Intravenous Anaesthetic Agents
Properties of the Ideal Intravenous Anaesthetic Agent
 Rapid onset – this is achieved by an agent which is mainly un-ionized at blood pH and which is highly soluble in lipid; these properties permit penetration of the blood–brain barrier
Rapid onset – this is achieved by an agent which is mainly un-ionized at blood pH and which is highly soluble in lipid; these properties permit penetration of the blood–brain barrier

 Analgesia at subanaesthetic concentrations
Analgesia at subanaesthetic concentrations
 Minimal cardiovascular and respiratory depression
Minimal cardiovascular and respiratory depression

 No excitatory phenomena (e.g. coughing, hiccup, involuntary movement) on induction
No excitatory phenomena (e.g. coughing, hiccup, involuntary movement) on induction
 No emergence phenomena (e.g. nightmares)
No emergence phenomena (e.g. nightmares)
 No interaction with neuromuscular blocking drugs
No interaction with neuromuscular blocking drugs


 Safe if injected inadvertently into an artery
Safe if injected inadvertently into an artery






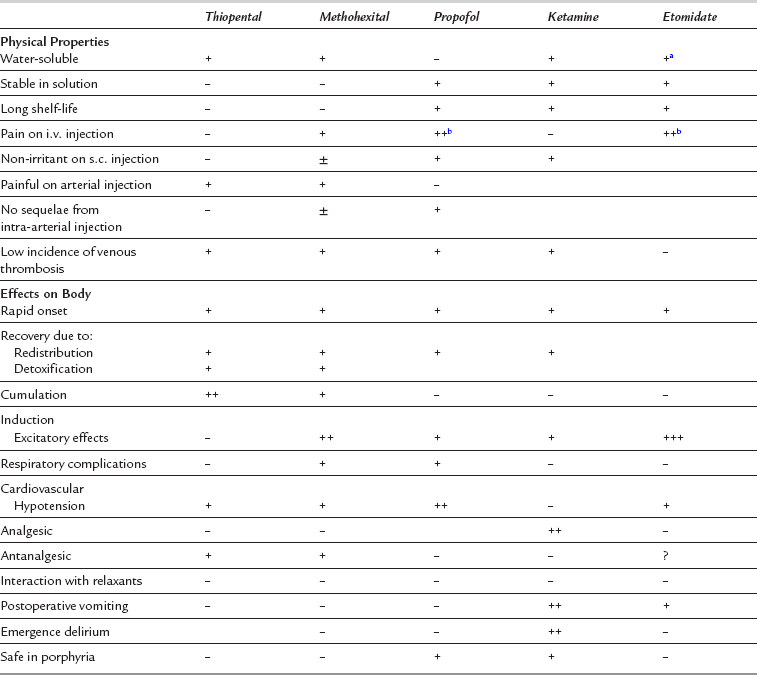
None of the agents available at present meets all these requirements. Features of the commonly used i.v. anaesthetic agents are compared in Table 3.1, and a classification of i.v. anaesthetic drugs is shown in Table 3.2.
TABLE 3.1
Main Properties of Intravenous Anaesthetics
aAqueous solution not commercially available.
bPain may be reduced when emulsion with medium-chain triglycerides is used.
TABLE 3.2
Classification of Intravenous Anaesthetics
Rapidly Acting (Primary Induction) Agents
Barbiturates:
Methohexital
Thiobarbiturates – thiopental, thiamylal
Imidazole compounds – etomidate
Sterically hindered alkyl phenols – propofol
Steroids – eltanolone, althesin, minaxolone (none currently available)
Eugenols – propanidid (not currently available)
Slower-Acting (Basal Narcotic) Agents
Ketamine
Benzodiazepines – diazepam, flunitrazepam, midazolam
Large-dose opioids – fentanyl, alfentanil, sufentanil, remifentanil
Neuroleptic combination – opioid + neuroleptic
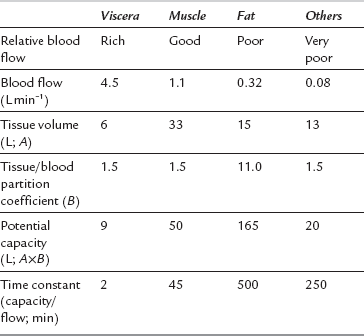
Pharmacokinetics of Intravenous Anaesthetic Drugs
Distribution to Other Tissues
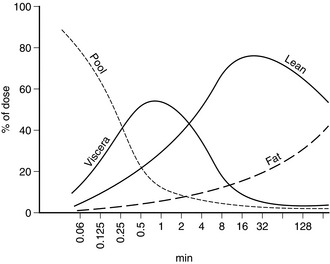
The anaesthetic effect of all i.v. anaesthetic drugs in current use is terminated predominantly by distribution to other tissues. Figure 3.1 shows this distribution for thiopental. The percentage of the injected dose in each of four body compartments as time elapses is shown after i.v. injection. A large proportion of the drug is distributed initially into well-perfused organs (termed the vessel-rich group, or viscera – predominantly brain, liver and kidneys). Distribution into muscle (lean) is slower because of its low lipid content, but it is quantitatively important because of its relatively good blood supply and large mass. Despite their high lipid solubility, i.v. anaesthetic drugs distribute slowly to adipose tissue (fat) because of its poor blood supply. Fat contributes little to the initial redistribution or termination of action of i.v. anaesthetic agents, but fat depots contain a large proportion of the injected dose of thiopental at 90 min, and 65–75% of the total remaining in the body at 24 h. There is also a small amount of redistribution to areas with a very poor blood supply, e.g. bone. Table 3.3 indicates some of the properties of the body compartments in respect of the distribution of i.v. anaesthetic agents.
BARBITURATES
Amobarbital and pentobarbital were used i.v. to induce anaesthesia in the late 1920s, but their actions were unpredictable and recovery was prolonged. Manipulation of the barbituric acid ring (Fig. 3.2) enabled a short duration of action to be achieved by:
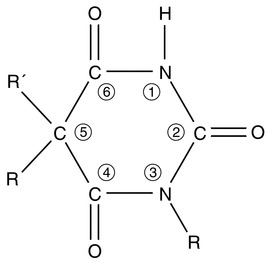


The anaesthetically active barbiturates are classified chemically into four groups (Table 3.4). The methylated oxybarbiturate hexobarbital was moderately successful as an i.v. anaesthetic agent, but was superseded by the development in 1932 of thiopental. Although propofol has become very popular in a number of countries, thiopental remains one of the most commonly used i.v. anaesthetic agents throughout the world. Its pharmacology is therefore described fully in this chapter. Many of its effects are shared by other i.v. anaesthetic agents and consequently the pharmacology of these drugs is described more briefly.

Thiopental Sodium
Physical Properties and Presentation
Central Nervous System: Thiopental produces anaesthesia usually less than 30 s after i.v. injection, although there may be some delay in patients with a low cardiac output. There is progressive depression of the CNS, including spinal cord reflexes. The hypnotic action of thiopental is potent, but its analgesic effect is poor, and surgical anaesthesia is difficult to achieve unless large doses are used; these are associated with cardiorespiratory depression. The cerebral metabolic rate is reduced and there are secondary decreases in CBF, cerebral blood volume and intracranial pressure. Recovery of consciousness occurs at a higher blood concentration if a large dose is given, or if the drug is injected rapidly; this has been attributed to acute tolerance, but may represent only altered redistribution. Consciousness is usually regained in 5–10 min. At subanaesthetic blood concentrations (i.e. at low doses or during recovery), thiopental has an antanalgesic effect and reduces the pain threshold; this may result in restlessness in the postoperative period. Thiopental is a very potent anticonvulsant.
Cardiovascular System: Myocardial contractility is depressed and peripheral vasodilatation occurs, particularly when large doses are administered or if injection is rapid. Arterial pressure decreases, and profound hypotension may occur in the patient with hypovolaemia or cardiac disease. Heart rate may decrease, but there is often a reflex tachycardia (see above).
Respiratory System: Ventilatory drive is decreased by thiopental as a result of reduced sensitivity of the respiratory centre to carbon dioxide. A short period of apnoea is common, frequently preceded by a few deep breaths. Respiratory depression is influenced by premedication and is more pronounced if opioids have been administered; assisted or controlled ventilation may be required. When spontaneous ventilation is resumed, ventilatory rate and tidal volume are usually lower than normal, but they increase in response to surgical stimulation. There is an increase in bronchial muscle tone, although frank bronchospasm is uncommon.
Skeletal Muscle: Skeletal muscle tone is reduced at high blood concentrations, partly as a result of suppression of spinal cord reflexes. There is no significant direct effect on the neuromuscular junction. When thiopental is used as the sole anaesthetic agent, there is poor muscle relaxation, and movement in response to surgical stimulation is common.
Uterus and Placenta: There is little effect on resting uterine tone, but uterine contractions are suppressed at high doses. Thiopental crosses the placenta readily, although fetal blood concentrations do not reach the same levels as those observed in the mother.
Adverse Effects
Laryngeal spasm. The causes have been discussed above.
Bronchospasm. This is unusual, but may be precipitated in asthmatic patients.
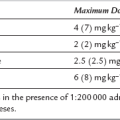
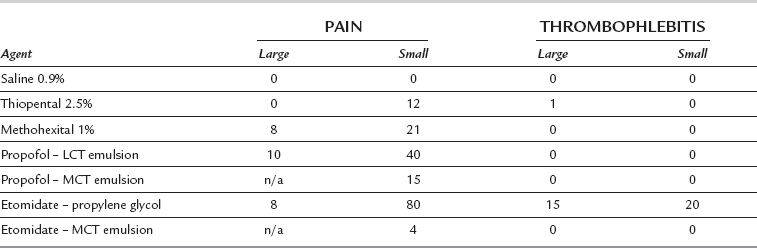
Thrombophlebitis. This is uncommon (Table 3.5) when the 2.5% solution is used.




Absolute Contraindications



Methohexital Sodium
Pharmacology
Central Nervous System: Unconsciousness is usually induced in 15–30 s. Recovery is more rapid with methohexital than with thiopental, and occurs after 2–3 min; it is caused predominantly by redistribution. Drowsiness may persist for several hours until blood concentrations are decreased further by metabolism. Epileptiform activity has been demonstrated by EEG in epileptic patients. However, in sufficient doses, methohexital acts as an anticonvulsant.
Cardiovascular System: In general, there is less hypotension in otherwise healthy patients than occurs after thiopental; the decrease in arterial pressure is mediated predominantly by vasodilatation. Heart rate may increase slightly because of a decrease in baroreceptor activity. The cardiovascular effects are more pronounced in patients with cardiac disease or hypovolaemia.
Adverse Effects
Epileptiform activity on EEG in epileptic subjects.
Pain on injection (Table 3.5).
Tissue damage after perivenous injection is rare with 1% solution.
NON-BARBITURATE INTRAVENOUS ANAESTHETIC AGENTS
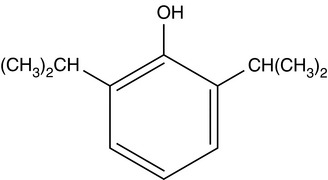
Pharmacology
Central Nervous System: Anaesthesia is induced within 20–40 s after i.v. administration in otherwise healthy young adults. Transfer from blood to the sites of action in the brain is slower than with thiopental, and there is a delay in disappearance of the eyelash reflex, normally used as a sign of unconsciousness after administration of barbiturate anaesthetic agents. Overdosage of propofol, with exaggerated side-effects, may result if this clinical sign is used; loss of verbal contact is a better end-point. EEG frequency decreases, and amplitude increases. Propofol reduces the duration of seizures induced by ECT in humans. However, there have been reports of convulsions following the use of propofol and it is recommended that caution be exercised in the administration of propofol to epileptic patients. Normally cerebral metabolic rate, CBF and intracranial pressure are reduced.
Cardiovascular System: In healthy patients, arterial pressure decreases to a greater degree after induction of anaesthesia with propofol than with thiopental; the reduction results predominantly from vasodilatation although there is a slight negative inotropic effect. In some patients, large decreases (> 40%) occur. The degree of hypotension is substantially reduced by decreasing the rate of administration of the drug and by appreciation of the kinetics of transfer from blood to brain (see above). The pressor response to tracheal intubation is attenuated to a greater degree by propofol than thiopental. Heart rate may increase slightly after induction of anaesthesia with propofol. However, there have been occasional reports of severe bradycardia and asystole during or shortly after administration of propofol, and it is recommended that a vagolytic agent (e.g. glycopyrronium or atropine) should be considered in patients with a pre-existing bradycardia or when propofol is used in conjunction with other drugs which are likely to cause bradycardia.
Respiratory System: After induction, apnoea occurs more commonly, and for a longer duration, than after thiopental. During infusion of propofol, tidal volume is lower and respiratory rate higher than in the conscious state. There is decreased ventilatory response to carbon dioxide. As with other agents, ventilatory depression is more marked if opioids are administered.
Gastrointestinal System: Propofol has no effect on gastrointestinal motility in animals. Its use is associated with a low incidence of postoperative nausea and vomiting.
Uterus and Placenta: Propofol has been used extensively in patients undergoing gynaecological surgery, and it does not appear to have any clinically significant effect on uterine tone. Propofol crosses the placenta. Its safety to the neonate has not been established and its use in pregnancy (except for termination), in obstetric practice and in breast-feeding mothers is not recommended by the manufacturers.
Adverse Effects
Pain on injection. This occurs in up to 40% of patients (Table 3.5). The incidence is greatly reduced if a large vein is used, if a small dose (10 mg) of lidocaine is injected shortly before propofol, or if lidocaine is mixed with propofol in the syringe (10–20 mg, i.e. 1–2 mL of 1% lidocaine per 20 mL of propofol). A preparation of propofol in an emulsion of medium-chain triglycerides and soya (Propofol-Lipuro®) causes a lower incidence of pain, and less severe pain in those who still experience it, than other formulations (which use long-chain triglycerides) and may obviate the need for lidocaine. Propofol 0.5% causes less pain than higher concentrations. Accidental extravasation or intra-arterial injection of propofol does not appear to result in adverse effects.
Etomidate
This carboxylated imidazole compound was introduced in 1972.
Adverse Effects
Suppression of synthesis of cortisol. See above.
Pain on injection. This occurs in up to 80% of patients if the propylene glycol preparation is injected into a small vein, but in less than 10% when the drug is injected into a large vein in the antecubital fossa (Table 3.5). The incidence is reduced by prior injection of lidocaine 10 mg. The incidence of pain on injection has been reported to be as low as 4% when the emulsion formulation is injected.
Venous thrombosis is more common than with other agents if the propylene glycol preparation is used.




Ketamine Hydrochloride
Pharmacology
Central Nervous System: Ketamine is extremely lipid-soluble. After i.v injection, it induces anaesthesia in 30–60 s. A single i.v. dose produces unconsciousness for 10–15 min. Ketamine is also effective within 3–4 min after i.m. injection and has a duration of action of 15–25 min. It is a potent somatic analgesic at subanaesthetic blood concentrations. Amnesia often persists for up to 1 h after recovery of consciousness. Induction of anaesthesia is smooth, but emergence delirium may occur, with restlessness, disorientation and agitation. Vivid and often unpleasant nightmares or hallucinations may occur during recovery and for up to 24 h. The incidences of emergence delirium and hallucinations are reduced by avoidance of verbal and tactile stimulation during the recovery period, or by concomitant administration of opioids, butyrophenones, benzodiazepines or physostigmine; however, unpleasant dreams may persist. Nightmares are reported less commonly by children and elderly patients.
Cardiovascular System: Arterial pressure increases by up to 25% and heart rate by approximately 20%. Cardiac output may increase, and myocardial oxygen consumption increases; the positive inotropic effect may be related to increased calcium influx mediated by cyclic adenosine monophosphate. There is increased myocardial sensitivity to adrenaline. Sympathetic stimulation of the peripheral circulation is decreased, resulting in vasodilatation in tissues innervated predominantly by α-adrenergic receptors, and vasoconstriction in those with β-receptors.
Respiratory System: Transient apnoea may occur after i.v. injection, but ventilation is well maintained thereafter and may increase slightly unless high doses are given. Pharyngeal and laryngeal reflexes and a patent airway are maintained well in comparison with other i.v. agents; however, their presence cannot be guaranteed, and normal precautions must be taken to protect the airway and prevent aspiration. Bronchial muscle is dilated.
Skeletal Muscle: Muscle tone is usually increased. Spontaneous movements may occur, but reflex movement in response to surgery is uncommon.
Adverse Effects








Other Drugs
Opioids and benzodiazepines may also be used to induce general anaesthesia. However, very large doses are required, and recovery is prolonged. Their use is confined to specialist areas, e.g. cardiac anaesthesia. The pharmacology of these drugs is described in Chapters 5 and 7, respectively.
INTRAVENOUS MAINTENANCE OF ANAESTHESIA
Techniques of Administration
Manual Infusion Techniques
A fixed-rate infusion is inappropriate because the serum concentration of the drug increases only slowly, taking four to five times the elimination half-life of the drug to reach steady state (Fig. 3.4). A bolus injection followed by a continuous infusion results initially in achievement of an excessive concentration (with an increased incidence of side-effects), and this is followed by a prolonged dip below the intended plasma concentration (Fig. 3.5). In order to achieve a reasonably constant plasma concentration (other than in very long procedures), it is necessary to use a multistep infusion regimen, a concept similar to that of overpressure for inhaled agents. A commonly used scheme for propofol is injection of a bolus dose of 1 mg kg–1 followed by infusion initially at a rate of 10 mg kg–1 h1 for 10 min, then 8 mg kg–1 h1 for the next 10 min, and a maintenance infusion rate of 6 mg kg–1 h1 thereafter. This achieves, on average, a plasma concentration of propofol of 3 μg mL–1, and this is effective in achieving satisfactory anaesthesia in unparalysed patients who also receive nitrous oxide and fentanyl; higher infusion rates are required if nitrous oxide and fentanyl are not administered. These infusion rates must be regarded only as a guide and must be adjusted as necessary according to clinical signs of anaesthesia.
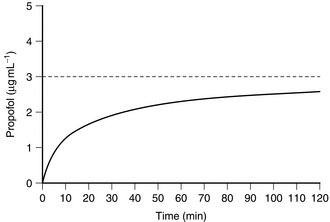
FIGURE 3.4 Average blood concentration during the first 2 h of a continuous infusion of propofol at a rate of 6 mg kg–1 h–1. Note that, even after 2 h, the equilibrium concentration of 3 μg mL–1 has not been achieved.
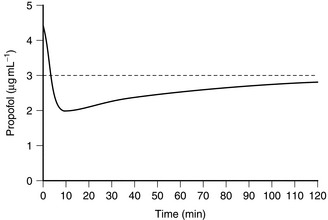
FIGURE 3.5 Average blood propofol concentration following a bolus dose of propofol followed by a continuous infusion of 6 mg kg–1 h–1. Note that the target concentration is initially exceeded, but that the blood concentration then decreases below the target concentration, which is not achieved within 2 h.
Target-Controlled Infusion (TCI) Techniques
By programming a computer with appropriate pharmacokinetic data and equations, it is possible at frequent intervals (several times a minute) to calculate the appropriate infusion rate required to produce a preset target plasma concentration of drug. The drug is infused by a syringe driver. To produce a step increase in plasma concentration, the syringe driver infuses drug very rapidly (a slow bolus) and then delivers drug at a progressively decreasing infusion rate (Fig. 3.6). To decrease the plasma concentration, the syringe driver stops infusing until the computer calculates that the target concentration has been achieved, and then infuses drug at an appropriate rate to maintain a constant level. The anaesthetist is required only to enter the desired target concentration and to change it when clinically indicated, in the same way as a vaporizer might be manipulated according to clinical signs of anaesthesia.
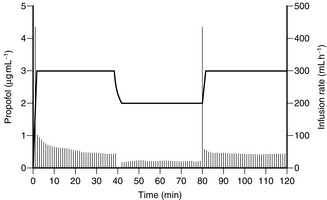
FIGURE 3.6 Average blood concentrations of propofol achieved using a target-controlled infusion system. The narrow vertical lines represent the infusion rate calculated by the computer to achieve, and then to maintain, the target concentration in blood. A target concentration of 3 μg mL–1 was programmed initially. When the target concentration is reset to 2 μg mL–1, the infusion is stopped and then restarted at a rate calculated to maintain that concentration. The target concentration is then increased to 3 μg mL–1; the infusion pump delivers a rapid infusion rate to achieve the target concentration, and then gradually decreases the infusion rate to maintain a constant blood concentration.
Closed-Loop Systems
Target-controlled infusion systems may be used as part of a closed-loop system to control depth of anaesthesia. Because there is no method of measuring blood concentrations of i.v. anaesthetics on-line, it is necessary to use some type of monitor of depth of anaesthesia (such as the auditory evoked response; see Chapter 16) on the input side of the system.
Adverse Reactions to Intravenous Anaesthetic Agents
Predisposing Factors
Age. In general, adverse reactions are less common in children than in adults.
Pregnancy. There is an increased incidence of adverse reactions in pregnancy.
Incidence
The incidences of hypersensitivity reactions associated with i.v. anaesthetic agents are shown in Table 3.6.
TABLE 3.6
Incidences of Adverse Reactions to Intravenous Anaesthetic Agents
| Drug | Incidence |
| Thiopental | 1:14 000–1:20 000 |
| Methohexital | 1:1600–1:7000 |
| Etomidate | 1:450 000 |
| Propofol | 1:50 000–100 000 (estimated) |
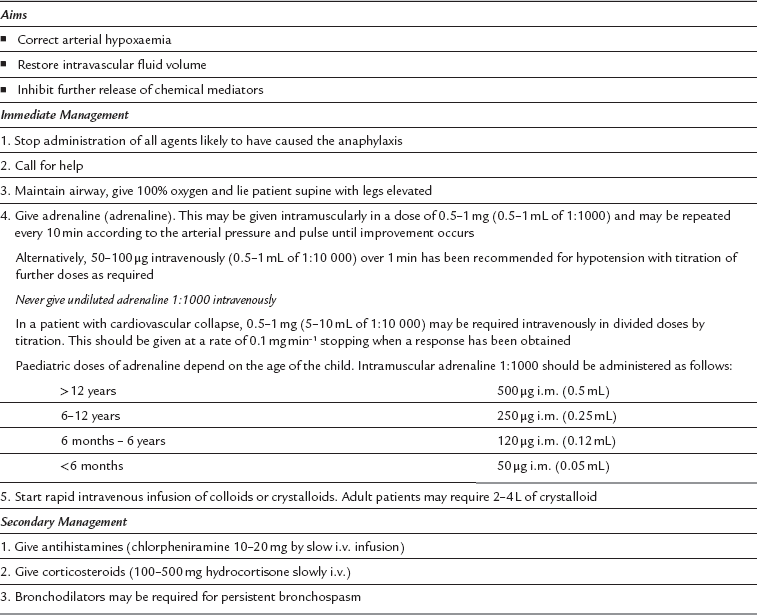
Association of Anaesthetists of Great Britain and Ireland. Suspected anaphylactic reactions associated with anaesthesia. Anaesthesia. 2009;64:199–211.
Calvey, N., Williams, N. Principles and practice of pharmacology for anaesthetists, fifth ed. Oxford: Wiley-Blackwell; 2008.
Sneyd, J.R. Recent advances in intravenous anaesthesia. Br. J. Anaesth. 2004;93:725–736.