Keratinization and blistering syndromes
Common skin disorders, e.g. atopic eczema or psoriasis, have a genetic component that is often subject to environmental influences. The genodermatoses differ in being single gene defects and include keratinization, blistering and neurocutaneous syndromes.
The ichthyoses
The ichthyoses are inherited disorders of keratinization and epidermal differentiation. They are characterized by dry scaly skin and vary from mild and asymptomatic to severe and incompatible with life (Table 1). Keratinization is abnormal. Some of the biochemical defects have been identified, e.g. steroid sulphatase is deficient in X-linked ichthyosis.
| Disorder | Inheritance | Clinical features |
|---|---|---|
| Ichthyosis vulgaris | Autosomal dominant | Common (1 in 250). Onset 1–4 years. It occurs with atopic eczema. Often mild. Small bran-like scale seen. Flexures spared. Defect in filaggrin, needed for keratin assembly |
| X-linked ichthyosis | X-linked recessive | 1 in 2000 males. Generalized involvement with large brown scale. Onset in first week of life. Improves in summer. Due to a deficiency of steroid sulphatase |
| Non-bullous ichthyosiform erythroderma* | Autosomal recessive | Rare (1 in 300 000). At birth may present as collodion baby. Red scaly skin and ectropion may follow. Erythema improves with age |
| Bullous ichthyosiform erythroderma† | Autosomal dominant | Rare (<1 in 100 000). Redness and blisters occur after birth but fade. Warty rippled hyperkeratosis appears in childhood |
* Lamellar ichthyosis is similar but rarer.
† Also called epidermolytic hyperkeratosis.
Clinical presentation
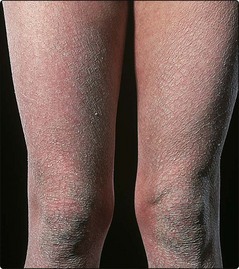
Ichthyosis vulgaris is common and unrecognized if mild. Small branny scales are seen on the extensor aspects of the limbs and the back (Fig. 1). The flexures are often spared.
The other types of ichthyosis are uncommon or rare and can usually be identified by their clinical features, onset and inheritance. Autosomal dominant conditions tend to improve with age, whereas recessive ichthyoses may worsen. Collodion baby describes a newborn infant with a tight shiny skin that causes feeding problems and ectropion. It is mainly due to non-bullous ichthyosiform erythroderma. Acquired ichthyosis (p. 88) usually starts in adulthood.
Keratoderma
Keratoderma describes gross hyperkeratosis of the palms and soles, and may be acquired or inherited.
Clinical presentation
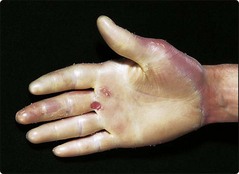
The degrees of involvement and modes of inheritance vary. A common pattern, tylosis, shows diffuse hyperkeratosis of the palms and soles (Fig. 2) and is usually of autosomal dominant inheritance. In a few families, tylosis is associated with carcinoma of the oesophagus. Other keratodermas may give punctate papular lesions on the palms and soles or, in ‘mutilating’ types, fibrous bands that strangulate digits.
Acquired palmoplantar keratoderma is seen in pityriasis rubra pilaris (p. 44) and lichen planus, and may develop in women at the menopause, particularly around the heels. Corns and callosities are different from keratoderma. Callosities are painless localized thickenings of the keratin layer and are seen as a protective response, induced by friction or pressure, which is often occupational in origin. Corns are painful and develop at areas of high local pressure on the feet, where shoes squeeze against bony points.
Keratosis pilaris
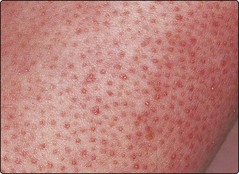
Keratosis pilaris is a common, sometimes inherited condition in which multiple small horny follicular plugs affect the upper thigh, upper arm and face (Fig. 3). It is occasionally associated with ichthyosis vulgaris. Application of 5% salicylic acid ointment or 10% urea cream lessens, but does not cure, the problem.
Darier’s disease
Darier’s disease (keratosis follicularis) is a rare autosomal dominant condition characterized by brownish scaly papules. The abnormal gene is sited on chromosome 12q and encodes calcium adenosine triphosphatase. Keratinocyte tonofilaments and desmosomes are dissociated on electron microscopy.
Clinical presentation
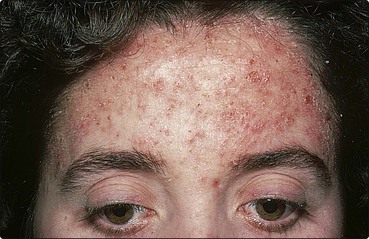
The disease presents in teenagers or young adults, with small brown greasy scaled papules, typically on the flexures, upper back, chest and forehead (Fig. 4). Its onset may follow sunburn. The severity varies from being mild and almost unnoticed to being extensive and severe. Nail changes (p. 68) and palmar pits or keratoses are found. Bacterial infection or eczema herpeticum (p. 36) may occur.
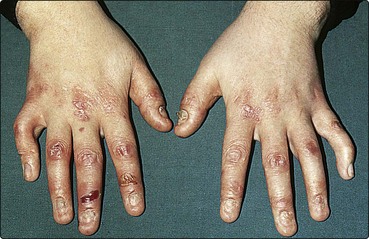
Epidermolysis bullosa
Epidermolysis bullosa (EB) defines a group of genetically inherited diseases characterized by skin fragility and blistering on minimal trauma. They range from being mild and trivial to being incompatible with life (Table 2).
| Disease | Inheritance | Clinical features |
|---|---|---|
| Simple EB | Autosomal dominant | Commonest type. Often mild and limited to hands and feet. Blisters caused by friction. Nails and mouth unaffected |
| Junctional EB | Autosomal recessive | Rare and often lethal. At birth, large erosions seen around mouth and anus. Slow to heal. No effective treatment |
| Dystrophic EB | Autosomal dominant | Hands, knees and elbows are affected. Scarring with milia is found. Deformity of the nails may occur |
| Dystrophic EB | Autosomal recessive | Starts in infancy. Severe blistering results in fusion of fingers and toes, with mucosal lesions and oesophageal stricture |
Aetiopathogenesis
Keratin synthesis is defective in simple EB (genes mapped to chromosomes 12 and 17). Collagen VII is abnormal in dystrophic EB (gene sited on chromosome 3). Anchoring fibrils (p. 2) are defective in certain types of EB.
Clinical presentation and management
Simple EB is fairly common and requires avoidance of trauma. The more severe forms (Table 2; Fig. 5) need to be managed in specialized centres. Avoidance of trauma, supportive measures and the control of infection are important. Treatment with various drugs has given disappointing results. Acquired EB, with an onset in adult life, shows trauma-induced blistering and resembles pemphigoid on immunofluorescent studies.
Prenatal diagnosis of inherited disorders
Inherited keratinization and blistering disorders







