[level-membership-for-cardiovascular-category]17
Ion Channel Trafficking in the Heart
Ion Channel Trafficking versus Quality Control
Anterograde Trafficking of Channels in the Heart
At Their Destination: Channels in the Myocyte Membrane
Retrograde Trafficking of Channels in the Heart
Regulation of Channel Trafficking
Alterations of Channel Trafficking in Pathophysiological States
Pharmacologic Manipulation of Channel Trafficking and Implications for Cardiac/Antiarrhythmic Therapy
Ion Channel Trafficking versus Quality Control
The steady-state cell surface density of ion channel proteins is determined by the balance between the anterograde and retrograde trafficking pathways. Anterograde trafficking ensues only after proper protein synthesis and processing in the endoplasmic reticulum and Golgi apparatus, including quality control mechanisms, glycosylation, and posttranslation modification (Figure 17-1).1 Often, channel trafficking, synthesis, and quality control are grouped together in common discussion. However, these processes can be distinct, using unique cellular machinery and subject to differential regulation. Retrograde movement initiates with endocytosis, after which internalized proteins can follow multiple routes to different intracellular fates (see Figure 17-1).2 One well-recognized fate is the targeting of internalized proteins to lysosomes, or proteasomes, followed by degradation (see Figure 17-1). Alternatively, trafficking through recycling endosomes allows proteins to return to the plasma membrane and protects them from degradation (see Figure 17-1).3 Sorting at early endosomes to Rab-GTPase–specific compartments is now established as an important event in determining the intracellular fate of internalized proteins.4–6 Another important component of the endocytic machinery regulating protein surface levels is the coordinated movement of molecular motors. In general, protein trafficking is highly coordinated between long-range events involving the microtubule-based kinesin and dynein motors, and short-range events using unconventional myosin motors.7–10 There is a significant and growing body of literature concerning ion channel trafficking from synthesis to sorting to degradation in multiple tissues and cells systems that has been reviewed previously.11–15 Our discussion will center mainly on recent work focused on the control of ion channel density at the plasma membrane, in the heart, where relatively little is known about protein trafficking. We have chosen to organize this chapter around the major transport events in ion channel movement within myocytes. Given that numerous and different ion channels play an essential role in all chambers of the heart and throughout the cardiovascular system, it is impossible to include a comprehensive review of all the literature. Rather, it is our intention to use select examples to highlight the major themes that have been recently uncovered in ion channel trafficking.
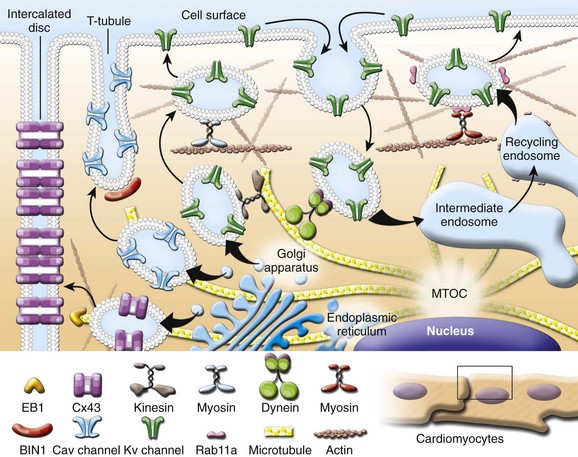
Figure 17-1 Ion channel trafficking pathways in the heart. Anterograde and retrograde transport using kinesin motors on microtubules and myosin motors on actin filaments is illustrated. Channel trafficking to subcellular compartments including intercalated disc, T-tubule, and lateral membrane is highlighted.
Anterograde Trafficking of Channels in the Heart
The Directed Targeting Paradigm
Posttranscription and posttranslation, the proteins that form cardiac ion channels are modified and usually oligomerize in the Golgi apparatus, where they are inserted into membrane vesicles for delivery to the plasma membrane. These channel-laden vesicles are transported using the cytoskeleton as a path to the plasma membrane, with most studies focusing on the role of microtubule-based forward transport.16–21 A critical aspect of forward transport is localization to membrane subdomains. It is entirely possible that channels may be delivered to random regions of the plasma membrane, only to then laterally diffuse within the membrane to their appropriate subdomain.17 However, the temporal and stochastic inefficiency of random channel insertion, together with unexplained mechanisms of subsequent lateral localization other than chance interaction with a subdomain-specific anchor protein, suggests that specificity of delivery from the Golgi apparatus to the surface submembrane may also occur. Still to be fully explored, this directed targeting paradigm of ion channel delivery may be generalizable to all cardiac ion channels and explored in terms of other cytoskeletal elements and anchor proteins (discussed next).
This section of the chapter focuses on mechanisms that govern the forward trafficking of two major cardiac ion channels that are indispensable for cellular excitability, cell-cell coupling, and excitation-contraction coupling: (1) the L-type calcium channel Cav1.2 and (2) the dominant cardiac ventricular gap junction, connexin43 (Cx43).22–26 We also discuss in detail a protein with a newly described role in the heart that is essential for Cav1.2 trafficking and delivery, bridging integrator 1 (BIN1). BIN1 is rapidly emerging as a multifunctional cardiac player beyond its previously understood function as a membrane scaffold.
Forward Trafficking of Cx43
Gap junctions are intercellular channels that form low-resistance pathways, allowing ions and metabolites to flow from cell to cell. In the heart, gap junctions electrically couple cardiomyocytes to orchestrate spatial propagation of action potentials.27 A gap junction consists of a pair of abutting hexameric hemichannels or connexons in adjacent cell membranes. Each connexon consists of six connexin proteins. More than 20 Cxs have been identified and each is usually named according to its respective molecular mass.28 All connexins contain four transmembrane domains. Connexin43 is the most abundant connexin in the ventricular myocardium. In individual ventricular cardiomyocytes, Cx43 is localized at the intercalated disc at longitudinal ends of the cell, where Cx43 gap junctions provide rapid action potential transmission and synchronized cardiac excitation.29,30
Cardiac-specific conditional deletion models with greater than 50% Cx43 loss develop increased arrhythmia susceptibility and sudden cardiac death.31–33 In general, these studies reveal that severe arrhythmia phenotypes can manifest when Cx43 coupling drops to less than 20% in the heart, which correlates with theoretic predictions.34 Thus, maintenance of proper Cx43 expression and localization has a critical role in appropriate electrical coupling and ventricular function.
Data exist for multiple, but not incompatible, models of Cx43 trafficking to cell-cell borders of cardiac intercalated discs.17,18,20,25 There is almost universal agreement that microtubules help deliver Cx43 to the plasma membrane. A landmark series of two papers in 2002 found evidence that newly-formed Cx43 appear at the perimeter of the Cx43 plaques and then diffuse into central plaque regions.17,20 Taken together with microtubule delivery, the model developed that Cx43 hemichannels are inserted into the general plasma membrane, rapidly diffuse to the edge of dense plaques, and then more slowly diffuse into the plaque center. Subsequent to these studies, it has been observed that Cx43 can be inserted directly at the edge and into plaques, and membrane fluidity exits within the plaque region.18,35 It has also been found that the plaques are internalized not necessarily from the center, but that different segments of plaque can be internalized at any time35 and full plaque internalization can occur in one step.36 More recently, it has been found that Cx43 occurs in regions surrounding Cx43 plaques, the “perinexus,” at higher density than in general membrane where Cx43 proteins may interact with scaffolding proteins and other ion channels.37 These studies are providing evidence that the gap junction plaque and surrounding regions are highly dynamic with complex behavior of targeted insertion and internalization.
Given the low density of Cx43 hemichannels in membrane well away from Cx43 plaque regions, and the technical difficulty of distinguishing Cx43 inserted in membrane from submembranous and still cytoplasmic collections of protein, it is difficult to find studies that can quantify the lateral diffusion coefficient of membrane-bound Cx43. It may be that free hemichannels rapidly diffuse within the plasma membrane before stopping at plaque regions, but direct evidence for this phenomenon is lacking. The directed targeting paradigm is based on the observation that de novo intracellular Cx43 hemichannels can arrive directly in the gap junction plaque region. The hemichannels are targeted to plaque regions with specificity obtained from the channel protein (Cx43), microtubule plus-end tracking proteins (EB1 and p150 [glued], and a membrane anchor [adherens junction structure]).18 By this model, the dynamic microtubule highways are anchored and terminate at adherens junction structures, allowing directed delivery of Cx43 hemichannels to adherens junction–containing membrane to occur. It is probable that once inserted into plaque regions, local hemichannel diffusion occurs within the plaque and between the plaque and the plaque perinexus.
Although most studies of Cx43 forward trafficking focus on the microtubule cytoskeleton, the actin cytoskeleton is also involved in delivery of membrane proteins and ion channels. Dye transfer studies revealed the dependence of Cx43 insertion into plaque on actin, as tested by pharmacologic actin disruption and anti-actin antibodies.38,39 Actin is also important for Cx43 plaque internalization, although we recently found that when internalization is blocked, forward delivery is still actin dependent.26 Furthermore, a remarkable greater than 80% of cytoplasmic post-Golgi Cx43 is slow-moving or stationary and actin-associated. These findings indicate that microtubules work in concert with actin to deliver Cx43 to the plasma membrane. It may be that post-Golgi Cx43 vesicles exist in actin-associated reservoirs within the cytoplasm, waiting for either the right microtubule to permit membrane delivery or mass acute delivery in the case of a metabolic stress.
Forward Trafficking of Cav1.2
Although connexins electrically couple cardiomyocytes, the membrane ion channel most responsible for calcium entry and excitation-contraction coupling is the α1C pore-forming subunit (Cav1.2) of the L-type voltage-gated (Cav) Ca2+ channel (LTCC). In response to membrane depolarization by sodium currents, LTCCs open to allow inward Ca2+ entry during the plateau phase of the cardiac action potential. The L-type currents (ICa,L) that are generated initiate calcium-induced Ca2+ release from the sarcoplasmic reticulum (SR) via ryanodine receptors, which leads to muscle contraction. The Cav1.2 protein is large and consists of 24 transmembrane segments organized into four homologous domains.40 Auxiliary subunits of the LTCC include the β-subunit, the α2δ-subunit, and the γ subunit, which help regulate trafficking of Cav1.2 to the cell membrane and regulate the voltage dependence of channel gating.41
T-tubule invaginations of ventricular cardiomyocyte plasma membrane are enriched with LTCCs. This enrichment is necessary for calcium-induced calcium release with nearby ryanodine receptors, which is important for beat-to-beat excitation-contraction coupling in the heart. Trafficking of the LTCCs is not as extensively studied as that of Cx43, probably owing to a combination of the large size of the α-subunit, Cav1.2, the multiple β-subunits, the difficulty of manipulating cardiomyocytes with T-tubules in culture, and less dense enrichment of the channel on the plasma membrane, presenting difficulty to clearly characterize by cytochemistry a distinct phenotype. Subunits are important for LTCC trafficking. For instance, the β-subunit is necessary for surface expression.42 Moreover, the α2δ-subunit synergizes with the β-subunit to promote surface channel expression.43 These auxiliary subunits enhance surface expression by promoting channel export from the endoplasmic reticulum and overall channel stability.44
Despite the necessary localization of LTCC to T-tubules, it was not understood until recently how the localization occurs. In 2010, it was found that Cav1.2-based channels adhere to the directed targeted paradigm.23 In particular, the membrane-scaffolding protein BIN1 was found to provide a membrane anchor that attaches dynamic microtubules, allowing delivery of Cav1.2 channels directly to T-tubule membranes. Using HL-1 cells that express Cav1.2 but do not form T-tubules, and non-muscle cell lines that neither express Cav1.2 nor naturally form T-tubules, it was found that exogenous BIN1 caused the formation of deep invaginations in the cell membrane enriched with endogenous or overexpressed Cav1.2, suggesting that BIN1-containing membrane is sufficient to recruit Cav1.2 channels. To test the possibility that BIN1 serves as an anchoring site for microtubules on which Cav1.2 channels are trafficked, the study tracked growing microtubules extending toward BIN1 clusters and found that the microtubule plus end-tracking proteins pause and associate with BIN1 clusters at the cell periphery. Moreover, it was determined that the non-BAR cytoskeleton–anchoring domain of BIN1 is required for this activity because truncation mutants lacking this domain failed to cluster Cav1.2 at cell surface invaginations. The microtubule plus end-tracking protein that may aid in microtubule anchoring to BIN1, analogous to EB1 binding to adherens junctions,18,25 has not yet been identified.
At Their Destination: Channels in the Myocyte Membrane
Subcellular Localization of Kv Channels Within Myocytes
Historically, Kv localization is believed to primarily involve protein-protein interactions among channel proteins and PDZ-domain–containing scaffolding proteins or the actin cytoskeleton. Recent work shows that Kv2.1 channels are immobilized on the lateral membrane of atrial myocytes, where their diffusion is likely limited by actin cytoskeletal corrals as in hippocampal neurons.45 In addition, Kv1.5 has been shown to directly couple to α-actinin-2 in HEK 293 cells via a specific sequence in the amino terminus of the channel.46 In addition, Kv1.5-cytoskeleton interactions appear to play a role in modulation of the channel by protein kinase A in oocytes.47 Further, the actin binding protein, cortactin, regulates Kv1.5./N-cadherin interactions at the intercalated disc. Kvβ-subunits are also likely to interact with the cytoskeleton and thus immobilize channels on the cell surface.48 In addition, some Kvβ-subunits behave as chaperone proteins, promoting the cell surface expression of a subset of co-expressed α-subunits and perhaps influencing localization.49,50 A role for PDZ domain–containing scaffolding proteins, such as SAP97, in the regulation of Kv1.5 surface localization has also been described.51,52 Furthermore, it has been shown that the K+ channel interacting protein, KChIP2, contributes to the formation of functional mouse Kv1.5-encoded ventricular channels.53 It is also believed that membrane microdomains, perhaps formed by protein-lipid interactions, are an important and novel mechanism of Kv channel localization54,55 in the heart. This is furthered by the finding that Kv1.5, caveolin, and SAP97 exist as part of a tripartite complex in the heart56; however, other reports indicate little co-localization of Kv1.5 with caveolin in myocytes. Other work shows that KCNQ1-KCNE1 channels, which encode for the K+ current in the heart, are localized by interactions with the A-kinase anchoring protein, Yotiao.57 Although progress has been made in identifying elements involved in Kv channel localization, clustering, and anchoring, less is known regarding the molecular machinery controlling their plasma membrane targeting or the regulation of surface expression.
Retrograde Trafficking of Channels in the Heart
At the plasma membrane, localization to specific membrane microdomains, and association with scaffolding proteins into macromolecular signaling complexes, likely contribute to the stability and biological function of Kv channels.58–64 Despite association with scaffolding proteins, Kv channels have been shown to undergo dynamic trafficking at the plasma membrane through constitutive internalization and recycling.65 Internalization of Kv1.5 occurs via a dynein-mediated, microtubule-dependent pathway.65,66 After internalization, sorting of Kv1.5 into specific Rab-dependent endocytic compartments determines the intracellular fate of the channel. Specifically, association of Kv1.5 with Rab4- or Rab11-containing endocytic vesicles is associated with recycling of the channel back to the plasma membrane, whereas association with rab7-containing vesicles denotes channel degradation.65–67 In addition, KCNQ1/KCNE1 potassium channels also undergo constitutive recycling involving Rab-specific compartments.68 Specifically, Rab11 is implicated in exocytic trafficking, whereas endocytosis of KCNQ1/KCNE1 is dependent on Rab5. Finally, data indicate that hERG channels, in heterologous expression systems, undergo endoplasmic reticulum export in COPII vesicles and endosomal recycling before being processed in the Golgi. This process involves the GTPases Sar1 and Rab11B, with a potential minor contribution from ARF1.69
Regulation of Channel Trafficking
Regulation of Cx43 Trafficking
Histone deacetylases, which modify histone tails to repress gene transcription, were shown to regulate Cx43 expression in mouse embryonic stem cells.70 In mdx hearts, global elevation of histone acetylase activity is associated with lateralized Cx43 and increased physical interaction between the acetylase P300/CBP-associated factor with Nε-lysine acetylated Cx43. Addition of the histone acetylase inhibitor anacardic acid reduced Cx43 Nε-lysine acetylation and restored Cx43 localization to the intercalated disc. Correspondingly, lateralization of Cx43 was achieved by a short drug treatment that increased total protein acetylation. These studies and others have revealed multiple new functions for proteins normally associated with gene expression regulation in directly affecting connexin trafficking.
Alterations of Channel Trafficking in Pathophysiological States
Kv Channel Trafficking in the Diseased Heart
Alterations in the cell surface expression of functional Kv channels occur in numerous cardiovascular disease states and undoubtedly contribute to their pathophysiology. One example is Kv1.5 and its role in paroxysmal and persistent atrial fibrillation,71,72 as well as chronic hypoxic pulmonary hypertension.73 Overexpression of Kv1.5 in rat cardiomyocytes dramatically shortens action potential duration, producing a phenotype similar to that observed in short QT syndrome.74 Conversely, intracellular sequestration of Kv1.5 in mouse cardiac myocytes, by overexpression of a dominant negative channel fragment, results in reduced surface levels of Kv1.5 and long QT arrhythmias.75 In patients with persistent or paroxysmal atrial fibrillation, reduced outward potassium current is observed. This reduction in current is caused, in part, by diminished Kv1.5 protein expression, whereas other potassium channels, such as Kv2.1, remain unaffected.71 Interestingly, Kv1.5 mRNA levels are unchanged, indicating that alterations in steady-state protein levels may be responsible for the aberrant phenotype; however, these changes have not been likened directly to channel trafficking. Nevertheless, these vicissitudes are intriguing, given that several other K+ channel trafficking defects lead to the development of disease. Of significance are changes that occur with long QT syndrome.
Pioneering work from January and colleagues first reported that hERG channel mutations altered protein trafficking, causing long QT syndrome type 2.44 These trafficking defects reduce the number of functional channels expressed on the myocyte surface, contributing to a reduction in repolarizing current. These observations have been confirmed by multiple groups and expanded to include numerous long QT syndrome type 2 mutations. Recently, it was shown that trafficking-deficient hERG K+ channels linked to long QT syndrome are regulated by a microtubule-dependent quality control event. This highlights the possibility that many of these reported alterations in surface density reflect changes in protein folding/stability events versus the surface transport of channels.
One recent example that directly links long QT syndrome mutants to channel trafficking involves KCNQ1 and KCNE1 subunits. In this report, it was shown that specific disease-associated mutations in these channel subunits disrupt normal endosomal recycling of potassium current channels.68 This work provides novel mechanistic insight into potentially fatal cardiac arrhythmias and highlights that trafficking pathways may represent important therapeutic intervention points for the treatment of cardiac arrhythmias.
Cx43 Regulation in the Diseased Heart
The density and composition of gap junctions determine cell-cell coupling efficiency and ensure orchestrated current flow. Changes in Cx43 expression and trafficking can alter conduction and impair heart function. Many types of ventricular remodeling that occur in humans as a result of cardiac overload are characterized by changes in the expression and distribution of Cx43. Early myocardial infarction studies revealed decreased Cx43 at the intercalated disc and lateralization of remaining channels in the infarct boarder zone.76–78 Altered gap junction plaque size exists in most forms of heart failure, where changes in the cellular distribution of Cx43 affects the spread of excitation in the heart, which can be arrhythmogenic. More recent studies have explored the mechanisms of altered Cx43 distribution in disease.
Reduced cardiac cell-cell coupling in ischemic and nonischemic hearts is strongly associated with Cx43 dephosphorylation, which generally has been inferred from Western blot band shifts and phosphospecific antibodies. For instance, Cx43 dephosphorylation leads to cell-cell uncoupling in the setting of ischemia, which can be rescued by direct suppression of Cx43 dephosphorylation.79,80 The specific Cx43 residues that undergo posttranslational modification during disease are still being explored and there is likely a rich balance of disease-related phosphorylation and dephosphorylation on the Cx43 C-terminus. An elegant study recently found that inhibiting three specific casein kinase sites (S325, S328, S330) from dephosphorylation protected cardiomyocytes from Cx43 remodeling and arrhythmias during ischemia. It is not known whether diminished cell-cell coupling is a result of increased rate of plaque internalization or diminished rate of Cx43 delivery, or both. We recently found that hearts with end-stage ischemic cardiomyopathy were characterized by specific disruption of the cytoskeleton-based Cx43 forward trafficking machinery without changes in total expression.25
Internalization of Cx43 is clathrin-dependent36 and may also involve Zonula Occludens-1 (ZO-1), which is a 220-kDa scaffolding protein that tethers transmembrane proteins such as connexins directly, or via associated adaptor proteins, to the cytoskeleton.81 ZO-1 associates with Cx43 via its second PDZ domain at the perimeter of the gap junctional plaque.82–85 In patients with end-stage congestive heart failure caused by idiopathic dilated cardiomyopathy DCM and ischemic cardiomyopathy, decreased Cx43 expression at the cell surface is accompanied by increased association with ZO-1.86 These studies suggest that ZO-1 may regulate cell surface gap junction availability by promoting Cx43 endocytosis.
Functional Significance of Cav1.2 and BIN1 in the Human Heart
Calcium influx in the working myocardium, which critically depends on the α1C pore-forming subunit, plays a central role in converting electrical impulses into mechanical activation of the contractile machinery. Gain-of-function mutations in Cav1.2, which cause near complete loss of voltage-dependent channel inactivation and calcium overload in multiple tissues, are linked to Timothy syndrome.87,88 Prolonged sodium currents resulting from disrupted channel inactivation delay and repolarization significantly increases QT interval duration and the risk of arrhythmias and sudden death. Interestingly, the disease-causing mutations G406R and G402S were found within exons 8 and 8a, which are alternatively spliced in a mutually exclusive fashion and are present in different relative amounts in various tissues. It was suggested that the multitude of severities of symptoms across multiple organs reflects tissue-specific expression of splice variants. In patients with hypertrophic heart failure, aberrant splicing of the mutually exclusive exons 31 and 32 was detected such that re-expression of the fetal exon contributed to disease progression.89
Loss-of-function mutations in Cav1.2 in patients with Brugada syndrome are characterized by a short QT interval and sudden cardiac death.90 When missense mutations at G490R and A39V were co-expressed with other LTCC subunits in CHO cells, a clear reduction of ICa,L was observed. Confocal microscopy studies revealed that channel trafficking was unaffected. Thus, it was hypothesized that the G490R mutation, which is located in a linker region, interferes with β-subunit binding to inhibit current density. Another study revealed that a V2041I mutation in the C-terminus also reduces ICa,L amplitude by decreasing channel conductance and altering current inactivation.90
Abnormal excitation-contraction coupling, resulting from deregulation at the onset of Ca2+ influx through the T-tubule network, is increasingly implicated in heart failure progression and sudden cardiac death. Heart content of Cav1.2, typically analyzed by biochemical assay of heart muscle lysates, is not altered in failing hearts.24 However, although it was recently confirmed that total cellular Cav1.2 content is unchanged in heart failure, we also found that the channels are internalized.24 In testing the forward trafficking machinery of Cav1.2 in failing hearts, we found that human heart failure involves a reduction in BIN1 at both the protein and mRNA message level, implying that the reduction is transcriptional. Subsequent studies in adult cardiomyocytes confirmed that decreased BIN1 reduces forward trafficking of Cav1.2, also diminishing intracellular calcium transients (in isolated cells and zebrafish hearts) and contractility (of zebrafish hearts). Recent rat studies confirmed that BIN1 is decreased in failing hearts and recovers with successful treatment.91 It is therefore possible that reduced contractility of progressively failing heart can be traced, in part, to decreased transcription of BIN1. Understanding the regulation of BIN1 transcription is currently an active investigation.
In keeping with the cell to bedside theme of this book, it is worth mentioning that the potential for BIN1 as a blood-available biomarker for arrhythmogenic right ventricular cardiomyopathy (ARVC) was also recently identified.92 ARVC is a primary myocardial disorder with a high incidence of ventricular arrhythmias.93 In a retrospective study of 24 patients, plasma BIN1 predicted cardiac function status and incidence of ventricular arrhythmias. BIN1 has high specificity and sensitivity in distinguishing ARVC patients with severe disease from those with milder symptoms. In addition, BIN1 levels correlated inversely with disease progression in serial blood draws and predicted future arrhythmias in patients without severe heart failure with 82% accuracy.92 Current studies are focused on why BIN1, which is an intracellular yet membrane-attached protein, is also available in the blood.
Pharmacologic Manipulation of Channel Trafficking and Implications for Cardiac/Antiarrhythmic Therapy
New therapeutic strategies that focus on the regulation of ion channel surface density are emerging.94,95 Traditional antiarrhythmic drugs target the ion permeability of channels; however, this approach has not yet yielded a satisfactory outcome. There are two ways to decrease channel current: either through a direct effect on the conduction properties (classically pore block) of channel subunits or through alterations in surface density of the protein. The concept of drugs modulating ion conduction and/or surface density of channels it not new. Research into the therapeutic potential of antiarrhythmic drugs that alter channel trafficking has focused primarily on hERG K+ channels. Alterations in hERG-mediated IKr current, whether drug-induced or a result of the more than 200 naturally occurring mutations of this channel, may induce or contribute to the development of long QT syndrome. Nearly 70% of these mutant channels can be rescued to the plasma membrane by antiarrhythmic drugs such as E4031.96,97 The precise mechanism by which drugs decrease the cell surface expression of the hERG channel protein is uncertain. These drugs likely act chronically to stabilize misfolded protein through facilitation of quality control machinery in the endoplasmic reticulum to facilitate its maturation and export from the endoplasmic reticulum.98,99 Importantly, these studies give credence to the idea that antiarrhythmic drugs may be developed to manipulate specific ion channel trafficking pathways as a novel therapeutic approach for treating cardiac arrhythmias.
Recently, a previously unrecognized mechanism of antiarrhythmic drug action in the acute modulation of Kv1.5 channel trafficking was reported.94 Using quinidine, an antiarrhythmic agent that has both class Ia actions100,101 and class III actions in mammalian atrium and ventricle, it demonstrated that channel blockers can both inhibit ion conduction and regulate the stability of the channel protein within the membrane. In this study, quinidine resulted in a dose- and time-dependent internalization of Kv1.5, concomitant with channel block. Interestingly, this quinidine-induced internalization of Kv1.5 was found to be subunit-dependent and stereospecific,94 which highlights the possibility for the development of atrial selective agents that specifically modulate surface density. Although much work needs to be done to identify the precise cellular trafficking machinery and the mechanisms regulating ion channel surface density, the recent studies highlighted in this chapter suggest that modulation of ion channel trafficking pathways may be an alternative or complementary strategy for treating cardiac arrhythmias.
References
1. Ellgaard, L, Helenius, A. Quality control in the endoplasmic reticulum. Nat Rev. 2003; 4:181–191.
2. Olson, TM, et al. Kv1. 5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006; 15:2185–2191.
3. London, B, et al. Targeted replacement of KV1. 5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of I(K,slow) and resistance to drug-induced qt prolongation. Circ Res. 2001; 88:940–946.
4. Marks, DL, Pagano, RE. Endocytosis and sorting of glycosphingolipids in sphingolipid storage disease. Trends Cell Biol. 2002; 12:605–613.
5. Maxfield, FR, McGraw, TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004; 5:121–132.
6. Prekeris, R. Rabs, Rips, FIPs, and endocytic membrane traffic. Sci World J. 2003; 3:870–880.
7. Brown, SS. Cooperation between microtubule- and actin-based motor proteins. Annu Rev Cell Dev Biol. 1999; 15:63–80.
8. Dantzig, JA, Liu, TY, Goldman, YE. Functional studies of individual myosin molecules. Ann N Y Acad Sci. 2006; 1080:1–18.
9. Langford, GM. Myosin-V, a versatile motor for short-range vesicle transport. Traffic. 2002; 3:859–865.
10. Seabra, MC, Coudrier, E. Rab GTPases and myosin motors in organelle motility. Traffic. 2004; 5:L393–399.
11. Steele, DF, Eldstrom, J, Fedida, D. Mechanisms of cardiac potassium channel trafficking. J Physiol. 2007; 582:17–26.
12. Steele, DF, Zadeh, AD, Loewen, ME, et al. Localization and trafficking of cardiac voltage-gated potassium channels. Biochem Soc Trans. 2007; 35:1069–1073.
13. Deutsch, C. The birth of a channel. Neuron. 2003; 40:265–276.
14. Deutsch, C. Potassium channel ontogeny. Annu Rev Physiol. 2002; 64:19–46.
15. Staudacher, I, Schweizer, PA, Katus, HA, et al. hERG: protein trafficking and potential for therapy and drug side effects. Curr Opin Drug Disc Dev. 2010; 13:23–30.
16. Hamm-Alvarez, SF, Sheetz, MP. Microtubule-dependent vesicle transport: modulation of channel and transporter activity in liver and kidney. Physiol Rev. 1998; 78:1109–1129.
17. Gaietta, G, et al. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002; 296:503–507.
18. Shaw, RM, et al. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007; 128:547–560.
19. Johnson, RG, et al. Gap junctions assemble in the presence of cytoskeletal inhibitors, but enhanced assembly requires microtubules. Exp Cell Res. 2002; 275:67–80.
20. Lauf, U, et al. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci U S A. 2002; 99:10446–10451.
21. Zadeh, AD, et al. Kif5b is an essential forward trafficking motor for the Kv1. 5 cardiac potassium channel. J Physiol. 2009; 587:4565–4574.
22. Zhang, SS, et al. Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network. Proc Natl Acad Sci U S A. 2011; 108:13576–13581.
23. Hong, TT, et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 2010; 8:e1000312.
24. Hong, TT, et al. BIN1 is reduced and Cav1. 2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 2012; 9:812–820.
25. Smyth, JW, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. 2010; 120:266–279.
26. Smyth, JW, et al. Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circ Res. 2012; 110:978–989.
27. Smyth, JW, Shaw, RM. The gap junction life cycle. Heart Rhythm. 2012; 9:151–153.
28. Sohl, G, Willecke, K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes. 2003; 10:173–180.
29. van Kempen, MJ, Fromaget, C, Gros, D, et al. Spatial distribution of connexin43, the major cardiac gap junction protein, in the developing and adult rat heart. Circ Res. 1991; 68:1638–1651.
30. Fromaget, C, el Aoumari, A, Gros, D. Distribution pattern of connexin 43, a gap junctional protein, during the differentiation of mouse heart myocytes. Differentiation. 1992; 51:9–20.
31. Eckardt, D, et al. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J Mol Cell Cardiol. 2004; 36:101–110.
32. Danik, SB, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004; 95:1035–1041.
33. Gutstein, DE, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001; 88:333–339.
34. Shaw, RM, Rudy, Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997; 81:727–741.
35. Falk, MM, Baker, SM, Gumpert, AM, et al. Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell. 2009; 20:3342–3352.
36. Piehl, M, et al. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007; 18:337–347.
37. Rhett, JM, Jourdan, J, Gourdie, RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011; 22:1516–1528.
38. Theiss, C, Meller, K. Microinjected anti-actin antibodies decrease gap junctional intercellular commmunication in cultured astrocytes. Exp Cell Res. 2002; 281:197–204.
39. Thomas, T, Jordan, K, Laird, DW. Role of cytoskeletal elements in the recruitment of Cx43-GFP and Cx26-YFP into gap junctions. Cell Commun Adhes. 2001; 8:231–236.
40. Dai, S, Hall, DD, Hell, JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009; 89:411–452.
41. Benitah, JP, Alvarez, JL, Gomez, AM. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010; 48:26–36.
42. Fang, K, Colecraft, HM. Mechanism of auxiliary beta-subunit-mediated membrane targeting of L-type (Ca(V)1. 2) channels. J Physiol. 2011; 589:4437–4455.
43. Yasuda, T, et al. Auxiliary subunit regulation of high-voltage activated calcium channels expressed in mammalian cells. Eur J Neurosci. 2004; 20:1–13.
44. Simms, BA, Zamponi, GW. Trafficking and stability of voltage-gated calcium channels. Cell Mol Life Sci. 2012; 69:843–856.
45. O’Connell, KM, Whitesell, JD, Tamkun, MM. Localization and mobility of the delayed-rectifer K+ channel Kv2. 1 in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008; 294:H229–H237.
46. Maruoka, ND, et al. Alpha-actinin-2 couples to cardiac Kv1. 5 channels, regulating current density and channel localization in HEK cells. FEBS Lett. 2000; 473:188–194.
47. Mason, HS, Latten, MJ, Godoy, LD, et al. Modulation of Kv1. 5 currents by protein kinase A, tyrosine kinase, and protein tyrosine phosphatase requires an intact cytoskeleton. Mol Pharmacol. 2002; 61:285–293.
48. Nakahira, K, Matos, MF, Trimmer, JS. Differential interaction of voltage-gated K+ channel beta-subunits with cytoskeleton is mediated by unique amino terminal domains. J Mol Neurosci. 1998; 11:199–208.
49. Shi, G, et al. Beta subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron. 1996; 16:843–852.
50. Manganas, LN, Trimmer, JS. Subunit composition determines Kv1 potassium channel surface expression. J Biol Chem. 2000; 275:29685–29693.
51. Mathur, R, et al. A specific N-terminal residue in Kv1. 5 is required for upregulation of the channel by SAP97. Biochem Biophys Res Commun. 2006; 342:1–8.
52. Murata, M, et al. SAP97 interacts with Kv1. 5 in heterologous expression systems. Am J Physiol Heart Circ Physiol. 2001; 281:H2575–H2584.
53. Li, H, Guo, W, Mellor, RL, et al. KChIP2 modulates the cell surface expression of Kv 1. 5-encoded K(+) channels. J Mol Cell Cardiol. 2005; 39:121–132.
54. McEwen, DP, Li, Q, Jackson, S, et al. Caveolin regulates kv1. 5 trafficking to cholesterol-rich membrane microdomains. Mol Pharmacol. 2007; 73:678–685.
55. Abi-Char, J, et al. Membrane cholesterol modulates Kv1. 5 potassium channel distribution and function in rat cardiomyocytes. J Physiol. 2007; 582:1205–1217.
56. Folco, EJ, Liu, GX, Koren, G. Caveolin-3 and SAP97 form a scaffolding protein complex that regulates the voltage-gated potassium channel Kv1. 5. Am J Physiol Heart Circ Physiol. 2004; 287:H681–H690.
57. Chen, L, Kass, RS. A-kinase anchoring protein 9 and IKs channel regulation. J Cardiovasc Pharmacol. 58, 2011. [459–413].
58. Abi-Char, J, et al. The anchoring protein SAP97 retains Kv1. 5 channels in the plasma membrane of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2008; 294:H1851–H1861.
59. Abi-Char, J, et al. Membrane cholesterol modulates Kv1. 5 potassium channel distribution and function in rat cardiomyocytes. J Physiol. 2007; 582:1205–1217.
60. Eldstrom, J, Choi, WS, Steele, DF, et al. SAP97 increases Kv1. 5 currents through an indirect N-terminal mechanism. FEBS Lett. 2003; 547:205–211.
61. Folco, EJ, Liu, GX, Koren, G. Caveolin-3 and SAP97 form a scaffolding protein complex that regulates the voltage-gated potassium channel Kv1. 5. Am J Physiol Heart Circ Physiol. 2004; 287:H681–690.
62. Martens, JR, Sakamoto, N, Sullivan, SA, et al. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1. 5 to caveolae. J Biol Chem. 2001; 276:8409–8414.
63. Maruoka, ND, et al. Alpha-actinin-2 couples to cardiac Kv1. 5 channels, regulating current density and channel localization in HEK cells. FEBS Lett. 2000; 473:188–194.
64. Mathur, R, et al. A specific N-terminal residue in Kv1. 5 is required for upregulation of the channel by SAP97. Biochem Biophys Res Commun. 2006; 342:1–8.
65. McEwen, DP, et al. Rab-GTPase-dependent endocytic recycling of Kv1. 5 in atrial myocytes. J Biol Chem. 2007; 282:29612–29620.
66. Choi, WS, et al. Kv1. 5 surface expression is modulated by retrograde trafficking of newly endocytosed channels by the dynein motor. Circ Res. 2005; 97:363–371.
67. Balse, E, et al. Cholesterol modulates the recruitment of Kv1. 5 channels from Rab11-associated recycling endosome in native atrial myocytes. Proc Natl Acad Sci U S A. 2009; 106:14681–14686.
68. Seebohm, G, et al. Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ Res. 2007; 100:686–692.
69. Delisle, BP, et al. Small GTPase determinants for the Golgi processing and plasmalemmal expression of human ether-a-go-go related (hERG) K+ channels. J Biol Chem. 2009; 284:2844–2853.
70. Zupkovitz, G, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol. 2006; 26:7913–7928.
71. Van Wagoner, DR, Pond, AL, McCarthy, PM, et al. Outward K+ current densities and Kv1. 5 expression are reduced in chronic human atrial fibrillation. Circ Res. 1997; 80:772–781.
72. Brundel, BJ. Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: differential regulation of protein and mRNA levels for K+ channels. J Am Coll Cardiol. 2001; 37:926–932.
73. Michelakis, ED, Weir, EK. The pathobiology of pulmonary hypertension. Smooth muscle cells and ion channels. Clin Chest Med. 2001; 22:419–432.
74. Tanabe, Y, et al. Over-expression of Kv1. 5 in rat cardiomyocytes extremely shortens the duration of the action potential and causes rapid excitation. Biochem Biophys Res Commun. 2006; 345:1116–1121.
75. London, B, et al. Long QT and ventricular arrhythmias in transgenic mice expressing the N terminus and first transmembrane segment of a voltage-gated potassium channel. Proc Natl Acad Sci U S A. 1998; 95:2926–2931.
76. Green, CR, Severs, NJ. Robert Feulgen Prize Lecture. Distribution and role of gap junctions in normal myocardium and human ischaemic heart disease. Histochemistry. 1993; 99:105–120.
77. Peters, AM, Bertram, P, Gahr, M, et al. Reduced secretion of interleukin-1 and tumor necrosis factor-alpha by neonatal monocytes. Biol Neonate. 1993; 63:157–162.
78. Smith, JH, Green, CR, Peters, NS, et al. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991; 139:801–821.
79. Beardslee, MA, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000; 87:656–662.
80. Ai, X, Pogwizd, SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res. 2005; 96:54–63.
81. Miyoshi, J, Takai, Y. Structural and functional associations of apical junctions with cytoskeleton. Biochim Biophys Acta. 2008; 1778:670–691.
82. Giepmans, BN, Moolenaar, WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998; 8:931–934.
83. Toyofuku, T, et al. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem. 1998; 273:12725–12731.
84. Zhu, C, et al. Quantitative analysis of ZO-1 colocalization with Cx43 gap junction plaques in cultures of rat neonatal cardiomyocytes. Microsc Microanal. 2005; 11:244–248.
85. Hunter, AW, Barker, RJ, Zhu, C, et al. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005; 16:5686–5698.
86. Bruce, AF, Rothery, S, Dupont, E, et al. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc Res. 2008; 77:757–765.
87. Splawski, I, et al. Ca(V)1. 2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004; 119:19–31.
88. Splawski, I, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005; 102:8089–8096.
89. Yang, Y, et al. L-type Ca2+ channel alpha 1c subunit isoform switching in failing human ventricular myocardium. J Mol Cell Cardiol. 2000; 32:973–984.
90. Antzelevitch, C, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007; 115:442–449.
91. Lyon, AR, et al. Plasticity of surface structures and beta2-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail. 2012; 5(3):357–365.
92. Hong, TT, et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2012; 9:961–967.
93. Sen-Chowdhry, S, Morgan, RD, Chambers, JC, et al. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010; 61:233–253.
94. Schumacher, SM, et al. Antiarrhythmic drug-induced internalization of the atrial-specific k+ channel kv1. 5. Circ Res. 2009; 104:1390–1398.
95. van der Heyden, MA, Smits, ME, Vos, MA. Drugs and trafficking of ion channels: a new pro-arrhythmic threat on the horizon? Br J Pharmacol. 2008; 153:406–409.
96. Ficker, E, Dennis, A, Kuryshev, Y, et al. HERG channel trafficking. Novartis Found Symp. 2005; 266:57–69.
97. Ficker, E, Obejero-Paz, CA, Zhao, S, et al. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J Biol Chem. 2002; 277:4989–4998.
98. Ficker, E, Dennis, AT, Wang, L, et al. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. 2003; 92:e87–e100.
99. Walker, VE, Atanasiu, R, Lam, H, et al. Co-chaperone FKBP38 promotes HERG trafficking. J Biol Chem. 2007; 282:23509–23516.
100. Colatsky, TJ. Mechanisms of action of lidocaine and quinidine on action potential duration in rabbit cardiac Purkinje fibers. An effect on steady state sodium currents? Circ Res. 1982; 50:17–27.
101. Slawsky, MT, Castle, NA. K+ channel blocking actions of flecainide compared with those of propafenone and quinidine in adult rat ventricular myocytes. J Pharmacol Exp Ther. 1994; 269:66–74.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]17
Ion Channel Trafficking in the Heart
Ion Channel Trafficking versus Quality Control
Anterograde Trafficking of Channels in the Heart
At Their Destination: Channels in the Myocyte Membrane
Retrograde Trafficking of Channels in the Heart
Regulation of Channel Trafficking
Alterations of Channel Trafficking in Pathophysiological States
Pharmacologic Manipulation of Channel Trafficking and Implications for Cardiac/Antiarrhythmic Therapy
Ion Channel Trafficking versus Quality Control
The steady-state cell surface density of ion channel proteins is determined by the balance between the anterograde and retrograde trafficking pathways. Anterograde trafficking ensues only after proper protein synthesis and processing in the endoplasmic reticulum and Golgi apparatus, including quality control mechanisms, glycosylation, and posttranslation modification (Figure 17-1).1 Often, channel trafficking, synthesis, and quality control are grouped together in common discussion. However, these processes can be distinct, using unique cellular machinery and subject to differential regulation. Retrograde movement initiates with endocytosis, after which internalized proteins can follow multiple routes to different intracellular fates (see Figure 17-1).2 One well-recognized fate is the targeting of internalized proteins to lysosomes, or proteasomes, followed by degradation (see Figure 17-1). Alternatively, trafficking through recycling endosomes allows proteins to return to the plasma membrane and protects them from degradation (see Figure 17-1).3 Sorting at early endosomes to Rab-GTPase–specific compartments is now established as an important event in determining the intracellular fate of internalized proteins.4–6 Another important component of the endocytic machinery regulating protein surface levels is the coordinated movement of molecular motors. In general, protein trafficking is highly coordinated between long-range events involving the microtubule-based kinesin and dynein motors, and short-range events using unconventional myosin motors.7–10 There is a significant and growing body of literature concerning ion channel trafficking from synthesis to sorting to degradation in multiple tissues and cells systems that has been reviewed previously.11–15 Our discussion will center mainly on recent work focused on the control of ion channel density at the plasma membrane, in the heart, where relatively little is known about protein trafficking. We have chosen to organize this chapter around the major transport events in ion channel movement within myocytes. Given that numerous and different ion channels play an essential role in all chambers of the heart and throughout the cardiovascular system, it is impossible to include a comprehensive review of all the literature. Rather, it is our intention to use select examples to highlight the major themes that have been recently uncovered in ion channel trafficking.
Figure 17-1 Ion channel trafficking pathways in the heart. Anterograde and retrograde transport using kinesin motors on microtubules and myosin motors on actin filaments is illustrated. Channel trafficking to subcellular compartments including intercalated disc, T-tubule, and lateral membrane is highlighted.
Anterograde Trafficking of Channels in the Heart
The Directed Targeting Paradigm
Posttranscription and posttranslation, the proteins that form cardiac ion channels are modified and usually oligomerize in the Golgi apparatus, where they are inserted into membrane vesicles for delivery to the plasma membrane. These channel-laden vesicles are transported using the cytoskeleton as a path to the plasma membrane, with most studies focusing on the role of microtubule-based forward transport.16–21 A critical aspect of forward transport is localization to membrane subdomains. It is entirely possible that channels may be delivered to random regions of the plasma membrane, only to then laterally diffuse within the membrane to their appropriate subdomain.17 However, the temporal and stochastic inefficiency of random channel insertion, together with unexplained mechanisms of subsequent lateral localization other than chance interaction with a subdomain-specific anchor protein, suggests that specificity of delivery from the Golgi apparatus to the surface submembrane may also occur. Still to be fully explored, this directed targeting paradigm of ion channel delivery may be generalizable to all cardiac ion channels and explored in terms of other cytoskeletal elements and anchor proteins (discussed next).
This section of the chapter focuses on mechanisms that govern the forward trafficking of two major cardiac ion channels that are indispensable for cellular excitability, cell-cell coupling, and excitation-contraction coupling: (1) the L-type calcium channel Cav1.2 and (2) the dominant cardiac ventricular gap junction, connexin43 (Cx43).22–26 We also discuss in detail a protein with a newly described role in the heart that is essential for Cav1.2 trafficking and delivery, bridging integrator 1 (BIN1). BIN1 is rapidly emerging as a multifunctional cardiac player beyond its previously understood function as a membrane scaffold.
Forward Trafficking of Cx43
Gap junctions are intercellular channels that form low-resistance pathways, allowing ions and metabolites to flow from cell to cell. In the heart, gap junctions electrically couple cardiomyocytes to orchestrate spatial propagation of action potentials.27 A gap junction consists of a pair of abutting hexameric hemichannels or connexons in adjacent cell membranes. Each connexon consists of six connexin proteins. More than 20 Cxs have been identified and each is usually named according to its respective molecular mass.28 All connexins contain four transmembrane domains. Connexin43 is the most abundant connexin in the ventricular myocardium. In individual ventricular cardiomyocytes, Cx43 is localized at the intercalated disc at longitudinal ends of the cell, where Cx43 gap junctions provide rapid action potential transmission and synchronized cardiac excitation.29,30
Cardiac-specific conditional deletion models with greater than 50% Cx43 loss develop increased arrhythmia susceptibility and sudden cardiac death.31–33 In general, these studies reveal that severe arrhythmia phenotypes can manifest when Cx43 coupling drops to less than 20% in the heart, which correlates with theoretic predictions.34 Thus, maintenance of proper Cx43 expression and localization has a critical role in appropriate electrical coupling and ventricular function.
Data exist for multiple, but not incompatible, models of Cx43 trafficking to cell-cell borders of cardiac intercalated discs.17,18,20,25 There is almost universal agreement that microtubules help deliver Cx43 to the plasma membrane. A landmark series of two papers in 2002 found evidence that newly-formed Cx43 appear at the perimeter of the Cx43 plaques and then diffuse into central plaque regions.17,20 Taken together with microtubule delivery, the model developed that Cx43 hemichannels are inserted into the general plasma membrane, rapidly diffuse to the edge of dense plaques, and then more slowly diffuse into the plaque center. Subsequent to these studies, it has been observed that Cx43 can be inserted directly at the edge and into plaques, and membrane fluidity exits within the plaque region.18,35 It has also been found that the plaques are internalized not necessarily from the center, but that different segments of plaque can be internalized at any time35 and full plaque internalization can occur in one step.36 More recently, it has been found that Cx43 occurs in regions surrounding Cx43 plaques, the “perinexus,” at higher density than in general membrane where Cx43 proteins may interact with scaffolding proteins and other ion channels.37 These studies are providing evidence that the gap junction plaque and surrounding regions are highly dynamic with complex behavior of targeted insertion and internalization.
Given the low density of Cx43 hemichannels in membrane well away from Cx43 plaque regions, and the technical difficulty of distinguishing Cx43 inserted in membrane from submembranous and still cytoplasmic collections of protein, it is difficult to find studies that can quantify the lateral diffusion coefficient of membrane-bound Cx43. It may be that free hemichannels rapidly diffuse within the plasma membrane before stopping at plaque regions, but direct evidence for this phenomenon is lacking. The directed targeting paradigm is based on the observation that de novo intracellular Cx43 hemichannels can arrive directly in the gap junction plaque region. The hemichannels are targeted to plaque regions with specificity obtained from the channel protein (Cx43), microtubule plus-end tracking proteins (EB1 and p150 [glued], and a membrane anchor [adherens junction structure]).18 By this model, the dynamic microtubule highways are anchored and terminate at adherens junction structures, allowing directed delivery of Cx43 hemichannels to adherens junction–containing membrane to occur. It is probable that once inserted into plaque regions, local hemichannel diffusion occurs within the plaque and between the plaque and the plaque perinexus.
Although most studies of Cx43 forward trafficking focus on the microtubule cytoskeleton, the actin cytoskeleton is also involved in delivery of membrane proteins and ion channels. Dye transfer studies revealed the dependence of Cx43 insertion into plaque on actin, as tested by pharmacologic actin disruption and anti-actin antibodies.38,39 Actin is also important for Cx43 plaque internalization, although we recently found that when internalization is blocked, forward delivery is still actin dependent.26 Furthermore, a remarkable greater than 80% of cytoplasmic post-Golgi Cx43 is slow-moving or stationary and actin-associated. These findings indicate that microtubules work in concert with actin to deliver Cx43 to the plasma membrane. It may be that post-Golgi Cx43 vesicles exist in actin-associated reservoirs within the cytoplasm, waiting for either the right microtubule to permit membrane delivery or mass acute delivery in the case of a metabolic stress.
Forward Trafficking of Cav1.2
Although connexins electrically couple cardiomyocytes, the membrane ion channel most responsible for calcium entry and excitation-contraction coupling is the α1C pore-forming subunit (Cav1.2) of the L-type voltage-gated (Cav) Ca2+ channel (LTCC). In response to membrane depolarization by sodium currents, LTCCs open to allow inward Ca2+ entry during the plateau phase of the cardiac action potential. The L-type currents (ICa,L) that are generated initiate calcium-induced Ca2+ release from the sarcoplasmic reticulum (SR) via ryanodine receptors, which leads to muscle contraction. The Cav1.2 protein is large and consists of 24 transmembrane segments organized into four homologous domains.40 Auxiliary subunits of the LTCC include the β-subunit, the α2δ-subunit, and the γ subunit, which help regulate trafficking of Cav1.2 to the cell membrane and regulate the voltage dependence of channel gating.41
T-tubule invaginations of ventricular cardiomyocyte plasma membrane are enriched with LTCCs. This enrichment is necessary for calcium-induced calcium release with nearby ryanodine receptors, which is important for beat-to-beat excitation-contraction coupling in the heart. Trafficking of the LTCCs is not as extensively studied as that of Cx43, probably owing to a combination of the large size of the α-subunit, Cav1.2, the multiple β-subunits, the difficulty of manipulating cardiomyocytes with T-tubules in culture, and less dense enrichment of the channel on the plasma membrane, presenting difficulty to clearly characterize by cytochemistry a distinct phenotype. Subunits are important for LTCC trafficking. For instance, the β-subunit is necessary for surface expression.42 Moreover, the α2δ-subunit synergizes with the β-subunit to promote surface channel expression.43 These auxiliary subunits enhance surface expression by promoting channel export from the endoplasmic reticulum and overall channel stability.44
Despite the necessary localization of LTCC to T-tubules, it was not understood until recently how the localization occurs. In 2010, it was found that Cav1.2-based channels adhere to the directed targeted paradigm.23 In particular, the membrane-scaffolding protein BIN1 was found to provide a membrane anchor that attaches dynamic microtubules, allowing delivery of Cav1.2 channels directly to T-tubule membranes. Using HL-1 cells that express Cav1.2 but do not form T-tubules, and non-muscle cell lines that neither express Cav1.2 nor naturally form T-tubules, it was found that exogenous BIN1 caused the formation of deep invaginations in the cell membrane enriched with endogenous or overexpressed Cav1.2, suggesting that BIN1-containing membrane is sufficient to recruit Cav1.2 channels. To test the possibility that BIN1 serves as an anchoring site for microtubules on which Cav1.2 channels are trafficked, the study tracked growing microtubules extending toward BIN1 clusters and found that the microtubule plus end-tracking proteins pause and associate with BIN1 clusters at the cell periphery. Moreover, it was determined that the non-BAR cytoskeleton–anchoring domain of BIN1 is required for this activity because truncation mutants lacking this domain failed to cluster Cav1.2 at cell surface invaginations. The microtubule plus end-tracking protein that may aid in microtubule anchoring to BIN1, analogous to EB1 binding to adherens junctions,18,25 has not yet been identified.
At Their Destination: Channels in the Myocyte Membrane
Subcellular Localization of Kv Channels Within Myocytes
Historically, Kv localization is believed to primarily involve protein-protein interactions among channel proteins and PDZ-domain–containing scaffolding proteins or the actin cytoskeleton. Recent work shows that Kv2.1 channels are immobilized on the lateral membrane of atrial myocytes, where their diffusion is likely limited by actin cytoskeletal corrals as in hippocampal neurons.45 In addition, Kv1.5 has been shown to directly couple to α-actinin-2 in HEK 293 cells via a specific sequence in the amino terminus of the channel.46 In addition, Kv1.5-cytoskeleton interactions appear to play a role in modulation of the channel by protein kinase A in oocytes.47 Further, the actin binding protein, cortactin, regulates Kv1.5./N-cadherin interactions at the intercalated disc. Kvβ-subunits are also likely to interact with the cytoskeleton and thus immobilize channels on the cell surface.48 In addition, some Kvβ-subunits behave as chaperone proteins, promoting the cell surface expression of a subset of co-expressed α-subunits and perhaps influencing localization.49,50 A role for PDZ domain–containing scaffolding proteins, such as SAP97, in the regulation of Kv1.5 surface localization has also been described.51,52
[/not-level-membership-for-cardiovascular-category]
