[level-membership-for-neurology-category]
Chapter 18 Involuntary Movement Disorders
The Basal Ganglia
Five subcortical gray-matter macroscopic nuclei (Figs 18-1 and 18-2) constitute the basal ganglia:
• The caudate nucleus and putamen, which together constitute the striatum
• The globus pallidus, which, together with the putamen, constitutes the lenticular nuclei
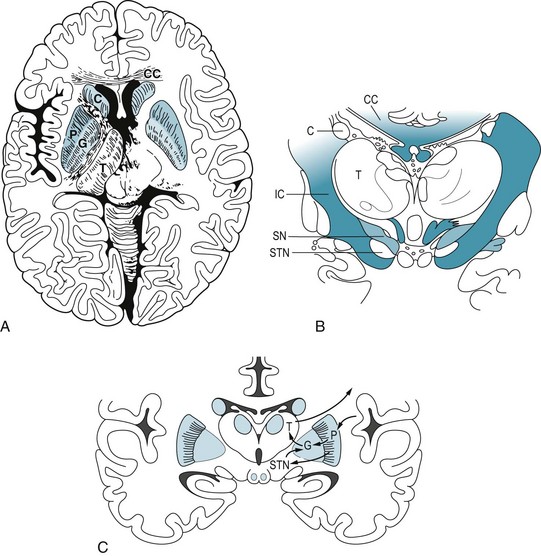
FIGURE 18-1 A, This axial view, the one used in computed tomography and magnetic resonance imaging studies, shows the basal ganglia in relation to other brain structures. The heads of the caudate nuclei (C) indent the lateral undersurface of the anterior horns of the lateral ventricles. The caudate and putamen (P) constitute the striatum. The globus pallidus (G), which has internal and external segments, and the putamen form the lenticular nucleus, named for its resemblance to an old-fashioned lens (see Fig. 18-1, C). The posterior limb of the internal capsule (IC) separates the lenticular nucleus from the thalamus (T), which is not a member of the basal ganglia family. B, In this coronal view of the diencephalon, the substantia nigra (SN), as well as the subthalamic nuclei (STN), sits below the thalamus. The substantia nigra, because of its characteristic shape and pigmentation, serves as a landmark. The lateral ventricles are bounded laterally by the heads of the caudate nuclei (C) and superiorly by the corpus callosum (CC). This myelin stain has blackened the heavily myelinated fibers of the internal capsule (IC) and corpus callosum (CC). C, A coronal view shows extrapyramidal circuits. The putamen sends a direct and an indirect dopamine tract to the internal segment of the globus pallidus (GPi). Dopaminergic neurons in the substantia nigra project to the putamen, where neurons with D1 receptors project directly to the GPi. Putaminal neurons with D2 receptors project through the globus pallidus external segment (GPe) and subthalamic nucleus and thence to GPi. The GPi innervates the ventrolateral nucleus of the thalamus, which projects to the motor cortex. The cortex, completing a circuit, innervates the putamen.
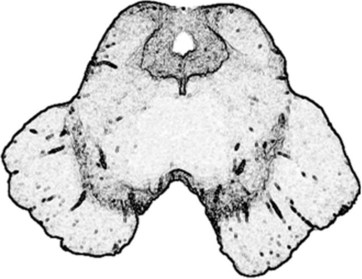
FIGURE 18-2 This computer-generated rendition of the midbrain should be compared to a photograph (see Fig. 2-9), functional drawings (see Figs 4-5 and 4-9), and an idealized sketch (see Fig. 21-1). The lower third of the midbrain, which lies just caudal to the diencephalon, contains the pair of horizontal but gently curved, elongated, pigmented nuclei – the substantia nigra. In Parkinson disease, the substantia nigra and other pigmented nuclei lose their pigment and, compared to normal, thus appear blanched. The midbrain also houses the prominent aqueduct of Sylvius, which is the dorsal heart-shaped hole surrounded by the periaqueductal gray matter. Cerebrospinal fluid passes from the third ventricle, through the aqueduct, to the fourth ventricle.
Although the extrapyramidal tract seems to play merely a supportive role – indirectly influencing the corticospinal tract by acting on thalamocortical connections and projecting only within the brain – it comprises several clinically important tracts. The most important one, from a clinical viewpoint, is the nigrostriatal tract, which, as its name suggests, extends from the substantia nigra to the striatum (Fig. 18-3). This tract provides dopamine innervation directly and indirectly, via the subthalamic nucleus, to the globus pallidus’ internal segment (GPi).
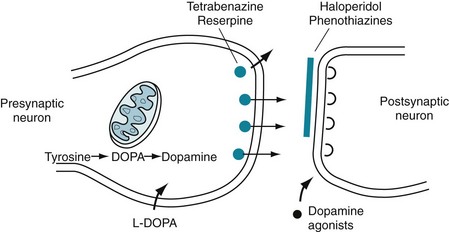
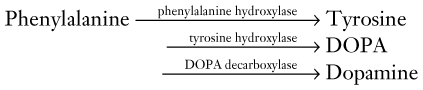
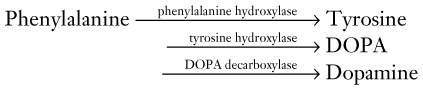
FIGURE 18-3 A succession of enzymes normally converts tyrosine to dopamine in the presynaptic nigrostriatal neuron. In Parkinson disease, the nigrostriatal tract degenerates, but the postsynaptic dopamine receptors remain intact. As a result of the degeneration, an absence of tyrosine hydroxylase leads to insufficient dopa and then markedly reduced synthesis of dopamine. L-dopa, which is a standard oral medication for Parkinson disease, penetrates the blood–brain barrier and substitutes for the deficient endogenous dopa in dopamine synthesis. D-dopa, the dextro-isomer, does not cross the blood–brain barrier and cannot enter the synthetic pathway. It is therefore useless as a treatment for Parkinson disease.
One of the main differences between dopamine receptors is that, in the striatum, dopamine binding to dopamine 1 (D1) receptors stimulates adenyl cyclase activity, but dopamine binding to dopamine 2 (D2) receptors inhibits adenyl cyclase activity (see Table 21-1).
General Considerations
The involuntary movement disorders share several clinical features. Anxiety, exertion, fatigue, and stimulants (including caffeine) increase the movements, but willful concentration and sometimes biofeedback may suppress them, at least transiently. Most movements disappear during sleep. The exceptions – hemifacial spasm, myoclonus, palatal tremor, tics, and specific sleep-related disorders, such as restless legs syndrome (RLS) and periodic movements – persist without regard to sleep stage (see Chapter 17).
Neurologists typically measure four parameters of involuntary movements:
• Hypokinesia or hyperkinesia. (For most clinical purposes, Parkinson disease and parkinsonism [see later] constitute the sole hypokinetic involuntary movement disorder.)
• Continuous or intermittent, discrete movements
Another error may occur when patients, at first glance, appear to have a psychogenic movement disorder (see later and Chapter 3). In many situations, the lack of a definitive confirmatory laboratory test forces neurologists to rely exclusively on their clinical judgment.
Parkinson Disease
The initial and ultimately most disabling physical feature of Parkinson disease is usually bradykinesia or, in the extreme, akinesia. Slow or absent movement produces the classic masked face (Fig. 18-4), paucity of trunk and limb movement (Figs 18-5 and 18-6), and impairment of activities of daily living. Parkinson patients sometimes liken their slow movements to slogging through hip-deep mud, wearing lead clothing, or driving a car with an engaged emergency brake.

FIGURE 18-4 Compared to normal individuals of the same age, Parkinson disease patients blink less frequently, offer fewer facial expressions, and less often move their head. Neurologists have called patients’ facial appearance a “stare” or “masked facies” (Latin, face or countenance), but now they usually describe the face as masked. Even when subtle, the masked face gives the appearance of apathy or depression.
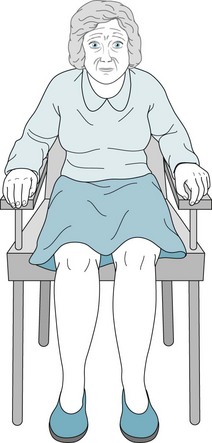
FIGURE 18-5 Parkinson disease patients typically sit motionless with their legs uncrossed and their feet flat. Their arms remain on the chair or in their lap and rarely participate in normal gestures or repositioning movements. In contrast to normal individuals and especially those with chorea, Parkinson disease patients do not shift their weight from one hip to another or make any unnecessary movements.
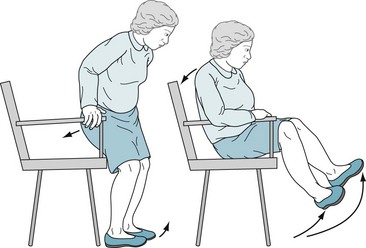
FIGURE 18-6 Patients with akinesia and rigidity cannot rapidly flex their spine, hips, or knees. When sitting, they tend to fall slowly and solidly backward into a chair. Unable to bend rapidly, their feet rise several inches off the floor. Sitting and turning en bloc signal early parkinsonism.
Rigidity typically accompanies bradykinesia (Fig. 18-7). Although rigidity is one of the cardinal features of Parkinson disease, it often appears as a manifestation of other extrapyramidal disorders. No matter the context, physicians should not confuse rigidity with spasticity, which signals corticospinal tract disease (see Chapter 2).
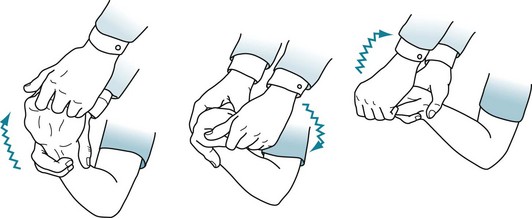
FIGURE 18-7 Physicians typically elicit rigidity by rotating the patient’s wrist. When present, rigidity causes an increased tone in all directions of movement. A superimposed tremor adds a ratchet-like, cogwheel rigidity resistance.
Tremor is often the most conspicuous feature of Parkinson disease; however, it is the least specific sign, least debilitating symptom, and least associated with dementia and depression. In Parkinson disease, the affected body part usually oscillates in a single plane with a regular rate, although with a variable amplitude. It primarily involves the hands. Even more characteristically, the tremor appears when patients rest quietly with their arms supported. Neurologists call it a resting tremor (Fig. 18-8) and can show that it differs from cerebellar and essential tremors. When patients have tremor as their primary symptom, neurologists refer to them as having “tremor-predominant” Parkinson disease.
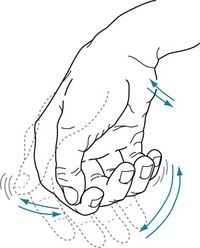
FIGURE 18-8 The resting tremor – a cardinal feature of Parkinson disease – consists of a relatively slow (4–6-Hz) to-and-fro flexion movement of the wrist, hand, thumb, and fingers most apparent when patients sit comfortably. Its similarity to shaking pills in a cupped hand gave rise to the description “pill-rolling” tremor. The tremor is exaggerated or sometimes apparent only when patients are anxious. However, voluntary movement or intense concentration may momentarily reduce the tremor, and sleep abolishes it. Rigidity and akinesia almost always accompany the Parkinson disease resting tremor.
Additional symptoms and signs may emerge as Parkinson disease advances. Patients lose their postural reflexes, which are neurologic compensatory mechanisms that adjust muscle tone in response to change in position. Loss of these reflexes, in combination with akinesia and rigidity, results in a characteristic gait impairment, marche à petit pas or festinating gait, which consists of a tendency to lean forward and accelerate the pace (Fig. 18-9, A). In a test of postural reflexes, the pull test, the examiner pulls the patient from the shoulders (Fig. 18-9, B). Normal individuals merely sway. Patients who have mild impairment of their postural reflexes take a few steps back, i.e., have retropulsion. More severely affected ones rock stiffly backwards without flexion or other compensatory movement and topple, en bloc, into the examiner’s arms.
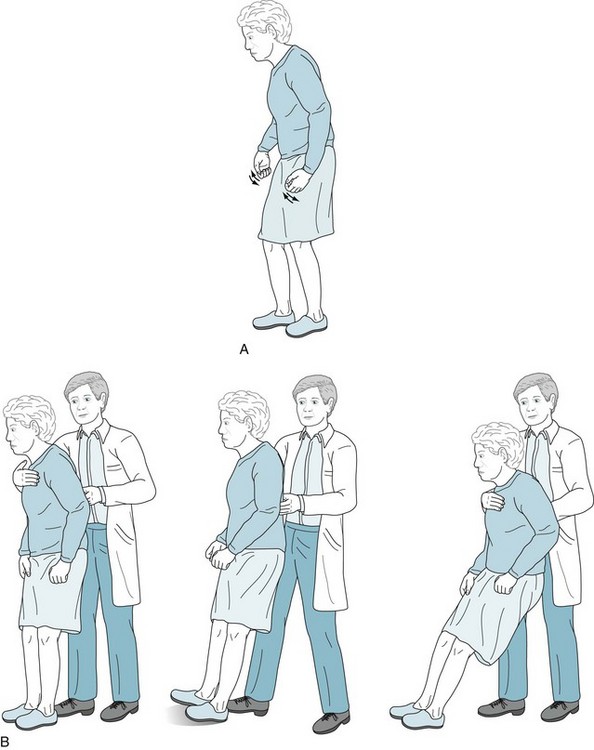
FIGURE 18-9 A, Parkinson disease often produces a festinating gait, in which patients take short, shuffling steps and accelerate their pace. Their neck and lower spine are typically flexed. When walking, they fail to swing their arms, look about, or have other normal accessory movements. Likewise, while turning en bloc, they simultaneously move their head, trunk, and legs. B, The pull test consists of the physician gently but rapidly pulling the patient’s shoulders. Unaffected individuals will compensate by taking one or two steps backward. Parkinson patients, who generally have impaired postural reflexes, will take many steps backward (i.e., have retropulsion), because they are unable to stop through reflexly weight-shifting compensatory maneuvers. In pronounced cases, as the one pictured here, patients unable to alter their posture will tilt backwards en bloc and fall into the physician’s arms. (Progressive supranuclear palsy patients have similar bradykinesia, lack of accessory movements, and a positive pull test, but their posture is extended or erect rather than flexed [see Fig. 7-7].)
Even at the onset of the illness, patients’ handwriting deteriorates to a small and tremulous script, micrographia (Fig. 18-10). In a parallel fashion, their voice loses both volume and normal fluctuations in pitch and cadence, i.e., their speech becomes hypophonic and monotonous. Also, because of their illness or the medications used to treat it, Parkinson disease patients develop sleep disturbances, characteristically rapid eye movement sleep behavior disorder (see Chapter 17).

FIGURE 18-10 The handwriting in this sample from a Parkinson disease patient shows progressive decrease in height (micrographia) and a superimposed tremor. These abnormalities in signatures, such as those on checks, can often date the onset of Parkinson disease. Although essential tremor may also cause tremor in handwriting samples, micrographia indicates that the underlying illness is Parkinson disease.
Nonmotor physical problems also emerge. For example, Parkinson patients characteristically lose their sense of smell. The anosmia, which also occurs in Alzheimer disease, reflects the neurodegenerative nature of these illnesses (see Chapters 4 and 7). They also routinely develop problems with their autonomic nervous system, including dysphagia, constipation, urinary incontinence, and abnormal sweating.
Psychiatric Conditions Comorbid with Parkinson Disease
Treatment of Comorbid Depression
Although SSRIs carry fewer side effects than TCAs in the population of depressed Parkinson disease patients, they may cause a unique problem. Prescribing SSRIs in conjunction with selegiline (deprenyl [Eldepryl]) can theoretically cause the serotonin syndrome because SSRIs prevent serotonin reuptake while selegiline, a monoamine oxidase (MAO) inhibitor, prevents its breakdown (see Chapter 6). The actual incidence of serotonin syndrome is very low because selegiline, in the doses used for Parkinson disease treatment, selectively inhibits only MAO-B, which metabolizes dopamine; whereas the serotonin syndrome is mostly a complication of inhibition of MAO-A, which metabolizes serotonin. Similarly, when physicians prescribe the selegiline patch (Emsam) for depression, as long as the dose remains below 12 mg/day, which is standard, it inhibits MAO-B; however, selegiline patch doses greater than 12 mg/day inhibit MAO-A as well as MAO-B, and leave patients at risk of tyramine-induced hypertension.
Dementia
However, as the illness progresses and patients age, dementia routinely complicates the illness. Dementia affects about 20% of all Parkinson disease patients, 40% of those older than 70 years, and more than 80% who have had the illness for 20 years. Its prevalence increases in proportion to the patient’s age, duration of the illness, and physical impairments. Dementia is more frequent when akinesia and rigidity rather than tremor predominate in Parkinson disease. Unlike the dementia in Alzheimer disease, the dementia in Parkinson disease is not associated with apolipoprotein E4 alleles (see Chapter 7).
Parkinson disease dementia, which differs clinically from that of Alzheimer disease dementia, is distinguished by inattention, poor memory, difficulty shifting mental sets, and bradyphrenia (slowed thinking, the cognitive counterpart of bradykinesia), as well as hallucinations. With its almost invariable gait impairment and preserved language function, Parkinson disease dementia serves as a prime example of “subcortical dementia” (see Chapter 7). The Mini-Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MoCA) are each valid screening tests for cognitive impairment in Parkinson disease; however, the MoCA is more sensitive.
A special diagnostic hazard when evaluating a patient with parkinsonism and dementia is failing to recognize dementia with Lewy bodies disease. This illness and Parkinson disease share rigidity and bradykinesia, delusions and hallucinations, and sleep disturbances as well as cognitive impairment. One major clinical difference is that in dementia with Lewy bodies disease, dementia constitutes an initial, not a late-developing, manifestation (see Chapter 7), but it is at least a relatively late development in Parkinson’s disease. On a histologic level, dementia with Lewy bodies disease features Lewy bodies in the cerebral cortex, not just confined to the basal ganglia.
Treatment of Comorbid Psychosis
Because administration of antiparkinson medication before bedtime tends to cause nightmares and hallucinations, neurologists advance the last daily dose to the early evening. Also, they avoid suddenly stopping these medicines because their abrupt withdrawal may lead to irreversible motor deterioration, complications of immobility, or even the neuroleptic-malignant syndrome (NMS) (see Chapter 6).
Pathology of Parkinson Disease
The loss of tyrosine hydroxylase represents the critical failure in Parkinson disease because this enzyme is the rate-limiting enzyme in dopamine synthesis (see Fig. 18-3). With the tyrosine hydroxylase deficit, the ever-shrinking pool of remaining nigrostriatal tract neurons cannot sustain the essential synthetic pathway. Once approximately 30% of these neurons degenerate, the nigrostriatal tract cannot synthesize adequate dopamine and Parkinson disease symptoms appear.
On a microscopic level, neurons in these locations characteristically accumulate Lewy bodies, which contain a core of α-synuclein (see Chapter 7). In contrast, Lewy bodies located in the cerebral cortex, as well as the basal ganglia, constitute the histologic hallmark of dementia with Lewy bodies disease. With their abundance of Lewy bodies, both Parkinson disease and dementia with Lewy bodies disease fall under the rubric of synucleinopathies.
Parkinsonism
Medication-Induced Parkinsonism
When medicines, as opposed to illicit drugs, induce parkinsonism, rigidity is the most prominent feature, all three cardinal features occur in only about one-third of cases, and individuals older than 60 years and women are particularly susceptible. Typical and most atypical antipsychotic agents – because to a greater or lesser degree they block D2 receptors – routinely cause parkinsonism. Tetrabenazine (Xenazine) induces parkinsonism because it depletes dopamine (see later and Figs 18-3 and 18-12). Similarly, nonpsychiatric medicines that block D2 receptors, such as metoclopramide (Reglan), trimethobenzamide (Tigan), prochlorperazine (Compazine), and promethazine (Phenergan), produce the same problem. Case reports have also implicated valproate (Depakote), lithium, amiodarone, and calcium channel blockers – medicines with no direct connection to D2 receptors.
Medication-induced parkinsonism, once physicians discontinue the offending drug, should fully resolve in a few weeks. Nevertheless, signs often persist for 3 months and occasionally for 1 year. However, physicians must be careful with persistent parkinsonism patients because approximately 10% harbor Parkinson disease and some probably have dementia with Lewy bodies disease (see Chapter 7). In children and young adults, persistent parkinsonism following antipsychotic treatment suggests several neurologic illnesses (see later).
Therapy of Parkinson Disease
Medications
L-Dopa.
Treatments for Parkinson disease alleviate motor symptoms for many years, but do not reverse the neurodegeneration. Medicines maintain normal dopamine activity by enhancing dopamine synthesis, retarding its metabolism, or acting as agonists at dopamine receptors. Enhancing dopamine synthesis, usually the initial treatment, consists of substituting orally administered L-dopa for endogenous but deficient DOPA in the synthetic pathway (Fig. 18-3).
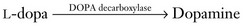
Dopa- and Dopamine-Preserving Medications
Several medications increase nigrostriatal dopa by inhibiting two enzymes – dopa decarboxylase and catechol-O-methyltransferase (COMT) – that metabolize it in the systemic circulation (Fig. 18-11). With a relative increase in nigrostriatal dopa, nigrostriatal dopamine concentration increases.
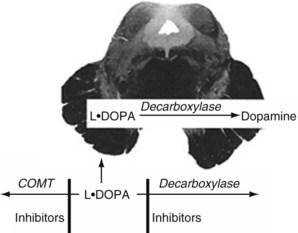
FIGURE 18-11 Medicines that inhibit catechol-O-methyltransferase (COMT), such as entacapone, and those that inhibit decarboxylase, such as carbidopa, slow the metabolism of L-dopa. Because of the blood–brain barrier, they do not affect dopamine synthesis in the nigrostriatal tract. These enzymes, combined with L-dopa, permit smaller L-dopa doses, which minimizes systemic side effects.
On the other hand, although they ameliorate some symptoms and provide a modicum of antidepressant effect, antiparkinson MAO-B inhibitors carry a caveat. At high doses, they inhibit MAO-A as well as MAO-B. Because MAO-A is important in both serotonin and catecholamine metabolism, MAO-B inhibitors place patients in a position where they are vulnerable to the serotonin syndrome or a hypertensive crisis (see previously and Chapters 6, 9, and 21).
Dopamine Agonists
As an initial medication or when dopamine production eventually falls to inadequate levels, dopamine agonists – pramipexole and ropinirole – provide stimulation of postsynaptic dopamine receptors (see Fig. 18-3). Bypassing degenerated presynaptic neurons, dopamine agonists act directly on D2 and, to a lesser extent, on other postsynaptic dopamine receptors.
Other Medications
Anticholinergics reduce tremor in Parkinson disease and other forms of parkinsonism. By reducing cholinergic activity, these medicines seem to act by maintaining the balance with the diminished dopamine activity (Fig. 18-12). On the other hand, anticholinergics routinely produce mental and physical side effects, especially in the elderly.
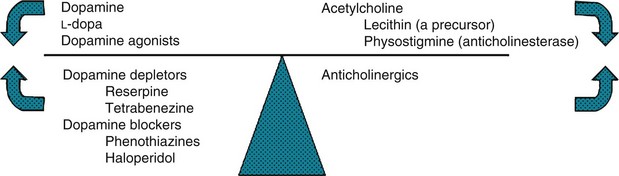
FIGURE 18-12 In a classic but limited model, dopamine and acetylcholine (cholinergic) activity normally balance each other. When Parkinson disease reduces dopamine activity, the left side of the scale rises. Dopamine precursors, dopamine agonists, and anticholinergics – Parkinson disease treatments – restore the balance. Conditions characterized by excessive dopamine activity, such as chorea, push the left side downward. Substances that either antagonize or deplete dopamine, or enhance cholinergic activity, restore the balance.
Athetosis
Athetosis consists of involuntary slow, continually changing, twisting movements predominantly affecting the face, neck, and distal limbs (Fig. 18-13). It sits at the beginning of a sequence – athetosis, choreoathetosis, chorea, and hemiballismus – of progressively larger and more irregular involuntary movements. In a potentially confusing aspect, additional involuntary movements may coexist with athetosis. For example, rapid jerks of chorea may punctuate slow movements of athetosis, and powerful twists of dystonia may interrupt and override athetosis’ slow fluctuations.
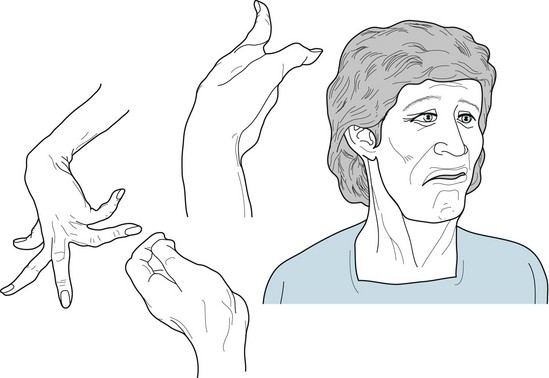
FIGURE 18-13 In athetosis, the face grimaces incessantly. Fragments of smiles alternate with frowns. Pulling of one or the other side distorts the entire appearance. In addition, neck muscles contract and rotate the head. Laryngeal contractions and irregular chest and diaphragm muscle movements cause an irregular speech cadence, nasal pitch, and dysarthria. Fingers writhe constantly and tend to assume hyperextension postures. At the same time, wrists rotate, flex, and extend. Involuntary limb activity prevents writing, buttoning, and other fine tasks, but it usually allows deliberate, larger-scale shoulder, trunk, and hip movement.
Usually encountered as a variety of cerebral palsy (see Chapter 13), athetosis is usually not apparent until early childhood. Most often athetosis results from combinations of perinatal hyperbilirubinemia (kernicterus), hypoxia, and prematurity. Genetic factors are unimportant.
Because athetosis originates in brain injuries that occur during the first 30 days after birth as well as in utero, which neurologists consider congenital brain injuries, seizures and mental retardation frequently accompany this movement disorder. However, with damage confined to the basal ganglia, as in many cases of athetosis, patients have normal intelligence despite disabling movements and garbled speech. Physicians, schoolteachers, family members, and friends should recognize that these patients retain cognitive and emotional capacities despite devastating physical neurologic disabilities.*
Chorea
Huntington Disease
Of the many causes of chorea (Box 18-1), Huntington disease, previously called “Huntington’s chorea,” remains pre-eminent. Chorea, dementia, and behavioral abnormalities characterize this autosomal dominantly inherited disorder. Symptoms first emerge when patients are, on the average, approximately 37 years. However, approximately 10% of patients develop symptoms in childhood or adolescence and 25% when they are older than 50 years. Adults with the illness usually succumb to aspiration and inanition one to two decades after the diagnosis.
Clinical Features
In contrast to the slow, writhing, continuous movements of athetosis, chorea consists of random, discrete, rapid movements that jerk the pelvis, trunk, and limbs (Fig. 18-14). Chorea also includes involuntary facial movements that produce brief, meaningless expressions (Fig. 18-15). When walking, the chorea characteristically interrupts patients’ cadence and stability (Fig. 18-16). In fact, the abnormal gait gave rise to the term chorea, (Greek, chorea, dance).
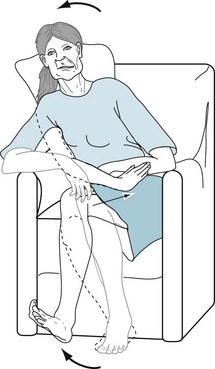
FIGURE 18-14 As in this woman with Huntington disease, chorea consists of intermittent, random involuntary movements, such as brisk pelvic, trunk, and limb jerks. It also encompasses a mere flick of a finger or wrist, forward jutting of the leg, or shrugging of the shoulder.
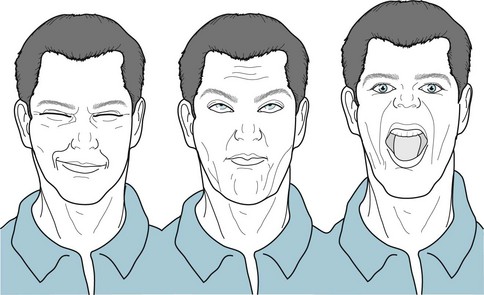
FIGURE 18-15 Huntington disease patients characteristically make unexpected, inappropriate, and incomplete facial expressions. Without provocation, they frown, raise eyebrows, and smirk. Because their tongue frequently protrudes, this chorea occasionally mimics tardive dyskinesia.
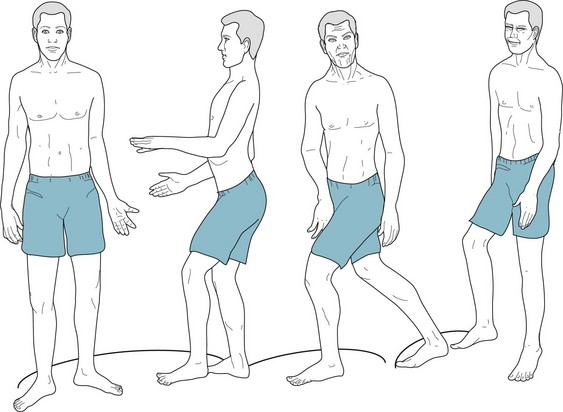
FIGURE 18-16 The signature of Huntington disease is a jerky gait that results from intermittent, unexpected trunk and pelvic motions, spontaneous knee flexion and extension, lateral swaying, variable cadence, and unequal stride length.
Chorea in its earliest stage merely resembles nonspecific “fidgety” movements seen with anxiety, restlessness, discomfort, or clumsiness. It may then consist of only excessive face or hand gestures, weight shifting, leg crossing, or finger twitching (Fig. 18-17). Chorea also impairs the ability to sustain a voluntary muscle contraction, which causes motor impersistence. Because of it, patients cannot either hold a firm grasp or extend their hands or tongue for more than 10 seconds. For example, when asked by the examiner to squeeze two fingers, patients exert irregular, variable pressure that neurologists call the “milkmaid’s sign.” Patients also show motor impersistence by intermittently, involuntarily withdrawing the tongue when neurologists ask them to protrude it for 30 seconds. Neurologists refer to that finding as a “Jack-in-the-box tongue.” The motor impersistence also prevents patients from sustaining postures. For example, when “standing at attention,” patients’ fingers twitch outward and their torso bends.
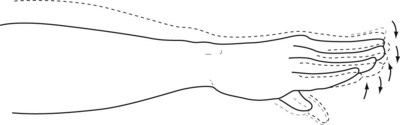
FIGURE 18-17 With their arms and hands extended, Huntington disease patients fidget with their fingers and wrists, in movements that neurologists label “piano playing.”
Huntington disease also characteristically interferes with normal eye movements (see Fig. 12-13). In particular, Huntington disease impairs saccades, which are the rapid, almost reflexive, conjugate eye movements that people normally use to glance from one object to another. In Huntington disease, patients cannot make a rapid, smooth, and accurate shift of their gaze toward an object that suddenly enters their visual field. They routinely first blink or jerk their head to initiate the saccade. Although characteristic, impaired saccades are not peculiar to Huntington disease. Patients with schizophrenia, for example, also have abnormal saccades.
Juvenile Huntington Disease
Sometimes symptoms appear in children and adolescents – individuals younger than 21 years of age. This variant, juvenile Huntington disease, comprises 10% of all cases and produces some different manifestations than the common, adult form. Their schoolwork declines and behavioral disturbances develop as the first signs. Also, rather than causing chorea as its initial physical symptom, juvenile Huntington disease presents with rigidity, dystonia, and akinesia (Fig. 18-18). Many affected children and adolescents look as though they have Parkinson disease. Another difference is that, unlike the adult variety, juvenile Huntington disease causes seizures. More importantly, juvenile Huntington disease progresses much more quickly and leads to death twice as rapidly as the adult form.
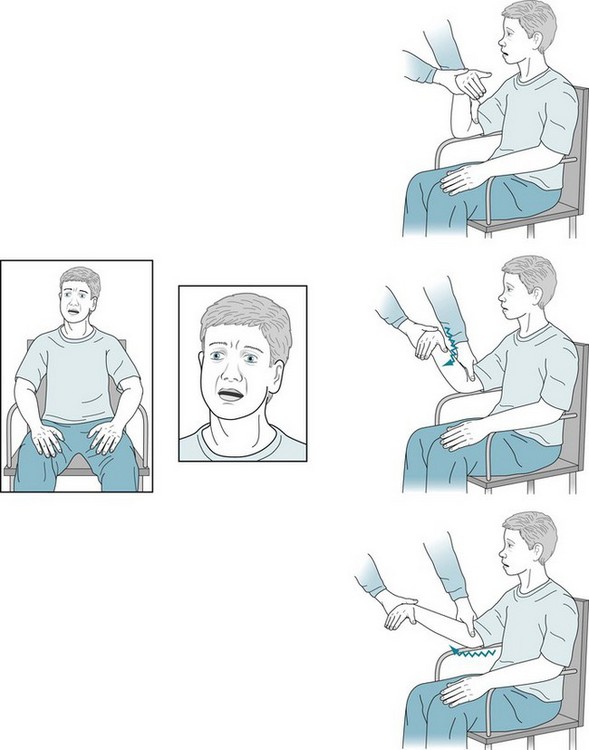
FIGURE 18-18 Having had deteriorating grades and progressively greater isolation from his family and friends, this 18-year-old high-school student came for evaluation. During the interview, he sat motionlessly, had few facial or limb gestures, and moved slowly; however, he had no tremor. He was apathetic and lacked insight. He did not fully cooperate with routine mental status testing, but it seemed to show deficits. When the neurologist flexed and extended his right arm, she detected rigidity. After a routine evaluation, she ordered tests for conditions that cause parkinsonism accompanied by mental deterioration in teenagers – Wilson disease, drug abuse, and the juvenile variety of Huntington disease. After the neurologist received the report that the genetic testing showed 85 CAG trinucleotide repeats, she asked to evaluate the father. He displayed subtle but definite chorea and cognitive impairment. His genetic testing also revealed 50 CAG repeats.
Genetics
The abnormality underlying juvenile and adult varieties of Huntington disease – as with myotonic dystrophy, several spinocerebellar ataxias, and fragile X syndrome – is a genetic mutation consisting of excessive trinucleotide repeats (see Chapter 6 and Appendix 3D). This gene, located on the short arm of chromosome 4, normally consists of 11–35 repeats of the trinucleotide base cytosine-adenine-guanine (CAG). Individuals with 36–39 trinucleotide repeats, whom neurologists classify as “indeterminate,” have no clinical signs or merely a forme fruste of the disease; however, because of the gene’s tendency to enlarge (see later), indeterminate individuals’ children may have unequivocal disease. Individuals with 40 or more trinucleotide repeats invariably develop all manifestations of the illness and the age of appearance of their symptoms correlates inversely with the number of their repeats.
Pathology
In Huntington disease, the mutation causes excessive polyglutamine synthesis, which leads to production of huntingtin, a cytoplasmic protein, by neurons vulnerable to neurodegeneration. Thus, neurologists designate Huntington disease and other illnesses transmitted by excessive CAG repeats as polyglutamine diseases. In another aspect of the pathology, glutamate and other excitatory amino acids overstimulate N-methyl-D-aspartate (NMDA) receptors. This pathologic interaction, excitotoxicity, allows a fatal influx of calcium into neurons (see Chapter 21).
Atrophy of the caudate nuclei, almost a pathognomonic macroscopic finding, correlates roughly with the severity of dementia. The atrophy of the caudate nuclei permits the lateral ventricles to balloon outward, i.e., develop a convex outline. They expand so much that neurologists call them “bat wing ventricles.” CT and MRI readily show the caudate atrophy and enlarged, convex ventricles (see Fig. 20-5). As Huntington disease progresses, the cerebral cortex also undergoes atrophy. PET studies demonstrate caudate hypometabolism even early in the illness.
By way of contrast, while normal aging and Alzheimer disease also cause cerebral atrophy, their atrophy does not preferentially affect the caudate nuclei. In these non-Huntington conditions, the caudate nuclei still bulge into the lateral ventricles. The ventricles enlarge, but they maintain a concave contour (see Figs 20-2, 20-3, and 20-18).
Other Varieties of Chorea
Sydenham Chorea
The chorea begins insidiously with grimaces and limb movements (Fig. 18-19). Sometimes the movements take on an urgency that makes the child seem willfully hyperactive. Sydenham chorea is one of the commonest neurologic causes of hyperactivity in children. A list of those causes would also include attention deficit hyperactivity disorder (ADHD), side effects from medications or illicit drugs, tics and Tourette disorder, and withdrawal-emergent syndrome (WES).
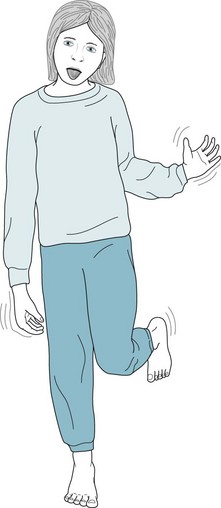
FIGURE 18-19 Children with Sydenham chorea may appear to have coy smiles and brief grimaces, and walk with a playful sashay. However, the pathologic nature of their movements – chorea – can be made obvious if the children attempt to maintain a fixed position, such as standing at attention, standing on the ball of one foot, or protruding their tongue. The involuntary movements cause dysarthria that is occasionally so severe that children refuse to speak. Surprisingly, the chorea may be asymmetric or unilateral in at least 20% of cases. It usually lasts between several weeks and 2 months.
Estrogen-Related Chorea
Many women affected by oral contraceptive-induced chorea or chorea gravidarum have had previous episodes of either condition or Sydenham chorea. Also, their close relatives often have had one of these conditions. Nevertheless, if chorea develops in a woman who is pregnant or taking oral contraceptives, physicians must consider conditions in addition to the estrogen-related choreas, including Sydenham chorea, systemic lupus erythematosus, antiphospholipid syndrome (see Chapter 11), hyperthyroidism, and even Huntington disease.
Hemiballismus
Hemiballismus consists of intermittent, large-scale movements of one side of the body. The movements resemble chorea, except that they are unilateral, less predictable, and more forceful. They resemble flinging (ballistic) thrusts (Fig. 18-20). It sits at the end of the sequence of progressively larger and more irregular involuntary movements.
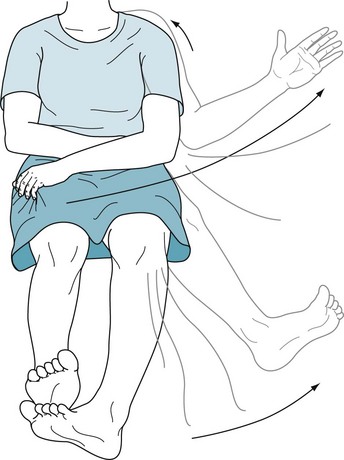
FIGURE 18-20 Beset by classic hemiballismus, this woman has sudden and large-scale movements of the limbs on one side of the body. Even when hemiballismus has a more modest amplitude, patients use a variety of strategies to suppress it. In this case, the patient typically uses her left hand to grip her skirt to anchor her moving left arm. Sometimes patients press their body or unaffected limb against an involuntarily moving limb. They also attempt to camouflage the involuntary movements by converting them into apparently purposeful movements. For example, if her arm were to fly upward, she might incorporate the movement into a gesture, such as waving to someone, or smoothing her hair.
Whatever the location of the lesion, the most common etiology is an occlusion of one of the small perforating branches of the middle cerebral artery, which perfuse the basal ganglia. Similarly, vasculitis can affect those arteries. In individuals infected with HIV, toxoplasmosis lesions have a tendency to develop in the basal ganglia and produce hemiballismus (see Fig. 20-11).
Wilson Disease
The insidious development of psychiatric disturbances, cognitive impairment, and a variety of involuntary movements in adolescents or young adults characterizes Wilson disease (hepatolenticular degeneration). Because early diagnosis and treatment can reverse its manifestations, neurologists often describe Wilson disease as a classic “reversible cause of dementia.” They also cite it as a cause of “dementia in adolescents” (see Box 7-2) and “parkinsonism in children and young adults.”
Due to an autosomal recessive mutation of a copper-transporting gene carried on chromosome 13, Wilson disease leads to insufficient serum copper binding and hepatic excretion. The resulting surplus of unbound serum copper, in turn, leads to destructive copper deposits in the brain, liver, cornea, and other organs. As its formal name implies, the illness primarily causes destruction of the liver and the lenticular nuclei of the basal ganglia (see Fig. 18-1, A).
The neurologic signs are dysarthria, dysphagia, gait impairment, anosmia, and ones reflecting basal ganglia damage: rigidity, akinesia, dystonia, nonspecific tremor, and the characteristic wing-beating tremor (Fig. 18-21). These movements tend to occur in combination and be accompanied by corticospinal or corticobulbar tract signs.
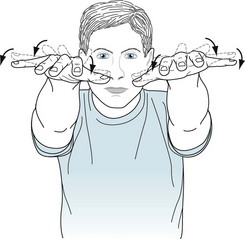
FIGURE 18-21 Wilson disease characteristically induces parkinsonism, dystonia, dysarthria, various tremors, and the wing-beating tremor. This particular tremor, which is a hallmark of the disease but occurs in only about one-third of cases, consists of rhythmic, coarse, up-and-down arm movements based at the shoulders. It gives patients the appearance of flapping their arms as though they were attempting to fly.
Especially in adolescents, nonneurologic manifestations often overshadow neurologic ones. For example, liver involvement leads to cirrhosis, which is sometimes so severe that it causes hepatic encephalopathy and then liver failure. Also, deposits of copper in the cornea produce the signature Kayser–Fleischer ring (Fig. 18-22).

FIGURE 18-22 The Kayser–Fleischer ring, which appears bilaterally, consists of green, brown, or orange copper pigment deposits in the periphery of the cornea. Most obvious at the superior and inferior margins of the cornea, the ring usually obscures the fine structure of the iris. Ophthalmologists must use a slit lamp to see the Kayser–Fleischer ring in its early stages. Although the ring develops in about 70% of cases when Wilson disease affects only the liver, it develops in almost 100% of cases when the disease affects the brain. In those cases, the ring’s size and density correlate with the duration of the disease. However, because the Kayser–Fleischer ring also develops in primary biliary cirrhosis and several other liver diseases, it is not pathognomonic of Wilson disease. With successful treatment of Wilson disease, the ring dissolves.
Dystonia
Generalized Dystonias
Early-Onset Primary Dystonia
Early-onset primary dystonia or idiopathic torsion dystonia develops predominantly among Ashkenazi Jews. The illness typically appears in 9–11-year-old children, but almost always by the age of 26 years. The dystonia usually begins with torsion of one hand or foot (Fig. 18-23) and subsequently the other limbs, pelvis (tortipelvis), trunk, and neck (torticollis). Untreated, it eventually incapacitates its victims (Fig. 18-24).
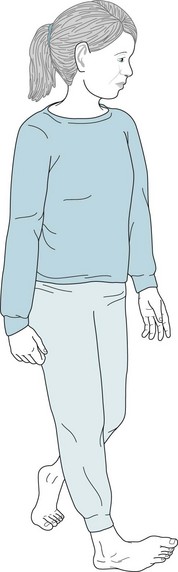
FIGURE 18-23 When the girl with early-onset primary dystonia walks, the foot twists slowly inward and on to its side. Even though incomplete, the twisting causes her to limp. Using one of her “sensory tricks,” she can correct her limp by skipping, dancing, or walking backward.
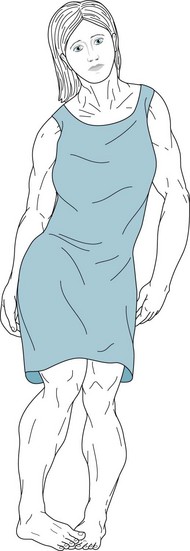
FIGURE 18-24 As early-onset primary dystonia encompasses the remaining limb and axial musculature, patients develop generalized dystonic postures. Because muscles continually contract, they hypertrophy. Patients lose their subcutaneous fat from the incessant exertion. Patients with Wilson disease or tardive dystonia may resemble those with early-onset primary dystonia because all these illnesses may cause generalized dystonia.
Dopa-responsive dystonia
In an illness first described by Segawa and his Japanese colleagues and often named after him, dopa-responsive dystonia (DRD) gives rise to dystonia that appears in children and follows a distinctive diurnal pattern. The predominant symptom, dystonia, becomes evident in children, on the average, when they are 8 years old. The dystonia is typically absent in the morning but pronounced by the late afternoon and evening (Fig. 18-25). As with early-onset dystonia, DRD first affects children’s legs, interfering with walking, and then progresses to become generalized. In a related sign, DRD sometimes superimposes parkinsonism on the dystonia. Neurologists include DRD in the DYT family as DYT5.
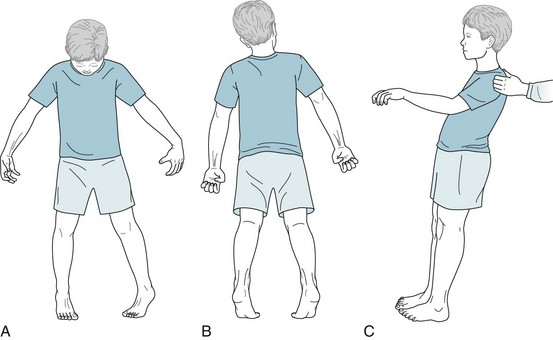
FIGURE 18-25 During the previous year, this 8-year-old boy began to have leg and trunk movements that forced him to walk on his toes after returning from school in the afternoon. In the evening, his arms and hands showed dystonic posturing and his legs assumed straightened positions until he went to sleep. When he awoke in the morning, he walked and ran normally. His parents initially thought that he had developed cerebral palsy (see Fig. 13-3) and a psychologic disturbance. Pediatric neurologists detected dystonia, more so in his legs than his arms, and elicited a positive response to the pull test (see Fig. 18-9, B). Cognitive testing showed no abnormality. For many reasons, including that cerebral palsy does not develop after age 5 years or show a diurnal fluctuation, pediatric neurologists administered a therapeutic trial of a daily small dose of L-dopa. The boy immediately reverted to normal. Genetic tests later confirmed the diagnosis of dopa-responsive dystonia.
Secondary Generalized Dystonia
One of these dystonia-producing illnesses, Lesch–Nyhan syndrome, an X-linked recessive genetic disorder, produces one of neurology’s most bizarre symptoms: 2–13-year-old boys and, extraordinarily rarely, girls develop ferocious self-mutilation (Fig. 18-26). This symptom, the quintessential neuropsychiatric abnormal behavior, consists of children biting their own lips and fingers. This self-injurious behavior begins abruptly and furiously. In addition to the self-mutilation, they have mental retardation, spasticity, and seizures, as well as dystonia. By way of contrast, in mental retardation and autism, self-injurious behavior usually consists of head banging and hitting that begin insidiously and remain relatively mild.
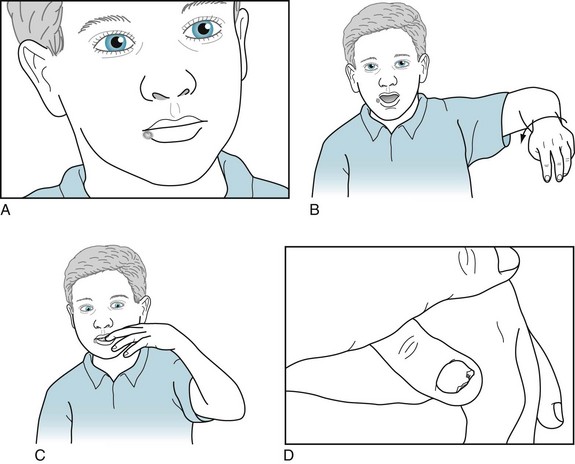
FIGURE 18-26 This mentally retarded boy shows the self-mutilation, as well as dystonia, that characterizes Lesch–Nyhan syndrome. A, His right lower lip has the scars of where he gnawed through it, even after his parents and physicians had his front teeth removed to prevent him from continuously biting himself. B, His limbs writhe and his thumb curls into his palm. C, In an apparent compulsion, he uncontrollably chews his fingers and already has amputated the tip of the smallest one. D, Prying away his left thumb exposes deep excoriations on his palm.
Focal Dystonias
Injections of botulinum into affected muscles may almost completely, but temporarily, eliminate focal dystonia (Fig. 18-27). Botulinum may also treat particularly troublesome isolated muscle groups in DYT dystonia and tardive dystonia (see later).
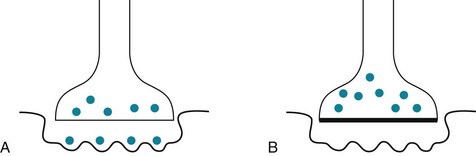
FIGURE 18-27 A, At the normal neuromuscular junction, the presynaptic membrane releases packets of acetylcholine (ACh: the black dots) that cross the cleft and bind to postsynaptic membrane receptors, triggering muscle contractions. Focal dystonias are characterized by involuntary segmental muscle contractions. B, Botulinum A toxin, injected directly into affected muscles, binds irreversibly to the presynaptic membrane. It interferes with the release of the ACh packets and thereby weakens the injected muscle. The treatment reduces abnormal contractions for about 3 months. During that period, injected muscles may be weak but they function normally and have little involuntary movement. To maintain the improvement, physicians must repeat the botulinum injections. For cosmetic purposes, physicians sometimes inject botulinum to relax the facial muscles that cause undesirable furrows of the forehead, eyebrow (“frown lines”), lateral canthus (“crow’s feet”), and other areas of the face and neck.
Cranial Dystonias
Blepharospasm, an easily recognizable and frequently occurring focal dystonia, consists of bilateral, simultaneous contractions of the orbicularis oculi (eyelid) and sometimes the frontalis (forehead) muscles (Fig. 18-28). The muscle spasms force the eyelids closed and tend to render patients functionally blind, as well as causing disfiguring facial expressions. Although neurologists cannot establish the cause in most cases, various eye diseases, including “dry eye,” represent powerful risk factors.

FIGURE 18-28 This man with blepharospasm has unprovoked, bilateral, and prolonged (average duration, 5 seconds) contractions (spasms) of his orbicularis oculi (eyelid) muscles. Because the spasms block his vision, he often resorts to prying open his eyelids. In contrast to eyelid closure as a tic, the closures in blepharospasm are longer, more forceful, and not provoked by a “need” to close the eyes.
To overcome the involuntary contractions, patients unconsciously learn tricks that temporarily suppress the contractions (Fig. 18-29). As with other focal dystonias, botulinum injections reduce or abolish blepharospasm, at least on a temporary basis, with each set of injections. If excessive botulinum causes ptosis from weakness of the upper eyelid levator muscles, apraclonidine, a weak alpha-adrenergic stimulant, will retract the eyelid. (Like any other sympathomimetic agent, apraclonidine will also dilate the pupil.)
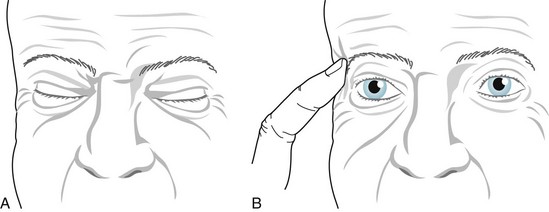
FIGURE 18-29 A, During periods of blepharospasm, this man cannot open his eyelids. He attempts, perhaps unconsciously, to open them by elevating his forehead muscles. B, He has instinctively learned that the sensory trick of pressing one eyebrow (a geste antagoniste) alleviates the spasms for several minutes.
When the contractions involve the entire face, as though blepharospasm joined oromandibular dystonia, neurologists call the condition Meige syndrome (Fig. 18-30). This disorder mimics tardive dyskinesia except that, like oromandibular dystonia, it lacks tongue involvement. Preliminary studies have shown that DBS of the GPi suppresses the movements of Meige syndrome.

FIGURE 18-30 This patient with Meige syndrome (cranial dystonia) has contractions of her entire facial musculature.
Hemifacial spasm, a completely different cranial dystonia, consists of spasms of the muscles on only one side of the face – all those innervated by the ipsilateral facial nerve (the seventh cranial nerve) (Fig. 18-31). In this disorder, spasms occur irregularly at 1–10-per-minute, often in flurries, disfigure the face, and close the eyelids. Unlike dystonia and almost all other movement disorders, hemifacial spasm routinely persists during sleep.
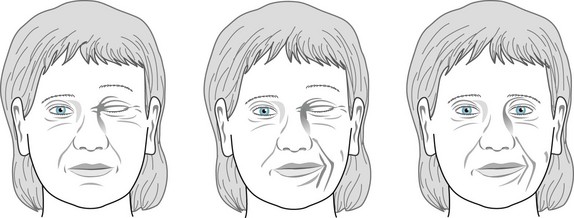
FIGURE 18-31 Beset by hemifacial spasm for several years, this 53-year-old woman has bursts of left-sided facial muscle contractions. The contractions repetitively squeeze shut her left eyelids and pull her mouth to her left side, but the upper and lower face muscles do not contract synchronously. As a compensatory mechanism to uncover her left eye, she elevates her left eyebrow.
Cervical Dystonias
In spasmodic torticollis (Latin, tortus twisted + collum, neck), the most frequently occurring focal dystonia, the sternocleidomastoid and adjacent neck muscles involuntarily contract to rotate and tilt the head and neck (Figs 18-32 and 18-33). The movements initially persist for several seconds to several minutes. As the disease progresses, the abnormal postures become continuous and a superimposed tremor often complicates the picture. Unlike facial dystonias, spasmodic torticollis causes pain because the muscle contractions forcefully compress and rotate the cervical vertebra on each other and irritate the cervical nerve roots that emerge between them.

FIGURE 18-32 Spasmodic torticollis consists of contraction of neck muscles that causes rotation, tilting, flexion (anterocollis), or extension (retrocollis) – usually in a combination – of the head and neck. Shoulder elevation, as though the head and shoulder pull toward each other, accompany the other movements. Continuous contractions result in muscle hypertrophy and pain.
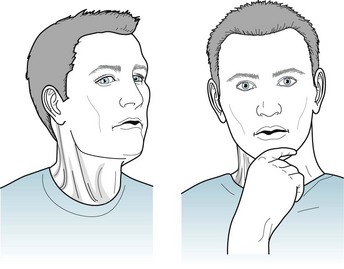
FIGURE 18-33 As with blepharospasm and other focal dystonias, patients instinctively learn sensory tricks, such as applying slight counter-rotational pressure against the chin, which is a classic geste antagoniste. In spasmodic torticollis, these maneuvers not only temporarily stop the movement, but they also camouflage it by allowing the patient to strike a studious pose.
Other cranial and cervical dystonias and head tremors often accompany spasmodic dysphonia. Clinical evaluation can distinguish it from related conditions, such as anxiety-induced vocal tremor, vocal cord tumors, and pseudobulbar palsy (see Chapter 4). Electromyography-guided botulinum injections through the anterior of the throat directly into the laryngeal muscles reduce or eliminate patients’ involuntary contractions and restore their voice.
Limb Dystonias
In writer’s cramp, a clear example of an occupational dystonia, shortly after authors begin to write, spasmodic contractions seize their finger and hand muscles. The contractions distort their hand and prevent it from properly grasping a pen or pencil (Fig. 18-34). However, writer’s cramp does not prevent eating, buttoning clothing, or manipulating small objects – movements that require dexterity equal to handwriting.

FIGURE 18-34 This patient is an author with writer’s cramp. She develops finger and hand muscle spasms several minutes after starting to write. The spasms force her hand into a fist and flex her wrist, which prevents her from holding a pen. They also cause moderate pain. However, when she uses the same hand to type, eat, or button clothing, she does not develop the cramps.
Essential Tremor
Patients’ performing particular actions or holding their arms in certain postures elicits or accentuates the tremor (Fig. 18-35). The affected body region oscillates usually in only one plane. Thus, tremor is the only involuntary movement disorder that is rhythmic and limited to one plane.
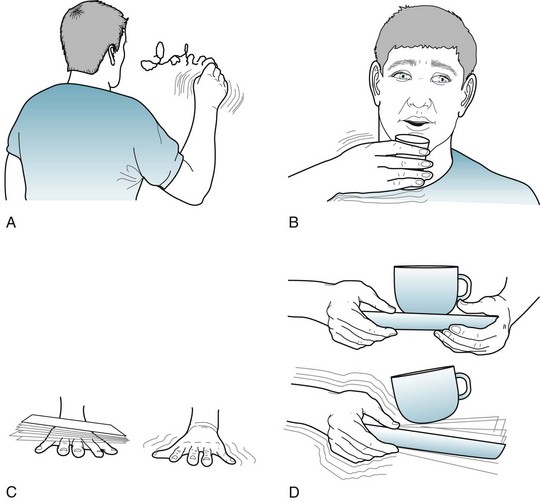
FIGURE 18-35 Physicians elicit this 34-year-old man’s essential tremor by having him (A) write his name; (B) drink from a filled glass; (C) support an envelope on his outstretched and pronated hand; or (D) transfer a cup and saucer from one hand to the other. When his hands rest in his lap, the tremor completely subsides. Its presence only when hands and arms are held in fixed positions against gravity has led to alternate, descriptive terms, such as postural or positional tremor.
No completely effective pharmacologic treatment is available. However, β-adrenergic blockers, such as propranolol (Inderal), can help many cases of essential tremor and usually at doses that do not cause depression.* The positive response that these medicines produce supports the hypothesis that excessive β-adrenergic activity causes or exacerbates essential tremor. Primidone (Mysoline), an antiepileptic drug closely related to phenobarbital, alone or in combination with propranolol, often reduces the tremor. Medicines with less efficacy include benzodiazepines and other β-blockers.
Other Tremors
Other fine, rapid tremors, because they also respond to β-blockers, probably originate in excessive adrenergic system activity. These tremors often represent a physical manifestation of anxiety, as in stage fright (performance anxiety), or the preliminary DSM-5 diagnosis Social Anxiety Disorder (Social Phobia). They may also result from hyperthyroidism and other medical illnesses. Caffeine, the world’s most popular drug, induces tremors (see Table 17-2). Also, tremors are often medication-induced – by steroids, the popular antiarrhythmic drug amiodarone, β-adrenergic stimulating agents, such as isoproterenol and epinephrine, and psychotropics, including amitriptyline, lithium, valproate, SSRIs, and dopamine-blocking antipsychotics. Lithium and valproate may induce tremor even at therapeutic concentrations. From another perspective, tremor is a symptom of withdrawal from alcohol, benzodiazepines, opiates, or many other substances.
By way of contrast, the Parkinson disease tremor occurs so characteristically at rest that it is the quintessential “resting tremor.” It also differs from essential tremor by its “pill-rolling” appearance (see Fig. 18-8). Cerebellar dysfunction causes a coarse, irregular tremor elicited by movement (see Fig. 2-11). Except for the characteristic wing-beating pattern, tremors induced by Wilson disease remain difficult to categorize, especially because they can appear similar to those of Parkinson disease, cerebellar disease, or essential tremor. Nevertheless, in young adults who develop a tremor, physicians should probably first consider Wilson disease. The fragile X syndrome includes mental retardation and tremor resembling essential tremor among its manifestations (see Chapter 13).
Palatal tremor, which neurologists until recently called palatal myoclonus, consists of uninterrupted symmetric, rhythmic contractions of the soft palate. The frequency – 120–140 times per minute – is consistent from patient to patient. It too persists during sleep or coma. Most cases are caused by small brainstem infarctions that involve the medulla’s inferior olivary nucleus or its connections (see Fig. 2-9).
Tics
Gilles de la Tourette (Tourette) Disorder
For a diagnosis of Tourette disorder, the preliminary DSM-5 requires the presence of both vocal and multiple motor tics, although not necessarily concurrently, for longer than 1 year, and the onset to have occurred before 18 years of age (Fig. 18-36). Neurologists, who may use the term Tourette syndrome, practice using the similar criteria, except that they do not insist on an onset before age 18 years.
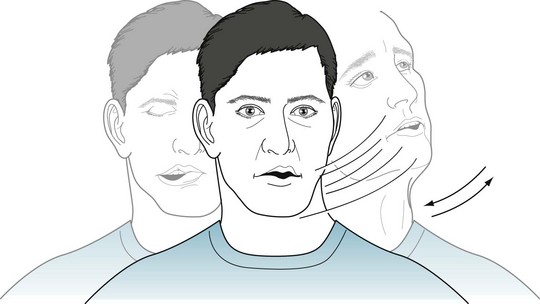
FIGURE 18-36 This young man with Tourette disorder has multiple motor tics, including head jerking (head toss), grimacing of the right side of his mouth (half-smile), and depression of his forehead (frowning). Vocal tics of throat clearing and a short blowing sound accompany his motor tics. All of his tics continue throughout the day and briefly during sleep. Conversation, eating, and social situations have little influence, but he can suppress his tics for several minutes by intense concentration.
Vocal tics, an essential feature of Tourette disorder, consist of irresistible, repetitive, stereotyped utterances – sounds, words, or, in the extreme, coprolalia. Vocal tics usually arise several years after the onset of motor tics and remain simple. They typically consist of only inarticulate sounds, such as sniffing, throat clearing, or clicks. However, many vocal tics rise to loud and disconcerting noises, such as grunting, snorting, or honking. Complex vocal tics consist of formed words that can culminate in unprovoked outbursts of obscene words, coprolalia. Although most coprolalia consists of only fractions of scatological words, such as “shi” or “fu,” some consist of strings of unequivocal obscenities. Sometimes coprolalia is merely socially reprehensible but occasionally dangerous, such as when a young Chinese girl’s vocal tic belittled Chairman Mao, a Bronx teenager endlessly and uncontrollably repeated two words disparaging the New York Yankees, or an otherwise lovely devoutly Catholic adolescent girl incessantly damned a particular saint. Equivalents of coprolalia, such as intrusions of obscene thoughts (mental coprolalia) or involuntary obscene movements or gestures (copropraxia; Fig. 18-37), may represent a form of coprolalia. When coprolalia complicates Tourette disorder, it does not appear until about 6 years after the onset of motor tics.
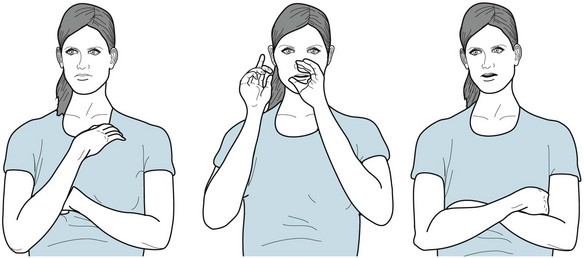
FIGURE 18-37 This woman has compulsive furtive obscene gestures – copropraxia. Similar to coprolalia, they have no affective or sexual component.
Myoclonus
A wide variety of disorders, although rarely structural lesions, produce myoclonus. Cerebral cortex damage is most often the cause of myoclonus. For example, myoclonus is one of the most prominent physical manifestations of subacute sclerosing panencephalitis (SSPE) (see Chapter 7), Creutzfeldt–Jakob disease (see Chapter 7), and paraneoplastic limbic encephalitis (see Chapter 19). In these and other similar conditions, dementia, delirium, or epilepsy accompanies the myoclonus.
In toxic-metabolic aberrations, myoclonus commonly complicates delirium. For example, uremic encephalopathy and toxic levels of medications, including penicillin, meperidine (Demerol), bismuth, and cyclosporine, induce myoclonus. Survivors of cerebral anoxia often show postanoxic myoclonus. Psychiatrists may encounter myoclonus in their patients taking clozapine, SSRIs, or lithium. When myoclonus develops in patients taking an SSRI, it suggests the serotonin syndrome (see Chapter 6). If physicians remedy an underlying toxic-metabolic disturbance, the myoclonus will usually resolve. Treatment with clonazepam or sometimes valproate may suppress myoclonus, but, of course, does not affect the underlying disorder.
Movement Disorders From Dopamine-Blocking Medications
As previously discussed, antipsychotic agents and other medicines that block dopamine potentially may cause NMS, lower the seizure threshold, alter the EEG (see Chapter 10), and produce retinal abnormalities (see Chapter 12). They may also cause dramatic involuntary movements as well as parkinsonism.
Neurologists usually divide these dopamine-blocking-induced movement disorders, dyskinesias, into two groups based on the interval between either initiating or increasing the dose of the medication and their onset. In this classification, acute dyskinesias develop within days and tardive (late) dyskinesias at 6 months or longer, but almost always within 12 months of starting the medication (Box 18-2).
Acute Dyskinesias
Acute dyskinesias fall into four categories:
Oculogyric Crisis and Other Acute Dystonias
Acute dystonic reactions consist of the abrupt development of limb or trunk dystonic postures, repetitive jaw and face muscle contractions, tongue protrusion, torticollis, or, in a special case, oculogyric crisis (Fig. 18-38). These dyskinesias may occur alone or in combinations, but typically cervical and jaw dystonia accompany oculogyric crisis. Physicians must keep in mind that several serious neurologic disorders – seizures, tetanus, drugs of abuse, and strychnine poisoning – can cause similar movements. For example, in large, urban hospitals, patients who present with oculogyric crisis or other acute dystonia and do not respond to anticholinergics and antihistaminics are often experiencing phencyclidine (PCP) intoxication.

FIGURE 18-38 During an oculogyric crisis, the eyes forcefully roll upward but sometimes sideways. Whichever direction, the eyes move conjugately. Simultaneously, in a mandibular and cervical dystonia component, the jaw opens or closes and the neck bends backwards (extends). Parenteral anticholinergics almost always abort this frightening dyskinesia.
Akathisia
In akathisia, almost regular limb and trunk movements continually plague the patient (Fig. 18-39). Although akathisia can involve the trunk and arms, it predominantly affects the legs. Most importantly, it forces patients to move about and prohibits their sitting still or lying quietly in bed. When standing, patients tend to shuffle, march in place, or rock on their heels and toes. When sitting, which they find difficult, patients squirm, sway back and forth, or rub their feet on the floor; hence, the origin of the term “akathisia” (Greek, a + kathisis, without sitting).
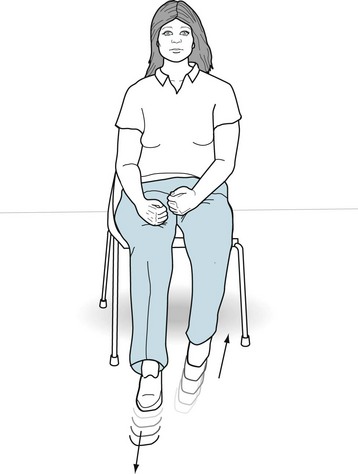
FIGURE 18-39 Beset by akathisia, this woman, who just began a regimen of antipsychotic medicines, continually shuffles her legs in regular to-and-fro sliding movements. Although it can lead to repetitive semi-purposeful arm movements, such as scratching, hair smoothing, and rubbing, akathisia predominantly affects the legs. In another characteristic of akathisia, she feels driven to move her legs.
Tardive Dyskinesias
Oral-Buccal-Lingual Variety
Neurologists refer to the common antipsychotic-induced oral-buccal-lingual (also called choreic or orofacial) dyskinesia as one of several varieties of tardive dyskinesia (see Box 18-2). In a potential nomenclature conflict, many psychiatrists apply the term “tardive dyskinesia” to all varieties of the disorder.
Oral-buccal-lingual dyskinesia consists of stereotyped tongue, jaw, and face movements (Fig. 18-40); however, tardive dyskinesia often involves movements of the extremities and trunk. Unlike tics and RLS, urges do not provoke any of these movements.
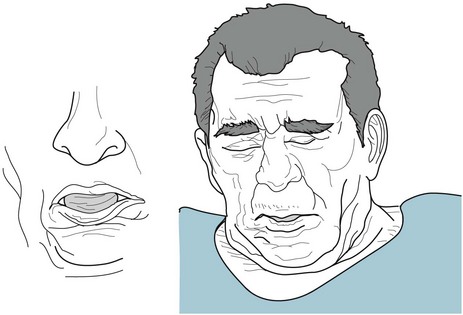
FIGURE 18-40 The oral-buccal-lingual variety of tardive dyskinesia consists of repetitive tongue movements accompanied by continual jaw and facial muscle contractions. These movements typically include tongue darting, lip smacking, kissing, lip puckering, chewing, and sometimes blepharospasm. Unlike the tongue movements in chorea and cranial dystonias, tongue movements in oral-buccal-lingual tardive dyskinesia are not only prominent, but they may lead to tongue enlargement (macroglossia).
The Abnormal Involuntary Movement Scale (AIMS) allows recording of the presence, locations, and severity of abnormal movements (Fig. 18-41). It provides a global rating scale that allows physicians to assess pretreatment status and subsequent changes. However, even assuming interrater reliability, the AIMS has several drawbacks. It quantifies dyskinesias that vary in intensity during the day. The measurements are gross. The AIMS does not recognize akinesia (lack of movement), which should carry as much diagnostic weight as hyperkinesia. It does not distinguish between chorea, dystonia, tics, and stereotypies. Finally, it omits several important movements, such as tremor, dysarthria, and respiratory tics.

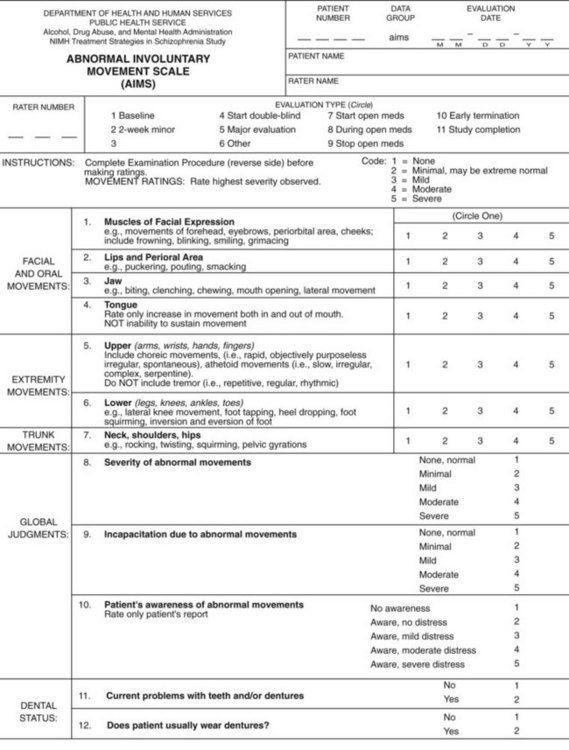
FIGURE 18-41 The Abnormal Involuntary Movement Scale (AIMS) guides the examiner through inspection for dyskinesias of the face, jaw, tongue, trunk, and limbs. The examiner records observations when the patient is at rest, extending the tongue or limbs, or performing certain activities, such as finger tapping, standing, or walking. In addition, the examiner checks for rigidity. This revision of the original (Guy, 1976) requests that patients remove their shoes and socks, and it does not rate as less severe those movements that are activated by voluntary movements.
Etiology
Despite its limitations, the classic dopamine receptor hypersensitivity theory remains consistent with several of the condition’s major clinical features (Fig. 18-42). Tardive dyskinesia begins only after 6 months from the start of dopamine-blocking medicines. The dyskinesias resemble movements produced by excessive L-dopa. Maneuvers that expose the postsynaptic neuron to increased dopamine activity – reducing the dosage of the dopamine receptor blocker, stopping it, or adding L-dopa – intensify the movements. Likewise, increasing the medication dosage, which reduces dopamine receptor activity, suppresses the movements.
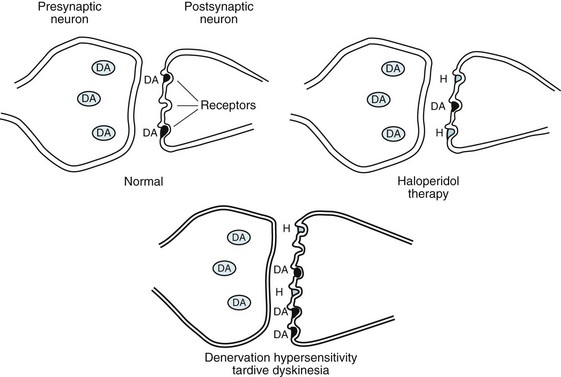
FIGURE 18-42 The denervation hypersensitivity theory proposes that when antipsychotic agents, such as haloperidol (H), bind to postsynaptic dopamine receptors, the remaining receptors become particularly sensitive and new ones develop. Then, minute quantities of dopamine (DA) – either released from the presynaptic neuron or present in the ambient fluid – evade the blockade and trigger these hypersensitive receptors.
Tardive Dystonia
Another tardive dyskinesia, tardive dystonia, consists of sustained, powerful, twisting, predominantly extensor movements of the neck, upper arms, and trunk – the axial musculature (Fig. 18-43). Unlike the rotation (torsion) and tilting that characterize common, idiopathic spasmodic torticollis, the increased tone of the extensor neck muscles in tardive dystonia produces its characteristic feature, retrocollis. Related movements, including oral-buccal-lingual dyskinesia, blepharospasm, and akathisia, often accompany tardive dystonia. Otherwise, tardive dystonia resembles other secondary dystonias, as would be observable in Wilson, juvenile Huntington, and DYT1 diseases.
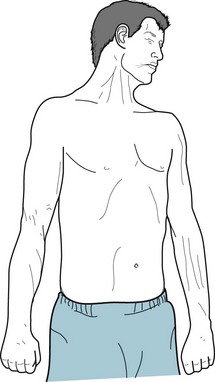
FIGURE 18-43 After receiving neuroleptic treatment for 6 years, this 25-year-old man began to develop prolonged, forceful twisting (torsion) and extension of his arms, extension of his head and neck (retrocollis), and exaggerated arching of his back. Subsequently, subtle oral-buccal-lingual choreiform movements, blepharospasm, or other varieties of tardive dyskinesia arose. After excluding Wilson disease and DYT1 and other varieties of dystonia, his physicians diagnosed his condition as tardive dystonia.
Movement Disorders From Other Psychiatric Medications
As SSRIs elevate a patient’s mood, they may also increase motor activity to abnormal levels, inducing myoclonus, tremor, or akathisia-like leg movements. As discussed previously, SSRIs may cause the serotonin syndrome (see Chapter 6). Despite these caveats, serious adverse reactions to SSRIs occur in only a small proportion of patients. Serious reactions usually occur only when SSRIs are administered in extraordinarily high doses or in combination with other medications, or in patients with a pre-existing neurologic disorder.
Psychogenic Movements
Neurologists’ diagnosis of psychogenic movements usually relies on several factors: (1) the movements’ abrupt onset and subsequent intermittent occurrence; (2) incongruency, inconsistency, and multiplicity of the movements (variability in location, pattern, and number); (3) presence of astasia-abasia (see Fig. 3-4); and (4) certain characteristics of particular movements (see later). Psychogenic movements and other psychogenic neurologic disorders appear to cause greater disability than the neurologic versions. Yet, despite patients’ apparent disability, they occasionally perform certain tasks with the limb beset by the movement. Neurologists describe their choosiness as “selective disability.”
Psychogenic gaits feature slowness and knee buckling. In addition, trembling and dystonic posturing sometimes disrupt normal walking. However, patients may merely exaggerate their effort or rely on uneconomic movements. Whatever the pattern, the impairment often “forces” patients into wheelchairs, keeps them at bedrest, or allows them to ambulate, but only with grimacing and apparently great effort. Their unsteadiness threatens to topple them (see astasia-abasia, Fig. 3-4). Some neurologists point out that crutches, neck braces, and lumbar supports do not speed recovery and serve only to advertise patients’ pain disability.
Movements as a Manifestation of Psychiatric Illnesses
Neurologists diagnose catatonia when patients with psychosis or major depression remain uncommunicative and staring vacantly, motionless, and in fixed natural or unnatural postures, but amenable to examiners’ placing them in different positions, i.e., show waxy flexibility. Despite patients’ mutism and immobility, their consciousness persists. They should have a normal or near-normal EEG. Unlike psychiatrists, neurologists exclude patients who have this appearance due to medical or neurologic conditions, especially NMS (see Chapter 6), PCP intoxication, parkinsonism, or dystonia. When catatonia occurs as a manifestation of a psychosis or mood disorder, an intravenously administered benzodiazepine may interrupt it and provide diagnostic as well as therapeutic help. Administration of ECT also reportedly aborts it.
Caveats
Even in the presence of an obvious psychiatric explanation, making a diagnosis of psychogenic movement disorder carries a risk. As discussed previously (see above and Chapter 3), neurologists and other physicians may easily misdiagnose movements as psychogenic when they are either bizarre or respond to tricks, concentration, anxiety-producing situations, or solitude. In another problem, psychotropic medications may induce unusual yet partly controllable side effects, such as acute dystonic reactions, tardive dystonia, tremors, and akathisia. Moreover, medication-induced movements, such as akathisia or oculogyric crisis, may exacerbate psychiatric disturbances they should have been treating. Finally, a caveat about mass hysteria is that Sydenham chorea tends to appear in miniepidemics among adolescent women living in the same community. The illness among some members may set off an exaggerated psychologic response and elicit movements in healthy adolescents throughout the entire community.
Summary
Physicians may diagnose many involuntary movement disorders exclusively on the basis of their clinical features, family history, patient’s age at onset (Box 18-3), presence or absence of dementia (Box 18-4), or exposure to neuroleptics (see Box 18-2). Recent studies have defined their psychiatric comorbidities. Tests can confirm a clinical diagnosis of Huntington and Wilson diseases, SSPE, Lesch–Nyhan syndrome, early-onset (DYT1) dystonia, Rett syndrome, and several familial forms of Parkinson disease. DBS has been a major advance in the treatment of Parkinson disease, DYT1 dystonia, and essential tremor. After researchers resolve technical issues and conduct clinical trials, DBS will probably also help alleviate cervical and other focal dystonias, Tourette disorder, and some forms of tardive dyskinesia.
Box 18-3
Commonly Cited Movement Disorders that may Begin in Childhood or Adolescence
Parkinson Disease and Parkinsonism
Appleby BS, Duggan PS, Regenberg A, et al. Psychiatric and neuropsychiatric adverse events associated with deep brain stimulation: A meta-analysis of ten years’ experience. Mov Disord. 2007;22:1722–1728.
Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22:1689–1707.
Forsaa EB, Larsen JP, Wentzel-Larsen T, et al. A 12-year population-based study of psychosis in Parkinson disease. Arch Neurol. 2010;67:996–1001.
Fox SH, Katzenschlanger R, Lim SY, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2011;26(Suppl 3):S2–41.
Lee AH, Weintraub D. Psychosis in Parkinson’s disease without dementia: Common and comorbid with other non-motor symptoms. Mov Disord. 2012;27:858–863.
Marras C, Lang A. Changing concepts in Parkinson disease: Moving beyond the decade of the brain. Neurology. 2008;70:1996–2003.
Martinez-Martin P, Arroyo S, Rojo-Abuin JM, et al. Burden, perceived health status, and mood among caregivers of Parkinson’s disease patients. Mov Disord. 2008;23:1673–1680.
Menza M, Dobkin RD, Marin H, et al. The impact of treatment of depression on quality of life, disability and relapse in patients in Parkinson’s disease. Mov Disord. 2009;24:1325–1332.
Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology. 2009;72:886–892.
Merola A, Zibetti M, Angrisano S, et al. Parkinson’s disease progression at 30 years: A study of subthalamic deep brain-stimulated patients. Brain. 2011;134:2074–2084.
Miyasaki JM, Shannon K, Voon V, et al. Practice parameter: Evaluation and treatment of depression, psychosis, and dementia in Parkinson disease (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66:996–1002.
Moro E, Lozano AM, Pollak P, et al. Long-term results of a multicenter study on subthalamic and pallidal stimulation in Parkinson’s disease. Mov Disord. 2010;25:578–586.
Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: Report of an NINDS, NIMH work group. Mov Disord. 2007;22:1061–1068.
Ravina B, Elm J, Camicioli R, et al. The course of depressive symptoms in early Parkinson’s disease. Mov Disord. 2009;24:1306–1311.
Soulas T, Gurrucha JM, Palfi S, et al. Attempted and completed suicide after subthalamic nucleus stimulation for Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2008;79:952–954.
Spencer AH, Rickards H, Fasano A, et al. The prevalence and clinical characteristics of punding in Parkinson’s disease. Mov Disord. 2011;26:578–586.
Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease. Arch Neurol. 2010;67:589–595.
Willis AW, Evanoff BA, Lian M, et al. Metal emissions and urban incident Parkinson disease: A community health study of Medicare beneficiaries by using geographic information systems. Am J Epidemiol. 2010;172:1357–1363.
Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11:1509–1523.
Kranick SM, Mowry EM, Colcher A, et al. Movement disorders and pregnancy: A review of the literature. Mov Disord. 2010;25:665–671.
Maia DP, Teixeria AL, Cunningham MCQ, et al. Obsessive compulsive behavior, hyperactivity, and attention deficit disorder in Sydenham chorea. Neurology. 2005;64:1799–1801.
Paulsen J. Cognitive impairment in Huntington disease: Diagnosis and treatment. Curr Neurol Neurosci Rep. 2011;11:474–483.
Paulsen JS, Hoth KF, Nehl C, et al. Critical periods of suicide risk in Huntington’s disease. Am J Psychiatry. 2005;162:725–731.
Philips W, Shannon KM, Barker RA. The current clinical management of Huntington’s disease. Mov Disord. 2008;23:1491–1504.
Machado A, Chien HF, Deguti MM, et al. Neurologic manifestations in Wilson’s disease: Report of 119 cases. Mov Disord. 2006;21:2192–2196.
Portala K, Westermark K, von Knorring L, et al. Psychopathology in treated Wilson’s disease determined by means of CPRS expert and self-ratings. Acta Psychiatr Scand. 2000;101:104–109.
Soltanzadeh A, Soltanzadeh P, Nafissi S, et al. Wilson’s disease: A great masquerader. Eur Neurol. 2007;57:80–85.
Dystonia (Non-Medication-Induced)
Altenmüeller E, Jabusch HC. Focal dystonia in musicians: phenomenology, pathophysiology, triggering factors, and treatment. Med Probl Perform Art. 2010;25:3–9.
Conti AM, Pullman S, Frucht SJ. The hand that has forgotten its cunning – lessons from musicians’ hand dystonia. Mov Disord. 2008;23:1398–1406.
Geyer HL, Bressman SB. The diagnosis of dystonia. Lancet Neurol. 2006;5:780–790.
Hallett M, Evinger C, Jankovic J, et al. Update in blepharospasm. Neurology. 2008;71:1275–1282.
Heiman GA, Ottman R, Saunders-Pullman RJ, et al. Increased risk for recurrent major depression in DYT1 dystonia mutation carriers. Neurology. 2004;63:631–637.
Isaias IU, Alterman RL, Tagliati M. Deep brain stimulation for primary generalized dystonia: Long term outcomes. Arch Neurol. 2009;66:465–470.
Jhanshahi M, Czernecki V, Zurowski AM. Neuropsychological, neuropsychiatric, and quality of life issues in DMS for dystonia. Mov Disord. 2011;26(Suppl 1):S63–S78.
Jinnah HA, Visser JE, Harris JC, et al. Delineation of the motor disorder of Lesch–Nyhan disease. Brain. 2006;129:1201–1217.
Schretlen DJ, Ward J, Meyer SM, et al. Behavioral aspects of Lesch–Nyhan disease and its variants. Dev Med Child Neurol. 2005;47:673–677.
Schuele S, Lederman RJ. Long-term outcome of focal dystonia in string instrumentalists. Mov Disord. 2003;19:43–48.
Yaltho TC, Jankovic J. The many faces of hemifacial spasm: Differential diagnosis of unilateral facial spasms. Mov Disord. 2011;26:1582–1592.
Deuschl G, Raethjen J, Hellriegel H, et al. Treatment of patients with essential tremor. Lancet Neurol. 2011;10:148–161.
Lorenz D, Poremba C, Papengut F, et al. The psychological burden of essential tremor in an outpatient- and a community-based cohort. Eur J Neurol. 2011;18:972–979.
Sadeghi R, Ondo WG. Pharmacological management of essential tremor. Drugs. 2010;70:2215–2228.
Zhang K, Bhatia S, Oh MY, et al. Long-term results of thalamic deep brain stimulation for essential tremor. J Neurosurg. 2010;112:1271–1276.
Tics, Tourette Disorder, and Related Disorders
Ackerman L, Duits A, van der Linden C, et al. Double-blind clinical trial of thalamic stimulation in patients with Tourette syndrome. Brain. 2011;134:832–844.
Bernard BA, Stebbins GT, Siegel S, et al. Determinants of quality of life in children with Gilles de la Tourette syndrome. Mov Disord. 2009;24:1070–1073.
Cubo E, Chmura T, Goetz C. Comparison of tic characteristics between children and adults. Mov Disord. 2008;23:2407–2411.
Jankovic J, Gelineau-Kattner R, Davidson A. Tourette’s syndrome in adults. Mov Disord. 2010;25:2171–2175.
Kurlan R. Tourette’s syndrome. N Engl J Med. 2010;363:2332–2338.
Kurlan R, Johnson D, Kaplan EL. Streptococcal infection and exacerbations of childhood tics and obsessive-compulsive symptoms: A prospective blinded cohort study. Pediatrics. 2008;121:1188–1197.
Martin-Fernandez R, Zrinzo L, Aviles-Olmos I, et al. Deep brain stimulation for Gilles de la Tourette syndrome. Move Dis. 2011;26:1922–1930.
Muller-Vahl KR, Krueger D. Does Tourette syndrome prevent tardive dyskinesia? Mov Disord. 2011;26:2442–2444.
Piacentini J, Woods DW, Scahill L, et al. Behavior therapy for children with Tourette disorder: A randomized controlled trial. JAMA. 2010;303:1929–1937.
Schrag A, Gilbert R, Giovannoni G, et al. Streptococcal infection, Tourette syndrome, and OCD. Neurology. 2009;73:1256–1263.
Shprecher D, Kurlan R. The management of tics. Mov Disord. 2009;24:15–24.
Swedo SE, Garvey M, Snider L, et al. The PANDAS subgroup: Recognition and treatment. CNS Spectrums. 2001;6:419–426.
Medication-Induced and -Related Movement Disorders
Bondon-Guitton E, Perez-Lloret S, Bagheri H, et al. Drug-induced parkinsonism: A review of 17 years’ experience in a regional pharmacovigilance center in France. Mov Disord. 2011;26:2226–2231.
Brust JCM. Substance abuse and movement disorders. Mov Disord. 2010;25:2010–2020.
Capelle HH, Blahak C, Schrader C, et al. Chronic deep brain stimulation in patients with tardive dystonia without a history of major psychosis. Mov Disord. 2010;25:1477–1481.
Chang EF, Schrock LE, Starr PA, et al. Long-term benefit sustained after bilateral pallidal deep brain stimulation in patients with refractory tardive dyskinesia. Stereotact Funct Neurosurg. 2010;88:304–310.
Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21:151–156.
Guy W. Abnormal Involuntary Movement Scale (AIMS). In: Rockville MD, ed. ECDEU Assessment Manual for Psychopharmacology. U.S.: Department of Health, Education, and Welfare; 1976:534–537.
Kane JM, Fleischhacker WW, Hansen L, et al. Akathisia: An updated review focusing on second-generation antipsychotics. J Clin Psychiatry. 2009;70:627–643.
Mejia NI, Jankovic J. Tardive dyskinesia and withdrawal emergent syndrome in children. Expert Rev Neurother. 2010;10:893–901.
Niethammer M, Ford B. Permanent lithium-induced cerebellar toxicity. Mov Disord. 2007;22:570–573.
Psychogenic Movement Disorders
Baik JS, Lang AE. Gait abnormalities in psychogenic movement disorders. Mov Dis. 2007;22:395–399.
Bartlesman M, Eckhardt PP. Mental illness in the former Dutch Indies – four psychiatric syndromes: amok, latah, koro, and neurasthenia. Ned Tijdschr Geneeskd. 2007;151:2845–2851.
Daniels J. Catatonia: Clinical aspects and neurobiological correlates. J Neuropsychiatry Clin Neurosci. 2009;21:371–380.
Espay AJ, Goldenhar LM, Voon V, et al. Opinions and clinical practices related to diagnosing and managing patients with psychogenic movement disorders. Mov Disord. 2009;24:1366–1374.
Ferrara J, Jankovic J. Psychogenic movement disorders in children. Mov Disord. 2008;23:1875–1881.
Fink M, Taylor MA. The catatonia syndrome. Arch Gen Psychiatry. 2009;66:1173–1177.
Hallett M, Fahn S, Jankovic J, et al. Psychogenic Movement Disorders. Philadelphia: Lippincott Williams & Wilkins; 2006.
Jankovic J, Vuong KD, Thomas M. Psychogenic tremor: Long-term outcome. CNS Spectr. 2006;11:501–508.
Lees A. Jumpers. Mov Disord. 2001;16:403–404.
Massey EW. Goosey patients: Relationship to jumping Frenchmen, Myriachit, Latah, and tic convulsif. N C Med J. 1984;45:556–558.
McKeon A, Ahlskog JE, Bower JH, et al. Psychogenic tremor. Mov Disord. 2009;24:72–76.
Saint-Hilaire MH, Saint-Hilaire JM. Jumping Frenchmen of Maine. Mov Disord. 2001;16:530.
Schwingenschuh P, Pont-Sunyer C, Surtees R, et al. Psychogenic movement in children: A report of 15 cases and a review of the literature. Mov Disord. 2008;13:1882–1888.
Shill H, Gerber P. Evaluation of clinical diagnostic criteria for psychogenic movement disorders. Mov Disord. 2006;21:1163–1168.
Tanner CM, Chamberland J. Latah in Jakarta, Indonesia. Mov Disord. 2001;16:526–529.
Chapter 18Questions and Answers
1. Which of the following statements concerning obsessive-compulsive symptoms and obsessive-compulsive disorder (OCD) in Tourette disorder is false?
17. A psychiatrist is asked to evaluate an immobile, expressionless, mute, 21-year-old woman who stares straight ahead. The psychiatrist cannot engage her but is able to position her limbs in a variety of positions that she holds for at least 10 minutes. The patient’s records reveal a history of a mood disorder. While awaiting a neurology consultation, which of the following is the best initial management of this patient?
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 18 Involuntary Movement Disorders
The Basal Ganglia
Five subcortical gray-matter macroscopic nuclei (Figs 18-1 and 18-2) constitute the basal ganglia:
• The caudate nucleus and putamen, which together constitute the striatum
• The globus pallidus, which, together with the putamen, constitutes the lenticular nuclei
FIGURE 18-1 A, This axial view, the one used in computed tomography and magnetic resonance imaging studies, shows the basal ganglia in relation to other brain structures. The heads of the caudate nuclei (C) indent the lateral undersurface of the anterior horns of the lateral ventricles. The caudate and putamen (P) constitute the striatum. The globus pallidus (G), which has internal and external segments, and the putamen form the lenticular nucleus, named for its resemblance to an old-fashioned lens (see Fig. 18-1, C). The posterior limb of the internal capsule (IC) separates the lenticular nucleus from the thalamus (T), which is not a member of the basal ganglia family. B, In this coronal view of the diencephalon, the substantia nigra (SN), as well as the subthalamic nuclei (STN), sits below the thalamus. The substantia nigra, because of its characteristic shape and pigmentation, serves as a landmark. The lateral ventricles are bounded laterally by the heads of the caudate nuclei (C) and superiorly by the corpus callosum (CC). This myelin stain has blackened the heavily myelinated fibers of the internal capsule (IC) and corpus callosum (CC). C, A coronal view shows extrapyramidal circuits. The putamen sends a direct and an indirect dopamine tract to the internal segment of the globus pallidus (GPi). Dopaminergic neurons in the substantia nigra project to the putamen, where neurons with D1 receptors project directly to the GPi. Putaminal neurons with D2 receptors project through the globus pallidus external segment (GPe) and subthalamic nucleus and thence to GPi. The GPi innervates the ventrolateral nucleus of the thalamus, which projects to the motor cortex. The cortex, completing a circuit, innervates the putamen.
FIGURE 18-2 This computer-generated rendition of the midbrain should be compared to a photograph (see Fig. 2-9), functional drawings (see Figs 4-5 and 4-9), and an idealized sketch (see Fig. 21-1). The lower third of the midbrain, which lies just caudal to the diencephalon, contains the pair of horizontal but gently curved, elongated, pigmented nuclei – the substantia nigra. In Parkinson disease, the substantia nigra and other pigmented nuclei lose their pigment and, compared to normal, thus appear blanched. The midbrain also houses the prominent aqueduct of Sylvius, which is the dorsal heart-shaped hole surrounded by the periaqueductal gray matter. Cerebrospinal fluid passes from the third ventricle, through the aqueduct, to the fourth ventricle.
Although the extrapyramidal tract seems to play merely a supportive role – indirectly influencing the corticospinal tract by acting on thalamocortical connections and projecting only within the brain – it comprises several clinically important tracts. The most important one, from a clinical viewpoint, is the nigrostriatal tract, which, as its name suggests, extends from the substantia nigra to the striatum (Fig. 18-3). This tract provides dopamine innervation directly and indirectly, via the subthalamic nucleus, to the globus pallidus’ internal segment (GPi).
FIGURE 18-3 A succession of enzymes normally converts tyrosine to dopamine in the presynaptic nigrostriatal neuron. In Parkinson disease, the nigrostriatal tract degenerates, but the postsynaptic dopamine receptors remain intact. As a result of the degeneration, an absence of tyrosine hydroxylase leads to insufficient dopa and then markedly reduced synthesis of dopamine. L-dopa, which is a standard oral medication for Parkinson disease, penetrates the blood–brain barrier and substitutes for the deficient endogenous dopa in dopamine synthesis. D-dopa, the dextro-isomer, does not cross the blood–brain barrier and cannot enter the synthetic pathway. It is therefore useless as a treatment for Parkinson disease.
One of the main differences between dopamine receptors is that, in the striatum, dopamine binding to dopamine 1 (D1) receptors stimulates adenyl cyclase activity, but dopamine binding to dopamine 2 (D2) receptors inhibits adenyl cyclase activity (see Table 21-1).
General Considerations
The involuntary movement disorders share several clinical features. Anxiety, exertion, fatigue, and stimulants (including caffeine) increase the movements, but willful concentration and sometimes biofeedback may suppress them, at least transiently. Most movements disappear during sleep. The exceptions – hemifacial spasm, myoclonus, palatal tremor, tics, and specific sleep-related disorders, such as restless legs syndrome (RLS) and periodic movements – persist without regard to sleep stage (see Chapter 17).
Neurologists typically measure four parameters of involuntary movements:
• Hypokinesia or hyperkinesia. (For most clinical purposes, Parkinson disease and parkinsonism [see later] constitute the sole hypokinetic involuntary movement disorder.)
• Continuous or intermittent, discrete movements
Another error may occur when patients, at first glance, appear to have a psychogenic movement disorder (see later and Chapter 3). In many situations, the lack of a definitive confirmatory laboratory test forces neurologists to rely exclusively on their clinical judgment.
Parkinson Disease
The initial and ultimately most disabling physical feature of Parkinson disease is usually bradykinesia or, in the extreme, akinesia. Slow or absent movement produces the classic masked face (Fig. 18-4), paucity of trunk and limb movement (Figs 18-5 and 18-6), and impairment of activities of daily living. Parkinson patients sometimes liken their slow movements to slogging through hip-deep mud, wearing lead clothing, or driving a car with an engaged emergency brake.
FIGURE 18-4 Compared to normal individuals of the same age, Parkinson disease patients blink less frequently, offer fewer facial expressions, and less often move their head. Neurologists have called patients’ facial appearance a “stare” or “masked facies” (Latin, face or countenance), but now they usually describe the face as masked. Even when subtle, the masked face gives the appearance of apathy or depression.
FIGURE 18-5 Parkinson disease patients typically sit motionless with their legs uncrossed and their feet flat. Their arms remain on the chair or in their lap and rarely participate in normal gestures or repositioning movements. In contrast to normal individuals and especially those with chorea, Parkinson disease patients do not shift their weight from one hip to another or make any unnecessary movements.
FIGURE 18-6 Patients with akinesia and rigidity cannot rapidly flex their spine, hips, or knees. When sitting, they tend to fall slowly and solidly backward into a chair. Unable to bend rapidly, their feet rise several inches off the floor. Sitting and turning en bloc signal early parkinsonism.
Rigidity typically accompanies bradykinesia (Fig. 18-7). Although rigidity is one of the cardinal features of Parkinson disease, it often appears as a manifestation of other extrapyramidal disorders. No matter the context, physicians should not confuse rigidity with spasticity, which signals corticospinal tract disease (see Chapter 2).
FIGURE 18-7 Physicians typically elicit rigidity by rotating the patient’s wrist. When present, rigidity causes an increased tone in all directions of movement. A superimposed tremor adds a ratchet-like, cogwheel rigidity resistance.
Tremor is often the most conspicuous feature of Parkinson disease; however, it is the least specific sign, least debilitating symptom, and least associated with dementia and depression. In Parkinson disease, the affected body part usually oscillates in a single plane with a regular rate, although with a variable amplitude. It primarily involves the hands. Even more characteristically, the tremor appears when patients rest quietly with their arms supported. Neurologists call it a resting tremor (Fig. 18-8) and can show that it differs from cerebellar and essential tremors. When patients have tremor as their primary symptom, neurologists refer to them as having “tremor-predominant” Parkinson disease.
FIGURE 18-8 The resting tremor – a cardinal feature of Parkinson disease – consists of a relatively slow (4–6-Hz) to-and-fro flexion movement of the wrist, hand, thumb, and fingers most apparent when patients sit comfortably. Its similarity to shaking pills in a cupped hand gave rise to the description “pill-rolling” tremor. The tremor is exaggerated or sometimes apparent only when patients are anxious. However, voluntary movement or intense concentration may momentarily reduce the tremor, and sleep abolishes it. Rigidity and akinesia almost always accompany the Parkinson disease resting tremor.
Additional symptoms and signs may emerge as Parkinson disease advances. Patients lose their postural reflexes, which are neurologic compensatory mechanisms that adjust muscle tone in response to change in position. Loss of these reflexes, in combination with akinesia and rigidity, results in a characteristic gait impairment, marche à petit pas or festinating gait, which consists of a tendency to lean forward and accelerate the pace (Fig. 18-9, A). In a test of postural reflexes, the pull test, the examiner pulls the patient from the shoulders (Fig. 18-9, B). Normal individuals merely sway. Patients who have mild impairment of their postural reflexes take a few steps back, i.e., have retropulsion. More severely affected ones rock stiffly backwards without flexion or other compensatory movement and topple, en bloc, into the examiner’s arms.
FIGURE 18-9 A, Parkinson disease often produces a festinating gait, in which patients take short, shuffling steps and accelerate their pace. Their neck and lower spine are typically flexed. When walking, they fail to swing their arms, look about, or have other normal accessory movements. Likewise, while turning en bloc, they simultaneously move their head, trunk, and legs. B, The pull test consists of the physician gently but rapidly pulling the patient’s shoulders. Unaffected individuals will compensate by taking one or two steps backward. Parkinson patients, who generally have impaired postural reflexes, will take many steps backward (i.e., have retropulsion), because they are unable to stop through reflexly weight-shifting compensatory maneuvers. In pronounced cases, as the one pictured here, patients unable to alter their posture will tilt backwards en bloc and fall into the physician’s arms. (Progressive supranuclear palsy patients have similar bradykinesia, lack of accessory movements, and a positive pull test, but their posture is extended or erect rather than flexed [see Fig. 7-7].)
Even at the onset of the illness, patients’ handwriting deteriorates to a small and tremulous script, micrographia (Fig. 18-10). In a parallel fashion, their voice loses both volume and normal fluctuations in pitch and cadence, i.e., their speech becomes hypophonic and monotonous. Also, because of their illness or the medications used to treat it, Parkinson disease patients develop sleep disturbances, characteristically rapid eye movement sleep behavior disorder (see Chapter 17).
FIGURE 18-10 The handwriting in this sample from a Parkinson disease patient shows progressive decrease in height (micrographia) and a superimposed tremor. These abnormalities in signatures, such as those on checks, can often date the onset of Parkinson disease. Although essential tremor may also cause tremor in handwriting samples, micrographia indicates that the underlying illness is Parkinson disease.
Nonmotor physical problems also emerge. For example, Parkinson patients characteristically lose their sense of smell. The anosmia, which also occurs in Alzheimer disease, reflects the neurodegenerative nature of these illnesses (see Chapters 4 and 7). They also routinely develop problems with their autonomic nervous system, including dysphagia, constipation, urinary incontinence, and abnormal sweating.
Psychiatric Conditions Comorbid with Parkinson Disease
Treatment of Comorbid Depression
Although SSRIs carry fewer side effects than TCAs in the population of depressed Parkinson disease patients, they may cause a unique problem. Prescribing SSRIs in conjunction with selegiline (deprenyl [Eldepryl]) can theoretically cause the serotonin syndrome because SSRIs prevent serotonin reuptake while selegiline, a monoamine oxidase (MAO) inhibitor, prevents its breakdown (see Chapter 6). The actual incidence of serotonin syndrome is very low because selegiline, in the doses used for Parkinson disease treatment, selectively inhibits only MAO-B, which metabolizes dopamine; whereas the serotonin syndrome is mostly a complication of inhibition of MAO-A, which metabolizes serotonin. Similarly, when physicians prescribe the selegiline patch (Emsam) for depression, as long as the dose remains below 12 mg/day, which is standard, it inhibits MAO-B; however, selegiline patch doses greater than 12 mg/day inhibit MAO-A as well as MAO-B, and leave patients at risk of tyramine-induced hypertension.
Dementia
However, as the illness progresses and patients age, dementia routinely complicates the illness. Dementia affects about 20% of all Parkinson disease patients, 40% of those older than 70 years, and more than 80% who have had the illness for 20 years. Its prevalence increases in proportion to the patient’s age, duration of the illness, and physical impairments. Dementia is more frequent when akinesia and rigidity rather than tremor predominate in Parkinson disease. Unlike the dementia in Alzheimer disease, the dementia in Parkinson disease is not associated with apolipoprotein E4 alleles (see Chapter 7).
Parkinson disease dementia, which differs clinically from that of Alzheimer disease dementia, is distinguished by inattention, poor memory, difficulty shifting mental sets, and bradyphrenia (slowed thinking, the cognitive counterpart of bradykinesia), as well as hallucinations. With its almost invariable gait impairment and preserved language function, Parkinson disease dementia serves as a prime example of “subcortical dementia” (see Chapter 7). The Mini-Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MoCA) are each valid screening tests for cognitive impairment in Parkinson disease; however, the MoCA is more sensitive.
A special diagnostic hazard when evaluating a patient with parkinsonism and dementia is failing to recognize dementia with Lewy bodies disease. This illness and Parkinson disease share rigidity and bradykinesia, delusions and hallucinations, and sleep disturbances as well as cognitive impairment. One major clinical difference is that in dementia with Lewy bodies disease, dementia constitutes an initial, not a late-developing, manifestation (see Chapter 7), but it is at least a relatively late development in Parkinson’s disease. On a histologic level, dementia with Lewy bodies disease features Lewy bodies in the cerebral cortex, not just confined to the basal ganglia.
Treatment of Comorbid Psychosis
Because administration of antiparkinson medication before bedtime tends to cause nightmares and hallucinations, neurologists advance the last daily dose to the early evening. Also, they avoid suddenly stopping these medicines because their abrupt withdrawal may lead to irreversible motor deterioration, complications of immobility, or even the neuroleptic-malignant syndrome (NMS) (see Chapter 6).
Pathology of Parkinson Disease
The loss of tyrosine hydroxylase represents the critical failure in Parkinson disease because this enzyme is the rate-limiting enzyme in dopamine synthesis (see Fig. 18-3). With the tyrosine hydroxylase deficit, the ever-shrinking pool of remaining nigrostriatal tract neurons cannot sustain the essential synthetic pathway. Once approximately 30% of these neurons degenerate, the nigrostriatal tract cannot synthesize adequate dopamine and Parkinson disease symptoms appear.
On a microscopic level, neurons in these locations characteristically accumulate Lewy bodies, which contain a core of α-synuclein (see Chapter 7). In contrast, Lewy bodies located in the cerebral cortex, as well as the basal ganglia, constitute the histologic hallmark of dementia with Lewy bodies disease. With their abundance of Lewy bodies, both Parkinson disease and dementia with Lewy bodies disease fall under the rubric of synucleinopathies.
Parkinsonism
Medication-Induced Parkinsonism
When medicines, as opposed to illicit drugs, induce parkinsonism, rigidity is the most prominent feature, all three cardinal features occur in only about one-third of cases, and individuals older than 60 years and women are particularly susceptible. Typical and most atypical antipsychotic agents – because to a greater or lesser degree they block D2 receptors – routinely cause parkinsonism. Tetrabenazine (Xenazine) induces parkinsonism because it depletes dopamine (see later and Figs 18-3 and 18-12). Similarly, nonpsychiatric medicines that block D2 receptors, such as metoclopramide (Reglan), trimethobenzamide (Tigan), prochlorperazine (Compazine), and promethazine (Phenergan), produce the same problem. Case reports have also implicated valproate (Depakote), lithium, amiodarone, and calcium channel blockers – medicines with no direct connection to D2 receptors.
Medication-induced parkinsonism, once physicians discontinue the offending drug, should fully resolve in a few weeks. Nevertheless, signs often persist for 3 months and occasionally for 1 year. However, physicians must be careful with persistent parkinsonism patients because approximately 10% harbor Parkinson disease and some probably have dementia with Lewy bodies disease (see Chapter 7). In children and young adults, persistent parkinsonism following antipsychotic treatment suggests several neurologic illnesses (see later).
Therapy of Parkinson Disease
Medications
L-Dopa.
Treatments for Parkinson disease alleviate motor symptoms for many years, but do not reverse the neurodegeneration. Medicines maintain normal dopamine activity by enhancing dopamine synthesis, retarding its metabolism, or acting as agonists at dopamine receptors. Enhancing dopamine synthesis, usually the initial treatment, consists of substituting orally administered L-dopa for endogenous but deficient DOPA in the synthetic pathway (Fig. 18-3).
Dopa- and Dopamine-Preserving Medications
Several medications increase nigrostriatal dopa by inhibiting two enzymes – dopa decarboxylase and catechol-O-methyltransferase (COMT) – that metabolize it in the systemic circulation (Fig. 18-11). With a relative increase in nigrostriatal dopa, nigrostriatal dopamine concentration increases.
FIGURE 18-11 Medicines that inhibit catechol-O-methyltransferase (COMT), such as entacapone, and those that inhibit decarboxylase, such as carbidopa, slow the metabolism of L-dopa. Because of the blood–brain barrier, they do not affect dopamine synthesis in the nigrostriatal tract. These enzymes, combined with L-dopa, permit smaller L-dopa doses, which minimizes systemic side effects.
On the other hand, although they ameliorate some symptoms and provide a modicum of antidepressant effect, antiparkinson MAO-B inhibitors carry a caveat. At high doses, they inhibit MAO-A as well as MAO-B. Because MAO-A is important in both serotonin and catecholamine metabolism, MAO-B inhibitors place patients in a position where they are vulnerable to the serotonin syndrome or a hypertensive crisis (see previously and Chapters 6, 9, and 21).
Dopamine Agonists
As an initial medication or when dopamine production eventually falls to inadequate levels, dopamine agonists – pramipexole and ropinirole – provide stimulation of postsynaptic dopamine receptors (see Fig. 18-3). Bypassing degenerated presynaptic neurons, dopamine agonists act directly on D2 and, to a lesser extent, on other postsynaptic dopamine receptors.
Other Medications
Anticholinergics reduce tremor in Parkinson disease and other forms of parkinsonism. By reducing cholinergic activity, these medicines seem to act by maintaining the balance with the diminished dopamine activity (Fig. 18-12). On the other hand, anticholinergics routinely produce mental and physical side effects, especially in the elderly.
FIGURE 18-12 In a classic but limited model, dopamine and acetylcholine (cholinergic) activity normally balance each other. When Parkinson disease reduces dopamine activity, the left side of the scale rises. Dopamine precursors, dopamine agonists, and anticholinergics – Parkinson disease treatments – restore the balance. Conditions characterized by excessive dopamine activity, such as chorea, push the left side downward. Substances that either antagonize or deplete dopamine, or enhance cholinergic activity, restore the balance.
Athetosis
Athetosis consists of involuntary slow, continually changing, twisting movements predominantly affecting the face, neck, and distal limbs (Fig. 18-13). It sits at the beginning of a sequence – athetosis, choreoathetosis, chorea, and hemiballismus – of progressively larger and more irregular involuntary movements. In a potentially confusing aspect, additional involuntary movements may coexist with athetosis. For example, rapid jerks of chorea may punctuate slow movements of athetosis, and powerful twists of dystonia may interrupt and override athetosis’ slow fluctuations.
FIGURE 18-13 In athetosis, the face grimaces incessantly. Fragments of smiles alternate with frowns. Pulling of one or the other side distorts the entire appearance. In addition, neck muscles contract and rotate the head. Laryngeal contractions and irregular chest and diaphragm muscle movements cause an irregular speech cadence, nasal pitch, and dysarthria. Fingers writhe constantly and tend to assume hyperextension postures. At the same time, wrists rotate, flex, and extend. Involuntary limb activity prevents writing, buttoning, and other fine tasks, but it usually allows deliberate, larger-scale shoulder, trunk, and hip movement.
Usually encountered as a variety of cerebral palsy (see Chapter 13), athetosis is usually not apparent until early childhood. Most often athetosis results from combinations of perinatal hyperbilirubinemia (kernicterus), hypoxia, and prematurity. Genetic factors are unimportant.
Because athetosis originates in brain injuries that occur during the first 30 days after birth as well as in utero, which neurologists consider congenital brain injuries, seizures and mental retardation frequently accompany this movement disorder. However, with damage confined to the basal ganglia, as in many cases of athetosis, patients have normal intelligence despite disabling movements and garbled speech. Physicians, schoolteachers, family members, and friends should recognize that these patients retain cognitive and emotional capacities despite devastating physical neurologic disabilities.*
Chorea
Huntington Disease
Of the many causes of chorea (Box 18-1), Huntington disease, previously called “Huntington’s chorea,” remains pre-eminent. Chorea, dementia, and behavioral abnormalities characterize this autosomal dominantly inherited disorder. Symptoms first emerge when patients are, on the average, approximately 37 years. However, approximately 10% of patients develop symptoms in childhood or adolescence and 25% when they are older than 50 years. Adults with the illness usually succumb to aspiration and inanition one to two decades after the diagnosis.
