Family history
1992–2000
2001–2010
With GN
95/908 (10.5 %)
77/822 (9.5 %)
With dialysis
42/908 (4.6 %)
58/822 (7.1 %)
Clinical phenotype of familial IgAN is generally similar to that of sporadic IgAN. There have been some reports of families clustering with Henoch-Schonlein purpura or thin basement membrane disease [18, 19], whereas clinical and pathological findings specific to familial IgAN have not been described.
3.3 Genetic Studies for the Pathogenesis of IgA Nephropathy
From decades ago, genetic analysis in sporadic IgAN had been performed. Initially, the majority of them were case-control association studies by using classical techniques, such as restriction fragment length polymorphism (RFLP) to measure allele frequencies of one or a few genetic polymorphisms of functional candidate [20–26]. Although these studies may have contributed to our better understanding of the mechanisms of the initiation, as well as progression of the disease, not all the results had been confirmed by replication studies, and majority of them were not convincing enough to influence the clinical diagnosis and practice of this disease.
Recently, using microarray DNA analysis methods, genome-wide association studies (GWAS) of sporadic IgAN have been performed using large cohorts of more than thousands of cases with IgAN and controls. These studies have identified multiple susceptibility loci, providing an insight into the genetic architecture of this disease [27–29]. They demonstrated strong associations of the major histocompatibility (MHC) locus and four non-HLA loci, including chromosome 1q32, a common deletion of the complement factor H-related CFHR3 and CFHR1 genes (CFHR3,1–delta); 8p23, the α-defensin (DEFA) gene cluster; 17p23 (including TNFSF13); and 22q12 (including HORMAD2 and several other genes).
More recently, by international collaboration, the largest GWAS has been performed in 2,747 biopsy-confirmed cases and 3,952 controls as the discovery cohorts (stage I), and subsequently, top signals, defined by P < 5 × 10−5, were genotyped in an additional 4,911 cases and 9,002 controls (stage II), followed by meta-analysis to identify genome-wide significant signals across the combined cohorts of 20,612 individuals. This two-stage design was adequately powered to detect ORs as small as 1.15–1.25. This study has identified six novel genome-wide significant associations including genes as follows: four in ITGAM–ITGAX (encoding leukocyte-specific integrin αX, a component of complement receptor 4 (CR4)), VAV3 (encoding a guanine nucleotide exchange factor for Rho GTPases that is important for B and T lymphocyte development and antigen presentation), and CARD9 (encoding caspase recruitment domain-containing protein 9, an adapter protein that promotes activation of NF-κB in macrophages) and two new independent signals at HLA–DQB1 and DEFA, replicating the nine previously reported signals, including known SNPs in the HLA–DQB1 and DEFA loci (Table 3.2). The cumulative burden of risk alleles was strongly associated with age at disease onset. Interestingly, most loci are either directly associated with risk of inflammatory bowel disease (IBD) or maintenance of the intestinal epithelial barrier and response to mucosal pathogens. The geospatial distribution of risk alleles is highly suggestive of multi-locus adaptation, and genetic risk correlates strongly with variation in local pathogens, particularly helminth diversity, suggesting a possible role for host-intestinal pathogen interactions in shaping the genetic landscape of IgAN [8, 28, 30].
Table 3.2
GWAS loci reported to be associated with IgAN
|
Nat Genet 2014;46:1187–96 [30]
|
||||||
|---|---|---|---|---|---|---|
|
CHR
|
SNP
|
Type of SNP
|
Risk allele
|
Locus
|
OR
|
Function
|
|
1
|
rs17019602
|
Intronic
|
G
|
VAV3
|
1.17
|
Chemokine signaling pathway
|
|
1
|
rs6677604
|
Intronic
|
G
|
CFHR3,1-delta
|
1.35
|
Innate immunity system (alternative pathway)
|
|
6
|
rs7763262
|
Intergenic
|
C
|
HLA-DR/DQ
|
1.41
|
Antigen presentation
|
|
6
|
rs9275224
|
Intergenic
|
G
|
HLA-DR/DQ
|
1.36
|
|
|
6
|
rs2856717
|
Intergenic
|
G
|
HLA-DR/DQ
|
1.27
|
|
|
6
|
rs9275596
|
Intergenic
|
T
|
HLA-DR/DQ
|
1.44
|
|
|
6
|
rs9357155
|
Intronic
|
G
|
TAP2/PSMB9
|
1.13
|
Antigen digestion and processing
|
|
6
|
rs1883414
|
ncRNA
|
G
|
HLA-DP
|
1.22
|
Antigen presentation
|
|
8
|
rs2738048
|
Intergenic
|
A
|
DEFA
|
1.10
|
Innate immunity system (antimicrobial)
|
|
8
|
rs10086568
|
Intergenic
|
A
|
DEFA
|
1.16
|
|
|
9
|
rs4077515
|
Missense
|
T
|
CARD9
|
1.16
|
Innate immune system
|
|
16
|
rs11150612
|
Intergenic
|
A
|
ITGAM-ITGAX
|
1.18
|
Cell adhesion molecules
|
|
16
|
rs11574637
|
Intronic
|
T
|
ITGAM-ITGAX
|
1.32
|
|
|
17
|
rs3803800
|
Intronic
|
A
|
TNFSF13
|
1.12
|
Mucosal immunity and immunoglobulin class switching
|
|
22
|
rs2412971
|
Intronic
|
G
|
HORMAD2
|
1.20
|
Mucosal immunity and inflammation
|
An additional GWAS of IgAN in Han Chinese, which was comprising 8,313 cases and 19,680 controls, has most recently been reported [31]. The authors identified novel associations at ST6GAL1 and ACCS, and their risk variants were strongly associated with mRNA expression levels in blood cells. Many studies described aberrant glycosylation of IgA1 in sporadic IgAN, and in also familial IgAN, that defect was reported to be inherited [32]. The associations of ST6GAL1 elucidated by GWAS might explain the genetic basis of defect of glycosylation in IgAN.
Indeed, it is obvious that recent genome-wide association studies have identified hundreds of genetic variants associated with complex human diseases and traits including IgAN and that these studies have provided valuable insights into their genetic architecture. However in general, most reported variants confer relatively small increments in risk and explain only a small proportion of familial clustering, leading many to question how the remaining, “missing” heritability can be explained. The underlying rationale for GWAS is the “common disease, common variant” hypothesis, positing that common diseases are attributable in part to allelic variants present in more than 1–5 % of the population. It has been suggested that low-frequency variants of intermediate effect might also contribute to explaining missing heritability that should be tractable through large meta-analyses and/or imputation of genome-wide association data to illuminate the genetics of complex diseases and enhance its potential to enable effective disease prevention or treatment (Fig. 3.1) [33].
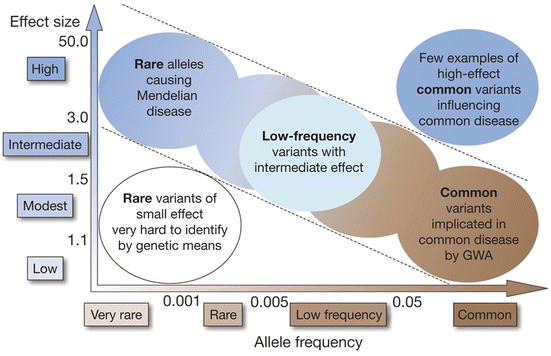
Fig. 3.1
Feasibility of identifying genetic variants by risk allele frequency and strength of genetic effect (From Ref. [33])
3.4 Genome-Wide Linkage and Whole Exome Sequencing Analysis of Familial IgAN
Apart from sporadic IgAN, researchers have intensely searched for causative genes for familial IgAN by positional cloning approach, as the other Mendelian diseases achieved success by that analysis. Gharavi et al. reported in 2000 a genome-wide analysis of linkage in 30 multiplex IgAN kindreds, demonstrating the remarkable linkage of IgAN to 6q22-23 under a dominant model of transmission with incomplete penetrance, with a LOD score of 5.6 and 60 % of kindreds linked. These findings for the first time indicated the existence of a locus with large effect on development of IgAN and identify the chromosomal location of this disease gene [34]. However, even by these expensive methods including positional cloning techniques as “genome-wide” microsatellite markers, the genes responsible for susceptibility to IgAN have not yet been identified. After that, two more genome-wide linkage analyses described other candidate loci for the development of familial IgAN [35, 36]. Until now, four loci have been reported, and locus heterogeneity is considered to exist in the genetic backgrounds of even in familial IgAN, which is one of the causes of difficulty in identifying the genes for IgAN.
With next-generation sequencing technology, whole exome sequencing has emerged as a powerful and cost-effective strategy for dissecting the genetic basis of diseases, and numerous causative genes for rare diseases have been identified so far. As described above, from the point of view of genetic heterogeneity of familial IgAN, an oligo-/polygenic and multiple susceptibility gene model for the disease has been proposed. Therefore, in order to identify the genetic causality of familial IgAN, sequencing analysis of each family would be necessary. Workflow of exome analysis is described in the following: (1) the whole exomes of affected and unaffected individuals were captured and subjected to massive parallel sequencing. (2) Variants identified by exome sequencing were filtered on the basis of variant annotation, functional expectation, and allele frequency. (3) The variants shared by the affected individuals in the family were selected and compared with public genetic variation database. Finally, causative gene variants must fulfill the following requirements: allele frequency of rare mutation, co-segregation in the family members, and demonstration of functional significance of the variants.
Recently, Italian researchers reported a genetic study of Sicilian family where IgAN segregates with an autosomal dominant pattern [37]. The exome and bioinformatic analysis identified p.Arg119Trp variant in the SPRY2 gene, and functional analysis of this variant demonstrated the inhibition of the MAPK/ERK1/2 pathway demonstrated only in the affected individuals. They suggested that downregulation of the MAPK/ERK1/2 pathway represents a common mechanism leading to IgAN. In other paper [38], Chinese researchers analyzed ten IgA families and described six deleterious variants in four genes co-segregated in the families. They mentioned causal relationships between the variants and IgAN; however, functional significance of these variants has not been demonstrated.
Rare variants and/or mutation for familial IgAN are considered as key genetic factors with moderate to strong effects. In the near future, identification of them would provide novel insights for the pathophysiology in the developments of IgAN.
3.5 Conclusion
Progress in technologies of genomic science has been uncovering the genetic diversity in patients with IgAN. From these studies, basic framework of pathogenesis of IgAN has been revealed. We speculate that the effects of genetic variants in each gene might influence the inheritance pattern of disease, sporadic or familial. To understand the precise pathogenic mechanism of IgAN in more detail, further elucidation of the role of genetic variants underlying IgAN, and hologenetic views of gene variants and environmental factors, would be necessary in the future.
Acknowledgment
This work was partly supported by Grant-in-Aid for Scientific Research (B) to I. Narita and (C) to S. Goto from the Japan Society for the Promotion of Science and also by MEXT KAKENHI (221S0002).
References
2.
3.
4.
D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med. 1987;64:709–27.PubMed
5.
6.
Wyatt RJ, Julian BA, Baehler RW, Stafford CC, McMorrow RG, Ferguson T, et al. Epidemiology of IgA nephropathy in central and eastern Kentucky for the period 1975 through 1994. Central Kentucky Region of the Southeastern United States IgA Nephropathy DATABANK Project. J Am Soc Nephrol. 1998;9:853–8.PubMed
7.
8.
Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8, e1002765.CrossRefPubMedPubMedCentral
9.
10.
11.
12.
Waldherr R, Rambausek M, Duncker WD, Ritz E. Frequency of mesangial IgA deposits in a non-selected autopsy series. Nephrol Dial Transplant. 1989;4:943–6.PubMed
13.
14.
Karnib HH, Sanna-Cherchi S, Zalloua PA, Medawar W, D’Agati VD, Lifton RP, et al. Characterization of a large Lebanese family segregating IgA nephropathy. Nephrol Dial Transpl. 2007;22:772–7.CrossRef
15.
16.
17.
18.
19.
Frasca GM, Soverini L, Gharavi AG, Lifton RP, Canova C, Preda P, et al. Thin basement membrane disease in patients with familial IgA nephropathy. J Nephrol. 2004;17:778–85.PubMed
20.
21.
22.
23.
24.
25.
Song J, Narita I, Goto S, Saito N, Omori K, Sato F, et al. Gender specific association of aldosterone synthase gene polymorphism with renal survival in patients with IgA nephropathy. J Med Genet. 2003;40:372–6.CrossRefPubMedPubMedCentral
26.
27.
Feehally J, Farrall M, Boland A, Gale DP, Gut I, Heath S, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. 2010;21:1791–7.CrossRefPubMedPubMedCentral
28.
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43:321–7.CrossRefPubMedPubMedCentral
29.
Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet. 2012;44:178–82.CrossRef
30.
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46:1187–96.CrossRefPubMedPubMedCentral
31.
Li M, Foo JN, Wang JQ, Low HQ, Tang XQ, Toh KY, et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat Commun. 2015;6:7270.CrossRefPubMedPubMedCentral
32.
Gharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, et al. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008;19:1008–14.CrossRefPubMedPubMedCentral
33.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53.CrossRefPubMedPubMedCentral
34.
35.
Bisceglia L, Cerullo G, Forabosco P, Torres DD, Scolari F, Di Perna M, et al. Genetic heterogeneity in Italian families with IgA nephropathy: suggestive linkage for two novel IgA nephropathy loci. Am J Human Genet. 2006;79:1130–4.CrossRef
36.
37.
Milillo A, La Carpia F, Costanzi S, D’Urbano V, Martini M, Lanuti P, et al. A SPRY2 mutation leading to MAPK/ERK pathway inhibition is associated with an autosomal dominant form of IgA nephropathy. Eur J Hum Genet: EJHG. 2015
38.
Liu R, Hu B, Li Q, Jing X, Zhong C, Chang Y et al. Novel genes and variants associated with IgA nephropathy by co-segregating with the disease phenotypes in 10 IgAN families. Gene 2015.
Conflict of Interest
The authors declare that they have no conflict of interest.