Lesions of The Liver
Hepatic tumors in children are relatively rare. The most common malignant hepatic neoplasms are not primary tumors, but rather metastatic lesions such as Wilms tumor, lymphoma, and neuroblastoma.1 Primary liver tumors comprise between 1–4% of all solid tumors in children. Malignant hepatic tumors occur at a rate of about 1 to 1.5 per million children per year.1,2 However, ten primary liver masses occur with some frequency in the pediatric age group Five of these occur only in children: infantile hepatic hemangiomas, hepatoblastoma, mesenchymal hamartoma, rhabdomyosarcoma of the biliary tract, and undifferentiated embryonal sarcoma (Table 67-1). The age distribution for hepatic masses is distinctive, with hepatoblastoma and infantile hepatic hemangioma occurring most commonly in the first two years of life, and hepatocellular carcinoma and focal nodular hyperplasia occurring most commonly after age 5 years (Table 67-2).1
TABLE 67-1
Hepatic Tumors in Pediatric Patients, Birth to 2 Years (AFIP 1970–1999)
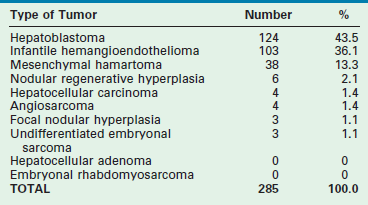
Reprinted from Stocker JT. Hepatic tumors in children. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 915.
TABLE 67-2
Hepatic Tumors in Pediatric Patients, 5 to 20 Years (AFIP 1970–1999)
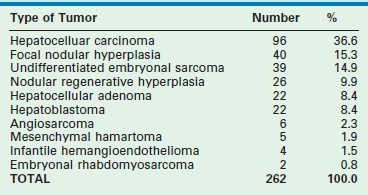
Reprinted from Stocker JT. Hepatic tumors in children. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2001.
Couinaud’s elegant description of the segmental anatomy of the liver has allowed hepatic operations to evolve to a level at which they can be performed with an acceptable morbidity and mortality (Fig. 67-1).3,4 The cumulative experience with hepatic resection and hepatic transplantation has allowed the development of techniques for both subsegmental and multisegmental resections of the liver in children. With the continued expansion of knowledge about these tumors, reasonable surgical and medical management plans can be devised.5
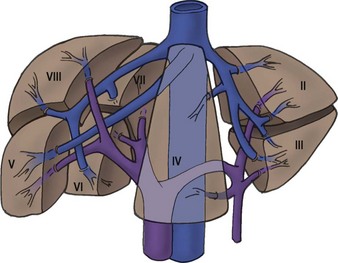
FIGURE 67-1 The segmental hepatic anatomy as defined by Couinaud. A comprehensive understanding of the hepatic segmental division is necessary for successful hepatic resection. (From Couinaud C. Surgical anatomy of the liver: Several new aspects. Chirurgie 1986;112:337–42; Couinaud C. The anatomy of the liver. Ann Ital Chir 1992;63:693–7.)
Benign Hepatic Tumors
Infantile Hepatic Hemangiomas
Incidence
Infantile hepatic hemangioma (IHH) is the most common benign solid hepatic tumor in children, comprising about 16% of all pediatric liver tumors.1 It also is the most common liver tumor in the first year of life. Almost all children with IHH are seen initially before age 6 months, and the majority are encountered in the first 2 months.6,7 Historically, a slight female predominance has been found, but this finding has not been uniformly seen.8
Clinical Presentation
IHHs can be either single lesions that can expand to a massive size or a multinodular infiltrative mass. Occasionally these lesions are asymptomatic and present as an abdominal mass or abdominal distention. Hepatic hemangiomas can also be found in association with congenital syndromes such as Osler–Weber–Rendu, Klippel–Trénaunay–Weber, and Ehlers–Danlos. IHH has also been reported in association with Beckwith–Wiedemann syndrome, diaphragmatic hernia, trisomy 21, transposition of the great arteries, and extranumerary digits.9,10
Infants with IHH can present with significant symptoms including hepatomegaly, high-output congestive heart failure, respiratory distress, and anemia. Occasionally these infants present with the Kasabach–Merritt syndrome that is characterized by acute thrombocytopenia, a microangiopathic hemolytic anemia, and a consumptive coagulopathy.11 This syndrome can be life threatening and requires aggressive supportive treatment, as well as treatment of the hemangioma itself. Fortunately, the Kasabach–Merritt syndrome occurs infrequently and is usually associated with IHH that have rapid growth to a diameter of 5 cm or more. No cases have been reported in association with smaller tumors.12
The Boston Children’s Hospital Vascular Anomalies Center has recently made a proposal to classify IHH as either focal, multifocal, or diffuse.13 Focal lesions tend to be asymptomatic, are rarely associated with cutaneous hemangiomas, and often are identified on prenatal ultrasound (US). Focal lesions can have high-flow shunts and can have an associated low grade anemia or thrombocytopenia. Multifocal lesions also can be asymptomatic. They are often found on visceral imaging that is performed when evaluating patients with multiple cutaneous hemangiomas. These extrahepatic hemangiomas can occur at multiple distant sites including the skin (45%), lung (10%), pancreas, lymph nodes, and bone.14,15 Some of these multifocal IHH lesions, however, are associated with high-output cardiac failure from either intralesional or portovenous shunting. The diffuse lesions present with near total replacement of the hepatic parenchyma with multiple hemangiomas which can result in massive hepatomegaly, abdominal compartment syndrome, or significant respiratory compromise. This lesion is often associated with severe hypothyroidism due to the overproduction of type III iodothronine deiodinase.16,17 The hypothyroidism can be severe enough to cause low-output cardiac failure or significant mental retardation. Interestingly, there has not been a patient reported with diffuse IHH who has developed high-output cardiac failure. Other symptoms that can occur in patients with IHH include jaundice, failure to thrive, respiratory difficulties, or poor feeding. Historically, as many as 50–60% of infants with IHH have symptoms of congestive heart failure that seems to be age related.18,19 Neonates with a focal hemangioma tend to present with high-output heart failure at birth, whereas infants with multifocal lesions tend to present with heart failure between the ages of 1 and 16 weeks.
In patients with IHH, the hepatic transaminase levels and occasionally the α-fetoprotein (AFP) level can be elevated. The cause of this AFP elevation is unclear. Moreover, an elevated AFP level in neonates must be interpreted with some caution. The AFP level is normally elevated in neonates and it does not decrease to adult levels until about 6 months of age.15 When there is a significant elevation of the AFP level, however, hepatoblastoma must be excluded by either imaging studies or biopsy of the lesion.20
A significant incidence of placental abnormalities has recently been reported in very low birth weight infants (<1500 g) who present with infantile hemangiomas.21 These researchers discovered a variety of anomalies that could potentially lead to the shedding of placental cells into the fetal circulation. If fetal hypoxic stress is also present, these placental cells could be exposed to an increased level of angiogenic factors that could then lead to increased endothelial cell or placental cell proliferation in the postnatal period with the subsequent development of an IHH.
Imaging
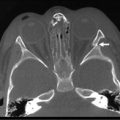
The ultrasound evaluation of IHH can be highly variable. Solitary lesions can have a heterogeneous echogenicity, and the Doppler spectral analysis can show a variety of flow patterns. Multifocal lesions, however, tend to be more uniform in their appearance and are seen as echolucent nodules associated with a high-flow vessel.22 On computed tomography (CT) with intravenous contrast, classically the lesions either enhance diffusely or show rim enhancement that is followed by gradual filling of the center of the lesion (Fig. 67-2).19,23,24 In one series, the CT enhancement pattern was correlated to the size of the lesions. Lesions less than 1 cm enhanced homogeneously in the arterial phase. Lesions greater than 2 cm demonstrated peripheral rim enhancement and those in between 1–2 cm showed a mixed enhancement pattern in the arterial phase (Fig. 67-3).25 Unfortunately, this ‘classic’ enhancement pattern is not always present in IHH which can make the radiologic diagnosis difficult.
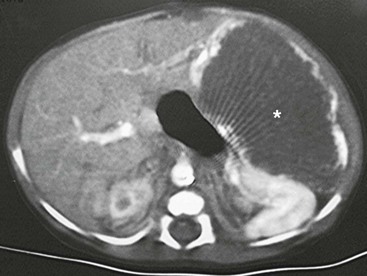
FIGURE 67-2 CT scan after intravenous administration of a contrast agent shows a large hemangioendothelioma (asterisk) with peripheral enhancement in the left lateral segment.
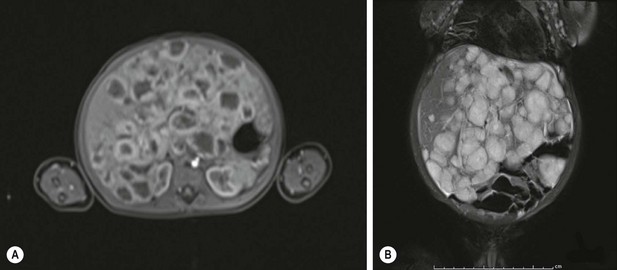
FIGURE 67-3 Infantile hepatic hemangioma: diffuse form. (A) An MRI in the arterial phase showing rim enhancement of the hemangiomas after injection with gadoxetate disodium (Eovist). (B) The T2-weighted image showing the same liver that is filled with hemangiomas.
Magnetic resonance imaging (MRI) is emerging as the single most useful modality to show both the location of the hemangioma and its flow pattern and structure.26,27 The addition of intravenous gadolinium and a gradient-recalled-echo sequence to the MRI enhances its utility.
Focal lesions on MRI are well defined and hypointense to the liver on T1 images and hyperintense on T2-weighted images. Gadolinium demonstrates peripheral enhancement with variable central enhancement that is related to the amount of hemorrhage or thrombosis that is present in the lesion. Multifocal and diffuse lesions tend to show similar MRI findings except that there are multiple lesions present. Recently, however, there has been a case report of a hypervascular hepatoblastoma that had an MRI pattern that was identical to a hemangioendothelioma.28 Therefore, again, radiology alone cannot reliably distinguish between benign and malignant lesions.
IHH also has been seen in conjunction with both focal nodular hyperplasia and hepatic mesenchymal hamartoma.29,30 These associations are important to remember during the radiologic evaluation of a child with multiple hepatic masses.
Histology
Microscopically, the histologic pattern has been divided into type 1 and type 2 lesions. A type 1 lesion consists of a single layer or, occasionally, several layers of flat endothelial cells on a supporting fibrous stroma.31 In a type 2 lesion (20% of cases), the endothelial cells are pleomorphic, larger, and more hyperchromatic than those seen in type 1 tumors. Also, there are poorly formed vascular spaces that often show tufting or branching. It is thought that the histologic picture of the type 2 lesion is more characteristic of a rapidly proliferating process. The histological differentiation between a type 2 lesion and an angiosarcoma can be difficult.14,32 The addition of a histologic stain for the marker Glut 1 can be helpful in making the diagnosis of IHH.9 Focal lesions do not stain positive for Glut 1, while multifocal lesions will be positive for this marker.33,34 Well-preserved bile ducts can frequently be seen near the periphery of a type 1 lesion, whereas bile ducts are absent in a type 2 lesion.
Treatment
The life cycle of infantile cutaneous hemangiomas is divided into three stages. The first starts usually after birth and is a characterized by a rapid growth phase that lasts about 9 to 12 months. In the second phase, the hemangioma begins the process of involution. This process can take anywhere between 5 and 7 years. The last phase is the involuted phase where the hemangioma is permanently replaced by fibrofatty tissue.13 Hepatic hemangiomas also tend to follow a similar life cycle. The therapy for IHH depends on the severity of the presenting symptoms and the size of the mass(es).
Asymptomatic lesions are monitored, and no specific therapy is instituted until symptoms occur.11,35 As part of the evaluation of the asymptomatic patient with multifocal hemangiomas, imaging studies of the brain and chest should also be performed to make sure there are no associated intracranial or pulmonary lesions. In addition, all patients with multifocal lesions should be screened for hypothyroidism. Focal IHH are felt to follow a unique clinical course that is similar to the clinical course of a cutaneous rapidly involuting congenital hemangioma where involution is seen by 12 to 14 months of age.36
Infants who present with congestive heart failure, coagulopathy, or respiratory compromise require intervention. Mortality rates in these patients have been reported to range from 17–35%, with some reports of death in as many as 90% of those who are severely symptomatic.37–39 Risk factors for death include congestive heart failure, jaundice, multiple tumor nodules, and the histologic absence of cavernous differentiation.
If there is hemodynamic instability, respiratory compromise, or coagulopathy, then therapy directed toward the hemangioma is needed. The usual initial treatment for a symptomatic lesion is prednisone or prednisolone at a dose of 2–3 mg/kg/day. The symptomatic response rate to corticosteroids is reported to be about 45%.40,41 If steroids are ineffective, then some IHHs have shown a response to vincristine at a weekly dose of 1–2 mg/m2 for two weeks.12 Labreze et al. have reported on the use of propranolol (2 mg/kg/day) for the treatment of severe infantile capillary hemangiomas with excellent results.42 Marsciani et al. have successfully used the combination of propranolol (1 mg/kg/day followed by 2 mg/kg/day) and steroids in the treatment of a 2-month-old child with diffuse IHH.43 They noted a dramatic decrease in liver size only one week after the addition of the propranolol. Another agent that has been reported to be possibly effective in IHH is curcumin, which is derived from the spice tumeric. Curcumin has antiangiogenic activity and has been tested with some success in a variety of tumors.44 Hassell et al. described its apparent successful use in a 6 month old with a multifocal IHH.45
Resection should be considered if the hemangioma is confined to a single lobe. In this situation, a survival rate of 92% has been reported, even if the clinical situation is complicated by congestive heart failure.32,46 However, some authors feel that resection is rarely needed if both pharmacotherapy and embolization are aggressively pursued.13
Embolization is an important part of the therapy of symptomatic hemangiomas.47,48 In order to decrease the chance of the IHH recurring, it is important that both the arterial and portal vascular supply to the IHH are occluded as distally as possible.11,19 After successful embolization, a rapid improvement in the clinical course usually occurs within five days. However, there may not be any associated change in the size of the hemangioma itself.47,48 Embolization should be considered early in the treatment course in those infants who are in significant cardiac failure. Unfortunately, embolization has not been helpful in infants with diffuse disease because of the absence of significant shunting.
Finally, in infants in whom other modes of treatment have failed, liver transplantation has been used successfully for severe congestive heart failure or unremitting coagulopathy (or both).49 The infants that will most commonly need to be considered for a liver transplant will be those who have the diffuse IHH subtype. Infants who experience rapid clinical deterioration in the postnatal period have a high mortality after liver transplantation so careful consideration needs to be given to their candidacy.50
The Vascular Anomalies Center at Boston Children’s Hospital has suggested a treatment algorithm for IHH.13 All asymptomatic lesions should be monitored with ultrasound until resolution. Symptomatic lesions should be treated with steroids. If there is no response, then the hemangioma should be embolized. Infants with diffuse disease should have a TSH checked and be started on steroids. If abdominal compartment syndrome is present, then the infant needs to be evaluated for a liver transplant in case there is no response to steroids.
Malignant transformation of an IHH to an angiosarcoma has been reported in older children.18,51 For this reason, patients who are asymptomatic or who become asymptomatic after therapy must be monitored for complete anatomic resolution of their hemangioma. Resection of any residual lesion should be strongly considered.
Mesenchymal Hamartoma
Incidence
Mesenchymal hamartoma is reported to be the third most common hepatic tumor and the second most common benign tumor in children.1 Of all benign hepatic lesions, mesenchymal hamartomas account for between 18–29% of these tumors.52,53
Epidemiology
The reasons for the development of a mesenchymal hamartoma are unclear. One theory is that it results from abnormal development of the primitive mesenchyme, which appears to occur at the level of the hepatic ductal plate, causing abnormal bile ducts.54 This concept is supported by the histologic finding of a combination of cystic, anaplastic, and proliferating bile ducts, as well as the presence of multiple portal vein branches, within the tumor. It is postulated that the tumor then develops a cystic component as a result of obstruction and dilatation of lymphatics or from occluded bile ducts (or both). The tumor enlarges during infancy as the cystic areas increase in size. Most of the proliferative growth appears to occur before or just after birth because no observable mesenchymal mitotic activity is visible on histologic sections of the tumor.55
A second theory is that the lesions are reactive rather than developmental.56 It is hypothesized that an abnormal blood supply to an otherwise normal hepatic parenchyma causes ischemic necrosis, leading to reactive cystic changes within that portion of the liver. This theory is supported by the findings that hamartomas often have a necrotic center, are often attached to the liver by only a thin pedicle, and rarely are found centrally in the liver.
The third theory suggests that a mesenchymal hamartoma is a proliferative lesion. This theory is supported by several findings. Increased fibroblast growth factor-2 (FGF-2) staining has been noted in the proliferating hepatic stellate cells adjacent to the mesenchymal hamartoma.57 Both the stellate cells in the liver and the spindle cells in the mesenchymal hamartoma tissue strongly express molecules of the FGF-receptor family. It is speculated that a local increase of FGF-2 secretion could stimulate the growth of the spindle cells to form the mesenchymal hamartoma. FGF-2 also is a potent angiogenic factor that could contribute to the intense vascularization seen within some of these lesions. A recent study has identified that the spindle cells that make up the predominant cell line in a mesenchymal hamartoma appear to be derived from hepatic stellate cells.58 A possible genetic cause for these abnormalities has been proposed as well. Cytogenetic studies of these tumors have documented several abnormalities including translocation/deletion of chromosome19q13 or the loss of heterozygosity due to multiple monosomies. The resulting dysregulated imprinting may be an additional cause for a mesenchymal hamartoma.59–63
Clinical Presentation
The widespread use of prenatal imaging has led to the detection of hepatic masses before birth.15 Cases of hepatic mesenchymal hamartoma that were diagnosed prenatally have been described.64,65 One of the unique characteristics of a prenatal mesenchymal hamartoma is that it can be solid as well as cystic. Unfortunately, the prognosis for these antenatally diagnosed neonates is often poor with a reported mortality rate of 29%.66 This mortality is related to: (1) the development of congestive heart failure from compression of the inferior vena cava or the umbilical vein by the tumor mass; or (2) the development of hydrops that can occur from fluid losses into cysts or decreased albumin production by the liver. Thus, it may be best to deliver these fetuses before fetal hydrops develops.65 Another series reported the use of repeated intrauterine aspiration of the cysts until the infants could be delivered and definitive treatment initiated.67 While this therapy is appealing, there currently is insufficient evidence to recommend its widespread use.66
In the neonate, these lesions can have a varying presentation. High-output cardiac failure, pulmonary hypertension, and disseminated intravascular coagulopathy have been reported in neonates with highly vascular mesenchymal hamartomas.68–70 Respiratory distress secondary to a large hepatic mass impinging on the diaphragm has also been described.65 Unfortunately, the prognosis for infants with a perinatal diagnosis is also poor with a reported mortality of 35%.66
The presentation of a mesenchymal hamartoma in the older child is usually that of progressive abdominal distention or an abdominal mass (or both). There is a significant right-sided predilection for these masses, and they tend to be somewhat more common in males.1 Occasionally, the associated symptoms of nausea and vomiting can occur that are secondary to the compression of the stomach and intestine by the expanding mass.
On physical examination, abdominal distention or a palpable abdominal mass is most common. The mass tends to be nontender and fixed. Laboratory studies almost always are normal, including liver function studies. The AFP level can be moderately elevated and it returns to normal after resection. However, it is important to remember that patients have been treated with chemotherapy for hepatoblastoma until a tumor biopsy was performed and a mesenchymal hamartoma was subsequently diagnosed.71,72
Imaging
CT, ultrasound, and MRI have all been used for diagnosis. On CT and ultrasound, a multiseptated, multicystic, anechoic mass is usually located either in the periphery or scattered throughout the liver.73,74 Occasionally the mass is pedunculated. Calcification within the tumor is unusual, but has been reported so the presence of calcifications within a tumor does not exclude a mesenchymal hamartoma.75,76 MR angiography has proven useful both in the diagnosis and planning of resection.77,78 The finding of a small round hyperechoic parietal nodule on ultrasound is usually highly sensitive for making the diagnosis.79
Histology
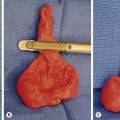
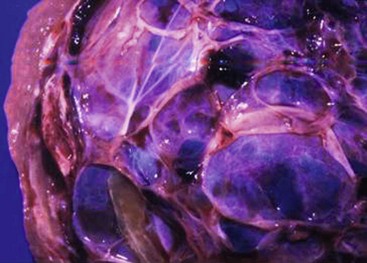
Mesenchymal hamartomas typically are large, well-circumscribed tumors that measure at least 8–10 cm in diameter (Fig. 67-4). Seventy-five per cent of these tumors occur in the right lobe of the liver, and only 3% are seen in both lobes of the liver. On cut section, multiple cysts can measure from a few millimeters to 15 cm in diameter. These cysts are filled with either serous or viscous fluid separated by loose fibrous and myxoid tissue (Fig. 67-5). The surrounding tissue is yellow-tan to brown and is loose to moderately dense.
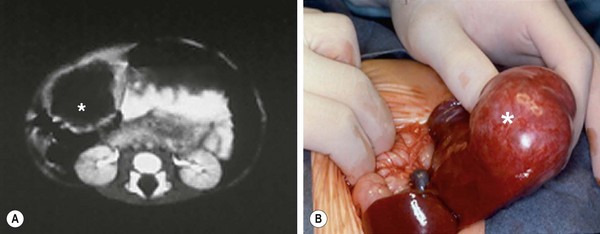
FIGURE 67-4 This young child presented with a palpable right upper abdominal mass. (A) CT scan shows an anechoic mass (asterisk) in the periphery of the liver. (B) At operation, the mass (asterisk) was found to be pedunculated and emanating from the right lobe of the liver. This hamartoma was easily removed.
Microscopically, the tissue consists of a mixture of bile ducts, liver cell cysts, and mesenchyme. The cysts may be dilated bile ducts, dilated lymphatics, or amorphous cysts surrounded by mesenchyme. In older patients, the cysts may be lined with cuboidal epithelium (Fig. 67-6). Elongated or tortuous bile ducts surrounded by connective tissue are unevenly distributed throughout the mesenchyme. Typically the hepatocytes appear normal, and they are not a predominant part of the pathologic process. The bile ducts in the periphery of the lesion seem to be undergoing active proliferation.1
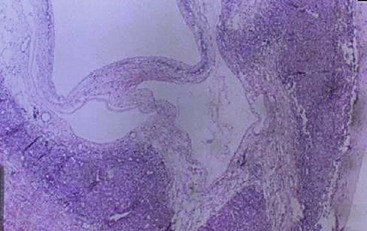
FIGURE 67-6 Light microscopy of mesenchymal hamartoma showing a cyst lined with cuboidal epithelium.
Despite the fact that the majority of these tumors are localized, there have been reports of these tumors being multifocal.63 This may account for the occasional recurrences that are seen after resection of the primary tumor.
Treatment
Various management strategies have been used for these lesions. Because they are sometimes encapsulated, enucleation may be possible. Very large, bilobar tumors that are not amenable to resection can be marsupialized into the peritoneal cavity, but recurrence after marsupialization can occur. If marsupialization is performed or the resection is incomplete, careful, long-term follow-up is important because of the risk of recurrence or the development of undifferentiated embryonal sarcoma.80 Complete excision of the lesion with a margin of normal liver is curative (including the use of liver transplantation for large, bilobar lesions) and is the recommended therapy.81,82 For massive lesions with multiple cysts, one group reported the use of ultrasound-guided, intraoperative aspiration of the cysts.83 This technique substantially reduced the size of the mass and made the resection much easier. Spontaneous involution of these lesions has been reported, but is unusual.84
There is now strong evidence that an undifferentiated embryonal sarcoma of the liver can develop within a preexisting mesenchymal hamartoma.63,85–88 This association has occurred both synchronously and metachronously. The evidence for a direct link between a mesenchymal hamartoma and an undifferentiated embryonal hepatic sarcoma comes from the simultaneous finding of both tumors arising within the same mass. Moreover, aneuploidy and similar chromosomal abnormalities involving chromosome 19q13 have been reported in both a hepatic mesenchymal hamartoma and an undifferentiated embryonal sarcoma.60,61,89,90
Focal Nodular Hyperplasia
Incidence
Focal nodular hyperplasia (FNH) accounts for about 10% of the hepatic tumors in children.1 The reported age range is between 7 months and 16 years, with a mean of 7 years, and there is a slight female predominance.91 The majority of these tumors are discovered incidentally.91,92 The most common symptom is abdominal pain, but some patients describe decreased appetite, an abdominal mass, weight loss, or a combination. Hepatomegaly is a common finding, and liver function abnormalities are often present.
FNH has been seen in association with a variety of different conditions and situations, including previous liver trauma, other liver tumors, hemochromatosis, Klinefelter syndrome, the use of itraconazole, after a successful Kasai procedure, after liver transplantation, and with cigarette smoking.93–100
The etiology of FNH is not certain, but the evidence suggests that it may be a congenital vascular abnormality. These lesions have a single feeding artery and there is an absence of bile ducts or veins in the lesion. The large artery causes a hyperperfused area of the parenchyma with subsequent growth of the liver tissue around the artery.101 In addition, FNH has been associated with other vascular lesions such as hemangiomas, arteriovenous malformations, and hereditary hemorrhagic telangiectasia.102,103 Further evidence that FNH is a reactive lesion secondary to vascular anomalies comes from a study in which an increase in the angiopoietin ratio (ANGPT1/ANGPT2) was seen.104 The ANGPT1 and ANGPT2 genes are necessary for normal vascular development. In FNH, an overexpression of the ANGPT1 gene and an absence of the antagonistic ANGPT2 gene lead to uncontrolled and disorganized vascular development. Although it is not clear that this is the exact pathogenesis of FNH, it certainly suggests that this genetic imbalance may play a causative role. FNH can also develop from an acquired abnormality of the hepatic vasculature. Thrombosis of either a hepatic artery or portal venous branch can initially cause ischemia of a portion of the liver followed by recannulation and reperfusion, which leads to hepatocyte proliferation.105
Controversy exists about the relation between oral contraceptive use and the development of FNH. In a case–control study, it was noted that neither menstrual nor reproductive factors correlated with FNH risk. However, oral contraceptive use was a significant risk factor in the development of FNH.99 As the use of oral contraceptives also appears to be associated with hepatocellular adenomas, a history of oral contraceptive use does not help in distinguishing between these two entities.99
In children, an association has been noted between the congenital absence of the portal vein (Abernathy syndrome) and FNH.106–108 In addition, these patients have an increased incidence of other solid tumors such as hepatoblastoma, hepatocellular carcinoma, and hepatocellular adenoma.109,110 FNH also has been seen, albeit less frequently than hepatocellular adenoma, in patients with glycogen storage disease (GSD) type 1.108
Over the last six years there has been an increasing awareness of the occurrence of FNH in oncologic patients who have completed their therapy. The reported incidence of FNH in the general pediatric population is 0.02% but is 0.45% in the postoncology treatment population.111 As opposed to nononcologic patients where the FNH is usually solitary, post-treatment oncologic patients frequently have multiple FNHs. The cause of these post oncologic FNHs is not known, but is felt to be secondary to alterations in liver perfusion that occur as the result of the chemotherapy or radiation that these patients receive.105,112 The development of hepatic mass(es) in an oncology patient leads to a diagnostic dilemma between an FNH or a recurrent tumor. One of the higher risk groups for developing an FNH after therapy are patients who were treated for neruoblastoma.111–116 There also seems to be an increase in the development of FNHs in children who have received a hematopoietic stem cell transplant.117–119 Therefore, a liver lesion that occurs after treatment of a previous cancer does not always mean that there has been a recurrence of the primary tumor. A biopsy of the lesion should always be done before the initiation of further therapy.120
Imaging
The diagnosis of FNH often requires the use of multiple different imaging modalities. On CT, the classic findings are early enhancement of the lesion and the presence of a central scar (Fig. 67-7).121 Unfortunately, this pathognomonic association is not often seen.122,123 Additional imaging modalities include single-photon emission radionuclide scans with either radiolabeled sulfur colloid or hepatobiliary iminodiacetic acid (HIDA) imaging. These studies usually demonstrate hypervascularization, increased tumor tracer uptake, and a central cold area.124–126 Technetium-99m sulfur colloid scanning can also be useful in distinguishing between FNH and a hepatic adenoma. FNH lesions take up the tracer because the Kupffer cells in the FNH take up the colloid but hepatic adenomas do not take up tracer because they lack Kupffer cells.127 MRI also has been useful when coupled with either gadolinium enhancement or the use of liver-specific contrast agents such as mangafodipir trisodium or iron oxide. During the arterial phase of dynamic contrast enhancement, an FNH will demonstrate distinct hypervascularity.128 These contrast agents can also help diagnose an FNH by allowing better identification of the central scar.129,130
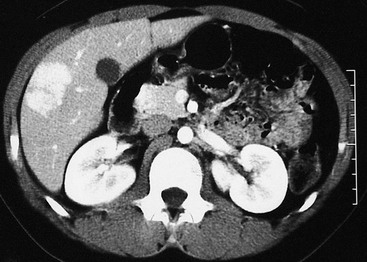
Histology
FNH classically is characterized by nodular architecture, a central or eccentric scar containing malformed vessels that resemble an arteriovenous malformation, and a variable amount of bile duct proliferation (Figs 67-8 and 67-9).131,132 FNHs always occur in the setting of a noncirrhotic liver.
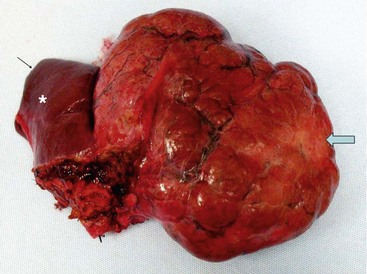
FIGURE 67-8 A large focal nodular hyperplasia lesion was resected. External scarring (arrow) is seen in the lesion. An asterisk marks normal liver.
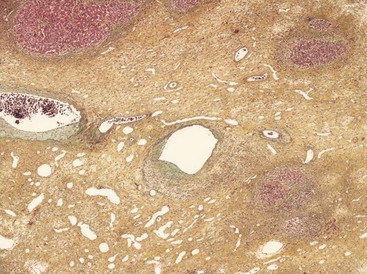
FIGURE 67-9 Light microscopy of a focal nodular hyperplasia showing a central scar containing abnormal blood vessels.
Histologically, the classic form of FNH with a central scar accounts for about 80% of the lesions. Twenty per cent are nonclassic where the FNH lacks either the nodular architecture or the presence of the malformed blood vessels. These nonclassic lesions are subdivided into three histologic categories: the telangiectatic form, the mixed hyperplastic form, and the adenomatous form.133 These nonclassic categories always lack a macroscopic scar and all three forms can be difficult to distinguish from and may be related to a hepatocellular adenoma.134 Immunohistochemical staining for cytokeratins 7 and 19 along with staining for neuronal cell adhesion molecule (CD56) has shown to be very helpful in distinguishing between FNH and hepatocellular adenoma.134 Hepatocellular adenomas have been reported to be present in association with FNH in about 4% of the cases.131
Recently, there has been a case report of what appears to be malignant transformation of an FNH to a fibrolamellar hepatocellular carcinoma.135 Also, FNH has been reported to occur in association with a well-differentiated fibrolamellar hepatocellular carcinoma.135,136 This observation is important to remember in patients who have multiple hepatic nodules.137,138
Treatment
The treatment of FNH depends on the clinical situation. If the diagnosis is certain, and the patient is asymptomatic, the consensus is that these patients can be followed with serial ultrasound to ensure that no progression of the lesion occurs.91,122 Percutaneous biopsy can be helpful in the diagnosis.139 However, if the patient is symptomatic, if the lesion is greater than 5 cm, if progression of the mass is seen, or if the diagnosis is unclear, then a biopsy or a resection of the lesion is recommended.140–143 Due to the association of FNH with hepatocellular carcinoma, patients who are expectantly managed must have serial evaluations to ensure that no progression occurs. Moreover, they need to be monitored for the development of other hepatic lesions.
Several reports have noted a 40–50% regression rate in cases of FNH that have been monitored.144,145 In the pediatric population, the true incidence of regression is not known due to the lack of long-term follow-up studies; however, it may be much lower than in the adult population.143 Regression of FNH is more likely if the use of oral contraceptives ceases.146,147
In two symptomatic patients in whom the FNH was in an area that was thought to be difficult for resection, arterial embolization either with Lipiodol and absorbable gelatin foam (Gelfoam) or iodized oil and polyvinyl alcohol resulted in a significant regression in the size of the mass.148–150
Hepatocellular Adenoma
Incidence
Hepatocellular adenoma is a very rare hepatic tumor in children, comprising only about 4% of all solid liver tumors.1 It is most commonly seen in women in their 20s and is associated with the use of oral contraceptives.
Imaging
Hepatic adenomas are solitary lesions in most cases, but occasionally two to three adenomas can be seen in one patient.149,151,152 On ultrasound, these lesions can have a variable appearance, depending on the tumor composition. They can have a hyperechoic, hypoechoic, or a mixed echoic pattern depending on whether it is a simple adenoma, an adenoma with fatty metamorphosis, or an adenoma with hemorrhagic necrosis.149
On CT, the adenoma can either be isoattenuating relative to the normal liver or hyperattenuating (due to the presence of fat). They are usually sharply marginated and nonlobular, but can be encapsulated or calcified in some patients.153 Hyperattenuated areas often correspond to areas of recent hemorrhage. On CT scan with intravenous contrast, a hypodense discrete lesion will show either arterial-phase enhancement or peripheral enhancement secondary to large subcapsular feeding vessels.154
On MRI, these lesions are either hypo- to hyperintense on T1-weighted images with uniform signal loss on out of phase T1-weighted views. On T2 images, the lesions are isointense to slightly hyperintense and gadolinium enhancement is maximal during the arterial phase with rapid fading.154 The finding of central hemorrhage or necrosis on CT scan helps differentiate hepatocellular adenoma from FNH.
Associated Conditions
Hepatocellular adenomas were extremely rare prior to 1960, which corresponds to the year in which oral contraceptives were first introduced.155 In women who have never used oral contraceptives, the annual incidence of hepatic adenoma is estimated to be about one per million. The duration of oral contraceptive use is directly related to the risk of developing a hepatic adenoma. The use of contraceptives for 5 to 7 years carries a fivefold increased risk, and use for 9 or more years has a 25-fold increased risk.156–158
Hepatocellular adenomas also have been associated with galactosemia, hypothyroidism, polycythemia, diabetes, Fanconi anemia, polycystic ovary syndrome, and the use of anabolic steroids.159–162
Hepatocellular adenomas are a significant complication in patients with type 1A GSD from their teenage years into adulthood.163,164 The estimated prevalence of adenomas in these patients is close to 50%.165,166 The pathogenesis of adenoma development is poorly understood in this group, but may be related in part to the tightness of the metabolic control.167 These adenomas are often multiple rather than solitary lesions. Unfortunately, in this patient population, hepatocellular carcinoma can occur in association with hepatocellular adenomas. The youngest reported patient with GSD was 6 years old at the time of the diagnosis of hepatocellular carcinoma.168 In several series, hepatocellular carcinoma has been found to develop in up to 18% of patients with a hepatocellular adenoma.169–173 Direct evidence for malignant transformation of a hepatocellular adenoma into a carcinoma has been confirmed with the reporting of a hepatocellular carcinoma within a hepatic adenoma in patients with GSD.174 Abnormalities in chromosome 6 have been also been identified in type 1A GSD adenomas and similar chromosome 6 alterations have been identified in hepatocellular carcinomas, suggesting a possible genetic link between these two diagnoses.175
Adenomatosis (the occurrence of more than ten simultaneous adenomas) is a rare disorder, with 38 cases reported in the literature through 2000.176 There is a massive form, characterized by multiple nodules measuring between 2–10 cm, and the multifocal form, in which most lesions are smaller than 1 cm, with only a few larger than 4 cm.177 Oral contraceptive use has been seen in about half of the female patients. Interestingly, diabetes and hepatic steatosis has been noted in these patients, but it is not clear if there is a causative relation.177,178 Adenomatosis has also been reported in patients seven to nine years after a Fontan procedure.179
Histology
Hepatocellular adenomas histologically consist of large plates or cords of cells that resemble normal hepatocytes. These plates are separated by dilated vascular sinusoids, which are equivalent to thin-walled capillaries perfused by arterial pressure. Adenomas do not have a portal venous supply and are fed solely by peripheral arterial vessels that account for the hypervascular nature of these lesions. Kupffer cells are found in reduced numbers and have little or no function. The absence of bile ducts serves as a key histologic feature that helps distinguish the hepatocellular adenoma from FNH. Lipid accumulation is responsible for the characteristic yellow appearance on the cut surface.1
The exact reason for their development is unclear. Two reports have cited the mutations of the Wnt/β-catenin pathway in patients with hepatocellular adenoma.180,181 This pathway mutation has been identified in many hepatocellular neoplasms, although its direct contribution to carcinogenesis is not completely understood. A second mutation has been found in the HNF1A gene that leads to the downregulation of hepatocyte nuclear factor-1α. This downregulation has been linked to the development of hepatic steatosis and hepatic adenomas.182 The significance of these findings in hepatocellular adenoma is also unknown.
Treatment
The treatment approach for these lesions depends on a variety of factors. In patients who are receiving oral contraceptives or androgenic steroid therapy, the first step should be withdrawal of these medications. Multiple case reports mention regression of the adenomas after withdrawal of these compounds. However, in other multiple reports, withdrawal of these agents has resulted in persistence of the adenoma.183–185 If discontinuation is not effective, then the adenoma should be resected. This removes the potential for future hemorrhage or malignant degeneration (10% of lesions).186 If the adenoma is larger than 5 cm or if the diagnosis of the hepatic lesion is uncertain, then excision is recommended.187,188
For patients with ruptured hepatocellular adenomas, the current suggested therapy in the hemodynamically stable patient is nonoperative monitoring and hemodynamic support. Once the hemorrhage has resolved and the patient has recovered, elective resection can be performed. In patients who continue to bleed actively, hepatic arterial embolization should be performed. This not only stops the hemorrhage but also can decrease the size of the adenoma.189 After resolution of the hemorrhage, resection is indicated. This management plan results in a decrease in size of the lesion and allows a more limited hepatic resection under controlled conditions.190,191
Malignant Hepatic Tumors
Hepatoblastoma
Most hepatoblastomas develop before age 3 years, with a median age of about 18 months.192 About 4% are present at the time of birth; 69% are present by the end of two years, and 90% develop by the end of 5 years. Only 3% of cases are noted in children older than 15 years.193 A definite male predominance (1.7 : 1) is seen.5
Epidemiology
Hepatoblastomas are associated with a variety of clinical conditions, syndromes, and malformations (Box 67-1). Beckwith–Wiedemann syndrome is associated most commonly with Wilms tumor, but other tumors such as hepatoblastomas, gonadoblastomas, and adrenal carcinomas are also seen.194,195 The association with hepatoblastoma is so strong that patients with Beckwith–Wiedemann syndrome must be monitored with serial AFP levels every three months until the age of 4 years and an abdominal ultrasound every three months until they reach age 8 years.195–197 Screening studies for hepatoblastoma are also recommended in patients with familial adenomatous polyposis. These patients should be screened for the APC tumor/suppressor gene. If this gene is present, then these patients are at increased risk for developing hepatoblastoma.198,199
Another interesting association exists between hepatoblastoma and extreme prematurity (<1000 g). In the Japanese Children’s Cancer Registry (JCCR), it was noted that hepatoblastomas accounted for 58% of the malignancies diagnosed in extremely low birth weight children.200 In recent epidemiologic studies, several factors were associated with an increased risk for the development of neonatal hepatoblastoma, including birth weight less than 1000 g, maternal age younger than 20 years, use of infertility treatment, maternal smoking, and a higher pre-pregnancy body mass index (BMI of 25–29).201,202 The time from birth to onset of hepatoblastoma in this population ranges from six months to six years.203 Unfortunately, the tumors that occurred in this group grew rapidly and had an unfavorable biologic behavior.204 Although the etiology for the predilection of hepatoblastomas to develop in very low birth weight infants is not known, oxygen therapy, furosemide use, and failure of the infant to grow appropriately were noted to be risk factors.204 The highest correlation was with the duration of oxygen therapy. The risk of hepatoblastoma increased by 20% if oxygen therapy was continued for 30 days, and the risk increased by 100% in children who were treated with oxygen for four months.
Cysteine has also been implicated as a contributing factor. Cysteine is an amino acid that is necessary for the production of glutathione and taurine, both of which are intracellular antioxidants. In premature infant livers, there appears to be impaired production of cysteine.205 Due to the complexities in treating ‘micropremies’ through their first few months, multiple other interventions could influence the development of a hepatoblastoma.
Histology
Hepatoblastomas tend to be unifocal lesions in most cases. Fifty per cent are isolated to the right lobe, 15% are in the left lobe, and 27% are centrally located to involve both lobes (Fig. 67-10).
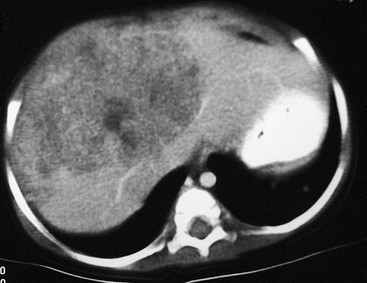
FIGURE 67-10 CT scan after intravenous administration of a contrast agent shows a large right lobe hepatoblastoma extending into the left lateral segment.
Histologically, the tumor can be divided into six different subtypes (Figs 67-11 and 67-12) based on light microscopy (Box 67-2). A correlation between clinical outcome and the histologic subtypes has been suggested. The pure fetal subtype appears to be associated with the better prognosis, whereas the small cell undifferentiated subtype appears to have a very poor prognosis.206–209 In several other studies, chemotherapy was initiated before surgical intervention and likely altered the accuracy of histologic definition of the resected tumor, making it difficult to correlate histology and outcome.210
Biology and Cytogenetics
Thrombocytosis is common in patients with hepatoblastoma. This may be related to increased thrombopoietin levels, which have been reported in hepatoblastoma cell extracts.210 Elevated interleukin-1b levels also have been noted in hepatoblastoma cell lines.211 This results in an increased production of interleukin-6, which is known to stimulate thrombopoiesis and thrombocytosis.212
Chromosomal abnormalities have been documented in patients with hepatoblastoma.213 The most common defects have been trisomy of chromosomes 20, 2, or 8, or a combination of these. Trisomy 18 also has been found.214 However, to date, no correlation has been noted between these cytogenetic abnormalities and either clinical outcome or tumor biology.
An association between hepatoblastoma and the APC gene was noted in patients with familial adenomatous polyposis and Gardner syndrome.212,215 A recent study found an association between the activation of the Wnt/β-catenin signaling pathway and the development of carcinogenesis in hepatoblastoma and hepatocellular carcinoma.216
Imaging
The first diagnostic test is usually an ultrasound. This usually differentiates between a renal and hepatic mass. An abdominal CT scan is then usually performed. In half of patients, calcification is noted within the mass.217 Spiral CT with an intravenous bolus of a contrast agent not only is helpful in the diagnosis, but also is useful in the staging of the tumor and in determining its resectability. With three-dimensional reconstruction, the location of the mass with respect to the vena cava, the hepatic veins, and the portal venous system can often be precisely delineated (Fig. 67-13). MRI is becoming an increasingly helpful modality for determining the relation of the tumor to the hepatic anatomy and in differentiating hepatoblastomas from other childhood hepatic tumors.218 However, no current noninvasive study can always differentiate between a benign or malignant liver lesion.28 If there is any concern about the diagnosis, a biopsy should be performed.
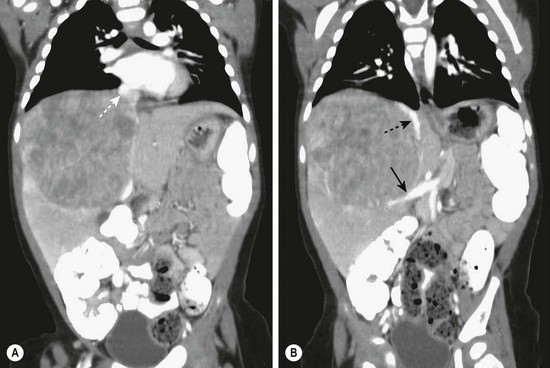
FIGURE 67-13 (A) This CT scan obtained after intravenous administration of a contrast agent in a 1-year-old child shows a large mass in the right hepatic lobe. On other images, the mass appears to invade or compress the medial segment of the left lobe. The middle hepatic vein (dotted arrow) is being compressed by the tumor. (B) The inferior vena cava (dotted arrow) is being displaced medially and anteriorly by the mass, and the portal vein (solid arrow) is markedly displaced inferiorly.
Laboratory Studies
Anemia and thrombocytosis (platelet count >500,000/mm3) are often found in patients with hepatoblastoma.219 However, the hallmark of hepatoblastoma is an elevated AFP level, which occurs in up to 90% of patients.220 The serum levels of AFP can sometimes exceed 1 million ng/mL. This can lead to the ‘hook’ effect in which the initially reported AFP level can be low despite the actual level being very high. If the lesion is suspicious for hepatoblastoma, a request should be made to dilute the AFP sample before retesting.221 Serum AFP has a half-life of between 4 and 9 days, and the levels usually decrease to normal by 4 to 8 weeks after complete removal of the tumor.222 It is important to remember that neonates normally have an elevated AFP level (25–50,000 ng/mL) at birth that does not decrease to ‘adult’ levels until age 6 months.15 This becomes important when evaluating a neonate with a hepatic mass or when monitoring the AFP after liver resection in a neonate or infant.
AFP also has been useful for monitoring purposes. In one case report, a radioimmunodetection method was used (technetium-labeled mouse antihuman monoclonal antibody to AFP).223 After an initial decline in the AFP following liver resection, it began to increase, and an anti-AFP nuclear medicine study accurately located an active tumor in the remaining liver.
Recently, a new marker has been found for hepatoblastoma. Glypican 3 (GPC3) has been detected in hepatic stem cells and has been identified as being expressed by fetal, embryonal, and small cell undifferentiated hepatoblastomas. This marker is also shed by the tumor cells and has the potential to be used as a serum marker for hepatoblastoma.224
Staging
Two staging systems are currently used. In the USA, a combined histologic and surgical staging system is used by the Children’s Oncology Group (COG) (Table 67-3).225 This tumor staging system is self-explanatory and is based on information gathered before any chemotherapy is started. The second staging system, used by the International Society of Pediatric Oncology (SIOP), is based on the radiologic location of the tumor before treatment and is called the PRETEXT (Pretreatment Extent of Disease) Grouping System (Fig. 67-14).226 Both of these staging systems are currently being used in ongoing studies so patient groups can be compared across different study groups.
TABLE 67-3
Children’s Oncology Group Staging System and Outcomes

From Ortega J, Siegel S. Biological markers in pediatric cancer. In: Pizzo P, Poplack D, editors. Principles and Practice of Pediatric Oncology. Philadelphia: Lippincott; 1989. p. 149–62, with permission.
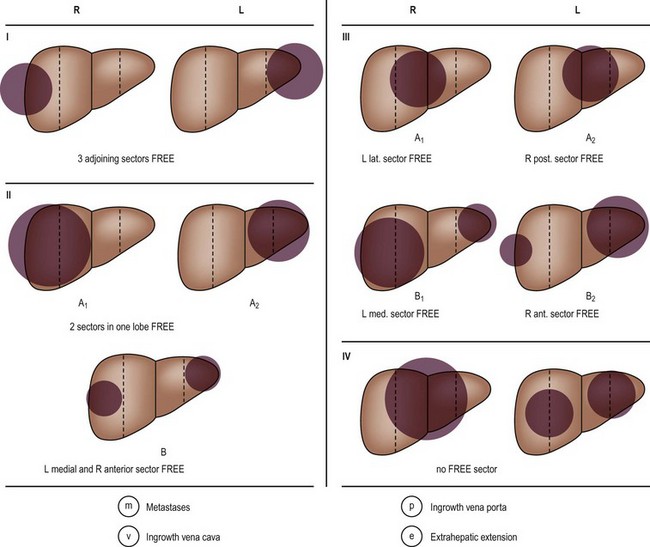
FIGURE 67-14 PRETEXT (Pretreatment Extent of Disease) staging system. Stage is determined by the number of liver sectors free of tumor.
In the PRETEXT system, the liver is divided into four sections: the anterior and posterior sectors on the right, and the medial and lateral sectors on the left. Therefore, based on the extent of the tumor, the patient is classified as follows: PRETEXT 1, with three adjoining sectors free (tumor in only one sector); PRETEXT 2, with two adjoining sectors free (two sectors involved); PRETEXT 3, in which only one sector is free or a total of three nonadjoining sectors are involved (tumor involves two or three sectors); and PRETEXT 4, in which no sector is free (tumor in all four sectors). It also takes into account hepatic vein or portal vein involvement, if extrahepatic spread has occurred (enlargement of the hilar lymph nodes), or if metastases are found. Both staging systems have been shown to have direct correlations with patient survival (Table 67-4).
TABLE 67-4
SIOP PRETEXT Staging and Outcome

SIOP, International Society of Pediatric Oncology.
From Brown J, Perilongo G, Shafford E, et al. Pretreatment prognostic factors for children with hepatoblastoma: Results from the International Society of Pediatric Oncology (SIOP) Study SIOPEL 1. Eur J Cancer 2000;36:1418–25.
In addition to the staging systems, in upcoming protocols, the patients will also be stratified according to risk. In the COG staging system, low-risk patients will be stage I/II without unfavorable biologic features, intermediate-risk patients will be stage III or stage I/II with small cell undifferentiated histology, and high-risk patients will be stage IV or stage I, II, III with an AFP less than 100 ng/mL at diagnosis. In the SIOPEL studies, the patients are stratified into standard risk (PRETEXT I, II, III) or high risk (PRETEXT IV or any tumor with metastases, vena cava or portal vein invasion, contiguous extrahepatic disease, or tumors with an AFP <100 ng/mL at presentation).227
Treatment
The treatment of hepatoblastoma requires a multidisciplinary approach. Except on very rare occasions, chemotherapy alone is unable to eradicate the tumor. The only chance for a long-term cure is complete resection of the primary tumor. This goal can be achieved by the use of either traditional hepatic resections (right lobectomy: segments 5–8; left lobectomy: segments 2–4; or left lateral segmentectomy: segments 2–3) or extended resections (right trisegmentectomy: segments 4–8; or left trisegmentectomies: segments 2–4, 5, 8). The resection can be performed using either open or laparoscopic techniques. There have been several case reports and two large series of laparoscopic liver resections in adults for both benign and malignant disease.228–231 The outcomes of these laparoscopic series have been identical to the results of resections by an open approach.
The resection should be planned so that there is an anticipated margin of normal liver around the tumor. Fortunately, this margin does not need to be large (2–3 mm).232 If it is doubtful that the tumor can be resected with a clear margin, then the patient should undergo preoperative chemotherapy to try to reduce the size of the tumor to the point where complete resection is possible. In several studies, only between 30% and 50% of the patients were able to undergo either a complete or gross total resection of the tumor at the initial procedure.233,234 In the COG trials, only 23% of the patients initially presented with stage I or stage II disease.235
If a difficult liver resection is elected, the conundrum exists as to what to do if the resection is unsuccessful and microscopic residual disease is left after a resection. The data from the COG studies, SIOPEL-1 and SIOPEL-2, indicate that microscopic residual disease after resection can be successfully treated with additional chemotherapy.208,236,237 On the other hand, several series have reported that the most common reason for tumor recurrence is either a gross or microscopically incomplete resection.238–241 If local recurrence of the tumor occurs, the prognosis is poor.222,242,243 ‘Rescue’ transplantation for local recurrence results in a survival rate of only 30%.244 This would suggest that there must be multiple, yet to be defined, variables that determine if microscopic disease can be eradicated or if it will result in recurrent disease. In any case, there must be careful thought before undertaking a ‘difficult’ liver resection when the possibility exists of leaving residual disease versus referring the patient directly for liver transplant evaluation.
Multiple chemotherapy regimens have been evaluated for hepatoblastoma.225,245 The most active chemotherapy agent for hepatoblastoma is cisplatin. This drug is then combined with either vincristine, doxorubicin, or 5-fluorouracil, and used in either an adjuvant or neoadjuvant fashion. The current recommended COG chemotherapy regimen for initially unresectable hepatoblastoma is doxorubicin, cisplatin, 5-fluorouracil, and vincristine.246 In the patients who have had complete resection of their tumor (without preoperative chemotherapy), either four to six postoperative courses of chemotherapy are given. In those children in whom the liver tumor was deemed unresectable at the initial operation (stage III disease), two rounds of chemotherapy are initially given, followed by repeat imaging.
If the tumor appears resectable, then resection should be done at this time. If the tumor is not resectable, then the patient undergoes an additional two rounds of chemotherapy. In addition, the patient also needs to be referred to a center where there is the option for transplantation. If after the additional two rounds of chemotherapy the tumor is resectable (total of four rounds), then resection is performed (Fig. 67-15). With chemotherapy and delayed or second-look surgery, the resection rate has been reported to increase to between 69% and 98% (Fig. 67-16).5,247–249 In patients with stage IV disease (initially unresectable), only 40% of these tumors were rendered resectable after four rounds of chemotherapy. If the tumor is still unresectable, the patient needs to be evaluated and listed for a liver transplant.
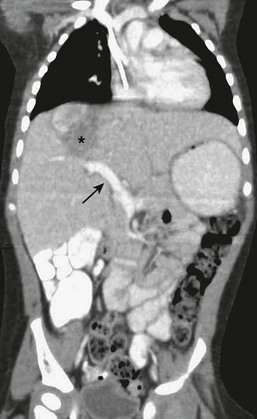
FIGURE 67-15 After four cycles of chemotherapy, the CT scan showed marked diminution in the size of the mass (asterisk). Note the portal vein (solid arrow) has returned to its normal anatomic position. Compare with the prechemotherapy CT scan seen in Figure 67-13.
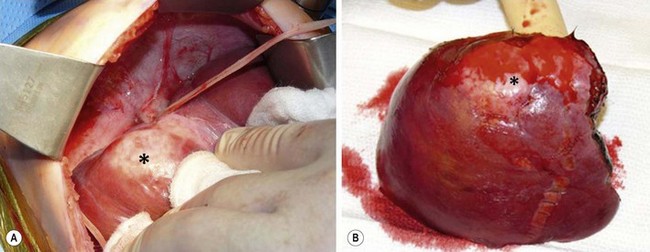
FIGURE 67-16 Operative findings of the patient whose CT scans are seen in Figures 67-13 and 67-15. (A) An umbilical tape has been used to encircle the middle hepatic vein. Note the residual tumor (asterisk) in the cephalad portion of the right hepatic lobe. (B) The right hepatic lobe containing the lesion (asterisk) is seen. It was possible to obtain a normal margin of liver in the left lobe, but a trisegmentectomy was needed.
Current United Network Organ Sharing (UNOS) policy allows hepatoblastoma patients to be listed as status 1B in order to maximize their chance of being transplanted within one month of listing. Preferably the time between the last round of chemotherapy and transplantation should be less than four weeks because it is now known that up to 80% of hepatoblastomas will develop drug resistance after four to five courses of chemotherapy.248,250 After transplant, the patient receives a final two rounds of chemotherapy.
Some newer chemotherapy agents have been investigated for hepatoblastoma. Topotecan inhibits growth and neovascularization in a mouse model.251 In addition, the suppressive effects of the topotecan lasted several weeks after discontinuation of the agent. Irinotecan appears to have some promise in salvaging patients who have recurrent disease. This drug could potentially be added to front-line chemotherapy regimens.252 High-dose chemotherapy with stem cell rescue has been attempted but has not been successful.
Another approach to the unresectable hepatoblastoma is the use of preoperative chemoembolization. In one study, transarterial catheterization with selective tumor chemoembolization was able to shrink the tumor by an average of 26%, which allowed subsequent complete tumor resection in every case.253 Interestingly, the surgical specimens showed only minimally viable or no viable tumor. It was postulated that this technique may be useful not only as a therapeutic modality for unresectable hepatoblastomas but also potentially for resectable tumors that could be made minimally viable to nonviable before surgical intervention.254
Another therapeutic dilemma occurs in the child first seen with a ruptured tumor. In one review, all three patients who survived the initial rupture had no evidence of recurrent disease, with a mean survival of 36 months.255 Even though tumor rupture and peritoneal soiling occurred, no peritoneal growths were subsequently identified in any patient.
Outcome
Patient-outcome studies have been based on histologic type, the extent of the original tumor, or tumor response to chemotherapy.206 Several studies have shown a good outcome with fetal histology and with complete resection of the tumor.51,194,209,256,257 All the studies that have consistently shown a good outcome based on fetal histology have limited this diagnosis to tumors with a mitotic activity less than two per ten high-power fields. Conversely, several studies have consistently reported a poor outcome for those patients who have small cell undifferentiated hepatoblastoma.207 Except for these data, no consistent correlation has been found with any of the other histologic patterns and patient outcomes.
The AFP level also has been found to have both prognostic and therapeutic implications. Patients with an AFP level less than 100 ng/mL or greater than 1 million ng/mL have a worse prognosis.258 The low AFP group comprised patients with small cell undifferentiated tumors, suggesting that a low AFP level could be related to a very primitive and poorly differentiated tumor that was unable to make AFP.259 Patients who had a slow decline in their AFP levels, either after resection or chemotherapy, had a worse long-term prognosis than did those who had an early, very rapid decline (>99% drop in AFP levels).260
The best survival rate in patients with hepatoblastoma is for those tumors with pure fetal histology. A recent COG study demonstrated that after complete surgical resection of a stage I tumor with pure fetal histology, the patient can be carefully monitored with no further therapy.261 However, if an area of undifferentiated small cell histology is noted within an otherwise pure fetal histology tumor, then aggressive chemotherapy should be instituted.207
In another COG study, the 3-year event free survival (EFS) was 90% for stage I/II tumors, 50% for stage III tumors, and only 20% for stage IV tumors.262 The European SIOPEL-2 study reported that the 3-year survival for standard risk tumors was 90% and was 50% for high-risk tumors. These data compare favorably with those of a large German series that noted an EFS of 100% for stage I, 80% for stage II, and 68% for stage III disease.249 None of the patients with stage IV disease in the German trial survived. In another prospective study, the German group showed that the important prognostic factors for survival appeared to be the tumor growth pattern, vascular invasion, and serum AFP levels.257,263
As the patients in the high-risk PRETEXT or COG trials have poor outcomes despite a complete surgical resection, additional or different chemotherapy regimens are necessary to improve patient survival in these groups.257 Currently there are ongoing studies that are evaluating the possibility of modifying different cellular or gene targets in hepatoblastoma cells that will make them more susceptible to chemotherapy.264
There are several treatment options currently under clinical evaluation by COG. For more information on ongoing studies, please check the COG website (www.childrensoncologygroup.org).
Transplantation
Once a patient has completed four rounds of chemotherapy, a decision is made to either perform a resection with the goal of complete tumor excision or to refer the patient for liver transplantation. Several characteristics of hepatoblastoma have recently been reported as indicators of unresectability. Patients with these indicators are possible candidates for liver transplantation. Patients who were younger than 3 years of age at presentation tend to respond better to chemotherapy with greater reductions in tumor size when compared with older children.265 Bilobar, multifocal tumors at presentation are candidates for transplantation because, despite apparent radiologic clearing of a lobe after chemotherapy, microscopic disease can persist in the liver leading to later recurrent disease.236,266,267 Patients who present with low AFP levels (<100 ng/mL) tend not to respond to chemotherapy, so they should be considered for either upfront resection or transplantation. Patients who have tumor extension into the inferior vena cava, all three hepatic veins, or the bifurcation of the portal vein are unlikely to sufficiently shrink their tumor with chemotherapy to allow a complete resection and therefore should be considered as possible candidates for transplantation.244
Orthotopic liver transplantation is a successful treatment for unresectable hepatoblastoma.268 A recurrence-free survival rate of between 79% and 92% has been reported.244,269,270 In several series, an important prognostic factor that predicted good results after transplantation was a good initial response to chemotherapy. In one series, only 60% of the patients who were poor responders are currently alive, with a follow-up of less than 1 year.269 As mentioned previously, liver transplantation for local tumor recurrence after resection is associated with a post-transplant recurrence rate of 30%.
Although two post-transplant rounds of chemotherapy are currently utilized, one multicenter review noted no significant difference in survival rates between those patients who received chemotherapy (77%) versus those who did not (70%).241,271–273
Patients with extrahepatic metastases found on initial evaluation can be successfully treated with liver transplantation if the metastatic disease is eradicated before the transplant. Hepatoblastoma commonly metastasizes to the lungs, and lung metastases are often present at diagnosis. Because this tumor tends to be chemoresponsive, these metastases will often disappear after the first several rounds of chemotherapy. If the pulmonary metastases do not resolve, then current data would suggest that these patients should undergo a metastasectomy for any remaining disease, either before or shortly after hepatic resection, and before consideration for transplantation. This approach has been successful in increasing patient survival.274,275 Unfortunately the data on resection of pulmonary recurrences are less optimistic. There may be a role for resection of late pulmonary metastasis in patients who presented with stage I disease. However, pulmonary relapse in stage III and IV disease portends a poor prognosis that is not changed by lung resection.
Undifferentiated Embryonal Sarcoma
Incidence
Undifferentiated embryonal sarcoma of the liver makes up about 7% of the solid liver tumors in children.1 Unfortunately, this is a very malignant tumor with a poor outcome.
Clinical Presentation
Undifferentiated embryonal sarcoma most commonly affects children between the ages of 6 and 10 years, but has been reported in a child as young as 19 months.276 A slight male predominance has been noted. The most common clinical presentation is either right upper quadrant or epigastric pain with or without a palpable abdominal mass. Occasionally, marked hepatomegaly is seen without a definite mass. Rarely, this tumor can even masquerade as a hepatic abscess or infection.277 Other nonspecific presenting complaints can include vomiting, anorexia, and lethargy. Laboratory studies, including AFP level, are usually normal.
Imaging
On ultrasound examination, the lesion appears predominantly solid.278 However, on CT and MRI, the lesion appears cystic without any significant solid component (Fig. 67-17). This same type of disparity has been reported only in Wilms’ tumor metastatic to the liver. Thus, it appears that such a discrepancy between the two imaging techniques would be highly suggestive of an undifferentiated embryonal sarcoma.
Histology
Undifferentiated embryonal sarcoma is a neoplasm with a very primitive mesenchymal phenotype. These tumors tend to occur predominantly in the right lobe of the liver and to be large, with an average diameter of 14–21 cm.278 In cross section, the tumors are often variegated, with white mucoid or gelatinous areas alternating with other areas of tumor necrosis and hemorrhage (Fig. 67-18). The tumor typically is well demarcated from the adjacent liver by a compressed, fibrous pseudocapsule.276

FIGURE 67-18 Note the variegated appearance on cut section of an undifferentiated embryonal sarcoma.
On microscopic section, these tumors are composed of medium to large spindle- to stellate-type cells in a variable amount of myxoid stroma (Fig. 67-19). The cells are usually densely arranged in a myxomatous background. In the periphery, entrapped bile ducts or hepatic cords have been noted.4,276 Mitoses are frequent and usually bizarre.
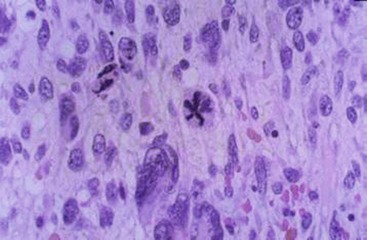
FIGURE 67-19 Histology of undifferentiated embryonal sarcoma shows large spindle cells with multiple mitoses.
No characteristic immunohistochemical stain pattern has been identified for embryonal sarcoma.279 The only consistent cell markers have been vimentin and the ‘histiocytic’ determinants α1-antitrypsin and α-antichymotrypsin.280,281
In a cytogenetic study, extensive chromosomal rearrangements were noted to be very similar to other soft tissue sarcomas such as leiomyosarcoma, osteosarcoma, and malignant fibrous histiocytoma.282 In only a few cytometric studies, the tumors have ranged from diploid to tetraploid to aneuploid.283,284
Treatment
In addition to the highly suggestive radiologic findings, these patients can be diagnosed by fine needle aspiration. Two separate reports have noted that the cytologic features of undifferentiated embryonal sarcoma are distinctive and different from other childhood tumors.285,286
The initial experience with undifferentiated embryonal sarcoma of the liver was poor. In a review of patients treated from 1950 to 1988, only 37% of the patients were noted to survive.284 This tumor usually proves fatal because of massive upper abdominal growth with secondary involvement of the diaphragm, stomach, abdominal wall, ribs, or pancreas rather than by metastases. Occasionally, intra-abdominal dissemination of the tumor can occur, causing diffuse matting of the small bowel. Pulmonary and pleural metastases have been noted but are much less common than the secondary involvement of the extrahepatic tissue by direct extension.276
The only chance for cure is radical excision.287,288 Unfortunately, despite complete resection of the tumor, many patients have recurrent disease, which suggests the need for postoperative chemotherapy.289
The chemotherapy regimens that have been used are based on nonrhabdomyosarcomatous soft tissue sarcoma-type protocols.290 With these regimens, survival rates have improved to 66%, because these tumors are very chemotherapy sensitive.291,292 This finding has led to the use of preoperative chemotherapy to shrink an unresectable tumor to a size at which a radical hepatic resection is possible.293 This is similar to the approach used to manage an initially nonresectable hepatoblastoma.
With the ongoing improvement in chemotherapy regimens for sarcomas, the previously bleak outlook for this tumor is now much more optimistic. The use of an aggressive chemotherapy regimen, along with complete resection of the primary tumor, has resulted in a 37% survival rate in patients whose tumors initially presented as free intraperitoneal rupture.292
Moreover, a prospective series from the Italian and German Soft Tissue Sarcoma Cooperative Groups reported a protocol that included surgery or biopsy followed by neoadjuvant chemotherapy for four cycles. This was followed by second-look surgery to try to remove residual tumor followed by additional chemotherapy. In their series, ten of 17 patients survived with complete remission.294
In patients in whom complete resection of the tumor is not possible despite chemotherapy, liver transplantation has been advocated as another possible means for complete excision.295,296 This aggressive approach is reasonable because these tumors are sensitive to chemotherapy.
Hepatocellular Carcinoma
Hepatocellular carcinoma (HCC) is a relatively rare, highly malignant tumor that is more commonly seen in adults than in children. It is the second most common pediatric liver tumor, occurring about 19% of the time, but it still comprises less than 1% of all pediatric cancers.297 Its peak incidence is between 10 and 15 years, and it is more common in boys.298
The predisposing factors for HCC are distinctly different between children and adults. In adults, cirrhosis seems to be the primary etiology. The cirrhosis is usually seen in patients with either hepatitis B, hepatitis C, genetic hemochromatosis, alcohol-related cirrhosis, or cirrhosis due to primary biliary cirrhosis. In a recent review, it was noted that patients in these groups were at a significantly increased risk for developing HCC.299 Hepatic ultrasound and serum AFP evaluations every 6 months were recommended in these populations to detect this tumor at an early stage.
In contrast, cirrhosis is often not part of the antecedent process in children. Moreover, a previous congenital or acquired disorder of the liver may be found (Box 67-3).300 HCC in children has been associated with a variety of metabolic, familial, and infectious disorders. Some of these metabolic disorders include tyrosinemia, α1-antitrypsin deficiency, and hemochromatosis.301 Patients with tyrosinemia seem to be at a particularly high risk for development of HCC. Because of this high prevalence rate, it has been suggested that liver transplantation be performed in this population before age 2 years.302,303 HCC also has been seen in patients with type 1 GSD. Most hepatic masses that develop in this population are hepatic adenomas, but carcinomas do present a real risk in this group304 A variety of other noncirrhotic liver diseases also have been associated with HCC, including familial polyposis, Gardner syndrome, Sotos syndrome, Blum syndrome, neurofibromatosis, Abernathy malformation, methotrexate therapy, and neonatal hepatitis. There is also an association with parenteral nutrition.305–310
Congenital and infectious disorders also are associated with this tumor, including extrahepatic biliary atresia, congenital hepatic fibrosis, Alagille syndrome, persistent familial intrahepatic cholestasis (PFIC),311 hepatitis B, and hepatitis C. In areas where hepatitis B is endemic, it ranks fifth in the causes of childhood malignancies and outnumbers hepatoblastoma by 3 : 1.312 The importance of hepatitis B and the subsequent development of HCC in children is highlighted by the aggressive hepatitis B vaccination program that began in 1984 in Taiwan.313 When the mortality from liver carcinoma in the group from birth to age 9 years was compared between the years 1984 and 1993, a substantial and statistically significant decrease in the mortality was seen by 1993. Similar results have been found in Gambia and the USA.313 Hepatitis C also has been linked to the development of hepatocellular carcinoma.314 In contrast to hepatitis B, the cirrhosis and the subsequent development of HCC in the hepatitis C population usually takes several decades to develop.315
Of particular interest is the association between HCC and biliary atresia.298,316 In a review, except for one patient first seen at age 5 months, all the other patients were older than 2 years with a mean of age 7.5 years when the HCC was discovered. These tumors were found either at autopsy or incidentally at the time of liver transplantation for biliary atresia. In those patients in whom HCC was identified at the time of transplantation, all of these patients are alive and well after transplant. This association between HCC and biliary atresia, or any other disease that leads to hepatic cirrhosis, suggests that a routine screening protocol every 6 months with hepatic ultrasound and AFP levels is warranted.317
Clinical Presentation
Most patients are initially seen with either an abdominal mass or abdominal pain. Other associated symptoms include nausea and vomiting, anorexia, malaise, and a significant weight loss. As many as 10% are seen initially with tumor rupture and hemoperitoneum.318 More than one-third of HCCs appear as multiple nodules rather than a single tumor.319
Laboratory studies can show mild elevations in the serum glutamic oxaloacetic transaminase (SGOT) and lactic dehydrogenase (LDH) levels. The AFP is elevated in about 85% of patients but can be normal or only mildly elevated with the fibrolamellar variant. 318, 320 An elevated AFP is associated with an increased risk for recurrence after resection and therefore reflects a worse prognosis.321,322
Imaging
CT and MRI are both helpful for delineating the mass and for determining resectability. With the advent of spiral CT with intravenous bolus contrast administration, the hepatic veins and portal venous system can be well delineated, and any tumor involvement can be adequately assessed. The American Association for the Study of Liver Diseases has published criteria for the noninvasive diagnosis of HCC. In nodules less than 2 cm in diameter in cirrhotic livers, the diagnosis of HCC can be made without biopsy of the lesion if two coincidental dynamic imaging studies (e.g., CT/MRI) reveal arterial-phase hypervascularity followed by wash-out in the portal venous phase.323 The fibrolamellar variant is notable as a hypodense, single or multilobed mass on CT that tends to be hypervascular as well as sometimes showing a central scar.324 This appearance could easily be confused with FNH, and care must be taken to distinguish between these two lesions.
Histology
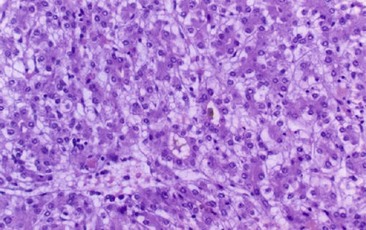
HCC can vary in size from 2–25 cm, and the surrounding liver can exhibit either micro- or macronodular cirrhosis in up to 60% of cases (Figs 67-20 and 67-21).1 Microscopically, trabeculae that are two to ten cell layers in thickness are seen with the larger trabeculae sometimes displaying central necrosis. The individual cells are usually larger than normal hepatocytes, with nuclear hypochromasia and frequent and bizarre mitosis (Fig. 67-22). Vascular invasion may be prominent. Tumors less than 2 cm generally are well differentiated. Over time, as the tumor grows, the original tumor cells are replaced by poorly differentiated cell clones.325 Moreover, as the tumor enlarges, its blood supply becomes more dependent on newly formed arterial vessels and less dependent on the portal circulation. This imbalance between the hepatic arterial and portal venous supply leads to the hypervascular pattern that is the radiologic hallmark for HCC.
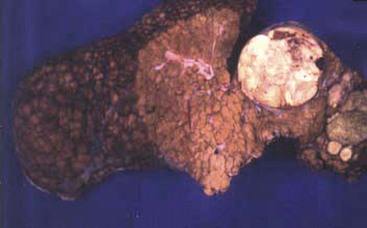
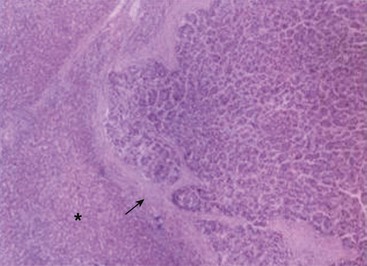
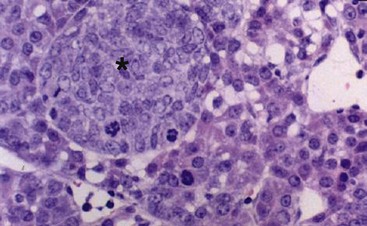
FIGURE 67-21 This histologic photomicrograph shows hepatocellular carcinoma with surrounding cirrhosis (arrow) and uninvolved liver (asterisk).
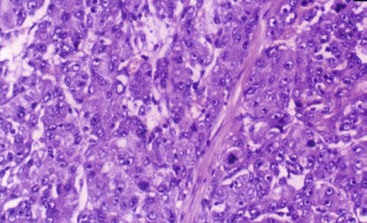
FIGURE 67-22 Histology of a hepatocellular carcinoma demonstrating enlarged hepatocytes and nuclear hypochromasia.
In the fibrolamellar variant, the hepatocytes are large, deeply eosinophilic, and embedded within a lamellar fibrosis. Clusters of cells are often separated by broad bands of laminated collagen.1,326 The presence of large amounts of fibrosis alone is not sufficient in itself for the diagnosis of fibrolamellar carcinoma.327
Treatment
The treatment of HCC for stages I and II is complete surgical resection followed by chemotherapy. A COG study in 2002 reported that seven of eight patients with stage I disease survived after adjuvant cisplatin-based chemotherapy.328 Moreover, it has been postulated that routine use of chemotherapy with cisplatin and doxorubicin may benefit children with completely resected HCC.5,329
In one pediatric report of 49 children, resection was possible in only 10%. Only two patients lived for more than 2 years.312 If a complete, microscopically free, radical resection is possible, the prognosis can be good, with an 80–90% survival.330 Unfortunately, the overall cure rate for this tumor in children is a dismal 15%. In the adult population, 3-year survival rates between 34–57% have been reported.331,332 Multiple studies have looked at prognostic factors that influence outcome and recurrence after resection for HCC. Multiple staging systems have been proposed based on multivariant analyses of various prognostic factors. Three factors that have been repeatedly associated with improved survival and decreased recurrence rates are small tumor size (<2 cm), the number of tumor nodules, and a histologic increase in tumor microvascular density. These findings are highly predictive of tumor relapse after resection.333 Unfortunately, it is rare to see patients initially with all three favorable variables. In most series, the tumors are usually larger than 5 cm in diameter at presentation.331,334
An important variant that should be mentioned is the fibrolamellar type. Only in the 1980s did this variant become established as a histologic and clinically distinct entity.335 This lesion is characterized by relatively slow growth and occurs almost exclusively in a noncirrhotic liver.326 The fibrolamellar variant usually occurs in adolescents and young adults, with a peak incidence in the second decade of life.336,337 It accounts for between 16–50% of HCC diagnosed in patients younger than 21 years of age.338 In contrast to conventional HCC, fibrolamellar carcinoma is not associated with risk factors such as cirrhosis or chronic hepatitis B infection.339,340 However, an association does exist between FNH and the fibrolamellar variant.
Radiologically, the fibrolamellar variant is often hypodense on noncontrast CT and can show variable perfusion, including hypervascularity, on contrast CT. In addition, a central hypodense or hypervascular area can be seen that can mimic a central scar.341 This can create confusion between the diagnosis of the fibrolamellar variant and FNH. MRI may be helpful in distinguishing between these two diagnoses.342 The results after resection for fibrolamellar carcinoma in adults are very good, with 50% 5-year survivals. However, after apparent curative resections, recurrences or metastases can occur after very long disease-free intervals.343,344 Unfortunately when the fibrolamellar variant was examined in children, there was no survival benefit to this diagnosis.262
Transplantation for Hepatocellular Carcinoma
Liver transplantation has been used as curative therapy for HCC.272,345 The early experience of liver transplantation in adults for HCC was discouraging.346 However, in a review of 344 patients who underwent liver transplantation for HCC (excluding the fibrolamellar type), three factors were associated with tumor-free survival: a unilobar tumor, a tumor smaller than 2 cm, and the absence of vascular invasion.347 Two different pretransplant selection criteria are currently being utilized in adults (Table 67-5).
TABLE 67-5
Milan and UCSF Transplant Criteria for Hepatocellular Carcinoma
| Milan Criteria | UCSF Criteria | |
| Single tumor maximum diameter | ≤5.0 cm | ≤6.5 cm |
| Maximum number | 3 | 3 |
| Largest tumor size | ≤3.0 cm | ≤4.5 cm |
| Total tumor size | NA | ≤8.0 cm |
NA, not applicable; UCSF, University of California at San Francisco.
The Milan criteria are widely recognized as excellent predictors of a low recurrence rate after transplantation.348 These criteria, however, have not been verified in the pediatric population, and their applicability in children is unclear.349 As HCC can be a rapidly growing tumor, it is common for the tumor to exceed these criteria by the time an organ becomes available and the patient is no longer a candidate for transplant. The University of California at San Francisco criteria are an attempt to allow an increased pretransplant tumor burden and still have a low post-transplant recurrence rate. Several studies have supported this hypothesis.350,351
Two absolute contraindications to transplantation for HCC in children are extrahepatic spread and macroscopic vascular invasion.272 The SIOPEL-1 study reported no long-term survivors among those who failed to respond to chemotherapy.352 Therefore, the lack of response to preoperative chemotherapy is a relative contraindication to transplantation. Despite these possible limitations, liver transplantation in children for HCC has a very good five-year survival rate between 63–89%.272,353 Both adult and child patients are given additional points in the UNOS matching system so they can be transplanted before the tumor progresses.
Interestingly, in contrast to patients with hepatoblastoma, several studies have shown that liver transplantation is an appropriate salvage procedure for patients with HCC who experience a recurrence after a resection. Their post-transplant survival is comparable to that of patients who underwent primary transplantation for HCC.354,355
Postresection chemotherapy has been uniformly ineffective in preventing or treating recurrences in the Pediatric Oncology Group (POG), SIOPEL, and German studies.328,352,356,357 New chemotherapeutic approaches are needed for this tumor, and currently studies are planned that will combine chemotherapeutic agents with antiangiogenic agents. However, in two small series, chemotherapy was given to children with unresectable HCC before transplant. Eighty per cent of these children had a dramatic decrease in their tumor size.297,358 At the time of transplant, all tumors still had viable cells but only one patient after transplant had a recurrence and subsequently died.
Patients who are not surgical or transplant candidates, patients who demonstrate recurrences, or patients who are on the transplant list and their tumors have grown to the point that they exceed the Milan criteria can potentially benefit from several nonsurgical strategies, including percutaneous ethanol injection, radiofrequency ablation, or chemoembolization of the tumor.359 All of these therapies have proved efficacious in decreasing or eradicating localized tumors, treating recurrent tumors, or shrinking tumors so that the patient again falls within the transplant criteria.360 These therapies also improved survival with this tumor, but unfortunately, none has been proved to be curative.299
A catastrophic presentation of HCC is spontaneous rupture. These patients present with acute right upper quadrant pain and hypovolemic shock. The diagnosis can be made with ultrasound or abdominal CT. In the hemodynamically stable patient, the treatment is conservative with correction of volume status and correction of any coagulopathy. Then a careful assessment of the tumor needs to be made with the goal of primary resection of the tumor. If the patient is unstable, then transarterial embolization of the tumor should be performed to control the hemorrhage. After the patient’s condition has stabilized, he or she needs to be evaluated for a resection.361
Rhabdomyosarcoma of the Biliary Tree
Incidence and Clinical Presentation
Even though rhabdomyosarcoma is the most common sarcoma in children, it accounts for only 1% of all liver tumors and only 0.8% of all rhabdomyosarcomas in children.1,362 Hepatobiliary rhabdomyosarcoma tends to be a disease of the young, with a median age of 3 years. It is rarely seen in children older than 15 years.363,364 This tumor most commonly arises in the intrahepatic biliary system and then extends into the liver parenchyma. It also has been reported to arise from a variety of other sites, including an intrahepatic cyst, the gallbladder, the cystic duct, the ampulla of Vater, a choledochal cyst, and the hepatic parenchyma itself.363–369
As it arises most commonly from the bile ducts, the most common presenting symptom is jaundice. The median tumor diameter at diagnosis is 8 cm. Therefore an abdominal mass is a common finding.362
Laboratory Findings and Differential Diagnosis
As jaundice is a common presenting symptom, elevated direct bilirubin, alkaline phosphatase, and γ-glutamyltransferase (GGT) levels are common. The differential diagnosis for an intraductal lesion in a child includes either an inflammatory pseudotumor or a cholangiocarcinoma, but these are extraordinarily rare.370 If the rhabdomyosarcoma is a predominantly hepatic mass with minimal bile duct involvement, then the differential diagnosis would be more dependent on the patient’s age, as noted in previous sections.
The pathology of the intraductal lesions is similar to that of rhabdomyosarcoma at extrabiliary sites. The intraductal tumor is usually either the botryoid or embryonal subtype, unless the lesion involves predominantly the hepatic parenchyma, in which the alveolar subtype predominates.371
Imaging
Multiple imaging modalities have been useful for diagnosis. Ultrasound typically reveals biliary dilatation and possibly a mass in the biliary system. Larger lesions may have cystic areas within them, possibly reflecting areas of partial tumor necrosis (Fig. 67-23).217 CT will often show an intraductal mass in association with areas of low attenuation within the tumor. MRI may have an advantage over CT in that not only can it show the anatomic source and location of the mass but, with the advent of MR cholangiography, also can evaluate the bile ducts by demonstrating biliary dilatation and intraductal irregularity.372 Percutaneous transhepatic cholangiography (PTC) also can be useful in patients who have a dilated biliary system. PTC can demonstrate multiple filling defects that correspond to the intraductal tumor.373 PTC also has the advantage of providing external drainage of the biliary system in those patients with obstructive jaundice.
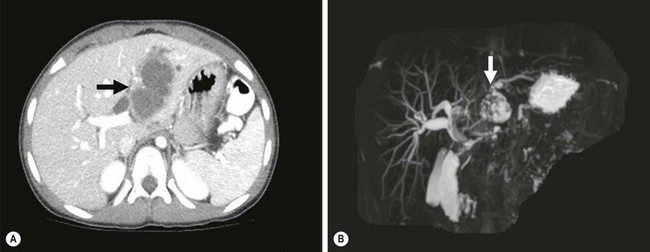
Treatment and Prognosis
The best treatment for biliary rhabdomyosarcoma is a multidisciplinary approach using operation, chemotherapy, and, potentially, radiation therapy. Unfortunately, resection alone is not usually possible because of spread of the tumor into the liver parenchyma or direct local extension into the duodenum, stomach, or pancreas. It is common to find lymphatic spread at the initial operation. Because of these problems, adequate resection is usually possible in only 20–40% of the patients.362–364 In patients in whom primary resection is not possible, the initial approach should be biopsy and lymph node sampling, followed by standard rhabdomyosarcoma chemotherapy protocols and a second-look procedure. In a study of biliary rhabdomyosarcoma that used this multimodality approach, four of ten children remained disease free after a mean of four years.362
References
1. Stocker, JT. Hepatic Tumors in Children. In: Suchy FJ, Sokol RJ, Balistreri WF, eds. Liver Disease in Children. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:915.
2. Silverberg, E. Cancer statistics. 1986. CA Cancer J Clin. 1986; 36:9–25.
3. Couinaud, C. Surgical anatomy of the liver. Several new aspects. Chirurgie. 1986; 112:337–342.
4. Couinaud, C. The anatomy of the liver. Ann Ital Chir. 1992; 63:693–697.
5. Exelby, PR, Filler, RM, Grosfeld, JL. Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American Academy of Pediatrics Surgical Section Survey–1974. J Pediatr Surg. 1975; 10:329–337.
6. Boon, LM, Burrows, PE, Paltiel, HJ, et al. Hepatic vascular anomalies in infancy: A twenty-seven-year experience. J Pediatr. 1996; 129:346–354.
7. Iyer, CP, Stanley, P, Mahour, GH. Hepatic hemangiomas in infants and children: A review of 30 cases. Am Surg. 1996; 62:356–360.
8. Chen, CC, Kong, MS, Yang, CP, et al. Hepatic hemangioendothelioma in children: Analysis of thirteen cases. Acta Paediatr Taiwan. 2003; 44:8–13.
9. Drut, RM, Drut, R. Extracutaneous infantile haemangioma is also Glut1 positive. J Clin Pathol. 2004; 57:1197–1200.
10. Riley, MR, Garcia, MG, Cox, KL, et al. Hepatic infantile hemangioendothelioma with unusual manifestations. J Pediatr Gastroenterol Nutr. 2006; 42:109–113.
11. Burrows, PE, Dubois, J, Kassarjian, A. Pediatric hepatic vascular anomalies. Pediatr Radiol. 2001; 31:533–545.
12. Perez, J, Pardo, J, Gomez, C. Vincristine–an effective treatment of corticoid-resistant life-threatening infantile hemangiomas. Acta Oncol. 2002; 41:197–199.
13. Christison-Lagay, ER, Burrows, PE, Alomari, A, et al. Hepatic hemangiomas: Subtype classification and development of a clinical practice algorithm and registry. J Pediatr Surg. 2007; 42:62–68.
14. Selby, DM, Stocker, JT, Waclawiw, MA, et al. Infantile hemangioendothelioma of the liver. Hepatology. 1994; 20:39–45.
15. Novaks, D, Suchy, FL, Balistreri, W. Disorders of the liver and biliary system relevant to clinical practice. In: Feigin RD, DeAngelis CD, Douglas Jones M, eds. Oski’s Principles and Practice of Pediatrics. Philadelphia: JB Lippincott, 1990.
16. Huang, SA, Tu, HM, Harney, JW, et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med. 2000; 343:185–189.
17. Mason, KP, Koka, BV, Eldredge, EA, et al. Perioperative considerations in a hypothyroid infant with hepatic haemangioma. Paediatr Anaesth. 2001; 11:228–232.
18. Daller, JA, Bueno, J, Gutierrez, J, et al. Hepatic hemangioendothelioma: Clinical experience and management strategy. J Pediatr Surg. 1999; 34:98–106.
19. Holcomb, GW, 3rd., O’Neill, JA, Jr., Mahboubi, S, et al. Experience with hepatic hemangioendothelioma in infancy and childhood. J Pediatr Surg. 1988; 23:661–666.
20. Sari, N, Yalcin, B, Akyuz, C, et al. Infantile hepatic hemangioendothelioma with elevated serum alpha-fetoprotein. Pediatr Hematol Oncol. 2006; 23:639–647.
21. Lopez Gutierrez, JC, Avila, LF, Sosa, G, et al. Placental anomalies in children with infantile hemangioma. Pediatr Dermatol. 2007; 24:353–355.
22. Paltiel, HJ, Patriquin, HB, Keller, MS, et al. Infantile hepatic hemangioma: Doppler ultrasound. Radiology. 1992; 182:735–742.
23. Keslar, PJ, Buck, JL, Selby, DM. From the archives of the AFIP. Infantile hemangioendothelioma of the liver revisited. Radiographics. 1993; 13:657–670.
24. Horton, KM, Bluemke, DA, Hruban, RH, et al. CT and MR imaging of benign hepatic and biliary tumors. Radiographics. 1999; 19:431–451.
25. Feng, ST, Chan, T, Ching, AS, et al. CT and MR imaging characteristics of infantile hepatic hemangioendothelioma. Eur J Radiol. 2010; 76:e24–e29.
26. Chung, T, Hoffer, FA, Burrows, PE, et al. MR imaging of hepatic hemangiomas of infancy and changes seen with interferon alpha-2a treatment. Pediatr Radiol. 1996; 26:341–348.
27. Mortele, KJ, Mergo, PJ, Urrutia, M, et al. Dynamic gadolinium-enhanced MR findings in infantile hepatic hemangioendothelioma. J Comput Assist Tomogr. 1998; 22:714–717.
28. Lu, M, Greer, ML. Hypervascular multifocal hepatoblastoma: Dynamic gadolinium-enhanced MRI findings indistinguishable from infantile hemangioendothelioma. Pediatr Radiol. 2007; 37:587–591.
29. Vilgrain, V, Uzan, F, Brancatelli, G, et al. Prevalence of hepatic hemangioma in patients with focal nodular hyperplasia: MR imaging analysis. Radiology. 2003; 229:75–79.
30. Behr, GG, Fishman, SJ, Caty, MG, et al. Hepatic mesenchymal hamartoma and infantile hemangioma: A rare association. J Pediatr Surg. 2012; 47:448–452.
31. Dehner, LP, Ishak, KG. Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol. 1971; 92:101–111.
32. Becker, JM, Heitler, MS. Hepatic hemangioendotheliomas in infancy. Surg Gynecol Obstet. 1989; 168:189–200.
33. Mo, JQ, Dimashkieh, HH, Bove, KE. GLUT1 endothelial reactivity distinguishes hepatic infantile hemangioma from congenital hepatic vascular malformation with associated capillary proliferation. Hum Pathol. 2004; 35:200–209.
34. Hernández, F, Navarro, M, Encinas, JL, et al. The role of GLUT1 immunostaining in the diagnosis and classification of liver vascular tumors in children. J Pediatr Surg. 2005; 40:801–804.
35. Prokurat, A, Kluge, P, Chrupek, M, et al. Hemangioma of the liver in children: Proliferating vascular tumor or congenital vascular malformation? Med Pediatr Oncol. 2002; 39:524–529.
36. Mulliken, JB, Enjolras, O. Congenital hemangiomas and infantile hemangioma: Missing links. J Am Acad Dermatol. 2004; 50:875–882.
37. Woltering, MC, Robben, S, Egeler, RM. Hepatic hemangioendothelioma of infancy: Treatment with interferon alpha. J Pediatr Gastroenterol Nutr. 1997; 24:348–351.
38. Wong, DC, Masel, JP. Infantile hepatic haemangioendothelioma. Australas Radiol. 1995; 39:140–144.
39. Davenport, M, Hansen, L, Heaton, ND, et al. Hemangioendothelioma of the liver in infants. J Pediatr Surg. 1995; 30:44–48.
40. Samuel, M, Spitz, L. Infantile hepatic hemangioendothelioma: The role of surgery. J Pediatr Surg. 1995; 30:1425–1429.
41. Wu, TJ, Teng, RJ, Tsou Yau, KI. Hepatic hemangioendothelioma: Successful treatment with steroid in a very-low-birth-weight infant. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1996; 37:56–58.
42. Léauté-Labrèze, C, Dumas de la Roque, E, Hubiche, T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008; 358(24):2649–2651.
43. Marsciani, A, Pericoli, R, Alaggio, R, et al. Massive response of severe infantile hepatic hemangioma to propanolol. Pediatr Blood Cancer. 2010; 54:176.
44. Sharma, RA, Euden, SA, Platton, SL, et al. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin Cancer Res. 2004; 10:6847–6854.
45. Hassell, LA, Roanh le, D. Potential response to curcumin in infantile hemangioendothelioma of the liver. Pediatr Blood Cancer. 2010; 55:377–379.
46. Isaacs, H, Jr. Fetal and neonatal hepatic tumors. J Pediatr Surg. 2007; 42:1797–1803.
47. Kullendorff, CM, Cwikiel, W, Sandstrom, S. Embolization of hepatic hemangiomas in infants. Eur J Pediatr Surg. 2002; 12:348–352.
48. Warmann, S, Bertram, H, Kardorff, R, et al. Interventional treatment of infantile hepatic hemangioendothelioma. J Pediatr Surg. 2003; 38:1177–1181.
49. Achilleos, OA, Buist, LJ, Kelly, DA, et al. Unresectable hepatic tumors in childhood and the role of liver transplantation. J Pediatr Surg. 1996; 31:1563–1567.
50. Grabhorn, E, Richter, A, Fischer, L, et al. Neonates with severe infantile hepatic hemangioendothelioma: Limitations of liver transplantation. Pediatr Transplant. 2009; 13:560–564.
51. Weinberg, AG, Finegold, MJ. Primary hepatic tumors of childhood. Hum Pathol. 1983; 14:512–537.
52. Luks, FI, Yazbeck, S, Brandt, ML, et al. Benign liver tumors in children: A 25-year experience. J Pediatr Surg. 1991; 26:1326–1330.
53. Ehren, H, Mahour, GH, Isaacs, H, Jr. Benign liver tumors in infancy and childhood. Report of 48 cases. Am J Surg. 1983; 145:325–329.
54. Caty, MG, Shamberger, RC. Abdominal tumors in infancy and childhood. Pediatr Clin North Am. 1993; 40:1253–1271.
55. Stocker, JT, Ishak, KG. Mesenchymal hamartoma of the liver: Report of 30 cases and review of the literature. Pediatr Pathol. 1983; 1:245–267.
56. Helal, A, Nolan, M, Bower, R, et al. Pathological case of the month. Mesenchymal hamartoma of the liver. Arch Pediatr Adolesc Med. 1995; 149:315–316.
57. von Schweinitz, D, Dammeier, BG, Gluer, S. Mesenchymal hamartoma of the liver–New insight into histogenesis. J Pediatr Surg. 1999; 34:1269–1271.
58. Shintaku, M, Watanabe, K. Mesenchymal hamartoma of the liver: A proliferative lesion of possible hepatic stellate cell (Ito cell) origin. Pathol Res Pract. 2010; 206:532–536.
59. Reed, RC, Kapur, RP. Hepatic mesenchymal hamartoma: A disorder of imprinting. Pediatr Dev Pathol. 2008; 11:264–265.
60. Mascarello, JT, Krous, HF. Second report of a translocation involving 19q13.4 in a mesenchymal hamartoma of the liver. Cancer Genet Cytogenet. 1992; 58:141–142.
61. Speleman, F, De Telder, V, De Potter, KR, et al. Cytogenetic analysis of a mesenchymal hamartoma of the liver. Cancer Genet Cytogenet. 1989; 40:29–32.
62. Otal, TM, Hendricks, JB, Pharis, P, et al. Mesenchymal hamartoma of the liver. DNA flow cytometric analysis of eight cases. Cancer. 1994; 74:1237–1242.
63. Stringer, MD, Alizai, NK. Mesenchymal hamartoma of the liver: A systematic review. J Pediatr Surg. 2005; 40:1681–1690.
64. Kamata, S, Nose, K, Sawai, T, et al. Fetal mesenchymal hamartoma of the liver: Report of a case. J Pediatr Surg. 2003; 38:639–641.
65. Dickinson, JE, Knowles, S, Phillips, JM. Prenatal diagnosis of hepatic mesenchymal hamartoma. Prenat Diagn. 1999; 19:81–84.
66. Cornette, J, Festen, S, van den Hoonaard, TL, et al. Mesenchymal hamartoma of the liver: A benign tumor with deceptive prognosis in the perinatal period. Case report and review of the literature. Fetal Diagn Ther. 2009; 25:196–202.
67. Tsao, K, Hirose, S, Sydorak, R, et al. Fetal therapy for giant hepatic cysts. J Pediatr Surg. 2002; 37:E31.
68. Balmer, B, Le Coultre, C, Feldges, A, et al. Mesenchymal liver hamartoma in a newborn; case report. Eur J Pediatr Surg. 1996; 6:303–305.
69. Ros, PR, Goodman, ZD, Ishak, KG, et al. Mesenchymal hamartoma of the liver: Radiologic-pathologic correlation. Radiology. 1986; 158:619–624.
70. Wan, P, Susman, J, Kandel, J, et al. Neonatal hepatic mesenchymal hamartoma causing cardiac failure and disseminated intravascular coagulopathy. Am J Perinatol. 2009; 26:601–604.
71. Justrabo, E, Martin, L, Yaziji, N, et al. Hepatic mesenchymal hamartoma in children. Immunohistochemical, ultrastructural and flow cytometric case study. Gastroenterol Clin Biol. 1998; 22:964–968.
72. Unal, E, Koksal, Y, Akcoren, Z, et al. Mesenchymal hamartoma of the liver mimicking hepatoblastoma. J Pediatr Hematol Oncol. 2008; 30:458–460.
73. Raffensperger, JG, Gonzalez-Crussi, F, Skeehan, T. Mesenchymal hamartoma of the liver. J Pediatr Surg. 1983; 18:585–587.
74. Ito, H, Kishikawa, T, Toda, T, et al. Hepatic mesenchymal hamartoma of an infant. J Pediatr Surg. 1984; 19:315–317.
75. Abramson, SJ, Lack, EE, Teele, RL. Benign vascular tumors of the liver in infants: Sonographic appearance. AJR Am J Roentgenol. 1982; 138:629–632.
76. Steiner, MA, Giles, HW. Mesenchymal hamartoma of the liver demonstrating peripheral calcification in a 12-year-old boy. Pediatr Radiol. 2008; 38:1232–1234.
77. Ye, BB, Hu, B, Wang, LJ, et al. Mesenchymal hamartoma of liver: Magnetic resonance imaging and histopathologic correlation. World J Gastroenterol. 2005; 11:5807–5810.
78. Moore, M, Anupindi, SA, Mattei, P, et al. Mesenchymal cystic hamartoma of the liver: MR imaging with pathologic correlation. J Radiol Case Rep. 2009; 3:22–26.
79. Koumanidou, C, Vakaki, M, Papadaki, M, et al. New sonographic appearance of hepatic mesenchymal hamartoma in childhood. J Clin Ultrasound. 1999; 27:164–167.
80. Shehata, BM, Gupta, NA, Katzenstein, HM, et al. Undifferentiated embryonal sarcoma of the liver is associated with mesenchymal hamartoma and multiple chromosomal abnormalities: A review of eleven cases. Pediatr Dev Pathol. 2011; 14:111–116.
81. Sharif, K, Ramani, P, Lochbuhler, H, et al. Recurrent mesenchymal hamartoma associated with 19q translocation. A call for more radical surgical resection. Eur J Pediatr Surg. 2006; 16:64–67.
82. Karpelowsky, JS, Pansini, A, Lazarus, C, et al. Difficulties in the management of mesenchymal hamartomas. Pediatr Surg Int. 2008; 24:1171–1175.
83. Anil, G, Fortier, M, Low, Y. Cystic hepatic mesenchymal hamartoma: The role of radiology in diagnosis and perioperative management. Br J Radiol. 2011; 84:e91–e94.
84. Barnhart, DC, Hirschl, RB, Garver, KA, et al. Conservative management of mesenchymal hamartoma of the liver. J Pediatr Surg. 1997; 32:1495–1498.
85. de Chadarevian, JP, Pawel, BR, Faerber, EN, et al. Undifferentiated (embryonal) sarcoma arising in conjunction with mesenchymal hamartoma of the liver. Mod Pathol. 1994; 7:490–493.
86. O’Sullivan, MJ, Swanson, PE, Knoll, J, et al. Undifferentiated embryonal sarcoma with unusual features arising within mesenchymal hamartoma of the liver: Report of a case and review of the literature. Pediatr Dev Pathol. 2001; 4:482–489.
87. Ramanujam, TM, Ramesh, JC, Goh, DW, et al. Malignant transformation of mesenchymal hamartoma of the liver: Case report and review of the literature. J Pediatr Surg. 1999; 34:1684–1686.
88. Millard, J, Fraser, N, Stewart, RJ. Mesenchymal hamartoma of the liver: Is biopsy always necessary? Pediatr Surg Int. 2006; 22:622–625.
89. Rajaram, V, Knezevich, S, Bove, KE, et al. DNA sequence of the translocation breakpoints in undifferentiated embryonal sarcoma arising in mesenchymal hamartoma of the liver harboring the t(11;19)(q11;q13.4) translocation. Genes Chromosomes Cancer. 2007; 46:508–513.
90. Talmon, GA, Cohen, SM. Mesenchymal hamartoma of the liver with an interstitial deletion involving chromosome band 19q13.4: A theory as to pathogenesis? Arch Pathol Lab Med. 2006; 130:1216–1218.
91. Reymond, D, Plaschkes, J, Luthy, AR, et al. Focal nodular hyperplasia of the liver in children: Review of follow-up and outcome. J Pediatr Surg. 1995; 30:1590–1593.
92. Stocker, JT, Ishak, KG. Focal nodular hyperplasia of the liver: A study of 21 pediatric cases. Cancer. 1981; 48:336–345.
93. Okugawa, Y, Uchida, K, Inoue, M, et al. Focal nodular hyperplasia in biliary atresia patient after Kasai hepatic portoenterostomy. Pediatr Surg Int. 2008; 24:609–612.
94. Ra, SH, Kaplan, JB, Lassman, CR. Focal nodular hyperplasia after orthotopic liver transplantation. Liver Transpl. 2010; 16:98–103.
95. Savoye-Collet, C, Herve, S, Koning, E, et al. Focal nodular hyperplasia occurring after blunt abdominal trauma. Eur J Gastroenterol Hepatol. 2002; 14:329–330.
96. Iordanidis, F, Hytiroglou, P, Drevelegas, A, et al. A 25-year-old man with a large hepatic tumor and multiple nodular lesions. Semin Liver Dis. 2002; 22:97–102.
97. Hohler, T, Lohse, AW, Schirmacher, P. Progressive focal nodular hyperplasia of the liver in a patient with genetic hemochromatosis–growth promotion by iron overload? Dig Dis Sci. 2000; 45:587–590.
98. Santarelli, L, Gabrielli, M, Orefice, R, et al. Association between Klinefelter syndrome and focal nodular hyperplasia. J Clin Gastroenterol. 2003; 37:189–191.
99. Scalori, A, Tavani, A, Gallus, S, et al. Risk factors for focal nodular hyperplasia of the liver: An Italian case-control study. Am J Gastroenterol. 2002; 97:2371–2373.
100. Wolf, R, Wolf, D, Kuperman, S. Focal nodular hyperplasia of the liver after intraconazole treatment. J Clin Gastroenterol. 2001; 33:418–420.
101. Knowles, DM, Wolff, M. Focal nodular hyperplasia of the liver: A clinicopathologic study and review of the literature. Hum Pathol. 1976; 7:533–545.
102. Mathieu, D, Zafrani, ES, Anglade, MC, et al. Association of focal nodular hyperplasia and hepatic hemangioma. Gastroenterology. 1989; 97:154–157.
103. Buscarini, E, Danesino, C, Plauchu, H, et al. High prevalence of hepatic focal nodular hyperplasia in subjects with hereditary hemorrhagic telangiectasia. Ultrasound Med Biol. 2004; 30:1089–1097.
104. Paradis, V, Bieche, I, Dargere, D, et al. A quantitative gene expression study suggests a role for angiopoietins in focal nodular hyperplasia. Gastroenterology. 2003; 124:651–659.
105. Kumagai, H, Masuda, T, Oikawa, H, et al. Focal nodular hyperplasia of the liver: Direct evidence of circulatory disturbances. J Gastroenterol Hepatol. 2000; 15:1344–1347.
106. Kinjo, T, Aoki, H, Sunagawa, H, et al. Congenital absence of the portal vein associated with focal nodular hyperplasia of the liver and congenital choledochal cyst: A case report. J Pediatr Surg. 2001; 36:622–625.
107. Altavilla, G, Guariso, G. Focal nodular hyperplasia of the liver associated with portal vein agenesis: A morphological and immunohistochemical study of one case and review of the literature. Adv Clin Path. 1999; 3:139–145.
108. Tanaka, Y, Takayanagi, M, Shiratori, Y, et al. Congenital absence of portal vein with multiple hyperplastic nodular lesions in the liver. J Gastroenterol. 2003; 38:288–294.
109. Osorio, MJ, Bonow, A, Bond, GJ, et al. Abernethy malformation complicated by hepatopulmonary syndrome and a liver mass successfully treated by liver transplantation. Pediatr Transplant. 2011; 15:E149–E151.
110. Lisovsky, M, Konstas, AA, Misdraji, J. Congenital extrahepatic portosystemic shunts (Abernethy malformation): A histopathologic evaluation. Am J Surg Pathol. 2011; 35:1381–1390.
111. Bouyn, CI, Leclere, J, Raimondo, G, et al. Hepatic focal nodular hyperplasia in children previously treated for a solid tumor. Incidence, risk factors, and outcome. Cancer. 2003; 97:3107–3113.
112. Towbin, AJ, Luo, GG, Yin, H, et al. Focal nodular hyperplasia in children, adolescents, and young adults. Pediatr Radiol. 2011; 41:341–349.
113. Benz-Bohm, G, Hero, B, Gossmann, A, et al. Focal nodular hyperplasia of the liver in longterm survivors of neuroblastoma: How much diagnostic imaging is necessary? Eur J Radiol. 2010; 74:e1–e5.
114. Gutweiler, JR, Yu, DC, Kim, HB, et al. Hepatoblastoma presenting with focal nodular hyperplasia after treatment of neuroblastoma. J Pediatr Surg. 2008; 43:2297–2300.
115. Gobbi, D, Dall’Igna, P, Messina, C, et al. Focal nodular hyperplasia in pediatric patients with and without oncologic history. Pediatr Blood Cancer. 2010; 551420–551422.
116. Sugito, K, Uekusa, S, Kawashima, H, et al. The clinical course in pediatric solid tumor patients with focal nodular hyperplasia of the liver. Int J Clin Oncol. 2011; 16:482–487.
117. Sudour, H, Mainard, L, Baumann, C, et al. Focal nodular hyperplasia of the liver following hematopoietic SCT. Bone Marrow Transplant. 2009; 43:127–132.
118. Anderson, L, Gregg, D, Margolis, D, et al. Focal nodular hyperplasia in pediatric allogeneic hematopoietic cell transplant: Case series. Bone Marrow Transplant. 2010; 45:1357–1359.
119. Masetti, R, Biagi, C, Kleinschmidt, K, et al. Focal nodular hyperplasia of the liver after intensive treatment for pediatric cancer: Is hematopoietic stem cell transplantation a risk factor? Eur J Pediatr. 2011; 170:807–812.
120. Citak, EC, Karadeniz, C, Oguz, A, et al. Nodular regenerative hyperplasia and focal nodular hyperplasia of the liver mimicking hepatic metastasis in children with solid tumors and a review of literature. Pediatr Hematol Oncol. 2007; 24:281–289.
121. Cheon, JE, Kim, WS, Kim, IO, et al. Radiological features of focal nodular hyperplasia of the liver in children. Pediatr Radiol. 1998; 28:878–883.
122. Somech, R, Brazowski, E, Kesller, A, et al. Focal nodular hyperplasia in children. J Pediatr Gastroenterol Nutr. 2001; 32:480–483.
123. Bioulac-Sage, P, Balabaud, C, Wanless, IR. Diagnosis of focal nodular hyperplasia: Not so easy. Am J Surg Pathol. 2001; 25:1322–1325.
124. Huang, YE, Wang, PW, Huang, HH, et al. A central scar in hepatic focal nodular hyperplasia detected on liver SPECT imaging. Clin Nucl Med. 2001; 26:367–369.
125. Swingle, CA, Fajman, WA, Alazraki, N. Early enhancing lesion seen on computed tomography consistent with focal nodular hyperplasia. Clin Nucl Med. 2003; 28:134–135.
126. Steiner, D, Klett, R, Puille, M, et al. Diagnosis of focal nodular hyperplasia with hepatobiliary scintigraphy using a modified SPECT technique. Clin Nucl Med. 2003; 28:136–137.
127. Casillas, VJ, Amendola, MA, Gascue, A, et al. Imaging of nontraumatic hemorrhagic hepatic lesions. Radiographics. 2000; 20:367–378.
128. Marin, D, Brancatelli, G, Federle, MP, et al. Focal nodular hyperplasia: Typical and atypical MRI findings with emphasis on the use of contrast media. Clin Radiol. 2008; 63:577–585.
129. Ba-Ssalamah, A, Schima, W, Schmook, MT, et al. Atypical focal nodular hyperplasia of the liver: Imaging features of nonspecific and liver-specific MR contrast agents. AJR Am J Roentgenol. 2002; 179:1447–1456.
130. Asbach, P, Klessen, C, Koch, M, et al. Magnetic resonance imaging findings of atypical focal nodular hyperplasia of the liver. Clin Imaging. 2007; 31:244–252.
131. Nguyen, BN, Flejou, JF, Terris, B, et al. Focal nodular hyperplasia of the liver: A comprehensive pathologic study of 305 lesions and recognition of new histologic forms. Am J Surg Pathol. 1999; 23:1441–1454.
132. Bioulac-Sage, P, Rebouissou, S, Thomas, C, et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology. 2007; 46:740–748.
133. Wanless, IR, Albrecht, S, Bilbao, J, et al. Multiple focal nodular hyperplasia of the liver associated with vascular malformations of various organs and neoplasia of the brain: A new syndrome. Mod Pathol. 1989; 2:456–462.
134. Ahmad, I, Iyer, A, Marginean, CE, et al. Diagnostic use of cytokeratins, CD34, and neuronal cell adhesion molecule staining in focal nodular hyperplasia and hepatic adenoma. Hum Pathol. 2009; 40:726–734.
135. Petsas, T, Tsamandas, A, Tsota, I, et al. A case of hepatocellular carcinoma arising within large focal nodular hyperplasia with review of the literature. World J Gastroenterol. 2006; 12:6567–6571.
136. Langrehr, JM, Pfitzmann, R, Hermann, M, et al. Hepatocellular carcinoma in association with hepatic focal nodular hyperplasia. Acta Radiol. 2006; 47:340–344.
137. Saul, SH, Titelbaum, DS, Gansler, TS, et al. The fibrolamellar variant of hepatocellular carcinoma. Its association with focal nodular hyperplasia. Cancer. 1987; 60:3049–3055.
138. Saxena, R, Humphreys, S, Williams, R, et al. Nodular hyperplasia surrounding fibrolamellar carcinoma: A zone of arterialized liver parenchyma. Histopathology. 1994; 25:275–278.
139. Fabre, A, Audet, P, Vilgrain, V, et al. Histologic scoring of liver biopsy in focal nodular hyperplasia with atypical presentation. Hepatology. 2002; 35:414–420.
140. Li, T, Qin, LX, Ji, Y, et al. Atypical hepatic focal nodular hyperplasia presenting as acute abdomen and misdiagnosed as hepatocellular carcinoma. Hepatol Res. 2007; 37:1100–1105.
141. Okada, T, Sasaki, F, Kamiyama, T, et al. Management and algorithm for focal nodular hyperplasia of the liver in children. Eur J Pediatr Surg. 2006; 16:235–240.
142. Yang, Y, Fu, S, Li, A, et al. Management and surgical treatment for focal nodular hyperplasia in children. Pediatr Surg Int. 2008; 24:699–703.
143. Lautz, T, Tantemsapya, N, Dzakovic, A, et al. Focal nodular hyperplasia in children: Clinical features and current management practice. J Pediatr Surg. 2010; 45:1797–1803.
144. Pain, JA, Gimson, AE, Williams, R, et al. Focal nodular hyperplasia of the liver: Results of treatment and options in management. Gut. 1991; 32:524–527.
145. Di Stasi, M, Caturelli, E, De Sio, I, et al. Natural history of focal nodular hyperplasia of the liver: An ultrasound study. J Clin Ultrasound. 1996; 24:345–350.
146. Ohmoto, K, Honda, T, Hirokawa, M, et al. Spontaneous regression of focal nodular hyperplasia of the liver. J Gastroenterol. 2002; 37:849–853.
147. Leconte, I, Van Beers, BE, Lacrosse, M, et al. Focal nodular hyperplasia: Natural course observed with CT and MRI. J Comput Assist Tomogr. 2000; 24:61–66.
148. Geschwind, JF, Degli, MS, Morris, JM, et al. Re: Treatment of focal nodular hyperplasia with selective transcatheter arterial embolization using iodized oil and polyvinyl alcohol. Cardiovasc Intervent Radiol. 2002; 25:340–341.
149. Hung, CH, Changchien, CS, Lu, SN, et al. Sonographic features of hepatic adenomas with pathologic correlation. Abdom Imaging. 2001; 26:500–506.
150. Wilhelm, L, Albrecht, L, Kirsch, M, et al. Preoperative application of selective angiographic embolization in the treatment of focal nodular hyperplasia. Surg Laparosc Endosc Percutan Tech. 2006; 16:177–181.
151. Ichikawa, T, Federle, MP, Grazioli, L, et al. Hepatocellular adenoma: Multiphasic CT and histopathologic findings in 25 patients. Radiology. 2000; 214:861–868.
152. Paulson, EK, McClellan, JS, Washington, K, et al. Hepatic adenoma: MR characteristics and correlation with pathologic findings. AJR Am J Roentgenol. 1994; 163:113–116.
153. Grazioli, L, Federle, MP, Brancatelli, G, et al. Hepatic adenomas: Imaging and pathologic findings. Radiographics. 2001; 21:877–894.
154. Das, CJ, Dhingra, S, Gupta, AK, et al. Imaging of paediatric liver tumours with pathological correlation. Clin Radiol. 2009; 64:1015–1025.
155. Edmonson, HA. Atlas of Tumor Pathology: Tumors of the liver and intrahepatic bile ducts. Washington, D.C.: Armed Forces Institute of Pathology; 1958.
156. Lizardi-Cervera, J, Cuellar-Gamboa, L, Motola-Kuba, D. Focal nodular hyperplasia and hepatic adenoma: A review. Ann Hepatol. 2006; 5:206–211.
157. Giannitrapani, L, Soresi, M, La Spada, E, et al. Sex hormones and risk of liver tumor. Ann N Y Acad Sci. 2006; 1089:228–236.
158. Reddy, KR, Schiff, ER. Approach to a liver mass. Semin Liver Dis. 1993; 13:423–435.
159. Bagia, S, Hewitt, PM, Morris, DL. Anabolic steroid-induced hepatic adenomas with spontaneous haemorrhage in a bodybuilder. Aust N Z J Surg. 2000; 70:686–687.
160. Toso, C, Rubbia-Brandt, L, Negro, F, et al. Hepatocellular adenoma and polycystic ovary syndrome. Liver Int. 2003; 23:35–37.
161. Adusumilli, PS, Lee, B, Parekh, K, et al. Hemoperitoneum from spontaneous rupture of a liver cell adenoma in a male with hyperthyroidism. Am Surg. 2002; 68:582–583.
162. Marie-Cardine, A, Schneider, P, Greene, V, et al. Erythrocytosis in a child with a hepatic adenoma. Med Pediatr Oncol. 2001; 36:659–661.
163. Howell, RR, Stevenson, RE, Ben-Menachem, Y, et al. Hepatic adenomata with type 1 glycogen storage disease. JAMA. 1976; 236:1481–1484.
164. Coire, CI, Qizilbash, AH, Castelli, MF. Hepatic adenomata in type Ia glycogen storage disease. Arch Pathol Lab Med. 1987; 111:166–169.
165. Mason, HH, Andersen, DH. Glycogen disease of the liver (von Gierke’s disease) with hepatomata; case report with metabolic studies. Pediatrics. 1955; 16:785–800.
166. Zangeneh, F, Limbeck, GA, Brown, BI, et al. Hepatorenal glycogenosis (type I glycogenosis) and carcinoma of the liver. J Pediatr. 1969; 74:73–83.
167. Wang, DQ, Fiske, LM, Carreras, CT, et al. Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr. 2011; 159:442–446.
168. Fraumeni, JF, Jr., Miller, RW, Hill, JA. Primary carcinoma of the liver in childhood: An epidemiologic study. J Natl Cancer Inst. 1968; 40:1087–1099.
169. Nakamura, T, Tamakoshi, K, Kitagawa, M, et al. A case of hepatocellular carcinoma in type I a glycogen storage disease. Nihon Shokakibyo Gakkai Zasshi. 1997; 94:866–870.
170. Labrune, P, Trioche, P, Duvaltier, I, et al. Hepatocellular adenomas in glycogen storage disease type I and III: A series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997; 24:276–279.
171. Kerlin, P, Davis, GL, McGill, DB, et al. Hepatic adenoma and focal nodular hyperplasia: Clinical, pathologic, and radiologic features. Gastroenterology. 1983; 84(5 Pt 1):994–1002.
172. Foster, JH, Berman, MM. The malignant transformation of liver cell adenomas. Arch Surg. 1994; 129:712–717.
173. Tao, LC. Oral contraceptive-associated liver cell adenoma and hepatocellular carcinoma. Cytomorphology and mechanism of malignant transformation. Cancer. 1991; 68:341–347.
174. Micchelli, ST, Vivekanandan, P, Boitnott, JK, et al. Malignant transformation of hepatic adenomas. Mod Pathol. 2008; 21:491–497.
175. Kishnani, PS, Chuang, TP, Bali, D, et al. Chromosomal and genetic alterations in human hepatocellular adenomas associated with type Ia glycogen storage disease. Hum Mol Genet. 2009; 18:4781–4790.
176. Chiche, L, Dao, T, Salame, E, et al. Liver adenomatosis: Reappraisal, diagnosis, and surgical management: eight new cases and review of the literature. Ann Surg. 2000; 231:74–81.
177. Musthafa, CP, Chettupuzha, AP, Syed, AA, et al. Liver adenomatosis. Indian J Gastroenterol. 2003; 22:30–31.
178. Vetelainen, R, Erdogan, D, de Graaf, W, et al. Liver adenomatosis: Re-evaluation of aetiology and management. Liver Int. 2008; 28:499–508.
179. Babaoglu, K, Binnetoglu, FK, Aydo an, A, et al. Hepatic adenomatosis in a 7-year-old child treated earlier with a Fontan procedure. Pediatr Cardiol. 2010; 31:861–864.
an, A, et al. Hepatic adenomatosis in a 7-year-old child treated earlier with a Fontan procedure. Pediatr Cardiol. 2010; 31:861–864.
180. Takayasu, H, Motoi, T, Kanamori, Y, et al. Two case reports of childhood liver cell adenomas harboring beta-catenin abnormalities. Hum Pathol. 2002; 33:852–855.
181. Chen, YW, Jeng, YM, Yeh, SH, et al. P53 gene and Wnt signaling in benign neoplasms: Beta-catenin mutations in hepatic adenoma but not in focal nodular hyperplasia. Hepatology. 2002; 36(4 Pt 1):927–935.
182. Rebouissou, S, Bioulac-Sage, P, Zucman-Rossi, J. Molecular pathogenesis of focal nodular hyperplasia and hepatocellular adenoma. J Hepatol. 2008; 48:163–170.
183. Aseni, P, Sansalone, CV, Sammartino, C, et al. Rapid disappearance of hepatic adenoma after contraceptive withdrawal. J Clin Gastroenterol. 2001; 33:234–236.
184. Steinbrecher, UP, Lisbona, R, Huang, SN, et al. Complete regression of hepatocellular adenoma after withdrawal of oral contraceptives. Dig Dis Sci. 1981; 26:1045–1050.
185. Ramseur, WL, Cooper, MR. Asymptomatic liver cell adenomas. Another case of resolution after discontinuation of oral contraceptive use. JAMA. 1978; 239:1647–1648.
186. Barthelmes, L, Tait, IS. Liver cell adenoma and liver cell adenomatosis. HPB (Oxford). 2005; 7:186–196.
187. Ault, GT, Wren, SM, Ralls, PW, et al. Selective management of hepatic adenomas. Am Surg. 1996; 62:825–829.
188. Leese, T, Farges, O, Bismuth, H. Liver cell adenomas. A 12-year surgical experience from a specialist hepato-biliary unit. Ann Surg. 1988; 208:558–564.
189. Stoot, JH, van der Linden, E, Terpstra, OT, et al. Life-saving therapy for haemorrhaging liver adenomas using selective arterial embolization. Br J Surg. 2007; 94:1249–1253.
190. Terkivatan, T, de Wilt, JH, de Man, RA, et al. Treatment of ruptured hepatocellular adenoma. Br J Surg. 2001; 88:207–209.
191. Heeringa, B, Sardi, A. Bleeding hepatic adenoma: Expectant treatment to limit the extent of liver resection. Am Surg. 2001; 67:927–929.
192. Ross, JA. Hepatoblastoma and birth weight: Too little, too big, or just right? J Pediatr. 1997; 130:516–517.
193. Stocker, J, Conran, R, Selby, D. Tumor and Pseudo Tumors of the Liver. London: Chapman & Hall; 1998.
194. Lack, EE, Neave, C, Vawter, GF. Hepatoblastoma. A clinical and pathologic study of 54 cases. Am J Surg Pathol. 1982; 6:693–705.
195. Martelli, C, Blandamura, S, Massaro, S, et al. A case study of Beckwith-Wiedemann syndrome associated with hepatoblastoma. Clin Exp Obstet Gynecol. 1993; 20:82–87.
196. McNeil, DE, Brown, M, Ching, A, et al. Screening for Wilms tumor and hepatoblastoma in children with Beckwith-Wiedemann syndromes: A cost-effective model. Med Pediatr Oncol. 2001; 37:349–356.
197. Tan, TY, Amor, DJ. Tumour surveillance in Beckwith-Wiedemann syndrome and hemihyperplasia: A critical review of the evidence and suggested guidelines for local practice. J Paediatr Child Health. 2006; 42:486–490.
198. Aretz, S, Koch, A, Uhlhaas, S, et al. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer. 2006; 47:811–818.
199. Hirschman, BA, Pollock, BH, Tomlinson, GE. The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr. 2005; 147:263–266.
200. Ikeda, H, Matsuyama, S, Tanimura, M. Association between hepatoblastoma and very low birth weight: A trend or a chance? J Pediatr. 1997; 130:557–560.
201. McLaughlin, CC, Baptiste, MS, Schymura, MJ, et al. Maternal and infant birth characteristics and hepatoblastoma. Am J Epidemiol. 2006; 163:818–828.
202. Reynolds, P, Urayama, KY, Von Behren, J, et al. Birth characteristics and hepatoblastoma risk in young children. Cancer. 2004; 100:1070–1076.
203. Ikeda, H, Hachitanda, Y, Tanimura, M, et al. Development of unfavorable hepatoblastoma in children of very low birth weight: Results of a surgical and pathologic review. Cancer. 1998; 82:1789–1796.
204. Maruyama, K, Ikeda, H, Koizumi, T, et al. Case-control study of perinatal factors and hepatoblastoma in children with an extremely low birthweight. Pediatr Int. 2000; 42:492–498.
205. Shew, SB, Keshen, TH, Jahoor, F, et al. Assessment of cysteine synthesis in very low-birth weight neonates using a 13C6 glucose tracer. J Pediatr Surg. 2005; 40:52–56.
206. Rowland, JM. Hepatoblastoma: assessment of criteria for histologic classification. Med Pediatr Oncol. 2002; 39:478–483.
207. Haas, JE, Feusner, JH, Finegold, MJ. Small cell undifferentiated histology in hepatoblastoma may be unfavorable. Cancer. 2001; 92:3130–3134.
208. Stringer, MD, Hennayake, S, Howard, ER, et al. Improved outcome for children with hepatoblastoma. Br J Surg. 1995; 82:386–391.
209. Haas, JE, Muczynski, KA, Krailo, M, et al. Histopathology and prognosis in childhood hepatoblastoma and hepatocarcinoma. Cancer. 1989; 64:1082–1095.
210. Nickerson, HJ, Silberman, TL, McDonald, TP. Hepatoblastoma, thrombocytosis, and increased thrombopoietin. Cancer. 1980; 45:315–317.
211. von Schweinitz, D, Hadam, MR, Welte, K, et al. Production of interleukin-1 beta and interleukin-6 in hepatoblastoma. Int J Cancer. 1993; 53:728–734.
212. Oda, H, Imai, Y, Nakatsuru, Y, et al. Somatic mutations of the APC gene in sporadic hepatoblastomas. Cancer Res. 1996; 56:3320–3323.
213. Surace, C, Leszl, A, Perilongo, G, et al. Fluorescent in situ hybridization (FISH) reveals frequent and recurrent numerical and structural abnormalities in hepatoblastoma with no informative karyotype. Med Pediatr Oncol. 2002; 39:536–539.
214. Maruyama, K, Ikeda, H, Koizumi, T. Hepatoblastoma associated with trisomy 18 syndrome: A case report and a review of the literature. Pediatr Int. 2001; 43:302–305.
215. Giardiello, FM, Petersen, GM, Brensinger, JD, et al. Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut. 1996; 39:867–869.
216. Yamaoka, H, Ohtsu, K, Sueda, T, et al. Diagnostic and prognostic impact of beta-catenin alterations in pediatric liver tumors. Oncol Rep. 2006; 15:551–556.
217. Miller, JH, Greenspan, BS. Integrated imaging of hepatic tumors in childhood. Part I: Malignant lesions (primary and metastatic). Radiology. 1985; 154:83–90.
218. Powers, C, Ros, PR, Stoupis, C, et al. Primary liver neoplasms: MR imaging with pathologic correlation. Radiographics. 1994; 14:459–482.
219. Shafford, EA, Pritchard, J. Extreme thrombocytosis as a diagnostic clue to hepatoblastoma. Arch Dis Child. 1993; 69:171.
220. Stocker, J, Conran, R. Hepatoblastoma. New York: Churchill Livingstone; 1997.
221. Jassam, N, Jones, CM, Briscoe, T, et al. The hook effect: A need for constant vigilance. Ann Clin Biochem. 2006; 43(Pt 4):314–317.
222. Ortega, J, Siegel, S. Biological Markers in Pediatric Cancer. Philadelphia: JB Lippincott; 1989.
223. Kairemo, KJ, Lindahl, H, Merenmies, J, et al. Anti-alpha-fetoprotein imaging is useful for staging hepatoblastoma. Transplantation. 2002; 73:1151–1154.
224. Zynger, DL, Gupta, A, Luan, C, et al. Expression of glypican 3 in hepatoblastoma: An immunohistochemical study of 65 cases. Hum Pathol. 2008; 39:224–230.
225. Stringer, MD. Liver tumors. Semin Pediatr Surg. 2000; 9:196–208.
226. MacKinlay, GA, Pritchard, J. A common language for childhood liver tumors. Pediatr Surg Int. 1992; 7:325–326.
227. Roebuck, DJ, Aronson, D, Clapuyt, P, et al. 2005 PRETEXT: A revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol. 2007; 37:123–132.
228. Cho, JY, Han, HS, Yoon, YS, et al. Experiences of laparoscopic liver resection including lesions in the posterosuperior segments of the liver. Surg Endosc. 2008; 22:2344–2349.
229. Koffron, AJ, Auffenberg, G, Kung, R, et al. Evaluation of 300 minimally invasive liver resections at a single institution: Less is more. Ann Surg. 2007; 246:385–394.
230. Yoon, YS, Han, HS, Choi, YS, et al. Total laparoscopic left lateral sectionectomy performed in a child with benign liver mass. J Pediatr Surg. 2006; 41:e25–e28.
231. Dutta, S, Nehra, D, Woo, R, et al. Laparoscopic resection of a benign liver tumor in a child. J Pediatr Surg. 2007; 42:1141–1145.
232. Dicken, BJ, Bigam, DL, Lees, GM. Association between surgical margins and long-term outcome in advanced hepatoblastoma. J Pediatr Surg. 2004; 39:721–725.
233. Ortega, JA, Douglass, EC, Feusner, JH, et al. Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children’s Cancer Group and the Pediatric Oncology Grou. J Clin Oncol. 2000; 18:2665–2675.
234. Carceller, A, Blanchard, H, Champagne, J, et al. Surgical resection and chemotherapy improve survival rate for patients with hepatoblastoma. J Pediatr Surg. 2001; 36:755–759.
235. Katzenstein, HM, Krailo, M, Malogolowkin, MH, et al, Biology and treatment of children with all stages of hepatoblastoma: COG proposal AHEP-0731, submitted to CTEP and NCI, 2007.
236. Perilongo, G, Shafford, E, Maibach, R, et al. Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology–SIOPEL 2. Eur J Cancer. 2004; 40:411–421.
237. Schnater, JM, Aronson, DC, Plaschkes, J, et al. Surgical view of the treatment of patients with hepatoblastoma: Results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Grou. Cancer. 2002; 94:1111–1120.
238. Casas-Melley, AT, Malatack, J, Consolini, D, et al. Successful liver transplant for unresectable hepatoblastoma. J Pediatr Surg. 2007; 42:184–187.
239. Chen, LE, Shepherd, RW, Nadler, ML, et al. Liver transplantation and chemotherapy in children with unresectable primary hepatic malignancies: Development of a management algorithm. J Pediatr Gastroenterol Nutr. 2006; 43:487–493.
240. Ang, JP, Heath, JA, Donath, S, et al. Treatment outcomes for hepatoblastoma: An institution’s experience over two decades. Pediatr Surg Int. 2007; 23:103–109.
241. Tiao, GM, Bobey, N, Allen, S, et al. The current management of hepatoblastoma: A combination of chemotherapy, conventional resection, and liver transplantation. J Pediatr. 2005; 146:204–211.
242. Raney, B. Hepatoblastoma in children: A review. J Pediatr Hematol Oncol. 1997; 19:418–422.
243. Feusner, JH, Krailo, MD, Haas, JE, et al. Treatment of pulmonary metastases of initial stage I hepatoblastoma in childhood. Report from the Childrens Cancer Group. Cancer. 1993; 71:859–864.
244. Otte, JB, de Ville de Goyet, J. The contribution of transplantation to the treatment of liver tumors in children. Semin Pediatr Surg. 2005; 14:233–238.
245. Perilongo, G, Shafford, EA. Liver tumours. Eur J Cancer. 1999; 35:953–959.
246. Malogolowkin, MH, Katzenstein, H, Krailo, MD, et al. Intensified platinum therapy is an ineffective strategy for improving outcome in pediatric patients with advanced hepatoblastoma. J Clin Oncol. 2006; 24:2879–2884.
247. Brown, J, Perilongo, G, Shafford, E, et al. Pretreatment prognostic factors for children with hepatoblastoma– results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer. 2000; 36:1418–1425.
248. von Schweinitz, D, Byrd, DJ, Hecker, H, et al. Efficiency and toxicity of ifosfamide, cisplatin and doxorubicin in the treatment of childhood hepatoblastoma. Study Committee of the Cooperative Paediatric Liver Tumour Study HB89 of the German Society for Paediatric Oncology and Haematology. Eur J Cancer. 1997; 33:1243–1249.
249. von Schweinitz, D, Burger, D, Bode, U, et al. Results of the HB-89 Study in treatment of malignant epithelial liver tumors in childhood and concept of a new HB-94 protocol. Klin Padiatr. 1994; 206:282–288.
250. Stringer, MD. The role of liver transplantation in the management of paediatric liver tumours. Ann R Coll Surg Engl. 2007; 89:12–21.
251. McCrudden, KW, Yokoi, A, Thosani, A, et al. Topotecan is anti-angiogenic in experimental hepatoblastoma. J Pediatr Surg. 2002; 37:857–861.
252. Katzenstein, HM, Rigsby, C, Shaw, PH, et al. Novel therapeutic approaches in the treatment of children with hepatoblastoma. J Pediatr Hematol Oncol. 2002; 24(9):751–755.
253. Tashjian, DB, Moriarty, KP, Courtney, RA, et al. Preoperative chemoembolization for unresectable hepatoblastoma. Pediatr Surg Int. 2002; 18:187–189.
254. Xuewu, J, Jianhong, L, Xianliang, H, et al. Combined treatment of hepatoblastoma with transcatheter arterial chemoembolization and surgery. Pediatr Hematol Oncol. 2006; 23:1–9.
255. Chan, KL, Fan, ST, Tam, PK, et al. Management of spontaneously ruptured hepatoblastoma in infancy. Med Pediatr Oncol. 2002; 38:137–138.
256. Kasai, M, Watanabe, I. Histologic classification of liver-cell carcinoma in infancy and childhood and its clinical evaluation. A study of 70 cases collected in Japan. Cancer. 1970; 25:551–563.
257. von Schweinitz, D, Hecker, H, Schmidt-von-Arndt, G, Harms, D. Prognostic factors and staging systems in childhood hepatoblastoma. Int J Cancer. 1997; 74(6):593–599.
258. von Schweinitz, D, Hecker, H, Schmidt-von-Arndt, G, et al. Complete resection before development of drug resistance is essential for survival from advanced hepatoblastoma–a report from the German Cooperative Pediatric Liver Tumor Study HB-89. J Pediatr Surg. 1995; 30:845–852.
259. De Ioris, M, Brugieres, L, Zimmermann, A, et al. Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: The SIOPEL group experience. Eur J Cancer. 2008; 44:545–550.
260. Van Tornout, JM, Buckley, JD, Quinn, JJ, et al. Timing and magnitude of decline in alpha-fetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: A report from the Children’s Cancer Group. J Clin Oncol. 1997; 15:1190–1197.
261. Malogolowkin, MH, Katzenstein, HM, Meyers, RL, et al. Complete surgical resection is curative for children with hepatoblastoma with pure fetal histology: A report from the Children’s Oncology Group. J Clin Oncol. 2011; 29:3301–3306.
262. Malogolowkin, MH, Katzenstein, HM, Krailo, M, et al. Redefining the role of doxorubicin for the treatment of children with hepatoblastoma. J Clin Oncol. 2008; 26:2379–2383.
263. von Schweinitz, D. Identification of risk groups in hepatoblastoma–another step in optimising therapy. Eur J Cancer. 2000; 36:1343–1346.
264. Warmann, SW, Fuchs, J. Drug resistance in hepatoblastoma. Curr Pharm Biotechnol. 2007; 8:93–97.
265. D’Antiga, L, Vallortigara, F, Cillo, U, et al. Features predicting unresectability in hepatoblastoma. Cancer. 2007; 110:1050–1058.
266. Dall’Igna, P, Cecchetto, G, Toffolutti, T, et al. Multifocal hepatoblastoma: Is there a place for partial hepatectomy? Med Pediatr Oncol. 2003; 40:113–117.
267. Czauderna, P, Otte, JB, Aronson, DC, et al. Guidelines for surgical treatment of hepatoblastoma in the modern era–recommendations from the Childhood Liver Tumour Strategy Group of the International Society of Paediatric Oncology (SIOPEL). Eur J Cancer. 2005; 41:1031–1036.
268. Tagge, EP, Tagge, DU, Reyes, J, et al. Resection, including transplantation, for hepatoblastoma and hepatocellular carcinoma: Impact on survival. J Pediatr Surg. 1992; 27:292–297.
269. Pimpalwar, AP, Sharif, K, Ramani, P, et al. Strategy for hepatoblastoma management: Transplant versus nontransplant surgery. J Pediatr Surg. 2002; 37:240–245.
270. Srinivasan, P, McCall, J, Pritchard, J, et al. Orthotopic liver transplantation for unresectable hepatoblastoma. Transplantation. 2002; 74:652–655.
271. Otte, JB, Pritchard, J, Aronson, DC, et al. Liver transplantation for hepatoblastoma: Results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer. 2004; 42:74–83.
272. Reyes, JD, Carr, B, Dvorchik, I, et al. Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr. 2000; 136:795–804.
273. Molmenti, EP, Wilkinson, K, Molmenti, H, et al. Treatment of unresectable hepatoblastoma with liver transplantation in the pediatric population. Am J Transplant. 2002; 2:535–538.
274. Meyers, RL, Katzenstein, HM, Krailo, M, et al. Surgical resection of pulmonary metastatic lesions in children with hepatoblastoma. J Pediatr Surg. 2007; 42:2050–2056.
275. Hacker, FM, von Schweinitz, D, Gambazzi, F. The relevance of surgical therapy for bilateral and/or multiple pulmonary metastases in children. Eur J Pediatr Surg. 2007; 17:84–89.
276. Lack, EE, Schloo, BL, Azumi, N, et al. Undifferentiated (embryonal) sarcoma of the liver. Clinical and pathologic study of 16 cases with emphasis on immunohistochemical features. Am J Surg Pathol. 1991; 15:1–16.
277. Aoyama, C, Hachitanda, Y, Sato, JK, et al. Undifferentiated (embryonal) sarcoma of the liver. A tumor of uncertain histogenesis showing divergent differentiation. Am J Surg Pathol. 1991; 15:615–624.
278. Buetow, PC, Buck, JL, Pantongrag-Brown, L, et al. Undifferentiated (embryonal) sarcoma of the liver: Pathologic basis of imaging findings in 28 cases. Radiology. 1997; 203:779–783.
279. Kiani, B, Ferrell, LD, Qualman, S, et al. Immunohistochemical analysis of embryonal sarcoma of the liver. Appl Immunohistochem Mol Morphol. 2006; 14:193–197.
280. Abramowsky, CR, Cebelin, M, Choudhury, A, et al. Undifferentiated (embryonal) sarcoma of the liver with alpha-1-antitrypsin deposits: Immunohistochemical and ultrastructural studies. Cancer. 1980; 45:3108–3113.
281. Keating, S, Taylor, GP. Undifferentiated (embryonal) sarcoma of the liver: Ultrastructural and immunohistochemical similarities with malignant fibrous histiocytoma. Hum Pathol. 1985; 16:693–699.
282. Iliszko, M, Czauderna, P, Babinska, M, et al. Cytogenetic findings in an embryonal sarcoma of the liver. Cancer Genet Cytogenet. 1998; 102:142–144.
283. Cho, HS, Park, YN, Lyu, CJ, et al. Embryonal sarcoma of the liver: Multiple recurrences and histologic dedifferentiation. Med Pediatr Oncol. 1999; 32(5):386–388.
284. Leuschner, I, Schmidt, D, Harms, D. Undifferentiated sarcoma of the liver in childhood: Morphology, flow cytometry, and literature review. Hum Pathol. 1990; 21:68–76.
285. Pollono, DG, Drut, R. Undifferentiated (embryonal) sarcoma of the liver: Fine-needle aspiration cytology and preoperative chemotherapy as an approach to diagnosis and initial treatment. A case report. Diagn Cytopathol. 1998; 19:102–106.
286. Krishnamurthy, SC, Datta, S, Jambhekar, NA. Fine needle aspiration cytology of undifferentiated (embryonal) sarcoma of the liver: A case report. Acta Cytol. 1996; 40:567–570.
287. Newman, KD, Schisgall, R, Reaman, G, et al. Malignant mesenchymoma of the liver in children. J Pediatr Surg. 1989; 24:781–783.
288. Harris, MB, Shen, S, Weiner, MA, et al. Treatment of primary undifferentiated sarcoma of the liver with surgery and chemotherapy. Cancer. 1984; 54:2859–2862.
289. Walker, NI, Horn, MJ, Strong, RW, et al. Undifferentiated (embryonal) sarcoma of the liver. Pathologic findings and long-term survival after complete surgical resection. Cancer. 1992; 69:52–59.
290. Nicol, K, Savell, V, Moore, J, et al. Distinguishing undifferentiated embryonal sarcoma of the liver from biliary tract rhabdomyosarcoma: A Children’s Oncology Group study. Pediatr Dev Pathol. 2007; 10:89–97.
291. Urban, CE, Mache, CJ, Schwinger, W, et al. Undifferentiated (embryonal) sarcoma of the liver in childhood. Successful combined-modality therapy in four patients. Cancer. 1993; 72:2511–2516.
292. Uchiyama, M, Iwafuchi, M, Yagi, M, et al. Treatment of ruptured undifferentiated sarcoma of the liver in children: A report of two cases and review of the literature. J Hepatobiliary Pancreat Surg. 2001; 8:87–91.
293. Baron, PW, Majlessipour, F, Bedros, AA, et al. Undifferentiated embryonal sarcoma of the liver successfully treated with chemotherapy and liver resection. J Gastrointest Surg. 2007; 11:73–75.
294. Bisogno, G, Pilz, T, Perilongo, G, et al. Undifferentiated sarcoma of the liver in childhood: A curable disease. Cancer. 2002; 94:252–257.
295. Okajima, H, Ohya, Y, Lee, KJ, et al. Management of undifferentiated sarcoma of the liver including living donor liver transplantation as a backup procedure. J Pediatr Surg. 2009; 44:e33–e38.
296. Kelly, MJ, Martin, L, Alonso, M, et al. Liver transplant for relapsed undifferentiated embryonal sarcoma in a young child. J Pediatr Surg. 2009; 44:e1–e3.
297. Broughan, TA, Esquivel, CO, Vogt, DP, et al. Pretransplant chemotherapy in pediatric hepatocellular carcinoma. J Pediatr Surg. 1994; 29:1319–1322.
298. Esquivel, CO, Gutierrez, C, Cox, KL, et al. Hepatocellular carcinoma and liver cell dysplasia in children with chronic liver disease. J Pediatr Surg. 1994; 29:1465–1469.
299. Ryder, SD. Guidelines for the diagnosis and treatment of hepatocellular carcinoma (HCC) in adults. Gut. 2003; 52(Suppl 3):iii1–iii8.
300. Stocker, J. Hepatic Tumors. New York: Taylor & Francis; 1990.
301. M, R, Gatzimos, CD. Primary carcinoma of the liver in infants and children. Cancer. 1957; 10:678–686.
302. Manowski, Z, Silver, MM, Roberts, EA, et al. Liver cell dysplasia and early liver transplantation in hereditary tyrosinemia. Mod Pathol. 1990; 3:694–701.
303. Kvittingen, EA. Tyrosinaemia–treatment and outcome. J Inherit Metab Dis. 1995; 18:375–379.
304. Fink, AS, Appelman, HD, Thompson, NW. Hemorrhage into a hepatic adenoma and type Ia glycogen storage disease: A case report and review of the literature. Surgery. 1985; 97:117–124.
305. Gruner, BA, DeNapoli, TS, Andrews, W, et al. Hepatocellular carcinoma in children associated with Gardner syndrome or familial adenomatous polyposis. J Pediatr Hematol Oncol. 1998; 20:274–278.
306. Sugarman, GI. Heuser ET and Reed WB: A case of cerebral gigantism and hepatocarcinoma. Am J Dis Child. 1977; 131:631–633.
307. Jain, D, Hui, P, McNamara, J, Schwartz, D, et al. Bloom syndrome in sibs: First reports of hepatocellular carcinoma and Wilms tumor with documented anaplasia and nephrogenic rests. Pediatr Dev Pathol. 2001; 4(6):585–589.
308. Ettinger, LJ, Freeman, AI. Hepatoma in a child with neurofibromatosis. Am J Dis Child. 1979; 133:528–531.
309. Ruymann, FB, Mosijczuk, AD, Sayers, RJ. Hepatoma in a child with methotrexate-induced hepatic fibrosis. JAMA. 1977; 238(24):2631–2633.
310. Moore, L, Bourne, AJ, Moore, DJ, et al. Hepatocellular carcinoma following neonatal hepatitis. Pediatr Pathol Lab Med. 1997; 17:601–610.
311. Bekassy, AN, Garwicz, S, Wiebe, T, et al. Hepatocellular carcinoma associated with arteriohepatic dysplasia in a 4-year-old girl. Med Pediatr Oncol. 1992; 20:78–83.
312. Ni, YH, Chang, MH, Hsu, HY, et al. Hepatocellular carcinoma in childhood. Clinical manifestations and prognosis. Cancer. 1991; 68:1737–1741.
313. Lee, CL, Ko, YC. Hepatitis B vaccination and hepatocellular carcinoma in Taiwan. Pediatrics. 1997; 99:351–353.
314. Yu, MW, Chen, CJ. Hepatitis B and C viruses in the development of hepatocellular carcinoma. Crit Rev Oncol Hematol. 1994; 17:71–91.
315. Strickland, DK, Jenkins, JJ, Hudson, MM. Hepatitis C infection and hepatocellular carcinoma after treatment of childhood cancer. J Pediatr Hematol Oncol. 2001; 23:527–529.
316. Kohno, M, Kitatani, H, Wada, H, et al. Hepatocellular carcinoma complicating biliary cirrhosis caused by biliary atresia: Report of a case. J Pediatr Surg. 1995; 30:1713–1716.
317. Wang, JD, Chang, TK, Chen, HC, et al. Pediatric liver tumors: Initial presentation, image finding and outcome. Pediatr Int. 2007; 49:491–496.
318. Brower, S, Hoff, P, Jones, D, et al, Pancreatic Cancer, Hepatobiliary Cancer, and Neuroendocrine Cancers of the GI Tract Huntington, 1998.
319. Bolondi, L. Screening for hepatocellular carcinoma in cirrhosis. J Hepatol. 2003; 39(6):1076–1084.
320. Berman, MA, Burnham, JA, Sheahan, DG. Fibrolamellar carcinoma of the liver: An immunohistochemical study of nineteen cases and a review of the literature. Hum Pathol. 1988; 19:784–794.
321. Kumada, T, Nakano, S, Takeda, I, et al. Patterns of recurrence after initial treatment in patients with small hepatocellular carcinoma. Hepatology. 1997; 25:87–92.
322. A new prognostic system for hepatocellular carcinoma: A retrospective study of 435 patients: The Cancer of the Liver Italian Program (CLIP) investigators. Hepatology. 1998; 28:751–755.
323. Bruix, J, Sherman, M. Management of hepatocellular carcinoma. Hepatology. 2005; 42:1208–1236.
324. Soyer, P, Roche, A, Levesque, M, et al. CT of fibrolamellar hepatocellular carcinoma. J Comput Assist Tomogr. 1991; 15:533–538.
325. Trevisani, F, Cantarini, MC, Wands, JR, et al. Recent advances in the natural history of hepatocellular carcinoma. Carcinogenesis. 2008; 29(7):1299–1305.
326. Debray, D, Pariente, D, Fabre, M, et al. Fibrolamellar hepatocellular carcinoma: Report of a case mimicking a liver abscess. J Pediatr Gastroenterol Nutr. 1994; 19:468–472.
327. El-Gazzaz, G, Wong, W, El-Hadary, MK, et al. Outcome of liver resection and transplantation for fibrolamellar hepatocellular carcinoma. Transpl Int. 2000; 13(Suppl 1):S406–S409.
328. Katzenstein, HM, Krailo, MD, Malogolowkin, MH, et al. Hepatocellular carcinoma in children and adolescents: Results from the Pediatric Oncology Group and the Children’s Cancer Group intergroup study. J Clin Oncol. 2002; 20:2789–2797.
329. Czauderna, P, Mackinlay, G, Perilongo, G, et al. Hepatocellular carcinoma in children: Results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol. 2002; 20:2798–2804.
330. von Schweinitz, D. Management of liver tumors in childhood. Semin Pediatr Surg. 2006; 15:17–24.
331. Lau, H, Fan, ST, Ng, IO, et al. Long term prognosis after hepatectomy for hepatocellular carcinoma: A survival analysis of 204 consecutive patients. Cancer. 1998; 83:2302–2311.
332. Arii, S, Okamoto, E, Imamura, M. Registries in Japan: Current status of hepatocellular carcinoma in Japan. Liver Cancer Study Group of Japan. Semin Surg Oncol. 1996; 12:204–211.
333. Poon, RT, Ng, IO, Lau, C, et al. Tumor microvessel density as a predictor of recurrence after resection of hepatocellular carcinoma: A prospective study. J Clin Oncol. 2002; 20:1775–1785.
334. Izumi, R, Shimizu, K, Ii, T, et al. Prognostic factors of hepatocellular carcinoma in patients undergoing hepatic resection. Gastroenterology. 1994; 106:720–727.
335. Craig, JR, Peters, RL, Edmondson, HA, et al. Fibrolamellar carcinoma of the liver: A tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer. 1980; 46:372–379.
336. Farhi, DC, Shikes, RH, Murari, PJ, et al. Hepatocellular carcinoma in young people. Cancer. 1983; 52:1516–1525.
337. Nagorney, DM, Adson, MA, Weiland, LH, et al. Fibrolamellar hepatoma. Am J Surg. 1985; 149:113–119.
338. Lack, EE, Neave, C, Vawter, GF. Hepatocellular carcinoma. Review of 32 cases in childhood and adolescence. Cancer. 1983; 52:1510–1515.
339. Friedman, AC, Lichtenstein, JE, Goodman, Z, et al. Fibrolamellar hepatocellular carcinoma. Radiology. 1985; 157:583–587.
340. Paradinas, FJ, Melia, WM, Wilkinson, ML, et al. High serum vitamin B12 binding capacity as a marker of the fibrolamellar variant of hepatocellular carcinoma. Br Med J (Clin Res Ed). 1982; 285:840–842.
341. Brandt, DJ, Johnson, CD, Stephens, DH, et al. Imaging of fibrolamellar hepatocellular carcinoma. AJR Am J Roentgenol. 1988; 151:295–299.
342. Titelbaum, DS, Hatabu, H, Schiebler, ML, et al. Fibrolamellar hepatocellular carcinoma: MR appearance. J Comput Assist Tomogr. 1988; 12:588–591.
343. Ang, PT, Evans, H, Pazdur, R. Fibrolamellar hepatocellular carcinoma. Therapeutic implications of a ten-year disease-free interval. Am J Clin Oncol. 1991; 14:175–178.
344. O’Grady, JG, Polson, RJ, Rolles, K, et al. Liver transplantation for malignant disease. Results in 93 consecutive patients. Ann Surg. 1988; 207:373–379.
345. Bilik, R, Superina, R. Transplantation for unresectable liver tumors in children. Transplant Proc. 1997; 29:2834–2835.
346. Iwatsuki, S, Gordon, RD, Shaw, BW, Jr., Starzl, TE. Role of liver transplantation in cancer therapy. Ann Surg. 1985; 202:401–407.
347. Iwatsuki, S, Dvorchik, I, Marsh, JW, et al. Liver transplantation for hepatocellular carcinoma: A proposal of a prognostic scoring system. J Am Coll Surg. 2000; 191:389–394.
348. Mazzaferro, V, Regalia, E, Doci, R, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med. 1996; 334:693–699.
349. Beaunoyer, M, Vanatta, JM, Ogihara, M, et al. Outcomes of transplantation in children with primary hepatic malignancy. Pediatr Transplant. 2007; 11:655–660.
350. Yao, FY, Ferrell, L, Bass, NM, et al. Liver transplantation for hepatocellular carcinoma: Expansion of the tumor size limits does not adversely impact survival. Hepatology. 2001; 33:1394–1403.
351. Leung, JY, Zhu, AX, Gordon, FD, et al. Liver transplantation outcomes for early-stage hepatocellular carcinoma: Results of a multicenter study. Liver Transpl. 2004; 10:1343–1354.
352. Czauderna, P, Mackinlay, G, Perilongo, G, et al. Hepatocellular carcinoma in children: Results of the first prospective study of the International Society of Pediatric Oncology grou. J Clin Oncol. 2002; 20:2798–2804.
353. Austin, MT, Leys, CM, Feurer, ID, et al. Liver transplantation for childhood hepatic malignancy: A review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg. 2006; 41:182–186.
354. Lau, WY, Lai, EC. Hepatocellular carcinoma: Current management and recent advances. Hepatobiliary Pancreat Dis Int. 2008; 7:237–257.
355. Scatton, O, Zalinski, S, Terris, B, et al. Hepatocellular carcinoma developed on compensated cirrhosis: Resection as a selection tool for liver transplantation. Liver Transpl. 2008; 14:779–788.
356. Von Schweinitz, D, et al, Treatment of liver tumors in chldren, in Liver Tumors: Current and Emerging Therapies. Clavien PA, Fong Y, Lyerly HK. Jones and Barlett, Boston, 2004:409–426.
357. Katzenstein, HM, Krailo, MD, Malogolowkin, MH, et al. Fibrolamellar hepatocellular carcinoma in children and adolescents. Cancer. 2003; 97:2006–2012.
358. Ahn, SI, Seo, JM, Shin, SH, et al. Hepatocellular carcinoma with lung metastasis in a 9-year-old-boy. J Pediatr Surg. 2001; 36:1599–1601.
359. Yeh, CN, Chen, MF, Jeng, LB. Resection of peritoneal implantation from hepatocellular carcinoma. Ann Surg Oncol. 2002; 9:863–868.
360. Salmi, A, Turrini, R, Lanzani, G, et al. Long-term effectiveness of radiofrequency ablation for hepatocellular carcinoma of 3.5 cm or less. Hepatogastroenterology. 2008; 55:191–196.
361. Wang, B, Lu, Y, Zhang, XF, et al. Management of spontaneous rupture of hepatocellular carcinoma. ANZ J Surg. 2008; 78:501–503.
362. Ruymann, FB, Raney, RB, Jr., Crist, WM, et al. Rhabdomyosarcoma of the biliary tree in childhood. A report from the Intergroup Rhabdomyosarcoma Study. Cancer. 1985; 56:575–581.
363. Pollono, DG, Tomarchio, S, Berghoff, R, et al. Rhabdomyosarcoma of extrahepatic biliary tree: Initial treatment with chemotherapy and conservative surgery. Med Pediatr Oncol. 1998; 30:290–293.
364. Sanz, N, de Mingo, L, Florez, F, et al. Rhabdomyosarcoma of the biliary tree. Pediatr Surg Int. 1997; 12:200–201.
365. Schweizer, PSM, Wehrmann, M. Major resection for embryonal rhabdomyosarcoma of the biliary tree. Pediatr Surg Int. 1994; 268–273.
366. Mihara, S, Matsumoto, H, Tokunaga, F, et al. Botryoid rhabdomyosarcoma of the gallbladder in a child. Cancer. 1982; 49:812–818.
367. Horowitz, ME, Etcubanas, E, Webber, BL, et al. Hepatic undifferentiated (embryonal) sarcoma and rhabdomyosarcoma in children. Results of therapy. Cancer. 1987; 59:396–402.
368. Isaacson, C. Embryonal rhabdomyosarcoma of the ampulla of Vater. Cancer. 1978; 41:365–368.
369. Patil, KK, Omojola, MF, Khurana, P, et al. Embryonal rhabdomyosarcoma within a choledochal cyst. Can Assoc Radiol J. 1992; 43:145–148.
370. Haith, EE, Kepes, JJ, Holder, TM. Inflammatory pseudotumor involving the common bile duct of a six-year-old boy: Successful pancreaticoduodenectomy. Surgery. 1964; 56:436–441.
371. Huang, FC, Eng, HL, Chen, CL, et al. Primary pleomorphic rhabdomyosarcoma of the liver: A case report. Hepatogastroenterology. 2003; 50:73–76.
372. Hirohashi, S, Hirohashi, R, Uchida, H, et al. Pancreatitis: Evaluation with MR cholangiopancreatography in children. Radiology. 1997; 203:411–415.
373. Friedburg, H, Kauffmann, GW, Bohm, N, et al. Sonographic and computed tomographic features of embryonal rhabdomyosarcoma of the biliary tract. Pediatr Radiol. 1984; 14:436–438.