior aspect of the duodenal anlage. The duodenum is encircled and often obstructed by normal pancreatic tissue.9 Abnormal expression patterns of endodermal hedgehog may be responsible for the formation of annular and ectopic pancreas.8 Duodenal atresia and stenosis, intestinal malrotation, and trisomy 21 can often be found in combination with an annular pancreas.10 The clinical significance relates primarily to intrinsic duodenal obstruction, typically with bilious vomiting. Radiographic studies may reveal the classic finding of the ‘double-bubble’ sign. Management consists of bypass of the obstructing lesion with a duodenoduodenostomy or gastrojejunostomy, depending on the anatomy. Resection or division of the annular pancreas should not be performed due to the variable and complex ductal drainage system. Occasionally, patients with complex ductal anatomy may require reoperation for pancreatobiliary anomalies not apparent at the time of the initial surgery.11
Cystic fibrosis (CF) is an autosomal recessive condition, seen primarily in Caucasians which occurs in about 1 of 2,500 births.12 It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that encodes a protein expressed on the apical membrane of exocrine epithelial cells. CF leads to significant pancreatic insufficiency. The pancreatic secretions generally have a reduced amount of bicarbonate, a lower pH, and a lower overall fluid volume. The inspissated secretions lead to blockage of the ducts with dilatation. This may lead to acinar cell degeneration, acute and chronic pancreatitis, and pancreatic fibrosis. The result is impaired digestion of fats and proteins from loss of these digestive enzymes.13 See Chapter 32 for more information about CF.
Pancreatitis
Acute Pancreatitis
Acute pancreatitis is an acute inflammation of the pancreas, varying in severity from mild abdominal pain to fulminant necrotizing pancreatitis and death. It has an incidence between 3.6 and 13.2 cases per 100,000 children. If episodes of acute inflammation completely resolve and then recur, it is termed acute recurrent pancreatitis. It is thought that complete interval resolution of morphology and function occurs between episodes, unlike in cases of chronic pancreatitis.14
The causes of acute pancreatitis include trauma, biliary tract stone disease, choledochal cyst, ductal developmental anomalies, drugs, metabolic derangements, and infections. Most commonly, the cause is not apparent and termed idiopathic. As the pancreas is fixed against the lumbar spine, trauma to the upper abdomen can fracture the pancreas or injure the major duct at that point (Fig. 46-3). Biliary stone disease, increasing in frequency in children, may lead to pancreatitis from transient pancreatic duct obstruction. Endoscopic retrograde cholangiopancreatography (ERCP) is safe and effective in children and is the preferred method for stone retrieval.15 Drugs that are thought to induce pancreatitis include asparaginase and valproic acid.16 Systemic illnesses and metabolic conditions, such as CF, Reye syndrome, Kawasaki disease, hyperlipidemias, and hypercalcemia as well as viral infections (e.g., coxsackievirus and rotavirus) and generalized bacterial sepsis, can also cause pancreatitis.17
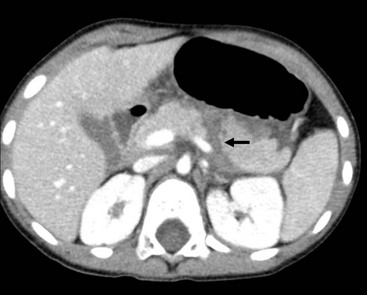
FIGURE 46-3 This abdominal contrast-enhanced CT shows a complete transection (arrow) of the body of the pancreas following a bicycle handlebar injury to the epigastrium. The patient underwent laparoscopic distal pancreatectomy and recovered uneventfully.
Pancreas divisum is an anomaly of the pancreas present in 10% of the population and is thought to result from failure of the dorsal duct to fuse with the ventral duct. In pancreas divisum, the majority of exocrine pancreatic secretions, including those from the entire body and tail, must drain through the small minor duct of Santorini. The resulting relative obstruction may cause recurring episodes of pancreatitis. Symptomatic patients should undergo sphincteroplasty of the minor papilla. Endoscopic stenting, with or without sphincterotomy, is the preferred treatment, with operation reserved for recurrent cases. Choledochal cysts produce pancreatitis by pancreatic duct compression or bile reflux resulting from a long common biliary-pancreatic duct within the head of the pancreas. Other rare ductal anomalies may result in obstruction and recurring bouts of pancreatitis.18
Though acute pancreatitis has many etiologies, they all appear to share a common pathway of nonphysiologic calcium signaling in the pancreas, followed by the premature activation of acinar proenzymes. These enzymes, especially trypsin, lead to acinar cell injury and cytokine release. The cytokines, along with vascular dissemination of activated enzymes, free radical formation, and release of vasoactive substances, such as kallikreins and histamine, together mediate extrapancreatic inflammation.19
Diagnostic criteria for acute pancreatitis include at least two of the following: acute abdominal pain (especially in the epigastric region), serum amylase or lipase more than three times the upper limit of normal, and imaging findings characteristic or compatible with acute pancreatitis.14 The abdomen is diffusely tender with signs of peritonitis, and distention occurs with a paucity of bowel sounds. In severe cases of necrotizing or hemorrhagic pancreatitis, hemorrhage may spread away from the pancreas along tissue planes, appearing as ecchymosis either in the flanks (Grey Turner’s sign) or at the umbilicus (Cullen’s sign) (Fig. 46-4). These ecchymoses generally take 1 to 2 days to develop.
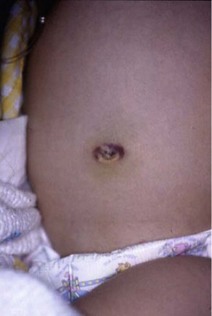
FIGURE 46-4 Positive Cullen’s sign, with periumbilical ecchymosis, in a patient with hemorrhagic pancreatitis.
Elevated amylase levels are helpful in the diagnosis, although a normal serum amylase level does not exclude pancreatitis. Hyperamylasemia may also be caused by salivary inflammation/trauma, intestinal disease (such as perforation, ischemia, necrosis, or inflammation), and macroamylasemia. Lipase has been suggested as an alternative marker, but can be falsely elevated in pancreatic cancer, macrolipasemia, renal insufficiency, cholecystitis, esophagitis, intestinal perforation, and hypertriglyceridemia. Elevated lipase tends to be more sensitive in infants and toddlers, and may also help differentiate pancreatic from salivary trauma. The degree of enzyme elevation does not correlate with disease severity.19,20
Abdominal ultrasonography (US) is useful in the evaluation of the patient with pancreatitis, but has limited applications. It is well established in the evaluation of biliary stone disease as the etiology for pancreatitis, and can detect choledochal cysts and pancreatic pseudocysts as well. Advanced techniques such as contrast-enhanced ultrasound and ultrasound elastography have also been shown to be useful in the diagnosis of pancreatitis and its complications, but their availability is limited to a few experienced centers.21
Abdominal computed tomography (CT) provides much better resolution of the pancreas than ultrasound. Its primary role is in the detection of early and late complications, such as pancreatic necrosis, pseudocysts, and fluid collections, and should be reserved for patients with more severe pathology, or recurrent symptomatology with an equivocal ultrasound. If necessary, CT can be combined with interventional procedures to drain fluid collections.22
Magnetic resonance cholangiopancreatography (MRCP) is a newer, noninvasive technique for evaluating the biliary tree and pancreatic duct (Fig. 46-5). It is the initial imaging study of choice for the evaluation of pancreatic ductal anatomy in children with recurrent or unexplained pancreatitis. Studies comparing MRCP and ERCP show high concordance in diagnoses. Its disadvantages are that it does not allow for therapeutic intervention (though it may direct the type of intervention necessary), its poor spatial resolution limits the visualization of ducts in smaller children, and it usually requires anesthesia in the pediatric age group.21,23
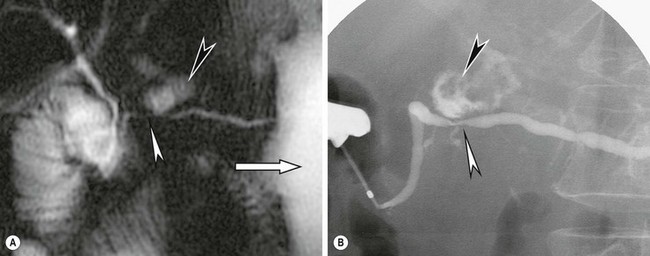
FIGURE 46-5 (A) This secretin-enhanced MRCP was performed in a patient with pancreatic ductal stenosis. The white arrowhead shows stenosis of the main pancreatic duct at the neck along with diffuse ascites (white arrow). A fluid collection communicating with the duct is also seen (black arrowhead). (B) An ERCP in the same patient demonstrates the ductal stenosis (white arrowhead), distal duct dilation, and the communicating fluid collection (black arrowhead). (From Akisik MF, Sandrasegaran K, Aisen AA, et al. Dynamic secretin-enhanced MR cholangiopancreatography. Radiographics: A review publication of the Radiological Society of North America, Inc. 2006;26:665–77.)
The most frequent indication for ERCP in children is in the diagnosis or treatment of acute, recurrent, or chronic pancreatitis. A large single-institution retrospective study found a low rate of post-ERCP complications and a high therapeutic success rate.15 ERCP was shown to be particularly useful in the diagnosis of recurrent pancreatitis, though in only 60% of patients an organic etiology was found. Sphincter of Oddi manometry was particularly useful in establishing a diagnosis when no anatomic abnormalities were present.
The treatment for pancreatitis has remained unchanged for decades. The mainstays of therapy are pain control, intravenous fluid resuscitation, pancreatic rest, and monitoring for complications. Fluid resuscitation and maintenance should be guided towards a goal urine output of 2 mL/kg/h measured by an indwelling urinary catheter. Because of circulating cytokines, activated digestive enzymes, and other pro-inflammatory molecules, extracellular fluid losses can be enormous. Constant monitoring is necessary to avoid the development of severe hypovolemia. Patients with severe acute pancreatitis may require nasogastric decompression. Most patients receive histamine-2 (H2) receptor antagonists to reduce exposure of the duodenal secretin-producing cells to gastric acid, a potent stimulator of pancreatic secretion. This therapeutic regimen is logical but empirical, because no studies have shown improvement in outcomes with these interventions. The effectiveness of somatostatin in the treatment of pancreatitis is equivocal and probably serves more to mitigate complications of pancreatitis rather than to treat the disease itself. Further studies are needed to clearly define its role in both adults and children.24
Nutrition is critically important in patients with pancreatitis. The past decade has seen a paradigm shift in the nutritional management of patients with pancreatitis. Early nutrition, within the first 72 hours, is still recommended, however enteral nutrition (EN) has become the preferred method over total parenteral nutrition (TPN). Patients with mild to moderate cases of acute pancreatitis often resolve prior to requiring EN or TPN. More severe cases should be treated with EN via a nasojejunal tube, though studies have demonstrated tolerance of nasogastric feeding in severe pancreatitis as well. TPN use, especially early in the disease course during the peak inflammatory response, has been associated with increased length of stay and delayed advancement of diet. When EN is contraindicated, some advocate waiting as long as 5 days prior to starting TPN. Compared to TPN, EN has been shown to decrease length of stay, reduce the need for surgery, and reduce the risk of infection.25 When restarting a diet, conservatively determined by resolution of symptoms, there appears to be no difference between clear liquid or solid food as the initial meal.26 Unfortunately, these data come almost exclusively from the adult population as pediatric trials are lacking.17,27
Adequate analgesia is critical to minimizing the physiologic stress that develops from pain. While meperidine (Demerol) was once advocated because morphine was thought to cause spasm of the Sphincter of Oddi, no clinical trials have shown superiority of meperidine over other narcotic analgesics. Large doses of meperidine, however, are associated with the risk of seizure, euphoria, and drug interactions, suggesting other narcotics such as morphine and fentanyl may be safer alternatives. The diagnosis of pancreatitis must be certain prior to initiating treatment with high doses of narcotics as these may mask signs of serious nonpancreatic pathology, such as intestinal or gastric perforations.28
As pancreatitis progresses in severity, patients need to be monitored closely for signs of multisystem organ failure. Pleural effusions, pulmonary edema, and tense abdominal distention may lead to hypoxia requiring intubation and adult respiratory distress syndrome. Hypocalcemia, hypomagnesemia, anemia from hemorrhage, hyperglycemia, renal failure, and late sepsis can also be seen in these patients. Disagreement exists regarding the use of prophylactic antibiotics in severe cases of pancreatitis. The most recent adult data suggests a trend towards decreased mortality and infection with prophylactic antibiotics, but this study failed to reach statistical significance.29 Imipenem is the antibiotic therapy of choice when necessary.
Operative exploration is usually not necessary in acute pancreatitis. However, exploration is needed in patients with infected necrotic pancreatitis or a pancreatic abscess. Infected pancreatic necrosis increases mortality significantly. Diagnosis is typically by CT pancreatography, with confirmation of infection by fine needle aspiration, clinical deterioration, or positive cultures. The latest adult data suggests that infected necrosis or peripancreatic abscesses are best treated in a stepwise manner from least to most invasive. When feasible, percutaneous drainage should be followed by minimally invasive necrosectomy if the patient fails to improve. Delayed operative therapy demonstrates improved mortality compared to primary necrosectomy.30,31
Pancreatic pseudocyst is a complication of trauma or pancreatitis that forms after injury to the pancreatic ductal system. The extravasated pancreatic enzymes and digested tissue are contained by the formation of a cavity composed from a fibroblastic reaction and inflammation that lacks an epithelial lining. The acute pseudocyst has an irregular wall on CT scan, is tender, and usually develops shortly after an episode of acute pancreatitis or trauma (Fig. 46-6). Chronic pseudocysts are usually spherical with a thick wall, and are commonly seen in patients with chronic pancreatitis. The distinction is important because half of acute pseudocysts resolve without treatment, while chronic pseudocysts rarely spontaneously resolve. An acute pseudocyst matures and forms a thick fibrous wall in four to six weeks, allowing for drainage. Those smaller than 5 cm in diameter usually spontaneously regress. When compared to those in adults, pseudocysts in children tend to resolve more frequently with medical therapy alone.32 There are anecdotal reports of pseudocyst resolution in children after treatment with long-acting somatostatin analogs.33
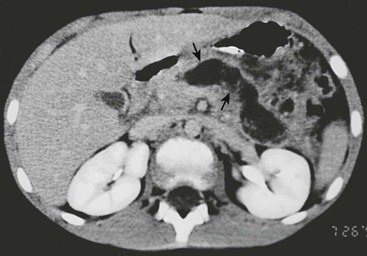
FIGURE 46-6 CT scan of an acute pseudocyst in a patient after a severe motor vehicle accident. The wall (arrows) is irregular with nonloculated fluid inside.
The pancreatic pseudocysts that persist, or are symptomatic, require either a drainage procedure or excision. Endoscopic treatment, well established in adults, has been reported to be safe and efficacious in children as well.34 This should only be performed at centers with sufficient experience with these techniques. Other options include laparoscopic transgastric and intragastric drainage either into the stomach or jejunum (Fig. 46-7). Percutaneous drainage is the preferred approach for infected pseudocysts because these cysts typically have thin, weak walls that are not amenable to internal drainage.
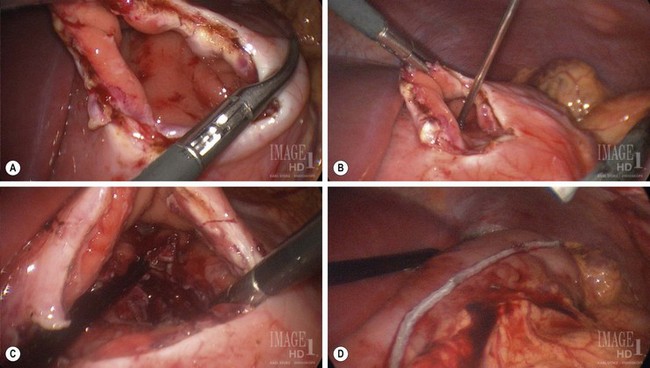
FIGURE 46-7 This adolescent sustained blunt trauma from a motor vehicle accident which resulted in a pancreatic pseudocyst several weeks later. After six weeks, laparoscopic cyst gastrostomy was performed. (A) The stomach has been opened. (B) A needle is introduced into the pseudocyst through the posterior wall of the stomach. (C) The common wall between the pseudocyst and stomach has been excised and is opened widely for adequate drainage of the pseudocyst. (D) The resulting gastrotomy has been closed with an endoscopic stapler. The patient recovered uneventfully and has not developed any postoperative problems.
Ascites in children usually follows trauma or pancreatic surgery.35 Free fluid results from the uncontained leakage of a major pancreatic duct. Treatment initially consists of bowel rest with hyperalimentation and the use of long-acting somatostatin analogs. In many cases, the ascites resolves spontaneously with this treatment. When suspected, CT, ERCP, or MRCP should be performed to assess for duct injury. A growing body of evidence supports nonoperative management of pediatric pancreatic trauma even in the presence of ductal injury. When operative treatment is necessary, drainage alone may suffice. Distal duct injuries can be treated with distal resection. Proximal injuries require Roux-en-Y jejunal onlay anastomosis to preserve pancreatic tissue. Pseudocyst formation is common in this patient population.36,37
Pancreatic fistula can occur postoperatively. Most low-output fistulas close spontaneously but may drain for several months. Long-acting somatostatin analogs decrease fistula output and accelerate the rate of closure, but do not appear to induce closure of fistulas that would not otherwise have closed. Managing a pancreatic fistula centers around maintaining nutrition, with hyperalimentation necessary if enteral feeding increases fistula output, and ensuring the fistula tract does not become obstructed. Operative intervention with a Roux-en-Y jejunostomy anastomosed to the fistula site is usually curative if the fistula fails conservative management.38
Chronic Pancreatitis
Chronic pancreatitis is distinguished from acute pancreatitis by the irreversibility of the changes associated with the inflammation.14 Chronic pancreatitis can be classified as either calcifying or noncalcifying. The calcifying form, most common in hereditary or idiopathic pancreatitis, is more prevalent than the obstructive form in children, and is associated with intraductal pancreatic stones, pseudocysts, and more aggressive scar formation. The obstructive type of chronic pancreatitis, which is associated with an anatomic or functional obstruction, is generally less severe with less scarring than calcifying pancreatitis. Single institutional experiences vary as to distributions in etiology, though the predominant causes are hereditary/genetic, obstructive, or idiopathic.39,40
The discovery of a genetic basis for certain forms of chronic pancreatitis represents a major breakthrough in our understanding of its pathogenesis. Current thinking is that familial pancreatitis results from a combination of environmental triggers, genetic susceptibility, and an inappropriate immune response leading to chronic inflammation and fibrosis. Hereditary pancreatitis, the most common genetic cause of pancreatitis, can result from abnormalities in the cationic trypsinogen gene PRSS1.41 This autosomal dominant condition most commonly results from one of two mutations that either prevent degradation of prematurely activated trypsin or make trypsin resistant to inactivation. Penetrance in this condition is approximately 80%. Other gene mutations implicated in chronic pancreatitis are SPINK1 and CFTR. Genetic testing is recommended for children with recurrent, idiopathic pancreatitis, with or without a family history of pancreatitis. Notably, patients with hereditary pancreatitis have a markedly increased risk of pancreatic cancer after age 50 years.
Obstructive pancreatitis is most often due to an anatomic or functional obstruction of the pancreatic duct. The most common anatomic causes are pancreas divisum followed by choledochal cysts. Uncertainty exists as to why a minority of patients with pancreas divisum develop chronic pancreatitis while most do not. The literature suggests that potential causes are the anatomic variant, structural narrowing of the minor papilla, sphincter of Oddi dysfunction, or the relatively high association with CFTR mutations.18 Many patients with ductal dilatation and anatomic or functional obstruction clearly improve with endoscopic sphincterotomy and/or stents. Pancreatic dyskinesia, though not well studied in children, may be improved by endoscopic sphincterotomy and sometimes temporary stent placement.42 For those in whom endoscopic treatment is not feasible or fails, individualized surgical treatment is reasonable based on the patient’s ductal anatomy, level of obstruction, and severity of pancreatic fibrosis.
Chronic pancreatitis can manifest with a characteristic pain, diminished pancreatic function, and radiographic abnormalities. Increased stool fat, diabetes, and steatorrhea are signs of pancreatic insufficiency. Frequently on a CT scan, the pancreas has microcalcifications throughout the parenchyma and calcified stones in the duct (Fig. 46-8). Additionally, pancreatic pseudocysts or inflammation may be seen on the CT scan. ERCP or MRCP can evaluate the ductal anatomy and can identify anatomic causes of chronic pancreatitis. Only ERCP provides a means for evaluating sphincter pressure measurements for functional obstruction.42
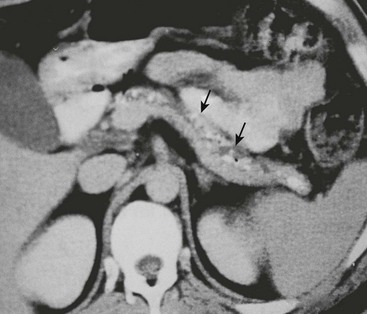
FIGURE 46-8 CT scan of a pancreas with chronic calcifying pancreatitis. A dilated duct can be seen within the pancreas, further supporting the diagnosis of chronic pancreatitis. Calcified stones (arrows) can be seen in the dilated duct.
Therapy for chronic pancreatitis is directed toward palliation of symptoms. Initial management for acute exacerbations is pain control and hydration. Steatorrhea indicates the need for pancreatic enzyme replacement. In general, these patients respond better with small, frequent meals. The diabetes that results from chronic pancreatitis tends to be unusually brittle, with a propensity for severe hypoglycemic episodes after even low doses of insulin. This hypersensitivity to insulin may be due to the loss of entire islets, which includes the glucagon-producing alpha cells that normally oppose the glucose-lowering effect of insulin.
In patients with chronic pancreatitis who have severe, intractable pain, ERCP or MRCP may help locate correctable problems such as large stones or a stricture with distal duct dilatation. Surgical options in chronic pancreatitis include sphincteroplasty, excision of localized pancreatitis, subtotal pancreatectomy, lateral pancreaticojejunostomy (modified Puestow procedure), and the duodenum or pylorus-preserving Whipple. Individualization of the operative approach and maximization of pancreatic ductal drainage are key. Patients failing more conservative surgical management may require a more definitive procedure to achieve symptomatic relief.43 Although the operative results in patients with hereditary pancreatitis are generally disappointing, evidence exists that complicated patients treated with a modified Puestow procedure may experience an improved quality of life with subsequent improvement in pancreatic function and nutritional status (Fig. 46-9).44 Unlike adults with hereditary pancreatitis, some reversal of the steatorrhea may be seen in children.
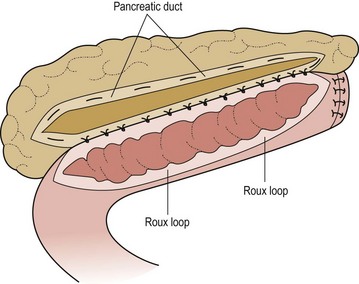
FIGURE 46-9 Modified Puestow procedure. The pancreatic duct is opened longitudinally and a side-to-side anastomosis to a Roux loop of jejunum is performed. (Adapted from Mayo-Smith WW, Iannitti DA, Dupuy DE. Intraoperative sonographically guided wire cannulation of the pancreatic duct for patients undergoing a Puestow procedure. AJR Am J Roentgenol 2000;175:1639–40.)
Functional Pancreatic Disorders
Congenital Hyperinsulinism
Nesidioblastosis, the original term for what is now called congenital hyperinsulinism of infancy (CHI), comes from the Greek nesidio, meaning ‘island,’ and blast, meaning ‘new formation.’ Nesidioblasts were originally thought to be progenitor cells in the wall of the pancreatic ducts which overproliferate in patients with this condition.45 With the advent of genetic analysis, this pathogenesis was proven to be incorrect.
Mutations in seven genes are currently known to lead to CHI, though roughly half of cases are caused by genetic malformations not yet understood. Initially discovered was the loss of functioned mutations in the SUR1 and Kir6.2 components of the ATP-sensitive potassium channel (KATP) found in the cell membrane of the pancreatic β-cell. These mutations either impair the ability of Mg-adenosine diphosphate (ADP) to stimulate channel activity or affect the expression of the KATP channels at the surface membrane, resulting in continuous depolarization of the β-cell membrane and dysregulated insulin secretion.46 Heterozygous gain-of-function mutations in GULD1, the mitochondrial gene encoding glutamate dehydrogenase, leads to CHI caused by insensitivity of the enzyme to inhibition by guanosine-5′-triphosphate. This mutation results in a milder form of CHI, which may be diagnosed later in childhood, and is associated with hyperammonemia and occasionally epilepsy. Heterozygous loss-of-function mutations in HNF4A, an islet transcription factor gene, are also associated with CHI through unknown mechanisms. More rare are cases of CHI caused by mutations in glucokinase (GCK), hydroxyacyl-coenzyme A dehydrogenase (HADH), and the solute carrier SLC16A1.47
CHI occurs in a focal and diffuse type, the differentiation of which is critical for operative management. However, the clinical presentation is identical. Patients with SUR1 mutations often have the focal type. The focal type is actually a focal adenomatous hyperplasia, different from an adenoma. Histologically, it is seen as a confluence of hyperplastic but otherwise normal-appearing islets. There is little insulin present within the lesion due to excess secretion, while uninvolved islets outside are small with high insulin content. The diffuse type is macroscopically normal, but careful evaluation of the islets reveals enlarged β-cells with abnormally large nuclei, a large Golgi apparatus, and weak insulin staining due to hypersecretion.48
CHI patients typically develop hypoglycemia shortly after birth, though it may present at a later age. Infants with CHI are often macrosomic. Symptoms may be subtle, such as lethargy and irritability, or severe with apnea, seizures, and coma. Simultaneous insulin and glucose measurements show a high ratio of insulin to glucose, keeping in mind that insulin levels may be normal, but inappropriate in the presence of hypoglycemia. These patients differ from insulinoma patients, who usually have high absolute insulin levels. Another powerful indicator of CHI is a glucose requirement greater than 8 mg/kg/min.49 Owing to the much higher incidence of insulinoma, patients older than 1 year at the onset of hypoglycemia should be evaluated for both conditions.
Treatment of CHI begins with medical management. Diazoxide remains a mainstay of therapy. Diazoxide binds the SUR1 component of the KATP channel and maintains it in a persistently open state, preventing insulin secretion. Patients with diazoxide-sensitive CHI who can tolerate fasting can be managed medically until they outgrow their condition. Those unresponsive to diazoxide should be managed with frequent feedings, glucose infusions, and somatostatin analogs. Somatostatin analogs inhibit pancreatic insulin secretion and can be administered subcutaneously either intermittently or continuously by a pump. Somatostatin analogs are associated with gallstones and biliary sludge, and have been implicated in cases of necrotizing enterocolitis.50 Medical failure to control hypoglycemia necessitates surgical intervention.49
Distinguishing diffuse versus focal-type CHI is critical for operative planning. The recent development of an 18F-DOPA PET-CT scanner has replaced pancreatic venous sampling as the optimal method for delineating focal versus diffuse disease with a sensitivity of 94% and specificity of 100% (Fig. 46-10).51 Intraoperative ultrasound can provide additional anatomic detail to help avoid injury to the biliary tree.52 For the focal type, resection of the hypermetabolic focus is curative. Focal lesions in the head may require duodenum-preserving pancreatic head resection and distal pancretaticojejunostomy while distal lesions are excised via spleen-preserving distal pancreatectomy. In patients with diffuse disease, a near-total pancreatectomy is needed, leaving only a rim of pancreatic tissue along the common bile duct (Fig. 46-11). The laparoscopic approach for both procedures has been reported but data are limited.53,54 Operative complications include bile duct injury, pancreatic insufficiency, and the need for repeat pancreatic resection due to persistent hypoglycemia.55
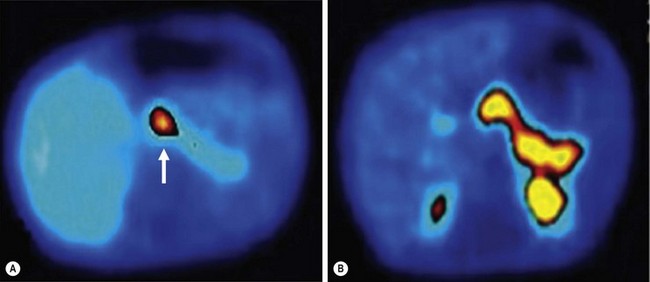
FIGURE 46-10 18F-DOPA PET-CT scans of two patients with congenital hyperinsulinism of infancy. (A) A focal lesion (arrow) is detected in the head of the pancreas. (B) The more diffuse uptake of the tracer along the entirety of the pancreas indicates the diffuse type of this condition in this patient. (From Arnoux JB, Verkarre V, Saint-Martin C, et al. Congenital hyperinsulinism: Current trends in diagnosis and therapy. Orphanet Journal of Rare Diseases 2011;6:63.)
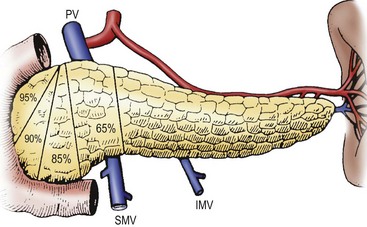
FIGURE 46-11 Various degrees of pancreatectomy may be indicated for persistent hyperinsulinemic hypoglycemia of infancy. Typically, a 95% pancreatectomy, as shown here, leaves behind a cuff of pancreas along the C-loop of the duodenum. IMV, inferior mesenteric vein; PV, portal vein; SA, splenic artery; SMV, superior mesenteric vein; SV, splenic vein.
Alternate forms of hyperinsulinemic hypoglycemia can arise secondary to maternal diabetes, birth asphyxia, or intrauterine growth retardation (IUGR). In these patients, the condition is transient, resolving for most patients within several days. A subgroup of patients with IUGR and prenatal asphyxia will have a prolonged condition that requires diazoxide for several months prior to resolution. Approximately 50% of children with Beckwith–Wiedemann syndrome have a CHI-like condition that tends to resolve spontaneously, but 5% will eventually require pancreatectomy.49
The long-term outlook for these patients depends primarily on the age at onset which reflects severity of disease, and on an expeditious diagnosis because a late diagnosis results in a higher incidence of neurologic sequelae.56 Most patients seem to ‘grow out of the disease’ after several years, implying diminished activity of the beta cells. Near-total pancreatectomy in one series was associated with a 91% incidence of insulin-dependent diabetes by age 14, while no patient with focal resection required long-term insulin therapy, emphasizing the need for a conservative operative approach when possible.57
Glycogen Storage Disease
Glycogen storage disease type Ia (GSD-Ia) and Ib classically appear as severe hypoglycemia in infants caused by the inability to dephosphorylate hepatic glycogen subunits into glucose. GSD-Ia is caused by inactivating mutations of the glucose-6-phosphatase enzyme itself, while GSD-Ib results from inactivating mutations of the glucose-6-phosphate transporter. Hypoglycemia becomes apparent when the time between feedings increases and the liver fails to generate glucose from glycogen stores. It is clinically diagnosed by hypoglycemia, hypoinsulinemia, hepatomegaly, nephromegaly, ketosis, and hyperlipidemia. Central venous access is required for continuous infusion of highly concentrated glucose. Survivors into adulthood have an increased incidence of hepatic adenoma after age 25 and have a 10% risk of malignant transformation. Liver transplantation ultimately becomes necessary in these patients.58
Pancreatic Tumors and Cysts
Pancreatic Endocrine Tumors
Insulinoma is the most common of the three, though is still quite rare amongst pediatric tumors in general.59 Only 10% of insulinomas are malignant, and these tend to spread to the liver and peripancreatic lymph nodes. Insulinomas cause symptoms of hypoglycemia, including dizziness, headaches, confusion, sweating, and seizures. The classic Whipple’s triad was described in patients with insulinoma and consists of the following: symptoms of hypoglycemia with fasting, glucose levels less than half of normal when fasting, and relief of symptoms with glucose administration. Patients are typically in their adolescence, though younger children have been described with insulinoma. The lesions are usually solitary, except in multiple endocrine neoplasia type I (MEN I) in which multiple insulinomas may be found.60
The gold standard test for insulinoma is the 72-hour fast, though studies have shown that a positive result is achieved in 80% and 90% of patients with insulinoma after a shorter 24-hour or 48-hour fast, respectively. While fasting, periodic blood glucose levels are obtained. When the patient’s blood glucose falls below 50 mg/dL and symptoms are present, blood is drawn for plasma glucose, C-peptide, proinsulin, insulin, β-hydroxybutarate, and sulfonylureas. Administration of exogenous insulin, followed by measurement of the C-peptide level, can be suggestive of an insulinoma, but is not completely reliable. Measuring the insulin : glucose ratio is no longer needed.61
Preoperative localization is challenging, but can be extremely helpful. Extrapancreatic insulinomas are rare. Most experts advocate for both transabdominal ultrasound and CT for initial localization, which identifies more than half of tumors. Centers experienced with endoscopic ultrasound also have reported good success in detecting insulinomas,62 but expertise is not widely available. MRI is most useful for detecting liver metastases. If noninvasive studies fail to identify the tumor, intra-arterial calcium stimulation via a catheter in several visceral arteries with parallel venous sampling from the right hepatic vein has been reported to regionally localize insulinomas in 80–94% of cases.63 Intraoperative ultrasound is strongly advocated for identification of adjacent biliary and vascular structures, and as a method of last resort if nonoperative localization fails.60,61
All patients with insulinoma should undergo resection. Insulinomas are pink, firm, encapsulated, and are usually amenable to enucleation. In most cases, through preoperative and intraoperative analysis, the tumors can be localized, but in patients in whom they cannot be localized, blind distal pancreatic resection is no longer advised. The distinction between benign and malignant lesions is difficult, and is based on tumor size (<2 cm tend to be benign) and the presence of metastases. Insulinomas that are hard, cause puckering of surrounding tissues, appear infiltrating, or cause distal pancreatic duct dilation should be assumed malignant, and resected with a margin instead of enucleated. Malignant tumors can be treated with chemotherapy, biotherapy (such as octreotide), hepatic artery embolization/chemoembolization, radiation therapy, or radiofrequency ablation.60
Fetal gastrin-producing cells in the pancreas are believed to give rise to pancreatic gastrinoma. In the fetus, the primary source of gastrin is the pancreas. After birth, the gastric antrum becomes the principal source. Zollinger–Ellison syndrome (ZES) consists of gastric hypersecretion with severe peptic ulcer disease and a gastrin-producing tumor, classically in the pancreas. ZES may occur as part of the MEN I syndrome or sporadically. Gastrinomas are now understood to be frequently malignant, especially with spread to the liver, and their removal is strongly advocated.64,65
Medically, patients should initially be treated with omeprazole to control peptic ulcer disease and prevent bleeding. All disease should be excised if possible to control symptoms and help prevent metastases. Patients cured by resection should be followed closely as recurrence is common. Medical treatment for unresectable disease includes proton pump inhibitors and octreotide. Tumor debulking in unresectable cases is recommended to improve the patient’s quality of life and increase the life expectancy.65 Chemotherapy has been utilized in a few cases with good results reported.66 There is a report of a patient with a multiple gastrinomas managed medically who survived 26 years before succumbing to unrelated causes.67
Non-Neoplastic Cysts
Although most cystic lesions of the pancreas are pseudocysts and are acquired, congenital cysts may be seen at an early age as a symptomatic mass with compression of surrounding structures. Alternatively, these congenital cysts may be noted incidentally on physical examination or radiographic studies. Congenital cysts contain cloudy, straw-colored fluid. The cysts are most often found in the distal pancreas and are amenable to local resection with a rim of normal pancreas. Lesions in the pancreatic head should be internally drained with a Roux-en-Y cystjejunostomy.68
Congenital duplications of the foregut may also present as pancreatic cysts. They have a gastric or intestinal mucosal lining and maintain pancreatic ductal communication. Gastric acid secretion from the cyst may cause episodes of pancreatitis. Surgical resection is necessary, either in the form of enucleation, distal pancreatectomy, or even pancreaticoduodenectomy.69
Acquired non-neoplastic cysts of the pancreas are called retention cysts, which appear to result from obstruction of the smaller pancreatic ducts. The preoperative distinction of a retention cyst from other types of cysts or pseudocysts may be difficult, but is supported by finding a small cystic lesion in communication with the pancreatic duct that has a proximal stricture. These can be excised if symptomatic, or otherwise observed. However, they are often confused with neoplastic cysts and are resected for this reason.70
Pancreatic Exocrine Tumors
The pancreatic exocrine system includes the pancreatic ducts, centroacinar cells, and acini. Tumors from this system include pseudopapillary tumors, ductal adenocarcinomas, acinar cell carcinomas, or pancreatoblastomas. Cystic tumors of the pancreas, including serous and mucinous cystadenomas and cystadenocarcinoma, are well described in adults, but are exceedingly rare in the pediatric population. Several case reports exist of serous and mucinous cystadenomas, but one review suggests that no confirmed reports of cystadenocarcinoma in children exist in the literature.59 Management of serous cystadenoma is debated based on its benign nature, but excision appears curative.
Adenocarcinoma/Pancreatoblastoma
Exocrine pancreatic cancers account for roughly half of pancreatic neoplasms in children. While ductal adenocarcinoma is most common in adults, its embryonic counterpart pancreatoblastoma is more common in children. Pancreatoblastoma is believed to result from the persistence of embryonic pancreatic progenitor cells beyond the eighth week of gestation. It tends to be diagnosed in early childhood and is more common in boys and those of Asian descent. An allelic loss on 11p is often associated and suggests a similar pathogenesis with Beckwith–Wiedemann syndrome and other embryonic tumors. A recent large multicenter review demonstrated that the tumor distribution was homogenous throughout the pancreas, most were >5 cm at presentation, and over half had distant metastases.71 Elevation of serum alpha-fetoprotein was an inconsistent feature. The prognosis is relatively good with complete resection and with appropriate neoadjuvant and/or adjuvant chemotherapy and radiation therapy. Relapse is common, so continued monitoring is essential. 72
Acinar cell carcinoma and ductal adenocarcinoma are less common pancreatic exocrine tumors. Ductal adenocarcinoma is infrequently reported in the pediatric literature and no definitive recommendations are possible.73 Acinar cell carcinoma is comparatively more common. Complete resection for both tumor types appears necessary with appropriate provision of neoadjuvant or adjuvant chemotherapy based on pre-treatment staging. Long-term survival improves with an earlier stage at presentation.74,75
Occurring slightly less frequently than pancreatoblastoma is the solid pseudopapillary tumor, also known as a papillary-cystic tumor or Frantz tumor. It has a female preponderance and is derived from exocrine cells, but without acinar or ductal structures. Presenting symptoms often include a palpable abdominal mass and abdominal pain. These tumors can be very large at the time of diagnosis (Fig. 46-12).76 They are very slow growing, and there are reports of patients surviving 20 years after diagnosis without treatment.77 While rarely metastatic, excision of regional and distal metastases greatly improves survival. Even with its indolent nature, an aggressive approach with complete resection is recommended as one retrospective study of patients with incomplete resections showed poor long-term survival.78
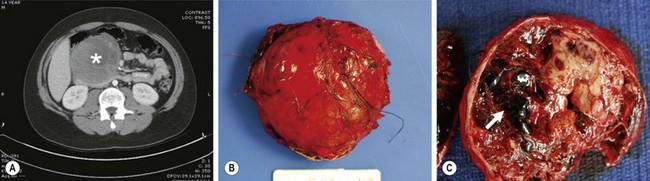
FIGURE 46-12 This solid pseudopapillary tumor was found in a 14-year-old boy. (A) CT scan of the abdomen demonstrates a large, heterogeneous mass (asterisk) in the head of the pancreas. (B) The tumor is seen at excision. Note that the tumor appears encapsulated and well circumscribed. (C) Cut section of the mass demonstrates solid and cystic regions as well as an area of hemorrhagic necrosis (arrow). (From Speer AL, Barthel ER, Patel MM, et al. Solid pseudopapillary tumor of the pancreas: A single-institution 20-year series of pediatric patients. J Pediatr Surg 2012;47:1217–22.)
References
1. Gittes, GK. Developmental biology of the pancreas: A comprehensive review. Dev Biol. 2009; 326:4–35.
2. Dawson, W, Langman, J. An anatomical-radiological study on the pancreatic duct pattern in man. Anat Record. 1961; 139:59–68.
3. Bertelli, E, Di Gregorio, F, Bertelli, L, et al. The arterial blood supply of the pancreas: A review I. The superior pancreaticoduodenal and the anterior superior pancreaticoduodenal arteries. An anatomical and radiological study. Surg Radiol Anat. 1995; 17:97–106.
4. Nakajima, H, Kambayashi, M, Okubo, H, et al. Annular pancreas accompanied by an ectopic pancreas in the adult: A case report. Endoscopy. 1995; 27:713.
5. Heller, RS, Tsugu, H, Nabeshima, K, et al. Intracranial ectopic pancreatic tissue. Islets. 2010; 2:65–71.
6. Tilson, MD, Touloukian, RJ. Mediastinal enteric sequestration with aberrant pancreas: A formes frustes of the intralobar sequestration. Ann Surg. 1972; 176:669–671.
7. Ozcan, C, Celik, A, Guclu, C, et al. A rare cause of gastric outlet obstruction in the newborn: Pyloric ectopic pancreas. J Pediatr Surg. 2002; 37:119–120.
8. van den Brink, GR. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol Rev. 2007; 87:1343–1375.
9. Skandalakis, JE, Gray, SW. Embryology for surgeons: The embryological basis for the treatment of congenital anomalies, 2nd ed. Baltimore: Williams & Wilkins; 1994.
10. Sencan, A, Mir, E, Gunsar, C, et al. Symptomatic annular pancreas in newborns. Med Sci Mont. 2002; 8:CR434–CR437.
11. Urushihara, N, Fukumoto, K, Fukuzawa, H, et al. Recurrent pancreatitis caused by pancreatobiliary anomalies in children with annular pancreas. J Pediatr Surg. 2010; 45:741–746.
12. Naruse, S, Kitagawa, M, Ishiguro, H, et al. Cystic fibrosis and related diseases of the pancreas. Best Pract Res Cl Ga. 2002; 16:511–526.
13. Taylor, CJ, Aswani, N. The pancreas in cystic fibrosis. Paediatr Respir Rev. 2002; 3:77–81.
14. Morinville, VD, Husain, SZ, Bai, H, et al. Definitions of pediatric pancreatitis and survey of current clinical practices: Report from INSPPIRE (International Study Group Of Pediatric Pancreatitis: In search for a cure). J Pediatr Gastroenterol Nutr. 2012; 55:261–265.
15. Otto, AK, Neal, MD, Slivka, AN, et al. An appraisal of endoscopic retrograde cholangiopancreatography (ERCP) for pancreaticobiliary disease in children: Our institutional experience in 231 cases. Surg Endosc. 2011; 25:2536–2540.
16. Trivedi, CD, Pitchumoni, CS. Drug-induced pancreatitis: An update. J Clin Gastroentrol. 2005; 39:709–716.
17. Lowe, ME, Greer, JB. Pancreatitis in children and adolescents. Curr Gastroenterol Rep. 2008; 10:128–135.
18. Schneider, L, Muller, E, Hinz, U, et al. Pancreas divisum: A differentiated surgical approach in symptomatic patients. World J Surg. 2011; 35:1360–1366.
19. Bai, HX, Lowe, ME, Husain, SZ. What have we learned about acute pancreatitis in children? J Pediatr Gastroenterol Nutr. 2011; 52:262–270.
20. Herman, R, Guire, KE, Burd, RS, et al. Utility of amylase and lipase as predictors of grade of injury or outcomes in pediatric patients with pancreatic trauma. J Pediatr Surg. 2011; 46:923–926.
21. Darge, K, Anupindi, S. Pancreatitis and the role of ultrasound, MRCP and ERCP. Pediatr Radiol. 2009; 39(Suppl 2):S153–S157.
22. Kim, DH, Pickhardt, PJ. Radiologic assessment of acute and chronic pancreatitis. Surg Clin N Am. 2007; 87:1341–1358.
23. Neblett, WW, 3rd., O’Neill, JA, Jr. Surgical management of recurrent pancreatitis in children with pancreas divisum. Ann Surg. 2000; 231:899–908.
24. Li, J, Wang, R, Tang, C. Somatostatin and octreotide on the treatment of acute pancreatitis—basic and clinical studies for three decades. Curr Pharm Design. 2011; 17(16):1594–1601.
25. Marick, PE, Zaloga, GP. Meta-analysis of parenteral nutrition versus enteral nutirion in patients with acute pancreatitis. BMJ. 2004; 328:1407–1412.
26. Moraes, JM, Felga, GE, Chebli, LA, et al. A full solid diet as the initial meal in mild acute pancreatitis is safe and result in a shorter length of hospitalization: Results from a prospective, randomized, controlled, double-blind clinical trial. J Clin Gastroenterol. 2010; 44:517–522.
27. McClave, SA, Chang, WK, Dhaliwal, R, et al. Nutrition support in acute pancreatitis: A systematic review of the literature. JPEN-Parenter Enter. 2006; 30:143–156.
28. Thompson, DR. Narcotic analgesic effects on the sphincter of Oddi: A review of the data and therapeutic implications in treating pancreatitis. Am J Gastroenterol. 2001; 96:1266–1272.
29. Villatoro, E, Mulla, M, Larvin, M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev. 2010.
30. Beger, HG, Rau, B, Mayer, J, Pralle, U. Natural course of acute pancreatitis. World J Surg. 1997; 21:130–135.
31. van Santvoort, HC, Bakker, OJ, Bollen, TL, et al. A conservative and minimally invasive approach to necrotizing pancreatitis improves outcome. Gastroenterol. 2011; 141:1254–1263.
32. Kisra, M, Ettayebi, F, Benhammou, M. Pseudocysts of the pancreas in children in Morocco. J Pediatr Surg. 1999; 34:1327–1329.
33. Wensil, AM, Balasubramanian, SA, Bell, TL. Resolution of a posttraumatic pancreatic pseudocyst with octreotide acetate in a pediatric patient. Pharmacotherapy. 2011; 31:924.
34. Sharma, SS, Maharshi, S. Endoscopic management of pancreatic pseudocyst in children-a long-term follow-up. J Pediatr Surg. 2008; 43:1636–1639.
35. D’Cruz, AJ, Kamath, PS, Ramachandra, C, et al. Pancreatic ascites in children. Acta Paediatr Jpn. 1995; 37:630–633.
36. de Blaauw, I, Winkelhorst, JT, Rieu, PN, et al. Pancreatic injury in children: Good outcome of nonoperative treatment. J Pediatr Surg. 2008; 43:1640–1643.
37. Juric, I, Pogorelic, Z, Biocic, M, et al. Management of blunt pancreatic trauma in children. Surg Today. 2009; 39:115–119.
38. Butturini, G, Daskalaki, D, Molinari, E, et al. Pancreatic fistula: Definition and current problems. J Hepato-Biliary-Pan. 2008; 15:247–251.
39. Clifton, MS, Pelayo, JC, Cortes, RA, et al. Surgical treatment of childhood recurrent pancreatitis. J Pediatr Surg. 2007; 42:1203–1207.
40. Lucidi, V, Alghisi, F, Dall’Oglio, L, et al. The etiology of acute recurrent pancreatitis in children: A challenge for pediatricians. Pancreas. 2011; 40:517–521.
41. Kandula, L, Whitcomb, DC, Lowe, ME. Genetic issues in pediatric pancreatitis. Curr Gastroenterol Rep. 2006; 8:248–253.
42. Halata, MS, Berezin, SH. Biliary dyskinesia in the pediatric patient. Curr Gastroenterol Rep. 2008; 10:332–338.
43. Chromik, AM, Seelig, MH, Saewe, B, et al. Tailored resective pancreatic surgery for pediatric patients with chronic pancreatitis. J Pediatr Surg. 2008; 43:634–643.
44. DuBay, D, Sandler, A, Kimura, K, et al. The modified Puestow procedure for complicated hereditary pancreatitis in children. J Pediatr Surg. 2000; 35:343–348.
45. Laidlaw, GF. Nesidioblastoma, the islet tumor of the pancreas. Am J Pathol. 1938; 14:125–134.
46. Saint-Martin, C, Arnoux, JB, de Lonlay, P, et al. KATP channel mutations in congenital hyperinsulinism. Semin Pediatr Surg. 2011; 20:18–22.
47. Flanagan, SE, Kapoor, RR, Hussain, K. Genetics of congenital hyperinsulinemic hypoglycemia. Semin Pediatr Surg. 2011; 20:13–17.
48. Rahier, J, Guiot, Y, Sempoux, C. Morphologic analysis of focal and diffuse forms of congenital hyperinsulinism. Semin Pediatr Surg. 2011; 20:3–12.
49. Kapoor, RR, Flanagan, SE, James, C, et al. Hyperinsulinaemic hypoglycaemia. Arch Dis Child. 2009; 94:450–457.
50. Laje, P, Halaby, L, Adzick, NS, et al. Necrotizing enterocolitis in neonates receiving octreotide for the management of congenital hyperinsulinism. Pediatr Diabetes. 2010; 11:142–147.
51. Mohnike, W, Barthlen, W, Mohnike, K, et al. Positron emission tomography/computed tomography diagnostics by means of fluorine-18-L-dihydroxyphenylalanine in congenital hyperinsulinism. Semin Pediatr Surg. 2011; 20:23–27.
52. von Rohden, L, Mohnike, K, Mau, H, et al. Visualization of the focus in congenital hyperinsulinism by intraoperative sonography. Semin Pediatr Surg. 2011; 20:28–31.
53. Al-Shanafey, S, Habib, Z, Al Nassar, S. Laparoscopic pancreatectomy for persistent hyperinsulinemic hypoglycemia of infancy. J Pediatr Surg. 2009; 44:134–138.
54. Bax, KN, van der Zee, DC. The laparoscopic approach toward hyperinsulinism in children. Sem Pediatr Surg. 2007; 16:245–251.
55. Pierro, A, Nah, SA. Surgical management of congenital hyperinsulinism of infancy. Semin Pediatr Surg. 2011; 20:50–53.
56. Leibowitz, G, Glaser, B, Higazi, AA, et al. Hyperinsulinemic hypoglycemia of infancy (nesidioblastosis) in clinical remission: High incidence of diabetes mellitus and persistent beta-cell dysfunction at long-term follow-up. J Clin Endocr Metab. 1995; 80:386–392.
57. Beltrand, J, Caquard, M, Arnoux, JB, et al. Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care. 2012; 35:198–203.
58. Chou, JY, Jun, HS, Mansfield, BC. Glycogen storage disease type I and G6Pase-beta deficiency: Etiology and therapy. Nat Rev Endocrinol. 2010; 6:676–688.
59. Shorter, NA, Glick, RD, Klimstra, DS, et al. Malignant pancreatic tumors in childhood and adolescence: The Memorial Sloan-Kettering experience, 1967 to present. J Pediatr Surg. 2002; 37:887–892.
60. Mathur, A, Gorden, P, Libutti, SK. Insulinoma. Surg Clin N Am. 2009; 89:1105–1121.
61. Grant, CS. Insulinoma. Best Pract Res Cl Ga. 2005; 19:783–798.
62. Patel, KK, Kim, MK. Neuroendocrine tumors of the pancreas: Endoscopic diagnosis. Curr Opin Gastroent. 2008; 24:638–642.
63. Guettier, JM, Kam, A, Chang, R, et al. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: The NIH experience. J Clin Endocrinol Metab. 2009; 94:1074–1080.
64. Jensen, RT. Gastrointestinal endocrine tumours. Gastrinoma. Bailliere Clin Gastr. 1996; 10:603–643.
65. Schettini, ST, Ribeiro, RC, Facchin, CG, et al. Gastrinoma in childhood: Case report and update on diagnosis and treatment. Eur J Pediatr Surg. 2009; 19:38–40.
66. Ohshio, G, Hosotani, R, Imamura, M, et al. Gastrinoma with multiple liver metastases: Effectiveness of dacarbazine (DTIC) therapy. J Hepatobiliary Pancreat Surg. 1998; 5:339–343.
67. Quatrini, M, Castoldi, L, Rossi, G, et al. A follow-up study of patients with Zollinger-Ellison syndrome in the period 1966–2002: Effects of surgical and medical treatments on long-term survival. J Clin Gastroenterol. 2005; 39:376–380.
68. Castellani, C, Zeder, SL, Spuller, E, et al. Neonatal congenital pancreatic cyst: Diagnosis and management. J Pediatr Surg. 2009; 44:e1–e4.
69. Fujishiro, J, Kaneko, M, Urita, Y, et al. Enteric duplication cyst of the pancreas with duplicated pancreatic duct. J Pediatr Surg. 2011; 46:e13–e16.
70. Goh, BK, Tan, YM, Chung, YF, et al. Non-neoplastic cystic and cystic-like lesions of the pancreas: May mimic pancreatic cystic neoplasms. ANZ J Surg. 2006; 76:325–331.
71. Bien, E, Godzinski, J, Dall’igna, P, et al. Pancreatoblastoma: A report from the European cooperative study group for paediatric rare tumours (EXPeRT). Eur J Cancer. 2011; 47:2347–2352.
72. Lee, YJ, Hah, JO. Long-term survival of pancreatoblastoma in children. J Pediat Hematol Onc. 2007; 29:845–847.
73. Luttges, J, Stigge, C, Pacena, M, et al. Rare ductal adenocarcinoma of the pancreas in patients younger than age 40 years. Cancer. 2004; 100:173–182.
74. Brecht, IB, Schneider, DT, Kloppel, G, et al. Malignant pancreatic tumors in children and young adults: Evaluation of 228 patients identified through the Surveillance, Epidemiology, and End Result (SEER) database. Klinische Padiatrie. 2011; 223:341–345.
75. Ellerkamp, V, Warmann, SW, Vorwerk, P, et al. Exocrine pancreatic tumors in childhood in Germany. Pediatric Blood Cancer. 2012; 58:366–371.
76. Speer, AL, Barthel, ER, Patel, MM, et al. Solid pseudopapillary tumor of the pancreas: A single-institution 20-year series of pediatric patients. J Pediatr Surg. 2012; 47:1217–1222.
77. Soloni, P, Cecchetto, G, Dall’igna, P, et al. Management of unresectable solid papillary cystic tumor of the pancreas. A case report and literature review. J Pediatr Surg. 2010; 45:e1–e6.
78. Campanile, M, Nicolas, A, LeBel, S, et al. Frantz’s tumor: Is mutilating surgery always justified in young patients? Surg Oncol. 2011; 20:121–125.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Lesions of The Pancreas
The pancreas originates during week 4 of gestation as dual evaginations from the foregut endoderm. The dorsal pancreatic bud gives rise to the body and tail of the pancreas, its minor duct (Santorini) and papilla, and the continuation of the major duct (Wirsung) into the body and tail. The ventral pancreatic bud arises from the biliary diverticulum and swings around the dorsal aspect of the duodenal anlage during gut rotation to give rise to the head of the pancreas as well as the proximal portion of Wirsung’s duct (Fig. 46-1).1
FIGURE 46-1 Pancreatic embryology. (A) Stomach (a), gallbladder (b), and ventral (c) and dorsal (d) pancreatic buds develop separately at embryologic week 4. The pancreas develops as an evagination of the developing foregut. The dorsal bud evaginates directly off of the duodenal anlage. (B) The ventral bud evaginates from the biliary bud and then swings around to the left, with gut rotation occurring simultaneously. (C) The main pancreatic duct of Wirsung and the minor accessory duct of Santorini are shown.
The two pancreatic buds fuse to form one pancreas at approximately 7 weeks’ gestation, although it appears that complete fusion of the two ducts to form the main pancreatic duct is delayed until the perinatal period.2 The endocrine component of the pancreas, the islets of Langerhans, starts to differentiate before evagination of the pancreatic buds from the wall of the foregut. The islets comprise 10% of the pancreas during early embryonic and fetal life, but this contribution decreases to less than 1% in adulthood. Pancreatic acini begin to form at 12 weeks gestation and begin to accumulate organelles and zymogen granules at this stage, but do not secrete appreciable amounts of enzyme until birth.1
The pancreas is convex and its midportion is reflected over the anterior surface of the upper lumbar vertebrae and aorta. Its lateral aspects falls posteriorly toward each kidney (Fig. 46-2). The arterial supply of the pancreas is from the celiac and SMA, which form the pancreaticoduodenal arcade. The pancreas also has anastomoses from the splenic artery.3
FIGURE 46-2 CT scan showing the cross-sectional anatomy of the pancreas. The pancreas (P) lies convexly across the lumbar spine with the tail of the pancreas next to the spleen (Sp) and the hilum of the left kidney. The head of the pancreas lies to the right of the spine near the hilum of the right kidney. L, Liver; S, stomach; C, colon. (From Maher MM, Hahn PF, Gervais DA, et al. Portable abdominal CT: Analysis of quality and clinical impact in more than 100 consecutive cases. AJR Am J Roentgenol 2004;183:663–70.)
Congenital Anomalies
Ectopic pancreatic rests are frequently encountered along foregut derivatives such as stomach, duodenum, jejunum, and colon, but are also infrequently encountered in the thorax and other sites.4–6 These lesions are found in approximately 2% of autopsy series and represent the most common anomaly of the gastric antrum. Moreover, they may cause gastric outlet obstruction.7 Their origin is unknown, but may be the result of aberrant epithelial–mesenchymal interactions leading to the transdifferentiation of embryonic epithelium into pancreatic epithelium. Several studies have implicated defects in hedgehog signaling and Notch signaling as the cause of ectopic pancreatic rests.8 Ectopic rests are typically asymptomatic and are encountered incidentally at laparotomy or during endoscopy. They can be identified as pancreatic tissue visually, because the surface has the same granular acinar appearance as the normal pancreas. These ectopic pancreatic rests usually do not become inflamed, possibly because they contain numerous small drainage ducts that usually do not obstruct; however, they can occasionally cause intestinal obstruction or bleeding. When encountered at laparotomy, ectopic rests should probably be excised, unless the excision would entail significant risk.
An annular pancreas is thought to result from faulty rotation of the ventral pancreatic bud in its course around the posterior aspect of the duodenal anlage. The duodenum is encircled and often obstructed by normal pancreatic tissue.9 Abnormal expression patterns of endodermal hedgehog may be responsible for the formation of annular and ectopic pancreas.8 Duodenal atresia and stenosis, intestinal malrotation, and trisomy 21 can often be found in combination with an annular pancreas.10 The clinical significance relates primarily to intrinsic duodenal obstruction, typically with bilious vomiting. Radiographic studies may reveal the classic finding of the ‘double-bubble’ sign. Management consists of bypass of the obstructing lesion with a duodenoduodenostomy or gastrojejunostomy, depending on the anatomy. Resection or division of the annular pancreas should not be performed due to the variable and complex ductal drainage system. Occasionally, patients with complex ductal anatomy may require reoperation for pancreatobiliary anomalies not apparent at the time of the initial surgery.11
Cystic fibrosis (CF) is an autosomal recessive condition, seen primarily in Caucasians which occurs in about 1 of 2,500 births.12 It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that encodes a protein expressed on the apical membrane of exocrine epithelial cells. CF leads to significant pancreatic insufficiency. The pancreatic secretions generally have a reduced amount of bicarbonate, a lower pH, and a lower overall fluid volume. The inspissated secretions lead to blockage of the ducts with dilatation. This may lead to acinar cell degeneration, acute and chronic pancreatitis, and pancreatic fibrosis. The result is impaired digestion of fats and proteins from loss of these digestive enzymes.13 See Chapter 32 for more information about CF.
Pancreatitis
Acute Pancreatitis
Acute pancreatitis is an acute inflammation of the pancreas, varying in severity from mild abdominal pain to fulminant necrotizing pancreatitis and death. It has an incidence between 3.6 and 13.2 cases per 100,000 children. If episodes of acute inflammation completely resolve and then recur, it is termed acute recurrent pancreatitis. It is thought that complete interval resolution of morphology and function occurs between episodes, unlike in cases of chronic pancreatitis.14
The causes of acute pancreatitis include trauma, biliary tract stone disease, choledochal cyst, ductal developmental anomalies, drugs, metabolic derangements, and infections. Most commonly, the cause is not apparent and termed idiopathic. As the pancreas is fixed against the lumbar spine, trauma to the upper abdomen can fracture the pancreas or injure the major duct at that point (Fig. 46-3). Biliary stone disease, increasing in frequency in children, may lead to pancreatitis from transient pancreatic duct obstruction. Endoscopic retrograde cholangiopancreatography (ERCP) is safe and effective in children and is the preferred method for stone retrieval.15 Drugs that are thought to induce pancreatitis include asparaginase and valproic acid.16 Systemic illnesses and metabolic conditions, such as CF, Reye syndrome, Kawasaki disease, hyperlipidemias, and hypercalcemia as well as viral infections (e.g., coxsackievirus and rotavirus) and generalized bacterial sepsis, can also cause pancreatitis.17
FIGURE 46-3 This abdominal contrast-enhanced CT shows a complete transection (arrow) of the body of the pancreas following a bicycle handlebar injury to the epigastrium. The patient underwent laparoscopic distal pancreatectomy and recovered uneventfully.
Pancreas divisum is an anomaly of the pancreas present in 10% of the population and is thought to result from failure of the dorsal duct to fuse with the ventral duct. In pancreas divisum, the majority of exocrine pancreatic secretions, including those from the entire body and tail, must drain through the small minor duct of Santorini. The resulting relative obstruction may cause recurring episodes of pancreatitis. Symptomatic patients should undergo sphincteroplasty of the minor papilla. Endoscopic stenting, with or without sphincterotomy, is the preferred treatment, with operation reserved for recurrent cases. Choledochal cysts produce pancreatitis by pancreatic duct compression or bile reflux resulting from a long common biliary-pancreatic duct within the head of the pancreas. Other rare ductal anomalies may result in obstruction and recurring bouts of pancreatitis.18
Though acute pancreatitis has many etiologies, they all appear to share a common pathway of nonphysiologic calcium signaling in the pancreas, followed by the premature activation of acinar proenzymes. These enzymes, especially trypsin, lead to acinar cell injury and cytokine release. The cytokines, along with vascular dissemination of activated enzymes, free radical formation, and release of vasoactive substances, such as kallikreins and histamine, together mediate extrapancreatic inflammation.19
Diagnostic criteria for acute pancreatitis include at least two of the following: acute abdominal pain (especially in the epigastric region), serum amylase or lipase more than three times the upper limit of normal, and imaging findings characteristic or compatible with acute pancreatitis.14 The abdomen is diffusely tender with signs of peritonitis, and distention occurs with a paucity of bowel sounds. In severe cases of necrotizing or hemorrhagic pancreatitis, hemorrhage may spread away from the pancreas along tissue planes, appearing as ecchymosis either in the flanks (Grey Turner’s sign) or at the umbilicus (Cullen’s sign) (Fig. 46-4). These ecchymoses generally take 1 to 2 days to develop.
FIGURE 46-4 Positive Cullen’s sign, with periumbilical ecchymosis, in a patient with hemorrhagic pancreatitis.
Elevated amylase levels are helpful in the diagnosis, although a normal serum amylase level does not exclude pancreatitis. Hyperamylasemia may also be caused by salivary inflammation/trauma, intestinal disease (such as perforation, ischemia, necrosis, or inflammation), and macroamylasemia. Lipase has been suggested as an alternative marker, but can be falsely elevated in pancreatic cancer, macrolipasemia, renal insufficiency, cholecystitis, esophagitis, intestinal perforation, and hypertriglyceridemia. Elevated lipase tends to be more sensitive in infants and toddlers, and may also help differentiate pancreatic from salivary trauma. The degree of enzyme elevation does not correlate with disease severity.19,20
Abdominal ultrasonography (US) is useful in the evaluation of the patient with pancreatitis, but has limited applications. It is well established in the evaluation of biliary stone disease as the etiology for pancreatitis, and can detect choledochal cysts and pancreatic pseudocysts as well. Advanced techniques such as contrast-enhanced ultrasound and ultrasound elastography have also been shown to be useful in the diagnosis of pancreatitis and its complications, but their availability is limited to a few experienced centers.21
Abdominal computed tomography (CT) provides much better resolution of the pancreas than ultrasound. Its primary role is in the detection of early and late complications, such as pancreatic necrosis, pseudocysts, and fluid collections, and should be reserved for patients with more severe pathology, or recurrent symptomatology with an equivocal ultrasound. If necessary, CT can be combined with interventional procedures to drain fluid collections.22
Magnetic resonance cholangiopancreatography (MRCP) is a newer, noninvasive technique for evaluating the biliary tree and pancreatic duct (Fig. 46-5). It is the initial imaging study of choice for the evaluation of pancreatic ductal anatomy in children with recurrent or unexplained pancreatitis. Studies comparing MRCP and ERCP show high concordance in diagnoses. Its disadvantages are that it does not allow for therapeutic intervention (though it may direct the type of intervention necessary), its poor spatial resolution limits the visualization of ducts in smaller children, and it usually requires anesthesia in the pediatric age group.21,23
FIGURE 46-5 (A) This secretin-enhanced MRCP was performed in a patient with pancreatic ductal stenosis. The white arrowhead shows stenosis of the main pancreatic duct at the neck along with diffuse ascites (white arrow). A fluid collection communicating with the duct is also seen (black arrowhead). (B) An ERCP in the same patient demonstrates the ductal stenosis (white arrowhead), distal duct dilation, and the communicating fluid collection (black arrowhead). (From Akisik MF, Sandrasegaran K, Aisen AA, et al. Dynamic secretin-enhanced MR cholangiopancreatography. Radiographics: A review publication of the Radiological Society of North America, Inc. 2006;26:665–77.)
The most frequent indication for ERCP in children is in the diagnosis or treatment of acute, recurrent, or chronic pancreatitis. A large single-institution retrospective study found a low rate of post-ERCP complications and a high therapeutic success rate.15 ERCP was shown to be particularly useful in the diagnosis of recurrent pancreatitis, though in only 60% of patients an organic etiology was found. Sphincter of Oddi manometry was particularly useful in establishing a diagnosis when no anatomic abnormalities were present.
The treatment for pancreatitis has remained unchanged for decades. The mainstays of therapy are pain control, intravenous fluid resuscitation, pancreatic rest, and monitoring for complications. Fluid resuscitation and maintenance should be guided towards a goal urine output of 2 mL/kg/h measured by an indwelling urinary catheter. Because of circulating cytokines, activated digestive enzymes, and other pro-inflammatory molecules, extracellular fluid losses can be enormous. Constant monitoring is necessary to avoid the development of severe hypovolemia. Patients with severe acute pancreatitis may require nasogastric decompression. Most patients receive histamine-2 (H2) receptor antagonists to reduce exposure of the duodenal secretin-producing cells to gastric acid, a potent stimulator of pancreatic secretion. This therapeutic regimen is logical but empirical, because no studies have shown improvement in outcomes with these interventions. The effectiveness of somatostatin in the treatment of pancreatitis is equivocal and probably serves more to mitigate complications of pancreatitis rather than to treat the disease itself. Further studies are needed to clearly define its role in both adults and children.24
Nutrition is critically important in patients with pancreatitis. The past decade has seen a paradigm shift in the nutritional management of patients with pancreatitis. Early nutrition, within the first 72 hours, is still recommended, however enteral nutrition (EN) has become the preferred method over total parenteral nutrition (TPN). Patients with mild to moderate cases of acute pancreatitis often resolve prior to requiring EN or TPN. More severe cases should be treated with EN via a nasojejunal tube, though studies have demonstrated tolerance of nasogastric feeding in severe pancreatitis as well. TPN use, especially early in the disease course during the peak inflammatory response, has been associated with increased length of stay and delayed advancement of diet. When EN is contraindicated, some advocate waiting as long as 5 days prior to starting TPN. Compared to TPN, EN has been shown to decrease length of stay, reduce the need for surgery, and reduce the risk of infection.25 When restarting a diet, conservatively determined by resolution of symptoms, there appears to be no difference between clear liquid or solid food as the initial meal.26 Unfortunately, these data come almost exclusively from the adult population as pediatric trials are lacking.17,27
Adequate analgesia is critical to minimizing the physiologic stress that develops from pain. While meperidine (Demerol) was once advocated because morphine was thought to cause spasm of the Sphincter of Oddi, no clinical trials have shown superiority of meperidine over other narcotic analgesics. Large doses of meperidine, however, are associated with the risk of seizure, euphoria, and drug interactions, suggesting other narcotics such as morphine and fentanyl may be safer alternatives. The diagnosis of pancreatitis must be certain prior to initiating treatment with high doses of narcotics as these may mask signs of serious nonpancreatic pathology, such as intestinal or gastric perforations.28
As pancreatitis progresses in severity, patients need to be monitored closely for signs of multisystem organ failure. Pleural effusions, pulmonary edema, and tense abdominal distention may lead to hypoxia requiring intubation and adult respiratory distress syndrome. Hypocalcemia, hypomagnesemia, anemia from hemorrhage, hyperglycemia, renal failure, and late sepsis can also be seen in these patients. Disagreement exists regarding the use of prophylactic antibiotics in severe cases of pancreatitis. The most recent adult data suggests a trend towards decreased mortality and infection with prophylactic antibiotics, but this study failed to reach statistical significance.29 Imipenem is the antibiotic therapy of choice when necessary.
Operative exploration is usually not necessary in acute pancreatitis. However, exploration is needed in patients with infected necrotic pancreatitis or a pancreatic abscess. Infected pancreatic necrosis increases mortality significantly. Diagnosis is typically by CT pancreatography, with confirmation of infection by fine needle aspiration, clinical deterioration, or positive cultures. The latest adult data suggests that infected necrosis or peripancreatic abscesses are best treated in a stepwise manner from least to most invasive. When feasible, percutaneous drainage should be followed by minimally invasive necrosectomy if the patient fails to improve. Delayed operative therapy demonstrates improved mortality compared to primary necrosectomy.30,31
Pancreatic pseudocyst is a complication of trauma or pancreatitis that forms after injury to the pancreatic ductal system. The extravasated pancreatic enzymes and digested tissue are contained by the formation of a cavity composed from a fibroblastic reaction and inflammation that lacks an epithelial lining. The acute pseudocyst has an irregular wall on CT scan, is tender, and usually develops shortly after an episode of acute pancreatitis or trauma (Fig. 46-6). Chronic pseudocysts are usually spherical with a thick wall, and are commonly seen in patients with chronic pancreatitis. The distinction is important because half of acute pseudocysts resolve without treatment, while chronic pseudocysts rarely spontaneously resolve. An acute pseudocyst matures and forms a thick fibrous wall in four to six weeks, allowing for drainage. Those smaller than 5 cm in diameter usually spontaneously regress. When compared to those in adults, pseudocysts in children tend to resolve more frequently with medical therapy alone.32 There are anecdotal reports of pseudocyst resolution in children after treatment with long-acting somatostatin analogs.33
FIGURE 46-6 CT scan of an acute pseudocyst in a patient after a severe motor vehicle accident. The wall (arrows) is irregular with nonloculated fluid inside.
The pancreatic pseudocysts that persist, or are symptomatic, require either a drainage procedure or excision. Endoscopic treatment, well established in adults, has been reported to be safe and efficacious in children as well.34
[/not-level-membership-for-pediatrics-category]
