Chapter 1 Introduction
Formulation of the Problem
Prenatal diagnoses have been possible for more than 25 years.2 At about embryonic day 20 in the human fetus, progenitor cells within the mesoderm become committed to a cardiogenic destination.3 Most malformations compatible with 6 months of intrauterine life permit live offspring at term. A given malformation may exist in relative harmony with the fetal circulation, only to be modified considerably, at least physiologically, by dramatic circulatory changes at birth.4 Weeks, months, or years may then elapse before an anomaly reveals itself as characteristic or typical. A functionally normal aortic valve that is congenitally bicuspid at birth may take decades to fibrose, calcify, and present as overt aortic stenosis. Conversely, a malformation may disappear, as is the case with spontaneous closure of a ventricular septal defect.
Congenital malformations of the heart and circulation are not fixed anatomic defects that appear at birth but instead are anomalies in flux that originate in the early embryo, evolve during gestation, survive the dramatic circulatory alterations at birth, and change considerably during the course of extrauterine life. Congenital heart disease was once the exclusive and legitimate domain of pediatrics, but survival patterns have changed appreciably.5,6 In the United States today, there are more adults with congenital heart disease than there are infants and children; these new generations of patients are best cared for by specially trained cardiologists.7 Clinical presentations, especially in adults,8,9 can be exceptionally complex.10–12
Furthermore, certain defects that are actually or potentially of functional significance are not gross structurally, such as congenital complete heart block, either isolated (see Chapter 4) or with congenitally corrected transposition of the great arteries (see Chapter 6) or left isomerism (see Chapter 3); Wolff-Parkinson-White preexcitation, either isolated or complicated (see Chapter 13); absence of a sinus node, as with a superior vena caval sinus venosus atrial septal defect (see Chapter 15) or left isomerism (see Chapter 3); ventricular tachycardia with the long QT syndromes13; or arrhythmogenic right ventricular dysplasia. Still other abnormalities, such as Marfan syndrome, which is the result of mutations in the gene that encodes fibrillin-1, are actually or potentially of functional significance but are not necessarily manifest at birth as gross structural malformations and, as a matter of convention, are usually not classified as congenital.14 And a handful of odd defects tend to escape inclusion, such as congenital kinking of the internal carotid artery.15
Because of the intimate interplay between congenital malformations of the heart and the fetal substrate, we now turn to the fetal circulation per se and the remarkable circulatory changes that occur at birth.4,16 In utero patterns of blood flow have been defined in fetal lambs after insertion of catheters into limb vessels,4 and in utero left and right ventricular outputs and flow distributions through the foramen ovale, ductus arteriosus, and pulmonary bed have been measured in human fetuses with high-resolution color Doppler ultrasound scan.16,17 The right and left ventricles do not function in series in the fetus as they do in the extrauterine circulation. Gas exchange occurs in the placenta, not in the lungs. About two thirds of the right ventricular output is diverted away from the lungs through a widely patent ductus arteriosus into the descending thoracic aorta, from which a large portion enters the umbilical circulation for oxygenation in the placenta. Pulmonary blood flow is low but meets nutritional requirements for growth of the lungs. Inferior vena cava blood, which is a composite from the umbilical vein, the left and right hepatic veins, the ductus venosus, and the distal inferior vena cava, streams in the direction of the foramen ovale into the left atrium and then into the left ventricle, assisted by the eustachian valve. About 60% of the umbilical venous return that is oxygenated in the placenta bypasses the liver through the ductus venosus and enters the inferior vena cava and right atrium. Blood from the superior vena cava is deflected within the right atrium toward the tricuspid valve, across which it reaches the right ventricle. About 70% of the relatively oxygen-rich blood ejected from the left ventricle is distributed to the coronary circulation (and thus the myocardium), to the head and neck (and thus the brain), and to the forelimbs. Almost 90% of the relatively oxygen-poor blood ejected by the right ventricle passes through the ductus arteriosus into the descending aorta to be oxygenated in the placenta. Accordingly, the fetal circulation is designed so that blood with higher oxygen saturation preferentially reaches the myocardium and brain and less saturated blood preferentially reaches the placenta.
The profound circulatory changes at birth occur almost simultaneously. The umbilical/placental circulation is eliminated, the lungs expand, rhythmic ventilation commences, oxygen tension rises as alveolar fluid is eliminated and ambient air is breathed, pulmonary vascular resistance falls, and pulmonary blood flow increases eight to 10 fold. Venous return to the left atrium increases, left atrial pressure rises, the valve of the foramen ovale closes, and the ductus arteriosus functionally closes (constricts) within hours after birth, effectively separating the pulmonary and systemic circulations.4
A number of important delayed events complete the transition from the fetal to the maturing circulation after birth. During the course of gestation, pulmonary vascular resistance falls progressively as new resistance arterioles develop. Regulation of the perinatal pulmonary circulation reflects a complex interplay between vasoconstrictor and vasodilator factors, but the net effect is a dramatic increase in pulmonary blood flow initiated by expansion of the lungs and rhythmic ventilation that reflects a shift from active pulmonary vasoconstriction in the fetus to active vasodilation in the neonate.4 Failure of this shift to occur results in persistent fetal circulation of the newborn. Thick-walled fetal pulmonary arterioles are designed to meet the full force of systemic right ventricular pressure the instant the lungs expand. As respiration commences, a marked increase in alveolar and systemic arterial oxygen tension occurs, to which pulmonary arterioles are exquisitely sensitive, setting the stage for dilation and involution. Large pulmonary arteries also play a role, although much lesser, in determination of the total drop in pressure across the lungs.
Maturational changes have an impact on the neonatal disparity in size between the main and branch pulmonary arteries and on the angulation at the origins of the right and left pulmonary arterial branches (see Chapter 11). Both of these factors are responsible for a physiologic drop in pressure distal to the pulmonary trunk. Another important change relates to the fetal right ventricle, which slowly loses its relative mass. With the stimulus of afterload eliminated, a gradual reduction is seen in right ventricular wall thickness relative to the ventricular septum and left ventricle. The neonatal right ventricle does not undergo regression but instead does not increase its mass as rapidly as the left ventricle. Normal physiologic adaptations at birth are remarkable in their own right. Therefore, that congenital malformations of the heart or circulation will, to varying degrees, interact with and be modified by extrauterine life is not surprising.
“One of the most wondrous features of animate nature must surely be the perfect harmony existing between structure and function.”17 No organ system exemplifies this harmony as well as the heart and circulation. In the presence of congenital heart disease, both the morphology and the physiology of the heart and circulation change with the passage of time from the fetus to the neonate to the infant, to the child, to the adolescent, and to the adult. Some of these changes result in neonatal death, and others express themselves gradually over weeks, months, years, or even decades. The clinical manifestations of congenital heart disease are dealt with herein in terms of anatomic and physiologic mechanisms. The method of assessment used in the process of clinical recognition is a syllogism of applied logic. When conclusions are drawn from accurate observations, correct diagnoses emerge with gratifying frequency, even when the malformations are complex. Logic is encouraged; memorization is minimized. In each chapter, the gross morphology is first established to shed light on the physiologic derangements. The question is then asked: What clinical manifestations might result from these anatomic and physiologic derangements? The stage is now set for clinical recognition—the thesis of this book—which depends on a synthesis of information from the history, the physical examination, the electrocardiogram, and the chest x-ray. Contemporary imaging techniques serve as important supplements.
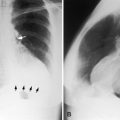
The history, which is an interview, is designed to secure background information. The physical examination includes physical appearance, arterial pulse, jugular venous pulse, precordial percussion, movement and palpation, and auscultation.18 The stethoscope is the oldest cardiovascular diagnostic instrument in continuous clinical use. The electrocardiogram, developed in 1903 by Willem Einthoven, when read closely and interpreted in clinical context, provides gratifying diagnostic insights, even in complex congenital heart disease.19 Meticulous attention should be devoted to the P wave, the P duration, the PR interval, the QRS, the QT interval, and the T wave. The electrocardiographic waves are thought to have been so-named by Rene Descartes, an 18th century French philosopher and scientist20 who was surely known to Einthoven. The x-ray was developed in 1895 by Wilhelm Konrad Roentgen. Chest x-rays must be read closely and interpreted in clinical context. Posteroanterior and lateral views should be read according to a planned sequence: namely, penetration, rotation, degree of inhalation, age and sex, right/left orientation, thoracic and abdominal situs, positions and malpositions above and below the diaphragm, the bones, extrapulmonary soft tissue densities, vascular and parenchymal intrapulmonary soft tissue densities, the great arteries and great veins, the atria, and the ventricles. Much can be said for learning interpretation by reading chest x-rays with a trained chest radiologist. As with the electrocardiogram, so too with the chest x-ray—the amount of information that can be extracted is gratifying, even in complex congenital heart disease.
Decades ago, Paul Wood proposed that we answer five clinical questions (Box 1-1). When approached according to this proposal, the clinical recognition of congenital heart disease becomes a stimulating challenge, a satisfying discipline in logical thinking, and a constant source of self education. The intensity of enquiry and the analytic standards used at the bedside should be the same as those in diagnostic laboratories.
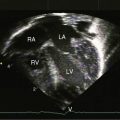
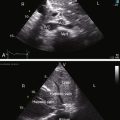
The Preface to the first edition stated, “I hope to stimulate clinicians to use the basic tools readily at their disposal—the history, the physical examination, the electrocardiogram, and the chest x-ray.” The Preface to the third edition stated, “Two-dimensional echocardiography was in its infancy when the second edition appeared, but now is a routine laboratory procedure.” Cardiac catheterization and angiography were followed by transthoracic and transesophageal echocardiography, and magnetic resonance imaging, magnetic resonance angiography, and computed tomography now provide exquisite anatomic detail and refined physiologic information.21–24
Given these advanced imaging techniques, and with more in the offing, has emphasis on the history, physical examination, electrocardiogram, chest x-ray, and echocardiogram become irrelevant? Intelligent selection of investigative procedures from an ever-increasing array requires far more sophisticated decision making than was necessary when choices were limited to the electrocardiogram and chest x-ray. The basic clinical assessment provides the information necessary for most of these decisions. With increasing emphasis on the cost of medical care, a resurgence of interest in the inexpensive and safe clinical examination is likely.25
Some additional points
The term natural history is not used. Julien Hoffman’s definition is relevant: “The natural history of any disease is a description of what happens to people with the disease who do not receive treatment for it.”26 Natural history is therefore not synonymous with unoperated, and unnatural history is not synonymous with postoperative. Nonsurgical therapeutic interventions can hardly be considered natural. A second point: differential diagnoses are not used. Instead, each chapter ends with a summary that permits clinical recognition of the congenital anomaly covered in the chapter. These summaries bring together clinical highlights and serve as reminders for the reader’s convenience.
Finally, a comment on terminology is needed. The First International Summit on Nomenclature for Congenital Heart Disease was held in 2001 in Toronto and ended with virtually unanimous support for the development of a unified system for describing congenial cardiac malformations.27 The terms used herein to describe congenital malformations of the heart and circulation include older Latinized forms and more recent Anglicized forms.28–30 The vocabulary of congenital heart disease is replete with abbreviations understood only by the initiated. Accordingly, when abbreviations are used, they are first defined for the reader’s convenience, but full terms are subsequently used. New terms are not used when old terms suffice. Acronyms are avoided.
1 Williams J.R. The life of Goethe. Oxford: Blackwell Publishers; 1998.
2 Allan L. Prenatal diagnosis of structural cardiac defects. Am J Med Genet C Semin Med Genet. 2007;145C:73-76.
3 Olson E.N., Srivastava D. Molecular pathways controlling heart development. Science. 1996;272:671-676.
4 Moller J.H., Hoffman J.I.E., editors. Pediatric cardiovascular medicine. New York: Churchill Livingstone, 2000.
5 Marelli A.J., Mackie A.S., Ionescu-Ittu R., Rahme E., Pilote L. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation. 2007;115:163-172.
6 Khairy P., Ionescu-Ittu R., Mackie A.S., Abrahamowicz M., Pilote L., Marelli A.J. Changing mortality in congenital heart disease. J Am Coll Cardiol. 2010;56:1149-1157.
7 Child J.S., Freed M.D., Mavroudis C., Moodie D.S., Tucker A.L. Task force 9: training in the care of adult patients with congenital heart disease. J Am Coll Cardiol. 2008;51:389-393.
8 Sommer R.J., Hijazi Z.M., Rhodes J.F.Jr. Pathophysiology of congenital heart disease in the adult: part I: shunt lesions. Circulation. 2008;117:1090-1099.
9 Rhodes J.F., Hijazi Z.M., Sommer R.J. Pathophysiology of congenital heart disease in the adult, part II. Simple obstructive lesions. Circulation. 2008;117:1228-1237.
10 Sommer R.J., Hijazi Z.M., Rhodes J.F. Pathophysiology of congenital heart disease in the adult: part III: complex congenital heart disease. Circulation. 2008;117:1340-1350.
11 Ellis C.R., Graham T.P.Jr, Byrd B.F.3rd. Clinical presentations of unoperated and operated adults with congenital heart disease. Curr Cardiol Rep. 2005;7:291-298.
12 Perloff J.K., Child J.S., Aboulhosn J., editors. Congenital heart disease in adults, 3rd ed, Philadelphia: Saunders/Elsevier, 2009.
13 Chiang C.E., Roden D.M. The long QT syndromes: genetic basis and clinical implications. J Am Coll Cardiol. 2000;36:1-12.
14 Milewicz D.M. Molecular genetics of Marfan syndrome and Ehlers-Danlos type IV. Curr Opin Cardiol. 1998;13:198-204.
15 Le Bret E., Pineau E., Folliguet T., et al. Congenital kinking of the internal carotid artery in twin brothers. Circulation. 2000;102:E173-E174.
16 Mielke G., Benda N. Cardiac output and central distribution of blood flow in the human fetus. Circulation. 2001;103:1662-1668.
17 Alcazar J.L., Rovira J., Ruiz-Perez M.L., Lopez-Garcia G. Transvaginal color Doppler assessment of fetal circulation in normal early pregnancy. Fetal Diagn Ther. 1997;12:178-184.
18 Perloff J.K. Physical examination of the heart and circulation, 4th ed. Shelton, Connecticut: People’s Medical Publishing House; 2009.
19 Khairy P., Marelli A.J. Clinical use of electrocardiography in adults with congenital heart disease. Circulation. 2007;116:2734-2746.
20 Hurst J.W. Naming of the waves in the ECG, with a brief account of their genesis. Circulation. 1998;98:1937-1942.
21 Hlavacek A.M., Baker G.H., Shirali G.S. Innovation in three-dimensional echocardiography and cardiac computed tomographic angiography. Cardiol Young. 2009;19(suppl 2):35-42.
22 Crean A. Cardiovascular MR and CT in congenital heart disease. Heart. 2007;93:1637-1647.
23 Valente A.M., Powell A.J. Clinical applications of cardiovascular magnetic resonance in congenital heart disease. Cardiol Clin. 2007;25:97-110.
24 Marx G.R., Su X. Three-dimensional echocardiography in congenital heart disease. Cardiol Clin. 2007;25:357-365.
25 Braunwald E. Foreword. In Perloff J.K., editor: Physical examination of the heart and circulation, 4th ed, Shelton, Connecticut: People’s Medical Publishing House, 2009.
26 Hoffman J.I.E. Reflections on the past, present and future of pediatric cardiology. Cardiol Young. 1994;4:208-223.
27 Beland M.J., Franklin R.C.G., Jacobs J.P., et al. Update from the International Working Group for Mapping and Coding of Nomenclatures for Paediatric and Congenital Heart Disease. Cardiol Young. 2004;14:225-229.
28 Botto L.D., Lin A.E., Riehle-Colarusso T., Malik S., Correa A., National Birth Defects Prevention S. Seeking causes: classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Research. 2007;79:714-727.
29 Carr C. Congenital nomenclature: a cause of confusion during literature search. Ann Thorac Surg. 2007;84:716.
30 Jacobs J.P., Anderson R.H., Weinberg P.M., et al. The nomenclature, definition and classification of cardiac structures in the setting of heterotaxy. Cardiol Young. 2007;17(suppl 2):1-28.