KCNQ1/KCNE1 Macromolecular Signaling Complex
Channel Microdomains and Human Disease
Autonomic nervous system control of heart rate and cardiac contractility, through sympathetic and parasympathetic activity, is a fundamental property of the cardiovascular system. Exercise or emotional stress stimulates the sympathetic nervous system (SNS), resulting in a rapid and dramatic increase in heart rate. To ensure adequate diastolic filling time, the increase in heart rate is accompanied by a concomitant reduction in the ventricular action potential duration (APD) and the corresponding QT interval on the electrocardiogram (ECG). Defective regulation of cardiac electrical activity in the face of sympathetic nervous system activity can lead to arrhythmias.1
SNS control of cardiac electrical activity is mediated by the activation of β-adrenergic receptors (β-ARs) that regulate the function of select ion channels via phosphorylation by cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA). PKA-dependent phosphorylation up-regulates the activity of L-type calcium channels, leading to enhanced calcium entry that contributes to action potential prolongation, as well as an increase in intracellular calcium available for subsequent uptake by the cardiac sarcoplasmic reticulum (SR). PKA phosphorylation also activates the major intracellular calcium release channel on the SR, the type 2 ryanodine receptor (RyR2),2 which is responsible for releasing calcium to trigger muscle contraction.
Sympathetic stimulation also leads to a PKA-dependent increase in a slowly activating potassium channel current, IKs (Figure 11-1). IKs channels consist of the pore-forming α-subunit KCNQ1 and the auxiliary β-subunit KCNE13,4 and contribute to cardiac repolarization. PKA-dependent modulation increases the repolarization current to counter the stimulatory effects of PKA on L-type calcium channels5 to achieve a balance of inward and outward membrane currents. This balance of modulated currents is thought of as a necessary mechanism to regulate calcium homeostasis in the face of sympathetic activity.
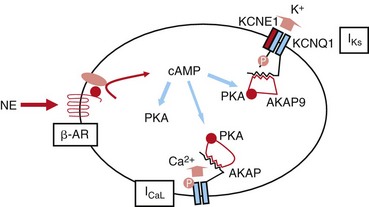
Figure 11-1 SNS regulation of IKs currents. Activation of the β-adrenergic receptor (β-AR) by norepinephrine (NE) results in an increase in the intracellular cAMP concentration, which, in turn, turns on PKA. IKs channels are phosphorylated by PKA. This regulation is mediated by a macromolecular complex coordinated by AKAP9. SNS, Sympathetic nervous system; NE, norepinephrine; β-AR, β-adrenergic receptor; PKA, protein kinase A; AKAP, A-kinase anchoring protein; ICaL, L-type Calcium channel.
Inherited mutations in ion channels have been associated with disorders that are exacerbated by SNS activity. For example, the genes that encode KCNQ1 and KCNE13,4 have been linked to the congenital long QT syndrome (LQTS). LQTS, a rare disease in which the QT interval of the ECG is prolonged as the result of dysfunctional ventricular repolarization, can precipitate lethal polymorphic ventricular tachycardia and is associated with syncope, seizures, and sudden death.6 Mutations in KCNQ1 cause LQT-1, and mutations in KCNE1 cause LQT-5.7 In affected patients, triggers of arrhythmias are gene-specific, and those with mutations in KCNQ1 or KCNE1 are at greatest risk of experiencing fatal cardiac arrhythmias in the face of elevated SNS activity.8 Unraveling the molecular links between the SNS and regulation of the KCNQ1/KCNE1 channel has direct implications for the mechanistic basis of triggers of arrhythmias in LQTS.
β-AR Signaling: Coordination of Localized Regulation of Channel Proteins by A-Kinase Anchoring Proteins
Key to the complex regulation of ion channels by β-AR stimulation is the spatiotemporal control of local cAMP concentration. This is mediated by the A-kinase anchoring proteins (AKAPs). AKAPs are a group of structurally diverse proteins with the common function of binding to, but not being limited to, PKA regulatory subunits.9–16 AKAPs provide structural scaffolding to integrate various enzymes and their substrates to form a compartmentalized environment. This enables spatiotemporal control of the regulatory enzymes (i.e., to present the enzymes at high concentrations at the site of their substrates when needed).17 Disruption of local complexes can unbalance the response and may have pathophysiological consequences.
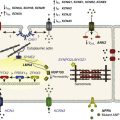
The IKs channel forms a macromolecular signaling complex that is coordinated by the binding of AKAP9, also known as yotiao,18, 19 via a leucine zipper (LZ) motif in the KCNQ1 carboxy (C)-terminus domain. Recent studies suggest that AKAP9 associates not only with the PKA regulatory subunit (RII), but also with protein phosphatase 1 (PP1),18 phosphodiesterase (PDE),20 and adenylyl cyclase (AC)21–23 (Figure 11-2). Together these enzymes control the phosphorylation state of the channel via the cAMP/PKA pathway.
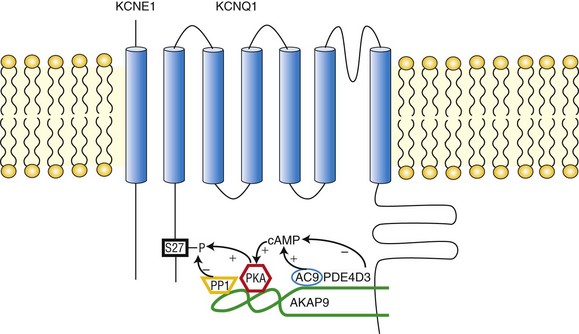
Figure 11-2 The KCNQ1/KCNE1/AKAP9 macromolecular complex. AKAP9 recruits PKA, phosphatase 1 (PP1), adenylyl cyclase 9 (AC9), and phosphodiesterase 4D3 (PDE4D3) to regulate the phosphorylation state of IKs channels.
Role of Leucine Zipper in Protein-Protein Interaction
Marks and colleagues were the first to show that the cardiac calcium release channel/RyR2 is regulated by a macromolecular signaling complex in which kinases and phosphatases are targeted to the channel via AKAP and leucine/isoleucine zipper (LZ) motifs.2,24,25 They suggested that this may be a common motif for coordination of ion channel signaling complexes.25 Subsequent investigations have shown that this is, in fact, the case for at least two other ion channels that are regulated by PKA: L-type calcium channels26, 27 and KCNQ1/KCNE1 potassium channels.18 The LZ domain is an α-helical structure that forms coiled coils and was originally identified as highly conserved motifs mediating the binding of transcription factors to DNA.28 Coiled coils are composed of heptad repeats (abcdefg)n in which hydrophobic residues occur at positions “a” and “d” and form the hydrophobic face of the helix, while “b, c, e, f, and g” are hydrophilic residues that form the solvent-exposed part of the coiled coil.29 LZs classically contain a leucine in position “d” because of its flexible side chain, although the canonical leucine residue can be replaced by an isoleucine or valine. Electrostatic interactions between side chains in the “e” and “g” sites from neighboring helices are believed to help specify binding partners.30 In vitro site-directed mutagenesis studies have successfully revealed the sequence specificity of interacting helices in proteins such as the GCN4 DNA binding domain,31 phospholamban,32 the myosin binding subunit/cGKIα,33 and ryanodine receptor types 1 and 2.25 In the case of the IKs channel (KCNQ1/KCNE1), Marx et al. identified an LZ motif in the C-terminus of the KCNQ1 subunit critical to AKAP9 interaction.18 Substitution of an alanine for one or more of the “d” position leucines or isoleucines in the LZ motif disintegrated the IKs/AKAP9 complex and rendered the channel unable to be regulated by PKA.
Molecular Components of the Iks/AKAP9 Complex
Associated with the IKs/AKAP9 complex are two pairs of enzymes with opposing effects on channel phosphorylation state. The first pair, which involves PKA and PP1, phosphorylates or de-phosphorylates the channel, respectively. The second pair involves AC and PDE, which increase and decrease the local cAMP gradient, respectively (see Figure 11-2). Working in concert, these enzymes fine-tune the IKs channel function.34
• PKA. AKAP9-bound PKA regulates IKs channel function by phosphorylation of a single amino acid residue (Ser27) on the N-terminus of KCNQ1.18 The biophysical consequence of PKA phosphorylation of the IKs channel is a profound increase in current amplitude, an accelerated onset of activation, a hyperpolarizing shift in the voltage dependence of channel activation, and a slowing of deactivation (the return of activated [open] to resting [closed] channels during diastole).35 The combined effect ensures that during voltage depolarization, KCNQ1/KCNE1 channel activity is increased in the presence of SNS stimulation. Consequently, the substantial repolarization reserve is activated in the face of SNS-mediated activity, and this reserve potassium channel current can offset SNS-mediated increases in calcium channel currents, which would prolong APD.5 It is interesting to note that AKAP9 itself was shown to be a substrate of PKA. Indeed, the phosphorylation of AKAP9 seemed to participate in the regulation of IKs channels.36 Early works identified a region on AKAP9 (residues 1440-1457: LEEEVAKVIVSMSIAFAQ) as the primary binding site for PKA RII subunits.37,38
• PP1. PP1 is a nonspecific serine/threonine phosphatase that dephosphorylates its substrate. AKAP9 provides a platform that allows PP1 to specifically target the IKs channel.18 Thus, PP1 can reverse the effect of PKA on the channel and attenuate the channel activity. This was evidenced by an experiment in which it was found that addition of okadaic acid, a PP1 inhibitor, enhanced the effect of PKA-dependent IKs channel regulation.18 The PP1 binding site on AKAP9 was shown to be located in a region that comprises residues 1171-1229.37
• AC. Upstream in the PKA pathway, ACs are activated by Gs-coupled receptors, such as the β-ARs. ACs are responsible for cAMP synthesis, which then activates PKA. Evidence now suggests that ACs are associated with various AKAPs, such as AKAP79/150, mAKAP, and AKAP9.21,39–41 AKAP9 associates with AC1, 2, 3, and 9 but not 4, 5, and 6. Residues 808-957 of AKAP9 bind directly to the AC2 N-terminus. Expression of AKAP9 inhibited the activity of AC2 and 3, but not AC1 or 9.22 It has been demonstrated that AC9 was a member of the IKs/AKAP9 complex in the cardiac myocytes of both IKs transgenic mice and guinea pigs. AC9 association with the complex sensitizes PKA phosphorylation of KCNQ1 to SNS stimulation.23
• PDE. Phosphodiesterases (PDEs) constitute the sole route for degrading cyclic nucleotides in cells. In the mammalian heart, the temporal and spatial dynamics of cAMP gradients are controlled mainly by PDE3s and PDE4s with a prevailing role of PDE4s, which are considered the cAMP-specific PDEs.42–44 In transgenic murine cardiac myocytes expressing IKs channels, PDE4s were shown to regulate the basal phosphorylation level of IKs channels. Two PDE4D isoforms, likely PDE4D3 and PDE4D5, were found to interact with the channels.20 However, in the heterologous expression system, only PDE4D3, which is known to interact with AKAPs (AKAP-18,45 AKAP-250,46,47 and AKAP-45047,48), was shown to be specifically recruited to KCNQ1 by AKAP9 and to regulate the amplitude of channels in response to cAMP stimulation.20 The binding site for PDE4D3 on AKAP9 currently is not known.
Iks Channel Regulation and Human Diseases
Phosphorylation of the IKs channel, stimulated by SNS and mediated by AKAP9, causes an increase in current density during depolarization, as well as a slowing of channel deactivation, which results in an accumulation of open channels on a beat-by-beat basis.18,35,49 The net result is an increased outward current reserve to counterbalance the increased activities of L-type calcium channels and RYR in the face of β-AR stimulation. Mutations that occur within the IKs/AKAP9 complex have been reported to disrupt the physical and functional integrity of the macromolecular complex, leading to an imbalance in intracellular calcium homeostasis and predisposing cells to two types of calcium-mediated rhythm disturbances: early afterdepolarizations (EADs)50,51 and delayed after depolarizations (DADs).5 It is now well established that disturbance of IKs channel regulation causes various cardiac arrhythmic disorders (Figure 11-3).
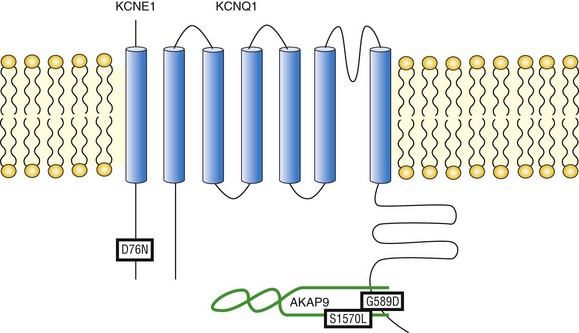
Figure 11-3 Mutations compromising the physical and functional integrity of the IKs/AKAP9 complex are associated with long QT syndrome (LQTS). Illustrated are the KCNQ1 mutation G589D, the KCNE1 mutation D76N, and the AKAP9 mutation S1570L. All are associated with LQTS.
KCNQ1 G589D and LQT-1
The inherited G589D mutation has been linked to the long QT syndrome variant 1 (LQT-1) in Finnish families.52 The naturally occurring G589D mutation occupies the “e” position in the KCNQ1 LZ motif that targets AKAP9.18 As has been noted, residues occupying the “e” and “g” positions in the LZ motif exhibit a limited range of substitutions based on the volume occluded by adjacent structures. Indeed, the G589D mutation disrupts the interaction between the IKs channel and AKAP9, and dislocates PKA and PP1 from the macromolecular complex. Functionally, the G589D mutation renders the IKs channel unresponsive to cAMP stimulation.18 Affected LQTS patients with this mutation suffer from dysfunctional regulation of QT duration during mental and physical stress53 and are at risk of arrhythmia and sudden cardiac death during exercise.52 Thus an inherited mutation of a single residue on the KCNQ1 channel disrupts the IKs/AKAP9 signaling complex and raises the risk of arrhythmia in affected patients.
KCNE1 D76N Mutation and LQT-5
Kurokawa et al demonstrated that cAMP-mediated functional regulation of KCNQ1/KCNE1 channels requires the expression of KCNQ1 with its auxiliary subunit KCNE1.54 Compared with the twofold increase in KCNQ1/KCNE1 current amplitude caused by cAMP and okadaic acid, KCNQ1 alone did not respond to stimulation.54 This suggests that the auxiliary subunit KCNE1 might play a role in transducing protein phosphorylation into channel functional regulation and represents a new paradigm that disruption of channel regulation by mutations on the auxiliary subunit may result in disease. This proved to be true in the case of a naturally occurring LQT-5 mutation KCNE1 D76N. Kurokawa et al. demonstrated that the D76N mutation ablated functional regulation of the channels by cAMP,54 resulting in an expected delay in the onset of repolarization that is more pronounced in the face of SNS stimulation.
AKAP9 S1570L Mutation and LQT-11
Given its critical role in the regulation of IKs channel function, it is conceivable to postulate that AKAP9 might be a candidate gene for LQTS. Chen et al.55 characterized the molecular mechanism of KCNQ1/AKAP9 interaction. The interaction between the two molecules is a three-way binding that involves both AKAP9 N- and C-termini as well as the LZ on the KCNQ1 C-terminus. Of particular interest, the AKAP9 C-terminal binding site (residues 1574-1643) contains an LZ motif that potentially matches the reciprocal counterpart in KCNQ1.55 Identification of AKAP9 binding motifs enabled quick screening in specific exons of the AKAP9 gene that encode for these important binding modules. In 50 LQTS patients with a strong clinical diagnosis (QTc ≥480 or Schwartz score ≥3.0) but negative for mutations in all LQTS genes, including KCNQ1 and KCNE1, a missense mutation (S1570L) was identified in one patient but was absent in 1320 ethnically matched reference alleles. The patient had experienced episodes of syncope and a prolonged QTc of 485 milliseconds, as well as a positive family history. AKAP9 S1570L mutation is located close to its C-terminal binding site for KCNQ1 and has a negative impact on the formation of the IKs/AKAP9 complex. The mutation also reduces PKA-induced phosphorylation of KCNQ1, rendering the IKs channels insensitive to PKA regulation. Moreover a computational modeling study revealed that the AKAP9 S1570L mutation significantly prolonged the cardiac action potential duration, especially under the impact of SNS innervations.55 These findings not only established AKAP9 as a novel causal gene for LQTS but also reinforced the idea that AKAPs play a critical role in many physiological processes.
IKs Channel and Atrial Fibrillation
The role of IKs channels in the pathophysiology of human cardiac arrhythmia is not limited to the LQTS. Recent findings suggest that mutations of the channel may also lead to short QT syndrome56 and familial atrial fibrillation (AF).57–60 Of particular interest are two adjacent KCNQ1 mutations (S140G57 and V141M58) that are associated with AF. Both mutations are located in the first transmembrane helix (S1) that interfaces with KCNE1.61 In intact IKs channels, these two mutations disrupt channel deactivation.62 With channels unable to close properly, currents accumulate with each heartbeat, giving rise to a gain-of-function phenotype and shortened APD. Chan et al63 observed that the phenotype of S140G and V141M mutations showed differential dependence on KCNE1. Although V141M requires the presence of KCNE1 to confer its deleterious effect on channel closing, S140G does not. This is thought to reflect the different physical distances between the two KCNQ1 residues and KCNE1.63 The functional consequences of the AF mutations resemble the regulatory response of the IKs channel to SNS stimulation. Both require the co-assembly of KCNQ1 and KCNE1, and both result in a beat-dependent increase in outward current. Is it possible that adrenergic regulation of the IKs channel may play a role in AF? This was illustrated in transgenic mice expressing IKs channels.64 Compared with wild type mice, which do not express functional IKs channels, these transgenic mice showed increased susceptibility to atrial arrhythmia upon β-AR stimulation. Computational simulation demonstrated that the stimulated IKs channels accumulate significantly at a fast rate as a result of slow deactivation and fast activation caused by phosphorylation, suggesting a large role of this slow channel in APD shortening and arrhythmia susceptibility in the face of β-adrenergic stimulation.64 These findings raise the possibility that drugs that target IKs channel regulation may be used to treat AF.
Using Stem Cells as a New Tool to Study Role of Iks Channels in Diseases
Until recently, little was known regarding the biophysical properties of native IKs channels in isolated human cardiomyocytes, as recordings of IKs in human cardiac cells had proved very difficult to obtain.65–75 As a result, most of our current knowledge of the cardiac IKs channel complex and its pathologic role had been obtained from studies using heterologous expression systems,76,77 genetically altered mice,18 and rabbits.78 Over the past few years, a new cellular model has emerged with the development of cardiac myocytes derived from human embryonic stem cells (hESC-CMs)79 and human induced pluripotent stem cells (hiPSC-CMs).80,81 The biophysical properties of IKs channels in these cells were found to be remarkably similar to those recorded in myocytes isolated from adult human heart but significantly different from KCNQ1/KCNE1 channels in established heterologous systems, including mammalian cell lines18,61,82,83: (1) Endogenous IKs currents are small in human cardiac myocytes74,84 but are much larger in heterologous systems as the result of overexpression of recombinant channels; and (2) the midpoint of activation of IKs channels in human cardiac myocytes is significantly less depolarized than in heterologous systems.73,84 Quantitative polymerase chain reaction (qPCR) measurements showed that KCNE1 is the major KCNE isoform expressed in hESC-CMs. Functional experiments suggest that KCNE1 is expressed at moderate levels in these cells such that IKs channels have variable α- and β-subunit stoichiometry that may be modulated further with changes in KCNE1 expression that occur in the developing heart or with disease.84 hiPSC-CMs are a potential source of functional human cardiac tissue that can be used as a model system; however, they also offer the possibility of investigating the mechanistic basis of heritable cardiac rhythm disorders—channelopathies—in genetic backgrounds specific for individual patients. Recently, hiPSC-CMs derived from LQT-1 patients were characterized and showed a phenotype consistent with the clinical manifestations.85
References
1. Wit, AL, Hoffman, BF, Rosen, MR. Electrophysiology and pharmacology of cardiac arrhythmias. IX. Cardiac electrophysiologic effects of beta adrenergic receptro stimulation and blockade. Part C. Am Heart J. 1975; 90:795–803.
2. Marx, SO, Reiken, S, Hisamatsu, Y, et al. PKA phosphorylation dissociates FKBP12. 6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000; 101:365–376.
3. Sanguinetti, MC, Curran, ME, Zou, A, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996; 384:80–83.
4. Barhanin, J, Lesage, F, Guillemare, E, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996; 384:78–80.
5. Kass, RS, Wiegers, SE. The ionic basis of concentration-related effects of noradrenaline on the action potential of calf cardiac purkinje fibres. J Physiol. 1982; 322:541–558.
6. Keating, MT, Sanguinetti, MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001; 104:569–580.
7. Splawski, I, Shen, J, Timothy, KW, et al. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000; 102:1178–1185.
8. Schwartz, PJ, Priori, SG, Spazzolini, C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001; 103:89–95.
9. Carnegie, GK, Means, CK, Scott, JD. A-kinase anchoring proteins: From protein complexes to physiology and disease. IUBMB Life. 2009; 61:394–406.
10. Beene, DL, Scott, JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007; 19:192–198.
11. McConnachie, G, Langeberg, LK, Scott, JD. AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med. 2006; 12:317–323.
12. Smith, FD, Langeberg, LK, Scott, JD. The where’s and when’s of kinase anchoring. Trends Biochem Sci. 2006; 31:316–323.
13. Dodge-Kafka, KL, Langeberg, L, Scott, JD. Compartmentation of cyclic nucleotide signaling in the heart: The role of A-kinase anchoring proteins. Circ Res. 2006; 98:993–1001.
14. Wong, W, Scott, JD. AKAP signalling complexes: Focal points in space and time. Nat Rev Mol Cell Biol. 2004; 5:959–970.
15. Michel, JJ, Scott, JD. AKAP mediated signal transduction. Annu Rev Pharmacol Toxicol. 2002; 42:235–257.
16. Colledge, M, Scott, JD. AKAPs: From structure to function. Trends Cell Biol. 1999; 9:216–221.
17. Gray, PC, Scott, JD, Catterall, WA. Regulation of ion channels by cAMP-dependent protein kinase and A-kinase anchoring proteins. Curr Opin Neurobiol. 1998; 8:330–334.
18. Marx, SO, Kurokawa, J, Reiken, S, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002; 295:496–499.
19. Potet, F, Scott, JD, Mohammad-Panah, R, et al. AKAP proteins anchor cAMP-dependent protein kinase to KvLQT1/IsK channel complex. Am J Physiol Heart Circ Physiol. 2001; 280:H2038–H2045.
20. Terrenoire, C, Houslay, MD, Baillie, GS, et al. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem. 2009; 284:9140–9146.
21. Dessauer, CW. Adenylyl cyclase—A-kinase anchoring protein complexes: The next dimension in cAMP signaling. Mol Pharmacol. 2009; 76:935–941.
22. Piggott, LA, Bauman, AL, Scott, JD, et al. The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci U S A. 2008; 105:13835–13840.
23. Li, Y, Chen, L, Kass, RS, Dessauer, CW. The A-kinase anchoring protein Yotiao facilitates complex formation between type 9 adenylyl cyclase and the IKs potassium channel in heart. J Biol Chem. 2012; 287:29815–29824.
24. Kass, RS, Kurokawa, J, Marx, SO, et al. Leucine/isoleucine zipper coordination of ion channel macromolecular signaling complexes in the heart: Roles in inherited arrhythmias. Trends Cardiovasc Med. 2003; 13:52–56.
25. Marx, SO, Reiken, S, Hisamatsu, Y, et al. Phosphorylation-dependent regulation of ryanodine receptors: A novel role for leucine/isoleucine zippers. J Cell Biol. 2001; 153:699–708.
26. Hulme, JT, Lin, TW, Westenbroek, RE, et al. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1. 2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003; 100:13093–13098.
27. Hulme, JT, Ahn, M, Hauschka, SD, et al. A novel leucine zipper targets AKAP15 and cyclic AMP-dependent protein kinase to the C terminus of the skeletal muscle Ca2+ channel and modulates its function. J Biol Chem. 2002; 277:4079–4087.
28. Landschulz, WH, Johnson, PF, McKnight, SL. The leucine zipper: A hypothetical structure common to a new class of DNA binding proteins. Science. 1988; 240:1759–1764.
29. Lupas, A. Coiled coils: New structures and new functions. Trends Biochem Sci. 1996; 21:375–382.
30. Walshaw, J, Shipway, JM, Woolfson, DN. Guidelines for the assembly of novel coiled-coil structures: Alpha-sheets and alpha-cylinders. Biochem Soc Symp. 2001; 111–123.
31. Harbury, PB, Zhang, T, Kim, PS, Alber, T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993; 262:1401–1407.
32. Simmerman, HK, Kobayashi, YM, Autry, JM, et al. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J Biol Chem. 1996; 271:5941–5946.
33. Surks, HK, Mochizuki, N, Kasai, Y, et al. Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science. 1999; 286:1583–1587.
34. Chen, L, Kass, RS. A-kinase anchoring protein 9 and IKs channel regulation. J Cardiovasc Pharmacol. 58, 2011. [459–413].
35. Terrenoire, C, Clancy, CE, Cormier, JW, et al. Autonomic control of cardiac action potentials: Role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005; 96:e25–e34.
36. Chen, L, Kurokawa, J, Kass, RS. Phosphorylation of the A-kinase-anchoring protein Yotiao contributes to protein kinase A regulation of a heart potassium channel. J Biol Chem. 2005; 280:31347–31352.
37. Westphal, RS, Tavalin, SJ, Lin, JW, et al. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999; 285:93–96.
38. Feliciello, A, Cardone, L, Garbi, C, et al. Yotiao protein, a ligand for the NMDA receptor, binds and targets cAMP-dependent protein kinase II(1). FEBS Lett. 1999; 464:174–178.
39. Bauman, AL, Soughayer, J, Nguyen, BT, et al. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006; 23:925–931.
40. Kapiloff, MS, Piggott, LA, Sadana, R, et al. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem. 2009; 284:23540–23546.
41. Efendiev, R, Samelson, BK, Nguyen, BT, et al. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem. 2010; 285:14450–14458.
42. Mongillo, M, McSorley, T, Evellin, S, et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004; 95:67–75.
43. Rochais, F, Abi-Gerges, A, Horner, K, et al. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006; 98:1081–1088.
44. Houslay, MD, Baillie, GS, Maurice, DH. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res. 2007; 100:950–966.
45. Stefan, E, Wiesner, B, Baillie, GS, et al. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J Am Soc Nephrol. 2007; 18:199–212.
46. Willoughby, D, Wong, W, Schaack, J, et al. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J. 2006; 25:2051–2061.
47. McCahill, A, McSorley, T, Huston, E, et al. In resting COS1 cells a dominant negative approach shows that specific, anchored PDE4 cAMP phosphodiesterase isoforms gate the activation, by basal cyclic AMP production, of AKAP-tethered protein kinase A type II located in the centrosomal region. Cell Signal. 2005; 17:1158–1173.
48. Tasken, KA, Collas, P, Kemmner, WA, et al. Phosphodiesterase 4D and protein kinase A type II constitute a signaling unit in the centrosomal area. J Biol Chem. 2001; 276:21999–22002.
49. Walsh, KB, Kass, RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988; 242:67–69.
50. January, CT, Moscucci, A. Cellular mechanisms of early afterdepolarizations. Ann N Y Acad Sci. 1992; 644:23–32.
51. January, CT, Riddle, JM. Early afterdepolarizations: Mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989; 64:977–990.
52. Piippo, K, Swan, H, Pasternack, M, et al. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J Am Coll Cardiol. 2001; 37:562–568.
53. Paavonen, KJ, Swan, H, Piippo, K, et al. Response of the QT interval to mental and physical stress in types LQT1 and LQT2 of the long QT syndrome. Heart. 2001; 86:39–44.
54. Kurokawa, J, Chen, L, Kass, RS. Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc Natl Acad Sci U S A. 2003; 100:2122–2127.
55. Chen, L, Marquardt, ML, Tester, DJ, et al. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007; 104:20990–20995.
56. Bellocq, C, van Ginneken, AC, Bezzina, CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004; 109:2394–2397.
57. Chen, YH, Xu, SJ, Bendahhou, S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003; 299:251–254.
58. Hong, K, Piper, DR, Diaz-Valdecantos, A, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005; 68:433–440.
59. Lundby, A, Ravn, LS, Svendsen, JH, et al. KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart Rhythm. 2007; 4:1532–1541.
60. Das, S, Makino, S, Melman, YF, et al. Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart Rhythm. 2009; 6:1146–1153.
61. Chung, DY, Chan, PJ, Bankston, JR, et al. Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc Natl Acad Sci U S A. 2009; 106:743–748.
62. Restier, L, Cheng, L, Sanguinetti, MC. Mechanisms by which atrial fibrillation-associated mutations in the S1 domain of KCNQ1 slow deactivation of IKs channels. J Physiol. 2008; 586:4179–4191.
63. Chan, PJ, Osteen, JD, Xiong, D, et al. Characterization of KCNQ1 atrial fibrillation mutations reveals distinct dependence on KCNE1. J Gen Physiol. 2012; 139:135–144.
64. Sampson, KJ, Terrenoire, C, Cervantes, DO, et al. Adrenergic regulation of a key cardiac potassium channel can contribute to atrial fibrillation: Evidence from an I Ks transgenic mouse. J Physiol. 2008; 586:627–637.
65. Beuckelmann, DJ, Nabauer, M, Erdmann, E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993; 73:379–385.
66. Varro, A, Nanasi, PP, Lathrop, DA. Potassium currents in isolated human atrial and ventricular cardiocytes. Acta Physiol Scand. 1993; 149:133–142.
67. Konarzewska, H, Peeters, GA, Sanguinetti, MC. Repolarizing K+ currents in nonfailing human hearts: Similarities between right septal subendocardial and left subepicardial ventricular myocytes. Circulation. 1995; 92:1179–1187.
68. Veldkamp, MW, van Ginneken, AC, Opthof, T, et al. Delayed rectifier channels in human ventricular myocytes. Circulation. 1995; 92:3497–3504.
69. Amos, GJ, Wettwer, E, Metzger, F, et al. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol. 1996; 491(Pt 1):31–50.
70. Iost, N, Virag, L, Opincariu, M, et al. Delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc Res. 1998; 40:508–515.
71. Bosch, RF, Gaspo, R, Busch, AE, et al. Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc Res. 1998; 38:441–450.
72. Li, GR, Yang, B, Feng, J, et al. Transmembrane ICa contributes to rate-dependent changes of action potentials in human ventricular myocytes. Am J Physiol. 1999; 276:H98–H106.
73. Li, GR, Feng, J, Yue, L, et al. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res. 1996; 78:689–696.
74. Jost, N, Virag, L, Bitay, M, et al. Restricting excessive cardiac action potential and QT prolongation: A vital role for IKs in human ventricular muscle. Circulation. 2005; 112:1392–1399.
75. Virag, L, Iost, N, Opincariu, M, et al. The slow component of the delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc Res. 2001; 49:790–797.
76. Charpentier, F, Merot, J, Loussouarn, G, et al. Delayed rectifier K(+) currents and cardiac repolarization. J Mol Cell Cardiol. 2010; 48:37–44.
77. Lu, JT, Kass, RS. Recent progress in congenital long QT syndrome. Curr Opin Cardiol. 2010; 25:216–221.
78. Brunner, M, Peng, X, Liu, GX, et al. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest. 2008; 118:2246–2259.
79. Vidarsson, H, Hyllner, J, Sartipy, P. Differentiation of human embryonic stem cells to cardiomyocytes for in vitro and in vivo applications. Stem Cell Rev. 2010; 6:108–120.
80. Zhang, J, Wilson, GF, Soerens, AG, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009; 104:e30–e41.
81. Yoshida, Y, Yamanaka, S. Recent stem cell advances: Induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation. 2010; 122:80–87.
82. Imredy, JP, Penniman, JR, Dech, SJ, et al. Modeling of the adrenergic response of the human IKs current (hKCNQ1/hKCNE1) stably expressed in HEK-293 cells. Am J Physiol Heart Circ Physiol. 2008; 295:H1867–H1881.
83. Wang, W, Xia, J, Kass, RS. MinK-KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J Biol Chem. 1998; 273:34069–34074.
84. Wang, K, Terrenoire, C, Sampson, KJ, et al. Biophysical properties of slow potassium channels in human embryonic stem cell derived cardiomyocytes implicate subunit stoichiometry. J Physiol. 2011; 589:6093–6104.
85. Moretti, A, Bellin, M, Welling, A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010; 363:1397–1409.