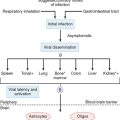
CHAPTER 42 ISCHEMIC STROKE: MECHANISMS, EVALUATION, AND TREATMENT
Stroke is a major public health problem. An estimated 500,000 new and 200,000 recurrent strokes occur in the United States annually, and the number of stroke survivors is estimated to be about 4.8 million. Stroke is the leading cause of disability and the third leading cause of death in the United States (after coronary heart disease and cancer) and accounts for a health care cost burden estimated at $53 billion spent in the year 2004.1
TRANSIENT ISCHEMIC ATTACK
Transient ischemic attacks (TIAs) have traditionally been distinguished from ischemic stroke on the basis of symptom duration, with the assumption, largely justified, that otherwise the two entities share risk factors and causes and that evaluation findings and secondary prophylaxis are similar for the two. TIA has traditionally been defined as a focal neurological deficit of abrupt onset, referable to a vascular territory, that lasts less than 24 hours. However, the 24-hour definition is arbitrary and is not based on any plausible biological mechanism. In fact, most TIAs resolve in less than an hour. In addition, studies have demonstrated that a significant proportion of clinically defined TIAs are not necessarily transient at the tissue level; there is evidence of tissue infarction on magnetic resonance imaging (MRI) performed early in the course of the syndrome.2,3 Indeed, in view of the advances in neuroimaging and epidemiological studies, a panel of experts has proposed that TIA be defined as a brief episode of neurological dysfunction caused by retinal or brain ischemia with symptoms lasting less than 1 hour and without evidence of acute infarction on neuroimaging (diffusion-weighted imaging [DWI] or computed tomography [CT] scan).4 When the symptoms and signs do not fit criteria for a TIA—that is, they persist for more than 1 hour and/or there is evidence on neuroimaging of tissue infarction—the syndrome is classified as an ischemic stroke. The modified duration for TIA is of practical benefit, in view of the new time window–dependent strategies for acute stroke management, when a decision for acute intervention should not be contingent on resolution of a patient’s signs and symptoms within 24 hours. However, the neuroimaging component of the proposed new definition is problematic because it sidesteps what is arguably most important about clinically defined TIAs: their reversibility. If symptoms resolve in under an hour but a lesion is identified with DWI, then what is the difference between this entity and ischemic stroke whose symptoms do not resolve? In addition, a definition of TIA based on the absence of a DWI lesion has the odd consequence of eliminating duration as an important distinguishing feature, inasmuch as studies have shown that a substantial proportion of patients with symptoms lasting less than an hour have DWI lesions. Indeed, one DWI study revealed that symptom duration could not distinguish TIA from ischemic stroke.5
Study findings support the contention that it is symptom duration or, more specifically, the rate of symptom reversal from time of onset, that is the essential characteristic of TIA; data indicate that risk of recurrent stroke is substantially higher in patients with rapid recovery than in those with fixed deficits and subsequent slow recovery.6–8 The largest study of risk of recurrent stroke after TIA showed that 50% of all ischemic strokes after TIA occur within 48 hours of the TIA.9 Thus, the presence or absence of lesions on DWI should not be required for a definition of TIA, only that the deficit substantially reverses, not necessarily fully, in less than an hour. This is not to say that DWI does not provide useful information about TIA mechanisms. For example, it has been shown that patients with deficits that reverse in less than 24 hours but who have DWI lesions have the highest risk of in-hospital recurrent stroke. This subgroup may have made up a substantial proportion of the patients in the large study mentioned previously.
The critical implication of the spate of studies on TIA is that TIA reflects a more unstable condition than does stroke and merits immediate attention. A TIA is to the brain what unstable angina or non–Q-wave myocardial infarction is to the heart, a condition of fluctuating tissue perfusion that is unstable and threatens tissue viability. TIA should be viewed as a medical emergency that necessitates immediate diagnostic workup and treatment. The clinical seriousness of TIA makes accurate diagnosis very important, but because the symptoms have nearly always resolved by the time the patient is seen by a physician, careful history taking is essential. This can be a challenge because a large number of nonischemic conditions, such as migraine, seizures, and multiple sclerosis, cause transient neurological symptoms. TIAs themselves vary in their mode of presentation, variation attributable primarily to the underlying cause. Different TIA presentations are therefore discussed as follows as they relate to each stroke subtype.
ISCHEMIC STROKE
In order to salvage penumbral tissue, CBF must be restored in the critical time window between electrical and membrane failure. There is good evidence, obtained mainly with positron emission tomographic (PET) studies,10 that ischemic penumbra exists in human and nonhuman primates and that it remains viable for longer and more variable periods of time than was suggested by initial small mammal experiments. Advances in MRI have made significant inroads in identifying the ischemic penumbra in patients after stroke and making it a target for therapeutic intervention (see “Stroke Evaluation” section). TIA might represent a form of penumbral phenomenon without an infarct core, and new data on the instability of TIA reflect the high risk of proceeding to infarction in this penumbral tissue.
STROKE CLASSIFICATION BY MECHANISM (STROKE SUBTYPES)
Large-Artery Atherosclerosis
This subtype is characterized by evidence for atherosclerosis (anterior or posterior circulation) in the large-vessel vascular distribution of a brain infarction, in the absence of a cardiac source of embolism. The most common location for atherosclerotic changes is at the bifurcation or proximal takeoff of the large vessels, where the shear stress on the wall, from turbulent flow usually related to long-standing hypertension, is maximal. The arterial narrowing can be either extracranial (in the carotid or vertebral arterial system) or intracranial (in the many tributaries of the circle of Willis and their branches). The pattern of parenchymal involvement can be small or large, cortical or subcortical, and is determined largely by the status of pial-pial collaterals and the circle of Willis. There are two other infarction patterns associated with proximal large vessel disease: border zone and watershed. Border zone infarction corresponds to involvement of areas at the junction of the distal fields of two nonanastomosing arterial systems. Watershed infarctions occur at a zone of pial-pial artery anastomoses between two large vessels: for example, the middle cerebral artery (MCA)–posterior cerebral artery (PCA) watershed. There are internal border zones (misleadingly also referred to as internal watersheds) in the centrum semiovale and paraventricular corona radiata, regions at the junction of subcortical and medullary penetrators off the MCA, and in the subinsular zone, a region between small insular penetrating arteries and the lateral lenticulostriate vessels.11 Thus, for example, critical internal carotid artery (ICA) stenosis or occlusion can cause watershed infarction at (1) the boundary of the anterior cerebral artery (ACA) and MCA, manifesting as a thin wedge extending from the anterior horn of the lateral ventricle to the frontal cortex or a string of infarction at the medial convexity surface; (2) the MCA/ACA/PCA boundary, often more difficult to distinguish from MCA branch occlusion, but again appearing as a cortical wedge extending from the occipital horn of the lateral ventricle to parieto-occipital cortex; and (3) a confluent or discontinuous long lesion in the centrum semiovale running parallel to the lateral ventricle.
Interpretation of patterns of infarction from large vessel disease is based on assumptions about underlying mechanisms: (1) large-vessel stenosis or occlusion by in situ disease or artery-to-artery embolism and (2) flow-failure distally from a more proximal occlusion or hypotension. However, these mechanisms are likely to coexist. A high-grade ICA or MCA stenosis may be both emboligenic and cause a low-flow state. The low-flow state reduces the chances of clearing small embolic particles from distal arterial beds, leading to a border zone or watershed pattern of infarction.12 In addition to producing cortical, subcortical, and watershed patterns of infarction, LAA can occlude the os of a single penetrating small vessel, causing a lacunar infarct (see later discussion). Thus, the variety of infarct patterns possible with LAA makes diagnosis based on clinical examination or infarct pattern alone difficult. A bruit on examination and the presence of other atherosclerotic risk factors or markers such as hypertension, elevated cholesterol, diabetes, smoking history, peripheral arterial disease, and coronary artery disease, are marginally helpful at best. Indeed, it is debatable whether there is any significant difference in modifiable risk factor profile and predisposition to LAA versus small-vessel disease.13 Information regarding race/ethnicity can be helpful, inasmuch as white persons have a higher incidence of extracranial atherosclerosis, whereas Asian, black, and Hispanic persons have a higher incidence of intracranial atherosclerosis.
Interestingly, unlike completed infarction, LAA-associated TIAs do have distinctive features that help make the diagnosis. Intermittent flow-failure through a critical stenosis, often brought on when the patient stands up suddenly or by overly aggressive antihypertensive therapy, can cause focal neurological deficits that reverse when the patient lies down. More rarely, patients manifest a “limb-shaking” TIA, consisting of brief periods of shaking of an extremity brought on by standing or sitting up, easily confused with a focal motor seizure. Another strong clue to LAA is an unstable course in the first 24 hours after stroke onset, manifested by deterioration after improvement.14
Determination of LAA as the underlying cause of a stroke through ancillary testing (see “Stroke Evaluation” section) is important because there is a high rate of early stroke recurrence in this subgroup of patients,15 both for extracranial and intracranial atherosclerosis. This is also true for the long term because patients with symptomatic ICA stenosis with greater than 70% diameter reduction have a stroke recurrence risk of 26% in 2 years.16 The recurrence rate for patients with symptomatic MCA stenosis is about 10% a year.17,18 Because of this relatively high risk of early recurrence and the established benefit of early endarterectomy in long-term stroke prevention for ICA stenosis, identification of this subtype of stroke is critical.19
Small-Vessel Atherosclerosis (Lacunar Strokes)
The definition of lacunar infarction has become quite confused. We believe that it should be a combined clinical and radiological diagnosis that is based on a presentation with one of a handful of typical syndromes (see “Lacunar Syndromes” section) and imaging evidence for a small infarct (2 to 20 mm in diameter) in the deep cerebral white matter, basal ganglia, thalamus, or pons. Evidence suggests that in situ single perforator disease is the cause in the majority of cases. Exclusion of LAA and a cardiac source of embolism is important, because lacunar infarction is estimated to originate from emboli or LAA in about 10% to 15% of cases.20 This may be true particularly for patients who have a lacunar syndrome with evidence of multiple subacute lesions on neuroimaging.21
However, despite these concerns, evidence favors preserving the lacunar infarction stroke category from a pathophysiological standpoint. First, the presence of lacunar infarcts is correlated with leukoaraiosis and with subcortical microhemorrhages.22 These correlations suggest that lacunar infarcts are manifestations of a more diffuse abnormality of small cerebral arterioles.23 Second, the proportion of embolic sources identified in patients with lacunar infarction is lower than that of hemispherical ischemic strokes. Third, after a lacunar infarction, a recurrent stroke is more likely to be lacunar than nonlacunar.23a This would not be expected if lacunar infarcts shared the same mechanism with larger cortical strokes. In addition, the early stroke recurrence rate is lower than those for LAA and cardioembolic stroke. Fourth, in a primate model, only 6% of even the smallest particles injected in the carotid artery ended up in the lenticulostriate vessels.23b
The pathology underlying lacunar infarction is still debated, mainly because lacunar infarcts are seldom fatal and cases are thus rarely subjected to autopsy. Nevertheless, it is assumed that lacunar infarction results from occlusion of a small penetrator by atheroma blocking its origin, by embolus, or by an intrinsic process, lipohyalinosis (narrowing the lumen at points along its length). Lenticulostriate pathology after lacunar infarction has been visualized with MRI.24 These images show a linear structure with signal features consistent with perforator occlusion by thrombus or leakage of vessel contents into the surrounding parenchyma. These findings support the idea of a pathological process unique to deep perforating arteries, which can cause lacunar infarction. Occlusion of the vessel itself might cause infarction, or blood vessel contents might be toxic to surrounding parenchyma. A spectrum of perforator disease with luminal thrombus and leakage of contents into the blood vessel wall and then into the perivascular tissue might explain the previously mentioned correlation among lacunar infarcts, leukoaraiosis, and deep microhemorrhages. In addition, a leakage mechanism might also explain an interesting feature of TIAs associated with lacunar infarction: the “capsular warning syndrome.” This consists of a stuttering cluster of stereotypical events over a period of about 72 hours. These events are brief bursts of typical lacunar phenomena that can come and go over minutes. Many of these patients progress to a fixed deficit; it is possible that the capsular warning syndrome evolves because a single penetrator undergoes occlusion or leakage damages surrounding tissue directly.23
Cardioembolic Strokes
Embolism of cardiac origin accounts for about 20% to 40% of ischemic strokes. Atrial fibrillation is the best established cause of cardioembolic stroke, and its identification is extremely important, in view of the relatively high recurrence rates (about 10% a year) and the effective prophylaxis (approximately 60% absolute risk reduction) achieved with chronic oral anticoagulation. Other known emboligenic sources, treated with anticoagulation, are valvular disease (especially prosthetic valves), documented intraventricular thrombus present in severe cardiomyopathies, and recent myocardial infarction. In one study, transesophageal echocardiography (TEE) was used to assess 151 consecutive patients, 1 week after ischemic stroke or TIA.24a Intracardiac thrombus was identified in 26% of the patients (70% in the left atrial appendage). Multivariate analysis showed an association with large stroke, symptomatic coronary artery disease, and evidence for ischemia on electrocardiogram.
Interatrial septal abnormalities, such as patent foramen ovale (PFO) and atrial septal aneurysm (ASA), result from failure of the septum to close at birth and occur in about 25% of the general population.24b Interatrial septal abnormalities are thought to be an important cause of embolic stroke in patients younger than 55. A PFO is an intact interatrial connection through the two overlapping septa that form the interatrial septum. An ASA is a hypermobile piece of the atrial septum that can protrude through the PFO into the left atrium during the cardiac cycle. PFOs can serve as a conduit for embolism originating from the venous circulation (lower extremity or pelvic venous thromboses) to the arterial circulation through right-to-left shunting of blood during the cardiac cycle and especially during Valsalva maneuvers. ASAs seem to enhance the stroke risk of a PFO, possibly by directing flow through the PFO or acting as a nidus for thrombus formation itself. A metaanalysis revealed a 24-fold increased risk of stroke in patients younger than 55 who had both a PFO and an ASA, in comparison with a fivefold risk in patients with only a PFO.25 The high frequency of interatrial septal abnormalities in the general population in comparison with the relatively low incidence of stroke in persons younger than 55 suggests that a second factor needs to combine with PFO in order for stroke to occur. Studies have shown increased frequency of two inherited hypercoagulable disorders, the factor V Leiden and the prothrombin 20210 mutations, in young patients with stroke and PFO.25a,25b Thus, perhaps a combination of PFO and an underlying hypercoagulable state, either acquired or inherited, is required in order for a PFO-related stroke to occur. In contrast to patients younger than 55, it does not seem that interatrial septal abnormalities are a substantial stroke risk in older patients, possibly because left-sided atrial pressures increase with aging. In all patients with stroke and PFO, it is important to emphasize the need for a thorough search for the other potential causes of stroke before attributing it to the PFO.
Prospective studies have shown that aortic arch atheroma is found more often in patients with stroke and that the presence of aortic arch atheroma, detected by TEE, is associated with increased risk of future stroke. A metaanalysis of these prospective studies gave an odds ratio for recurrent stroke of 3.76, similar in magnitude to those for atrial fibrillation and high-grade carotid stenosis.26 Aortic arch atheroma, occurring with increasing age and in people with vascular risk factors,27 may be just a marker for atherosclerosis. However, a number of observations suggest that aortic arch atheroma causes embolic stroke. First, aortic arch atheroma is often present in patients with stroke but without concomitant carotid disease. Second, stroke risk is highest for atheroma with mobile components. Third, stroke is more common in the presence of aortic arch atheroma than with atheroma in the thoracic aorta.28 Fourth, left hemisphere events are more common than right hemisphere events, and most atheroma is found in the middle to distal arch, after takeoff of the innominate artery.29
A number of clinical features and radiographic features suggest cardioembolic stroke: (1) sudden onset with rapid progression to maximal focal neurological deficit (<5 minutes); (2) simultaneous or sequential strokes in multiple arterial territories, such as a left homonymous hemianopia and a right hemiparesis; (3) Wernicke’s aphasia (inferior division of the left MCA) and visual field cuts (distal PCA); (4) a large deficit that then rapidly regresses, probably as a result of recanalization of a large proximal vessel; (5) appearance on imaging, especially DWI, of bihemispherical, both anterior and posterior territory, or bilateral or multilevel posterior circulation infarcts (the typical pattern of stroke on MRI is a wedge-shaped lesion with its base at the cortex and the apex located subcortically); (6) hemorrhagic transformation of an ischemic infarct as a result of recanalization and irrigation of infarcted tissue or, less likely, dissection at the site of thrombus impact; and (7) the presence of single or multiple small subcortical infarcts in the absence of cortical infarcts, which makes the diagnosis of embolic stroke less likely.30
Cryptogenic Strokes
The patient with cryptogenic stroke lacks a documented cardioembolic source despite investigation with transthoracic echocardiography (TTE) or TEE; does not show evidence of extracranial or intracranial large-artery pathology on ultrasonographic or angiographic studies; and does not have the expected risk factor profile, clinical syndrome, and imaging characteristics for a diagnosis of a lacunar stroke. The neuroimaging features can be variable and may include cortical or subcortical pattern or even the presence of infarcts in multiple arterial territories not explained by a readily identifiable embolic source. It is likely that in the near future, as diagnostic accuracy improves, the overall frequency of diagnosis of cryptogenic strokes will decrease.
Stroke of Other Determined Cause
Dissection is a tear in the intima or the media that allows luminal blood to be redirected into a false lumen within the blood vessel wall, with formation of an intramural hematoma, which may limit flow (Fig. 42-1) or cause aneurysmal dilatation. Arterial dissection of the extracranial portions of the carotid and vertebral arteries accounts for only about 2% of all ischemic strokes but for up to 25% of ischemic strokes in patients younger than 55. Cervical artery dissection can occur after clear-cut neck trauma (motor vehicle accidents, attempted strangulation, fall with neck injury), after relatively trivial mobilization of the neck (hair washing, chiropractic manipulation),31 or spontaneously, without any obvious precipitant. The latter two scenarios probably reflect an underlying structural weakness of the arterial wall, inasmuch as dermal connective tissue abnormalities have been detected in up to a third of these cases. Approximately 5% of spontaneous dissections can be attributed to inherited disorders of collagen structure, the most common of which is Ehlers-Danlos syndrome type IV. Others include Marfan’s syndrome, autosomal dominant polycystic kidney disease, and osteogenesis imperfecta type I. In addition, about 5% of patients have a family history of dissection. Approximately 15% of patients have angiographic evidence of fibromuscular dysplasia. Infection, migraine, and elevated homocysteine have also been associated with dissection, but these risk factors have mainly been assessed with case-control studies, which are subject to information and selection bias, as well as confounding.32
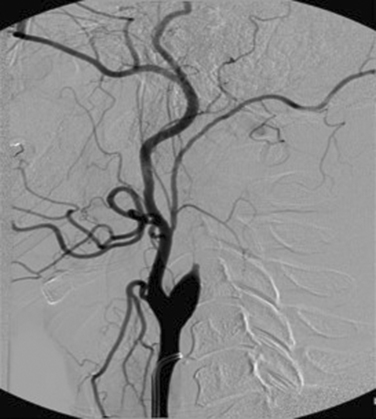
Moyamoya disease occurs in younger patients and is characterized by a progressive intracranial occlusive noninflammatory vasculopathy of the distal ICA and its bifurcation into the middle and anterior cerebral arteries. Its hallmark is the development of deep intracranial collateral vessels, usually in the lenticulostriate vessels, giving its peculiar characteristic defined by Japanese investigators as “puff-of-smoke” (Fig. 42-2). Its cause is unclear but does not seem to be an inflammatory process, as in the primary CNS vasculitides. A similar pattern can occur with other conditions such as prior pituitary tumor irradiation, Down syndrome, neurofibromatosis, and sickle cell disease. In these situations, the pattern appears secondary to the underlying condition and is described as moyamoya syndrome rather than moyamoya disease. It should be suspected in young patients with deep hemispherical hemorrhage but without the associated risk factors for intracerebral hemorrhage (ICH) and in young patients with cryptogenic ischemic stroke, especially if one of the commonly associated conditions listed previously is present.
CLINICAL SYNDROMES
Lacunar Syndromes
There are five classical lacunar syndromes (Table 42-1): pure motor hemiparesis, pure sensory syndrome, sensorimotor syndrome, dysarthria–clumsy hand syndrome, and ataxic hemiparesis. It should be emphasized however, that many additional, albeit rarer, lacunar syndromes almost certainly exist and that nonlacunar subcortical and cortical strokes can cause the classical syndromes.32a For example, an embolus to the rolandic branch of the MCA could affect the primary sensorimotor cortex and cause sensorimotor syndrome. Another example is occlusion of a paramedian pontine penetrator by basilar artery atheroma, causing pure motor hemiparesis. However, studies have shown an excellent positive predictive value of the lacunar syndromes for the presence of lacunar infarction on brain imaging.
TABLE 42-1 Classic Lacunar Syndromes with Vascular Territories and Anatomical Structures Most Commonly Affected
| Lacunar Syndromes (Structures Affected) | Vessel Distribution |
|---|---|
| Pure Sensory Syndrome | |
| Thalamus | Thalamic perforators |
| Pure Motor Hemiparesis | |
| Basis pontis | Pontine penetrators (basilar branch) |
| Posterior limb of internal capsule | Lenticulostriate vessels (MCA branches) |
| Cerebral peduncle | Basilar or PCA penetrators |
| Dysarthria–Clumsy Hand Syndrome | |
| Anterior limb or genu of internal capsule | Lenticulostriate vessels (MCA branches) |
| Basis pontis | Pontine penetrators (basilar branch) |
| Ataxic Hemiparesis | |
| Contralateral basis pontis | Pontine penetrators |
| Contralateral thalamus | Thalamic penetrators |
| Posterior limb of internal capsule | Lenticulostriate branches |
| Sensorimotor Syndrome | |
| Posterior limb of internal capsule and thalamus | Lenticulostriate branches |
MCA, middle cerebral artery; PCA, posterior cerebral artery.
Pure Motor Hemiparesis
This is the most common manifestation of a lacunar stroke. In this syndrome, there is motor involvement of the face, arm, and leg, sometimes with more involvement of one than the other, but with absence of sensory, visual, language, or other cortical symptoms. There are often varying degrees of dysarthria and dysphagia, as a result of involvement of corticobulbar tracts. Structures commonly involved in this syndrome reflect the descending course of the corticospinal tract: the corona radiata, the posterior limb of the internal capsule (Fig. 42-3), and the basis pontis. Less often, a midbrain peduncular or medullary pyramidal infarct (with sparing of the face) can also cause this syndrome. The diagnosis of brainstem pure motor hemiparesis requires the absence of all the following: vertigo, deafness, tinnitus, diplopia, nystagmus, and ataxia. Traditionally, it has been taught that isolated monoparesis, usually brachial, is rarely caused by lacunar infarct but instead indicates a cortical or centrum semiovale lesion, regions where the motor homunculus is more spatially separated. However, with the advent of MRI, this has been shown not to be the case. Isolated monoparesis is compatible with small-vessel disease: for example, in the corona radiata and pons.33
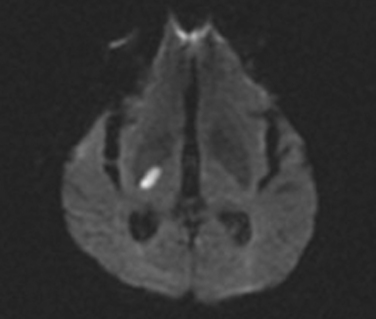
Pure Sensory Syndrome
Although this is considered the sensory analogue of pure motor hemiparesis, it occurs far less frequently than pure motor hemiparesis. It is usually caused by infarction of the ventro posteromedial and ventroposterolateral nuclei of the thalamus as a result of involvement of the inferolateral artery off the P2 segment of the PCA, but it can also result from corona radiata infarction through interruption of thalamocortical projections. Both spinothalamic and lemniscal modalities are usually affected, but selective sensory impairment can also occur. Perfect splitting of the midline on sensory testing can be observed, and involvement of midline structures such as tongue and genitalia may also be present. The hemisensory deficits may be complete or incomplete with a cheiro-oral predominance. Some patients may develop a chronic pain syndrome, especially with right thalamic lesions, with pronounced dysesthesias and paresthesias on the side contralateral to the thalamic involvement (Dejerine-Roussy syndrome). This syndrome usually appears in the subacute or chronic period and can be difficult to manage.
Sensorimotor Syndrome
This condition is a combination of sensory and motor deficits. Although single penetrator disease can probably cause this syndrome, there have been only a few autopsy studies, and the specificity of this syndrome for a lacunar mechanism is probably lower than for the other four syndromes. Nevertheless, most authors postulate predominant involvement of the thalamus with impingement on the adjacent posterior limb of the internal capsule. An alternative explanation, however, is that both the sensory loss and hemiparesis result from thalamic involvement alone, inasmuch as the inferolateral artery also supplies the ventrolateral nucleus, which projects to motor cortex.34 It is likely that MRI will help to better define the anatomical basis of this syndrome.
Ataxic Hemiparesis
The classic syndrome, caused by basis pontis infarction, was described as limb ataxia ipsilateral to distal leg paresis with minimal or no facial or arm weakness. This diagnosis can be difficult to establish, because limb dysmetria must be out of proportion to the hemiparesis. Other typical locations of infarction are the corona radiata, the anterior or posterior limb of the internal capsule, and the thalamus. The cerebellum has been implicated in some cases. Larger cortical strokes involving the ACA territory may manifest in a similar way, but the more pronounced leg involvement and the many behavioral abnormalities occurring with ACA-distribution strokes may help differentiate the two (see Table 42-1).
Cortical Syndromes
The most commonly encountered clinical syndromes (Table 42-2) are briefly reviewed according to vessel distribution and associated stroke causes.
TABLE 42-2 Clinical Stroke Syndromes with Corresponding CNS Structures and Arterial Territory Affected
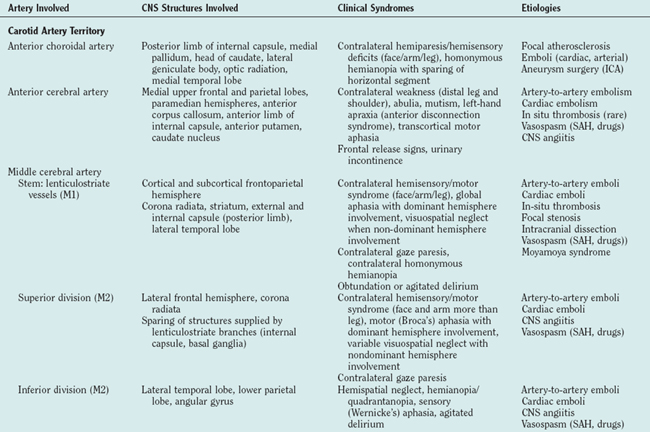


Anterior Circulation
Middle Cerebral Arteries
Middle Cerebral Artery Stem Occlusion
The proximal syndrome is usually dramatic and reflects damage to the basal ganglia and internal capsule, supplied by the medial and lateral lenticulostriate branches that arise from the dorsal surface of the MCA stem, as well as large areas of cortical infarction in the territories of the superior and inferior divisions of the MCA (Fig. 42-4). The typical patient presents with contralateral hemiplegia with equal involvement of the arm and leg, variable degrees of primary sensory abnormality, dysphagia, and hemianopia. There is forced eye deviation toward the side of the affected hemisphere, sometimes with accompanying ipsilateral head deviation. With dominant hemispherical involvement, there is usually global aphasia, buccofacial apraxia, and ideomotor apraxia. In the first few days there may be frank mutism. With nondominant hemispherical involvement, there is usually contralateral hemineglect, contralateral anosognosia, and delirium. Less frequently, syndromes more often associated with bilateral hemispherical damage, such as prosopagnosia and the reduplicative paramnesias, can be present with unilateral nondominant hemisphere damage. Individuals with an MCA stem occlusion are the most likely to develop massive hemispherical swelling with midline shift, and subfalcine and transtentorial herniation. The prognosis for these patients is guarded, and in complete MCA-distribution strokes, the mortality rate can be as high as 80% despite neurointensive care efforts.
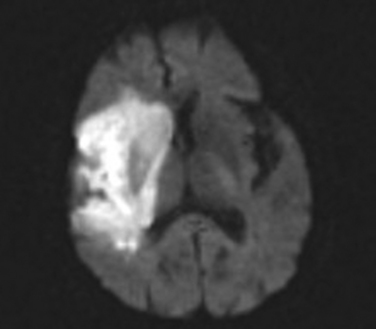
Middle Cerebral Artery Upper Division Occlusion
Isolated involvement is uncommon because the superior trunk is short, but when it occurs, it is usually caused by ICA or MCA atherosclerosis (Fig. 42-5). This division supplies most of the frontal convexity and anterior parietal lobe, and the syndrome resembles stem occlusion with a contralateral hemiparesis, forced eye and head deviation toward the side of the lesion, and variable degrees of aphasia and hemineglect. In contrast to stem occlusion, motor deficits are characterized by a gradient of weakness with the contralateral side of the face and arm (brachiofacial pattern) more severely affected than the leg, reflecting the involvement of the corresponding cortical structures rather than the internal capsule. A visual field defect is usually absent.
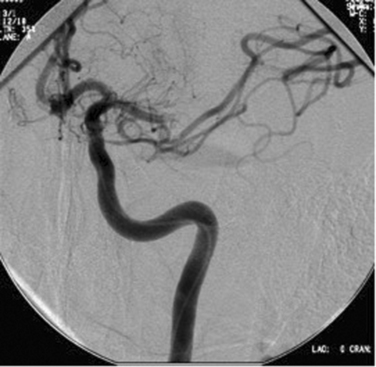
With dominant hemispherical involvement, there is language disturbance characterized in the acute phase by global aphasia, which tends to reduce to predominantly Broca’s aphasia and speech apraxia. This highlights the fact that in the acute stroke setting, vascular aphasias are often global and nonclassic in their presentation.35 With nondominant involvement, there may be some degree of visuospatial neglect but usually not as pronounced as with a larger territorial infarct. Acute agitated delirium is usually not present, because this requires infarction of the right middle temporal gyrus and inferior parietal lobule, supplied by the inferior division of the MCA (see later discussion). Because of the smaller volume of tissue infarction, upper-division MCA-distribution strokes do not cause the same high rate of mortality observed with holo-MCA strokes but carry a significant degree of long-term disability that necessitates intensive rehabilitation.
Anterior Cerebral Arteries
In comparison with strokes in the MCA distribution, ACA-distribution strokes are uncommon and are most often secondary to embolism from a proximal source such as the carotid artery or the heart. Less frequent causes of an ACA-distribution stroke include vasospasm from a ruptured saccular aneurysm (anterior communicating artery), in which case the strokes may be bilateral, and inflammatory vasculopathy involving the intracranial vessels. The most common manifestations of an infarct in the distal territory of the ACA are a function of the territories supplied: anterior and medial frontal lobes, including the motor-sensory cortex for the contralateral foot and leg; the supplementary motor area; and the central bladder representation, and also of lesion side36 (Fig. 42-6).
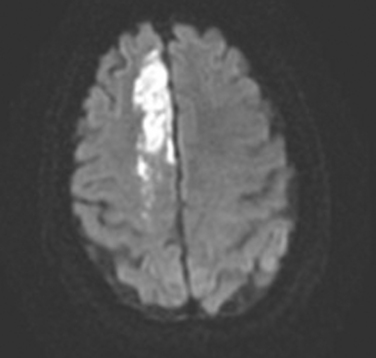
Left-sided infarction causes transient akinetic mutism (abulia), transcortical motor aphasia, contralateral leg and shoulder weakness with sparing of the distal upper extremity and face, and contralateral deficits in higher order sensory functions such as stereognosis (ability to discriminate two simultaneous stimuli) and joint-position sense discrimination. Right-sided infarction causes acute confusional state, motor hemineglect, transient akinetic mutism, contralateral hemiparesis, and sensory deficits in the pattern described for left-sided infarction. Predominant leg weakness is not unique to ACA infarction; it is also present with MCA-territory cortical infarction. It can also be present with capsular and pontine infarcts. In general, lesions that affect the medial premotor cortex, the supplementary motor area, and the rear portion of the medial part of the precentral gyrus, or their projections, can cause leg-predominant hemiparesis.37
In addition to abulia and predominant leg weakness, callosal disconnection syndromes can help distinguish ACA from MCA infarcts. The three main syndromes are left unilateral ideomotor apraxia, agraphia, and tactile anomia. All three syndromes affect the left hand in right-handed patients. Left unilateral ideomotor apraxia, also called the anterior disconnection syndrome, is the inability to perform overlearned skilled movements in response to verbal command. Patients with left-hand agraphia have severely impaired handwriting with their left hands but not their right. Patients with unilateral tactile anomia are unable to name objects placed in their left hands. All three syndromes are probably caused by interruption of transcallosal information to or from the language areas in the left hemisphere. They usually result from infarcts in different regions of the corpus callosum caused by interruption of pericallosal branches of the ACA. However, these syndromes, although of phenomenological interest, are overemphasized, in view of their low frequency of occurrence. They are relatively rare probably because the anterior corpus callosum is supplied by both ACAs.
Internal Carotid Artery
Many times, it is difficult to differentiate an ICA-territory infarction, especially when the onset is abrupt, from embolic MCA strokes. The hallmark of a proximal ICA disease is a history of a preceding TIA, either retinal (transient monocular blindness or amaurosis fugax) or hemispherical. Its presence is strongly suggestive of ICA disease proximal to the origin of the ophthalmic artery and should prompt rapid investigation of the extracranial carotid system. ICA dissection is another possible cause (see Table 42-2).
Posterior Circulation
Vertebrobasilar Territory Strokes
Basilar Artery Branches
Superior Cerebellar Arteries.
Traditionally, it was thought that isolated superior cerebellar artery territory infarcts were rare but instead occurred in combination with midbrain, thalamic, and PCA territory infarcts, as a result of embolism to the top of the basilar artery. However, modern MRI suggests that the occurrence of isolated superior cerebellar artery infarcts has been underestimated.38 These infarcts can be small or territorial, both most commonly caused by embolism. The superior cerebellar artery supplies most of the cerebellar cortex, the cerebellar nuclei, and the superior cerebellar peduncle. Superior cerebellar artery infarcts result in a combination of the following signs and symptoms: vertigo and dizziness, nystagmus, limb ataxia, gait ataxia, and mild hemiparesis. Clinical brainstem signs are usually absent.
Basilar Artery and Its Penetrators.
The posterior wall of the basilar artery gives rise to small paramedian and short circumferential arteries that supply the paramedian and lateral pontine regions. These arteries can be affected by either microatheroma at their origins or lipohyalinosis along their length. These are believed to be distinct pathological processes. Stroke from basilar artery disease is very variable and can range from a mild lacunar stroke, resulting from a small penetrator disease, to a large, devastating syndrome with extensive destruction of the brainstem from complete or almost complete basilar occlusion (Fig. 42-7). Pontine syndromes reflect the particular structures affected, with a basic division between strokes that predominantly affect the anterior pons and those that affect predominantly the pontine tegmentum. Anterior pontine infarctions can manifest as classic lacunar syndromes with pure motor hemiparesis or ataxic hemiparesis/dysarthria–clumsy hand syndrome, as a result of damage to the corticospinal tract and crossing corticopontocerebellar fibers. Strokes that predominantly affect the pontine tegmentum manifest with a constellation of brainstem signs as a result of involvement of the medial lemniscus, the medial longitudinal fasciculus, the cerebellar peduncles, the abducens nucleus, and the vestibular nuclei.
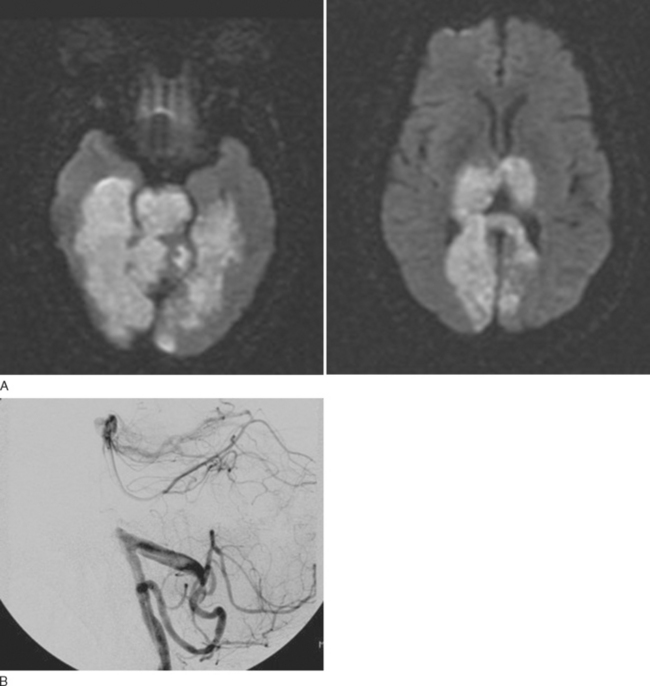
Other structures that can be affected by an embolus to the distal basilar artery include the occipital lobes, medial temporal lobes, and thalami (Fig. 42-8). Their involvement can be either unilateral or bilateral and gives rise to multiple signs or symptoms referable to the involvement of the PCA territory (see next section).39
Posterior Cerebral Arteries.
Balint’s syndrome can arise after unilateral or bilateral infarction of the superior parietal lobule at the parieto-occipital junction, supplied by the MCA-PCA watershed. The syndrome, also known as optic ataxia, is characterized by defective directional control of visually guided reaching movements, but motor execution is normal when other sensory modalities are used. On-line control is more impaired than feedforward planning, and errors in reaching are variable with no appreciable constant errors. Anton’s syndrome is denial of blindness despite clear physiological evidence for it after bilateral occipital infarction. This syndrome can be accompanied by agitation/delirium, possibly from the decreased visual input or concomitant involvement of the temporal lobes.
STROKE EVALUATION
Parenchymal Imaging
Critical in the evaluation of a presumed ischemic stroke, especially in the hyperacute period, is assessment of the extent and location of affected brain parenchyma, as well as ruling out stroke mimics such as ICH, subdural hematoma, subarachnoid hemorrhage, and brain tumor. CT scanning of the head should be the first step in such an evaluation, because of its widespread availability and short scanning time. CT scan is still considered the “gold standard” for detecting hyperacute hemorrhage, although one prospective study showed 100% sensitivity for hyperacute ICH with T2* MRI. Besides obvious intraparenchymal hemorrhage, abnormalities on CT scan related to hyperacute stroke, include blurring of the gray-white matter interface, effacement of the sulcal margins, loss of the normal sharp definition between the basal ganglia and surrounding white matter, and effacement of the insular cortex. These abnormalities confirm ischemia as the most likely cause of the acute syndrome and help predict not only the area at risk and ultimate infarct size but also which patients are likely to develop significant brain edema and require more aggressive monitoring in the intensive care unit. The extent of these early changes also has a direct relationship with the risk of symptomatic intracranial hemorrhage from systemic thrombolysis and is predictive of a poor outcome.40,41
MRI, increasingly available for the evaluation of ischemic stroke in the acute setting, is more sensitive than CT scanning for detecting acute ischemia and is the only reliable method for detecting ultra-early ischemic changes (<1 hour after stroke onset). Different magnetic resonance sequences are available, each emphasizing different tissue components and different degrees of tissue definition and contrast. The sequences of most utility in the evaluation of patients with stroke are T1-weighted imaging, T2-weighted imaging, fluid-attenuated inversion recovery, DWI, proton density imaging, and gradient-echo imaging, the last being the most sensitive for detection of hemorrhage in all stages of evolution. In the hyperacute evaluation, DWI identifies early ischemic lesions and distinguishes them from old infarcts. Ischemia is characterized by a hyperintense signal, which represents the restricted intracellular diffusion of hydrogen ions, which normally diffuse freely in interstitial fluid but are shifted intracellularly because of cytotoxic edema in the affected area. In humans, DWI is able to detect ischemia as early as 40 minutes after stroke onset and in one study, based on confirmation at autopsy, had a sensitivity of 88% and a specificity of 96.6%. Abnormalities observed on DWI can be short lived, resolving within 2 weeks of the ischemic insult; this feature is not seen with the other magnetic resonance sequences.42,43 However, hyperintense lesions on DWI do not distinguish reversible from irreversible ischemia, can be observed with other conditions (brain abscess, seizure, acute multiple sclerosis plaque, hypoglycemia, and brain tumors), and may result from T2 decay (T2 shine-through) rather than a diffusion abnormality. T2 shine-through can be ruled out with computation of a directionally averaged diffusion coefficient (ADC). On an ADC map, only ischemia appears dark, indicative of a low ADC. The utility of ADC values to reliably distinguish differing degrees of ischemia—a critical issue, inasmuch as the entire rationale for acute treatment of stroke is to rescue areas of reversible ischemia—is under active investigation but seems questionable. Currently, rather than reliance on differential ADC values, the favored approach with MRI is to identify areas of reduced perfusion beyond the boundaries of the DWI abnormality (see next section).
Vascular Imaging
Duplex Doppler and Transcranial Doppler Imaging
Duplex study of the extracranial circulation is most useful in the determination of the degree of stenosis of the ICA (Fig. 42-9). This determination is crucial because of the difference in the approach to patients with a recent ischemic stroke related to an ipsilateral ICA stenosis (see later discussion). Analysis of the ECVAs also provides valuable information in patients with posterior circulation strokes.
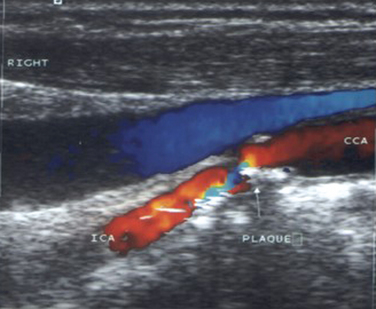
When duplex Doppler imaging is compared with the “gold standard” for anatomical definition (digital subtraction angiog raphy), the sensitivity and specificity for hemodynamically significant (>70% diameter narrowing) extracranial ICA stenosis varies between 85% to 90% and 75% to 80%, respectively.44 On the basis of these numbers, the best approach is to combine duplex Doppler study with another noninvasive imaging modality, such as MRA, because both tests, when concordant, have been shown to accurately predict significant extracranial ICA stenosis in more than 90% of the cases.45,46 Nonconcordance most often occurs because ultrasonography and MRA can differ in the degree of stenosis they reveal, with a tendency for MRA to overcall degree of stenosis. Another concern is whether noninvasive techniques can reliably differentiate high-grade stenosis from occlusion, a critical factor in surgical decision making. For example, Doppler imaging might demonstrate total ICA occlusion, whereas, in fact, a string sign is present; conversely, an ascending pharyngeal artery or muscular branch may be mistaken for residual flow through an occluded ICA. In such a situation, conventional angiography, CTA, or gadolinium-enhanced MRA should be considered.
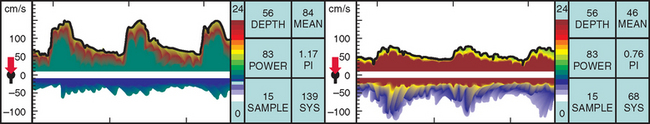
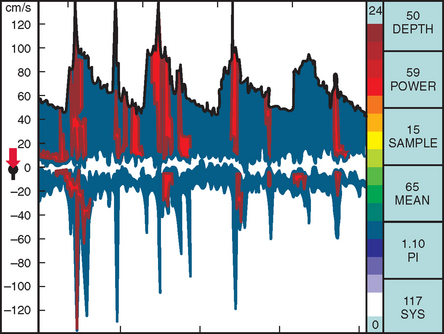
TCD imaging is a valuable tool in the assessment of the intracranial consequences of cervical carotid or vertebral stenosis. Specifically, TCD imaging can detect blunted waveforms and recruitment of collateral pathways (e.g., cross-filling from the anterior communicating artery) (Fig. 42-10). TCD imaging can also provide information on adequacy of collateral flow through the use of vasodilatory challenges such as carbon dioxide inhalation (see later discussion). Also, TCD imaging can be used to monitor the MCAs for evidence of intermittent embolization from a proximal arterial or cardiac (PFO, atrial fibrillation) source (Fig. 42-11).
Magnetic Resonance Angiography
Like CTA, this is a noninvasive modality to study the intracranial and extracranial circulation. The objective of any MRA technique is to maximize signal coming from the vessel while minimizing background signal. The most popular sequences are two- and three-dimensional time-of-flight (TOF) MRA. TOF MRA is based on flow-related enhancement of blood signal, which allows blood to be differentiated from stationary tissues. The data may be acquired as single slices (two-dimensional) or as a thick slab that is subsequently reconstructed into thin sections (three-dimensional). In the case of three-dimensional imaging, blood flow must be more rapid in order to traverse the whole slab before saturation occurs with attendant loss of signal. Thus, three-dimensional TOF is usually better for vessels in which flow velocity is high (i.e., cervical arteries and the circle of Willis), whereas two-dimensional TOF is better for assessing cerebral veins and sinuses. Phase-contrast MRA is another technique that is time-consuming and has not had much application in the workup of ischemic stroke so far. One scenario in which phase-contrast MRA can be helpful is when an arterial lumen is stenosed by subacute thrombus, which is bright on T1 images. Thus, a TOF MRA might misread a bright vessel as patent when it is in fact occupied by subacute thrombus. This high signal is subtracted out in phase-contrast imaging.
TOF MRA is helpful as a screening test for significant (>70% diameter reduction) ICA stenosis because it has comparable sensitivity to digital subtraction angiography. However, TOF images can be contaminated by artifacts in the presence of slow flow or turbulence. This is a problem mainly when slow flow caused by ICA stenosis leads to signal dropout, which is mistaken for very high-grade stenosis or complete ICA occlusion. Therefore, when no ICA stenosis is identified on MRA, no further testing is necessary, but when significant stenosis is detected, imaging with another noninvasive modality should be obtained (duplex Doppler imaging or CTA).46 In the case when MRA suggests complete occlusion, CTA, gadolinium-enhanced three-dimensional MRA, or conventional angiography should be used as confirmatory tests, despite the high sensitivity and specificity of MRA in complete ICA occlusion reported in some studies.47 MRA also has utility in detecting intracranial atherostenotic disease but, again, tends to overestimate the degree of stenosis and should be followed by digital subtraction angiography for better spatial definition and determination of whether the lesion is amenable to interventional radiological approaches. Other pathologies detectable by MRA are extracranial dissection, Takayasu’s arteritis, and moyamoya syndrome. MRA is usually followed by digital subtraction angiography when intervention is planned.
Assessing Cerebral Tissue Perfusion
A variety of imaging methods are available to address questions about perfusion. PET scans can quantitate CBF, cerebral blood volume, cerebral metabolic rate of oxygen and OEF, but are expensive, not widely available, and require injection of a radioactive tracer. Single photon emission computed tomography (SPECT) can only estimate relative CBF and also requires injection of a radionuclide agent. Neither PET scanning nor SPECT is practical for making decisions in the acute stroke setting, but they nonetheless have a role in decisions about revascularization procedures in patients with chronic occlusive disease. For example, PET scanning is being used in an ongoing study of extracranial-intracranial bypass surgery in patients with stroke and carotid occlusion (Carotid Occlusion Surgery Study, described later). Xenon CT scanning is another reliable test for quantifying CBF and can be performed at the time of the initial head CT scanning, but the technique has lost ground despite its reliability because of the need for xenon, its intolerance to head motion, and the development of perfusion MRI and CT scanning.
Other Laboratory Tests
The initial laboratory evaluation of an ischemic stroke patient serves three main purposes:
ACUTE TREATMENT
Emergency Room Evaluation
Although symptom recognition and stroke awareness are important limiting factors for timely stroke evaluation, studies have also shown that triage is often inadequate and have identified significant in-hospital delays for physician evaluation and for obtaining and interpreting initial CT scans.48,49 Dedicated acute stroke teams and an algorithm for emergency evaluation of acute stroke in the emergency department are necessary to overcome these obstacles.
Evaluation by emergency room physicians has been validated by multiple studies and should improve as more community hospitals assume an active role in treating acute stroke.50,51 In many smaller hospitals, where a consulting neurologist might not be immediately available, the emergency room physician ultimately evaluates and treats those patients in the crucial first few hours after ictus.
Adequate Oxygenation
Despite promising results with animal models52,53 and anecdotal reports of clinical efficacy, there is currently no definitive evidence to support the use of supplemental oxygen therapy, despite its widespread use in victims of acute stroke.54 When hypoxemia is present, it should be treated aggressively because it is extremely detrimental to penumbral brain tissue. All efforts in the emergency room should be geared toward avoiding further neuronal loss through transformation of the ischemic penumbra into infarct.
Blood Pressure Management
Many patients with acute stroke have an elevated blood pressure in the first few hours to days after the ictus. The levels may be above 200 mm Hg for systolic blood pressure but usually normalize without treatment within a few days. In view of the current notion of a viable ischemic penumbra and loss of local vessel tone in the area surrounding an acute infarction (where cerebral vascular resistance is less responsive to changes in CPP), it is imperative that no attempt is made to reduce the blood pressure in the acute peristroke period. Moreover, in chronically hypertensive individuals, the upper and lower limits of the autoregulatory curve are shifted upward, and therefore blood pressure reduction to within the normal range may extend infarction into penumbral regions. Two important exceptions are made: (1) for patients eligible for intravenous tissue-type plasminogen (tPA), because current guidelines require that systolic blood pressure be less than 185 mm Hg and diastolic pressure less than 110 mm Hg, and (2) patients with evidence of ongoing or imminent end-organ damage, such as pulmonary edema, acute myocardial infarction, acute aortic dissection, or acute renal insufficiency.55 In such patients, careful gradual blood pressure lowering can be attempted with parenteral medications such as β blockers (labetalol) or calcium channel blockers (nicardipine). When possible, these agents are preferred over direct venodilators such as nitroprusside or nitroglycerin. Research has focused on the dependence of peri-infarct brain perfusion on systemic blood pressure and its relevance for short- and long-term recovery. Investigators using SPECT found a negative relationship between blood pressure reduction and improvement in CBF,56 but the sample size was too small to extrapolate the results to the general stroke population. In two prospective studies, investigators found deleterious effects of acute blood pressure lowering on outcomes at 21 days57 and at 3 months, respectively.58
In the debate on how to better manage blood pressure in the peristroke period, a new concept has arisen: pharmacologically induced hypertension in patients in whom acute, reversible flow failure is suspected on clinical grounds or from perfusion MRI/CT scan. The approach seems most plausible for patients with acute large-vessel stenosis or occlusion, in whom penumbral tissue would benefit from increased perfusion and further recruitment of collateral channels. However, except for small pilot studies,59 no universally accepted protocol is in place, and this strategy cannot be recommended outside of a clinical trial.
Glycemic Control
The notion that hyperglycemia is detrimental to ischemic brain tissue is not new.60 Only since 2000, however, have the concepts of neuronal damage and infarct expansion been demonstrated for hyperglyamia in humans by neuroimaging61 and functional outcomes studies.62 In the National Institute of Neurological Disorders and Stroke (NINDS) recombinant tissue plasminogen activator (rt-PA) trial, hyperglycemia was identified as an independent predictor of worse outcomes as well as a risk factor for ICH complicating treatment with thrombolytics.63 Hyperglycemia also seems to reduce the effect of thrombolysis, probably by making the ischemic penumbra less amenable to the benefits of reperfusion.64 The effects of hyperglycemia are likely to predispose to acute clinical worsening65 and possibly play a significant role in long-term recovery and mortality.66 Although there is nearly consensus on the detrimental effects of hyperglycemia in the acute stroke period, more studies are needed to determine its optimal management. The only large multicenter trial to date to address the safety of aggressive glycemic control was the United Kingdom Glucose Insulin Trial (GIST-UK). The investigators concluded that continuous infusion of glucose/potassium/insulin solutions is feasible and safe in the peristroke period.67 A smaller group of patients has been reported in whom tight glycemic control was maintained with a continuous insulin infusion, but symptomatic, readily reversible hypoglycemia occurred in 20% of the patients.68 It is currently not known whether aggressive control of blood glucose translates into short- or long-term benefits or whether there is increased risk of hypoglycemic episodes. Despite the lack of current guidelines and formal recommendations, it seems reasonable that the patients with acute stroke, particularly those eligible for thrombolytic treatment, should undergo aggressive correction of hyperglycemia.
Temperature Control
Although hypothermia seems plausible as a neuroprotective strategy, studies are still scarce, and no clear recommendations can be made at this point. Animal studies have reliably demonstrated adverse outcomes associated with hyperthermia in the peristroke period, but only as recently as 2000 have objective data in humans shown an effect of elevated temperature on clinical outcomes.69 Two large studies demonstrated the benefit of induced hypothermia in hypoxic-ischemic brain damage after cardiac arrest, but there are no comparable studies on the effects of hypothermia on focal ischemic lesions.70,71 It is reasonable to recommend control of peristroke pyrexia with the usual measures available in current clinical practice, acetaminophen and cooling blankets, until more data become available.
Recanalization Strategies
Intravenous Thrombolysis
With the demonstration of efficacy of intravenous thrombolytics in acute myocardial infarction, it was only a matter of time until a similar approach was investigated in the setting of acute ischemic stroke. On the basis of successful animal studies, the first large trials to employ intravenous thrombolytics were conducted with streptokinase administered within 6 hours of stroke onset.72,73 Both studies revealed excess mortality in the groups treated with streptokinase, especially when streptokinase was combined with aspirin in the acute setting. In 1996, the Australian Streptokinase Study (ASK) investigated the efficacy of streptokinase within 3 hours and between 3 and 6 hours of stroke onset. The results showed worse outcomes for those receiving streptokinase within 3 and 6 hours of stroke onset and a significant decrease in mortality in patients treated within 3 hours in comparison with those treated between 3 and 6 hours. This, however, was not translated into a significant clinical benefit for thrombolysis at 3 months in comparison with placebo.74 In view of these results, streptokinase was abandoned, and attention shifted to rt-PA, a more selective, fibrin-dependent plasmin generator.
Results from multiple animal studies, which showed efficacy of rt-PA for recanalization and with lower rates of ICH than streptokinase, paved the way for clinical trials in humans. The first large, randomized, multicenter clinical trial to evaluate the use of rt-PA in acute ischemic stroke was the European Cooperative Acute Stroke Study (ECASS). Patients were enrolled within 6 hours of their ischemic stroke and were randomly assigned to received rt-PA (alteplase) at 1.1 mg/kg or placebo. The primary outcomes analyzed were the Barthel index (a measure of functional impairment in the activities of daily living) and the modified Rankin scale (a measure of neurological disability) at 3 months (Table 42-3).
| Grade | Description |
|---|---|
| 0 | No symptoms |
| 1 | No significant disability despite symptoms: able to carry out all usual duties and activities |
| 2 | Slight disability: unable to carry out all previous activities but able to look after own affairs without assistance |
| 3 | Moderate disability: requiring some help but able to walk without assistance |
| 4 | Moderate-severe disability: unable to walk without assistance and unable to attend to own bodily needs without assistance |
| 5 | Severe disability: bedridden, incontinent, requiring constant nursing care |
| 6 | Dead |
Even though the overall rate of ICH was not different between the two groups, the rate of large ICH was significantly higher in the treated group. No definite benefit was observed for the tPA group, but a post hoc analysis revealed a statistically insignificant treatment effect in patients treated within 3 hours, which suggested that this time window should be further explored.75
In 1995, the NINDS rt-PA Stroke Study group published the results of their trial.76 It was a randomized, placebo-controlled, multicenter trial that enrolled patients within 3 hours of the onset of an acute ischemic stroke who were then randomly assigned to receive either intravenous rt-PA (0.9 mg/kg) or placebo. There were strict exclusion criteria to ensure adequate patient selection and to improve the odds of a favorable result (Table 42-4).
TABLE 42-4 Inclusion and Exclusion Criteria for rt-PA Administration According to the National Institute of Neurological Disorders and Stroke Study
aPTT, activated partial thromboplastin time; CT, computed tomography; INR, international normalized ratio; rt-PA, recombinant tissue plasmin activator.
Adapted from Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med 1995; 333:1581-1587.
The dose of rt-PA chosen was lower than that for ECASS, and the blood pressure exclusion criteria were stricter (systolic blood pressure <185 mm Hg and diastolic blood pressure <110 mm Hg) (Table 42-5). Endpoints were defined in two parts: Part 1 required an improvement in the first 24 hours of more than 4 points in the National Institutes of Health Stroke Score (NIHSS) scale. Part 2 required improvement at 3 months on the Barthel Index (score = 95 to 100), the modified Rankin Scale (score = 1), and the Glasgow Outcome Scale (score = 1). The results of part 1 did not show a significant difference between the two groups. In part 2, benefit from tPA was observed in all outcome measures: Patients treated with tPA were 30% to 50% more likely to have no or minimal disability at 3 months than were those treated with placebo.76 The benefit was maintained regardless of stroke subtype and was still present 6 months and 1 year after stroke.77 The rate of symptomatic ICH was 6.4% in the tPA-treated individuals, in comparison with 0.6% in the placebo arm, but the mortality rates at 3 months and 1 year were not different between the two groups. Predictors of better outcome were treatment within 90 minutes78 (Fig. 42-12) and decrease in NIHSS score at 24 hours.79

Rights were not granted to include this table in electronic media. Please refer to the printed book.
From Adams, HP, Adams RJ, Brott T, et al: Guidelines for the early management of patients with ischemic stroke. Stroke 2003; 34:1056-1083.

Independent risk factors for symptomatic ICH were the severity of the stroke (measured by the NIHSS score), brain edema, or mass effect at baseline CT scan.80 Later studies showed that elevated blood glucose level at the time of treatment was also independently associated with higher risk of ICH.63 In 1996, on the basis of these favorable results, the U.S. Food and Drug Administration (FDA) approved the use of intra venous tPA for acute ischemic stroke within 3 hours of onset in patients who met inclusion/exclusion criteria.
After a post hoc analysis of ECASS demonstrated benefit in patients treated within 3 hours of stroke onset, a second trial (ECASS II) was conducted. The changes in the original protocol included a lower dose of tPA (0.9 mg/kg) and the exclusion of patients with CT evidence of large infarction (more than one third of the MCA territory involved, diffuse swelling). Other exclusion criteria were similar to those in the NINDS trial. The final analysis showed no significant difference between the treatment and the placebo recipients when the favorable outcome of a modified Rankin Scale score of 0 or 1 was considered (minimal or no disability). However, a post hoc analysis dichotomizing patients as either functionally dependent (modified Rankin Scale score > 2) or independent (modified Rankin Scale score = 2) found a significant 8.3% absolute difference in favor of the treatment recipients.41 As observed in the NINDS trial, the rate of symptomatic ICH was higher in the treatment recipients (8.8%) than in the placebo recipients (3.4%), but the 3-month mortality rates were similar.
Intravenous Thrombolysis Beyond the 3-Hour Period
The Alteplase Thrombolysis for Acute Noninterventional Therapy in Ischemic Stroke (ATLANTIS) study was designed in an attempt to expand the narrow window for intravenous tPA use.81 The study tested the safety and effectiveness of intravenous tPA (0.9 mg/kg) within 5 hours of stroke onset. The primary endpoint was excellent neurological recovery (NIHSS score = 1) at day 90. The tPA dose and the other inclusion and exclusion criteria were similar to those of the NINDS trial. The result was negative; tPA treated patients had higher rates of symptomatic ICH and mortality at 90 days. The delayed time window drove the negative result, as only 15% of patients were treated within 3 hours of onset.
A metaanalysis of pooled data from the NINDS, ATLANTIS, and ECASS trials has been performed. A total of 2775 patients treated with either placebo or intravenous thrombolysis within 360 minutes of stroke onset were analyzed. The odds ratios of a favorable outcome at 3 months were 2.81 for the patients treated within 90 minutes and 1.55 for those treated within 91 to180 minutes. There was no significant difference in mortality between patients treated within 270 minutes, but the mortality rate among patients given thrombolytics more than 270 minutes after stroke onset was in excess of 45%. Hemorrhage rate did not appear to be related to the time for thrombolysis but was related to age and the use of thrombolytics as opposed to placebo.82 Of note, this metaanalysis, like a Cochrane review,82a also suggested some benefit in patients treated between 180 and 270 minutes after stroke onset (odds ratio, 1.4). Nevertheless, intravenous thrombolysis is currently not recommended more than 3 hours after stroke onset. It is clear that 3 hours is not a magic number, and more physiological measures are sorely needed to optimally select patients for recanalization therapy. There are almost certainly patients in whom recanalization and completion of infarction have occurred within 3 hours and who would therefore not benefit from intravenous tPA and only be placed at unnecessary risk for hemorrhage. Conversely, there may be other patients, with persistent occlusion and large areas of misery perfusion, who could still benefit from recanalization more than 3 hours after stroke onset. The critical measures are likely to be the demonstration of both large-artery occlusion and the presence of a substantial volume of penumbral tissue beyond the infarct core.
A way forward is to use the combination of DWI, PWI, and MRA to provide a rapid assessment of the ischemic infarct core (DWI), the presence of arterial occlusion (MRA), and tissue at risk, defined as an area of reduced perfusion that extends beyond the boundaries of the diffusion abnormality. This is known as PWI-DWI mismatch. The concept of PWI-DWI mismatch has recently been modified for two reasons. First, as noted in the “Ischemic stroke” section, there is a spectrum of reversible ischemia from benign oligemia to impending necrosis. Second, DWI abnormalities are not always indicative of irreversible ischemia. Thus, the new definition of the penumbra (tissue at risk) excludes a rim of benign oligemia and includes some of the DWI core with high ADC values.83
A recent magnetic resonance study of the evolution of DWI- and PWI-observed lesion volumes, from acute to subacute time points, showed a contraction of the PWI-observed lesion by 85% and an expansion of the DWI-observed lesion by 136%.84 The Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET) pilot study showed improved recanalization, reduced DWI expansion, and penumbral salvage in patients who received intravenous tPA within 6 hours.61 The magnetic resonance surrogate measures were correlated with clinical outcome measures. These theoretical considerations, observational studies, and pilot studies have led to five ongoing studies (EPITHET; Diffusion-weighted imaging Evaluation For Understanding Stroke Evolution [DEFUSE]; Desmoteplase in Acute Ischemic Stroke Trial [DIAS]; MR and Recanalization of Stroke Clots Using Embolectomy [MR RESCUE]; and ReoPro Retavase Reperfusion of Stroke Safety Study [ROSIE]) evaluating tPA or mechanical thrombolysis on the basis of penumbral dynamics. Preliminary data are favorable,85 but it is necessary to wait until the trials are completed because many intuitively appealing ideas for stroke treatment have been shown to be untenable after completion of properly randomized trials.
Thrombolysis in the Community
With the approval of intravenous tPA for acute stroke treatment, multiple groups tried to ascertain whether the benefits observed in the NINDS trial were reproducible at the community care level. In 2000, Katzan and colleagues published the experience at 29 hospitals in the Cleveland area and determined the rate of use of thrombolytics, related complications, and protocol violations.86 Their results showed that only 1.8% of all patients admitted with a diagnosis of ischemic stroke received intravenous tPA. Moreover, of the treated patients, 15.7% had symptomatic ICH, and the NINDS protocol was violated in 50% of the cases. The analysis also showed higher in-hospital mortality rates in the tPA-treated patients than in the placebo-treated patients.86 Protocol violations were also high in another community-based study87; not surprisingly, there was a higher rate of symptomatic ICH than in the original NINDS trial.
With increased awareness of its indications and the importance to adhere strictly to the published protocol, tPA use was reanalyzed in the Cleveland area 2 years after the first study. A higher percentage (2.7%) of patients received treatment; they represented 18.8% of all eligible patients. Protocol violations occurred for 19% of patients, and symptomatic ICH occurred in 6.4%,88 a significant improvement from the original study. Familiarity with the use of thrombolytics and better adherence to published guidelines were likely reasons for the observed improvement. However, the overall tPA usage rates were not impressive, and this raises the possibility that specialized centers, analogous to designated trauma centers, might be a better option.
In 2001 Grotta and associates published the Houston experience with tPA for acute stroke. In a period of 4 years, 15% of all patients presenting with an acute ischemic stroke were treated with intravenous tPA; almost a third of those were treated within 2 hours after onset. The rate of symptomatic ICH was 4.5%, and protocol violations occurred for 13% of all treated patients. This study proved that a well-organized team approach and adherence to currently accepted protocols are key to successful implementation of intravenous thrombolysis.89
In 2002, a group from Canada evaluated their experience with over 1000 patients treated with intravenous tPA over a period of 30 months. Their results were similar to the NINDS trial, in which 30% of the treated patients had no or minimal deficits at 90 days. The ICH rate was 4.6%, and protocol violations were observed in 15% of cases.90 Other studies have yielded similar results.
Intra-Arterial Thrombolysis
Despite the inherent plausibility of an intra-arterial approach, only one placebo-controlled, double-blind multicenter trial has been completed. The Prolyse in Acute Cerebral Thromboembolism Trial (PROACT) was published in two parts. In PROACT I, recombinant prourokinase, a highly fibrin-selective agent, was administered intra-arterially, and its effects were compared with those of placebo in patients with an angiographically documented MCA (M1 or M2) occlusion within 6 hours of stroke onset. Inclusion criteria included a NIHSS score of at least 4, as well as the other NINDS tPA trial inclusion/exclusion criteria, including those for blood pressure. Heparin was administered concomitantly for 4 hours in both groups, and mechanical clot disruption was not allowed in either group. Forty patients were enrolled, and the rate of angiographically proven recanalization was 57.7% for the treatment recipients and 14.3% for the placebo recipients. Among patients who received higher doses of intravenous heparin together with the intra-arterial recombinant prourokinase, recanalization and symptomatic ICH rates were higher (81.8% and 27%, respectively). The recanalization rate was lower in the patients given lower dose heparin (40%), but the rate of symptomatic ICH was also significantly lower (6%). Overall, the rate of ICH was 15.4% among patients receiving thrombolytics and 14.3% among those receiving placebo. The trend toward effectiveness in this phase 2 trial prompted the second part of the trial (PROACT II), which tested clinical efficacy. The inclusion criteria in PROACT II were similar to those of PROACT I except, as in to ECASS, patients with CT hypodensity representing involvement of more than a third of the MCA territory were excluded. The baseline severity of strokes was higher than in the NINDS tPA trial (mean NIHSS scores = 17 versus 14) and the median time from onset to treatment was 5.3 hours. The primary outcome was the proportion of patients with a modified Rankin Scale score of 2 or less (independent) at 90 days. The study found a 15% absolute benefit in the patients who received treatment (intra-arterial recombinant prourokinase followed by intravenous heparin for 4 hours) in comparison with those who received placebo (only intravenous heparin for 4 hours). This translates into a number-needed-to-treat of seven. Symptomatic ICH occurred in 10% of recombinant prourokinase recipients and 2% of the control group, and recanalization rates at 2 hours were 66% for the treatment recipients and 18% for the control group. No difference in mortality rates was observed between the two groups.91,92
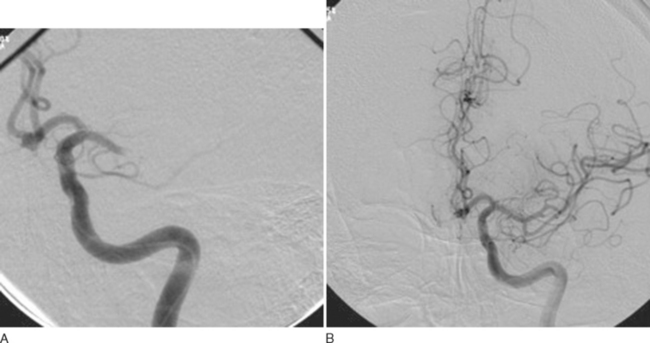
Data from the PROACT suggest that patients outside the currently accepted treatment window for intravenous tPA could benefit from an aggressive interventional approach. However, the results of PROACT II did not suffice for FDA approval. Nevertheless, this approach is likely to be particularly useful for those patients with either ICA or MCA occlusion (Fig. 42-13A and B), in whom the rate of recanalization with systemic thrombolysis appears to be much lower than that with locally delivered thrombolytics.
Combined Intravenous and Intra-Arterial Thrombolysis
The rate of complete recanalization of ICA or MCA occlusion after intravenous tPA appears to be low, ranging from 10% to 30%. Moreover, in many instances, recanalization is followed by reocclusion and neurological worsening. The Emergency Management of Stroke (EMS) pilot study aimed at establishing the safety and feasibility of lower dose intravenous thrombolysis (0.6 mg/kg) within 3 hours, followed by intra-arterial tPA for presumed MCA or ICA occlusion (NIHSS score > 5). The rationale behind this approach is to combine speed of delivery of intravenous tPA with the higher recanalization rate and extended time window of intra-arterial tPA. The study results showed that the combination of intravenous and intra-arterial thrombolysis led to higher recanalization rates and more bleeding complications, without appreciable effect on overall outcome.93 A follow-up study, the Interventional Management of Stroke (IMS) trial, enrolled 44 patients to the intravenous/intra-arterial tPA treatment group. The rate of symptomatic ICH was 6.3%. In comparison with the historical data from the NINDS intravenous tPA trial, in which 36% of patients with a NIHSS score higher than 10 achieved a good outcome, 56% of such patients in the IMS study had a good outcome, which is suggestive of added benefit from intra-arterial therapy in patients with probable large-vessel occlusion.94 Currently, however, such an approach is experimental and is not recommended outside a clinical trial setting.
SECONDARY PREVENTION
Antiplatelet Therapy
Aspirin
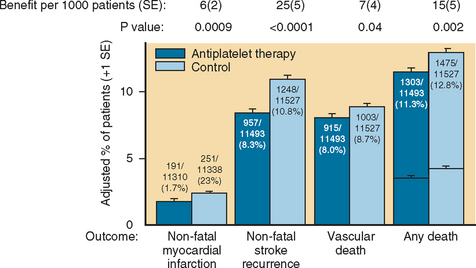
Aspirin use is a time-honored strategy for secondary prevention of ischemic stroke in both the acute and chronic settings. Multiple clinical trials have been conducted, the most important being the French, British, European, and Canadian trials. They revealed a benefit ranging from 20% to 30% relative risk reduction for stroke and vascular death. The Antithrombotic Trialists Collaboration addressed the role of aspirin among patients at high risk for vascular events, including those with a prior ischemic stroke. The benefit of aspirin was modest, with a 22% relative risk reduction for vascular events (stroke, myocardial infarction, vascular death) in patients with a prior TIA/stroke. The absolute benefit of aspirin, across all doses, was a modest 2.5% (Fig. 42-14).95
Whereas most studies agree on the benefit of aspirin, its optimal dose has been a matter of some controversy. The Dutch trial showed equal benefit for very low and moderate doses of salicylates (30 mg/day versus 283 mg/day),95a a finding similar to that in the British trial,95b in which very different dosing schedules were used (300 mg/day versus 1200 mg/day). In view of the potential side effects, mainly from gastrointestinal bleeding and ulceration, it is reasonable to prescribe low-dose aspirin (81 mg/day) for long-term secondary stroke prevention, although efficacy at this low dose of aspirin for long-term stroke prevention has not been determined. The International Stroke Trial (IST) evaluated patients within 48 hours of an acute stroke and established the benefit of 300 mg/day of aspirin in reducing recurrent strokes at 14 days and death or dependency at 6 months.96 The Chinese Acute Stroke Trial (CAST) evaluated the use of 160 mg/day of aspirin within 48 hours of suspected ischemic stroke (13% of patients had no confirmatory CT scan before randomization).97 Analysis of the results showed a 12% relative risk reduction of death or recurrent stroke within 4 weeks for the aspirin recipients. Together, the IST and CAST trials showed an improved outcome for patients receiving aspirin within 48 hours of a stroke, with 13 fewer dead or dependent patients in the months after the event.96,97
Aspirin is often discontinued for elective surgery or bleeding complications. There have been concerns for some time that discontinuation of aspirin, and other antiplatelet agents, might lead to a transient prothrombotic “rebound” effect that increases the risk of ischemic stroke. This concern is supported by a case-control study whose results suggest that aspirin discontinuation can increaset the risk of ischemic stroke, especially in patients with multiple cardiovascular risk factors.97a
Combination Antiplatelet Therapy
Clopidogrel and Aspirin
To specifically answer the question whether the clopidogrel-aspirin combination is more effective in reducing the risk of stroke recurrence than aspirin alone, the Management of Atherothrombosis with Clopidogrel in High-Risk Patients with Recent Transient Ischemic or Ischemic Stroke (MATCH) trial was conducted. The 7599 patients already taking clopidogrel (75 mg/day) who had had a recent TIA or stroke (and at least one other risk factor) were randomly assigned to receive aspirin or placebo. The duration of treatment and follow-up was 18 months. The primary endpoint was a composite of ischemic stroke, myocardial infarction, vascular death, or hospitalization for acute ischemic event (including TIA, angina, or peripheral arterial disease). The primary endpoint was reached by 15.7% of patients receiving clopidogrel/aspirin and by 16.7% of those receiving aspirin alone. This relative risk reduction of 6.4% was offset by the significantly higher rates of life-threatening bleeding episodes in the patients receiving combination therapy (2.6% versus 1.3%). The degree to which the MATCH results apply to secondary stroke prevention in general is questionable for a number of reasons. First, 70% of the patients enrolled had diabetes. The study was designed to enroll patients with multiple stroke risk factors to increase the background stroke recurrence rate, which was high in both groups. It is possible that this population has such severe small-vessel disease that inhibiting platelets was not sufficient to prevent lacunar infarction. Indeed, 54% of the recurrent strokes were lacunar. The question, extrapolated from CURE, of whether patients with TIA or stroke from LAA might benefit from aspirin and clopidogrel, was not really addressed by MATCH. For example, in the Clopidogrel and Aspirin for Reduction of Emboli in Symptomatic Carotid Stenosis (CARESS) trial, dual antiplatelet therapy with clopidogrel and aspirin was superior to aspirin alone for reduction of embolic signals in the MCA in patients with symptomatic carotid stenosis. This was admittedly a small study with a surrogate marker, but the results do tend to support the idea that LAA might be more amenable to combination therapy than is small-vessel disease.
Dipyridamole and Aspirin
The European Stroke Prevention Study II (ESPS-II) was a double-blind, randomized investigation of the different efficacy of placebo in comparison with aspirin (25 mg twice daily), extended-release dipyridamole (200 mg twice daily), and the combination of aspirin and extended-release dipyridamole. The outcomes of stroke and/or death were measured over a follow-up period of 2 years. The results showed a stroke rate of 15.8% in the placebo recipients, 13.2% in the extended-release dipyridamole recipients, 12.9% in the aspirin recipients, and 9.9% in the combination recipients. The combination therapy yielded a 23% relative risk reduction in comparison with either aspirin or dipyridamole alone and a 37% relative risk reduction in comparison with placebo.98 Moderate to severe bleeding events were almost twice as common in the aspirin recipients; mortality rates were not statistically different among the four groups. The combination is therefore a valid first-line option for secondary prevention after a noncardioembolic stroke and is being increasingly used in clinical practice. The main limitation of this therapy is the frequent headaches observed at the initiation of therapy (present in as many as 40% of patients). However, a slow dose-escalation schedule and use of over-the-counter pain medications usually lead to resolution of headaches within 2 weeks.
Anticoagulants
Heparin and Heparinoids
However, despite these arguments, the use of unfractionated or low-molecular-weight heparin (LMWH) in the acute stroke setting is not supported by current evidence. In 1997, the IST evaluated the use of subcutaneous unfractionated heparin in the acute stroke setting. Chosen outcomes measures were death at 14 days and death and dependency at 6 months. The investigators used a factorial design in which patients were given subcutaneous unfractionated heparin (5000 IU or 12,500 IU twice a day), placebo in addition to aspirin (300 mg/day), or placebo. Treatment was started within 48 hours of an acute stroke and continued for 14 days. Among heparin-treated patients, there were nonsignificantly fewer deaths within 14 days than among patients who received no heparin (9.0% versus 9.3%, respectively), corresponding to 3 fewer deaths for every 1000 patients treated. Also, there was no difference between the groups in dead or dependent patients at 6 months. Patients receiving heparin had significantly fewer recurrent ischemic strokes within 14 days (2.9% versus 3.8%), a finding offset by a similar increase in hemorrhagic strokes (1.2% versus 0.4%). Heparin use was also associated with a greater risk of any extracranial hemorrhage and hemorrhages necessitating transfusion; the higher risks were present with higher heparin doses. The nonsignificant decrease in deaths at 14 days was similar to that observed with the use of aspirin alone. Indeed, the reduction at 6 months in death or nonfatal recurrent stroke was greater in the aspirin group than in the heparin group, predominantly because of lower rates of ICH in the aspirin-treated group. The conclusion drawn from this study was that heparin is of no obvious benefit in the prevention of either stroke recurrence, death, or disability, and therefore there is no support for the use of high-dose subcutaneous heparin (exceeding 5000 IU twice daily) in the acute stroke setting.96 The major limitations of the IST study were the open-label nature of the study, absence of initial CT scan in up to one third of all patients, lack of anticoagulation parameter control in the heparin-treated patients, and lack of weight adjustment of the heparin dose.
In the late 1980s, Duke and colleagues investigated the role of intravenous unfractionated heparin or placebo up to 24 hours after an acute, stable, noncardioembolic ischemic stroke. Rates of stroke progression, death, and recovery were similar in the two groups.99 Many smaller studies and case series have been published since, with most results suggesting no benefit from acute use of intravenous unfractionated heparin.
The use of LMWH in acute ischemic stroke has also been investigated in small studies, the majority of which revealed no significant benefit of subcutaneous LMWH in the prevention of recurrence or death after an acute ischemic stroke. Most notable, the Tinzaparin in Acute Ischemic Stroke Trial (TAIST) evaluated the efficacy of two different doses of subcutaneous LMWH (tinzaparin), in comparison with aspirin, for 10 days after an acute ischemic stroke. The results revealed no differences in outcome of clinical deterioration and stroke recurrence in 15 days or in modified Rankin Scale score at 6 months between the two groups, in all subtypes of ischemic stroke, including cardioembolic stroke. There was a higher rate of symptomatic intracranial hemorrhage in the tinzaparin-treated patients and a higher rate of venous thromboembolism in the aspirin-treated patients.100
The Trial of ORG 10172 in Acute Stroke Treatment (TOAST) enrolled approximately 1300 patients to receive either placebo or intravenous danaparoid (LMWH) for 7 days after an acute ischemic stroke.101 The primary endpoint was a favorable outcome (based on scores on the Glasgow Outcome Scale and the Barthel index) at 3 months. The level of anticoagulation was corrected according to anti–factor Xa levels. The outcomes of neurological worsening and recurrent ischemic stroke at 7 days and the 3-month outcomes were similar in the two groups. When the subgroups were evaluated according to stroke subtype, the early recurrence rates for patients with cardioembolic stroke were similar in the two treatment groups. However, in contrast to the TAIST trial, the only patients who showed a trend toward benefit from intravenous danaparoid were those with LAA.
When embolic stroke subtypes are considered, the results of the Heparin in Acute Embolic Stroke (HAEST) trial provide some insight.102 It was a double-blind, randomized investigation of the effectiveness of LMWH (dalteparin) given subcutaneously in comparison with aspirin (160 mg/day) in patients with an acute stroke presumably secondary to atrial fibrillation. The patients were assigned to receive either dalteparin or aspirin within 30 hours of the ischemic stroke and were treated for 10 days. The primary outcome of recurrent stroke within 14 days occurred in 7.5% of the aspirin-treated patients and 8.5% of the dalteparin-treated patients, with a trend toward more ICH in the dalteparin-treated group. This study emphasizes that the recurrence after a cardioembolic stroke is lower than previously thought and that emergency anticoagulation may be more harmful than helpful.
In summary, the role for acute heparin use after an ischemic stroke, including those with a cardioembolic mechanism, is not supported by current evidence.103 In some cases, however, scenarios may arise that were not systematically investigated by existing trials. In such scenarios, the underlying pathophysiology (arterial dissection, “stuttering” TIAs, acutely symptomatic critical arterial stenosis, large left ventricular thrombus) might nonetheless justify anticoagulation. In these circumstances, a careful weighing of the (at best theoretical) benefit of acute anticoagulation against its risks should be the rule.
Oral Anticoagulation
Cardioembolic Strokes
In view of the modest benefit of aspirin in both primary and secondary stroke prevention in patients with atrial fibrillation, various studies investigated warfarin as a more effective prophylactic agent. Multiple randomized clinical trials established the effectiveness of warfarin for both primary and secondary stroke prevention in patients with chronic atrial fibrillation.104 When all results are considered, warfarin has shown relative risk reductions of 59% in primary prevention and 68% in secondary prevention, with overall acceptable bleeding risk ranging from 0.3% to 2.0% per year. These results are put into perspective when the absolute risk reductions for warfarin (3% per year for primary prevention and 8% per year for secondary prevention) are compared with those of aspirin (1.5% and 2.5%, respectively). The recommended intensity of anticoagulation in nonvalvular atrial fibrillation is an international normalized ratio (INR) between 2.0 and 3.0, given the increased risk of bleeding with higher levels.105 On the basis of the trial results, the choice to administer anticoagulants to patients with atrial fibrillation (either chronic or paroxysmal) should be based on stratification of their stroke risk. All patients with a prior cardioembolic stroke or TIA should undergo long-term anticoagulation unless a strong contraindication is present. For primary prevention, the patients who would benefit the most from long-term anticoagulation are the ones with the risk factors mentioned previously. For patients without risk factors (lone atrial fibrillation) and/or younger than 65, aspirin is justifiable, in view of the low incidence of ischemic stroke (1% per year).
Alternative oral anticoagulants are under investigation for stroke prevention in patients with atrial fibrillation. The Stroke Prevention Using an Oral Thrombin Inhibitor in Atrial Fibrillation (SPORTIF III) trial was a study of ximelagatran, a non–vitamin K-dependent direct thrombin inhibitor.106 It was an open-label study that randomly assigned patients with atrial fibrillation and one or more other stroke risk factors to receive either dose-adjusted warfarin (target INR of 2.0 to 3.0) or fixed-dose ximelagatran (36 mg twice daily). Results showed that patients receiving ximelagatran had fewer ischemic strokes or systemic emboli than did those receiving warfarin (1.6% versus 2.3% per year, respectively), with a relative risk reduction of 29%. Rates of disabling or fatal stroke, mortality, and major bleeding were similar in both groups, but combined minor and major hemorrhage rates were lower with ximelagatran than with warfarin (29.8% versus 25.8% per year). The major side effect from ximelagatran in this study was elevation in serum alanine aminotransferase level. The major advantages of this direct thrombin inhibitor over warfarin are the fixed dose with more predictive effects without the need for constant monitoring and the lack of significant food or drug interactions. Liver toxicity is the main limitation, and the use of ximelagatran is not currently approved in the United States.
There is less evidence for the use of chronic anticoagulation in other cardiac conditions such as valvular heart disease and severely reduced left ventricular function. Dilated cardiomyopathy predisposes to thrombus formation in as many as 50% of affected patients and is therefore a significant risk factor for cardioembolic stroke (3% to 4% per year). Currently, two large trials (Warfarin vs. Aspirin in Reduced Ejection Fraction [WARCEF] and Warfarin and Antiplatelet Trial in Chronic Heart Failure [WATCH]) are evaluating the benefits of chronic anticoagulation over the use of aspirin in the rates of stroke and death in patients with severely reduced ejection fraction (<35%).107 Previous myocardial infarction confers a risk of stroke of 1% to 2% per year, with higher rates in the first 3 months after the event and particularly with anterior wall myocardial infarction. The modest risk reduction offered by conventional dosages of warfarin balanced against the increased risk of hemorrhagic complications does not allow the recommendation of long-term anticoagulation in unselected patients after a myocardial infarction. In patients with an anterior wall myocardial infarction, in whom the risk of mural thrombus formation can be as high as 40%, and in patients with documented ventricular thrombus, in whom the annual risk of systemic embolization may be as high as 20%, anticoagulation with warfarin is indicated. Despite the lack of randomized studies, current practice is for patients to receive anticoagulants for 6 months after an anterior wall myocardial infarction with documented intraventricular thrombus. Resolution of the thrombus should be assessed by serial echocardiographic studies and the length of therapy adjusted accordingly. In cases of an anterior wall myocardial infarction but no documented thrombus, a shorter period of anticoagulation is reasonable.
Noncardioembolic Stroke
Traditionally, neurologists used long-term anticoagulation in patients with noncardioembolic stroke in whom secondary prevention with aspirin was deemed ineffective (“aspirin failure”). The poor prognosis associated with strokes secondary to intracranial atherosclerosis despite the use of antiplatelet agents has also been a common justification for long-term anticoagulation. Two studies have shed light onto these two issues. The WARSS was the first large trial to investigate whether warfarin was superior to aspirin for the secondary prevention of noncardioembolic ischemic stroke. It was a multicenter, double-blind, randomized trial that enrolled more than 2200 patients to receive either aspirin (325 mg/day) or warfarin for a target INR of 1.4 to 2.8. At the 2-year follow up, the primary endpoint of recurrent stroke or death was observed in 16% of patients receiving aspirin and 17.8% of those receiving warfarin; these percentages represented a statistically insignificant difference. The beneficial effects of warfarin seemed to be mostly in the INR range of 1.5 without significant additive benefits above that range. The endpoint was achieved in a similar proportion of patients in both groups regardless of the underlying stroke etiology, with a comparable rate of bleeding complications in the two groups (2.22%/year for warfarin and 1.49%/year for aspirin). The WARSS investigators concluded that aspirin and warfarin are equivalent in efficacy in the secondary prevention of noncardioembolic strokes, with a comparable relatively low risk of bleeding complications.108
The risk of recurrent stroke in large-vessel intracranial atherosclerosis can be as high as 10% per year. The retrospective portion of the Warfarin Aspirin for Symptomatic Intracranial Disease (WASID) trial evaluated the efficacy of anticoagulation in comparison with antiplatelet therapy for the secondary prevention of strokes attributable to posterior circulation intracranial atherosclerosis.109 The results of the retrospective analysis, showing recurrence rates of 10.4% per year in the aspirin recipients and 3.6%/year in the warfarin recipients, led to a prospective trial. The prospective part of the WASID study was a double-blind randomized trial that enrolled patients with symptomatic, angiographically proven, high-degree (>50% diameter reduction) intracranial stenosis. Patients were assigned to receive either warfarin (INR goal of 2.0 to 3.0) or aspirin (1300 mg/day). Determination of recurrent stroke or vascular death at 3-year follow-up was planned, but the study was prematurely interrupted after a mean follow-up of only 1.8 years because interim analysis showed higher rates of major hemorrhagic complications in the warfarin-treated group (8.3% for warfarin versus 3.2% for aspirin). The rates for all strokes or vascular death were similar in both groups (21.8% for warfarin versus 22.1% for aspirin). These results argue against the widespread practice of chronic anticoagulation for patients with symptomatic intracranial atherosclerosis.18 However, they also attest to the inadequacy of either medication.
Further evidence for the nonsuperiority of warfarin over aspirin was provided by a Cochrane review.110 A metaanalysis of five trials (>4000 patients) that compared anticoagulation with antiplatelet therapy for the secondary prevention of noncardioembolic strokes showed similar rates of recurrent stroke at all levels of anticoagulation (INR ranges 1.4 to 2.8, 2.1 to 3.6, and 3.0 to 4.5) in comparison with antiplatelet therapy. Major bleeding rates were significantly higher in patients receiving high-intensity anticoagulation but similar in the patients receiving low- and medium-intensity anticoagulation in comparison with antiplatelet therapy.
Strokes Associated with Antiphospholipid Antibodies
The best therapy, antiplatelet or anticoagulation, for patients with stroke who also have positive results on tests for antiphospholipid antibody is still a matter of debate. A WARSS substudy, the Antiphospholipid Antibodies and Stroke Study (APASS), sought to answer this question. Of the 1770 patients from WARSS who had blood tests for antiphospholipid antibodies, 41% were found to have positive results. These patients showed no differences in the rates of death or thrombotic events in comparison with those who had negative results for antibodies. Of more importance, there was no difference in outcome of recurrent stroke or death when the treatment with either aspirin or warfarin was considered.111 However, the WARSS population had an average age of more than 50, and the study was not powered to answer questions of antibody titer or subtype. It remains possible that patients younger than 45 with high titers of immunoglobulin G antiphospholipid antibody, who evidence suggests are at risk for arterial stroke as well as venous thrombosis, might benefit from anticoagulation.
Stroke Associated with a Patent Foramen Ovale
In patients with a cryptogenic stroke and a documented PFO, the best strategy for secondary prevention has been a matter of intense debate. The results of a prospective study evaluating young patients (<55 years old) with cryptogenic stroke and a PFO found the risk of recurrent stroke to be 15% over 4 years, with aspirin, when the PFO was associated with an ASA. This recurrence rate was dramatically higher than for isolated PFO112 and suggested that aspirin is inadequate. The Patent foramen ovale In Cryptogenic Stroke Study (PICSS), another substudy of WARSS, is the only randomized, double-blind study to investigate the best medical management of PFO.113 Six hundred and thirty patients from WARSS underwent a transesophageal examination; a PFO was found in 39% of patients with cryptogenic strokes and in 29.9% of patients with other known causes. Besides reinforcement of the notion of a PFO being a risk factor for a first-ever stroke, other aspects of the PICSS results are worth noting:
Blood Pressure Control for Secondary Stroke Prevention
Many studies have shown the benefit of blood pressure control in primary stroke prevention, but only a few studies have assessed the effect of blood pressure control on secondary stroke prevention. The Heart Outcomes Prevention Evaluation (HOPE) study was a double-blind, randomized trial that investigated the role of angiotensin-converting enzyme (ACE) inhibitor (ramipril) as a protective strategy for patients with evidence of coronary artery disease, peripheral vascular disease, or prior stroke. Diabetes and at least one vascular risk factor were required for inclusion in the study. The study had a 2 × 2 factorial design in which 9297 patients were randomly assigned to receive ramipril (10 mg/day), vitamin E (400 IU/day), or placebo. Patients were monitored for over 5 years. Primary endpoints were myocardial infarction, stroke, and vascular death. Approximately 11% of patients in both ramipril and placebo groups had a history of prior stroke, and approximately 80% had a history of coronary artery disease. There was a statistically significant reduction in all primary outcomes in the ramipril recipients, in comparison with the placebo recipients (14% versus 17.8%). Stroke was the primary outcome in 3.4% of patients receiving ramipril and 4.9% of those receiving placebo. Fatal strokes occurred in 0.4% and 1.0%, and ischemic strokes in 2.2% and 3.4%, of patients receiving ramipril and the placebo groups, respectively.114 Only a small part of the benefit was attributable to reduction in blood pressure, inasmuch as only a minority of patients were hypertensive at baseline. A benefit for ramipril was observed regardless of the degree of blood pressure reduction.115 These results, although more applicable to primary stroke prevention in patients at high risk, make a strong case for long-term ACE inhibitor therapy as part of secondary stroke prevention, perhaps independent of baseline blood pressure.
The Perindopril Protection Against Recurrent Stroke Study (PROGRESS) was conducted to investigate the effects of a blood-pressure regimen with the ACE inhibitor perindopril on hypertensive and nonhypertensive patients with prior stroke or TIA. The study enrolled 6105 patients with a history of a prior stroke or TIA in the preceding 5 years, and they were randomly assigned to receive placebo or active treatment consisting of perindopril (4 mg/day) with or without the addition of the thiazide diuretic indapamide (2 to 2.5 mg/day), at the discretion of the treating physician (58% received combination therapy and 42% received perindopril alone). More individuals with baseline hypertension were identified in PROGRESS than in the HOPE trial. The primary outcome was fatal or nonfatal stroke. After 4 years of follow-up, 10% of patients receiving perindopril and 14% of patients receiving placebo had had a stroke, with an annual rate of stroke of 2.7% and 3.8%, respectively. This reduction occurred with both ischemic and hemorrhagic strokes, and fewer patients receiving perindopril had had fatal or disabling strokes. The association of perindopril with indapamide conferred a 43% relative risk reduction in comparison with placebo, whereas the use of perindopril alone had no discernible effect in stroke risk reduction. This medication combination was also associated with a more significant blood pressure reduction than the one produced with perindopril alone (12/5 mm Hg versus 5/3 mm Hg), and the benefit was observed in patients with or without hypertension at baseline.116 Whether the results observed were secondary to blood pressure control or to a direct effect of both the ACE agent and the diuretic remains unknown. The PROGRESS and HOPE studies suggest that ACE inhibitors, together with antithrombotics, should be considered first-line agents for secondary stroke prevention.
Cholesterol-Reducing Strategies
Most prospective studies of cholesterol-lowering strategies have focused on ischemic heart disease. Only a few trials have focused on statin agents and secondary stroke prevention. The Heart Protection Study (HPS) was a randomized trial in which simvastatin (40 mg/day) was compared with placebo for prevention of vascular events (myocardial infarction, stroke, vascular death) in patients at high risk between 40 and 80 years old. High risk was defined as having prior occlusive arterial disease, diabetes, and elevated cholesterol. More than 3000 of the total 20,000 patients had a history of ischemic stroke or TIA. Over 5 years, the mean total cholesterol was reduced by 20%, and the odds for all ischemic strokes were reduced by 25% and for fatal strokes by 19%. All major vascular events were decreased by 30% in the simvastatin recipients. Of importance, ICH was observed with equal frequency in the two groups.117 In other smaller trials (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial [ALLHAT]; Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm [ASCOT-LLA]; Cholesterol and Recurrent Events [CARE]; Long-term Intervention with Pravastatin in Ischaemic Disease [LIPID]; and Pravastatin or Atorvastatin Evaluation and Infection Therapy [PROVE IT]), researchers also examined the effect of various statins on prevention of vascular events, including strokes. Most showed a 15% to 25% reduction in the incidence in TIA or stroke. However, although these results show that statins are effective as a primary prevention strategy for TIA/stroke in patients at high risk, their effects on secondary prevention cannot be fully appreciated, because most patients enrolled in such studies were stroke free. Another unexpected result from these studies is the possibility that statins might be as effective in patients with normal cholesterol levels as they are in patients at high risk with elevated total cholesterol, elevated low-density lipoprotein, and low high-density lipoprotein levels. Current recommendations are that all patients with prior stroke and elevated cholesterol levels should take a statin. For patients with a prior TIA/stroke and other vascular risk factors, the use of statins also seems indicated. More data are necessary to determine whether a statin is indicated for patients with TIA/stroke but without elevated cholesterol or other vascular risk factors.
Control of Other Potential Risk Factors
Elevated Homocysteine Level
Elevated blood homocysteine levels have been observed in patients with coronary artery disease, as well as in patients with a prior TIA/stroke. Whether these represent a marker of advanced atherosclerosis or have a causal role in the pathogenesis of vascular disease remains to be determined. Blood homocysteine levels are inversely related to the dietary intake of folic acid, vitamin B6, and vitamin B12. However, whether dietary supplementation with these vitamins to lower homocysteine levels decreases cardiovascular risk is uncertain. In the Vitamin Intervention for Stroke Prevention (VISP) trial, in which more than 10,000 patients with a prior TIA/stroke were given vitamin supplementation (folate, vitamin B6, and vitamin B12) or placebo, results showed that vitamin supplementation did not reduce the risk of recurrent stroke over 2 years.118 The Vitamins To Prevent Stroke (VITATOPS) study was also an evaluation of the effect of vitamin supplementation on a large population of patients with prior TIA/stroke. Preliminary results revealed no significant reduction in markers of vascular inflammation (C-reactive protein) or hypercoagulability (prothrombin fragments, D-dimer), despite reduction in total homocysteine levels.119,120 The benefits, if any, on stroke recurrence will be known only after completion of the study. Routine use of folic acid, cobalamin, and pyridoxine supplementation is not currently recommended for secondary stroke prevention.
Hormone Replacement Therapy
The use of hormone replacement therapy, with either estrogen or a combination of estrogen and progesterone, has increased dramatically since the mid-1990s. The main indication is prevention of postmenopausal osteoporosis. Accumulating evidence, however, points to an increased risk of cardiovascular disease. In the Women’s Estrogen for Stroke Trial (WEST), estrogen replacement was compared with placebo in more than 600 menopausal women with a history of a prior TIA/stroke. After a mean follow-up period of 2.8 years, estrogen was not shown to provide any protection against minor strokes (odds ratio = 1.0) but was shown to be associated with increased risk of fatal strokes in comparison with placebo (odds ratio = 2.8). Even though the numbers of minor strokes were comparable in both groups, strokes in the estrogen recipients were associated with slightly worse neurological and functional deficits.121 Those results were reinforced by the Women’s Health Initiative (WHI) study. The main goal of the WHI study was to investigate primary prevention of cardiovascular disease in postmenopausal women and hormone replacement therapy. Nevertheless, it demonstrated an odds ratio for ischemic stroke of 1.5, independent of all other stroke risk factors.122 On the basis of these results, hormone replacement therapy is not recommended as primary prevention for stroke and certainly should not be used in women with a history of prior TIA/stroke. In postmenopausal women without cardiovascular risk factors, its use may be considered if truly indicated.
Surgical and Interventional Strategies for Secondary Stroke Prevention
Carotid Endarterectomy
In the early 1980s, resection of the stenotic portion of the ICA, or carotid endarterectomy (CEA), was a common surgical procedure. The conviction that CEA was an optimal form of secondary stroke prevention arose from recognition of the fact that large-artery atherosclerotic strokes are associated with a high rate of early recurrence. However, in the middle to late 1980s, the uncertainty about the efficacy of CEA in comparison with optimal medical therapy and the failure of the extracranial-intracranial bypass trial led to a decrease in enthusiasm for the CEA. However, in 1991, the North American Symptomatic Carotid Endarterectomy (NASCET) trial published its results.123 There was an absolute risk reduction in the incidence of recurrent stroke of 17% for CEA in comparison with medical treatment in symptomatic patients with ICA stenosis of more than 70% diameter reduction. In absolute numbers, strokes recurred within 2 years in 26% of the medically treated patients and 9% of the surgically treated patients; this was a 66% relative risk reduction for CEA. The risks of major or fatal ipsilateral stroke in the surgically and medically treated patients were 2.5% and 13.1%, respectively.16 When all centers were considered, the overall surgical morbidity and mortality rate was 5.8%. In their latest results, the NASCET investigators found that in patients with a moderate degree of ICA stenosis (50% to 69% diameter reduction), the benefit was more modest, with a 5-year rate of stroke in the surgically and medically treated patients of 15.7% and 22.2%, respectively.124
The Veterans Affairs Cooperative Study yielded similar results, with an absolute risk reduction of 11.7% for surgery versus medical therapy at 1 year of follow-up. Patients had ICA stenosis that ranged from 50% to 99% diameter reduction, and, as in the NASCET trial, the results showed greater benefit for patients with higher degrees of stenosis. The perioperative morbidity and mortality rate was approximately 5%.125 The European Carotid Surgery Trial (ECST) was conducted around the same time as the two other CEA trials. There were some important differences between ECST and NASCET, which led to slightly different results. The main methodological differences were in the angiographic measurement of the degree of stenosis (the ECST method overestimated degree of stenosis in comparison with the NASCET method) and in the definition of the stroke outcome (ECST defined stroke as a deficit persistent for >7 days, and NASCET defined stroke as a neurological deficit persisting for >24 hours). The final ECST results for 3-year risk of stroke or death showed a 26.5% risk in medically treated patients with a symptomatic ICA stenosis of more than 80% diameter reduction, and a 14.9% risk for patients undergoing CEA; the absolute benefit for surgery was 11.6%. The risk of major stroke or death complicating surgery was 7%.126–128 Reanalysis of the ECST results, with degree of stenosis recalculated with the NASCET method, revealed an absolute risk reduction of 21.2% for 5-year risk of stroke or death after CEA in comparison with medical treatment in patients with ICA stenosis of 70% to 99% diameter reduction.128
Symptomatic complete ICA occlusion is a special case that is not amenable to CEA. No benefit was observed for extracranial-intracranial bypass surgery in unselected patients in a trial in 1985,129 which failed to show a benefit from the surgical procedure as a result of high complication rates. However, the elevated risks of recurrent stroke in this patient population (as high as 14% per year in some studies), justifies another trial with better selection of candidates based on preoperative evaluation of hemodynamic reserve, because it is thought that recurrent stroke in these patients is caused by flow failure rather than embolus. The Carotid Occlusion Surgery Study is currently under way and aims to determine whether extracranial-intracranial bypass is superior to medical therapy in patients with recently symptomatic unilateral ICA occlusion and with evidence on PET scan for misery perfusion with increased OEF. Although these results are not available, risk factor modification, antiplatelet medications, and aggressive cholesterol control are the recommended treatment modalities for these patients.
Angioplasty and Stenting
Angioplasty and stenting are currently considered preferable to surgery in cases of ICA stenosis secondary to prior radiation therapy in the neck, previous CEA with recurrent stenosis, contralateral ICA occlusion, and surgically inaccessible high bifurcation of the common carotid artery.130 In addition, angioplasty and stenting might be preferable in patients with multiple comorbid conditions who might not be ideal surgical candidates. The efficacy of angioplasty and stenting in comparison with CEA remains to be determined by ongoing trials (Carotid Revascularization Endarterectomy versus Stent Trial [CREST]; Stent-protected Percutaneous Angioplasty of the Carotid vs. Endarterectomy [SPACE]; International Carotid Stenting Study of the Carotid and Vertebral Artery Transluminal Angioplasty Study [CAVATAS-2]).
The first large study to compare the safety and efficacy of CEA and angioplasty was the Carotid and Vertebral artery Transluminal and Angioplasty Study (CAVATAS). About 500 patients were randomly assigned to undergo either CEA or angioplasty (with or without stent implantation) and were monitored for 1 year. Major perioperative morbidity, mortality, and stroke rates were similar in the two groups at 30 days, and stroke rates were similar at 1 year, although the rate of restenosis was greater for the patients who underwent angioplasty. Of importance, angioplasty was followed by stent implantation, a procedure thought necessary to maintain vessel patency, in only 26% of the patients undergoing endovascular treatment. In addition, no distal protection device to minimize embolization was used in this trial.131 CAVATAS-2 is currently under way and should answer some questions with regard to equivalency to CEA, when both stent deployment and distal protection devices are used during the endovascular procedure.
The recent Stenting and Angioplasty with Protection in Patients at High Risk for Endarterectomy (SAPPHIRE) trial was designed to determine the noninferiority of angioplasty with stent implantation with ICA stenosis in comparison with CEA. The SAPPHIRE study showed a 7.9% absolute benefit in the outcome of stroke/myocardial infarction/death at 30 days and ipsilateral stroke or death at 1 year in favor of angioplasty with stent implantation over CEA in patients considered to be at high risk (with associated medical comorbid conditions). For the patients undergoing angioplasty and stent implantation, the perioperative morbidity and mortality rate was about half of that observed after CEA. However, a major problem with this study is that almost 70% of the patients were asymptomatic. It is already known from the CEA trials for high-grade asymptomatic carotid stenosis (see Chapter 46) that patients with medical comorbid conditions should not undergo operation because they need to be alive at 5 years to show the modest benefit. Thus, it is not clear why asymptomatic patients with associated medical comorbid conditions should undergo angioplasty and stent implantation at all. What are needed are studies of angioplasty with stent implantation versus CEA as a secondary prevention strategy in patients without comorbid conditions and those at high risk. The ongoing CREST should answer the former question.
Angioplasty and stent implantation for symptomatic intracranial stenoses remain investigational and controversial. The seriousness of intracranial atherosclerosis was made plain by the WASID study (discussed previously), which showed, over a 1.8-year follow-up period, that ischemic stroke recurrence rate for intracranial stenoses of more than 50% diameter reduction ranged from 17% to 20%, almost half being fatal or disabling strokes. In view of this poor prognosis and the lack of better secondary prevention strategies, small studies have been performed to investigate safety, feasibility, and efficacy of interventional procedures in patients with symptomatic intracranial atherosclerosis. A review of a single center’s experience with intracranial stent implantation over a 6-year period revealed a very high overall periprocedural complication rate of 28% for patients in whom the procedure was deemed a last resort.132 Unfortunately, the idea of last resort assumes good natural history data for medically treated symptomatic intracranial stenoses; however, such data are not available. There have been two studies of elective stent implantation with symptomatic M1 stenosis. The rationale for pursuing this treatment is that symptomatic M1 stenosis carries an annual stroke recurrence rate of about 8%. One study enrolled 14 patients, and the other enrolled 40. The former had morbidity and mortality rates of 33.3% and 8.3%, respectively; the latter, 10% and 2.5%. In the larger study, M1 lesions were classified on the basis of their location, morphology, and access. All three factors were relevant to periprocedural complication rates and efficacy at 10-month median follow-up.
Algra A, De Schryver ELLM, van Gijn J, et al. Oral anticoagulants versus antiplatelet therapy for preventing further vascular events after transient ischemic attack or minor stroke of presumed arterial origin. Cochrane Database Syst Rev. 3, 2006. CD001342
Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med. 1991;325:445-453.
Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363:768-774.
Hart RG, Palacio S, Pearce LA. Atrial fibrillation, stroke, and acute antithrombotic therapy: analysis of randomized clinical trials. Stroke. 2002;33:2722-2727.
Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581-1587.
1 American Heart Association. Heart Diseases and Stroke Statistics—2005 Update. Dallas: American Heart Association, 2005;16-20.
2 Saver JL, Kidwell C. Neuroimaging in TIAs. Neurology. 2004;62(8, Suppl 6):S22-S25.
3 Rovira A, Rovira-Gols A, Pedraza S, et al. Diffusion-weighted MR imaging in the acute phase of transient ischemic attacks. AJNR Am J Neuroradiol. 2002;23:77-83.
4 Albers GW, Caplan LR, Easton JD, et al. TIA working group (2002). Transient ischemic attack—proposal for a new definition. N Engl J Med. 2002;347:1713-1716.
5 Ay H, Koroshetz W, Benner T, et al. Transient ischemic attack with infarction: a unique syndrome? Ann Neurol. 2005;57:679-686.
6 Hill MD, Yiannakoulias N, Jeerakathil T, et al. The high risk of stroke immediately after transient ischemic attack. Neurology. 2004;62:2015-2020.
7 Lovett JK, Dennis MS, Sandercock PAG, et al. Very early risk of stroke after a first transient ischemic attack. Stroke. 2003;34:e138-e142.
8 Lisabeth LD, Ireland JK, Risser JMH, et al. Stroke risk after transient ischemic attack in a population-based setting. Stroke. 2004;35:1842-1846.
9 Johnston SC, Gress DR, Browner WS, et al. Short-term prognosis after emergency department diagnosis of TIA. JAMA. 2000;284:2901-2906.
10 Baron JC. Mapping the ischaemic penumbra with PET: implications for acute stroke treatment. Cerebrovasc Dis. 1999;9:193-201.
11 Wong EW, Pullicino PM, Benedict R. Deep cerebral infarcts extending to the subinsular region. Stroke. 2001;32:2272-2277.
12 Wong KS, Gao S, Chan YL, et al. Mechanisms of acute cerebral infarctions in patients with middle cerebral artery stenosis: a diffusion-weighted imaging and microemboli monitoring study. Ann Neurol. 2002;52:74-81.
13 Jackson C, Sudlow C. Are lacunar strokes really different? A systematic review of differences in risk factor profiles between lacunar and nonlacunar infarcts. Stroke. 2005;36:891-901.
14 Alexandrov AV, Felberg RA, Demchuck AM, et al. Deterioration following spontaneous improvement: sonographic findings in patients with acutely resolving symptoms of cerebral ischemia. Stroke. 2000;31:915-919.
15 Eliasziw M, Kennedy J, Hill MD, et al. Early stroke risk after a transient ischemic attack in patients with internal carotid artery disease. CMAJ. 2004;170:1105-1109.
16 North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med. 1991;325:445-453.
17 Arenillas JF, Molina CA, Montaner J, et al. Progression and clinical recurrence of symptomatic middle cerebral artery stenosis: a long-term follow-up transcranial Doppler ultrasound study. Stroke. 2001;32:2898-2904.
18 Chimowitz MI, Lynn MJ, Howlett-Smith H, et al. Comparison of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med. 2005;352:1305-1316.
19 Gasecki AP, Ferguson GG, Eliasziw M, et al. Endarterectomy for severe carotid artery stenosis after a nondisabling stroke: results from the North American Symptomatic Carotid Endarterectomy Trial. J Vasc Surg. 1994;20:288-295.
20 Gan R, Sacco RL, Kargman DE, et al. Testing the validity of the lacunar hypothesis: the Northern Manhattan Stroke Study experience. Neurology. 1997;48:1204-1211.
21 Ay H, Oliveira-Filho J, Buonanno FS, et al. Diffusion weighted imaging identifies a subset of lacunar infarction associated with embolic source. Stroke. 1999;30:2644-2650.
22 Kato H, Izumiyama M, Izumiyama K, et al. Silent cerebral microbleeds on T2*-weighted MRI: correlation with stroke subtype, stroke recurrence, and leukoaraiosis. Stroke. 2002;33:1536-1540.
23 Wardlaw JM. What causes lacunar stroke? J Neurol Neurosurg Psychiatry. 2005;76:617-619.
23a Cerebrovasc Dis. 2004;17(Suppl 5):52.
23b Macdonald RL, Kowalczuk A, Johns L. Emboli enter penetrating arteries of monkey brain in relation to their size. Stroke. 1995;26:1247-1250.
24 Wardlaw JM, Dennis MS, Warlaw CP. Imaging appearance of the symptomatic perforating artery in patients with lacunar infarction: occlusion or other vascular pathology? Ann Neurol. 2001;50:208-215.
24a Sen S, Laowatana S, Lima J, Oppenheimer SM. Risk factors for intracardiac thrombus in patients with recent ischemic cerebrovascular events. J Neurol Neurosurg Psychiatry. 2004;75:1421-1425.
24b Hagen PT, Scholz DG, Edwards WD. Incidence and size of patient foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts. Mayo Clin Proc. 1984;59:17-20.
25 Overell JR, Bone I, Lees KR. Interatrial septal abnormalities and stroke: a metaanalysis of case-control studies. Neurology. 2000;55:1172-1179.
25a Karttunen V, Hiltunen L, Rasi V, et al. Factor V Leiden and prothrombin gene mutation may predispose to paradoxical embolism in subjects with patient foramen ovale. Blood Coagul Fibrinolysis. 2003;14:261-268.
25b Pezzini A, Del Zotto E, Magoni M. Inherited thrombophilic disorders in young adults with ischemic stroke and patent foramen ovale. Stroke. 2003;34:28-33.
26 Mcleod MR, Amarenco P, Davis SM, et al. Atheroma of the aortic arch: an important and poorly recognised factor in the aetiology of stroke. Lancet Neurol. 2004;3:408-414.
27 Agmon Y, Khanderia BK, Meissner I, et al. Relation of coronary artery disease and cerebrovascular disease with atherosclerosis of the thoracic aorta in the general population. Am J Cardiol. 2002;89:262-267.
28 Amarenco P, Cohen A, Tzourio C, et al. Atherosclerotic disease of the aortic arch and the risk of ischemic stroke. N Engl J Med. 1994;331:1474-1479.
29 Tunick PA, Perez JL, Kronzon I. Protruding atheromas in the thoracic aorta and systemic embolization. Ann Intern Med. 1991;115:423-427.
30 Chowdhury D, Wardlaw JM, Dennis MS. Are multiple acute small subcortical infarctions caused by embolic mechanisms? J Neurol Neurosurg Psychiatry. 2004;75:1416-1420.
31 Dziewas R, Konrad C, Drager B, et al. Cervical artery dissection—clinical features, risk factors, therapy and outcome in 126 patients. J Neurol. 2003;250:1179-1184.
32 Rubinstein SM, Peerdeman SM, van Tulder MW, et al. A systematic review of the risk factors for cervical artery dissection. Stroke. 2005;36:1575-1580.
32a Arboix A, Lopez-Grau M, Casasnovas C, et al. Clinical study of 39 patients with atypical lacunar syndrome. J Neurol Neurosurg Psychiatry. 2006;77:381-384.
33 Paciaroni M, Caso V, Milia P. Isolated monoparesis following stroke. J Neurol Neurosurg Psychiatry. 2005;76:805-807.
34 Schmahmann JD. Vascular syndromes of the thalamus. Stroke. 2003;34:2264-2278.
35 Godefroy O, Dubois C, Debachy B, et al. Vascular aphasias: main characteristics of patients hospitalized in acute stroke units. Stroke. 2002;33:702-705.
36 Kumral E, Bayulkem G, Evyapan D, et al. Spectrum of anterior cerebral artery territory infarction: clinical and MRI findings. Eur J Neurol. 2002;9:615-624.
37 Schneider R, Gautier JC. Leg weakness due to stroke. Site of lesions, weakness patterns and causes. Brain. 1994;117(Pt 2):347-354.
38 Kumral E, Kisabay A, Atac C. Lesion patterns and etiology of ischemia in superior cerebellar artery territory infarcts. Cerebrovasc Dis. 2005;19:283-290.
39 Caplan LR. “Top of the basilar” syndrome. Neurology. 1980;30:72-79.
40 Barber PA, Demchuk AM, Zhang J, et al. Validity and reliability of a quantitative computed tomography score in predicting outcome of hyperacute stroke before thrombolytic therapy. ASPECTS Study Group. Alberta Stroke Programme Early CT Score. Lancet. 2000;355:1670-1674.
41 Hacke W, Kaste M, Fieschi C, et al. Randomised double-blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II). Second European-Australasian Acute Stroke Study Investigators. Lancet. 1998;352:1245-1251.
42 Davis S, Fisher M, Warach S, editors. Magnetic Resonance Imaging in Stroke. Cambridge, UK: Cambridge University Press, 2003.
43 Li F, Silva MD, Sotak CH, et al. Temporal evolution of ischemic injury evaluated with diffusion, perfusion, and T2-weighted MRI. Neurology. 2000;54:689.
44 Buskens E, Nederkoorn P, Tineke BW, et al. Imaging of carotid arteries in symptomatic patients: cost-effectiveness of diagnostic strategies. Radiology. 2004;233:101-112.
45 Long A, Lepoutre A, Corbillon E, et al. Critical review of non- or minimally invasive methods (duplex ultrasonography, MR- and CT-angiography) for evaluating stenosis of the proximal internal carotid artery. Eur J Vasc Endovasc Surg. 2002;24:43-52.
46 Johnston DCC, Goldstein LB. Clinical carotid endarterectomy decision making: noninvasive vascular imaging versus angiography. Neurology. 2001;56:1009-1015.
47 El-Saden SM, Grant EG, Hathout GM, et al. Imaging of the internal carotid artery: the dilemma of total versus near total occlusion. Radiology. 2001;221:301-308.
48 Wester P, Radberg J, Lundgren B, et al. Factors associated with delayed admission to hospital and in-hospital delays in acute stroke and TIA: a prospective, multicenter study. Stroke. 1999;30:40-48.
49 Morris DL, Rosamond W, Madden K, et al. Prehospital and emergency department delays after acute stroke. The Genentech Stroke Presentation Survey. Stroke. 2000;31:2585-2590.
50 Kothari RU, Brott T, Broderick JP, et al. Emergency physicians: accuracy in the diagnosis of stroke. Stroke. 1995;26:2238-2241.
51 Ferro JM, Pinto AN, Falcao I, et al. Diagnosis of stroke by the nonneurologist. A validation study. Stroke. 1998;29:1106-1109.
52 Flynn EP, Auer RN. Eubaric hyperoxemia and experimental cerebral infarction. Ann Neurol. 2002;52:566-572.
53 Singhal AB, Dijkhuizen RM, Rosen B, et al. Normobaric hyperoxia reduces MRI diffusion abnormalities and infarct size in experimental stroke. Neurology. 2002;58:945-952.
54 Pancioli AM, Bullard MJ, Grulee ME, et al. Supplemental oxygen use in ischemic stroke patients: does utilization correspond to need for oxygen therapy? Arch Intern Med. 2002;162:49-52.
55 Goldstein L. Blood pressure management in patients with acute ischemic stroke. Hypertension. 2004;43:137-141.
56 Lisk DR, Grotta JC, Lamki LM, et al. Should hypertension be treated after acute stroke? A randomized controlled trial using single photon emission computed tomography. Arch Neurol. 1993;50:855-862.
57 Wahlgren NG, MacMahon DG, DeKeyser J, et al. Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischemic stroke. Cerebrovasc Dis. 1994;4:204-210.
58 Oliveira-Filho J, Silva SCS, Trabuco CC, et al. Detrimental effect of blood pressure reduction in the first 24 hours of acute stroke onset. Neurology. 2003;61:1047-1051.
59 Rordorf G, Koroshetz WJ, Ezzeddine MA, et al. A pilot study of drug-induced hypertension for treatment of acute stroke. Neurology. 2001;56:1210-1213.
60 Pulsinelli WA, Waldman S, Rawlinson D, et al. Moderate hyperglycemia augments ischemic brain damage: a neuropathologic study in the rat. Neurology. 1982:1239-1246.
61 Parsons MW, Barber A, Desmond PM, et al. Acute hyperglycemia adversely affects stroke outcome: a magnetic resonance imaging and spectroscopy study. Ann Neurol. 2002;52:20-28.
62 Baird TA, Parsons MW, Phanh T, et al. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke. 2003;34:2208-2214.
63 Bruno A, Levine SR, Frankel MR, et al. Admission glucose level and clinical outcomes in the NINDS rt-PA Stroke Trial. Neurology. 2002;59:669-674.
64 Alvarez-Sabin J, Molina C, Ribo M, et al. Impact of admission hyperglycemia on stroke outcome after thrombolysis: risk stratification in relation to time to reperfusion. Stroke. 2004;35:2493-2499.
65 Leigh R, Zaidat OO, Suri MF, et al. Predictors of hyperacute clinical worsening in ischemic stroke patients receiving thrombolytic therapy. Stroke. 2004;35:1903-1907.
66 Williams LS, Rotich J, Qi R, et al. Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke. Neurology. 2002;59:67-71.
67 Gray CS, Hildreth AJ, Alberti GKMM, et al. Poststroke hyperglycemia. Natural history and immediate management. Stroke. 2004;35:122-126.
68 Bruno A, Saha C, Williams LS, et al. IV Insulin during acute cerebral infarction in diabetic patients. Neurology. 2004;62:1441-1442.
69 Hajat CH, Hajat S, Sharma P. Effects of poststroke pyrexia on stroke outcome: a metaanalysis of studies in patients. Stroke. 2000;31:410-414.
70 Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
71 The Hypothermia After Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549-556.
72 Multicentre Acute Stroke Trial—Italy (MAST-I) Group. Randomised controlled trial of streptokinase, aspirin, and combination of both in treatment of acute ischaemic stroke. Lancet. 1995;346:1509-1514.
73 The Multicenter Acute Stroke Trial—Europe Study Group (MAST-E). Thrombolytic therapy with streptokinase in acute ischemic stroke. N Engl J Med. 1996;335:145-150.
74 Donnan GA, Davis SM, Chambers BR, et al. Streptokinase for acute ischemic stroke with relationship to time of administration: Australian Streptokinase (ASK) Trial Study Group. JAMA. 1996;276:961-966.
75 Hacke W, Kaste M, Fieschi C, et al. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. The European Cooperative Acute Stroke Study (ECASS). JAMA. 1995;274:1017-1025.
76 The NINDS rt-PA Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581-1587.
77 Kwiatkowski TG, Libman RB, Frankel M, et al. Effects of tissue plasminogen activator for acute ischemic stroke at one year. NINDS Recombinant Tissue Plasminogen Activator Stroke Study. N Engl J Med. 1999;10:1781-1787.
78 Marler JR, Tilley BC, Lu M, et al. Early stroke treatment associated with better outcome. The NINDS rt-PA Stroke Study. Neurology. 2000;55:1649-1655.
79 Broderick JP, Lu M, Kothari R, et al. Finding the most powerful measures of effectiveness of tissue plasminogen activator in the NINDS t-PA Stroke Trial. Stroke. 2000;31:2335-2341.
80 Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. NINDS rt-PA Stroke Study Group. Stroke. 1997;28:2109-2118.
81 Clark WM, Wissman S, Albers GW, et al. Recombinant tissue-type plasminogen activator (alteplase) for ischemic stroke 3 to 5 hours after symptom onset. The ATLANTIS study: a randomized controlled trial. JAMA. 1999;282:2019-2026.
82 The ATLANTIS, ECASS and NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363:768-774.
82a Wardlaw JM, Zoppo G, Yamaguchi T, Berge E. Thrombolysis for acute ischaemic stroke. Cochrane Database Syst Rev. (3):2003. CD000213
83 Kidwell CS, Alger JR, Saver JL. Beyond mismatch: evolving paradigms in imaging the ischemic penumbra with multimodal magnetic resonance imaging. Stroke. 2003;34:2729-2735.
84 Barber PA, Parsons MW, Desmond PM, et al. The use of PWI and DWI measures in the design of “proof-of-concept” stroke trials. J Neuroimaging. 2004;14:123-132.
85 Kidwell C, Starkman S, Jahan R, et al. Pretreatment MRI penumbral pattern predicts good clinical outcome following mechanical embolectomy [Abstract]. Stroke. 2004;35:294.
86 Katzan IL, Furlan AJ, Lloyd LE, et al. Use of tissue-type plasminogen activator for acute ischemic stroke: the Cleveland area experience. JAMA. 2000;283:1151-1158.
87 Lopez-Yunez AM, Bruno A, Williams L, et al. Protocol violations in community-based rTPA stroke treatment are associated with symptomatic intracerebral hemorrhage. Stroke. 2001;32:12-16.
88 Katzan IL, Hammer MD, Furlan AJ, et al. Quality improvement and tissue-type plasminogen activator for acute ischemic stroke. A Cleveland update. Stroke. 2003;34:799-800.
89 Grotta J, Burgin WS, El-Mitwalli A, et al. Intravenous tissue-type plasminogen activator therapy for ischemic stroke. Arch Neurol. 2001;58:2009-2013.
90 Hill MD, Buchan AM. Canadian Activase for Stroke Effectiveness Study (CASES). Can J Neurol Sci. 2001;28:232-238.
91 Del Zoppo GJ, Higashida RT, Furlan AJ, et al. PROACT: a phase II randomized trial of recombinant prourokinase by direct arterial delivery in acute middle cerebral artery stroke. Stroke. 1998;29:4-11.
92 Furlan AJ, Higashida RT Wechsler L, et al. Intra-arterial prourokinase for acute ischemic stroke—The PROACT II study: a randomized controlled trial. JAMA. 1999;282:2003-2011.
93 Lewandowski CA, Frankel M, Tomsick TA, et al. Combined intravenous and intra-arterial t-PA versus intra-arterial therapy for acute ischemic stroke: Emergency Management of Stroke (EMS) Bridging Trial. Stroke. 1999;30:2598-2605.
94 Tomsick T, Broderick JP, Pancioli AP, et al. Combined IV-IA t-PA treatment in major acute ischemic stroke [Abstract]. Stroke. 2002;33:359.
95 Antithrombotic Trialists’ Collaboration. Collaborative metaanalysis of randomized trials of antiplatelet therapy for the prevention of death, MI, and stroke in high risk patients. BMJ. 2002;324:71-86.
95a A comparison of two doses of aspirin (30 mg vs. 283 mg a day) in patients after a transient ischemic attack or minor ischemic stroke. The Dutch TIA Trial Study Group. N Engl J Med. 1991;325:1261-1266.
95b Farrell B, Godwin J, Richards S, Warlow C. The United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: final results. J Neurol Neurosurg Psychiatry. 1991;54:1044-1054.
96 International Stroke Trial Collaborative Group. The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19,435 patients with acute ischaemic stroke. Lancet. 1997;349:1569-1581.
97 Chinese Acute Stroke Trial collaborative group. CAST: randomised placebo-controlled trial of early aspirin use in 20,000 patients with acute ischaemic stroke. Lancet. 1997;349:1641-1649.
97a Maulaz AB, Bezerra DC, Michel P, Bogousslavsky J. Effect of discontinuing aspirin therapy on the risk of brain ischemic stroke. Arch Neurol. 2006;62:1217-1220.
98 Diener HC, Cunha L, Forbes C, et al. European Stroke Prevention Study 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci. 1996;143:1-13.
99 Duke RJ, Bloch RF, Fong HC, et al. Intravenous heparin for the prevention of stroke progression in acute partial stable stroke. Ann Intern Med. 1986;105:825-828.
100 Bath PM, Lindenstrom E, Boysen G, et al. Tinzaparin in Acute Ischemic Stroke (TAIST): a randomised aspirin-controlled trial. Lancet. 2001;358:702-710.
101 Low molecular weight heparinoid, ORG 10172 (danaparoid), and outcome after acute ischemic stroke: a randomized controlled trial. The Publications Committee for the Trial of ORG 10172 in Acute Stroke Treatment (TOAST) investigators. JAMA. 1998;279:1265-1272.
102 Berge E, Abdelnoor M, Nakstad PH, et al. Low molecular-weight heparin versus aspirin in patients with acute ischaemic stroke and atrial fibrillation: a double-blind randomised study. Lancet. 2000;355:1205-1210.
103 Hart RG, Palacio S, Pearce LA. Atrial fibrillation, stroke, and acute antithrombotic therapy: analysis of randomized clinical trials. Stroke. 2002;33:2722-2727.
104 Hart RG, Benavente O, McBride R, et al. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a metaanalysis. Ann Intern Med. 1999;131:492-501.
105 SPIRIT study: a randomized trial of anticoagulants versus aspirin after cerebral ischemia of presumed arterial origin. The Stroke Prevention in Reversible Ischemia Trial (SPIRIT) Study Group. Ann Neurol. 1997;42:857-865.
106 Olsson SB, Executive Steering Committee on behalf of the SPORTIF III Investigators. Stroke prevention with the oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with nonvalvular atrial fibrillation (SPORTIF III): randomised controlled trial. Lancet. 2003;362:1691-1698.
107 Pullicino PM, Halperin JL, Thompson JL. Stroke in patients with heart failure and reduced left ventricular ejection fraction. Neurology. 2000;54:288-294.
108 Mohr JP, Thompson JL, Lazar RM, et al. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med. 2001;345:1444-1451.
109 Chimowitz MI, Kokkinos J, Strong J, et al. The Warfarin-Aspirin Symptomatic Intracranial Disease Study. Neurology. 1995;45:1488-1493.
110 Algra A, De Schryver ELLM, van Gijn J, et al. Oral anticoagulants versus antiplatelet therapy for preventing further vascular events after transient ischemic attack or minor stroke of presumed arterial origin. Cochrane Database Syst Rev. 3, 2006. CD001342
111 Levine SR, Tilley BC, Thompson JL, et al. Antiphospholipid antibodies and subsequent thrombo-occlusive events in patients with ischemic stroke. JAMA. 2004;291:576-584.
112 Mas JL, Arquizan C, Lamy C, et al. Recurrent cerebrovascular events associated with patent foramen ovale, atrial septal aneurysms or both. N Engl J Med. 2001;345:1740-1746.
113 Homma S, Sacco RL, Di Tullio MR, et al. Effect of medical treatment in stroke patients with patent foramen ovale. Circulation. 2002;105:2625-2631.
114 Bosch J, Yusuf S, Pogue J, et al. Use of ramipril in preventing stroke: double blind randomized trial. BMJ. 2002;324:699-704.
115 Yusuf S, Sleight P, Pogue J, et al. Effects of angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145-153.
116 PROGRESS Collaborative group. Randomised trial of a perindopril-based blood pressure-lowering regimen among 6105 individuals with previous stroke or transient ischaemic attack. Lancet. 2001;358:1033-1041.
117 Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7-22.
118 Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004;291:565-575.
119 Dusitanond P, Eikelboom JW, Hankey GJ, et al. Homocysteine-lowering treatment with folic acid, cobalamin, and pyridoxine does not reduce blood markers of inflammation, endothelial dysfunction, or hypercoagulability in patients with previous transient ischemic attack or stroke: a randomized substudy of the VITATOPS trial. Stroke. 2005;36:144-146.
120 VITATOPS Trial Study Group. The VITATOPS (Vitamins to Prevent Stroke) Trial: rationale and design of an international, large, simple, randomised trial of homocysteine-lowering multivitamin therapy in patients with recent transient ischaemic attack or stroke. Cerebrovasc Dis. 2002;13:120-126.
121 Viscoli CM, Brass LM, Kernan WN, et al. A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med. 2001;345:1243-1249.
122 Wassertheil-Smoller S, Hendrix SL, Limacher M, et al. Effect of estrogen plus progestin on stroke in postmenopausal women. The Women’s Health Initiative: a randomized trial. JAMA. 2003;289:2673-2684.
123 North American Symptomatic Carotid Endarterectomy Trialist’s Collaborative Group. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med. 1998;339:1415-1425.
124 Mayberg MR, Wilson SE, Yatsu F, et al. Veterans Affairs Cooperative Studies Program 309 Trialist Group. Carotid endarterectomy and prevention of cerebral ischemia in symptomatic carotid stenosis. JAMA. 1991;266:3289-3294.
125 European Carotid Surgery Triallists Collaborative Group. MRC European Carotid Surgery Trial: interim results for symptomatic patients with severe (70–99%) or mild (0–29%) carotid stenosis. Lancet. 1991;337:1235-1243.
126 European Carotid Surgery Trialists’ Collaborative Group. Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Surgery Trial (ECST). Lancet. 1998;351:1379-1387.
127 Rothwell PM, Gutnikov SA, Warlow CP. Reanalysis of the final results of the European Carotid Surgery Trial. Stroke. 2003;34:514-523.
128 Rothwell PM, Eliasziw M, Gutnikov SA, et al. Sex difference in the effect of time from symptoms to surgery on benefit from carotid endarterectomy for transient ischemic attack and nondisabling stroke. Stroke. 2004;35:2855-2861.
129 Brott TG, Brown RD, Meyer FB, et al. Carotid revascularization for prevention of stroke: carotid endarterectomy and carotid artery stenting. Mayo Clin Proc. 2004;79:1197-1208.
130 Endovascular versus surgical treatment in patients with carotid stenosis in the Carotid and Vertebral Artery Transluminal Angioplasty Study (CAVATAS): a randomised trial. Lancet. 2001;357:1729-1737.
131 Yadav JS, Wholey MH, Kuntz RE, et al. Protected carotidartery stenting versus endarterectomy in high-risk patients. N Engl J Med. 2004;351:1493-1501.
132 SSYLVIA Study Investigators. Stenting of Symptomatic Atherosclerotic Lesions in the Vertebral or Intracranial Arteries (SSYLVIA): study results. Stroke. 2004;35:1388-1392.