Fig. 1.1
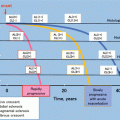
Geographical variations in the prevalence of IgAN. Percentages represent the proportion of cases of IgAN compared to all native kidney biopsies performed. The numbers in brackets represent minority racial groups, African-Americans in the USA and Polynesians in New Zealand (From Feehally and Floege [14])
Some of this variability can be attributable to differences in clinical practice and thresholds for renal biopsy. For example, in countries where screening programmes for urinary abnormalities exist, more patients will be identified, who may then proceed to having a renal biopsy. A correlation between the number of renal biopsies performed per country and the incidence of primary glomerulonephritis and IgAN has been observed (Fig. 1.2) [15]. The same study also showed that between two centres in Scotland, located only 100 km apart, there was a 70 % difference in the incidence of IgAN, which was associated with a marked increase in the number of renal biopsies performed per year in one of the centres [15]. This implies that increasing the range of people subjected to renal biopsy, presumable for minor urinary abnormalities, will increase the identification of IgAN.
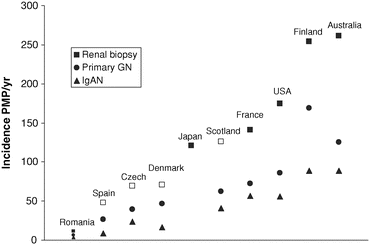
Fig. 1.2
Reported rates of biopsy incidence per million population (PMP) in published studies from national (closed squares) and regional (open squares) native renal biopsy registries. Renal biopsy incidence appears to correlate with the incidence of primary glomerulonephritis and IgAN (From McQuarrie et al. [15])
However, data from migrant populations suggest that true variations in susceptibility exist between people of different ethnicities. In Australia, East and Southeast Asian-born patients have higher rates of IgAN accounting for ESRD compared to those born in the Middle East, Southern Europe or Australia [16]. In the USA, patients of an Asian or Pacific Island ethnicity have higher rates of IgAN than other glomerular diseases [17], whereas IgAN is much less common in African-Americans than in other ethnic groups [7].
As well as overall prevalence, marked variation in gender distribution also exists in IgAN. In North American and European cohorts, a male to female predominance is reported, with the ratio being 2–3:1, whereas in Asia this approximates 1:1 [18–20]. These differences in incidence and gender bias do suggest that the ‘disease’ IgAN may be behaving very differently across the globe. It is likely that both genetic and environmental factors play contributory roles in the differences observed, although the relative importance of each is likely to vary in different populations. Emerging evidence supports an increasing gradient in currently understood risk alleles from West to East, from Africa [21]. Whether this constitutes different diseases in the West and East is open to debate.
In parallel, a number of studies have reported the importance of environmental factors in determining the prevalence of IgAN, although not all populations appear equally susceptible. Perhaps the best example of varying susceptibility to environmental antigens in IgAN is the subgroup of patients with IgAN who develop flares of nephritis, demonstrated by visible haematuria, in conjunction with stimulation of the mucosal immune system, for example, during an upper respiratory tract infection. There is also a well-described subgroup of patients where IgAN and coeliac disease overlap and where gluten avoidance leads to a reduction in severity of renal disease [22, 23]. Interestingly, a recent GWAS meta-analysis identified susceptibility loci for IgAN that are also related to inflammatory bowel disease and maintenance of the intestinal epithelial barrier [24]. On a global scale it has been suggested that an inverse relationship exists between IgAN and membranoproliferative disease, with IgAN being more prevalent in countries with a higher gross domestic product [25]. By contrast, a study from Scotland showed a higher number of renal biopsies were performed in patients from areas of socioeconomic deprivation, and of these patients, a higher proportion were diagnosed with IgAN [26]. Although most studies have reported a low prevalence of IgAN in African-Americans [14, 27], two studies conducted in specific regions of the USA found equivalent prevalence, leading some to suggest that environmental factors may have played a role [28, 29].
At present the exact role for environmental factors in IgAN has not been fully established, although there do certainly appear to be differences both across and within populations in terms of responsiveness to environmental triggers. This heterogeneity lends further support to the argument that IgAN may not be the same disease in all parts of the world. Importantly, these differences may have a direct impact on how we manage patients diagnosed with IgAN. Patients with IgAN in association with gluten-sensitive enteropathy are likely to respond to a simple antigen exclusion diet and not require immunosuppression, whereas most patients with IgAN will gain no benefit from such a change and will require more tailored therapy.
1.4 The Clinical Presentation of IgA Nephropathy Is Highly Variable
Patients with IgAN can present with the full spectrum of nephrological presentations from asymptomatic urine abnormalities through to malignant hypertension, nephrotic syndrome or a rapidly progressive glomerulonephritis. This is perhaps not surprising given the wide variety of histopathological changes that can be seen in association with mesangial IgA deposition. In part these differences in presentation may reflect differences in an individual’s willingness to access healthcare promptly, local public health screening strategies, different renal biopsy policies and differing access to specialist renal services. These factors do not, however, explain all the variation seen in patients presenting with IgAN. Within individual centres, such as our own in Leicester, UK, which has a single biopsy policy, serves a relatively homogeneous population and where there is universal primary care coverage, mesangial IgA deposition can present after detection of non-visible haematuria, with episodes of visible haematuria and normal renal function, as a rapidly progressive glomerulonephritis and as nephrotic syndrome. In each of these cases, the pattern and extent of mesangial IgA staining will be almost identical. Clearly, different pathogenic pathways are operating in these patients that require very different therapeutic interventions and have very different implications for long-term renal survival. Do these patients have the same disease?
Furthermore, accepting that there are external factors that may influence the mode of presentation of patients with IgAN, there do also appear to be global differences in the way patients with IgAN present. In a study of 711 adult patients with biopsy-proven IgAN from Glasgow, Helsinki, Sydney and Toronto, there were significant variations in age, proteinuria, mean arterial blood pressure and creatinine clearance at presentation between centres [30]. Patients from Helsinki had relatively milder disease at presentation, while patients from Glasgow and Toronto were slightly older at presentation, and there was a higher male-female ratio in Glasgow than other centres. In a separate study of 152 Asian patients from Canada and 76 Thai patients with IgAN, Thai patients were more likely to be female (63.2 vs. 44.1 %) and have less baseline proteinuria (1.2 vs. 1.7 g/day) at time of first presentation [31]. By contrast, in a study from India of 478 patients with IgAN, the mean age at presentation was 32 (±11) years, with a predominance of males (65 %) [32]. Patients presented with a much more severe clinical phenotype: nephrotic syndrome (55 %), hypertension (58 %), renal failure (serum creatinine >125 μmol/L in 56 %) and acute nephritis (28 %). Very few patients presented with asymptomatic urine abnormalities (5 %). The mean serum creatinine and 24 h urine protein were 205 μmol/L and 2.9 g/day, respectively.
The differences seen both within centres and across the globe in the way patients present, all of whom have almost identical patterns of mesangial IgA deposition, suggest that labelling all of these patients as having a single disease may be oversimplifying a complex group of nephrological conditions with very different natural histories.
1.5 The Clinical Course of IgA Nephropathy Varies Widely
Not only is there significant heterogeneity in the way patients with mesangial IgA deposition present to their clinician, but there is also enormous variation, both within and across populations, in the likelihood that this mesangial IgA deposition will be associated with progressive glomerular and tubulointerstitial injury and chronic kidney disease. Interestingly, clinical outcomes in IgAN do not correlate with the severity of mesangial IgA deposition, the defining feature of IgAN [33].
Spontaneous remission has been reported in a subgroup of patients with IgAN, and when a repeat biopsy has been performed, resolution of urinary abnormalities is associated with clearance of IgA immune deposits [34]. Equally most studies report that 20–30 % of patients with IgAN develop slowly progressive chronic kidney disease leading to ESRD over a period of 20 years [35]. Rates of progression have been shown to differ both within and between different ethnic groups, although accurate comparisons across populations have been difficult as most published data is retrospective and observational and may, therefore, be confounded by differences in the identification and management of patients. One additional difficulty in interpreting this type of data is the potential impact of lead-time bias (Fig. 1.3). In centres with a proactive urine screening policy, clinical presentation, renal biopsy and diagnosis are likely to be at an early stage of the disease and consequently prognosis is likely be better than that reported from a centre that only biopsies and diagnoses patients at a late stage of the disease. This is also a factor when comparing centres who have different renal biopsy policies and patient groups, who have differing access to specialist renal services.
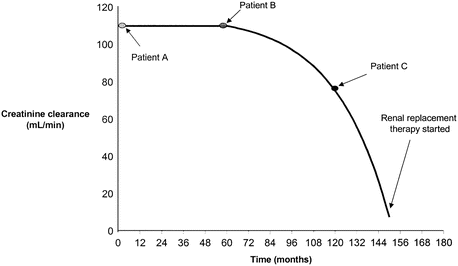
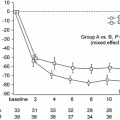
Fig. 1.3
The concept of lead-time bias. Three hypothetical patients with IgAN who have an identical clinical course are depicted. Marked differences in their prognosis are observed depending on when their disease is identified (From Geddes et al. [30])
While some studies in Asia have reported equivalent outcomes in IgAN cohorts to those in Western populations, this is not uniformly the case [19, 36]. Outcomes in 478 Indian patients from Vellore were significantly worse than those of European cohorts with a median renal survival of only 61 months from time of renal biopsy [32]. A recent study from Canada prospectively followed a multiracial cohort and, after adjusting for age, gender, eGFR, medication use, blood pressure and proteinuria, found that prognosis was worse in patients with IgAN of self-reported Pacific Asian origin compared to patients of all other backgrounds [37].
Traditionally patients with isolated non-visible haematuria, without proteinuria or renal dysfunction, are thought to have an excellent prognosis. Indeed, in a recent study from Spain, which included 141 Caucasian IgAN patients with non-visible haematuria, absent or minimal proteinuria, and no renal dysfunction, followed up for a median of 9 years, a rise in serum creatinine >50 % was observed in only 3.5 % of patients. Furthermore, 37 % of patients had complete resolution of their urinary abnormalities [38]. By contrast, in a series of 72 Chinese IgAN patients with non-visible haematuria, proteinuria <0.4 g/day and normal eGFR, 33 % developed proteinuria >1 g/day and 7 % developed impaired renal function (creatinine clearance <70 ml/min) during follow-up for up to 12 years. Only 14 % had complete resolution of their haematuria and had stable kidney function over this period [39].
If IgAN is a single disease, why do we see such varied clinical outcomes when the defining feature of mesangial IgA deposition is uniformly seen in all patients? Whether there are true global differences in clinical outcomes in IgAN is difficult to ascertain from current data due to the confounding effects of factors known to influence progression of chronic kidney disease irrespective of cause: socioeconomic status, differences in predisposing genetic backgrounds, prenatal and perinatal care and experiences, including low birth weight, inadequate diets, infectious diseases or exposure to toxins. Where it has been possible to correct as much as possible for these confounders, there does appear to be a difference both in the likelihood of spontaneous remission and the risk of developing progressive renal injury in different parts of the world. How this translates to different disease pathways in IgAN is unclear but should be the focus of future studies.
1.6 Differences in the Rates of IgA Nephropathy Recurrence in Renal Allografts
Recurrent mesangial IgA deposition in renal allografts occurs frequently, but not uniformly, in published IgAN series. Retrospective observational studies, where biopsies were performed due to allograft dysfunction, report recurrent IgA deposition in anywhere between 21 and 58 % of recipients [40]. It is clear not all patients with IgAN develop recurrent IgA deposits, and perhaps more interestingly, while recurrent IgA deposition is common, allograft failure due to recurrent disease occurs far less frequently [41]. The risk of allograft loss is greatest in younger recipients and the subgroup of patients who had an aggressive original disease course, e.g. crescentic glomerulonephritis [42]. A recent study from Japan of 29 IgAN transplant recipients with >10 years post-transplant follow-up and no urinary abnormalities showed that 11 of these patients had recurrent mesangial IgA deposition with no evidence of allograft dysfunction [43]. Compared to the 18 patients with no mesangial IgA recurrence, there were no significant differences in either histopathology or long-term allograft function between the groups.
The heterogeneity in rates of mesangial IgA deposition and translation of this deposition to allograft dysfunction reflect the varied picture we see in native kidneys. However, when reviewing the data on recurrence of IgAN post-transplant, it is impossible to remove the confounding effects of concomitant immunosuppression. Again, as in native kidneys, there appears to be little correlation between the pattern of mesangial IgA deposition in recurrent IgAN and the risk of allograft loss. It is possible, therefore, that these variations reflect underlying differences in disease processes in individual recipients and the varied susceptibility of these disease processes to the immunosuppression regimen used.
1.7 Do Differences in Treatment Responses Reflect Different Underlying Diseases?
When examining the response to pharmacological intervention in any disease, a number of factors will invariably affect the efficacy of that treatment. Drug absorption, metabolism and elimination vary and can be influenced by genetic polymorphisms (pharmacogenetics), age, body size, the use of other drugs, dietary supplements and medicinal herbs, the presence of other diseases and development of tolerance and resistance. There are also well-recognised ethnic differences in drug responses, perhaps the most well known being the differences in response between β-blockers and ACE inhibitors in hypertension [44]. Interpreting the significance of treatment effects from clinical trials in IgAN is further confounded by the different inclusion and exclusion criteria employed, the different treatment regimens used and variable endpoints chosen.
Accepting these relatively ‘fixed’ confounding factors, there is still marked heterogeneity in the response of patients with IgAN to a variety of immunomodulatory therapies. This variation in response could reflect underlying differences in the biochemical pathways operating in different subgroups of patients which are variably sensitive to the drug being studied. For example, the data supporting the use of corticosteroids in IgAN is mixed, and even in those studies reporting a beneficial effect of corticosteroids, the response to steroids varies greatly between trial participants, even though all met the same inclusion and exclusion criteria for the respective trial. The same is seen in trials of other immunosuppressants including azathioprine, mycophenolate mofetil (MMF) and cyclophosphamide. Perhaps more striking is the apparent variation in response to treatments in IgAN in different ethnic groups. Clinical trials of MMF suggest there is no benefit from MMF in Caucasian populations, while a beneficial effect has been observed in Chinese patients [45–48]. Tonsillectomy, a commonly used treatment in Japan, is believed to have a significant disease-modifying effect in Japanese patients; however, this is not the case in Caucasian patients [49–52].
With advances in pharmacogenetics and an improvement in clinical trial design in IgAN, it may be possible in the coming years to better understand the response to treatment in IgAN and identify subgroups of patients who may have discreet diseases and are more or less likely to respond to specific immune modulation strategies.
1.8 Does Our Knowledge of IgAN Pathogenesis Support IgA Nephropathy Being a Single Disease?
There is now convincing evidence that the production of poorly galactosylated IgA1 and O-glycan-specific IgG and IgA autoantibodies can lead to the formation of IgA-containing immune complexes and that these immune complexes deposit within the mesangium, where their consequent effects on mesangial cells, podocytes and proximal tubule cells are central to the development of IgAN [53]. Elevated levels of poorly galactosylated IgA1 and IgG and IgA autoantibodies against the IgA1 hinge region O-glycans are, however, only found in some, and not all, patients with IgAN. Elevated levels of total serum IgA, complement component C3 and other markers of alternative pathway activation may also be seen in some IgAN patients. Unlike other kidney diseases such as SLE (antinuclear antibodies), antiglomerular basement membrane (GBM) disease (anti-GBM autoantibodies), pauci-immune vasculitis (antineutrophil cytoplasmic antibodies) and primary membranous nephropathy (PLA2R autoantibodies), IgAN is not associated with a consistent serological profile. This may simply reflect the fact we have not yet made the necessary discoveries. Alternatively, it may be that we are not dealing with a simple, single disease with a predictable single serological phenotype.
The most consistently reported serological abnormality in IgAN is the increase in poorly galactosylated IgA1 in IgAN [54]. However, if you look at the distribution of IgA1 O-glycoforms in any of the reported case series from across the globe, including our own, you will see marked variation in the extent of serum IgA1 O-glycosylation in IgAN, with significant overlap between patients with IgAN and healthy subjects (Fig. 1.4). This suggests that serum IgA1 O-glycosylation may be pathogenically unimportant in some cases of IgAN. In the more recent studies reporting the existence of IgG and IgA hinge region-specific autoantibodies, it is clear that these are not universally found in IgAN [55]. Alternative mechanisms for immune complex formation may therefore operate in different patient groups. It has been proposed that there is a subgroup of patients who develop IgA immune complexes as a consequence of soluble CD89 shedding from circulating myeloid cells and that in these patients CD89 is essential for mesangial cell activation by deposited IgA immune complexes [56]. We have already discussed a further subgroup of patients with gluten-sensitive enteropathy, where the pathway to mesangial IgA deposition is likely to be different again. How these different pathogenic pathways correlate with the clinical phenotype is unclear at present.
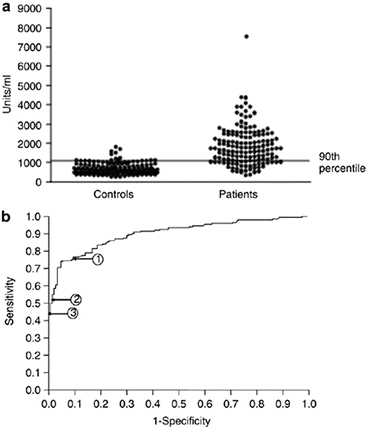
Fig. 1.4
Serum levels of galactose-deficient IgA in IgAN patients and healthy controls. Although the majority of patients display a higher serum galactose-deficient IgA level compared to the healthy controls, approximately one quarter had levels that overlapped, suggesting that other susceptibility factors play a role in the development of IgAN (From Moldoveanu et al. [54])
Finally, data from genetic studies is also beginning to help define potential pathogenic pathways and important disease-associated allelic variants in IgAN. While there are genetic associations common to all published studies, principally genetic variants in the HLA gene family, there is clear disparity in associations with other genetic loci. For example, associations between IgAN and variants of the angiotensin converting enzyme, TAP1/PSMB (transporters associated with antigen processing 1/proteosomal subunit) and DEFA (α-defensin) genes have only been reported in Asian but not in Western patients [57–59]. By contrast, the familial form of IgAN has been more frequently reported in Europe than in Asia, and within Europe, familial IgAN is more evident in Southern than in Northern populations [21, 60]. These genetic observations raise the possibility of involvement of different genes in the pathogenesis of IgAN in different parts of the world. In time we may be able to use this information to redefine IgAN on the basis of clear genotypic variants rather than on the basis of a generic renal biopsy appearance.
Current concepts of the pathogenesis of IgAN are discussed extensively in this book. We would argue strongly that the data presented in the following chapters supports the existence of multiple pathogenic pathways. These pathways operate, to variable extents, in different IgAN patients, but that ultimately all of them lead to a final common end point of mesangial IgA deposition. Currently we are using the umbrella of a diagnosis of IgAN to encompass all of these patients. In the future hopefully we will be able to link each of these pathogenic pathways to a specific disease genotype/phenotype and produce a tailored treatment paradigm and thereby fully embrace the era of ‘personalised medicine’ in IgAN.
1.9 Conclusion
In this chapter we have been deliberately provocative in the way we have interpreted the existing literature on IgAN. No matter whether you believe IgAN is or is not a single disease, it is hard to get away from the fact that IgAN is highly heterogeneous with marked genotypic and phenotypic variation. Until we have a clear serological marker akin to antinuclear or PLA2R autoantibodies, confirming a unifying pathogenic pathway, we will be continually confronted with the argument that mesangial IgA deposition alone is a poor way to define this complex nephrological disorder.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
1.
2.
Scully JL. What is a disease? EMBO Rep. 2004;5(7):650–3.CrossRefPubMedPubMedCentral
3.
World Health Organisation. Preamble to the constitution of the World Health Organization as adopted by the international health conference. New York: WHO; 1946.
4.
Berger J, Hinglais N. Les depots intercapillaries d’IgA-IgG. J Urol Nephrol. 1968;74:694–5.
5.
Sinniah R. Occurrence of mesangial IgA and IgM deposits in a control necropsy population. J Clin Pathol. 1983;36(3):276–9.CrossRefPubMedPubMedCentral
6.
Varis J, Rantala I, Pasternack A. Immunofluorescence of immunoglobulins and complement in kidneys taken at necropsy. J Clin Pathol. 1989;42(11):1211–4.CrossRefPubMedPubMedCentral
7.
Waldherr R, Rambausek M, Duncker WD, Ritz E. Frequency of mesangial IgA deposits in a non-selected autopsy series. Nephrol Dial Transplant. 1989;4(11):943–6.PubMed
8.
10.
11.
12.
Jennette JC, Olson JL, Schwartz MM, Silva FG. Heptinstall’s pathology of the kidney, vol. 1. 6th ed. Philadelphia: Lippincott Williams and Wilkins; 2007. p. 424–87.
13.
14.
Feehally J, Floege J. IgAN and Henoch-Schonlein nephritis. In: Feehally J, Floege J, Johnson R, editors. Comprehensive clinical nephrology. 3rd ed. Philadelphia: Mosby Elsevier; 2007. p. 253–64.
15.
16.
17.
Hall YN, Fuentes EF, Chertow GM, Olson JL. Race/ethnicity and disease severity in IgAN. BMC Nephrol. 2004;5:10.CrossRefPubMedPubMedCentral
18.
D’Amico G, Imbasciati E, Barbiano Di Belgioioso G, Bertoli S, Fogazzi G, Ferrario F, et al. Idiopathic IgA mesangial nephropathy. Clinical and histological study of 374 patients. Medicine (Baltimore). 1985;64(1):49–60.CrossRef
19.
20.
21.
Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, et al. Geographic differences in genetic susceptibility to IgAN: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6):e1002765.CrossRefPubMedPubMedCentral
22.
Woodrow G, Innes A, Boyd SM, Burden RP. A case of IgAN with coeliac disease responding to a gluten-free diet. Nephrol Dial Transplant. 1993;8(12):1382–3.PubMed
23.
Koivuviita N, Tertti R, Heiro M, Metsärinne K. A case report: a patient with IgAN and coeliac disease. Complete clinical remission following gluten-free diet. NDT Plus. 2009;2(2):161–3.PubMedPubMedCentral
24.
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgAN implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46(11):1187–96.CrossRefPubMedPubMedCentral
25.
26.
27.
28.
29.
Wyatt RJ, Julian BA, Baehler RW, Stafford CC, McMorrow RG, Ferguson T, et al. Epidemiology of IgAN in central and eastern Kentucky for the period 1975 through 1994. Central Kentucky Region of the Southeastern United States IgAN DATABANK Project. J Am Soc Nephrol. 1998;9(5):853–8.PubMed
30.
31.
Prakash S, Kanjanabuch T, Austin PC, Croxford R, Hsu CY, Choi AI, et al. Continental variations in IgA nephropathy among Asians. Clin Nephrol. 2008;70(5):377–84.CrossRefPubMedPubMedCentral
32.
33.
Working Group of the International IgAN Network, the Renal Pathology Society, Cattran DC, Coppo R, Cook HT, Feehally J, Roberts IS, Troyanov S, et al. The Oxford classification of IgAN: rationale, clinicopathological correlations, and classification. Kidney Int. 2009;76(5):534.CrossRef
34.
35.
36.
37.
38.
Gutiérrez E, Zamora I, Ballarín JA, Arce Y, Jiménez S, Quereda C, Grupo de Estudio de Enfermedades Glomerulares de la Sociedad Española de Nefrología (GLOSEN), et al. Long-term outcomes of IgAN presenting with minimal or no proteinuria. J Am Soc Nephrol. 2012;23(10):1753–60.CrossRefPubMedPubMedCentral
39.
40.
41.
42.
43.
44.
Johnson JA. Ethnic differences in cardiovascular drug response: potential contribution of pharmacogenetics. Circulation. 2008;118(13):1383–93.CrossRefPubMedPubMedCentral
45.
46.
Tang SC, Tang AW, Wong SS, Leung JC, Ho YW, Lai KN. Long-term study of mycophenolate mofetil treatment in IgAN. Kidney Int. 2010;77:543–9. doi:10.1038/ki.2009.499. Epub 2009 Dec 23.CrossRefPubMed
47.
48.
49.
50.
Komatsu H, Fujimoto S, Hara S, Sato Y, Yamada K, Kitamura K. Effect of tonsillectomy plus steroid pulse therapy on clinical remission of IgAN: a controlled study. CJASN. 2008;3(5):1301–7.PubMedPubMedCentral
51.
Rasche FM, Schwarz A, Keller F. Tonsillectomy does not prevent a progressive course in IgAN. Clin Nephrol. 1999;51(3):147–52.PubMed
52.
53.
Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22(10):1795–803.CrossRefPubMedPubMedCentral
54.
55.
Yanagawa H, Suzuki H, Suzuki Y, Kiryluk K, Gharavi AG, Matsuoka K, et al. A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLoS One. 2014;9(5):e98081.CrossRefPubMedPubMedCentral
56.
Launay P, Grossetête B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, et al. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J Exp Med. 2000;191(11):1999–2009.CrossRefPubMedPubMedCentral
57.
58.
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321–7.CrossRefPubMedPubMedCentral
59.