Kidney and urinary tract disorders
The spectrum of renal disease in children differs from that in adults:
• Many structural abnormalities of the kidneys and urinary tract are identified on antenatal ultrasound screening
• Urinary tract infection, vesicoureteric reflux and urinary obstruction have the potential to damage the growing kidney
• Nephrotic syndrome is usually steroid-sensitive and only rarely leads to chronic renal failure
• Chronic renal disorders and the drugs used to treat them may affect growth and development.
Assessment of the kidneys and urinary tract
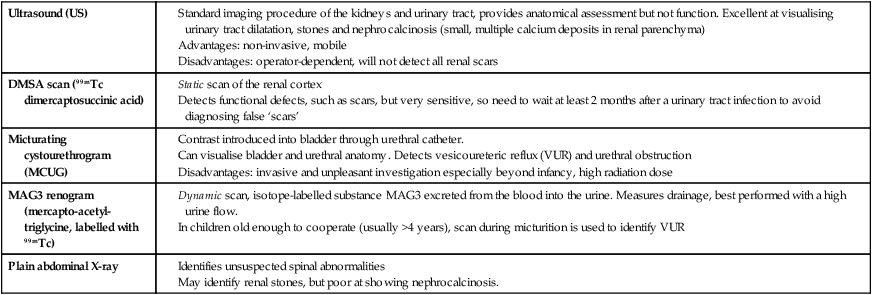
The glomerular filtration rate (GFR) is low in the newborn infant and is especially low in premature infants; the GFR at 28 weeks’ gestation is only 10% of the term infant. In term infants, the corrected GFR (15–20 ml/min per 1.73 m2) rapidly rises to 1–2 years of age when the adult rate of 80 to 120 ml/min per 1.73 m2 is achieved (Fig. 18.1). The assessment of renal function in children is listed in Table 18.1 and the radiological investigations of the kidneys and urinary tract in Table 18.2.
Table 18.1
Assessment of renal function in children
| Plasma creatinine concentration (PCr) | Main test of renal function. Rises progressively throughout childhood according to height and muscle bulk. May not be outside laboratory ‘normal range’ until renal function has fallen to less than half normal |
| Estimated glomerular filtration rate (eGFR) | The formula eGFR = k × height (cm) ÷ creatinine (µmol/L) provides estimate of GFR. Better measure of renal function than creatinine and useful to monitor renal function serially in children with renal impairment (k is 40 if creatinine measured using Jaffe method or 30 if measured enzymatically) |
| Inulin or EDTA glomerular filtration rate | More accurate as clearance from the plasma of substances freely filtered at the glomerulus, and is not secreted or reabsorbed by the tubules. Need for repeated blood tests limits use in children |
| Creatinine clearance | Requires timed urine collection and blood tests. Rarely done in children as inconvenient and inaccurate |
| Plasma urea concentration | Increased in renal failure, often before creatinine starts rising, and raised levels may be symptomatic. Urea levels also increased by high protein diet and if in a catabolic state. |
Table 18.2
Radiological investigation of the kidneys and urinary tract
(mercapto-acetyl-triglycine, labelled with 99mTc)
Congenital abnormalities
• be associated with abnormal renal development or function
• predispose to postnatal infection
• involve urinary obstruction which requires surgical treatment.
Anomalies detectable on antenatal ultrasound screening
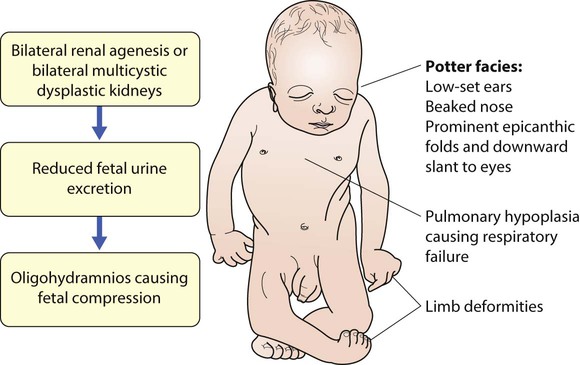
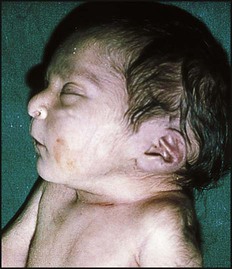
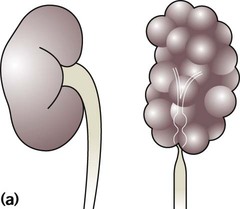
Absence of both kidneys (renal agenesis) – as amniotic fluid is mainly derived from fetal urine, there is severe oligohydramnios resulting in Potter syndrome (Fig. 18.2a, 18.2b), which is fatal.
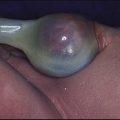
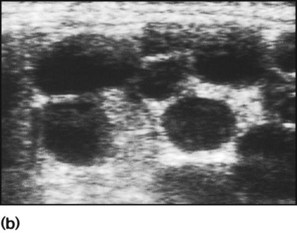
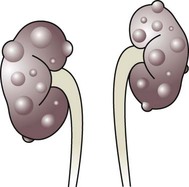
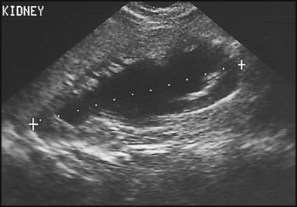
Multicystic dysplastic kidney (MCDK) – results from the failure of union of the ureteric bud (which forms the ureter, pelvis, calyces and collecting ducts) with the nephrogenic mesenchyme. It is a non-functioning structure with large fluid-filled cysts with no renal tissue and no connection with the bladder (Fig. 18.3). Half will have involuted by 2 years of age, and nephrectomy is indicated only if it remains very large or hypertension develops, but this is rare. Since they produce no urine, Potter syndrome will result if the lesion is bilateral. Other causes of large cystic kidneys are autosomal recessive polycystic kidney disease (ARPKD) (Fig. 18.4), autosomal dominant polycystic kidney disease (ADPKD) (Fig. 18.5) and tuberous sclerosis. In contrast to a multicystic dysplastic kidney, in these disorders some or normal renal function is maintained but both kidneys are always affected. Autosomal dominant polycystic kidney disease has an incidence of 1 in 1000; the main symptoms in childhood are hypertension and haematuria and it causes renal failure in late adulthood. It is associated with several extra-renal features including cysts in liver and pancreas, cerebral aneurysms and mitral valve prolapse.
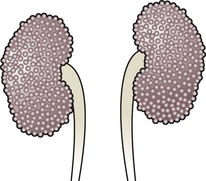
Abnormal caudal migration may result in a pelvic kidney or a horseshoe kidney (Fig. 18.6), when the lower poles are fused in the midline. The abnormal position may predispose to infection or obstruction to urinary drainage.
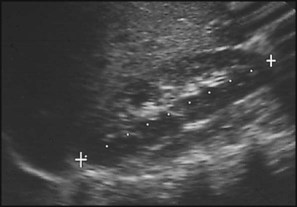
Premature division of the ureteric bud gives rise to a duplex system, which can vary from simply a bifid renal pelvis to complete division with two ureters. These ureters frequently have an abnormal drainage so that the ureter from the lower pole moiety often refluxes, whereas the upper pole ureter may drain ectopically into the urethra or vagina or may prolapse into the bladder (ureterocele) and urine flow may be obstructed (Fig. 18.7).
Failure of fusion of the infraumbilical midline structures results in exposed bladder mucosa (bladder extrophy). Absence or severe deficiency of the anterior abdominal wall muscles is frequently associated with a large bladder and dilated ureters (megacystis-megaureters) and cryptorchidism, the absent musculature syndrome (prune-belly syndrome) (Fig. 18.8).
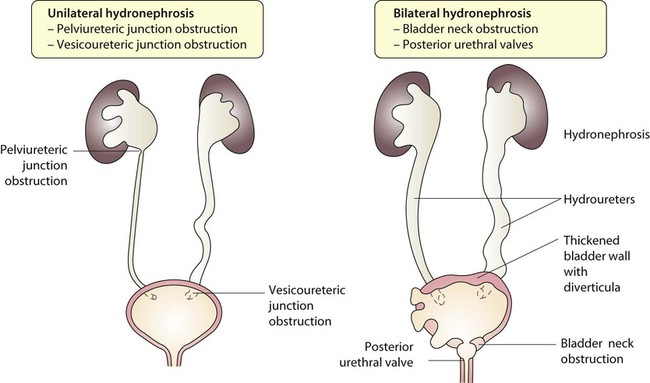
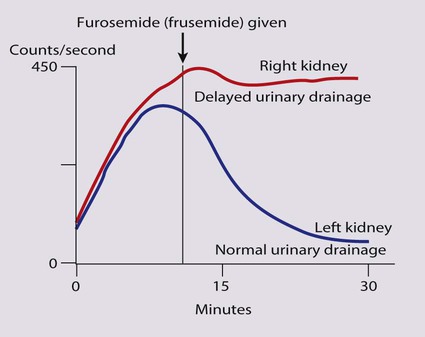
Obstruction to urine flow may occur at the pelvi-ureteric or vesicoureteric junction, at the bladder neck (e.g. due to disruption of the nerve supply, neuropathic bladder) or at the posterior urethra in a boy due to mucosal folds or a membrane, known as posterior urethral valves. The consequences of obstruction to urine flow are shown in Figures 18.9a–18.9d. At worst, this results in a dysplastic kidney which is small, poorly functioning and may contain cysts and aberrant embryonic tissue such as cartilage. In the most severe and bilateral cases Potter syndrome is present. Renal dysplasia can also occur in association with severe intrauterine vesicoureteric reflux, in isolation or in certain rare, inherited syndromes affecting multiple systems.
Some congenital abnormalities of the kidneys and urinary tract
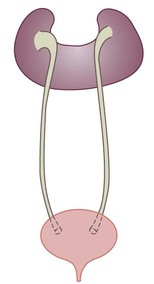
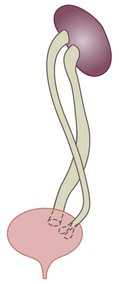
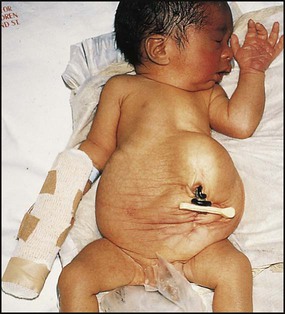
Postnatal management
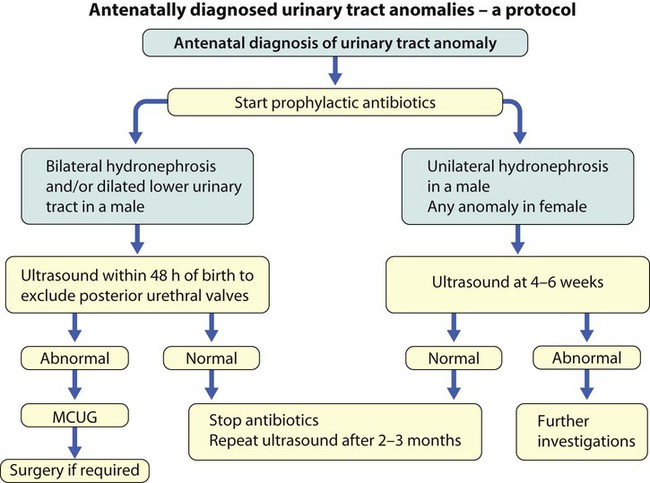
An example of a protocol for infants with antenatally diagnosed anomalies is shown in Figure 18.10. Prophylactic antibiotics may be started at birth to try to prevent urinary tract infection, although practice varies between centres. As the newborn kidney has a low GFR, urine flow is low and mild outflow obstruction may not be evident in the first few days of life. The ultrasound scan should therefore be delayed for several weeks. However, bilateral hydronephrosis in a male infant warrants an ultrasound shortly after birth to exclude posterior urethral valves, which always requires urological intervention such as cystoscopic ablation (Case History 18.1).
Case History
18.1 Posterior urethral valves
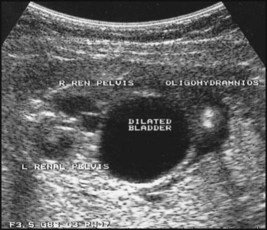
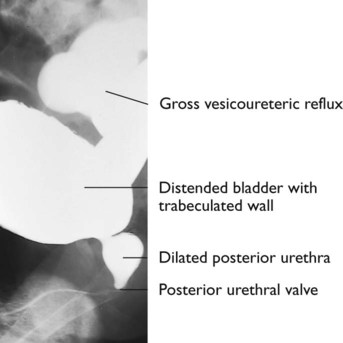
Bilateral hydronephrosis was noted on antenatal ultrasound at 20 weeks’ gestation in a male fetus. There was poor renal growth, progressive hydronephrosis and decreasing volume of amniotic fluid (Fig. 18.11a) on repeated scans. After birth, prophylactic antibiotics were started. An urgent ultrasound showed bilateral hydronephrosis with small dysplastic kidneys. The bladder and ureters were grossly distended. The plasma creatinine was raised. A micturating cystourethrogram (MCUG) (Fig. 18.11b) showed vesicoureteric reflux, a dilated posterior urethra and posterior urethral valves which was treated endoscopically. Renal function initially improved but then progressed to chronic renal failure. He had a renal transplant at 10 years of age.
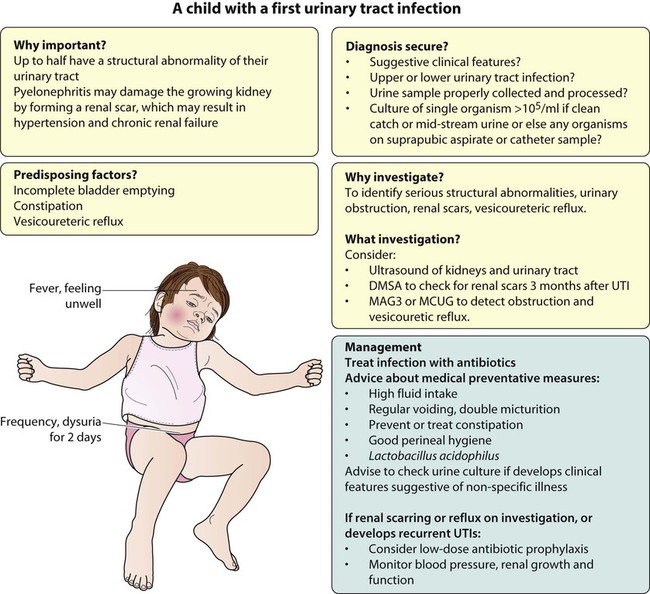
Urinary tract infection
• up to half of patients have a structural abnormality of their urinary tract
• pyelonephritis may damage the growing kidney by forming a scar, predisposing to hypertension and to chronic renal failure if the scarring is bilateral.
Clinical features
Presentation of UTI varies with age (Box 18.1). In infants, symptoms are non-specific; fever is usually but not always present, and septicaemia may develop rapidly. The classical symptoms of dysuria, frequency and loin pain become more common with increasing age. Serious illness from septicaemia is described in the child with a fever in Chapter 14. Dysuria alone is usually due to cystitis, or vulvitis in girls or balanitis in uncircumcised boys. Symptoms suggestive of a UTI may also occur following sexual abuse.
Collection of samples
For the child in nappies, urine can be collected by:
• A ‘clean-catch’ sample into a waiting clean pot when the nappy is removed. This is the recommended method
• An adhesive plastic bag applied to the perineum after careful washing, although there may be contamination from the skin
• A urethral catheter if there is urgency in obtaining a sample and no urine has been passed
• Suprapubic aspiration (SPA), when a fine needle attached to a syringe is inserted directly into the bladder just above the symphysis pubis under ultrasound guidance; it may be used in severely ill infants requiring urgent diagnosis and treatment, but it is an invasive procedure, and is increasingly replaced by urethral catheter sampling.
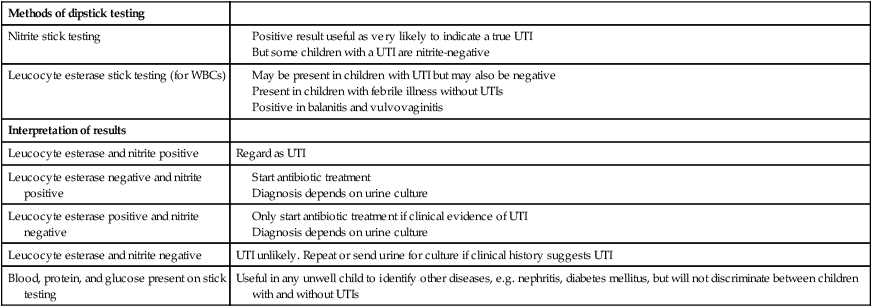
Ideally, the urine sample should be microscoped to identify organisms and cultured straight away. This is indicated in all infants and children <3 years old with a suspected UTI. If this is not possible, the urine sample should be refrigerated to prevent the overgrowth of contaminating bacteria. Urinary white cells are not a reliable feature of a UTI, as they may lyse during storage and may be present in febrile children without a UTI and in children with balanitis or vulvovaginitis. Dipsticks can be used as a screening test. Urine culture should still be performed unless both leucocyte esterase and nitrite are negative or if the clinical symptoms and dipstick tests do not correlate (Table 18.3).
Table 18.3
Methods and interpretation of dipstick testing in children
| Methods of dipstick testing | |
| Nitrite stick testing |
 A urine sample should be tested in all infants with an unexplained fever >38°C.
A urine sample should be tested in all infants with an unexplained fever >38°C.Bacterial and host factors that predispose to infection
Infecting organism
Vesicoureteric reflux
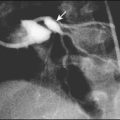
Vesicoureteric reflux (VUR) is a developmental anomaly of the vesicoureteric junctions. The ureters are displaced laterally and enter directly into the bladder rather than at an angle, with a shortened or absent intramural course. Severe cases may be associated with renal dysplasia. It is familial, with a 30–50% chance of occurring in first-degree relatives. It may also occur with bladder pathology, e.g. a neuropathic bladder or urethral obstruction, or temporarily after a UTI. Its severity varies from reflux into the lower end of an undilated ureter during micturition to the severest form with reflux during bladder filling and voiding, with a distended ureter, renal pelvis and clubbed calyces (Fig. 18.12). Mild reflux is unlikely to be of significance, but the more severe degrees of VUR may be associated with intrarenal reflux (IRR), the backflow of urine from the renal pelvis into the papillary collecting ducts; intrarenal reflux is associated with a particularly high risk of renal scarring if UTIs occur. The incidence of renal defects increases with increasing severity of reflux. There is considerable controversy as to whether renal scarring is a congenital abnormality already present in children with reflux and which predisposes to infection or if children with reflux have normal kidneys at birth which are damaged by UTIs and that preventing UTIs in these children prevents scars. Reflux tends to resolve with age especially lower grades of VUR.
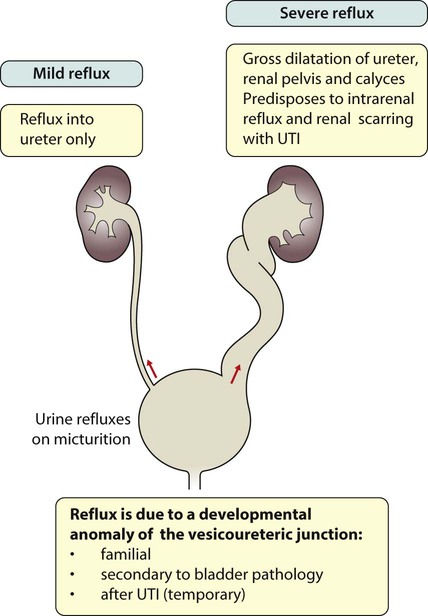
Reflux with associated ureteric dilatation is important, as:
• urine returning to the bladder from the ureters after voiding results in incomplete bladder emptying, which encourages infection
• the kidneys may become infected (pyelonephritis), particularly if there is intrarenal reflux
• bladder voiding pressure is transmitted to the renal papillae; this may contribute to renal damage if voiding pressures are high.
Investigation
An initial ultrasound will identify:
A suggested schema for investigation of the first proven UTI is shown in Figure 18.13, but varies between centres.
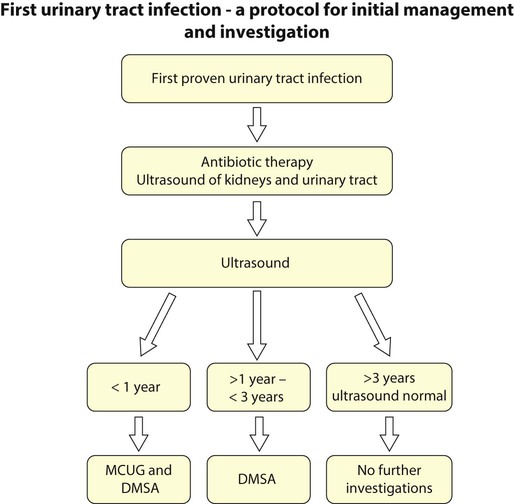
Case History
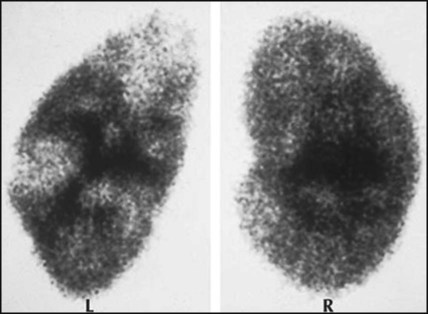
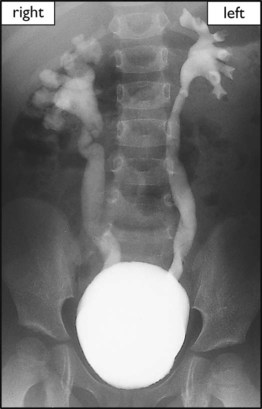
Jack, a 2-month-old infant, stopped feeding and had a high, intermittent fever. He was referred to hospital, where he had an infection screen. Urine examination showed >100 white blood cells, >105 E. coli/ml. He was treated with intravenous antibiotics. An ultrasound showed that the left kidney was smaller than the right kidney with dilated ureters. He was started on prophylactic antibiotics. A DMSA scan (Fig. 18.14) performed 3 months later confirmed bilateral renal scarring, with the left kidney contributing 33% of renal function. The MCUG (Fig. 18.15) showed bilateral vesicoureteric reflux. At 4 years of age, the reflux had resolved and antibiotic prophylaxis was stopped. His blood pressure and renal growth and function continue to be monitored.
Medical measures for the prevention of UTI
• High fluid intake to produce a high urine output
• Ensuring complete bladder emptying by encouraging the child to try a second time to empty his bladder after a minute or two, commonly known as double micturition; this empties any urine residue or refluxed urine returning to the bladder
• Prevention or treatment of constipation
• Lactobacillus acidophilus, a probiotic to encourage colonisation of the gut by this organism and reduce the number of pathogenic organisms that might potentially cause invasive disease
• Antibiotic prophylaxis, although this is controversial. It is often used in those under 2 years of age with a congenital abnormality of the kidneys or urinary tract or who have had an upper urinary tract infection and those with severe reflux. Trimethoprim (2 mg/kg at night) is used most often, but nitrofurantoin or cephalexin may be given. Broad-spectrum, poorly absorbed antibiotics such as amoxicillin should be avoided.
Follow-up of children with recurrent UTIs, renal scarring or reflux
• Urine culture should be checked with any non-specific illness in case it is caused by a UTI (urine should not be cultured routinely).
• Long-term, low-dose antibiotic prophylaxis can be used. There is no evidence for when antibiotic prophylaxis should be stopped.
• Circumcision in boys may sometimes be considered, as there is evidence that it reduces the incidence of urinary tract infection.
• Anti-reflux surgery may be indicated if there is progression of scarring with ongoing reflux, but it has not been shown to improve outcome.
• Blood pressure should be checked annually if renal defects are present.
• Regular assessment of renal growth and function is necessary if there are bilateral defects because of the risk of chronic renal failure.
Enuresis
Primary nocturnal enuresis
This is considered in Chapter 23.
Daytime enuresis
• Lack of attention to bladder sensation: a manifestation of a developmental or psychogenic problem, although it may occur in otherwise normal children who are too preoccupied with what they are doing to respond to the sensation of a full bladder
• Detrusor instability (sudden, urgent urge to void induced by sudden bladder contractions)
• A neuropathic bladder (bladder is enlarged and fails to empty properly, irregular thick wall and is associated with spina bifida and other neurological conditions)
• A urinary tract infection (rarely in the absence of other symptoms)
• An ectopic ureter, causes constant dribbling and child is always damp.
Secondary (onset) enuresis
The loss of previously achieved urinary continence may be due to:
• Emotional upset, the commonest cause
• Polyuria from an osmotic diuresis in diabetes mellitus or a renal concentrating disorder, e.g. sickle cell disease or chronic renal failure.
Proteinuria
A common cause is orthostatic (postural) proteinuria, where proteinuria is only found when the child is upright, i.e. during the day. It can be diagnosed by measuring the urine protein/creatinine ratio in a series of early morning urine specimens. The prognosis is excellent and further investigations are not necessary. Other causes of proteinuria, which needs further evaluation, are listed in Box 18.2.
Nephrotic syndrome
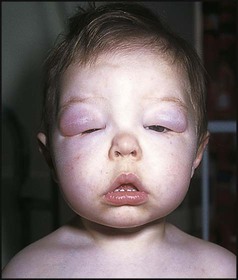
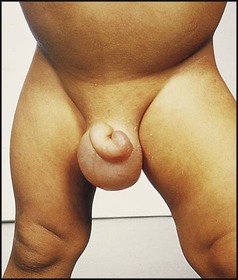
Clinical signs of the nephrotic syndrome are:
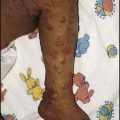
• Periorbital oedema (particularly on waking), the earliest sign (Fig. 18.16)
• Scrotal or vulval, leg and ankle oedema (Fig. 18.17)
• Breathlessness due to pleural effusions and abdominal distension.
The initial investigations are listed in Box 18.3.
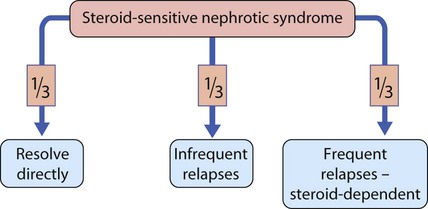
Steroid-sensitive nephrotic syndrome
Management
• Hypovolaemia. During the initial phase of oedema formation the intravascular compartment may become volume depleted. The child who becomes hypovolaemic characteristically complains of abdominal pain and may feel faint. There is peripheral vasoconstriction and urinary sodium retention. A low urinary sodium (<20 mmol/L) and a high packed cell volume of red blood cells are indications of hypovolaemia, which requires urgent treatment with intravenous albumin as the child is at risk of vascular thrombosis and shock. Increasing peripheral oedema, assessed clinically and by daily weight, may cause discomfort and respiratory compromise. If severe, this may need treatment with intravenous albumin. Care must be taken with the use of colloid, as it may precipitate pulmonary oedema and hypertension from fluid overload, and also with diuretics, which may cause or worsen hypovolaemia.
• Thrombosis. A hypercoagulable state, due to urinary losses of antithrombin, thrombocytosis which may be exacerbated by steroid therapy, increased synthesis of clotting factors and increased blood viscosity from the raised haematocrit, predisposes to thrombosis. This may affect the brain, limbs and splanchnic circulation with potentially catastrophic results.
• Infection. Children in relapse are at risk of infection with capsulated bacteria, especially Pneumococcus. Spontaneous peritonitis may occur. Pneumococcal and seasonal influenza vaccination is widely recommended. Chickenpox and shingles should be treated with aciclovir.
• Hypercholesterolaemia. This correlates inversely with the serum albumin, but the cause of the hyperlipidaemia is not fully understood.
Prognosis
This is summarised in Figure 18.18. Relapses are identified by parents on urine testing. The side-effects of corticosteroid therapy may be reduced by an alternate-day regimen. If relapses are frequent, or if a high maintenance dose is required, involvement of a paediatric nephrologist is advisable as other drug therapy may be considered to enable reduction in steroid use. Possible steroid-sparing agents include the immunomodulator levamisole, alkylating agents (e.g. cyclophosphamide), calcineurin inhibitors such as tacrolimus and ciclosporin A and the immunosuppressant mycophenolate mofetil.
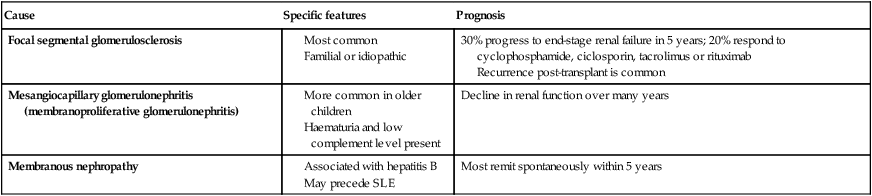
Steroid-resistant nephrotic syndrome (Table 18.4)
Table 18.4
Steroid-resistant nephrotic syndrome
| Cause | Specific features | Prognosis |
| Focal segmental glomerulosclerosis |
Recurrence post-transplant is common
 An oedematous child – test for proteinuria to diagnose nephrotic syndrome.
An oedematous child – test for proteinuria to diagnose nephrotic syndrome.Haematuria
Urinary tract infection is the most common cause of haematuria (Box 18.4), although seldom as the only symptom. The history and examination may suggest the diagnosis, e.g. a family history of stone formation or nephritis or a history of trauma. A plan of investigation is outlined in Box 18.5.
A renal biopsy may be indicated if:
Acute nephritis
The causes of acute nephritis in childhood are listed in Box 18.6. Increased glomerular cellularity restricts glomerular blood flow and therefore filtration is decreased. This leads to:
• decreased urine output and volume overload
• hypertension, which may cause seizures
Henoch–Schönlein purpura
Henoch–Schönlein purpura is the combination of some of the following features:
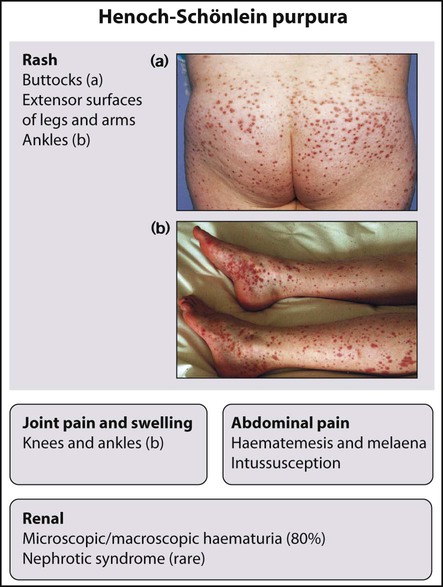
Hypertension
Blood pressure in children needs to be measured with a cuff over two-thirds the length of the upper arm (see Ch. 2). Blood pressure increases with age and height and readings should be plotted on a centile chart (see Appendix). Hypertension is blood pressure above 95th percentile for height, age and sex. Symptomatic hypertension in children is usually secondary to renal, cardiac or endocrine causes (Box 18.7).
Renal masses
An abdominal mass identified on palpating the abdomen should be investigated promptly by ultrasound scan. The causes of palpable kidneys are shown in Box 18.8. Bilaterally enlarged kidneys in early life are most frequently due to autosomal recessive polycystic kidney disease, which is associated with hypertension, hepatic fibrosis and progression to chronic renal failure. This form of polycystic kidney disease must be distinguished from the autosomal dominant adult-type polycystic kidney disease, which has a more benign prognosis in childhood with onset of renal failure in adulthood.
Renal tubular disorders
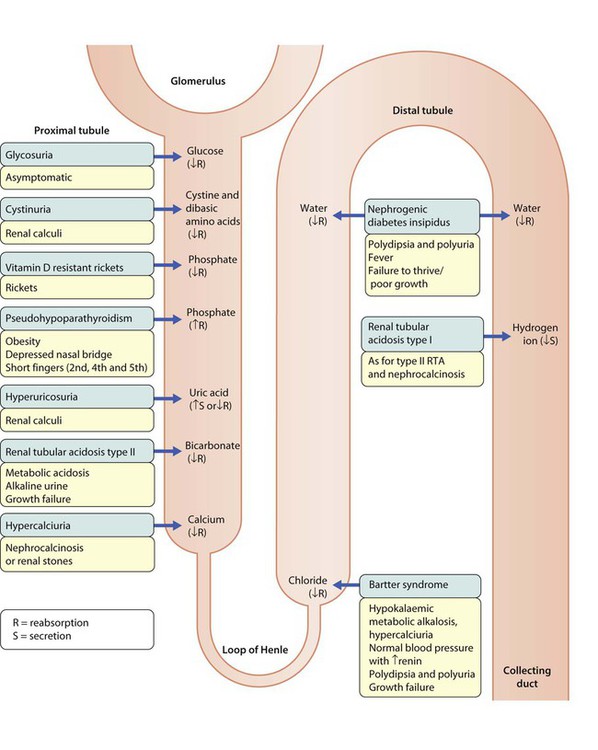
Generalised proximal tubular dysfunction (Fanconi syndrome)
Proximal tubule cells are among the most metabolically active in the body, so are especially vulnerable to cellular damage. The cardinal features are excessive urinary loss of amino acids, glucose, phosphate, bicarbonate, sodium, calcium, potassium and urate. The causes are listed in Box 18.9. Fanconi syndrome should be considered in a child presenting with:
Acute kidney injury
Acute kidney injury has acute renal failure at the most severe end of the spectrum where there is a sudden, potentially reversible, reduction in renal function. Oliguria (<0.5 ml/kg per hour) is usually present. It can be classified as (see Box 18.10):
• Prerenal: the commonest cause in children
• Renal: there is salt and water retention; blood, protein and casts are often present in the urine; and there may be symptoms specific to an accompanying disease (e.g. haemolytic uraemic syndrome)
Management
Renal failure
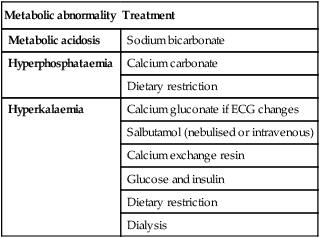
If there is circulatory overload, restriction of fluid intake and challenge with a diuretic may increase urine output sufficiently to allow gradual correction of sodium and water balance. A high-calorie, normal protein feed will decrease catabolism, uraemia and hyperkalaemia. Emergency management of metabolic acidosis, hyperkalaemia and hyperphosphataemia is shown in Table 18.5. If the cause of renal failure is not obvious, a renal biopsy should be performed to identify rapidly progressive glomerulonephritis, as this may need immediate treatment with immunosuppression. The two commonest renal causes of acute renal failure in children in the UK are the haemolytic uraemic syndrome and acute tubular necrosis, the latter usually in the setting of multisystem failure in the intensive care unit or following cardiac surgery.
Table 18.5
Some metabolic abnormalities in acute renal failure and their therapy
| Metabolic abnormality | Treatment |
| Metabolic acidosis | Sodium bicarbonate |
| Hyperphosphataemia | Calcium carbonate |
| Dietary restriction | |
| Hyperkalaemia | Calcium gluconate if ECG changes |
| Salbutamol (nebulised or intravenous) | |
| Calcium exchange resin | |
| Glucose and insulin | |
| Dietary restriction | |
| Dialysis |
 Haemolytic uraemic syndrome (HUS) – the triad of:
Haemolytic uraemic syndrome (HUS) – the triad of:Chronic kidney disease
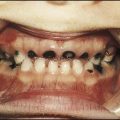
Chronic renal failure, with GFR <15 ml/min per 1.73 m2, is much less common in children than in adults, with an incidence of only 10 per million of the child population each year. Congenital and familial causes are more common in childhood than are acquired diseases (Table 18.6).
Table 18.6
Causes of chronic renal failure
| Structural malformations | 40% |
| Glomerulonephritis | 25% |
| Hereditary nephropathies | 20% |
| Systemic diseases | 10% |
| Miscellaneous/unknown | 5% |