[level-membership-for-internal-medicine-category]
Chapter 12 Kidney and urinary tract disease
Functional anatomy
Renal arteries and arterioles
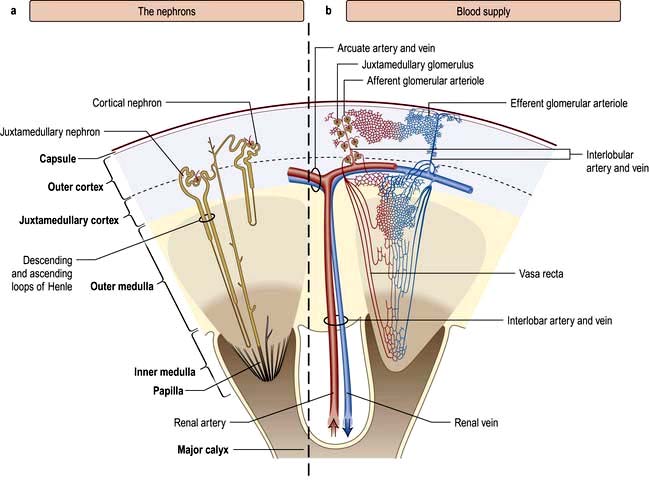
The renal artery undergoes a series of divisions within the kidney (Fig. 12.1) forming successively, the interlobar arteries, which run radially to the corticomedullary junction, arcuate arteries, which run circumferentially along the corticomedullary junction, and interlobular arteries, which run radially through the renal cortex towards the surface of the kidney. Afferent glomerular arterioles arise from the interlobular arteries to supply the glomerular capillary bed, which drains into efferent glomerular arterioles. Efferent arterioles from the outer cortical glomeruli drain into a peritubular capillary network within the renal cortex and then into increasingly large and more proximal branches of the renal vein. By contrast, blood from the juxtamedullary glomeruli passes via the vasa recta in the medulla and then turns back towards the area of the cortex from which the vasa recta originated.
The glomerulus
The glomerulus comprises four main cell types:
 Endothelial cells which are fenestrated with 500–1000 Å pores
Endothelial cells which are fenestrated with 500–1000 Å pores



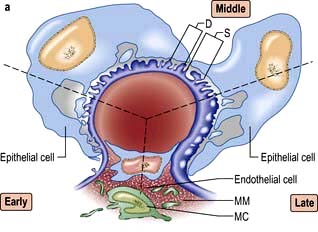
Mesangial cells are believed to be related to macrophages of the reticuloendothelial system and have a phagocytic function and contractile capabilities that can control blood flow and filtration surface area along the glomerular capillaries in response to a host of mediators. They also secrete the mesangial matrix, which provides a skeletal framework for the glomerular capillaries. The glomerular capillary basement membrane lies between the endothelial and the visceral epithelial cells. The latter put out multiple long foot processes which interdigitate with those of adjacent epithelial cells. Together the endothelial cells, basement membrane and epithelial cells form the filtration barrier or sieve (see Fig. 12.12a).
Renal tubules
The renal tubules are lined by epithelial cells, which are cuboidal except in the thin limb of the loop of Henle where they are flat. Proximal tubular cells differ from other cells of the system as they have a luminal brush border. The cortical portion of the collecting ducts contains two cell types with different functions, namely principal cells and intercalated cells (see p. 561). Fibroblast-like cells in the renal cortical interstitium have been shown to produce erythropoietin in response to hypoxia (p. 567).
The juxtaglomerular apparatus
The juxtaglomerular apparatus comprises the macula densa, the extraglomerular mesangium and the terminal portion of the afferent glomerular arteriole (which contains renin-producing granular cells) together with the proximal portion of the efferent arteriole. The macula densa is a plaque of cells containing large, tightly packed cell nuclei (hence the name macula densa; see Fig. 12.2) within the thick ascending limb of the loop of Henle. This anatomical arrangement is such as to allow changes in the renal tubule to influence behaviour of the adjacent glomerulus (tubuloglomerular feedback).
Renal function
Physiology
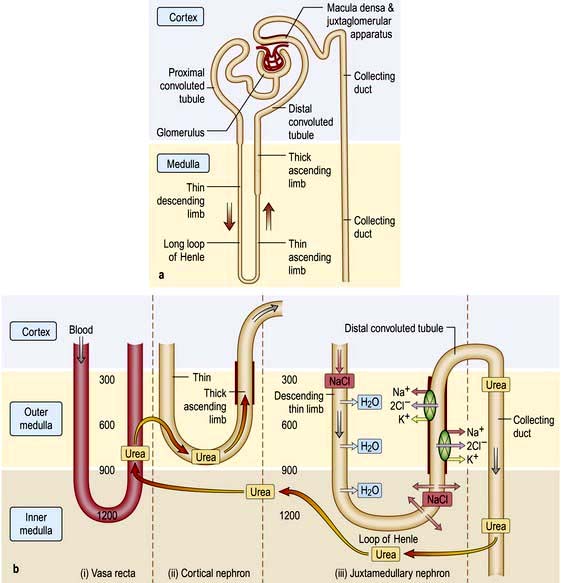
A conventional diagrammatic representation of the nephron is shown in Figure 12.2a and a physiological version in Figure 12.2b.
A hydrostatic pressure gradient of approximately 10 mmHg (a capillary pressure of 45 mmHg minus 10 mmHg of pressure within Bowman’s space and 25 mmHg of plasma oncotic pressure) provides the driving force for ultrafiltration of virtually protein-free and fat-free fluid across the glomerular capillary wall into Bowman’s space and so into the renal tubule (Fig. 12.3).
The ultrafiltration rate (glomerular filtration rate; GFR) varies with age and sex but is approximately 120–130 mL/min per 1.73 m2 surface area in adults. This means that, each day, ultrafiltration of 170–180 L of water and unbound small-molecular-weight constituents of blood occurs. If these large volumes of ultrafiltrate were excreted unchanged as urine, it would be necessary to ingest huge amounts of water and electrolytes to stay in balance. This is avoided by the selective reabsorption of water, essential electrolytes and other blood constituents, such as glucose and amino acids, from the filtrate in transit along the nephron. Thus, 60–80% of filtered water and sodium are reabsorbed in the proximal tubule along with virtually all the potassium, bicarbonate, glucose and amino acids (Fig. 12.2b). Additional water and sodium chloride are reabsorbed more distally, and fine tuning of salt and water balance is achieved in the distal tubules and collecting ducts under the influence of aldosterone and antidiuretic hormone (ADH). The final urine volume is thus 1–2 L daily. Calcium, phosphate and magnesium are also selectively reabsorbed in proportion to the need to maintain a normal electrolyte composition of body fluids.
Urine concentration and the countercurrent system
Urine is concentrated by a complex interaction between the loops of Henle, the medullary interstitium, medullary blood vessels (vasa recta) and the collecting ducts (see p. 640). The proposed mechanism of urine concentration is termed ‘the countercurrent mechanism’. The countercurrent hypothesis states that: ‘a small difference in osmotic concentration at any point between fluid flowing in opposite directions in two parallel tubes connected in a hairpin manner is multiplied many times along the length of the tubes’. Tubular fluid moves from the renal cortex towards the papillary tip of the medulla via the proximal straight tubule and the thin descending limb of the loop of Henle, which is permeable to water and impermeable to sodium. The tubule then loops back towards the cortex so that the direction of the fluid movement is reversed in the ascending limb, which is impermeable to water but permeable to sodium. This results in a large osmolar concentration difference between the corticomedullary junction and the hairpin loop at the tip of the papilla, and hence countercurrent multiplication. There is an analogy with heat exchangers.
Since the urine that emerges from the proximal tubule is iso-osmotic, the first nephron segment actually involved in urinary concentration is the descending limb of Henle’s loop. There are two types of descending limbs (Fig. 12.2b).


Glomerular filtration rate (GFR)
The concentration of urea or creatinine in plasma represents the dynamic equilibrium between production and elimination. In healthy subjects there is an enormous reserve of renal excretory function, and serum urea and creatinine do not rise above the normal range until there is a reduction of 50–60% in the GFR. Thereafter, the level of urea depends both on the GFR and its production rate (Table 12.1). The latter is heavily influenced by protein intake and tissue catabolism. The level of creatinine is much less dependent on diet but is more related to age, sex and muscle mass. Once it is elevated, serum creatinine is a better guide to GFR than urea and, in general, measurement of serum creatinine is a good way to monitor further deterioration in the GFR.
| Production | Elimination |
|---|---|
|
Increased by |
Increased by |
|
High-protein diet |
Elevated GFR, e.g. pregnancy |
|
Increased catabolism |
|
|
Surgery |
Decreased by |
|
Infection |
Glomerular disease |
|
Trauma |
Reduced renal blood flow |
|
Corticosteroid therapy |
Hypotension |
|
Tetracyclines |
Dehydration |
|
Gastrointestinal bleeding |
Urinary obstruction |
|
Cancer |
Tubulointerstitial nephritis |
|
Decreased by |
|
|
Low-protein diet |
|
|
Reduced catabolism, e.g. old age |
|
|
Liver failure |
|
GFR, glomerular filtration rate.
It must be re-emphasized that a normal serum urea or creatinine is not synonymous with a normal GFR.
Measurement of the glomerular filtration rate
Measurement of the GFR is necessary to define the exact level of renal function. It is essential when the serum (plasma) urea or creatinine is within the normal range. The most widely used measurement is the creatinine clearance (Fig. 12.4).
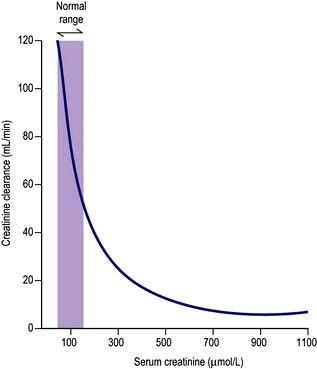
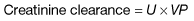
Calculated GFR. Measurement of true GFR is cumbersome, time-consuming and may be inaccurate if 24-hour urine collections are incomplete. Therefore, several formulae have been developed that allow a prediction of creatinine clearance or GFR from serum creatinine and demographics. The Cockcroft–Gault equation for creatinine clearance is shown in Box 12.1.
 Box 12.1
Box 12.1
Estimation of glomerular filtration rate (GFR)
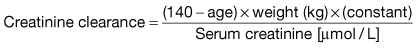

A prediction equation has been developed based on the data derived from the Modification of Diet in Renal Disease (MDRD) study in people with chronic kidney disease (CKD) (Box 12.1). This equation is based on age, sex, creatinine and ethnicity. A modification of MDRD equation is used by most chemical pathology laboratories to calculate eGFR but it is less reliable if actual GFR is >60 mL/min and can result in inappropriate referral to renal physicians.
Tubular function
The major function of the tubule is the selective reabsorption or excretion of water and various cations and anions to keep the volume and electrolyte composition of body fluid normal (see Ch. 13).
Conversely, inherited or acquired defects in tubular function may lead to incomplete absorption of a normal filtered load, with loss of the compound in the urine (a lowered ‘renal threshold’). This is seen in renal glycosuria, in which there is a genetically determined defect in tubular reabsorption of glucose. It is diagnosed by demonstrating glycosuria in the presence of normal blood glucose levels. Inherited or acquired defects in the tubular reabsorption of amino acids, phosphate, sodium, potassium and calcium also occur, either singly or in combination. Examples include cystinuria and Fanconi’s syndrome (see p. 1040 and Ch. 13). Tubular defects in the reabsorption of water result in nephrogenic diabetes insipidus (p. 992). Under normal circumstances, antidiuretic hormone induces an increase in the permeability of water in the collecting ducts by attachment to receptors with subsequent activation of adenyl cyclase. This then activates a protein kinase, which induces preformed cytoplasmic vesicles containing water channels (termed ‘aquaporins’) to move to and insert into the tubular luminal membrane. This allows water entry into tubular cells down a favourable osmotic gradient. Water then crosses the basolateral membrane and enters the bloodstream. When the effect of ADH wears off, water channels return to the cell cytoplasm (see Fig. 13.5).
Investigation of tubular function in clinical practice
Two tests of distal tubular function are commonly applied in clinical practice:


Endocrine function
Renin-angiotensin system
Juxtaglomerular apparatus
 Pressure changes in the afferent arteriole
Pressure changes in the afferent arteriole

 Chloride and osmotic concentration in the distal tubule via the macula densa (Fig. 12.2a)
Chloride and osmotic concentration in the distal tubule via the macula densa (Fig. 12.2a)

The renin-angiotensin-aldosterone system is illustrated in Figure 12.5.
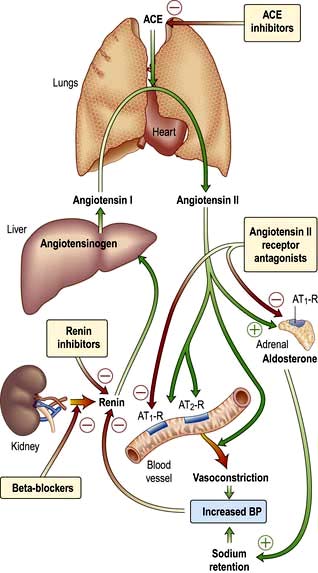


In addition to influencing systemic haemodynamics, angiotensin II also regulates GFR. Although it constricts both afferent and efferent arterioles, vasoconstriction of efferent arterioles is three times greater than that of afferent, resulting in increase of glomerular capillary pressure and maintenance of GFR. In addition, angiotensin II constricts mesangial cells, reducing the filtration surface area, and sensitizes the afferent arteriole to the constricting signal of tubuloglomerular feedback (see p. 562). The net result is that angiotensin II has opposing effects on the regulation of GFR: (a) an increase in glomerular pressure and consequent rise in GFR; (b) reduction in renal blood flow and mesangial cell contraction, reducing filtration (see Fig. 12.48). In renal artery stenosis with resultant low perfusion pressure, angiotensin II maintains GFR. However, in cardiac failure and hypertension, GFR may be reduced by angiotensin II.
The renin-angiotensin system can be blocked at several points with renin inhibitors, angiotensin-converting enzyme inhibitors (ACEI) and angiotensin II receptor antagonists (A-IIRA). These are useful agents in treatment of hypertension and heart failure (see p. 782 and p. 719) but have differences in action: ACEIs also block kinin production while A-IIRAs are specific for the AT-II receptors.
Erythropoietin
Erythropoietin (see also p. 374) is the major stimulus for erythropoiesis. It is a glycoprotein produced principally by fibroblast-like cells in the renal interstitium.
 Under hypoxic conditions both the α and β subunits of hypoxia inducible factor 2 (HIF-2) are expressed forming a heterodimer, causing erythropoietin gene transcription via the combined effects of hepatic nuclear factor 4 (HNF-4) and coactivator p300. Erythropoietin, once formed, binds to its receptors on erythroid precursor cells.
Under hypoxic conditions both the α and β subunits of hypoxia inducible factor 2 (HIF-2) are expressed forming a heterodimer, causing erythropoietin gene transcription via the combined effects of hepatic nuclear factor 4 (HNF-4) and coactivator p300. Erythropoietin, once formed, binds to its receptors on erythroid precursor cells.

 The hydroxylated HIF-2-α subunit binds to von Hippel-Lindau protein and a ubiquitin ligase E3 complex is activated. This leads to ubiquitination (p. 31) and subsequent degradation of HIF-2-α via proteosomes so that no erythropoietin is transcribed. In normoxic conditions HIF-2-α also undergoes asparaginyl hydroxylation which prevents HIF complex from recruiting coactivators. These hydroxylation steps have absolute requirement for molecular oxygen which forms the basis of oxygen sensing.
The hydroxylated HIF-2-α subunit binds to von Hippel-Lindau protein and a ubiquitin ligase E3 complex is activated. This leads to ubiquitination (p. 31) and subsequent degradation of HIF-2-α via proteosomes so that no erythropoietin is transcribed. In normoxic conditions HIF-2-α also undergoes asparaginyl hydroxylation which prevents HIF complex from recruiting coactivators. These hydroxylation steps have absolute requirement for molecular oxygen which forms the basis of oxygen sensing.
Loss of renal substance, with decreased erythropoietin production, results in a normochromic, normocytic anaemia. Conversely, erythropoietin secretion may be increased, with resultant polycythaemia, in people with polycystic renal disease, benign renal cysts or renal cell carcinoma. Recombinant human erythropoietin has been biosynthesized and is available for clinical use, particularly in people with chronic kidney disease (CKD) (see p. 623).
Vitamin D metabolism
Naturally occurring vitamin D (see also p. 622) (cholecalciferol) requires hydroxylation in the liver at position 25 and again by a 1α-hydroxylase enzyme (mitochondrial cytochrome P450) mainly in the distal convoluted tubule, the cortical and inner medullary part of the collecting ducts and the papillary epithelia of the kidney to produce the metabolically active 1,25-dihydroxycholecalciferol (1,25-(OH)2D3). The 1α-hydroxylase activity is increased by high plasma levels of parathyroid hormone (PTH), low phosphate and low 1,25-(OH)2D3. 1,25-dihydroxycholecalciferol and 25-hydroxycholecalciferol are degraded in part by being hydroxylated at position 24 by 24-hydroxylase. The activity of this enzyme is reduced by PTH and increased by 1,25-(OH)2D3 (which therefore promotes its own inactivation).
Autocrine function
Prostaglandins
Prostaglandins are unsaturated, oxygenated fatty acids, derived from the enzymatic metabolism of arachidonic acid, mainly by constitutively expressed cyclo-oxygenase-1 (COX-1) or inducible COX-2 (see Fig. 15.30). COX-1 is highly expressed in the collecting duct, while COX-2 expression is restricted to the macula densa. Both COX isoforms convert arachidonic acid to the same product, the bioactive but unstable prostanoid precursor, prostaglandin H2 (PGH2). PGH2 is converted to:
 PGE2 (formed by PDE2 synthase in the collecting duct, responsible for natriuretic and diuretic effects)
PGE2 (formed by PDE2 synthase in the collecting duct, responsible for natriuretic and diuretic effects)
 PGD2 (undetermined significance, produced in proximal tubule)
PGD2 (undetermined significance, produced in proximal tubule)
 prostacyclin (PGI2) (mainly synthesized in the interstitial and vascular compartment)
prostacyclin (PGI2) (mainly synthesized in the interstitial and vascular compartment)
 thromboxane A2 (vasoconstrictor, mainly synthesized in glomerulus).
thromboxane A2 (vasoconstrictor, mainly synthesized in glomerulus).
Nitric oxide and the kidney
Nitric oxide (see Fig. 16.18), a molecular gas, is formed by the action of three isoforms of nitric oxide synthase (NOS; p. 879). The most recognized cellular target of nitric oxide is soluble guanylate cyclase. The stimulation of this enzyme enhances the synthesis of cyclic GMP from GTP. All three isoforms are expressed in the kidney with eNOS in the vascular compartment, nNOS mainly in the macula densa and inner medullary collecting duct, and iNOS in several tubule segments. Nitric oxide mediates the following physiological actions in the kidney:



Investigations
Examination of the urine
Chemical (Stix) testing
Blood
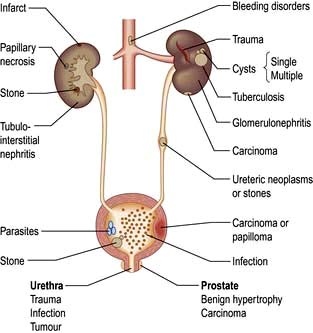
Haematuria may be overt, with bloody urine, or microscopic and found only on chemical testing. A positive Stix test must always be followed by microscopy of fresh urine (with the exception of menstruating women) to confirm the presence of red cells or red-cell casts and so exclude the relatively rare conditions of haemoglobinuria or myoglobinuria. Bleeding may come from any site within the urinary tract (Fig. 12.6):



Bacteriuria
Dipstick tests for bacteriuria are based on the detection of nitrite produced from the reduction of urinary nitrate by bacteria and also for the detection of leucocyte esterase, an enzyme specific for neutrophils. Although each test on its own has limitations, a positive reaction with both tests has a high predictive value for urinary tract infection (p. 593).
Microscopy
 White blood cells. The presence of ≥10 WBCs/mm3 in fresh unspun mid-stream urine samples is abnormal and indicates an inflammatory reaction within the urinary tract such as urinary tract infection (UTI), stones, tubulointerstitial nephritis, papillary necrosis, tuberculosis and interstitial cystitis.
White blood cells. The presence of ≥10 WBCs/mm3 in fresh unspun mid-stream urine samples is abnormal and indicates an inflammatory reaction within the urinary tract such as urinary tract infection (UTI), stones, tubulointerstitial nephritis, papillary necrosis, tuberculosis and interstitial cystitis.


 Bacteria, see page 593. Always culture urine prior to starting antibiotic therapy for sensitivities. Stix testing for blood or protein is of no value in the diagnosis of a UTI as both can be absent in the urine of many people with bacteriuria.
Bacteria, see page 593. Always culture urine prior to starting antibiotic therapy for sensitivities. Stix testing for blood or protein is of no value in the diagnosis of a UTI as both can be absent in the urine of many people with bacteriuria.
Blood and quantitative tests
The use of serum urea, creatinine and GFR as measures of renal function is discussed on page 564. Other quantitative tests of disturbed renal function are described under the relevant disorders, as are diagnostic tests, e.g. ANCA, immunofluorescence and complement.
Imaging techniques
Plain X-ray
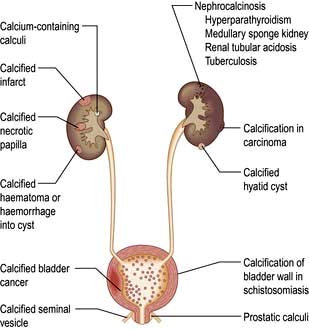
A plain radiograph of the abdomen is valuable to identify renal calcification or radiodense calculi in the kidney, renal pelvis, line of the ureters or bladder (Fig. 12.7).
Ultrasonography
 Renal measurement and for renal biopsy or other interventional procedures
Renal measurement and for renal biopsy or other interventional procedures

 Characterizing renal masses as cystic or solid
Characterizing renal masses as cystic or solid
 Diagnosing polycystic kidney disease
Diagnosing polycystic kidney disease
 Detecting intrarenal and/or perinephric fluid (e.g. pus, blood)
Detecting intrarenal and/or perinephric fluid (e.g. pus, blood)


The disadvantages of using ultrasonography to assess the urinary tract are:




Computed tomography (CT)
 Characterize renal masses which are indeterminate at ultrasonography
Characterize renal masses which are indeterminate at ultrasonography




 Visualize the renal arteries and veins by CT angiography
Visualize the renal arteries and veins by CT angiography
 Stage bladder and prostate tumours (MRI is increasingly used instead to stage prostate cancer).
Stage bladder and prostate tumours (MRI is increasingly used instead to stage prostate cancer).
Disadvantages include radiation and contrast nephrotoxicity (p. 614).
Magnetic resonance imaging (MRI)



Magnetic resonance urography is preferred over intravenous urography (IVU) in patients with chronic urolithiasis or intrinsic or extrinsic ureteric tumour, and in paediatric uroradiology. Gadolinium is used as contrast medium and is less nephrotoxic than iodine-containing agents used in IVU. However, the Federal Drug Administration (FDA) advises not using gadolinium in patients with renal insufficiency because of development of nephrogenic systemic fibrosis (pp. 1220 and 598).
Antegrade pyelography
Antegrade pyelography (Fig. 12.8) involves percutaneous puncture of a pelvicalyceal system with a needle and the injection of contrast medium to outline the pelvicalyceal system and ureter to the level of obstruction. It is used when ultrasonography has shown a dilated pelvicalyceal system in a patient with suspected obstruction. Antegrade pyelography is the preliminary to percutaneous placing of a drainage catheter or ureteric stent in the obstructed pelvicalyceal system (percutaneous nephrostomy).
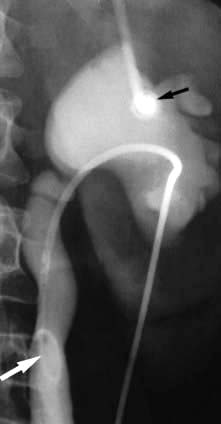
Micturating cystourethrography (MCU)
This involves catheterization and the instillation of contrast medium into the bladder. The catheter is then removed and the patient screened during voiding to check for vesicoureteric reflux and to study the urethra and bladder emptying. It is used in children with recurrent infection (see p. 592).
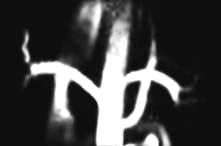
Aortography or renal arteriography
Conventional or digital subtraction angiography (DSA) is used. The latter allows the use of smaller doses of contrast medium which can be injected via a central venous catheter (venous DSA) or via a fine transfemoral arterial catheter (arterial DSA). Angiography is mainly used to define extrarenal or intrarenal arterial disease. Magnetic resonance angiography and multislice CT angiography are used (Fig. 12.9). Complications include cholesterol embolizations (p. 599) and contrast-induced kidney damage (contrast nephropathy).
Renal scintigraphy
Renal scintigraphy using a gamma camera is divided into:
 dynamic studies in which the function of the kidney is examined serially over a period of time, most often using a radiopharmaceutical excreted by glomerular filtration
dynamic studies in which the function of the kidney is examined serially over a period of time, most often using a radiopharmaceutical excreted by glomerular filtration
 static studies involving imaging of tracer that is taken up and retained by the renal tubule.
static studies involving imaging of tracer that is taken up and retained by the renal tubule.
Dynamic scintigraphy
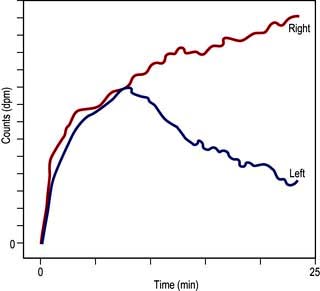
Investigation of obstruction. Renal scintigraphy provides functional evidence of obstruction. After injection usually of [99mTc]MAG3 a rise in resistance to flow in the pelvis or ureter prolongs the parenchymal transit of tracer and there is usually a delay in emptying the pelvis. On whole-kidney renograms, the time-activity curve fails to fall after an initial peak, or continues to rise (Fig. 12.10).
Transcutaneous renal biopsy (Practical Box 12.1)
 Practical Box 12.1
Practical Box 12.1The complications of transcutaneous renal biopsy are shown in Table 12.2.
Glomerular diseases
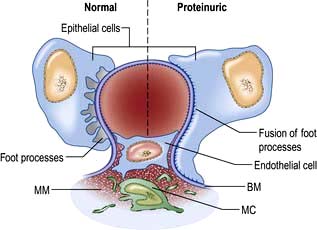
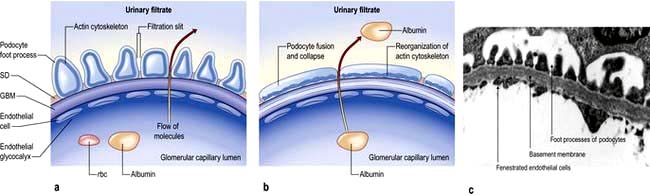
A glomerulus consists of a collection of capillaries which come from the afferent arteriole and are confined within the urinary space (Bowman’s capsule); this is continuous with the proximal tubule. The capillaries are partially attached to mesangium, a continuation of the arteriolar wall consisting of mesangial cells and the matrix. The free wall of glomerular capillaries (across which filtration takes place) consists of basement membrane covered by visceral epithelial cells with individual foot processes and lined by endothelial cells (Fig. 12.11). The normal thickness of the basement membrane is about 250–300 nm. The spaces between foot processes, with diameters of 20–60 nm, are called filtration pores, by which filtered fluid reaches the urinary space. The endothelial cells on the luminal aspect of the basement membrane are fenestrated (diameter 70–100 nm). The basement membrane is arranged in three zones: lamina rara externa, lamina rara densa and lamina rara interna, and is composed of type IV collagen and negatively charged proteoglycans (heparan sulphate).
Filtration barrier (slit diaphragm) (Fig. 12.12)


Glomerulopathies
Glomerulopathy (GN) is a general term for a group of disorders in which:
 there is primarily an immunologically mediated injury to glomeruli, although renal interstitial damage is a regular accompaniment
there is primarily an immunologically mediated injury to glomeruli, although renal interstitial damage is a regular accompaniment
 the kidneys are involved symmetrically
the kidneys are involved symmetrically

 renal lesions may be part of a generalized disease (e.g. systemic lupus erythematosus, SLE).
renal lesions may be part of a generalized disease (e.g. systemic lupus erythematosus, SLE).
Pathological terms in glomerular disease






Classification of glomerulopathies
 Nephrotic syndrome – massive proteinuria (>3.5 g/day), hypoalbuminaemia, oedema, lipiduria and hyperlipidaemia.
Nephrotic syndrome – massive proteinuria (>3.5 g/day), hypoalbuminaemia, oedema, lipiduria and hyperlipidaemia.



Certain types of GN, particularly those that are a part of a systemic disease, can present as more than one syndrome, e.g. lupus nephritis, cryoglobulinaemia, and Henoch–Schönlein purpura. They are usually associated with the nephrotic syndrome and will be discussed below. Investigation of glomerular diseases is shown in Table 12.3.
| Investigations | Positive findings |
|---|---|
|
Urine microscopy |
Red cells, red-cell casts |
|
Urinary protein |
Nephrotic or sub-nephrotic range proteinuria |
|
Serum urea |
May be elevated |
|
Serum creatinine |
May be elevated |
|
Culture (throat swab, discharge from ear, swab from inflamed skin) |
Nephritogenic organism (not always) |
|
Antistreptolysin-O titre |
Elevated in post-streptococcal nephritis |
|
C3 and C4 levels |
May be reduced |
|
Antinuclear antibody |
Present in significant titre in systemic lupus erythematosus |
|
ANCA |
Positive in some vasculitis |
|
Anti-GBM |
Positive in Goodpasture’s syndrome |
|
Cryoglobulins |
Increased in cryoglobulinaemia |
|
Creatinine clearance |
Normal or reduced |
|
Chest X-ray |
Cardiomegaly, pulmonary oedema (not always) |
|
Renal imaging |
Usually normal |
|
Renal biopsy |
Any glomerulopathy |
Nephrotic syndrome
Management
General measures
 Initial treatment should be with dietary sodium restriction and a thiazide diuretic (e.g. bendroflumethiazide 5 mg daily). Unresponsive patients require furosemide 40–120 mg daily with the addition of amiloride (5 mg daily), with the serum potassium concentration monitored regularly. Nephrotic patients may malabsorb diuretics (as well as other drugs) owing to gut mucosal oedema, and parenteral administration is then required initially. Patients are sometimes hypovolaemic, and moderate oedema may have to be accepted in order to avoid postural hypotension.
Initial treatment should be with dietary sodium restriction and a thiazide diuretic (e.g. bendroflumethiazide 5 mg daily). Unresponsive patients require furosemide 40–120 mg daily with the addition of amiloride (5 mg daily), with the serum potassium concentration monitored regularly. Nephrotic patients may malabsorb diuretics (as well as other drugs) owing to gut mucosal oedema, and parenteral administration is then required initially. Patients are sometimes hypovolaemic, and moderate oedema may have to be accepted in order to avoid postural hypotension.






Specific measures
Table 12.4 shows the glomerular lesions commonly associated with the nephrotic syndrome. These are divided into diseases with or without RBC casts (active or bland urine sediments). Each of these entities occurs as a primary renal lesion or as a secondary component of a systemic disease.
Table 12.4 Glomerulopathies associated with the nephrotic syndrome
Nephrotic syndrome with ‘bland’ urine sediments
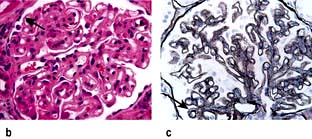
Minimal-change glomerular lesion (minimal-change nephropathy, minimal-change disease, MCD)
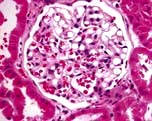
In this condition the glomeruli appear normal on light microscopy (Fig. 12.13). The only abnormality seen on electron microscopy is fusion of the foot processes of epithelial cells (podocytes) (Fig. 12.12b; p. 573). This is a nonspecific finding and is seen in many conditions associated with proteinuria, e.g. focal segmental glomerulosclerosis (FSGS). Neither immune complexes nor anti-GBM antibody can be demonstrated by immunofluorescence.
Focal segmental glomerulosclerosis (FSGS)
Pathology
This glomerulopathy is defined primarily by its appearance on light microscopy. Segmental glomerulosclerosis is seen, which later progresses to global sclerosis. The deep glomeruli at the corticomedullary junction are affected first. These may be missed on transcutaneous biopsy, leading to a mistaken diagnosis of a minimal-change glomerular lesion. A pathogenetic link may exist between minimal-change nephropathy (p. 575) and focal glomerulosclerosis, as a proportion of cases classified as having the former condition develop progressive CKD, which is unusual. Immunofluorescence shows deposits of C3 and IgM in affected portions of the glomerulus. The other glomeruli are usually enlarged but may be of normal size. In some patients, mesangial hypercellularity is a feature. Focal tubular atrophy and interstitial fibrosis are invariably present. Electron microscopic findings mirror light microscopic features with capillary obliteration by hyaline deposits (mesangial matrix and basement membrane material) and lipids. The other glomeruli exhibit primarily foot process effacement, occasionally in a patchy distribution.
Five histological variants of FSGS exist:
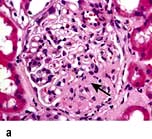
 In classic FSGS (Fig. 12.14a) the involved glomeruli show sclerotic segments in any location of the glomerulus.
In classic FSGS (Fig. 12.14a) the involved glomeruli show sclerotic segments in any location of the glomerulus.
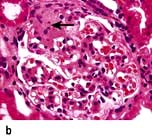
 The glomerular tip lesion is characterized by segmental sclerosis, at the tubular pole of all the affected glomeruli at a very early stage (tip FSGS) (Fig. 12.14b). Capillaries contain foam cells, and overlying visceral epithelial cells are enlarged and adherent to the most proximal portion of proximal tubules. These patients have a more favourable response to steroids and run a more benign course.
The glomerular tip lesion is characterized by segmental sclerosis, at the tubular pole of all the affected glomeruli at a very early stage (tip FSGS) (Fig. 12.14b). Capillaries contain foam cells, and overlying visceral epithelial cells are enlarged and adherent to the most proximal portion of proximal tubules. These patients have a more favourable response to steroids and run a more benign course.
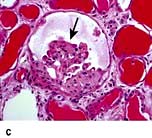
 In collapsing FSGS (Fig. 12.14c) the visceral cells are usually enlarged and coarsely vacuolated with wrinkled and collapsed capillary walls. These features indicate a severe lesion, with a corresponding progressive clinical course of the disease. Collapsing FSGS is commonly seen in young blacks with human immunodeficiency virus (HIV) infection or disease and is known as HIV-associated nephropathy (HIVAN) (see p. 93).
In collapsing FSGS (Fig. 12.14c) the visceral cells are usually enlarged and coarsely vacuolated with wrinkled and collapsed capillary walls. These features indicate a severe lesion, with a corresponding progressive clinical course of the disease. Collapsing FSGS is commonly seen in young blacks with human immunodeficiency virus (HIV) infection or disease and is known as HIV-associated nephropathy (HIVAN) (see p. 93).
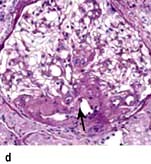
 The perihilar variant (Fig. 12.14d) consists of perihilar sclerosis and hyalinosis in more than 50% of segmentally sclerotic glomeruli. It is frequently observed with secondary FGS due to processes associated with increased glomerular capillary pressure and declining renal mass.
The perihilar variant (Fig. 12.14d) consists of perihilar sclerosis and hyalinosis in more than 50% of segmentally sclerotic glomeruli. It is frequently observed with secondary FGS due to processes associated with increased glomerular capillary pressure and declining renal mass.
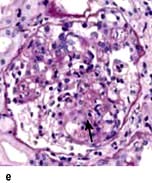
 The cellular variant (Fig. 12.14e) is characterized by at least one glomerulus with segmental endocapillary hypercellularity that occludes the capillary lumen. Other glomeruli may exhibit findings consistent with classic FGS.
The cellular variant (Fig. 12.14e) is characterized by at least one glomerulus with segmental endocapillary hypercellularity that occludes the capillary lumen. Other glomeruli may exhibit findings consistent with classic FGS.



Membranous glomerulopathy
Aetiopathogenesis
An identical glomerular histological picture is seen in the primary or idiopathic form (which comprises 75% of the cases) and also when membranous GN is secondary to drugs (e.g. penicillamine, gold, NSAIDs, probenecid, mercury, captopril), autoimmune disease (e.g. SLE, thyroiditis), infectious disease (e.g. hepatitis B, hepatitis C, schistosomiasis, Plasmodium malariae), neoplasia (e.g. carcinoma of lung, colon, stomach, breast and lymphoma) and other causes (e.g. sarcoidosis, kidney transplantation, sickle cell disease). At all stages, immunofluorescence shows the presence of uniform granular capillary wall deposits of IgG and complement C3. In the early stage the deposits are small and can be missed on light microscopy. Electron microscopy reveals small electron-dense deposits in the subepithelial aspects of the capillary walls. In the intermediate stage the deposits are encircled by basement membrane, which gives an appearance of spikes of basement membrane perpendicular to the basement membrane on silver staining. Late in the disease the deposits are completely surrounded by basement membrane and are undergoing resorption, which appears as uniform thickening of the capillary basement membrane on light microscopy (Fig. 12.15).
FURTHER READING
Fogō AB. Milk and membranous nephropathy. N Engl J Med 2011; 364:2154–2159.
Prunotto M, Carnevali ML, Candiano G et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol 2010; 21:507–519.
Stone JH et al. Mechanisms of disease: IgG4-related disease. N Engl J Med 2012; 366:539–551.





Amyloidosis (see p. 1042)
Pathology
On light microscopy, eosinophilic deposits are seen in the mesangium, capillary loops and arteriolar walls. Staining with Congo red renders these deposits pink and they show green birefringence under polarized light (Fig. 12.16). Immunofluorescence is unhelpful, but on electron microscopy the characteristic fibrils of amyloid can be seen. Amyloid consisting of immunoglobulin light chains (AL amyloid) can be identified by immunohistochemistry in only 40% of the cases as compared to almost 100% of patients with protein found in secondary amyloid (AA amyloid). Amyloid A (AA) amyloidosis, also referred to as secondary amyloidosis, is a rare but serious complication of chronic inflammatory diseases and chronic infections.
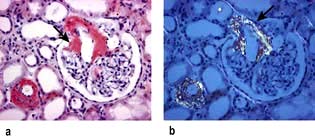
Diagnosis and treatment
The diagnosis can often be made clinically when features of amyloidosis are present elsewhere. On imaging, the kidneys are often large. Scintigraphy with radiolabelled serum amyloid P (SAP), a technique for quantitatively imaging amyloid deposits in vivo, is used to detect the rate of regression or progression of amyloidosis over a period of time (p. 1043). Renal biopsy is necessary in all suspected cases of renal involvement.
Treatment. Treatments that reduce production of the amyloidogenic protein can improve organ function and survival in immunoglobulin-light-chain-related (AL) amyloidosis and hereditary transthyretin-associated (ATTR) amyloidosis (see p. 1042).
In AA amyloidosis, production of serum amyloid A can sometimes be decreased by treatment of the underlying inflammatory condition but cannot be completely suppressed. A new class of drug, eprodisate (see p. 1043), has shown modest success in patients with renal amyloidosis.
Renoprotective measures should be started (p. 622). The success of dialysis and kidney transplantation is dependent upon the extent of amyloid deposition in extrarenal sites, especially the heart.
Diabetic nephropathy
Diabetic renal disease is the leading cause of ESKD in the western world. People with type 1 and type 2 diabetes (see p. 1025) have equivalent rates of proteinuria, azotaemia, and ultimately ESKD. Both types of diabetes show strong similarities in their rate of renal functional deterioration, and onset of co-morbid complications.
Pathology
The kidneys enlarge initially and there is glomerular hyperfiltration (GFR >150 mL/min). The major early histological lesions seen are glomerular basement membrane thickening and mesangial expansion. Moreover, progressive depletion of podocytes (p. 573) from the filtration barrier due to either apoptosis or detachment and resulting podocyturia appears to be a very early ultrastructural change. Later, glomerulosclerosis develops with nodules (Kimmelstiel–Wilson lesion) and hyaline deposits in the glomerular arterioles (Fig. 12.17). It has recently been shown that the mesangial expansion and the hyalinosis are partly due to amylin (beta islet specific amyloid protein) deposits. These later changes are associated with heavy proteinuria. The lesions seen in type 1 are also seen in type 2.
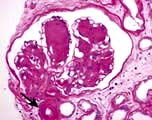
The Renal Pathology Society has developed a consensus classification combining type 1 and type 2 diabetic nephropathies (Table 12.5). This discriminates lesions by various degrees of severity for use in international clinical practice.
Table 12.5 Renal Pathology Society Classification of Type 1 and 2 diabetic nephropathy
| Class | Name |
|---|---|
|
I |
Isolated glomerular basement membrane thickening (>395 nm in females, >430 nm in males). No evidence of mesangial expansion, mesangial matrix increase, or global glomerulosclerosis involving >50% of glomeruli |
|
IIa |
Mild mesangial expansion |
|
IIb |
Severe mesangial expansion (in a severe lesion >25% of the total mesangium contains areas of expansion larger than the mean area of a capillary lumen) |
|
III |
Nodular intercapillary glomerulosclerosis (≥1 Kimmelstiel–Wilson lesion(s)) and <50% global glomerulosclerosis |
|
IV |
Advanced diabetic glomerulosclerosis and >50% global glomerulosclerosis |
Treatment
Lifestyle changes (cessation of smoking and increase in exercise), hypertension, poor metabolic regulation and hyperlidaemia should be addressed in every diabetic. Microalbuminuria is a reason to start treatment with ACE inhibitors or an angiotensin II receptor antagonist (AIIRA) in either type of diabetes, regardless of blood pressure elevation. Like other kidney diseases, however, nearly the entire course of renal injury in diabetes is clinically silent. The timing of medical intervention during this silent phase (see Box 12.6) is renoprotective, as judged by slowed loss of glomerular filtration. Despite intensified metabolic control and antihypertension treatment in patients with diabetes, a substantial number still go on to develop ESKD. In a randomized controlled trial, the addition of paricalcitol (a selective activator of the vitamin D receptor) to treatment with ACE inhibitors reduced albuminuria (a surrogate marker of progressive renal disease) in patients with type 2 diabetes. Paricalcitol worked best in patients with a high sodium intake in their diet, who respond poorly to ACE inhibitor and ARB therapy.
FURTHER READING
Tervaert TW, Mooyaart AL, Amann K et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol 2010; 21:556–563.
de Zeeuw D, Agarwal R, Amdahl M et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet 2010; 376:1543–1551.
Nephrotic syndrome with ‘active’ urine sediments (mixed nephrotic/nephritic)
Mesangiocapillary (membranoproliferative) glomerulonephritis (MCGN)
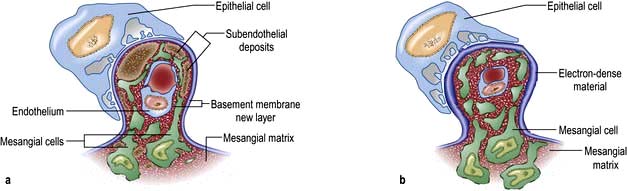
 Type 1 MCGN. There is mesangial cell proliferation, with mainly subendothelial immune deposition and apparent splitting of the capillary basement membrane, giving a ‘tram-line’ effect. It can be associated with persistently reduced plasma levels of C3 and normal levels of C4 due to activation of the complement cascade by the classical pathway. It is often idiopathic but occurs with chronic infection (abscesses, infective endocarditis, infected ventriculoperitoneal shunt) or cryoglobulinaemia secondary to hepatitis C infections (Fig. 12.18a).
Type 1 MCGN. There is mesangial cell proliferation, with mainly subendothelial immune deposition and apparent splitting of the capillary basement membrane, giving a ‘tram-line’ effect. It can be associated with persistently reduced plasma levels of C3 and normal levels of C4 due to activation of the complement cascade by the classical pathway. It is often idiopathic but occurs with chronic infection (abscesses, infective endocarditis, infected ventriculoperitoneal shunt) or cryoglobulinaemia secondary to hepatitis C infections (Fig. 12.18a).
 Type 2 MCGN. There is mesangial cell proliferation with electron-dense, linear intramembranous deposits that usually stain for C3 only (Fig. 12.18b). This type may be idiopathic or be associated with partial lipodystrophy (loss of subcutaneous fat on face and upper trunk). MCGN affects young adults. These patients have low C3 levels as in type 1 but this is due to the activation of the alternative pathway of the complement cascade; they also have autoantibodies to C3 convertase enzyme.
Type 2 MCGN. There is mesangial cell proliferation with electron-dense, linear intramembranous deposits that usually stain for C3 only (Fig. 12.18b). This type may be idiopathic or be associated with partial lipodystrophy (loss of subcutaneous fat on face and upper trunk). MCGN affects young adults. These patients have low C3 levels as in type 1 but this is due to the activation of the alternative pathway of the complement cascade; they also have autoantibodies to C3 convertase enzyme.

Management
In adults with the nephritic syndrome and/or renal impairment, aspirin (325 mg) or dipyridamole (75–100 mg) daily or a combination of the two, should be given for 6–12 months. Again, if no benefits are seen, the treatment should be stopped. Treatment to slow the rate of progression of CKD is instituted (p. 622).
Mesangial proliferative GN (IgM nephropathy, C1q nephropathy)
IgM nephropathy is characterized by increased mesangial cellularity in most of the glomeruli, associated with granular immune deposits (IgM and complement) in the mesangial regions. People present with episodic or persistent haematuria with the nephrotic syndrome. Unlike minimal-change disease, the prognosis is not uniformly good, as steroid response is only 50%. Between 10% and 30% develop progressive renal insufficiency with evidence of secondary FSGS (p. 576) on repeat biopsy. A trial of cyclophosphamide with prednisolone is used with persistent nephrotic syndrome, particularly with a rising plasma creatinine concentration.
Systemic lupus erythematosus (lupus glomerulonephritis)
Overt renal disease occurs in at least one-third of SLE patients and, of these, 25% reach end-stage CKD within 10 years (see also p. 536). Histologically, almost all patients will have changes. Box 12.2 shows the progression of histological findings and the clinical picture from classes I to VI.
 Box 12.2
Box 12.2
Classification of lupus nephritis
Class I – Minimal mesangial lupus nephritis (LN), with immune deposits but normal on light microscopy. Asymptomatic.
Class II – Mesangial proliferative LN with mesangial hypercellularity and matrix expansion. Clinically, mild renal disease.
Class III – Focal LN (involving <50% of glomeruli) with subdivisions for active or chronic lesions. Subepithelial deposits seen. Clinically have haematuria and proteinuria; 10–20% of all LN.
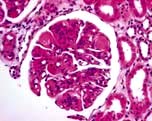
Class IV – Diffuse LN (involving >50% of glomeruli) (Fig. 12.19) classified by the presence of segmental and global lesions as well as active and chronic lesions. Subendothelial deposits are present. Clinically, there is progression to the nephrotic syndrome, hypertension and renal insufficiency. Most common and most severe form of LN.
Class V – Membranous LN affects 10–20% of patients. Can occur in combination with III or IV. Good prognosis.
Class VI – Advanced sclerosing LN (≥90% globally sclerosed glomeruli without residual activity). This represents the advanced stages of the above, as well as healing. Immunosuppressive therapy is unlikely to help as it is ‘inactive’. Progressive CKD.
Modified from International Society of Nephrology/Renal Pathological Society 2004.
Pathophysiology
SLE is now known to be an autoantigen-driven, T-cell-dependent and B-cell-mediated autoimmune disorder (p. 535). Lupus nephritis typically has circulating autoantibodies to cellular antigens (particularly anti-dsDNA, anti-Ro) and complement activation which leads to reduced serum levels of C3, C4, and particularly C1q. C1q is the first component of the classical pathway of the complement cascade (see p. 51) and is involved in the activation of complement and clearance of self-antigens generated during apoptosis. Anti-C1q antibodies may help in distinguishing a renal from a non-renal relapse. However, not all autoantibodies are pathogenic to the kidney. These nephrogenic antibodies have specific physicochemical characteristics and correlate well with the pattern of renal injury. DNA was thought to be the inciting autoantigen, but nucleosomes (structures comprising DNA and histone, generated during apoptosis) are the most likely autoantigen. Nucleosome-specific T cells, antinucleosome antibodies and nephritogenic immune complexes are generated. Positively charged histone components of the nucleosome bind to the negatively charged heparan sulphate (within the glomerular basement membrane) inciting an inflammatory reaction and resulting in mesangial cell proliferation, mesangial matrix expansion and inflammatory leucocytes. Other pathogenic mechanisms include infarction of glomerular segments, thrombotic microangiopathy, vasculitis and glomerular sclerosis.
The extraglomerular features of lupus nephritis include tubulointerstitial nephritis (75% of patients), renal vein thrombosis and renal artery stenosis. Thrombotic manifestations are associated with autoantibodies to phospholipids (anticardiolipin or lupus anticoagulant) (p. 538).
Management
 There have been a number of clinical trials with immunosuppressive agents have been trialled in types III, IV and V which are the most severe form of lupus nephritis. Outcomes of these types are affected by ethnicity, clinical characteristics, irreversible damage (on renal biopsy), initial response to treatment and the future frequency of renal flares.
There have been a number of clinical trials with immunosuppressive agents have been trialled in types III, IV and V which are the most severe form of lupus nephritis. Outcomes of these types are affected by ethnicity, clinical characteristics, irreversible damage (on renal biopsy), initial response to treatment and the future frequency of renal flares.




Cryoglobulinaemic renal disease
Glomerular disease is more common in type II than in type III cryoglobulinaemia. In approximately 30% of these ‘mixed’ cryoglobulinaemias, no underlying or associated disease is found (essential cryoglobulinaemia). Recognized associations include viral infections (hepatitis B and C, HIV, cytomegalovirus, Epstein–Barr infection), fungal and spirochaetal infections, malaria and infective endocarditis and autoimmune rheumatic diseases (SLE, rheumatoid arthritis and Sjögren’s syndrome). Glomerular pathological changes resemble MCGN (Fig. 12.18).
Acute glomerulonephritis (acute nephritic syndrome) (Table 12.6)
 Haematuria (macroscopic or microscopic) – red-cell casts are typically seen on urine microscopy
Haematuria (macroscopic or microscopic) – red-cell casts are typically seen on urine microscopy




Table 12.6 Diseases commonly associated with the acute nephritic syndrome
Post-streptococcal glomerulonephritis (PSGN)
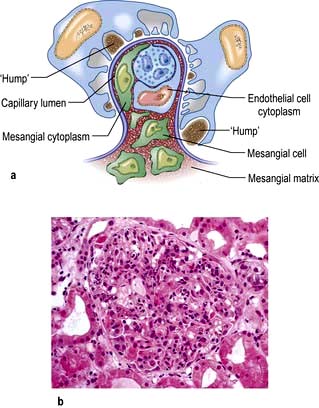
The patient, usually a child, suffers a streptococcal infection 1–3 weeks before the onset of the acute nephritic syndrome. Streptococcal throat infection, otitis media or cellulitis can all be responsible. The infecting organism is a Lancefield group A β-haemolytic streptococcus of a nephritogenic type. The latent interval between the infection and development of symptoms and signs of renal involvement reflects the time taken for immune complex formation and deposition and glomerular injury to occur. PSGN is now rare in developed countries. Renal biopsy shows diffuse, florid, acute inflammation in the glomerulus (without necrosis but occasionally cellular crescents), with neutrophils and deposition of immunoglobulin (IgG) and complement (Fig. 12.20). Ultrastructural findings are those of electron-dense deposits, characteristically but not solely in the subepithelial aspects of the capillary walls. Endothelial cells often are swollen. Similar biopsy findings may be seen in non-streptococcal post-infectious glomerulonephritis (Table 12.6).
Asymptomatic urinary abnormalities
Histology
A new Oxford histological classification for IgA nephropathy is shown in Table 12.7. The features have prognostic significance and it is recommended that they be taken into account for predicting outcome independent of the clinical features both at the time of presentation and during follow-up.
Table 12.7 Oxford histological classification for IgA nephropathy
| Histological variable | Class | |
|---|---|---|
|
Mesangial hypercellularity |
Average mesangial hypercellularitya >0.5 |
M1 |
|
Average mesangial hypercellularity <0.5 |
M0 |
|
|
Segmental glomerulosclerosis |
Part of the glomerular tuft is involved in sclerosis |
S1 |
|
No segmental glomerulosclerosis |
S0 |
|
|
Endocapillary hypercellularity |
Hypercellularity present and results in lumina narrowing |
E1 |
|
No hypercellularity |
E0 |
|
|
Tubular atrophy/interstitial fibrosis |
Percentage of cortical area involved: |
|
|
>50 |
T2 |
|
|
26–50 |
T1 |
|
|
0–25 |
T0 |
a Mesangial hypercellularity is scored zero (0) for glomeruli with <4 mesangial cells per mesangial area; 1 for those with 4–5 cells; 2 for 6–7 cells; and 3 for ≥8 cells. Scores obtained for all glomeruli are then averaged.
Clinical presentation
IgA nephropathy tends to occur in children and young males. They present with asymptomatic microscopic haematuria or recurrent macroscopic haematuria sometimes following an upper respiratory or gastrointestinal viral infection. Proteinuria occurs and 5% can be nephrotic. The prognosis is usually good, especially in those with normal blood pressure, normal renal function and absence of proteinuria at presentation. Surprisingly, recurrent macroscopic haematuria is a good prognostic sign, although this may be due to ‘lead-time bias’ (p. 436), as patients with overt haematuria come to medical attention at an earlier stage of their illness. The risk of eventual development of ESKD is about 25% in those with proteinuria of more than 1 g per day, elevated serum creatinine, hypertension, ACE gene polymorphism (DD isoform) and tubulointerstitial fibrosis on renal biopsy.
Alport’s syndrome
The primary glomerular filtration barrier of the glomerular capillary consists of the basement membrane and the outer slit diaphragm formed between adjacent podocytes. Loss of slit function causes massive proteinuria (congenital nephrotic syndrome, p. 576) but deterioration of glomerular basement membrane produces only mild proteinuria. The mild proteinuria in Alport’s syndrome is the result of glomerular sclerosis, rather than primary loss of slit pores. In pedigrees with a history of CKD, disease progresses from concomitant interstitial fibrosis, macrophage and lymphocyte infiltration secondary to tubular basement disruption and transdifferentiation of epithelial mesenchymal cells to fibroblasts. This fibrogenic response destroys renal architecture. The renal histology characteristically shows split basement membrane. In some patients with Alport’s syndrome and carriers, thin basement membrane, as seen in benign familial haematuria, is the only abnormality detected on histology. For this reason, the boundary between Alport’s and benign familial haematuria has become increasingly vague.
Rapidly progressive glomerulonephritis (RPGN)
RPGN is a syndrome with glomerular haematuria (RBC casts or dysmorphic RBCs), rapidly developing acute kidney failure over weeks to months and focal glomerular necrosis (Fig. 12.22) with or without glomerular crescent development on renal biopsy. The ‘crescent’ is an aggregate of macrophages and epithelial cells in Bowman’s space (Fig. 12.22). RPGN can develop with immune deposits (anti-GBM or immune complex type, e.g. SLE) or without immune deposits (pauci-immune, e.g. anti-PR3 and or anti-MPO-ANCA positive vasculitides). It can also develop as an idiopathic primary glomerular disease or can be superimposed on secondary glomerular diseases such as IgA nephropathy, membranous GN and post-infective GN. The classification used here is based on the immunofluorescence information obtained from renal histology (Table 12.8), viz linear, granular and negative immunofluorescent patterns.
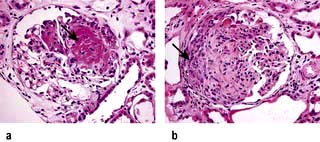
Table 12.8 Types of rapidly progressive glomerulonephritis (RPGN)
Anti-GBM glomerulonephritis (Fig. 12.23a)
Anti-GBM glomerulonephritis, characterized by linear capillary loop staining with IgG and C3 and extensive crescent formation, accounts for 15–20% of all cases of RPGN, although overall it accounts for less than 5% of all forms of glomerulonephritis. This condition is rare, with an incidence of 1 per 2 million in the general population. About two-thirds of these patients have Goodpasture’s syndrome with associated lung haemorrhage (p. 850). The remainder have a renal restricted anti-GBM RPGN, which is seen in patients over 50 years and affects both genders equally.
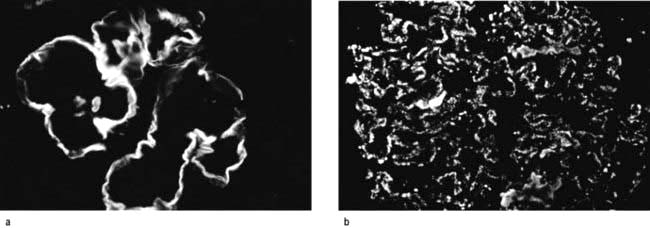
Anti-GBM antibodies (detected by ELISA) are present in serum and are directed against the non-collagenous (NCl) component of α3 (IV) collagen of basement membrane. This target antigen must be present as a component of the native α3, α4, α5 (IV) network of selected basement membrane in order for pulmonary and renal disease to develop. Consequently, there are no known cases of anti-GBM glomerulonephritis in patients with Alport’s syndrome (p. 584). However, the α3, α4, α5 (IV) network is also a target for anti-GBM alloantibodies in Alport’s syndrome (see this chapter) post-transplantation glomerulonephritis, which occurs in 3–5% of patients with Alport’s syndrome and results in allograft loss. Alport’s post-transplantation nephritis is mediated by the deposition of alloantibodies to the α3NC1 and α5NC1 domains in response to the ‘foreign’ α3, α4, α5 (IV) collagen network that is absent in the patient’s own kidneys with Alport’s syndrome but present in the renal allograft. Alloantibodies in Alport’s syndrome patients bind to epitopes in intact cross-linked α345NC1 hexamers. In contrast, autoantibodies in Goodpasture’s syndrome bind to native cross-linked α345NC1 hexamers only if dissociated first to unmask hidden epitopes.



ANCA-positive vasculitides (see also p. 544)
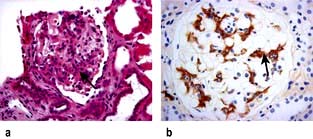
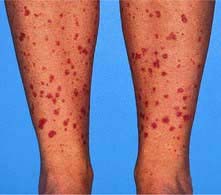
Inflammation and necrosis of the blood vessel wall occurs in many primary vasculitic disorders. Wegener’s granulomatosis, microscopic polyangiitis and Churg–Strauss syndrome are described as small vessel vasculitides and are commonly associated with antineutrophil cytoplasm antibodies (ANCA). These diseases share common pathology with focal necrotizing lesions, which affect many different vessels and organs; in the lungs, a capillaritis may cause lung haemorrhage; within the glomerulus of the kidney, crescentic GN and/or focal necrotizing lesions (FNGN) may cause acute kidney injury (Fig. 12.22); in the dermis, a purpuric rash or vasculitic (Fig. 12.24) ulceration. Wegener’s and Churg–Strauss syndrome may have additional granulomatous lesions.
 Focal ( ≥50% normal glomeruli that are not affected by the disease process)
Focal ( ≥50% normal glomeruli that are not affected by the disease process)
 Crescentic ( ≥50% of glomeruli with cellular crescents)
Crescentic ( ≥50% of glomeruli with cellular crescents)
 Mixed (a heterogeneous glomerular phenotype wherein no glomerular feature predominates)
Mixed (a heterogeneous glomerular phenotype wherein no glomerular feature predominates)

Pathogenesis
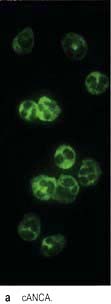
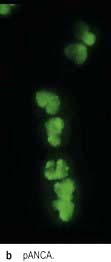
ANCA positive glomerulonephritis – immunofluorescence.
(From Schrier RW 1999. The Schrier Atlas of Diseases of the Kidney Vol IV: Systemic Diseases and the Kidney. Wiley-Blackwell, with permission).
There are two forms of ANCA (p. 544), viz PR3-ANCA (cANCA) and MPO-ANCA (pANCA). If ELISA and indirect immunofluorescence techniques are combined, diagnostic specificity is 99%. Testing for antineutrophil cytoplasmic antibodies should be accompanied by appropriate tests of autoantibodies directed against DNA and the glomerular basement membrane antigen. The simultaneous occurrence of ANCA and anti-GMB antibody is well documented; such patients tend to follow the natural history of Goodpasture’s syndrome. Variations in the ANCA titres have been used in the assessment of disease activity.
 PR3-ANCA positivity is found in the large majority (>90%) of patients with active Wegener’s granulomatosis and in up to 50% of patients with microscopic polyangiitis.
PR3-ANCA positivity is found in the large majority (>90%) of patients with active Wegener’s granulomatosis and in up to 50% of patients with microscopic polyangiitis.



 Autoimmunity. It is unclear how and why autoimmunity causes the formation of ANCA antibodies. Patients with anti-PR3 also have autoantibodies to a peptide translated from the antisense DNA strand of PR-3 (complementary PR-3; cPR-3) or to a mimetic of this peptide. This suggests that autoimmunity can be initiated through an immune response against a peptide that is antisense or complementary to the autoantigen, which then induces anti-idiotypic antibodies (autoantibodies) that cross-react with the autoantigen.
Autoimmunity. It is unclear how and why autoimmunity causes the formation of ANCA antibodies. Patients with anti-PR3 also have autoantibodies to a peptide translated from the antisense DNA strand of PR-3 (complementary PR-3; cPR-3) or to a mimetic of this peptide. This suggests that autoimmunity can be initiated through an immune response against a peptide that is antisense or complementary to the autoantigen, which then induces anti-idiotypic antibodies (autoantibodies) that cross-react with the autoantigen.

Treatment
The sooner treatment is instituted the more chance there is of recovery of renal function.
 Corticosteroids and cyclophosphamide are of benefit: high-dose oral prednisolone (maximum 80 mg/day reducing over time to 15 mg/day by 3 months) and cyclophosphamide (2 mg/kg per day, adjusted for age, renal function and prevailing WBC count). Intravenous pulse, rather than daily oral, cyclophosphamide is associated with an equivalent response with better side-effect profile but is associated with higher relapse rate. The best indicators of prognosis are pulmonary haemorrhage and severity of renal failure at presentation.
Corticosteroids and cyclophosphamide are of benefit: high-dose oral prednisolone (maximum 80 mg/day reducing over time to 15 mg/day by 3 months) and cyclophosphamide (2 mg/kg per day, adjusted for age, renal function and prevailing WBC count). Intravenous pulse, rather than daily oral, cyclophosphamide is associated with an equivalent response with better side-effect profile but is associated with higher relapse rate. The best indicators of prognosis are pulmonary haemorrhage and severity of renal failure at presentation.







FURTHER READING
Berden AE, Ferrario F, Hagen EC et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol 2010; 21:1628–1636.
Jayne D. Challenges in the management of microscopic polyangiitis: past, present and future. Curr Opin Rheumatol 2008; 20(1):3–9.
Jennette JC, Falk RJ. New insight into the pathogenesis of vasculitis associated with antineutrophil cytoplasmic autoantibodies. Curr Opin Rheumatol 2008; 20(1):55–60.
Other glomerular disorders
HIV-associated nephropathy (HIVAN)
A number of renal lesions have been described in association with HIV infection (see p. 177). These include glomerulonephritis of various histological types and the haemolytic uraemic syndrome. The most common (80–90%) histological abnormality is a focal glomerulosclerosis (FGS).
HIV-associated FGS
A characteristic ‘collapsed’ appearance of glomeruli is often seen on light microscopy similar to that seen in other causes of focal segmental glomerulosclerosis (see Fig. 12.14c). In HIVAN many visceral epithelial cells (podocytes) are enlarged, hyperplastic, coarsely vacuolated, contain protein absorption droplets and overlie capillaries with varying degrees of wrinkling and collapse of the walls. It is associated with loss of podocyte-specific markers such as Wilms’ tumour factor and synaptopodin due to HIV-1 infection of podocytes of patients with HIVAN. HIVAN has striking predilection; over 90% of patients are black. Clinically HIVAN presents with proteinuria in the nephrotic range, oedema and a ‘bland’ urine. Hypertension is unusual. If untreated, patients go on to CKD which can be rapid in progression.
Paraneoplastic glomerulonephritis
The pathology of paraneoplastic glomerulonephritis varies depending on types of malignancy.
 Thymoma or Hodgkin’s lymphoma: polarization of the immune response toward a T-helper-2 profile and possibly excessive production of interleukin-13 may lead to the development of minimal change disease or focal associated membranoproliferative glomerulonephritis and membranous nephropathy
Thymoma or Hodgkin’s lymphoma: polarization of the immune response toward a T-helper-2 profile and possibly excessive production of interleukin-13 may lead to the development of minimal change disease or focal associated membranoproliferative glomerulonephritis and membranous nephropathy


 Myelodysplastic syndromes: autoimmunity causes a variety of glomerulonephritides
Myelodysplastic syndromes: autoimmunity causes a variety of glomerulonephritides

[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 12 Kidney and urinary tract disease
Functional anatomy
Renal arteries and arterioles
The renal artery undergoes a series of divisions within the kidney (Fig. 12.1) forming successively, the interlobar arteries, which run radially to the corticomedullary junction, arcuate arteries, which run circumferentially along the corticomedullary junction, and interlobular arteries, which run radially through the renal cortex towards the surface of the kidney. Afferent glomerular arterioles arise from the interlobular arteries to supply the glomerular capillary bed, which drains into efferent glomerular arterioles. Efferent arterioles from the outer cortical glomeruli drain into a peritubular capillary network within the renal cortex and then into increasingly large and more proximal branches of the renal vein. By contrast, blood from the juxtamedullary glomeruli passes via the vasa recta in the medulla and then turns back towards the area of the cortex from which the vasa recta originated.
The glomerulus
The glomerulus comprises four main cell types:
Endothelial cells which are fenestrated with 500–1000 Å pores
Mesangial cells are believed to be related to macrophages of the reticuloendothelial system and have a phagocytic function and contractile capabilities that can control blood flow and filtration surface area along the glomerular capillaries in response to a host of mediators. They also secrete the mesangial matrix, which provides a skeletal framework for the glomerular capillaries. The glomerular capillary basement membrane lies between the endothelial and the visceral epithelial cells. The latter put out multiple long foot processes which interdigitate with those of adjacent epithelial cells. Together the endothelial cells, basement membrane and epithelial cells form the filtration barrier or sieve (see Fig. 12.12a).
Renal tubules
The renal tubules are lined by epithelial cells, which are cuboidal except in the thin limb of the loop of Henle where they are flat. Proximal tubular cells differ from other cells of the system as they have a luminal brush border. The cortical portion of the collecting ducts contains two cell types with different functions, namely principal cells and intercalated cells (see p. 561). Fibroblast-like cells in the renal cortical interstitium have been shown to produce erythropoietin in response to hypoxia (p. 567).
The juxtaglomerular apparatus
The juxtaglomerular apparatus comprises the macula densa, the extraglomerular mesangium and the terminal portion of the afferent glomerular arteriole (which contains renin-producing granular cells) together with the proximal portion of the efferent arteriole. The macula densa is a plaque of cells containing large, tightly packed cell nuclei (hence the name macula densa; see Fig. 12.2) within the thick ascending limb of the loop of Henle. This anatomical arrangement is such as to allow changes in the renal tubule to influence behaviour of the adjacent glomerulus (tubuloglomerular feedback).
Renal function
Physiology
A conventional diagrammatic representation of the nephron is shown in Figure 12.2a and a physiological version in Figure 12.2b.
A hydrostatic pressure gradient of approximately 10 mmHg (a capillary pressure of 45 mmHg minus 10 mmHg of pressure within Bowman’s space and 25 mmHg of plasma oncotic pressure) provides the driving force for ultrafiltration of virtually protein-free and fat-free fluid across the glomerular capillary wall into Bowman’s space and so into the renal tubule (Fig. 12.3).
The ultrafiltration rate (glomerular filtration rate; GFR) varies with age and sex but is approximately 120–130 mL/min per 1.73 m2 surface area in adults. This means that, each day, ultrafiltration of 170–180 L of water and unbound small-molecular-weight constituents of blood occurs. If these large volumes of ultrafiltrate were excreted unchanged as urine, it would be necessary to ingest huge amounts of water and electrolytes to stay in balance. This is avoided by the selective reabsorption of water, essential electrolytes and other blood constituents, such as glucose and amino acids, from the filtrate in transit along the nephron. Thus, 60–80% of filtered water and sodium are reabsorbed in the proximal tubule along with virtually all the potassium, bicarbonate, glucose and amino acids (Fig. 12.2b). Additional water and sodium chloride are reabsorbed more distally, and fine tuning of salt and water balance is achieved in the distal tubules and collecting ducts under the influence of aldosterone and antidiuretic hormone (ADH). The final urine volume is thus 1–2 L daily. Calcium, phosphate and magnesium are also selectively reabsorbed in proportion to the need to maintain a normal electrolyte composition of body fluids.
Urine concentration and the countercurrent system
Urine is concentrated by a complex interaction between the loops of Henle, the medullary interstitium, medullary blood vessels (vasa recta) and the collecting ducts (see p. 640). The proposed mechanism of urine concentration is termed ‘the countercurrent mechanism’. The countercurrent hypothesis states that: ‘a small difference in osmotic concentration at any point between fluid flowing in opposite directions in two parallel tubes connected in a hairpin manner is multiplied many times along the length of the tubes’. Tubular fluid moves from the renal cortex towards the papillary tip of the medulla via the proximal straight tubule and the thin descending limb of the loop of Henle, which is permeable to water and impermeable to sodium. The tubule then loops back towards the cortex so that the direction of the fluid movement is reversed in the ascending limb, which is impermeable to water but permeable to sodium. This results in a large osmolar concentration difference between the corticomedullary junction and the hairpin loop at the tip of the papilla, and hence countercurrent multiplication. There is an analogy with heat exchangers.
Since the urine that emerges from the proximal tubule is iso-osmotic, the first nephron segment actually involved in urinary concentration is the descending limb of Henle’s loop. There are two types of descending limbs (Fig. 12.2b).
Glomerular filtration rate (GFR)
The concentration of urea or creatinine in plasma represents the dynamic equilibrium between production and elimination. In healthy subjects there is an enormous reserve of renal excretory function, and serum urea and creatinine do not rise above the normal range until there is a reduction of 50–60% in the GFR. Thereafter, the level of urea depends both on the GFR and its production rate (Table 12.1). The latter is heavily influenced by protein intake and tissue catabolism. The level of creatinine is much less dependent on diet but is more related to age, sex and muscle mass. Once it is elevated, serum creatinine is a better guide to GFR than urea and, in general, measurement of serum creatinine is a good way to monitor further deterioration in the GFR.
| Production | Elimination |
|---|---|
|
Increased by |
Increased by |
|
High-protein diet |
Elevated GFR, e.g. pregnancy |
|
Increased catabolism |
|
|
Surgery |
Decreased by |
|
Infection |
Glomerular disease |
|
Trauma |
Reduced renal blood flow |
|
Corticosteroid therapy |
Hypotension |
|
Tetracyclines |
Dehydration |
|
Gastrointestinal bleeding |
Urinary obstruction |
|
Cancer |
Tubulointerstitial nephritis |
|
Decreased by |
|
|
Low-protein diet |
|
|
Reduced catabolism, e.g. old age |
|
|
Liver failure |
|
GFR, glomerular filtration rate.
It must be re-emphasized that a normal serum urea or creatinine is not synonymous with a normal GFR.
Measurement of the glomerular filtration rate
Measurement of the GFR is necessary to define the exact level of renal function. It is essential when the serum (plasma) urea or creatinine is within the normal range. The most widely used measurement is the creatinine clearance (Fig. 12.4).
Calculated GFR. Measurement of true GFR is cumbersome, time-consuming and may be inaccurate if 24-hour urine collections are incomplete. Therefore, several formulae have been developed that allow a prediction of creatinine clearance or GFR from serum creatinine and demographics. The Cockcroft–Gault equation for creatinine clearance is shown in Box 12.1.
Box 12.1
Estimation of glomerular filtration rate (GFR)
A prediction equation has been developed based on the data derived from the Modification of Diet in Renal Disease (MDRD) study in people with chronic kidney disease (CKD) (Box 12.1). This equation is based on age, sex, creatinine and ethnicity. A modification of MDRD equation is used by most chemical pathology laboratories to calculate eGFR but it is less reliable if actual GFR is >60 mL/min and can result in inappropriate referral to renal physicians.
Tubular function
The major function of the tubule is the selective reabsorption or excretion of water and various cations and anions to keep the volume and electrolyte composition of body fluid normal (see Ch. 13).
Conversely, inherited or acquired defects in tubular function may lead to incomplete absorption of a normal filtered load, with loss of the compound in the urine (a lowered ‘renal threshold’). This is seen in renal glycosuria, in which there is a genetically determined defect in tubular reabsorption of glucose. It is diagnosed by demonstrating glycosuria in the presence of normal blood glucose levels. Inherited or acquired defects in the tubular reabsorption of amino acids, phosphate, sodium, potassium and calcium also occur, either singly or in combination. Examples include cystinuria and Fanconi’s syndrome (see p. 1040 and Ch. 13). Tubular defects in the reabsorption of water result in nephrogenic diabetes insipidus (p. 992). Under normal circumstances, antidiuretic hormone induces an increase in the permeability of water in the collecting ducts by attachment to receptors with subsequent activation of adenyl cyclase. This then activates a protein kinase, which induces preformed cytoplasmic vesicles containing water channels (termed ‘aquaporins’) to move to and insert into the tubular luminal membrane. This allows water entry into tubular cells down a favourable osmotic gradient. Water then crosses the basolateral membrane and enters the bloodstream. When the effect of ADH wears off, water channels return to the cell cytoplasm (see Fig. 13.5).
Investigation of tubular function in clinical practice
Two tests of distal tubular function are commonly applied in clinical practice:
Endocrine function
Renin-angiotensin system
Juxtaglomerular apparatus
Pressure changes in the afferent arteriole
Chloride and osmotic concentration in the distal tubule via the macula densa (Fig. 12.2a)
The renin-angiotensin-aldosterone system is illustrated in Figure 12.5.
In addition to influencing systemic haemodynamics, angiotensin II also regulates GFR. Although it constricts both afferent and efferent arterioles, vasoconstriction of efferent arterioles is three times greater than that of afferent, resulting in increase of glomerular capillary pressure and maintenance of GFR. In addition, angiotensin II constricts mesangial cells, reducing the filtration surface area, and sensitizes the afferent arteriole to the constricting signal of tubuloglomerular feedback (see p. 562). The net result is that angiotensin II has opposing effects on the regulation of GFR: (a) an increase in glomerular pressure and consequent rise in GFR; (b) reduction in renal blood flow and mesangial cell contraction, reducing filtration (see Fig. 12.48). In renal artery stenosis with resultant low perfusion pressure, angiotensin II maintains GFR. However, in cardiac failure and hypertension, GFR may be reduced by angiotensin II.
The renin-angiotensin system can be blocked at several points with renin inhibitors, angiotensin-converting enzyme inhibitors (ACEI) and angiotensin II receptor antagonists (A-IIRA). These are useful agents in treatment of hypertension and heart failure (see p. 782 and p. 719) but have differences in action: ACEIs also block kinin production while A-IIRAs are specific for the AT-II receptors.
Erythropoietin
Erythropoietin (see also p. 374) is the major stimulus for erythropoiesis. It is a glycoprotein produced principally by fibroblast-like cells in the renal interstitium.
Under hypoxic conditions both the α and β subunits of hypoxia inducible factor 2 (HIF-2) are expressed forming a heterodimer, causing erythropoietin gene transcription via the combined effects of hepatic nuclear factor 4 (HNF-4) and coactivator p300. Erythropoietin, once formed, binds to its receptors on erythroid precursor cells.
The hydroxylated HIF-2-α subunit binds to von Hippel-Lindau protein and a ubiquitin ligase E3 complex is activated. This leads to ubiquitination (p. 31) and subsequent degradation of HIF-2-α via proteosomes so that no erythropoietin is transcribed. In normoxic conditions HIF-2-α also undergoes asparaginyl hydroxylation which prevents HIF complex from recruiting coactivators. These hydroxylation steps have absolute requirement for molecular oxygen which forms the basis of oxygen sensing.
Loss of renal substance, with decreased erythropoietin production, results in a normochromic, normocytic anaemia. Conversely, erythropoietin secretion may be increased, with resultant polycythaemia, in people with polycystic renal disease, benign renal cysts or renal cell carcinoma. Recombinant human erythropoietin has been biosynthesized and is available for clinical use, particularly in people with chronic kidney disease (CKD) (see p. 623).
Vitamin D metabolism
Naturally occurring vitamin D (see also p. 622) (cholecalciferol) requires hydroxylation in the liver at position 25 and again by a 1α-hydroxylase enzyme (mitochondrial cytochrome P450) mainly in the distal convoluted tubule, the cortical and inner medullary part of the collecting ducts and the papillary epithelia of the kidney to produce the metabolically active 1,25-dihydroxycholecalciferol (1,25-(OH)2D3). The 1α-hydroxylase activity is increased by high plasma levels of parathyroid hormone (PTH), low phosphate and low 1,25-(OH)2D3. 1,25-dihydroxycholecalciferol and 25-hydroxycholecalciferol are degraded in part by being hydroxylated at position 24 by 24-hydroxylase. The activity of this enzyme is reduced by PTH and increased by 1,25-(OH)2D3 (which therefore promotes its own inactivation).
Autocrine function
Prostaglandins
Prostaglandins are unsaturated, oxygenated fatty acids, derived from the enzymatic metabolism of arachidonic acid, mainly by constitutively expressed cyclo-oxygenase-1 (COX-1) or inducible COX-2 (see Fig. 15.30). COX-1 is highly expressed in the collecting duct, while COX-2 expression is restricted to the macula densa. Both COX isoforms convert arachidonic acid to the same product, the bioactive but unstable prostanoid precursor, prostaglandin H2 (PGH2). PGH2 is converted to:
PGE2 (formed by PDE2 synthase in the collecting duct, responsible for natriuretic and diuretic effects)
PGD2 (undetermined significance, produced in proximal tubule)
prostacyclin (PGI2) (mainly synthesized in the interstitial and vascular compartment)
thromboxane A2 (vasoconstrictor, mainly synthesized in glomerulus).
Nitric oxide and the kidney
Nitric oxide (see Fig. 16.18), a molecular gas, is formed by the action of three isoforms of nitric oxide synthase (NOS; p. 879). The most recognized cellular target of nitric oxide is soluble guanylate cyclase. The stimulation of this enzyme enhances the synthesis of cyclic GMP from GTP. All three isoforms are expressed in the kidney with eNOS in the vascular compartment, nNOS mainly in the macula densa and inner medullary collecting duct, and iNOS in several tubule segments. Nitric oxide mediates the following physiological actions in the kidney:
Investigations
Examination of the urine
Chemical (Stix) testing
Blood
Haematuria may be overt, with bloody urine, or microscopic and found only on chemical testing. A positive Stix test must always be followed by microscopy of fresh urine (with the exception of menstruating women) to confirm the presence of red cells or red-cell casts and so exclude the relatively rare conditions of haemoglobinuria or myoglobinuria. Bleeding may come from any site within the urinary tract (Fig. 12.6):
Bacteriuria
Dipstick tests for bacteriuria are based on the detection of nitrite produced from the reduction of urinary nitrate by bacteria and also for the detection of leucocyte esterase, an enzyme specific for neutrophils. Although each test on its own has limitations, a positive reaction with both tests has a high predictive value for urinary tract infection (p. 593).
Microscopy
White blood cells. The presence of ≥10 WBCs/mm3 in fresh unspun mid-stream urine samples is abnormal and indicates an inflammatory reaction within the urinary tract such as urinary tract infection (UTI), stones, tubulointerstitial nephritis, papillary necrosis, tuberculosis and interstitial cystitis.
Bacteria, see page 593. Always culture urine prior to starting antibiotic therapy for sensitivities. Stix testing for blood or protein is of no value in the diagnosis of a UTI as both can be absent in the urine of many people with bacteriuria.
Blood and quantitative tests
The use of serum urea, creatinine and GFR as measures of renal function is discussed on page 564. Other quantitative tests of disturbed renal function are described under the relevant disorders, as are diagnostic tests, e.g. ANCA, immunofluorescence and complement.
Imaging techniques
Plain X-ray
A plain radiograph of the abdomen is valuable to identify renal calcification or radiodense calculi in the kidney, renal pelvis, line of the ureters or bladder (Fig. 12.7).
Ultrasonography
Renal measurement and for renal biopsy or other interventional procedures
Characterizing renal masses as cystic or solid
Diagnosing polycystic kidney disease
Detecting intrarenal and/or perinephric fluid (e.g. pus, blood)
The disadvantages of using ultrasonography to assess the urinary tract are:
Computed tomography (CT)
Characterize renal masses which are indeterminate at ultrasonography
Visualize the renal arteries and veins by CT angiography
Stage bladder and prostate tumours (MRI is increasingly used instead to stage prostate cancer).
Disadvantages include radiation and contrast nephrotoxicity (p. 614).
