[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 20 Jaundice
BILIRUBIN METABOLISM AND MEASUREMENT
METABOLISM
Bilirubin, a hydrophobic and potentially toxic compound, is a tetrapyrrole that is an end product of heme degradation. Bilirubin metabolism has been reviewed in depth elsewhere1,2 and is summarized briefly in Figure 20-1. Each day, a healthy adult produces approximately 4 mg/kg of bilirubin (i.e., almost 0.5 mmol in a 70-kg person). Most bilirubin (70% to 80%) is derived from degradation of hemoglobin from senescent erythrocytes, and a minor component arises from premature destruction of newly formed erythrocytes in the bone marrow or circulation (i.e., ineffective erythropoiesis). Most of the remaining 20% to 30% is formed from breakdown of hemoproteins, such as catalase and cytochrome (CYP family) oxidases, in hepatocytes. Although nonhemoglobin heme-containing proteins are also present in extrahepatic tissues, their mass is so small or their turnover rate so slow (as for myoglobin) that their overall contribution to bilirubin production is minimal.
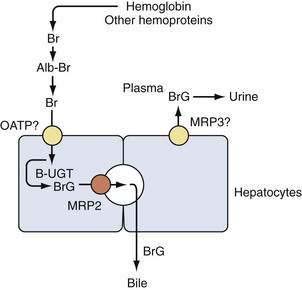
Bilirubin circulates in plasma tightly, but noncovalently, bound to albumin. Excretion of bilirubin requires conversion to water-soluble conjugates by hepatocytes and subsequent secretion into bile. Bilirubin metabolism and elimination is a multistep process for which several inherited disorders have been identified (see later). Bilirubin is taken up across the sinusoidal (basolateral) membrane of hepatocytes by a carrier-mediated mechanism. The uptake of bilirubin is inhibited competitively by certain organic anions such as sulfobromophthalein (BSP) and indocyanine. Bilirubin uptake has been suggested to be mediated by a liver-specific sinusoidal organic anion transport protein, (OATP1B1, SLC21A6), but this is not entirely certain.3,4 After uptake, bilirubin is directed by cytosolic binding proteins (e.g., glutathione S-transferase B, fatty acid binding protein) to the endoplasmic reticulum, where it is conjugated with uridine diphosphate (UDP)–glucuronic acid by the enzyme bilirubin UDP–glucuronyl transferase (B-UGT). Conjugation converts hydrophobic bilirubin into a water-soluble form suitable for excretion. Conjugated bilirubin is then directed primarily toward the canalicular (apical) membrane, where it is transported into the bile canaliculus by an adenosine triphosphate (ATP)-dependent export pump. The responsible protein, multidrug resistance–associated protein-2 (MRP2, ABCC2), appears to function as a multispecific transporter of various organic anions (including BSP, glutathione, and conjugated bile salts).5 Small amounts of bilirubin glucuronides are secreted across the sinusoidal membrane via a pathway postulated to be mediated by a distinct multispecific organic ion export pump, MRP3 (ABCC3)6; conjugated bilirubin in plasma undergoes renal elimination (see Fig. 20-1). This pathway may be up-regulated in disorders characterized by cholestasis (impaired bile flow). With prolonged cholestasis (or a metabolic disorder of conjugated hyperbilirubinemia; see later), an increasing proportion of conjugated bilirubin in plasma becomes covalently bound to albumin, and this covalently bound bilirubin cannot be excreted into urine.
MEASUREMENT
The normal bilirubin concentration in the serum of adults is lower than 1 to 1.5 mg/dL. In general, jaundice is not evident until the serum bilirubin concentration exceeds 3 mg/dL. In healthy persons, most bilirubin circulates in its unconjugated form; less than 5% of circulating bilirubin is present in conjugated form. In cholestatic conditions, the proportion of unconjugated bilirubin may increase as a consequence of upregulated MRP3 expression. The importance of accurate measurement of bilirubin is underscored by its incorporation as a critical variable in scoring systems such as the Model for End-stage Liver Disease (MELD), which provide estimates of survival in various acute and chronic liver disorders.7
Although the direct bilirubin concentration is influenced by changes in conjugated bilirubin levels, the two are not equivalent. Similarly, indirect bilirubin is not equivalent to unconjugated bilirubin. In particular, reliance on direct and indirect bilirubin measurements can lead to errors in the diagnosis of isolated disorders of bilirubin metabolism (e.g., suspected Gilbert’s syndrome; see later). Many clinical laboratories have abandoned measurements of direct and indirect bilirubin and instead use automated reflectance spectroscopic assays that provide more accurate estimates of conjugated and unconjugated bilirubin. These assays are useful clinically in the management of physiologic jaundice of the newborn (see later), in which neurotoxicity may result from the passage of unconjugated bilirubin across the blood-brain barrier (kernicterus). In disorders characterized by prolonged cholestasis, however, such assays may underestimate the conjugated bilirubin concentration, because they do not accurately detect albumin-bound conjugated bilirubin (so-called delta bilirubin). Indeed, if an isolated disorder of bilirubin metabolism is suspected, the diagnosis may require more sophisticated chromatographic techniques that precisely measure the concentrations of unconjugated, monoglucuronidated, and diglucuronidated bilirubin, as well as conjugated bilirubin-albumin complexes.2 In practice, these techniques are not widely used. Even with such accurate methods, measurements of conjugated and unconjugated bilirubin will not distinguish hepatic disorders from biliary obstruction. Therefore, in most cases, these tests are of limited use.
DIFFERENTIAL DIAGNOSIS
Jaundice can result from an increase in the formation of bilirubin or a decrease in the hepatobiliary clearance of bilirubin. From a practical standpoint, conditions that produce jaundice can be classified under the broad categories of isolated disorders of bilirubin metabolism, liver disease, and obstruction of the bile ducts (Table 20-1).
Table 20-1 Differential Diagnosis of Jaundice and Hyperbilirubinemia
| Isolated Disorders of Bilirubin Metabolism |
| Unconjugated Hyperbilirubinemia |
|
Increased bilirubin production (e.g., hemolysis, ineffective erythropoiesis, blood transfusion, resorption of hematomas)
|
AIDS, acquired immunodeficiency syndrome.
ISOLATED DISORDERS OF BILIRUBIN METABOLISM
Unconjugated Hyperbilirubinemia
Increased Bilirubin Production
Processes that can generate excessive bilirubin production include hemolysis, ineffective erythropoiesis, and resorption of a hematoma.2 Jaundice may thus complicate the clinical course of patients with hemolytic anemias, megaloblastic anemia from folate or vitamin B12 deficiency, iron deficiency anemia, sideroblastic anemia, and polycythemia vera. With these disorders, bilirubin concentration does not generally exceed 4 to 5 mg/dL. Jaundice can follow massive blood transfusions, because the foreshortened lifespan of transfused erythrocytes leads to excessive hemoglobin release. Hyperbilirubinemia resulting from resorption of hematomas and blood transfusions also may develop in patients who have experienced major trauma.8
Decreased Bilirubin Uptake
A decrease in hepatocellular uptake of bilirubin can be seen with certain drugs. For example, the antituberculosis agent rifampin has been shown to inhibit bilirubin uptake by hepatocytes competitively and may produce jaundice by inhibiting the transport protein OATP1B1 (SLC21A6); similar effects may be produced by the immunosuppressive drug cyclosporine A.9,10 Decreased bilirubin uptake also may contribute to phenotypic expression of the hereditary disorder Gilbert’s syndrome, in which the predominant abnormality is impaired bilirubin conjugation resulting from reduced B-UGT activity.11
Decreased Bilirubin Conjugation
Three autosomally inherited disorders of unconjugated hyperbilirubinemia are attributable to impaired bilirubin conjugation (Table 20-2). The most common of these disorders is Gilbert’s syndrome, which has a prevalence of approximately 10% in white populations. The disorder is entirely benign and rarely produces clinical jaundice. Serum bilirubin levels may rise two- to threefold with fasting or dehydration but are generally below 4 mg/dL. Patients with Gilbert’s syndrome typically present during or after adolescence, when isolated hyperbilirubinemia is detected as an incidental finding on routine multiphasic biochemical screening. The molecular basis of Gilbert’s syndrome has been linked to a reduction in transcription of the B-UGT gene UGT1A1 as a result of mutations in the promoter region and, less commonly, in the coding region.1
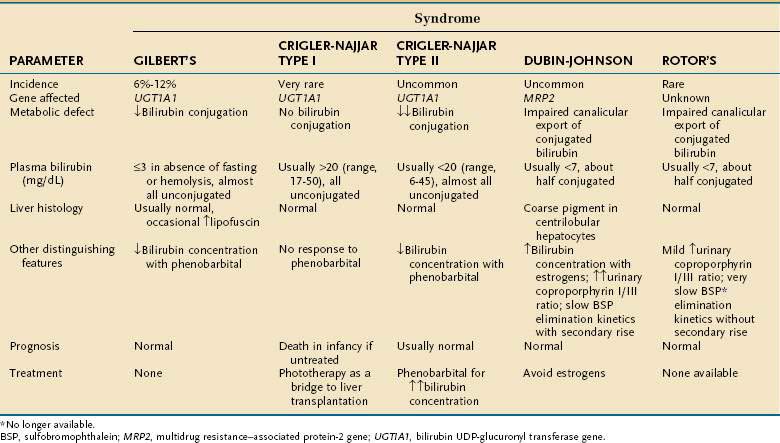
Mutations in the coding region of UGT1A1 appear to be responsible for Crigler-Najjar syndrome.12 In type I Crigler-Najjar syndrome, B-UGT activity is absent, and many patients die of kernicterus in the neonatal period (see Table 20-2). Phototherapy (see later) is required to prevent kernicterus, and liver transplantation can be lifesaving. Persons with type II Crigler-Najjar syndrome have markedly reduced B-UGT activity, with serum bilirubin levels between those of patients with Gilbert’s syndrome and those with type I Crigler-Najjar syndrome (see Table 20-2). In contrast to patients with type I Crigler-Najjar syndrome, those with type II Crigler-Najjar syndrome are not ill during the neonatal period and may not be diagnosed until early childhood. Although the degree of jaundice can wax and wane, most patients with type II Crigler-Najjar syndrome experience a fall in serum bilirubin levels to 2 to 5 mg/dL with phenobarbital, an agonist for the constitutive androstane receptor CAR, which increases expression of UGT1A1 and thus increases B-UGT activity.13 Such patients have normal life expectancies and do not manifest neurologic impairment.
A related disorder of bilirubin metabolism is physiologic jaundice of the newborn. This syndrome, which is believed to result from delayed developmental expression of B-UGT, is characterized by transient jaundice that generally resolves rapidly in the neonatal period. A brief course of phototherapy may be required to prevent kernicterus. B-UGT is inhibited competitively by the viral protease inhibitors indinavir and atazanavir, which produce hyperbilirubinemia in more than 25% of patients who receive these agents.14,15
Conjugated or Mixed Hyperbilirubinemia
A selective decrease in bilirubin secretion into the bile canaliculus may produce conjugated or mixed hyperbilirubinemia (i.e., an increase in conjugated and unconjugated bilirubin concentrations). Such a defect underlies two autosomally inherited disorders, Dubin-Johnson syndrome and Rotor’s syndrome. Each of these disorders is associated with a benign clinical course. In Dubin-Johnson syndrome, the molecular defect has been linked to an absence of expression of or impaired canalicular membrane targeting of the multispecific organic anion transporter MRP2.5 Interestingly, in Dubin-Johnson syndrome and in selected cholestatic disorders (e.g., primary biliary cirrhosis), compensatory up-regulation of the sinusoidal export protein MRP3 has been reported.16 Up-regulation of MRP3 may prevent hepatocellular overload by potentially toxic organic anions that are normally secreted by MRP2. The molecular basis of Rotor’s syndrome is unknown and does not appear to involve mutations in MRP2.17 In both Dubin-Johnson and Rotor’s syndromes, global hepatic function is preserved. Serum bilirubin levels are elevated, but serum levels of other commonly measured liver biochemical tests are normal.
Dubin-Johnson and Rotor’s syndromes can be distinguished biochemically and histologically (see Table 20-2). In Dubin-Johnson syndrome, hepatocytes contain a characteristic black pigment that is not seen in Rotor’s syndrome. This pigment is believed to result from lysosomal deposition of aromatic amino acid metabolites that are putative substrates for MRP2.5 Liver biopsy is generally unnecessary in the diagnostic evaluation of patients suspected to have Dubin-Johnson or Rotor’s syndrome, however, because neither disorder is associated with an adverse clinical outcome.
LIVER DISEASE
Acute Hepatocellular Dysfunction
Generalized impairment of hepatocellular function can result from acute or chronic liver injury. A clue to such disorders is the presence of elevated serum activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (see later and Chapter 73). Among conditions that produce acute or subacute hepatocellular injury are viral hepatitis, exposure to hepatotoxins, hepatic ischemia, and certain metabolic derangements. Acute viral hepatitis often is heralded by anorexia, malaise, myalgias, or discomfort in the epigastrium or right upper abdominal quadrant before jaundice develops (see Chapters 77 to 81). Five major hepatitis viruses have been isolated. Hepatitis A and E viruses are transmitted enterally. Each typically produces a self-limited illness that does not progress to chronic liver disease. By contrast, hepatitis B, C, and D viruses are transmitted parenterally, and illness produced by these agents can be prolonged and may lead to chronic disease. Major risk factors for hepatitis B, C, and D include injection drug use, exposure to blood products, and unprotected sexual exposures. The diagnosis of each of these disorders is aided by serologic testing (see later).
Many drugs and toxins produce hepatocellular injury (see Chapters 86 and 87). In particular, ingestion of acetaminophen (in large quantities) or of the mushroom Amanita phalloides may lead to hepatocellular necrosis and jaundice within several days after exposure. Toxic liver injury can have a fulminant course associated with a high mortality rate (see Chapter 93). In patients who survive, jaundice generally resolves and hepatic function recovers completely in those without preexisting liver disease. Certain drugs can produce idiosyncratic hepatocellular injury and jaundice, and these are discussed extensively elsewhere in this text (see Chapter 86). Alcoholic hepatitis should be a diagnostic consideration in the jaundiced patient with ethanol dependency, particularly when hepatomegaly and fever are present (see Chapter 84). Laboratory studies may help distinguish this entity from most other acute liver diseases (see later).
Jaundice related to hepatic ischemia may result from hypotension, hypoxia, hyperthermia, or hepatic venous outflow obstruction (see Chapter 83). Thrombosis of the hepatic vein (Budd-Chiari syndrome) or sinusoidal obstruction syndrome (hepatic veno-occlusive disease) should be suspected in a patient who presents with the rapid onset of ascites and hepatomegaly; the latter syndrome is more commonly associated with jaundice and is a complication of certain cytotoxic agents, particularly in the setting of hematopoietic cell transplantation (see also Chapter 34).
Wilson disease, an inherited disorder of hepatobiliary copper secretion, may manifest de novo with clinical features indistinguishable from those of acute viral hepatitis (see Chapter 75). The disease should be a diagnostic consideration in patients younger than 40 years, particularly when neurologic abnormalities are present or Kayser-Fleischer rings are seen on slit-lamp examination of the eye. Hemolytic anemia is a part of the spectrum of Wilson disease and contributes to the disproportionate hyperbilirubinemia often present in these patients. The diagnosis of Wilson disease is confirmed by biochemical testing and liver copper analysis (see later). Reye’s syndrome, a disorder of fatty infiltration of the liver associated with impaired mitochondrial metabolism of fatty acids, may produce jaundice as a manifestation of acute liver failure (see Chapter 86 and 93). It usually follows a viral illness in children, has been associated with the ingestion of aspirin, and is heralded by nausea and vomiting; its incidence has declined markedly as a result of public health campaigns advocating the avoidance of aspirin in children.
Chronic Hepatocellular Dysfunction
In contrast with acute liver disease, jaundice does not typically develop in chronic conditions associated with hepatocellular injury unless cirrhosis is present. A major cause of cirrhosis is chronic viral hepatitis, which should be a diagnostic consideration in patients with risk factors for parenteral exposure to causative agents. Diagnosis is aided by serologic testing (see later). Cirrhosis is part of the spectrum of nonalcoholic fatty liver disease, which is emerging as the most common cause of chronic hepatocellular injury in industrialized nations; major risk factors are obesity and diabetes mellitus (see Chapter 85). A similar histologic picture of steatohepatitis and sinusoidal fibrosis is found in the setting of alcoholic liver disease (see Chapter 84). Toxic injury by other compounds is less likely to produce cirrhosis, although cirrhosis has been described as a manifestation of industrial exposure to vinyl chloride and as a consequence of chronic ingestion of large quantities of vitamin A (see Chapter 87). Certain hereditary metabolic liver diseases may progress to cirrhosis. Hemochromatosis, a disorder characterized by excessive intestinal iron absorption with resulting hepatocellular iron accumulation and injury, is the most common of these (see Chapter 74). Although most affected persons are asymptomatic, the presence of diabetes mellitus, arthritis, or deep pigmentation in a jaundiced person should heighten suspicion for the disorder. The diagnosis is confirmed by detection of mutations in the HFE gene or by hepatic iron analysis. Hepatocellular copper overload and injury in Wilson disease also may progress to cirrhosis (see Chapter 75). As noted, the diagnosis should be suspected in younger persons, and the disease confirmed by biochemical testing and liver copper analysis. In a jaundiced patient with chronic obstructive pulmonary disease, α1-antitrypsin deficiency should be suspected (see Chapter 76). In this disorder, mutant α1-antitrypsin is misfolded and accumulates in the endoplasmic reticulum of hepatocytes, proteasomal degradation is impaired, and liver injury results. The diagnosis can be confirmed by laboratory testing and liver biopsy (see later). Autoimmune hepatitis, a disease that may be associated with systemic complaints such as malaise, fever, and arthralgias, is more common in women than in men (see Chapter 88). The diagnosis is aided by serologic testing and liver biopsy (see later). Celiac disease (see Chapter 104) may manifest as otherwise unexplained chronic liver disease—although rarely, if ever, with jaundice.
Hepatic Disorders with Prominent Cholestasis
Infiltrative Diseases
Infiltrative diseases of the liver disrupt the network of intrahepatic bile ductules and are often associated with striking cholestasis. Granulomatous diseases of the liver can be caused by the following: infections, such as tuberculosis, Mycobacterium avium complex infection (particularly in an immunocompromised host), leprosy, brucellosis, Q fever, syphilis, fungal diseases, parasitic diseases, and mononucleosis; toxins, such as beryllium, quinidine, allopurinol, and sulfonamides; and systemic disorders, including sarcoidosis, lymphoma (in particular, Hodgkin’s disease), and Wegener’s granulomatosis (see Chapters 35 and 82). The most common of these disorders that produce jaundice are tuberculosis and sarcoidosis.18 Granulomatous diseases should be suspected when jaundice accompanies fever of undetermined origin; other nonspecific symptoms include night sweats and weight loss. Physical examination usually reveals hepatosplenomegaly; right upper quadrant abdominal tenderness is uncommon. Lymphadenopathy often is seen in sarcoidosis and is also a clue to an infectious cause or lymphoma. The presence of erythema nodosum suggests mycobacterial disease, sarcoidosis, or syphilis. Elevations in the blood eosinophil count should heighten suspicion of sarcoidosis, parasitic disease, or drug toxicity. Radiographic chest abnormalities often provide a clue to the diagnosis of sarcoidosis or mycobacterial infection. Ultimately, diagnosis may require liver biopsy, if other tissue is not available. Jaundice is an unusual manifestation of amyloidosis but, when present, invariably is accompanied by marked hepatomegaly.19 The diagnosis of amyloidosis also may be suspected if clinical evidence of involvement of other organs is detected, such as macroglossia, malabsorption, congestive heart failure, or peripheral neuropathy. With renal involvement, proteinuria will be present. Otherwise, no specific biochemical clues may be found. Amyloidosis often can be detected on rectal valve or abdominal wall fat pad biopsy, but if the results are negative, liver biopsy specimens are diagnostic for hepatic amyloidosis. Jaundice resulting from extensive neoplastic replacement of hepatic parenchyma usually is heralded by anorexia and weight loss. Noninvasive imaging studies generally lead to the diagnosis (see later).
Disorders Involving Cholangiocyte Injury
A variety of disorders can lead to cholangiocyte damage and progressive loss of bile ductules. These encompass the differential diagnosis of vanishing bile duct syndrome, a term that has been employed to describe cholestatic conditions that are associated with a paucity of small intrahepatic bile ducts. In many of these disorders, the cholangiocyte is a target of an immune-mediated inflammatory response, as is characteristic of primary biliary cirrhosis (see Chapter 89) and is part of the spectrum of graft-versus-host disease encountered in organ transplant recipients (see Chapter 34). Primary biliary cirrhosis is a disease that occurs predominantly in women. In patients with jaundice, pruritus is also usually present and fatigue is common. The skin is often hyperpigmented in patients with advanced primary biliary cirrhosis, and detection of xanthelasma or xanthomata related to hypercholesterolemia is highly suggestive of the diagnosis. Serologic testing (antimitochondrial antibodies) is generally sufficient to establish a diagnosis of primary biliary cirrhosis, but liver biopsy may be necessary to confirm the diagnosis in selected cases (see later). Graft-versus-host disease is a complication of hematopoietic and solid organ transplantation; by contrast, hepatic involvement is rare after liver transplantation. Jaundice related to graft-versus-host disease develops in approximately 10% of hematopoietic cell transplant recipients.20 Certain drugs also can produce cholestasis with inflammation of the portal tracts (see Chapter 86). These include erythromycin (particularly the estolate salt), trimethoprim-sulfamethoxazole, amoxicillin–clavulanic acid, and terbinafine.21 Clinical features that may heighten suspicion of drug-induced cholestasis include arthralgias, rash, and peripheral eosinophilia; cholestasis generally resolves within several months following discontinuation of the causative drug. The most common inherited disorder of cholangiocyte injury is cystic fibrosis, a systemic disease that affects secretory epithelia and is linked to mutations in the CFTR gene, which encodes the cystic fibrosis transmembrane conductance regulator ion channel protein. Cholestatic hepatobiliary disease occurs in at least 30% of adults with this disorder (see Chapter 76).
Cholestasis with Minimal Histologic Abnormalities
Benign recurrent cholestasis is an autosomal recessive disorder associated with mutations in the genes that encode transport proteins involved in bile formation—the familial intrahepatic cholestasis 1 protein (FIC1, ATP8B1) and the bile salt export pump (BSEP, ABCB11; see Chapters 64 and 76).22,23 FIC1 (ATP8B1) is a P-type ATPase found in cholangiocytes as well as on the canalicular membrane of hepatocytes and is believed to function as a “flippase” for aminophospholipids such as phosphatidylserine. FIC1 dysfunction appears to alter the characteristic canalicular membrane lipid asymmetry and to increase cholesterol extraction into the canaliculus, thereby impairing BSEP activity.24 BSEP is an ATP-dependent bile salt export pump on the canalicular membrane of hepatocytes, and its activity provides a major driving force for bile formation.25 Factors that interfere with BSEP activity or expression lead to cholestasis.16 Mutations in FIC1 and BSEP also are responsible for progressive familial intrahepatic cholestasis types 1 and 2, two morbid pediatric cholestatic disorders (see Chapter 76).
Patients with benign recurrent cholestasis typically experience recurrent episodes of malaise and pruritus in association with jaundice; fever and abdominal pain are uncommon.26 The first episode of jaundice commonly occurs before the second decade of life. During periods of jaundice, laboratory abnormalities include elevations in serum alkaline phosphatase and aminotransferase levels but, as in other cholestatic disorders, the elevation in serum alkaline phosphatase characteristically predominates. When performed during an icteric episode, liver biopsy findings are generally confined to centrilobular cholestasis. Portal-based inflammatory cell infiltrates are uncommon (and, if present, are mild), and hepatocellular necrosis is not observed. Cholestatic episodes may last up to several months and are separated by periods of clinical remission. Although the patient’s quality of life may be affected adversely, the disease does not progress histologically, and liver failure does not occur.
A number of drugs produce histologically bland intrahepatic cholestasis (see Chapter 86). Estrogens reduce bile formation principally by inhibiting bile salt secretion.16 Estrogens down-regulate the sinusoidal bile salt uptake protein sodium taurocholate cotransporting peptide (NTCP, SLC10A1) and competitively inhibit BSEP. Other potential mechanisms of estrogen-induced cholestasis include inhibition of the hepatocellular plasma membrane sodium-potassium pump, an important modulator of solute transport from blood to bile, and impaired acidification of intracellular organelles, with disruption of the targeting of organic anion transporters to their proper membrane domains.27 Cholestasis related to the use of oral contraceptives usually develops within two months of the initiation of therapy. Jaundice is generally accompanied by pruritus, but fever, rash, and arthralgias are absent. Cholestasis resolves promptly with discontinuation of the drug. Anabolic steroids can produce a syndrome clinically indistinguishable from estrogen-induced cholestasis. The clinical features of cholestasis associated with total parenteral nutrition may also resemble those of estrogen and anabolic steroid-induced cholestasis, but progressive hepatic fibrosis has also been described.28 The syndrome is believed to be related, in part, to an alteration in the enterohepatic circulation and to diminished neuroendocrine stimulation of bile flow.
Cholestasis and jaundice also may develop during bacterial infections, likely because of the down-regulation of the transporters NTCP and MRP2 by tumor necrosis factor-α and interleukin (IL)-1β, as well as IL-1β–dependent down-regulation of BSEP.29 Sepsis-related cholestasis in the critically ill patient may be difficult to distinguish from obstruction of the bile ducts; abdominal pain and pruritus are generally absent in patients with sepsis-related cholestasis. However, depending on the severity of illness and response to antibiotic therapy, imaging studies may be required to exclude intrahepatic abscesses or biliary tract obstruction (see Chapter 82).
Jaundice resulting from intrahepatic cholestasis has been reported as a paraneoplastic phenomenon (i.e., in the absence of malignant infiltration of the liver) in patients with lymphoma and renal cell carcinoma. The latter, referred to as Stauffer’s syndrome, classically is associated with hepatosplenomegaly.30 Cholestasis and hepatosplenomegaly resolve after nephrectomy. The pathogenesis of this disorder is uncertain but may relate to tumor-derived secretion of cytokines such as IL-6,31 which down-regulates MRP2 and BSEP and reduces NTCP activity.29
Atypical Presentations of Cholestasis
Viral hepatitis rarely may cause profound cholestasis with marked pruritus.32 Unless the patient has risk factors for viral hepatitis, no features reliably distinguish this disorder from other cholestatic syndromes or biliary tract obstruction. A high level of suspicion and appropriate serologic tests will help establish the diagnosis. Alcoholic hepatitis manifesting as fever, jaundice, abdominal pain, and leukocytosis also may be difficult to distinguish from obstruction of the bile ducts. Occasionally, the increase in serum alkaline phosphatase levels is greater than the increase in aminotransferase levels.33 Liver biopsy may be required to confirm the diagnosis if there is a high clinical index of suspicion.
Jaundice in Pregnancy
Several cholestatic disorders are associated with pregnancy (see Chapter 38). Jaundice uncommonly may accompany hyperemesis gravidarum, a generally self-limited disorder of the first trimester, but liver failure is not a feature of this illness.34 Intrahepatic cholestasis of pregnancy typically occurs in the third trimester and manifests with pruritus; it occasionally is associated with jaundice. Cholestasis generally resolves within two weeks of delivery and tends to recur with subsequent pregnancies. Polymorphisms in the genes encoding the canalicular phospholipid transporters MDR3 (ABCB4), BSEP, FIC1, and MRP2s, and nuclear receptors that modulate their expression have been associated with this disorder,35–38 and these mutations may result in increased sensitivity to the inhibitory effects of estrogens on bile formation (see earlier). A far more serious syndrome is acute fatty liver of pregnancy, which typically occurs in the third trimester. The characteristic histologic features of microvesicular steatosis in hepatocytes resembles Reye’s syndrome. Jaundice, when present, usually is accompanied by nausea, abdominal pain, and encephalopathy. The disorder may be fatal unless obstetrical delivery is performed promptly. Preeclampsia, a microvascular disorder of the third trimester, is heralded by hypertension and proteinuria and affects the liver in approximately 10% of cases. A particularly severe form, the (HELLP) syndrome (hemolysis, elevated liver enzyme levels, and a low platelet count), is treated by prompt obstetric delivery.
Jaundice in the Critically Ill Patient
The diagnosis of jaundice in the critically ill patient often presents a major challenge to intensivists and their consultants. In this setting, concerned advocates for the patient, including family and friends, may regard jaundice as the cause rather than a manifestation of the underlying problems, and the persistence of jaundice can be a source of dismay and frustration. Indeed, under ideal conditions in which the patient recovers, resolution of jaundice may lag behind disease remission by days or weeks. Therefore, the management of such individuals requires a careful search for reversible causes of jaundice and a great deal of patience. Possible predisposing factors to jaundice in critically ill patients include hepatic ischemia, blood transfusions, massive trauma, hepatotoxic pharmacologic agents, parenteral nutrition, and occult sepsis.39 Moreover, jaundice can be exacerbated by renal insufficiency, which leads to decreased excretion of conjugated bilirubin into urine (see Fig. 20-1) (see Chapter 35).
OBSTRUCTION OF THE BILE DUCTS
Choledocholithiasis
The most common cause of biliary obstruction is choledocholithiasis. Cholesterol gallstones that obstruct the bile ducts typically originate in the gallbladder, migrate into the common bile duct, and occlude the ampulla of Vater or produce partial obstruction in a ball valve fashion (see Chapter 65). In patients with unconjugated hyperbilirubinemia, calcium bilirubinate stones, so-called black pigment gallstones, form in the gallbladder and also may form in situ at any level of the biliary tree. Brown pigment gallstones, a distinct type of bilirubinate stone, also form in situ within the biliary tree. Obstruction of the bile ducts by these stones leads to repeated bouts of cholangitis (recurrent pyogenic cholangitis) in patients from certain regions of Asia and in patients with prior biliary tract surgery (see Chapter 68).
Diseases of the Bile Ducts
Intrinsic narrowing of the bile ducts occurs in inflammatory, infectious, or neoplastic biliary disease. Congenital disorders of the bile ducts, including cysts and biliary atresia, are discussed in Chapter 62. Primary sclerosing cholangitis, an inflammatory disorder of the bile ducts, is characterized by focal and segmental biliary strictures and is discussed extensively in Chapter 68. Jaundice is an unusual complication of a similar disorder characterized by focal narrowing and localized obstruction of the bile ducts (AIDS [acquired immunodeficiency syndrome] cholangiopathy) in patients with AIDS (see Chapter 33). Biliary strictures also may follow hepatic arterial infusion of certain chemotherapeutic agents40 or result from surgical injury to the bile duct or hepatic artery. Neoplasms of the biliary tree, including cholangiocarcinoma, are discussed in detail in Chapter 69.
Extrinsic Compression
Extrinsic compression of the biliary tree may result from neoplastic involvement or inflammation of surrounding viscera. Rarely, marked enlargement of the surrounding vasculature (e.g., arterial aneurysms, cavernous transformation of the portal vein [portal cavernoma]) can compress the bile ducts (see Chapter 83).
Painless jaundice is a classic feature of carcinoma of the head of the pancreas (see Chapter 60). Occasionally, hepatocellular carcinoma or periportal lymph nodes enlarged by metastatic tumor or lymphoma obstructs the extrahepatic bile ducts. Pancreatitis may also produce extrinsic biliary compression as a result of edema, pseudocyst formation, or fibrosis (see Chapters 58 and 59). Rarely, gallstones in the cystic duct or infundibulum of the gallbladder compress the common hepatic duct (Mirizzi’s syndrome) and produce jaundice.41
DIAGNOSTIC APPROACH TO JAUNDICE
A general algorithm for evaluating the patient with jaundice is depicted in Figure 20-2. The sequential approach involves the following: (1) a carefully taken patient history, thorough physical examination, and screening laboratory studies; (2) formulation of a working differential diagnosis; (3) selection of specialized tests to narrow the diagnostic possibilities; and (4) development of a strategy for treatment or further testing if unexpected diagnostic possibilities arise.
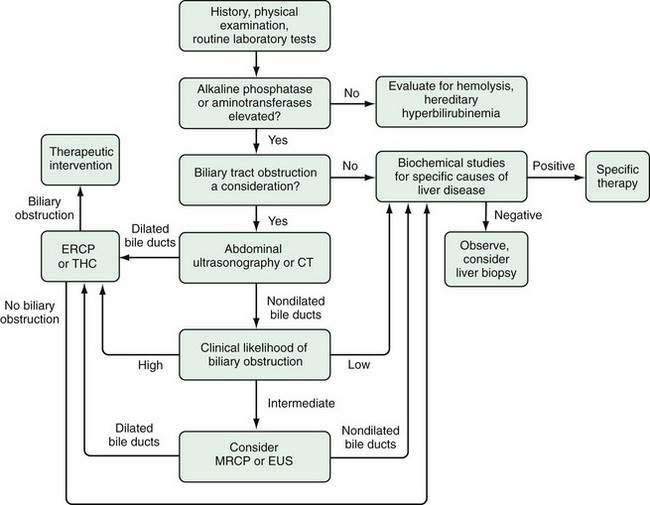
HISTORY AND PHYSICAL EXAMINATION
The patient’s history and physical examination provide important clues regarding the cause of jaundice (Table 20-3). A history of biliary surgery, fever, especially when accompanied by rigors, and abdominal pain, particularly in the right upper quadrant, is suggestive of biliary obstruction with cholangitis. Obstructive jaundice from gallstone disease or malignant neoplasms is more common in older adults than in younger persons. Symptoms compatible with a viral prodrome, such as anorexia, malaise, and myalgias, make viral hepatitis a strong diagnostic possibility, as does a history of a known infectious exposure, injection drug use, or prior transfusions of blood products. A carefully taken history may suggest that environmental hepatotoxins, ethanol, or medications underlie the patient’s cholestatic liver disease. Furthermore, a family history of jaundice or liver disease suggests the possibility of hereditary hyperbilirubinemia or genetic liver disease. All clues must be interpreted with caution, because fever and abdominal pain accompany diseases other than biliary obstruction, and viral hepatitis may occur coincidentally in patients with a history of prior biliary surgery. Conversely, anorexia and malaise are not specific for viral hepatitis, and gallstones frequently develop in patients with chronic liver disease. Nonetheless, when clues are evaluated in the context of the physical findings and routine laboratory tests, jaundice can be characterized correctly as obstructive or nonobstructive in at least 75% of cases.42
Table 20-3 Clues to the Differential Diagnosis of Jaundice: Biliary Obstruction versus Liver Disease
| PARAMETER | BILIARY OBSTRUCTION | LIVER DISEASE |
|---|---|---|
| History |
INR, international normalized ratio.
* Except early after acute obstruction when the opposite pattern may be seen transiently.
INITIAL LABORATORY STUDIES
Essential laboratory studies in the patient with jaundice include serum total bilirubin, alkaline phosphatase, ALT, and AST levels, a complete blood count, and the prothrombin time (see Chapter 73). Serum alkaline phosphatase activity reflects a number of related enzymes of overlapping substrate specificity. Alkaline phosphatase is associated predominantly with the apical domain of the plasma membrane of hepatocytes and cholangiocytes. Under physiologic conditions, this protein is cleaved enzymatically from a glycolipid anchor and released into bile, and small amounts are released from the sinusoidal (basolateral membrane) into plasma as well. Biliary obstruction and intrahepatic cholestasis increase the synthesis and basolateral release of alkaline phosphatase, and serum alkaline phosphatase activity increases. An increase in serum alkaline phosphatase activity, however, also may reflect release of alkaline phosphatase isoenzymes from extrahepatic tissues. Therefore, other more specific markers, such as the serum activities of the canalicular enzymes gamma glutamyl transpeptidase, or 5′-nucleotidase (or alternatively, alkaline phosphatase isoenzymes), are measured to confirm the hepatobiliary origin of an elevated serum alkaline phosphatase level when other liver biochemical test results (e.g., total bilirubin, ALT, AST) are normal. In a jaundiced patient, a predominant increase in (hepatic) alkaline phosphatase activity relative to levels of the serum aminotransferases suggests the possibility of biliary tract obstruction. Intrahepatic cholestatic disorders can produce an identical biochemical picture.
The aminotransferases—ALT, a cytosolic enzyme found predominantly in hepatocytes, and AST, isozymes of which are found in both the cytosol and mitochondria of parenchymal cells of liver and several other tissues—are ordinarily detected in serum in low concentrations. Hepatocellular injury caused by ischemia, toxins, or immune-mediated responses to foreign antigens such as viral proteins greatly increases serum aminotransferase activity. Predominant elevation of serum aminotransferase activity in comparison with alkaline phosphatase activity suggests that jaundice is the result of intrinsic hepatocellular disease. A serum activity of AST that is less than 10 times the upper limit of normal and that exceeds ALT activity by at least a factor of 2 is usually suggestive of alcoholic liver disease (see Chapter 84), but there are exceptions to these generalizations. For example, transient biliary obstruction from choledocholithiasis associated with cholangitis may cause a brief but dramatic elevation (exceeding 10 to 20 times normal) of serum aminotransferase activity.43
OVERALL APPROACH
Integration of the patient’s history, physical examination, and laboratory study results will provide an estimate of the likelihood that obstructive jaundice is present. For example, an asymptomatic patient with hyperbilirubinemia who has an unremarkable physical examination, normal serum alkaline phosphatase and aminotransferase levels, normal platelet count, and normal prothrombin time is unlikely to have liver disease or biliary obstruction. In this patient, further testing for specific disorders, such as isolated defects in bilirubin metabolism or hemolysis, is warranted (see Fig. 20-2). Alternatively, if the history, physical examination, and laboratory study results suggest the possibility of biliary obstruction, an imaging study of the biliary tree is appropriate. Selection of the appropriate imaging study depends on the likelihood of bile duct obstruction and the diagnostic accuracy, cost, complication rate, and availability of each test (see later), especially if therapeutic intervention at the time of the study is anticipated.
IMAGING STUDIES
Abdominal Ultrasonography
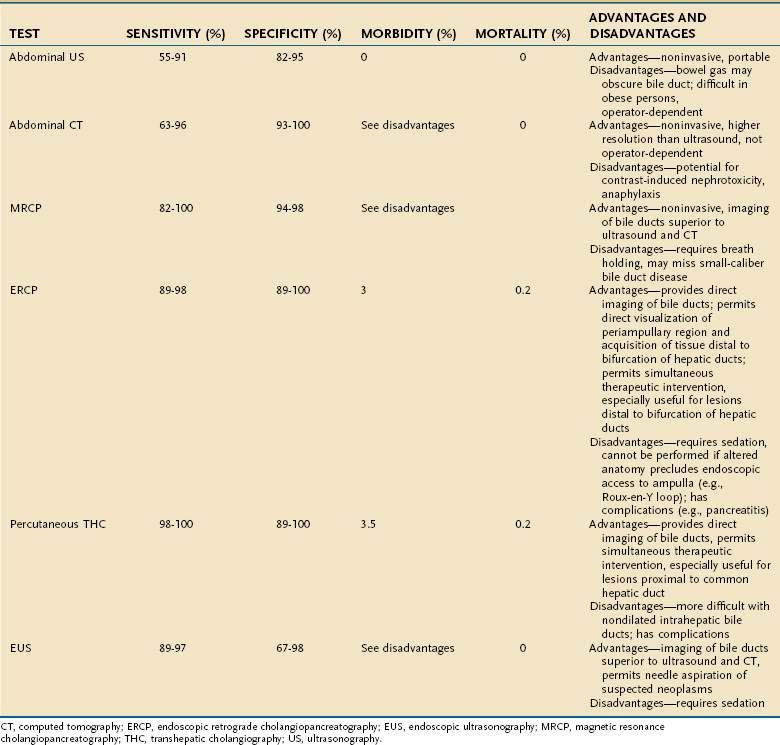
Abdominal ultrasonography is usually the initial imaging test in the evaluation of hepatobiliary disease because it determines the caliber of the extrahepatic biliary tree and reveals intra- or extrahepatic mass lesions. The sensitivity of abdominal ultrasonography for the detection of biliary obstruction in jaundiced patients ranges from 55% to 91%, and the specificity ranges from 82% to 95%.44–48 Ultrasonography also can demonstrate cholelithiasis, although bile duct stones may not be well seen, and intrahepatic space-occupying lesions more than 1 cm in diameter. Ultrasonography has the advantages of being noninvasive, portable (invaluable in the evaluation of the critically ill patient), and relatively inexpensive (Table 20-4). The major disadvantages are that the procedure is operator-dependent and interpretation may be difficult in obese patients or patients with overlying bowel gas. An additional caveat is that in patients with cirrhosis and other conditions associated with poorly compliant hepatic parenchyma, such as primary sclerosing cholangitis, intrahepatic ducts may not dilate with biliary obstruction.
Computed Tomography
Computed tomography (CT) of the abdomen with intravenous contrast is an alternative noninvasive means of evaluating the possibility of biliary tract obstruction. Abdominal CT permits accurate measurement of the caliber of the biliary tree, with sensitivity and specificity rates of 63% to 96% and 93% to 100%, respectively, for detecting biliary obstruction; these rates are comparable with those for ultrasonography.44–47 Abdominal CT detects intrahepatic space-occupying lesions as small as 5 mm, is not operator-dependent, and provides technically superior images in obese persons and in those in whom the biliary tree is obscured by bowel gas. The caveats that apply to the accuracy of ultrasonography for the diagnosis of biliary obstruction also apply to abdominal CT. Abdominal CT also lacks portability, it is more expensive than ultrasonography, and the requirement for the use of intravenous contrast is a potential contraindication in the setting of kidney failure (see Table 20-4).
Magnetic Resonance Cholangiopancreatography
Magnetic resonance cholangiopancreatography (MRCP) is a technical refinement of standard magnetic resonance imaging that permits rapid clear-cut delineation of the biliary tree without the need for intravenous contrast. MRCP appears to be superior to conventional ultrasound or CT for the detection of biliary tract obstruction and now plays a major role as a diagnostic test in this setting (see Table 20-4). Moreover, standard magnetic resonance imaging can be performed during the same examination if there is a question of a hepatobiliary or pancreatic mass or if a contrast allergy precludes CT. For detection of obstruction of the bile ducts, the sensitivity of MRCP is 82% to 100% and the specificity is 94% to 98%.49–52 Its expense is higher than that of ultrasound or CT and comparable with that of ERCP.53
Endoscopic Retrograde Cholangiopancreatography
Endoscopic retrograde cholangiopancreatography (ERCP) permits direct visualization of the biliary tree. ERCP is more invasive than ultrasonography and CT (see Table 20-4). The procedure involves passage of an endoscope into the duodenum, introduction of a catheter into the ampulla of Vater, and injection of contrast medium into the bile duct; sedation and analgesia are necessary. ERCP is highly accurate in the diagnosis of biliary obstruction, with sensitivities of 89% to 98% and specificities of 89% to 100%.47,54,55 In addition to providing radiographic images, ERCP permits biopsy and brushings for cytology of distal biliary and periampullary lesions. Moreover, if a focal cause of biliary obstruction is identified (e.g., choledocholithiasis, biliary stricture), maneuvers to relieve obstruction (e.g., sphincterotomy, stone extraction, stricture dilation, stent placement) can be performed during the same session (see Chapter 70). Acquisition of biopsy specimens and therapeutic interventions via ERCP are limited largely to lesions distal to the bifurcation of the right and left hepatic bile ducts. The technical success rate of ERCP is higher than 90%; the technique fails when the ampulla of Vater cannot be cannulated, as may be the case in patients with prior abdominal surgery and altered anatomy (e.g., gastric bypass, choledochojejunostomy). Rates of morbidity and mortality from untoward events, such as respiratory depression, aspiration, bleeding, perforation, cholangitis, and pancreatitis, are 3% and 0.2%, respectively, in patients undergoing ERCP.55 These rates are higher when interventional procedures are carried out.56
Percutaneous Transhepatic Cholangiography
Percutaneous transhepatic cholangiography (THC) is a procedure that complements ERCP. Percutaneous THC requires the passage of a needle through the skin and subcutaneous tissues into the hepatic parenchyma and advancement into a peripheral bile duct. When bile is aspirated, a catheter is introduced through the needle, and radiopaque contrast medium is injected. Sensitivity and specificity rates of percutaneous THC for the diagnosis of biliary tract obstruction are 98% to 100% and 89% to 100%, respectively, and are comparable with those for ERCP.57,58 Like ERCP, interventional procedures, such as balloon dilation and stent placement, can be performed at the time of percutaneous THC to relieve focal obstructions of the biliary tree (see Chapter 70). Percutaneous THC is potentially technically advantageous when the level of biliary obstruction is proximal to the common hepatic duct or altered anatomy precludes ERCP (see earlier). Percutaneous THC may be technically challenging in the absence of dilatation of the intrahepatic bile ducts; in this situation, multiple passes may be required, and visualization of the biliary tree may be unsuccessful in up to 10% of attempts.59 Rates of morbidity and mortality as a result of bleeding, perforation, and cholangitis are 3% and 0.2%, respectively, in patients undergoing percutaneous THC. Percutaneous THC is more expensive than abdominal ultrasonography and CT (see Table 20-4).
Endoscopic Ultrasonography
Endoscopic ultrasonography (EUS) also can detect obstruction of the bile duct and major intrahepatic bile ducts, with a sensitivity and specificity comparable with those of MRCP.49,60,61 EUS has the potential advantage of permitting biopsy of suspected malignant lesions, and under appropriate circumstances, the operator can proceed directly to ERCP for definitive biliary decompression (see Table 20-4). The risk of diagnostic EUS is comparable with that of diagnostic upper endoscopy; when needle biopsy is used, the mortality rate is approximately 0.1%.62 EUS may be most useful in circumstances in which the patient is thought to be at high risk for complications of ERCP or percutaneous THC.
Nuclear Imaging Studies
Nuclear scintigraphy of the biliary tree, although helpful in the diagnosis of cholecystitis, is not sufficiently sensitive to justify its routine use in the diagnostic evaluation of jaundice.45,46 Furthermore, hepatic uptake of radiolabeled derivatives of iminodiacetic acid (e.g., HIDA) is limited when the serum bilirubin level exceeds 7 to 10 mg/ dL.63 One exception to this generalization is in the evaluation of a potential bile leak, an uncommon cause of jaundice following biliary surgery, in which scintigraphy has an accuracy rate as high as 87%.64
Suggested Strategies for Imaging
The order of imaging studies depends largely on the clinical likelihood of obstructive jaundice (see Fig. 20-2). Several diagnostic strategies have been compared by clinical decision analysis.65 If the probability of biliary obstruction is approximately 20%, the positive and negative predictive values of a strategy that uses ultrasonography as the initial test is estimated to be 96% and 98%, respectively. This strategy compares favorably with one that uses ERCP as the initial test. Alternatively, if the probability of biliary obstruction is 60%, a strategy that uses ultrasonography as the first test would yield a positive predictive value of 99%, whereas the negative predictive value would fall to 89%. The implication is that if the level of suspicion for biliary tract obstruction is high and an ultrasound does not show dilated bile ducts, further studies to visualize the biliary tree should be pursued.
Therefore, in jaundiced patients in whom biliary obstruction is a possibility, abdominal ultrasonography (or CT) is an appropriate initial approach. If the bile ducts are dilated, the biliary tree should be imaged directly with ERCP (or percutaneous THC) and appropriate therapy undertaken if biliary obstruction is found. If the bile ducts are not dilated on abdominal ultrasonography (or CT), the next step depends on the clinical likelihood of biliary obstruction. If the likelihood of biliary obstruction is thought to be low, the patient should be evaluated for intrinsic liver disease (see later). If the likelihood of biliary obstruction is believed to be intermediate, MRCP or EUS is a reasonable option for imaging the biliary tree before an evaluation for a hepatic disorder is undertaken. Among patients in whom biliary obstruction is believed to be likely, ERCP (or percutaneous THC) should be considered as the next step. If ERCP or percutaneous THC does not show biliary obstruction, the patient should be evaluated for cholestatic liver disease. The decision to use ERCP versus percutaneous THC will be influenced by various factors (see Table 20-4), including the availability of each procedure at a particular facility, presence or absence of dilated bile ducts on initial imaging, and suspected level of biliary obstruction. Under most circumstances, ERCP should be the procedure of choice, because it is comparable with percutaneous THC in accuracy, technical success rate, and frequency of major complications; tends to be more widely available; and may offer better postprocedure tolerability (e.g., no need for an external biliary drainage tube).
OTHER STUDIES
Liver Biopsy
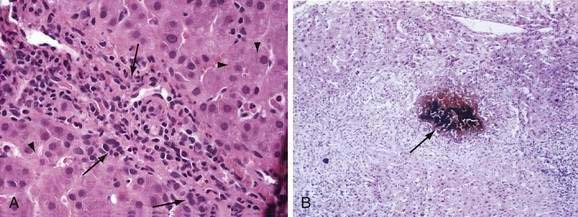
Liver biopsy provides precise information regarding hepatic lobular architecture and extent and pattern of fibrosis, and is most helpful for patients with persistent and undiagnosed jaundice. With special histologic stains and, if appropriate, quantification of copper or iron content, liver biopsy permits the diagnosis of viral hepatitis, fatty liver disease, hemochromatosis, Wilson disease, primary biliary cirrhosis, granulomatous hepatitis, and neoplasms. Occasionally, liver biopsy specimens provide clues to otherwise unsuspected biliary tract obstruction, the histologic features of which are shown in Figure 20-3; however, liver histology may be entirely normal in acute biliary obstruction. Liver biopsy is associated with a low but definite complication rate, predominantly from bleeding and perforation, and the need for hospitalization in 1% of cases; the mortality rate is approximately 0.01% (see Chapter 73).66
THERAPEUTIC APPROACHES
BILIARY OBSTRUCTION
In the patient with obstruction of the bile ducts, therapy is typically directed at relieving the obstruction. Interventional endoscopic or radiologic approaches include sphincterotomy, balloon dilation of focal strictures, and placement of drains or stents; the alternatives are surgical (see Chapter 70). The therapeutic strategy chosen will depend, in part, on the location and likely cause of the obstructing lesion. Focal intrahepatic strictures may be amenable to an interventional radiologic approach, whereas lesions distal to the bifurcation of the hepatic ducts may be more suitably managed endoscopically (e.g., sphincterotomy for choledocholithiasis); mass lesions may require surgery.
OTHER CONDITIONS
When jaundice is caused by liver disease, the optimal treatment is directed toward the underlying cause (e.g., cessation of ethanol, discontinuation of the offending drug, administration of antiviral agents, phlebotomy for hemochromatosis, immunosuppressive agents for autoimmune hepatitis). Therapy for hyperbilirubinemia per se is generally not necessary in adults, because the neurotoxicity of bilirubin is confined to disorders characterized by extreme elevations of unconjugated bilirubin in neonates and infants, such as physiologic jaundice of the newborn or type I Crigler-Najjar syndrome. In these special cases, the risk of neurotoxicity can be reduced with phototherapy, in which exposure to blue or green light produces photoisomerization of bilirubin to more water-soluble enantiomers that do not require conjugation for excretion in bile.67,68 Preliminary observations have suggested that orlistat, which increases intestinal fat excretion, may trap unconjugated bilirubin intraluminally and may augment phototherapy- or phenobarbital-induced reduction of unconjugated hyperbilirubinemia in children with type I or type II Crigler-Najjar syndrome, respectively.69
The choleretic bile acid ursodeoxycholic acid (ursodiol) has been studied as a treatment for several cholestatic disorders.70 Ursodeoxycholic acid improves biochemical indices and has been suggested to slow disease progression in primary biliary cirrhosis (see Chapter 89). Ursodeoxycholic acid has been shown to improve biochemical markers and clinical outcomes in patients with intrahepatic cholestasis of pregnancy,71,72 and pilot studies have suggested that it may be helpful in improving biochemical indices of cholestasis related to parenteral nutrition73 and cystic fibrosis74 and in preventing cholestasis following hematopoietic cell transplantation.75 By contrast, although initial pilot studies suggested that ursodeoxycholic acid reversed cholestasis in primary sclerosing cholangitis, a long-term benefit of this agent in this disorder has not been demonstrated to date in randomized controlled trials (see Chapter 68). In addition to the specific treatments outlined, cholestatic disorders may lead to impaired absorption of fat-soluble vitamins (A, D, E, and K), and supplementation is recommended. The management of pruritus caused by cholestasis is discussed in Chapter 89.
Borst P, de Wolf C, van de Wetering K. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 2007;453:661-73. (Ref 6)
Bosma PJ. Inherited disorders of bilirubin metabolism. J Hepatol. 2003;38:107-17. (Ref 1)
Brienza N, Dalfino L, Cinnella G, et al. Jaundice in critical illness: Promoting factors of a concealed reality. Intensive Care Med. 2006;32:267-74. (Ref 39)
Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. N Engl J Med. 2001;344:581-90. (Ref 67)
Fevery J. Bilirubin in clinical practice: A review. Liver Int. 2008;28:592-605. (Ref 2)
Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta. 2007;1773:283-308. (Ref 29)
Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126:322-42. (Ref 16)
Labori KJ, Bjornbeth BA, Raeder MG. Aetiology and prognostic implication of severe jaundice in surgical trauma patients. Scand J Gastroenterol. 2003;38:102-8. (Ref 8)
Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. 2004;8:133-49. (Ref 26)
Malchow-Moller A, Gronvall S, Hilden J, et al. Ultrasound examination in jaundiced patients. Is computer-assisted preclassification helpful? J Hepatol. 1991;12:321-6. (Ref 42)
Nies AT, Keppler D. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. 2007;453:643-59. (Ref 5)
Paulusma CC, Groen A, Kunne C, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44:195-204. (Ref 24)
Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525-31. (Ref 70)
Richter JM, Silverstein MD, Schapiro R. Suspected obstructive jaundice: A decision analysis of diagnostic strategies. Ann Intern Med. 1983;99:46-51. (Ref 65)
Stieger B, Meier Y, Meier PJ. The bile salt export pump. Pflugers Arch. 2007;453:611-20. (Ref 25)
1. Bosma PJ. Inherited disorders of bilirubin metabolism. J Hepatol. 2003;38:107-17.
2. Fevery J. Bilirubin in clinical practice: A review. Liver Int. 2008;28:592-605.
3. Cui Y, Konig J, Leier I, et al. Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J Biol Chem. 2001;276:9626-30.
4. Wang P, Kim RB, Chowdhury JR, Wolkoff AW. The human organic anion transport protein SLC21A6 is not sufficient for bilirubin transport. J Biol Chem. 2003;278:20695-9.
5. Nies AT, Keppler D. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. 2007;453:643-59.
6. Borst P, de Wolf C, van de Wetering K. Multidrug resistance–associated proteins 3, 4, and 5. Pflugers Arch. 2007;453:661-73.
7. Kamath PS, Kim WR. The model for end-stage liver disease (MELD). Hepatology. 2007;45:797-805.
8. Labori KJ, Bjornbeth BA, Raeder MG. Aetiology and prognostic implication of severe jaundice in surgical trauma patients. Scand J Gastroenterol. 2003;38:102-8.
9. Vavricka SR, Van Montfoort J, Ha HR, et al. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36:164-72.
10. Campbell SD, de Morais SM, Xu JJ. Inhibition of human organic anion transporting polypeptide OATP 1B1 as a mechanism of drug-induced hyperbilirubinemia. Chem Biol Interact. 2004;150:179-87.
11. Persico M, Persico E, Bakker CT, et al. Hepatic uptake of organic anions affects the plasma bilirubin level in subjects with Gilbert’s syndrome mutations in UGT1A1. Hepatology. 2001;33:627-32.
12. Kadakol A, Ghosh SS, Sappal BS, et al. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: Correlation of genotype to phenotype. Hum Mutat. 2000;16:297-306.
13. Sugatani J, Kojima H, Ueda A, et al. The phenobarbital response enhancer module in the human bilirubin UDP-glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CAR. Hepatology. 2001;33:1232-38.
14. Zucker SD, Qin X, Rouster SD, et al. Mechanism of indinavir-induced hyperbilirubinemia. Proc Natl Acad Sci U S A. 2001;98:12671-6.
15. Lankisch TO, Moebius U, Wehmeier M, et al. Gilbert’s disease and atazanavir: From phenotype to UDP-glucuronosyltransferase haplotype. Hepatology. 2006;44:1324-32.
16. Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126:322-42.
17. Hrebicek M, Jirasek T, Hartmannova H, et al. Rotor-type hyperbilirubinaemia has no defect in the canalicular bilirubin export pump. Liver Int. 2007;27:485-91.
18. Sartin JS, Walker RC. Granulomatous hepatitis: A retrospective review of 88 cases at the Mayo Clinic. Mayo Clin Proc. 1991;66:914-18.
19. Peters RA, Koukoulis G, Gimson A, et al. Primary amyloidosis and severe intrahepatic cholestatic jaundice. Gut. 1994;35:1322-25.
20. Hogan WJ, Maris M, Storer B, et al. Hepatic injury after nonmyeloablative conditioning followed by allogeneic hematopoietic cell transplantation: A study of 193 patients. Blood. 2004;103:78-84.
21. Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349:474-85.
22. Van Mil SW, Van Der Woerd WL, Van Der Brugge G, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379-84.
23. Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27-38.
24. Paulusma CC, Groen A, Kunne C, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44:195-204.
25. Stieger B, Meier Y, Meier PJ. The bile salt export pump. Pflugers Arch. 2007;453:611-20.
26. Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. 2004;8:133-49.
27. Van Dyke RW, Root KV. Ethinyl estradiol decreases acidification of rat liver endocytic vesicles. Hepatology. 1993;18:604-13.
28. Guglielmi FW, Regano N, Mazzuoli S, et al. Cholestasis induced by total parenteral nutrition. Clin Liver Dis. 2008;12:97-110.
29. Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta. 2007;1773:283-308.
30. Dourakis SP, Sinani C, Deutsch M, et al. Cholestatic jaundice as a paraneoplastic manifestation of renal cell carcinoma. Eur J Gastroenterol Hepatol. 1997;9:311-14.
31. Walther MM, Johnson B, Culley D, et al. Serum interleukin-6 levels in metastatic renal cell carcinoma before treatment with interleukin-2 correlates with paraneoplastic syndromes but not patient survival. J Urol. 1998;159:718-22.
32. Gordon SC, Reddy KR, Schiff L, Schiff ER. Prolonged intrahepatic cholestasis secondary to acute hepatitis A. Ann Intern Med. 1984;101:635-7.
33. Perrillo RP, Griffin R, DeSchryver-Kecskemeti K, et al. Alcoholic liver disease presenting with marked elevation of serum alkaline phosphatase. A combined clinical and pathological study. Am J Dig Dis. 1978;23:1061-6.
34. Orazi G, Dufour PH, Puech F. Jaundice induced by hyperemesis gravidarum. Int J Gynaecol Obstet. 1998;61:181-3.
35. Pauli-Magnus C, Lang T, Meier Y, et al. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p-glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics. 2004;14:91-102.
36. Mullenbach R, Bennett A, Tetlow N, et al. ATP8B1 mutations in British cases with intrahepatic cholestasis of pregnancy. Gut. 2005;54:829-34.
37. Sookoian S, Castano G, Burgueno A, et al. Association of the multidrug-resistance-associated protein gene (ABCC2) variants with intrahepatic cholestasis of pregnancy. J Hepatol. 2008;48:125-32.
38. Van Mil SW, Milona A, Dixon PH, et al. Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology. 2007;133:507-16.
39. Brienza N, Dalfino L, Cinnella G, et al. Jaundice in critical illness: Promoting factors of a concealed reality. Intensive Care Med. 2006;32:267-74.
40. Brown KT, Kemeny N, Berger MF, et al. Obstructive jaundice in patients receiving hepatic artery infusional chemotherapy: Etiology, treatment implications, and complications after transhepatic biliary drainage. J Vasc Interv Radiol. 1997;8:229-34.
41. Freeman ME, Rose JL, Forsmark CE, Vauthey J. Mirizzi syndrome: A rare cause of obstructive jaundice. Dig Dis. 1999;17:44-8.
42. Malchow-Moller A, Gronvall S, Hilden J, et al. Ultrasound examination in jaundiced patients. Is computer-assisted preclassification helpful? J Hepatol. 1991;12:321-6.
43. Nathwani RA, Kumar SR, Reynolds TB, Kaplowitz N. Marked elevation in serum transaminases: An atypical presentation of choledocholithiasis. Am J Gastroenterol. 2005;100:295-8.
44. Baron RL, Stanley RJ, Lee JK, et al. A prospective comparison of the evaluation of biliary obstruction using computed tomography and ultrasonography. Radiology. 1982;145:91-8.
45. Matzen P, Malchow-Moller A, Brun B, et al. Ultrasonography, computed tomography, and cholescintigraphy in suspected obstructive jaundice—a prospective comparative study. Gastroenterology. 1983;84:1492-7.
46. O’Connor KW, Snodgrass PJ, Swonder JE, et al. A blinded prospective study comparing four current noninvasive approaches in the differential diagnosis of medical versus surgical jaundice. Gastroenterology. 1983;84:1498-504.
47. Pasanen PA, Partanen KP, Pikkarainen PH, et al. A comparison of ultrasound, computed tomography and endoscopic retrograde cholangiopancreatography in the differential diagnosis of benign and malignant jaundice and cholestasis. Eur J Surg. 1993;159:23-9.
48. Pedersen OM, Nordgard K, Kvinnsland S. Value of sonography in obstructive jaundice. Limitations of bile duct caliber as an index of obstruction. Scand J Gastroenterol. 1987;22:975-81.
49. Materne R, Van Beers BE, Gigot JF, et al. Extrahepatic biliary obstruction: Magnetic resonance imaging compared with endoscopic ultrasonography. Endoscopy. 2000;32:3-9.
50. Stiris MG, Tennoe B, Aadland E, Lunde OC. MR cholangiopancreaticography and endoscopic retrograde cholangiopancreaticography in patients with suspected common bile duct stones. Acta Radiol. 2000;41:269-72.
51. Griffin N, Wastle ML, Dunn WK, et al. Magnetic resonance cholangiopancreatography versus endoscopic retrograde cholangiopancreatography in the diagnosis of choledocholithiasis. Eur J Gastroenterol Hepatol. 2003;15:809-13.
52. Vaishali MD, Agarwal AK, Upadhyaya DN, et al. Magnetic resonance cholangiopancreatography in obstructive jaundice. J Clin Gastroenterol. 2004;38:887-90.
53. Talwalkar JA, Angulo P, Johnson CD, et al. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology. 2004;40:39-45.
54. Kumar M, Prashad R, Kumar A, et al. Relative merits of ultrasonography, computed tomography and cholangiography in patients of surgical obstructive jaundice. Hepatogastroenterology. 1998;45:2027-32.
55. Mallery JS, Baron TH, Dominitz JA, et al. Complications of ERCP. Gastrointest Endosc. 2003;57:633-8.
56. Freeman ML. Adverse outcomes of ERCP. Gastrointest Endosc. 2002;56:S273-S82.
57. Pereiras RJr, Chiprut RO, Greenwald RA, Schiff ER. Percutaneous transhepatic cholangiography with the “skinny” needle. A rapid, simple, and accurate method in the diagnosis of cholestasis. Ann Intern Med. 1977;86:562-8.
58. Gold RP, Casarella WJ, Stern G, Seaman WB. Transhepatic cholangiography: The radiological method of choice in suspected obstructive jaundice. Radiology. 1979;133:39-44.
59. Funaki B, Zaleski GX, Straus CA, et al. Percutaneous biliary drainage in patients with nondilated intrahepatic bile ducts. AJR Am J Roentgenol. 1999;173:1541-4.
60. Rosch T, Meining A, Fruhmorgen S, et al. A prospective comparison of the diagnostic accuracy of ERCP, MRCP, CT, and EUS in biliary strictures. Gastrointest Endosc. 2002;55:870-6.
61. Ainsworth AP, Rafaelsen SR, Wamberg PA, et al. Is there a difference in diagnostic accuracy and clinical impact between endoscopic ultrasonography and magnetic resonance cholangiopancreatography? Endoscopy. 2003;35:1029-32.
62. Shah JN, Muthusamy VR. Minimizing complications of endoscopic ultrasound and EUS-guided fine needle aspiration. Gastrointest Endosc Clin N Am. 2007;17:129-43.
63. Krishnamurthy S, Krishnamurthy GT. Technetium-99m-iminodiacetic acid organic anions: Review of biokinetics and clinical application in hepatology. Hepatology. 1989;9:139-53.
64. Brugge WR, Rosenberg DJ, Alavi A. Diagnosis of postoperative bile leaks. Am J Gastroenterol. 1994;89:2178-83.
65. Richter JM, Silverstein MD, Schapiro R. Suspected obstructive jaundice: A decision analysis of diagnostic strategies. Ann Intern Med. 1983;99:46-51.
66. Rockey DC, Caldwell SH, Goodman ZD, et al. Liver biopsy. Hepatology. 2009;49:1017-44.
67. Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. N Engl J Med. 2001;344:581-90.
68. Maisels MJ, McDonagh AF. Phototherapy for neonatal jaundice. N Engl J Med. 2008;358:920-8.
69. Hafkamp AM, Nelisse-Haak R, Sinaasappel M, et al. Orlistat treatment of unconjugated hyperbilirubinemia in Crigler-Najjar disease: A randomized controlled trial. Pediatr Res. 2007;62:725-30.
70. Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525-31.
71. Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129:894-901.
72. Glantz A, Marschall HU, Lammert F, Mattsson LA. Intrahepatic cholestasis of pregnancy: A randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42:1399-405.
73. San Luis VA, Btaiche IF. Ursodiol in patients with parenteral nutrition-associated cholestasis. Ann Pharmacother. 2007;41:1867-72.
74. Nousia-Arvanitakis S, Fotoulaki M, Economou H, et al. Long-term prospective study of the effect of ursodeoxycholic acid on cystic fibrosis-related liver disease. J Clin Gastroenterol. 2001;32:324-8.
75. Ruutu T, Eriksson B, Remes K, et al. Ursodeoxycholic acid for the prevention of hepatic complications in allogeneic stem cell transplantation. Blood. 2002;100:1977-83.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 20 Jaundice
BILIRUBIN METABOLISM AND MEASUREMENT
METABOLISM
Bilirubin, a hydrophobic and potentially toxic compound, is a tetrapyrrole that is an end product of heme degradation. Bilirubin metabolism has been reviewed in depth elsewhere1,2 and is summarized briefly in Figure 20-1. Each day, a healthy adult produces approximately 4 mg/kg of bilirubin (i.e., almost 0.5 mmol in a 70-kg person). Most bilirubin (70% to 80%) is derived from degradation of hemoglobin from senescent erythrocytes, and a minor component arises from premature destruction of newly formed erythrocytes in the bone marrow or circulation (i.e., ineffective erythropoiesis). Most of the remaining 20% to 30% is formed from breakdown of hemoproteins, such as catalase and cytochrome (CYP family) oxidases, in hepatocytes. Although nonhemoglobin heme-containing proteins are also present in extrahepatic tissues, their mass is so small or their turnover rate so slow (as for myoglobin) that their overall contribution to bilirubin production is minimal.
Bilirubin circulates in plasma tightly, but noncovalently, bound to albumin. Excretion of bilirubin requires conversion to water-soluble conjugates by hepatocytes and subsequent secretion into bile. Bilirubin metabolism and elimination is a multistep process for which several inherited disorders have been identified (see later). Bilirubin is taken up across the sinusoidal (basolateral) membrane of hepatocytes by a carrier-mediated mechanism. The uptake of bilirubin is inhibited competitively by certain organic anions such as sulfobromophthalein (BSP) and indocyanine. Bilirubin uptake has been suggested to be mediated by a liver-specific sinusoidal organic anion transport protein, (OATP1B1, SLC21A6), but this is not entirely certain.3,4 After uptake, bilirubin is directed by cytosolic binding proteins (e.g., glutathione S-transferase B, fatty acid binding protein) to the endoplasmic reticulum, where it is conjugated with uridine diphosphate (UDP)–glucuronic acid by the enzyme bilirubin UDP–glucuronyl transferase (B-UGT). Conjugation converts hydrophobic bilirubin into a water-soluble form suitable for excretion. Conjugated bilirubin is then directed primarily toward the canalicular (apical) membrane, where it is transported into the bile canaliculus by an adenosine triphosphate (ATP)-dependent export pump. The responsible protein, multidrug resistance–associated protein-2 (MRP2, ABCC2), appears to function as a multispecific transporter of various organic anions (including BSP, glutathione, and conjugated bile salts).5 Small amounts of bilirubin glucuronides are secreted across the sinusoidal membrane via a pathway postulated to be mediated by a distinct multispecific organic ion export pump, MRP3 (ABCC3)6; conjugated bilirubin in plasma undergoes renal elimination (see Fig. 20-1). This pathway may be up-regulated in disorders characterized by cholestasis (impaired bile flow). With prolonged cholestasis (or a metabolic disorder of conjugated hyperbilirubinemia; see later), an increasing proportion of conjugated bilirubin in plasma becomes covalently bound to albumin, and this covalently bound bilirubin cannot be excreted into urine.
MEASUREMENT
The normal bilirubin concentration in the serum of adults is lower than 1 to 1.5 mg/dL. In general, jaundice is not evident until the serum bilirubin concentration exceeds 3 mg/dL. In healthy persons, most bilirubin circulates in its unconjugated form; less than 5% of circulating bilirubin is present in conjugated form. In cholestatic conditions, the proportion of unconjugated bilirubin may increase as a consequence of upregulated MRP3 expression. The importance of accurate measurement of bilirubin is underscored by its incorporation as a critical variable in scoring systems such as the Model for End-stage Liver Disease (MELD), which provide estimates of survival in various acute and chronic liver disorders.7
Although the direct bilirubin concentration is influenced by changes in conjugated bilirubin levels, the two are not equivalent. Similarly, indirect bilirubin is not equivalent to unconjugated bilirubin. In particular, reliance on direct and indirect bilirubin measurements can lead to errors in the diagnosis of isolated disorders of bilirubin metabolism (e.g., suspected Gilbert’s syndrome; see later). Many clinical laboratories have abandoned measurements of direct and indirect bilirubin and instead use automated reflectance spectroscopic assays that provide more accurate estimates of conjugated and unconjugated bilirubin. These assays are useful clinically in the management of physiologic jaundice of the newborn (see later), in which neurotoxicity may result from the passage of unconjugated bilirubin across the blood-brain barrier (kernicterus). In disorders characterized by prolonged cholestasis, however, such assays may underestimate the conjugated bilirubin concentration, because they do not accurately detect albumin-bound conjugated bilirubin (so-called delta bilirubin). Indeed, if an isolated disorder of bilirubin metabolism is suspected, the diagnosis may require more sophisticated chromatographic techniques that precisely measure the concentrations of unconjugated, monoglucuronidated, and diglucuronidated bilirubin, as well as conjugated bilirubin-albumin complexes.2 In practice, these techniques are not widely used. Even with such accurate methods, measurements of conjugated and unconjugated bilirubin will not distinguish hepatic disorders from biliary obstruction. Therefore, in most cases, these tests are of limited use.
DIFFERENTIAL DIAGNOSIS
Jaundice can result from an increase in the formation of bilirubin or a decrease in the hepatobiliary clearance of bilirubin. From a practical standpoint, conditions that produce jaundice can be classified under the broad categories of isolated disorders of bilirubin metabolism, liver disease, and obstruction of the bile ducts (Table 20-1).
Table 20-1 Differential Diagnosis of Jaundice and Hyperbilirubinemia
| Isolated Disorders of Bilirubin Metabolism |
| Unconjugated Hyperbilirubinemia |
|
Increased bilirubin production (e.g., hemolysis, ineffective erythropoiesis, blood transfusion, resorption of hematomas)
|
AIDS, acquired immunodeficiency syndrome.
ISOLATED DISORDERS OF BILIRUBIN METABOLISM
Unconjugated Hyperbilirubinemia
Increased Bilirubin Production
Processes that can generate excessive bilirubin production include hemolysis, ineffective erythropoiesis, and resorption of a hematoma.2 Jaundice may thus complicate the clinical course of patients with hemolytic anemias, megaloblastic anemia from folate or vitamin B12 deficiency, iron deficiency anemia, sideroblastic anemia, and polycythemia vera. With these disorders, bilirubin concentration does not generally exceed 4 to 5 mg/dL. Jaundice can follow massive blood transfusions, because the foreshortened lifespan of transfused erythrocytes leads to excessive hemoglobin release. Hyperbilirubinemia resulting from resorption of hematomas and blood transfusions also may develop in patients who have experienced major trauma.8
Decreased Bilirubin Uptake
A decrease in hepatocellular uptake of bilirubin can be seen with certain drugs. For example, the antituberculosis agent rifampin has been shown to inhibit bilirubin uptake by hepatocytes competitively and may produce jaundice by inhibiting the transport protein OATP1B1 (SLC21A6); similar effects may be produced by the immunosuppressive drug cyclosporine A.9,10 Decreased bilirubin uptake also may contribute to phenotypic expression of the hereditary disorder Gilbert’s syndrome, in which the predominant abnormality is impaired bilirubin conjugation resulting from reduced B-UGT activity.11
Decreased Bilirubin Conjugation
Three autosomally inherited disorders of unconjugated hyperbilirubinemia are attributable to impaired bilirubin conjugation (Table 20-2). The most common of these disorders is Gilbert’s syndrome, which has a prevalence of approximately 10% in white populations. The disorder is entirely benign and rarely produces clinical jaundice. Serum bilirubin levels may rise two- to threefold with fasting or dehydration but are generally below 4 mg/dL. Patients with Gilbert’s syndrome typically present during or after adolescence, when isolated hyperbilirubinemia is detected as an incidental finding on routine multiphasic biochemical screening. The molecular basis of Gilbert’s syndrome has been linked to a reduction in transcription of the B-UGT gene UGT1A1 as a result of mutations in the promoter region and, less commonly, in the coding region.1
Mutations in the coding region of UGT1A1 appear to be responsible for Crigler-Najjar syndrome.12 In type I Crigler-Najjar syndrome, B-UGT activity is absent, and many patients die of kernicterus in the neonatal period (see Table 20-2). Phototherapy (see later) is required to prevent kernicterus, and liver transplantation can be lifesaving. Persons with type II Crigler-Najjar syndrome have markedly reduced B-UGT activity, with serum bilirubin levels between those of patients with Gilbert’s syndrome and those with type I Crigler-Najjar syndrome (see Table 20-2). In contrast to patients with type I Crigler-Najjar syndrome, those with type II Crigler-Najjar syndrome are not ill during the neonatal period and may not be diagnosed until early childhood. Although the degree of jaundice can wax and wane, most patients with type II Crigler-Najjar syndrome experience a fall in serum bilirubin levels to 2 to 5 mg/dL with phenobarbital, an agonist for the constitutive androstane receptor CAR, which increases expression of UGT1A1 and thus increases B-UGT activity.13 Such patients have normal life expectancies and do not manifest neurologic impairment.
A related disorder of bilirubin metabolism is physiologic jaundice of the newborn. This syndrome, which is believed to result from delayed developmental expression of B-UGT, is characterized by transient jaundice that generally resolves rapidly in the neonatal period. A brief course of phototherapy may be required to prevent kernicterus. B-UGT is inhibited competitively by the viral protease inhibitors indinavir and atazanavir, which produce hyperbilirubinemia in more than 25% of patients who receive these agents.14,15
Conjugated or Mixed Hyperbilirubinemia
A selective decrease in bilirubin secretion into the bile canaliculus may produce conjugated or mixed hyperbilirubinemia (i.e., an increase in conjugated and unconjugated bilirubin concentrations). Such a defect underlies two autosomally inherited disorders, Dubin-Johnson syndrome and Rotor’s syndrome. Each of these disorders is associated with a benign clinical course. In Dubin-Johnson syndrome, the molecular defect has been linked to an absence of expression of or impaired canalicular membrane targeting of the multispecific organic anion transporter MRP2.5 Interestingly, in Dubin-Johnson syndrome and in selected cholestatic disorders (e.g., primary biliary cirrhosis), compensatory up-regulation of the sinusoidal export protein MRP3 has been reported.16 Up-regulation of MRP3 may prevent hepatocellular overload by potentially toxic organic anions that are normally secreted by MRP2. The molecular basis of Rotor’s syndrome is unknown and does not appear to involve mutations in MRP2.17 In both Dubin-Johnson and Rotor’s syndromes, global hepatic function is preserved. Serum bilirubin levels are elevated, but serum levels of other commonly measured liver biochemical tests are normal.
Dubin-Johnson and Rotor’s syndromes can be distinguished biochemically and histologically (see Table 20-2). In Dubin-Johnson syndrome, hepatocytes contain a characteristic black pigment that is not seen in Rotor’s syndrome. This pigment is believed to result from lysosomal deposition of aromatic amino acid metabolites that are putative substrates for MRP2.5 Liver biopsy is generally unnecessary in the diagnostic evaluation of patients suspected to have Dubin-Johnson or Rotor’s syndrome, however, because neither disorder is associated with an adverse clinical outcome.
LIVER DISEASE
Acute Hepatocellular Dysfunction
Generalized impairment of hepatocellular function can result from acute or chronic liver injury. A clue to such disorders is the presence of elevated serum activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (see later and Chapter 73). Among conditions that produce acute or subacute hepatocellular injury are viral hepatitis, exposure to hepatotoxins, hepatic ischemia, and certain metabolic derangements. Acute viral hepatitis often is heralded by anorexia, malaise, myalgias, or discomfort in the epigastrium or right upper abdominal quadrant before jaundice develops (see Chapters 77 to 81). Five major hepatitis viruses have been isolated. Hepatitis A and E viruses are transmitted enterally. Each typically produces a self-limited illness that does not progress to chronic liver disease. By contrast, hepatitis B, C, and D viruses are transmitted parenterally, and illness produced by these agents can be prolonged and may lead to chronic disease. Major risk factors for hepatitis B, C, and D include injection drug use, exposure to blood products, and unprotected sexual exposures. The diagnosis of each of these disorders is aided by serologic testing (see later).
Many drugs and toxins produce hepatocellular injury (see Chapters 86 and 87). In particular, ingestion of acetaminophen (in large quantities) or of the mushroom Amanita phalloides may lead to hepatocellular necrosis and jaundice within several days after exposure. Toxic liver injury can have a fulminant course associated with a high mortality rate (see Chapter 93). In patients who survive, jaundice generally resolves and hepatic function recovers completely in those without preexisting liver disease. Certain drugs can produce idiosyncratic hepatocellular injury and jaundice, and these are discussed extensively elsewhere in this text (see Chapter 86). Alcoholic hepatitis should be a diagnostic consideration in the jaundiced patient with ethanol dependency, particularly when hepatomegaly and fever are present (see Chapter 84). Laboratory studies may help distinguish this entity from most other acute liver diseases (see later).
Jaundice related to hepatic ischemia may result from hypotension, hypoxia, hyperthermia, or hepatic venous outflow obstruction (see Chapter 83). Thrombosis of the hepatic vein (Budd-Chiari syndrome) or sinusoidal obstruction syndrome (hepatic veno-occlusive disease) should be suspected in a patient who presents with the rapid onset of ascites and hepatomegaly; the latter syndrome is more commonly associated with jaundice and is a complication of certain cytotoxic agents, particularly in the setting of hematopoietic cell transplantation (see also Chapter 34).
Wilson disease, an inherited disorder of hepatobiliary copper secretion, may manifest de novo with clinical features indistinguishable from those of acute viral hepatitis (see Chapter 75). The disease should be a diagnostic consideration in patients younger than 40 years, particularly when neurologic abnormalities are present or Kayser-Fleischer rings are seen on slit-lamp examination of the eye. Hemolytic anemia is a part of the spectrum of Wilson disease and contributes to the disproportionate hyperbilirubinemia often present in these patients. The diagnosis of Wilson disease is confirmed by biochemical testing and liver copper analysis (see later). Reye’s syndrome, a disorder of fatty infiltration of the liver associated with impaired mitochondrial metabolism of fatty acids, may produce jaundice as a manifestation of acute liver failure (see Chapter 86 and 93). It usually follows a viral illness in children, has been associated with the ingestion of aspirin, and is heralded by nausea and vomiting; its incidence has declined markedly as a result of public health campaigns advocating the avoidance of aspirin in children.
Chronic Hepatocellular Dysfunction
In contrast with acute liver disease, jaundice does not typically develop in chronic conditions associated with hepatocellular injury unless cirrhosis is present. A major cause of cirrhosis is chronic viral hepatitis, which should be a diagnostic consideration in patients with risk factors for parenteral exposure to causative agents. Diagnosis is aided by serologic testing (see later). Cirrhosis is part of the spectrum of nonalcoholic fatty liver disease, which is emerging as the most common cause of chronic hepatocellular injury in industrialized nations; major risk factors are obesity and diabetes mellitus (see Chapter 85). A similar histologic picture of steatohepatitis and sinusoidal fibrosis is found in the setting of alcoholic liver disease (see Chapter 84). Toxic injury by other compounds is less likely to produce cirrhosis, although cirrhosis has been described as a manifestation of industrial exposure to vinyl chloride and as a consequence of chronic ingestion of large quantities of vitamin A (see Chapter 87). Certain hereditary metabolic liver diseases may progress to cirrhosis. Hemochromatosis, a disorder characterized by excessive intestinal iron absorption with resulting hepatocellular iron accumulation and injury, is the most common of these (see Chapter 74). Although most affected persons are asymptomatic, the presence of diabetes mellitus, arthritis, or deep pigmentation in a jaundiced person should heighten suspicion for the disorder. The diagnosis is confirmed by detection of mutations in the HFE gene or by hepatic iron analysis. Hepatocellular copper overload and injury in Wilson disease also may progress to cirrhosis (see Chapter 75). As noted, the diagnosis should be suspected in younger persons, and the disease confirmed by biochemical testing and liver copper analysis. In a jaundiced patient with chronic obstructive pulmonary disease, α1-antitrypsin deficiency should be suspected (see Chapter 76). In this disorder, mutant α1-antitrypsin is misfolded and accumulates in the endoplasmic reticulum of hepatocytes, proteasomal degradation is impaired, and liver injury results. The diagnosis can be confirmed by laboratory testing and liver biopsy (see later). Autoimmune hepatitis, a disease that may be associated with systemic complaints such as malaise, fever, and arthralgias, is more common in women than in men (see Chapter 88). The diagnosis is aided by serologic testing and liver biopsy (see later). Celiac disease (see Chapter 104) may manifest as otherwise unexplained chronic liver disease—although rarely, if ever, with jaundice.
