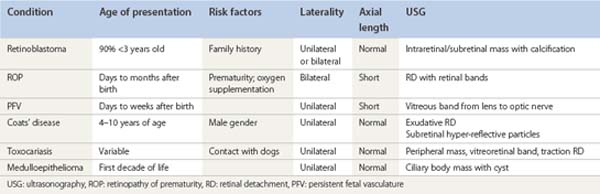
[level-membership-for-opthalmology-category]
Chapter 11 Intraocular Tumors

Retinoblastoma
Retinoblastoma is the most common intraocular malignancy of childhood and occurs with a frequency of approximately one in 14,000 to 20,000 live births.1 Ninety percent of cases are diagnosed in children under the age of 3 years. Ultrasonography along with other forms of imaging is invaluable in establishing the diagnosis of retinoblastoma.
Clinical features, symptoms, and signs
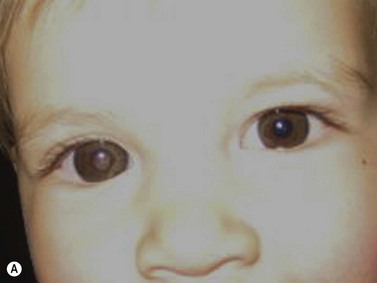
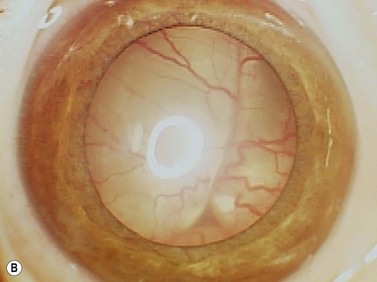
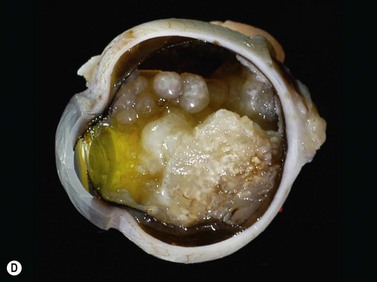
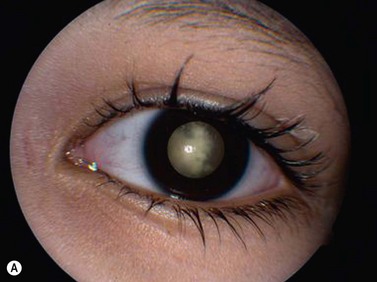
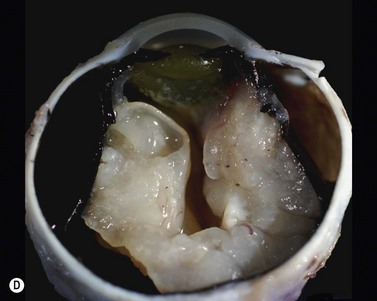
While leukocoria is the most common presenting symptom of retinoblastoma, strabismus, decreased vision, ocular inflammation, and other rarer symptoms have also been observed.1 In general, the presentation varies with the stage of the disease at the time of diagnosis. In its earliest clinical stage, retinoblastoma appears as a flat transparent to slightly whitish colored lesion in the sensory retina. Dilated and tortuous feeding retinal vessels may be evident. As the tumor enlarges, it loses its transparency and takes on a creamy yellow to whitish coloration with foci of chalk-like calcification. As it grows beyond the boundary of the sensory retina, retinoblastoma will typically follow either an endophytic or exophytic growth pattern (Figure 11.1). Other growth patterns including mixed and diffuse infiltrative forms (Figure 11.2) are less commonly observed. Necrosis may be a significant component of the tumor. Endophytic retinoblastomas grow from the retina inward towards the vitreous cavity. Vitreous seeding from these friable tumors as well as anterior chamber involvement can simulate endophthalmitis and other inflammatory conditions. In contrast, exophytic retinoblastomas grow from the retina outward into the subretinal space and can cause exudative retinal detachment, sometimes displacing the retina anteriorly behind the lens. Advanced retinoblastoma can present with neovascular glaucoma, corneal edema, spontaneous hyphema, vitreous hemorrhage, pseudohypopyon, and vitreitis.
Diagnostic evaluation
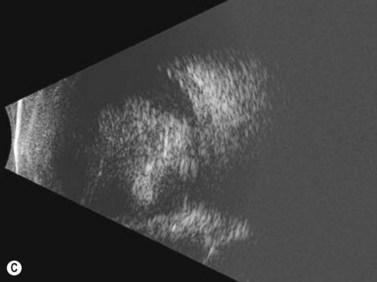
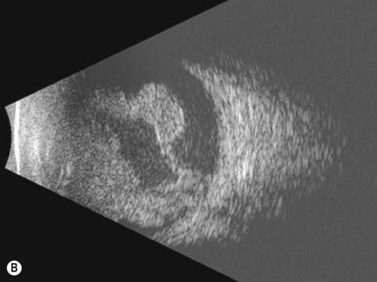
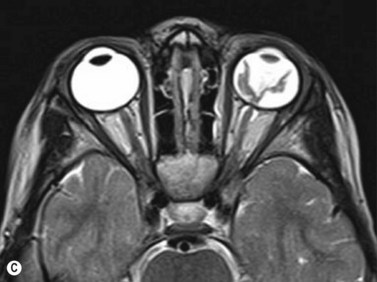
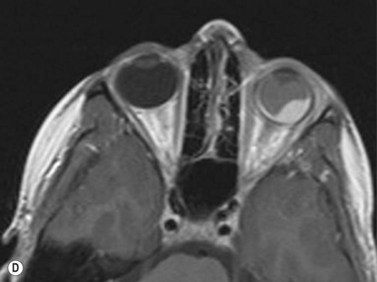
Ultrasonography is helpful in confirming the diagnosis of retinoblastoma and in differentiating the disease from other causes of leukocoria. This is particularly valuable when funduscopic examination is limited in advanced cases. On A-scan, the internal reflectivity of these lesions varies in accordance to the degree of calcification within the tumor. Non-calcified tumors exhibit low to medium internal reflectivity, whereas calcified lesions demonstrate high internal reflectivity. When a significant degree of calcification is present, shadowing of the adjacent sclera and orbit occurs. B-scan ultrasonography typically displays a rounded or irregular intraocular mass. It should be noted that mildly elevated and diffuse lesions have also been reported.2,3 Other associated ultrasonographic findings may include retinal detachment and vitreous opacities. When extraocular extension is present in cases of retinoblastoma, invasion of the optic nerve is the most common route. In cases where extensive calcification is present, tumor involvement of the optic nerve and extraocular extension can be difficult to detect secondary to the shadowing effect. CT and MRI imaging of the orbits should be used in combination with ultrasonography when optic nerve or extraocular invasion is suspected (Figure 11.2). MRI of the optic nerve, orbits, and brain is preferred as this modality offers superior soft tissue resolution and avoids potentially harmful exposure to radiation.
Salient diagnostic findings
The diagnosis of retinoblastoma can generally be suspected based upon the clinical findings observed in a complete ophthalmic examination in the office or an examination performed under anesthesia. The most commonly observed finding is an elevated intraocular mass with characteristic calcification demonstrating either an endophytic or exophytic growth pattern. Other causes of intraocular calcification are listed in Box 11.1.
Differential diagnosis
There are several pediatric ocular conditions that can cause leukocoria and should be considered in the differential diagnosis of retinoblastoma. The conditions that most commonly present a diagnostic challenge include retinopathy of prematurity (ROP), persistent fetal vasculature (PFV), Coats’ disease, toxocariasis, and medulloepithelioma (Table 11.1).
Retinopathy of prematurity
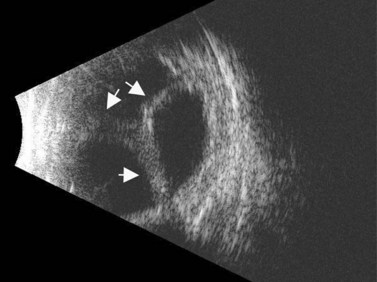
ROP occurs in the setting of known risk factors including: prematurity, low birth weight, and exposure to supplemental oxygenation in the neonatal period. While both ROP and retinoblastoma can present with leukocoria, in ROP the absence of the red reflex is caused by retinal dragging toward fibrovascular tissue in the retinal periphery. Eyes that develop retinoblastoma are usually of normal axial length. In contrast, in ROP it is more common for eyes to have some degree of the axial length shortening. Additionally, ROP is typically a bilateral condition whereas retinoblastoma can be either unilateral or bilateral. In the most advanced cases of ROP, the retina is detached in a funnel-like configuration, resulting in a hyper-reflective retrolental membrane on B-scan. The peripheral retina frequently exhibits a loop or trough-like appearance as a result of traction by the retrolental membrane (Figure 11.3).
Persistent fetal vasculature
The lens is often thin with irregularities in the posterior capsule (Figure 11.4). Eyes usually have some degree of axial length shortening. Calcification may be present, however in contrast to retinoblastoma, there is no discrete mass visualized clinically or with ultrasonography.
Coats’ disease
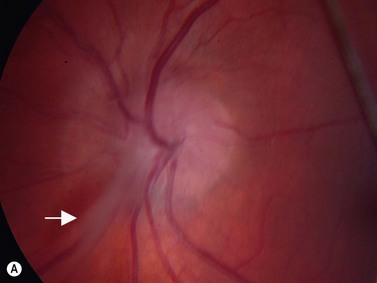
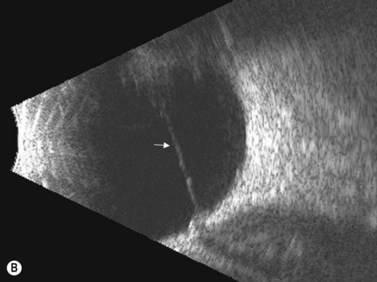
Coats’ disease is a retinal vascular disorder characterized by telangiectasia, intraretinal exudation, and exudative retinal detachment. Although Coats’ disease can present at any age, it usually is diagnosed in young males between 4 and 10 years of age.4 It is most commonly a unilateral disease process. In the early stages of Coats’ disease, localized, shallow retinal detachments may occur. In more advanced cases, total exudative detachments secondary to leakage from aneurysmal blood vessels are observed. This exudative process results in yellow cholesterol crystal deposition in the subretinal space that can be observed clinically as refractile bodies. These particles are much less reflective than the calcium particles in retinoblastoma. Ultrasonography is helpful in differentiating the two entities, in that in retinoblastoma a distinct tumor can be detected beneath the retinal detachment, whereas no distinct mass is seen in Coats’ disease (Figure 11.5).
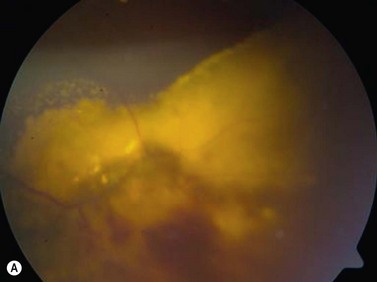
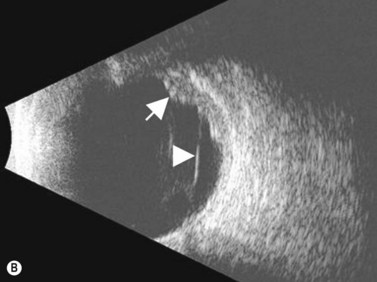
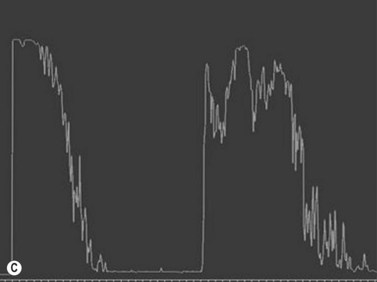
Medulloepithelioma
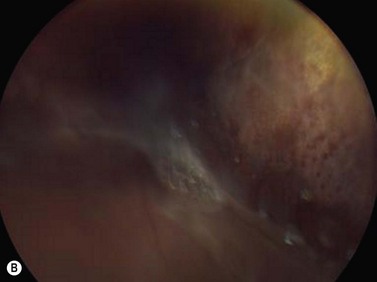
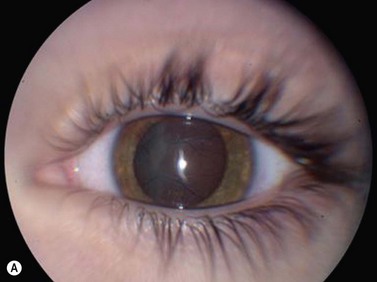
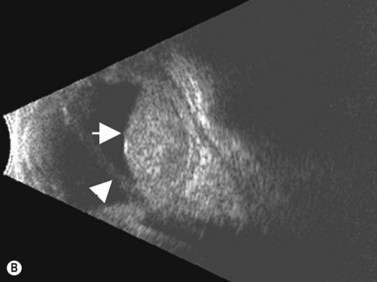
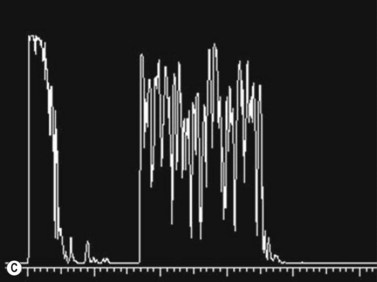
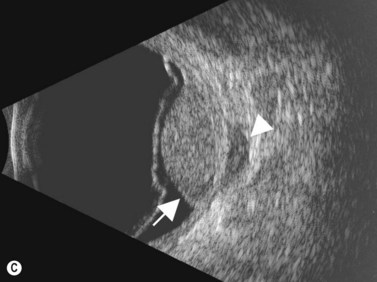
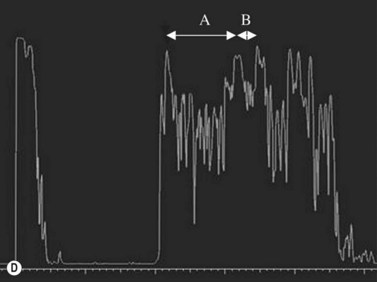
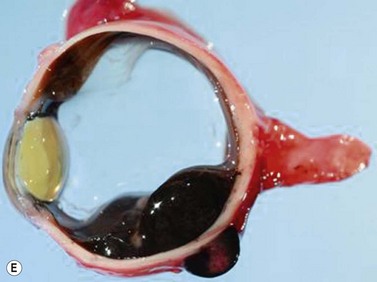
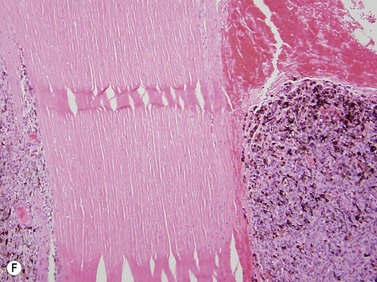
Medulloepithelioma is a congenital neuroepithelial tumor that typically manifests during the first decade of life. It most commonly arises from the ciliary body, however involvement of the iris and optic nerve has also been reported.5–10 On ophthalmic examination, the tumor appears as a lightly pigmented or amelanotic cystic mass. Large cysts may break off from the main tumor and float freely in the anterior chamber or vitreous cavity. Because of their appearance and because medulloepithelioma may present with leukocoria, these tumors are an important consideration in the differential diagnosis of retinoblastoma. A-scan of medulloepithelioma shows mainly high internal reflectivity with a medium spike corresponding to cystic regions of the tumor. On B-scan, medulloepitheliomas typically appear as a dome-shaped, highly reflective mass with irregular internal structures. Cystic spaces can be demonstrated in some lesions (Figure 11.6).
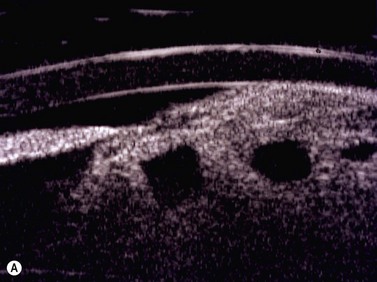
Benign uveal tumors
Circumscribed and diffuse choroidal hemangioma
Clinical features, symptoms, and signs
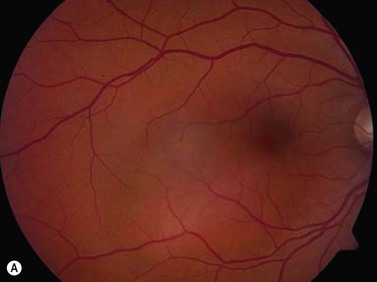
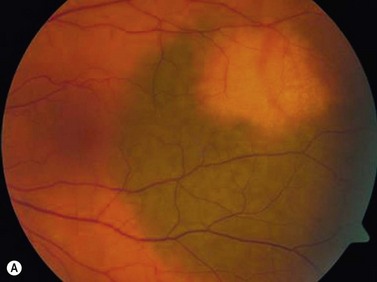
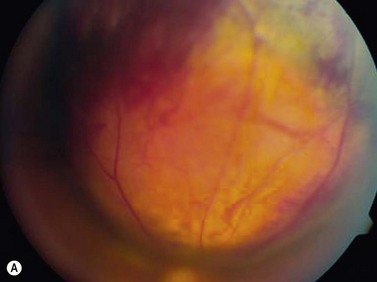
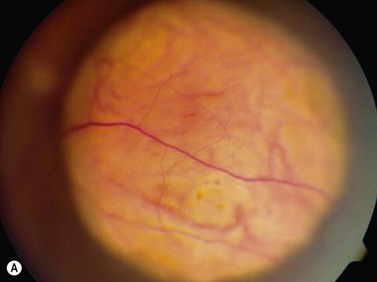
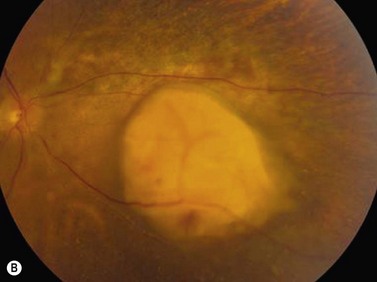
On ophthalmoscopic examination, circumscribed choroidal hemangiomas appear as an orange choroidal mass with indistinct margins that blend with the surrounding choroid. They are frequently located in the macular region of the posterior pole, and are not usually thicker than 6 mm.11 Although these tumors are highly vascular, dilated and tortuous feeder vessels are not typically observed. Surrounding subretinal fluid leading to exudative retinal detachment with macular involvement is common in symptomatic cases. Retinal hard exudates are minimal or absent. Diffuse choroidal hemangiomas appear as orange, diffuse choroidal thickening that has been likened to a “tomato-catsup fundus.” Focal regions of excessively thickened choroid within the diffuse hemangioma may simulate circumscribed choroidal hemangioma. As with circumscribed hemangiomas, there may be associated exudative retinal detachment that often does not become manifest until adolescence.
Diagnostic evaluation
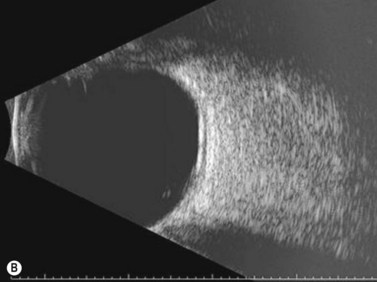
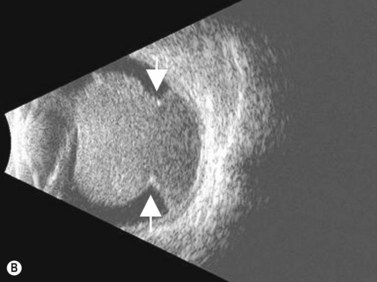
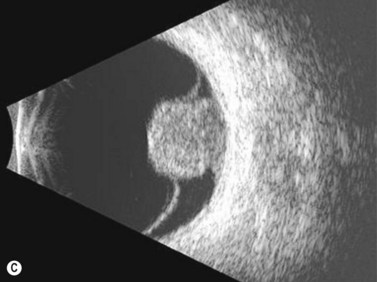
On A-scan, circumscribed choroidal hemangiomas demonstrate high internal reflectivity with negligible attenuation. This differs from other tumors in the differential diagnosis including malignant melanoma which classically demonstrates low to medium reflectivity on A-scan. On B-scan, circumscribed choroidal hemangioma appears as a dome-shaped choroidal mass with smooth contours. They are hyperechoic with regular internal structure and little internal blood flow. Serous retinal detachment at the tumor margins and calcification on the tumor surface may be present.11 Angiographic studies such as fluorescein and indocyanine green (ICG) can also be diagnostic. Fluorescein angiography demonstrates a hyperfluorescent mass with a fine lacy vascular network of intrinsic vessels in the early phases followed by increasing hyperfluorescence throughout the angiogram with variable leakage in late views.12 With ICG angiography, a rapid increase in hyperfluorescence is seen early on followed by a “washout” effect in the late phase.13 OCT can also be helpful in evaluating secondary changes in the overlying retina such as shallow subretinal fluid or cystoid macular edema (Figure 11.7).
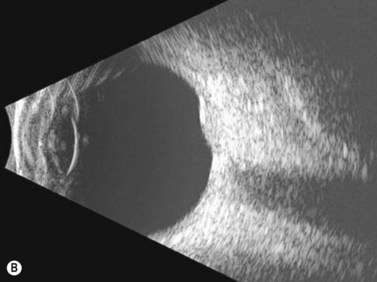
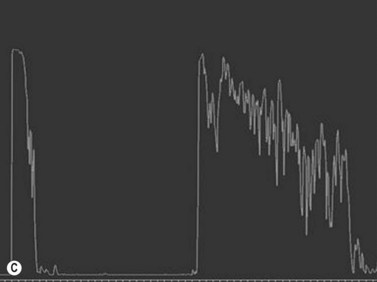
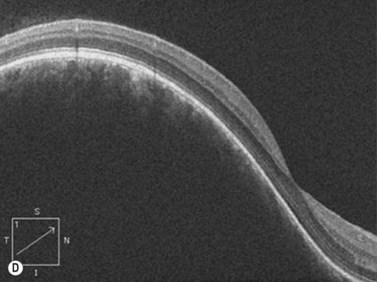
In the setting of Sturge–Weber syndrome with typical cutaneous features, the diagnosis of diffuse choroidal hemangioma is usually straightforward. In some cases, additional studies including ultrasonography may be useful. B-scan ultrasonography demonstrates a dome-shaped choroidal mass and diffusely thickened choroid (Figure 11.8). A-scan ultrasonography shows high internal reflectivity. Fluorescein angiography demonstrates early hyperfluorescence with persistence of diffuse hyperfluorescence through the late phases of the angiogram corresponding with the tumor margins. Leakage of fluorescein in the late phase of the angiogram is observed less commonly than with circumscribed choroidal hemangioma. ICG has a similar appearing pattern with rapid diffuse filling in early phases of the angiogram with intense persistence of hyperfluorescence into the late phases. A fine lacy intrinsic vascular pattern with a diffuse distribution is characteristic. The “washout” phenomenon observed in circumscribed choroidal hemangiomas is generally absent. OCT can also be used to identify the presence of and to assess the degree of subretinal fluid in appropriate cases.
Salient diagnostic findings
The characteristic diagnostic finding of circumscribed and diffuse choroidal hemangioma is as a dome-shaped choroidal mass or diffusely thickened choroid respectively on B-scan with high internal reflectivity on A-scan. While biopsy is not typically indicated to confirm the diagnosis, histopathology reveals that the tumor is composed of vascular channels lined with endothelium. The tumor involves the full thickness of the choroid with secondary changes of the overlying retinal pigment epithelium and the retina.14
Iris and ciliary body nevus
Iris nevi are more commonly solitary and circumscribed although a diffuse variant can also be observed. They have a predilection for the inferior half of the iris and are typically dark brown to black in coloration. Ciliary body nevi have rarely been reported in the literature, however their rate of occurrence is likely higher than that observed clinically.15 Ciliary body nevi typically appear as a dome-shaped mass with a smooth surface. These tumors are most appropriately evaluated using ultrasound biomicroscopy. This method provides quantitative measurement of tumor size, degree of posterior extension, and information related to internal consistency, which can differentiate solid from cystic lesions. Gonioscopy and anterior segment OCT can also be useful in characterizing these tumors.
Choroidal nevus
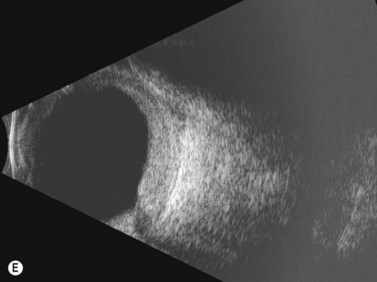
The Collaborative Ocular Melanoma Study (COMS) Group defined choroidal nevi as melanocytic lesions with a largest basal dimension of less than 5 mm and a height of less than 1 mm.16 Distinguishing choroidal nevi from small melanomas both clinically and with the aid of ultrasonography is difficult due to their small size. Choroidal nevi are often too flat to be accurately measured by ultrasonography. Suspicious nevi should therefore be closely followed in the clinical setting for changes in size and appearance. Nevi that are elevated sufficiently for detection by B-scan appear highly reflective (Figure 11.9). Nevi can be mistaken for other tumors with high reflectivity including choroidal hemangioma and metastatic carcinoma.
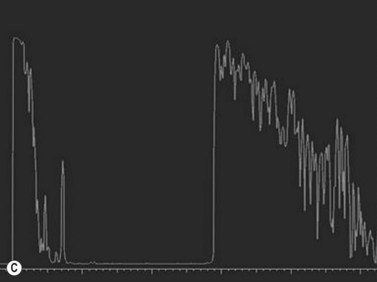
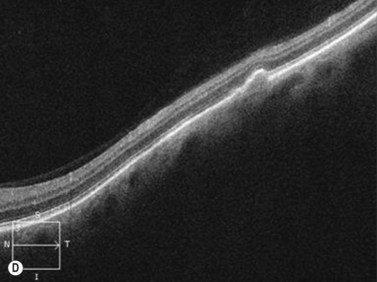
For nevi and other choroidal tumors that are too small to be accurately characterized with ultrasonography, spectral domain optical coherence tomography (SD-OCT) in combination with enhanced depth imaging (EDI) has recently been used.17 Increased speed, sensitivity, and resolution make SD-OCT superior to conventional time domain OCT. The technique of SD-OCT EDI improves the resolution of the deeper layers of the choroid and sclera thereby allowing for quantitative assessment of choroidal thickness.17 In a SD-OCT EDI study by Torres and colleagues, melanocytic tumors (nevi and melanoma) demonstrated a highly reflective band within the choriocapillaris layer with posterior shadowing. Melanocytic nevi were noted to contain choroidal vascular spaces of normal caliber within the lesion. In contrast, circumscribed choroidal hemangiomas showed low to medium reflectivity and a homogeneous signal with large intrinsic (possibly vascular) spaces. Choroidal metastases could be differentiated by the presence of a low reflective band in the deeper choroid with enlargement of the suprachoroidal space.18 EDI SD-OCT techniques are also advantageous as they have the ability to simultaneously describe choroidal tumors as well as their associated overlying retinal changes.
Uveal melanocytoma
Melanocytoma, also referred to as magnocellular nevus, is a darkly pigmented tumor that most commonly involves the optic disc and uveal tract. Although these tumors are considered to be congenital hamartomas, most go undetected throughout childhood and are discovered on routine ophthalmic examination. The mean age of diagnosis is 50 years.19 On ophthalmoscopic examination, melanocytoma appears as a dark-brown to black, well-circumscribed, flat to slightly elevated tumor. It is important to distinguish optic disc melanocytoma from malignant melanoma. In melanocytoma, features such as prominent intrinsic vasculature and subretinal fluid are generally absent. In addition, A-scan typically reveals high internal reflectivity for melanocytoma as opposed to low to medium reflectivity observed with choroidal melanoma. B-scan ultrasonography of melanocytoma demonstrates an acoustically solid mass (Figure 11.10). Fluorescein and ICG angiography are also useful for differentiating the lesions as melanocytoma characteristically has dense hypofluorescence corresponding to the location of the tumor. The majority of melanocytomas remain stable in size and appearance, however approximately 10% will display subtle growth over several years.20,21 Malignant transformation is rare and is reported in less than 2% of cases.21–23
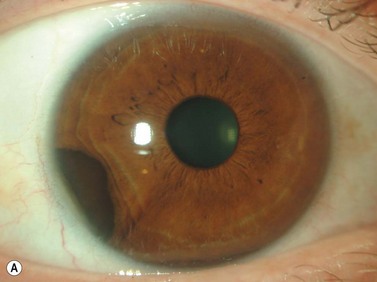
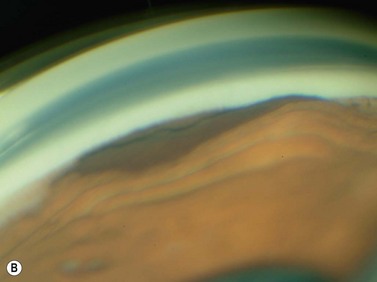
Schwannoma (neurilemoma)
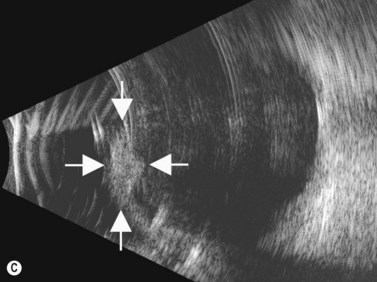
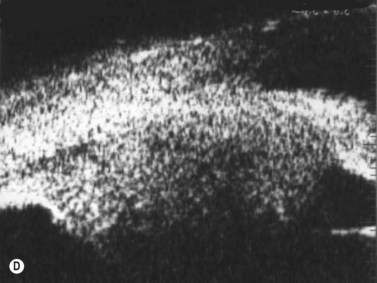
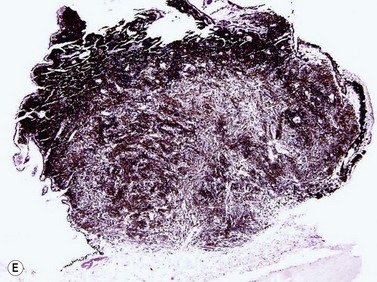
Schwannomas are rare, benign tumors that arise from the peripheral nerve sheaths. The majority of these tumors are diagnosed following enucleation, because the clinical and ultrasonographic features can closely simulate those seen in malignant melanoma. Clinically, a schwannoma appears as an elevated pigmented or amelanotic choroidal mass. B-scan ultrasonography reveals a choroidal mass and can be helpful in determining tumor dimensions. A-scan reveals a regular internal structure and variable reflectivity. Schwannomas are generally well circumscribed (Figure 11.11) and histopathology will demonstrate a spindled cell population with twisted nuclei and variable Antoni A and Antoni B patterns.
Malignant uveal tumors
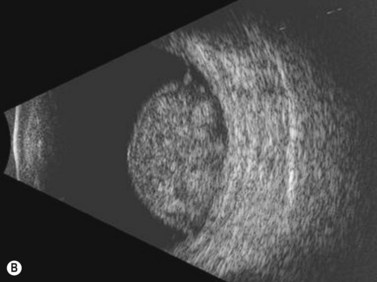
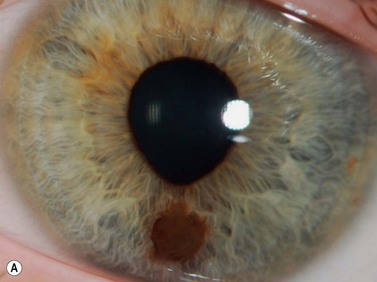
Iris and ciliary body melanoma
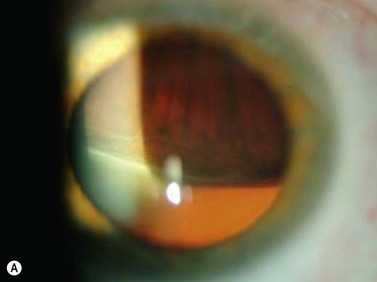
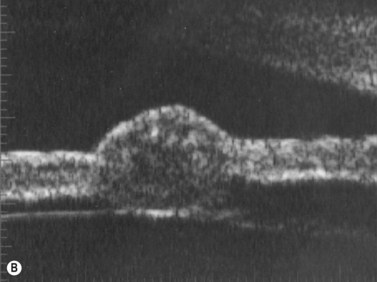
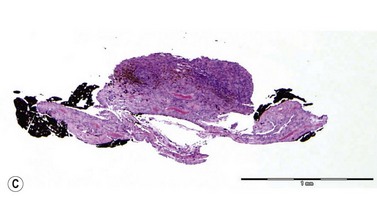
Iris melanomas can be circumscribed or diffuse. Circumscribed tumors are typically nodular, have variable pigmentation, and have a predilection for the inferior half of the iris. Diffuse iris melanoma can present in one of two patterns: either by primary infiltration of the iris stroma or by secondary seeding of tumor cells from a circumscribed iris or ciliary body melanoma.24,25 Slit lamp examination, gonioscopy, and immersion techniques with high frequency ultrasound biomicroscopy are best suited for evaluating the features of these tumors. Together, these techniques can be used to quantitatively assess tumor dimensions, presence of anterior chamber angle involvement, and posterior extension into the ciliary body (Figure 11.12).
Choroidal melanoma
The reported incidence of choroidal melanoma has ranged from 4.3 to 10.9 cases per million depending upon inclusion criteria and the methodology used to estimate the incidence.26 Ultrasonography is well suited for characterizing uveal melanoma and for differentiating these malignant tumors from other entities which may present with a similar clinical appearance.
Diagnostic evaluation
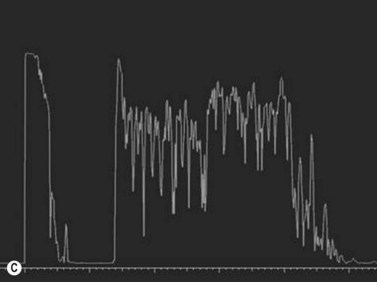
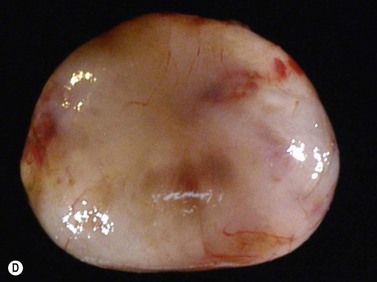
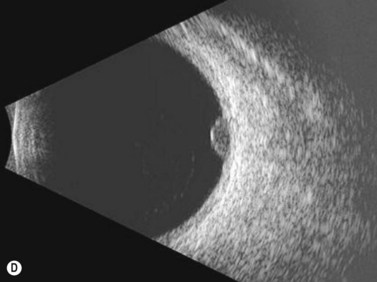
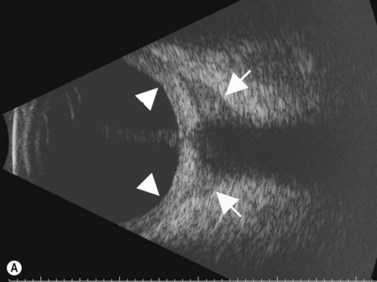
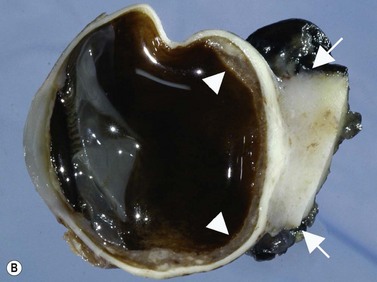
While their clinical presentation may be variable, choroidal melanomas demonstrate characteristic ultrasonographic features (Box 11.2). On A-scan, choroidal melanomas typically demonstrate low to medium internal reflectivity due to their homogeneous histologic architecture. For larger tumors, sound attenuation is often observed with lower reflectivity at the base of the tumor secondary to the more homogeneous nature of the melanoma in this region. When choroidal melanoma presents with features such as hemorrhage, necrosis, or dilated vessels, the A-scan may demonstrate irregular and internal reflectivity that is higher than anticipated. A-scan is also useful for evaluating internal blood flow. This acoustic property is represented by fast, spontaneous, low-amplitude flickering within the internal tumor spikes. On B-scan, the classic appearance of choroidal melanoma is a collar button configuration resulting from the tumor rupturing through Bruch’s membrane (Figure 11.13). In contrast, choroidal melanomas confined to the subretinal space, are dome-shaped, lobulated, or diffuse. On B-scan, choroidal melanoma appears as an echo-dense mass. The appearance is caused by internal acoustic interfaces between the cellular mass and varying degrees of vascularity. See Clip 11.1A,B At the tumor base, where the cellular composition is more homogeneous, and in relatively avascular lesions, an echolucent area can be seen. This region is called the acoustic quiet zone or acoustic hollowing.27 As choroidal melanoma infiltrates the surrounding choroid, it causes a bowl-shaped indentation surrounding the tumor base. This feature is called choroidal excavation and is not specific for melanoma as it has also been demonstrated in metastatic carcinoma.28 Posterior bowing of the sclera is a feature that has been reported in younger individuals and may represent extension into the sclera.29 Exudative retinal detachment, subretinal hemorrhage, and vitreous hemorrhage are all features that can be observed in choroidal melanoma. Dense subretinal and/or vitreous hemorrhages can potentially mask the underlying tumor. In these cases, serial examinations must be performed to rule out choroidal melanoma and other tumors. Extrascleral extension of choroidal melanoma appears as nodules near the base of the tumor. These nodules are often echolucent secondary to the sound attenuation from the primary tumor. Care must be taken, as congested blood vessels, the insertion site of the extraocular muscles, and inflammation in the sub-Tenon space can be mistaken for extrascleral extension (Figure 11.14). In cases where choroidal melanoma follows a diffuse growth pattern, diagnosis using ultrasonographic techniques presents a challenge. Internal reflectivity is often difficult to assess because of the shallow nature of the tumor. Moreover, on B-scan diffuse choroidal melanomas may have an irregular surface making them more difficult to quantitatively characterize. For this reason, in cases of diffuse choroidal melanoma in which extrascleral extension is suspected, alternate modalities of imaging such as MRI and fine needle aspiration biopsy should be considered.
See Clip 11.1A,B At the tumor base, where the cellular composition is more homogeneous, and in relatively avascular lesions, an echolucent area can be seen. This region is called the acoustic quiet zone or acoustic hollowing.27 As choroidal melanoma infiltrates the surrounding choroid, it causes a bowl-shaped indentation surrounding the tumor base. This feature is called choroidal excavation and is not specific for melanoma as it has also been demonstrated in metastatic carcinoma.28 Posterior bowing of the sclera is a feature that has been reported in younger individuals and may represent extension into the sclera.29 Exudative retinal detachment, subretinal hemorrhage, and vitreous hemorrhage are all features that can be observed in choroidal melanoma. Dense subretinal and/or vitreous hemorrhages can potentially mask the underlying tumor. In these cases, serial examinations must be performed to rule out choroidal melanoma and other tumors. Extrascleral extension of choroidal melanoma appears as nodules near the base of the tumor. These nodules are often echolucent secondary to the sound attenuation from the primary tumor. Care must be taken, as congested blood vessels, the insertion site of the extraocular muscles, and inflammation in the sub-Tenon space can be mistaken for extrascleral extension (Figure 11.14). In cases where choroidal melanoma follows a diffuse growth pattern, diagnosis using ultrasonographic techniques presents a challenge. Internal reflectivity is often difficult to assess because of the shallow nature of the tumor. Moreover, on B-scan diffuse choroidal melanomas may have an irregular surface making them more difficult to quantitatively characterize. For this reason, in cases of diffuse choroidal melanoma in which extrascleral extension is suspected, alternate modalities of imaging such as MRI and fine needle aspiration biopsy should be considered.
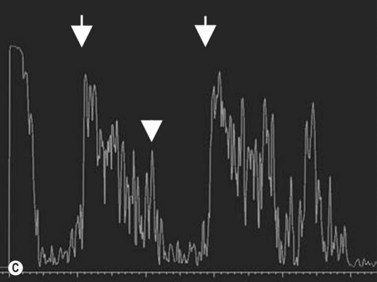
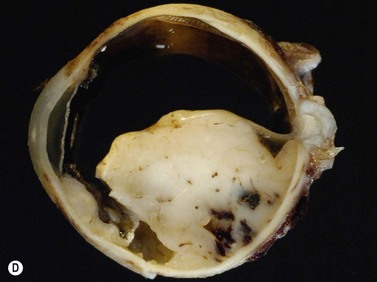
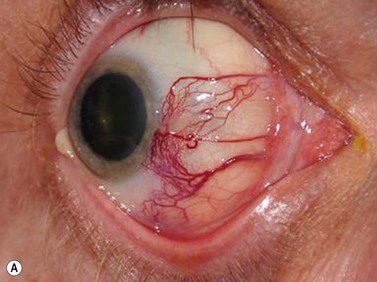
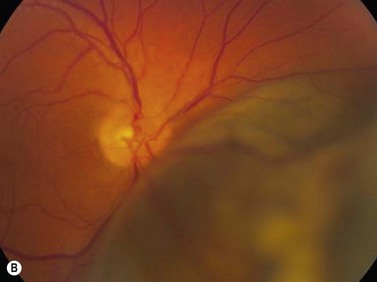
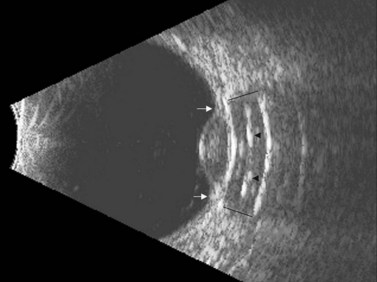
Ultrasonography can also be utilized to facilitate in the intraoperative management of choroidal melanoma, particularly in aiding in the precise confirmation of radioactive plaque placement. In cases where brachytherapy is the treatment of choice, localization of the radioactive plaque can be performed in the operating room under sterile conditions or postoperatively. Ultrasonography is particularly useful for posteriorly located tumors, because conventional methods of transillumination are often unable to clearly delineate the tumor margins adequately. The iodine-125 plaque produces an echolucent pattern with marked shadowing of the orbital tissues (Figure 11.15). Response to radiation therapy following treatment can also be assessed using ultrasonographic techniques. Following radiation treatment, choroidal melanoma becomes more irregular and reflective as tissue necrosis occurs. The tumor loses its internal vascularity and decreases in size, indicating successful treatment (Figure 11.16). Some lesions initially enlarge as a result of edema, but most will eventually decrease in size. Continued enlargement may signify true tumor growth. Additionally, long-term follow-up is recommended even in lesions that demonstrate initial regression, as delayed tumor growth following treatment has been reported.30,31
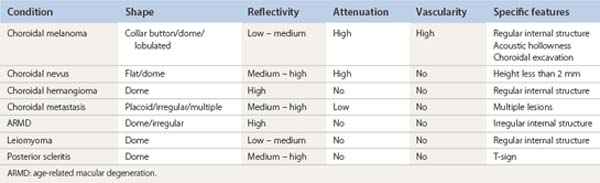
Differential diagnosis
Several pigmented and amelanotic lesions can resemble choroidal melanoma and should be considered in the differential diagnosis. These entities include: choroidal nevus, choroidal metastasis, choroidal hemangioma, posterior scleritis, age-related macular degeneration (ARMD) and leiomyoma. Ultrasonography can be helpful in differentiating some of the more common simulating lesions (Table 11.2). Choroidal nevi, choroidal hemangioma, and leiomyoma have been previously discussed in this chapter. A brief discussion of choroidal metastasis, ARMD, and posterior scleritis follows.
Choroidal metastasis
Choroidal metastases most frequently present in the posterior pole as focal or multifocal lesions. They may be flat or dome-shaped, pigmented or amelanotic, and unilateral or bilateral. Associated serous retinal detachment is a common feature. On A-scan, the internal reflectivity is usually medium to high with some degree of internal irregularity resulting from variability of the histologic architecture within the tumor. Internal vascularity is minimal or absent. On B-scan, choroidal metastases demonstrate an irregular surface and often display central excavations. Serous retinal detachments are frequently more extensive than those observed in choroidal melanomas of comparable size (Figure 11.17).Vitreous and subretinal hemorrhages are rarely associated with metastatic carcinoma. Some choroidal metastases present with atypical features such as low internal reflectivity and internal vascularity. The most common metastasis to produce these atypical findings is small cell carcinoma of the lung.
Intraocular calcification
Several intraocular tumors as well as various degenerative conditions can result in intraocular calcification. A complete list of entities is presented in Box 11.1. Several of these conditions have been previously discussed within this chapter. A discussion of astrocytic hamartoma, choroidal osteoma, sclerochoroidal calcification, and other rarer causes are discussed in this section.
Astrocytic hamartoma
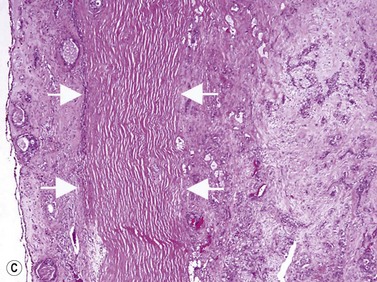
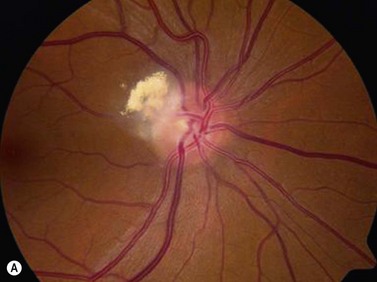
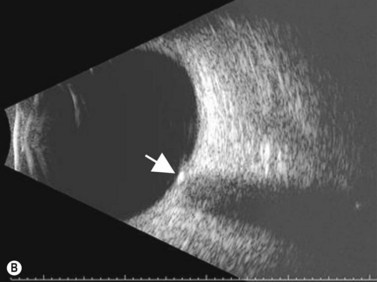
Astrocytic hamartoma of the retina and optic disc is a benign tumor that typically occurs in patients with tuberous sclerosis complex (TSC) and neurofibromatosis type 1 (NF1). Small, non-calcified tumors can be extremely subtle and appear as ill-defined translucent thickening of the nerve fiber layer. Larger lesions are more opaque and appear as a sessile white mass at the level of the nerve fiber layer. Some lesions contain characteristic dense yellow, refractile calcification that has been likened to a “mulberry,” “tapioca” or “fish egg” appearance. Ultrasonography is of little diagnostic value in small, flat, non-calcified lesions. Larger calcified lesions, however, appear as well demarcated oval masses with a high reflectivity, sharp anterior borders, and orbital shadowing on B-scan. A-scan demonstrates high internal reflectivity and attenuation of orbital echoes posterior to the tumor. OCT reveals an irregular and thickened nerve fiber layer (Figure 11.18).
Choroidal osteoma
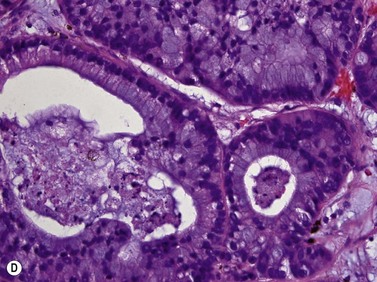
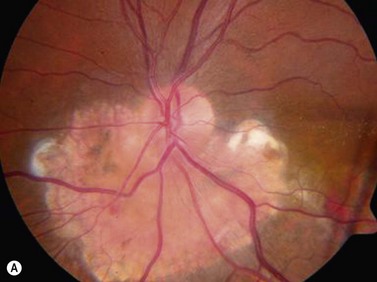
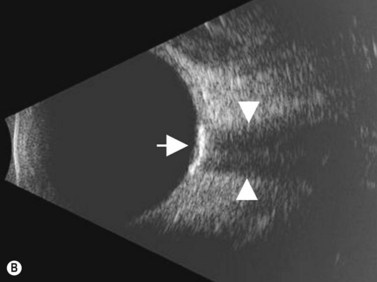
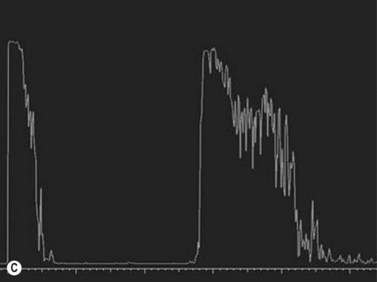
Choroidal osteomas are benign choristomas comprised of cancellous bone usually found in a juxtapapillary or a macular location. Approximately 90% of these tumors occur in females with a mean age of presentation of 21 years.32 Ophthalmoscopically, a choroidal osteoma appears as a flat to minimally elevated, yellow-white choroidal lesion. Ultrasonography may be useful in differentiating choroidal osteomas from similar lesions. A-scan typically shows a sharp high-intensity echo spike from the anterior surface of the tumor, along with high internal reflectivity. B-scan demonstrates high reflectivity at the level of the choroid and orbital shadowing consistent with calcium deposition (Figure 11.19). Additionally, CT scan may show the characteristic appearance of a radio-opaque lesion at the level of the choroid.
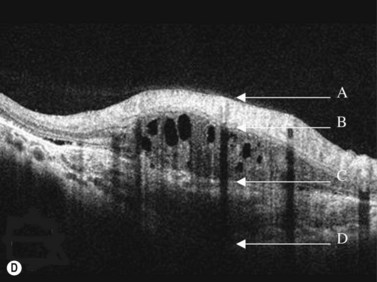
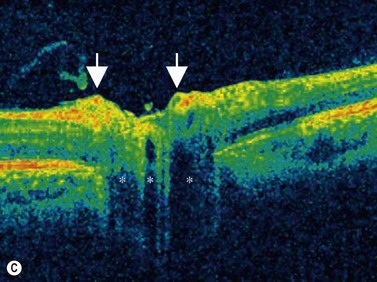
Sclerochoroidal calcification
In most cases sclerochoroidal calcification occurs as a degenerative process; however in a minority of individuals there is an association with hyperparathyroidism and other disorders related to calcium metabolism.33 Sclerochoroidal calcification can have a similar appearance to choroidal osteoma and the two can be difficult to differentiate using ultrasonographic techniques. On ultrasonography, sclerochoroidal calcification demonstrates high internal reflectivity and marked orbital shadowing (Figure 11.20). Distinction between these two entities is based on several clinical features.
1 Shetlar DJ, Chevez-Barrios P, et al. Basic and Clinical Science Course: Ophthalmic Pathology and Intraocular Tumors. San Francisco, CA: American Academy of Ophthalmology; 2007.
2 Shields CL, Shields JA, Shah P. Retinoblastoma in older children. Ophthalmology. 1991;98:395-399.
3 All-Ericsson C, Economou MA, Landau I, et al. Uveitis masquerade syndromes: diffuse retinoblastoma in an older child. Acta Ophthalmol Scand. 2007;85:569-570.
4 Ridley ME, Shields JA, Brown GC, et al. Coats’ disease. Evaluation of management. Ophthalmology. 1982;89:1381-1387.
5 Shields JA, Eagle RCJr, Shields CL, et al. Congenital neoplasms of the nonpigmented ciliary epithelium (medulloepithelioma). Ophthalmology. 1996;103:1998-2006.
6 Broughton WL, Zimmerman LE. A clinicopathologic study of 56 cases of intraocular medulloepitheliomas. Am J Ophthalmol. 1978;85:407-418.
7 Morris AT, Garner A. Medulloepithelioma involving the iris. Br J Ophthalmol. 1975;59:276-278.
8 Orellana J, Moura RA, Font RL, et al. Medulloepithelioma diagnosed by ultrasound and vitreous aspirate. Electron microscopic observations. Ophthalmology. 1983;90:1531-1539.
9 Green WR, Iliff WJ, Trotter RR. Malignant teratoid medulloepithelioma of the optic nerve. Arch Ophthalmol. 1974;91:451-454.
10 Biswas J, Bhushan B, Jayakumar N, et al. Teratoid malignant medulloepithelioma of the optic nerve: report of a case and review of the literature. Orbit. 1999;18:191-196.
11 Witschel H, Font R. Hemangioma of the choroid. A clinicopathologic study of 71 cases and a review of the literature. Surv Ophthalmol. 1976;20(6):415-431.
12 Singh A, Kaiser P, Sears J. Choroidal hemangioma. Ophthalmol Clin North Am. 2005;18(1):151-161.
13 Arevalo JF, Shields CL, Shields JA, et al. Circumscribed choroidal hemangioma: characteristic features with indocyanine green videoangiography. Ophthalmology. 2000;107(2):344-350.
14 Shields C, Honavar S, Shields JA, et al. Circumscribed choroidal hemangioma: clinical manifestations and factors predictive of visual outcome in 200 consecutive cases. Ophthalmology. 2001;108(12):2237-2248.
15 Cogan DG, Kuwabara T. Tumors of the ciliary body. Int Ophthalmol Clin. 1971;11(3):27-56.
16 Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol. 1997;115(12):1537-1544.
17 Spaide RF, Koizumi H, Pozzoni MC. Enhanced depth imaging spectral-domain optical coherence tomography. Am J Ophthalmol. 2008;146(4):496-500.
18 Torres VL, Brugnoni N, Kaiser PK, et al. Optical coherence tomography- enhanced depth imaging (EDI) of choroidal tumors. Invest Ophthalmol Vis Sci. 2010;5134:D1130. ARVO E-abstract
19 Shields JA, Demirci H, Mashayekhi A, et al. Melanocytoma of optic disc in 115 cases: the 2004 Samuel Johnson Memorial Lecture, part 1. Ophthalmology. 2004;111(9):1739-1746.
20 Mansour AM, Zimmerman L, La Piana FG, et al. Clinicopathological findings in a growing optic nerve melanocytoma. Br J Ophthalmol. 1989;73(6):410-415.
21 Roth AM. Malignant change in melanocytomas of the uveal tract. Surv Ophthalmol. 1978;22(6):404-412.
22 Apple DJ, Craythorn JM, Reidy JJ, et al. Malignant transformation of an optic nerve melanocytoma. Can J Ophthalmol. 1984;19(7):320-325.
23 Meyer D, Ge J, Blinder KJ, et al. Malignant transformation of an optic disc melanocytoma. Am J Ophthalmol. 1999;127(6):710-714.
24 Brown D, Boniuk M, Font RL. Diffuse malignant melanoma of iris with metastases. Surv Ophthalmol. 1990;34(5):357-364.
25 Char DH, Crawford JB, Gonzales J, et al. Iris melanoma with increased intraocular pressure. Differentiation of focal solitary tumors from diffuse or multiple tumors. Arch Ophthalmol. 1989;107(4):548-551.
26 Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology. 2003;110(5):956-961.
27 Goldberg MF, Hodes BL. Ultrasonographic diagnosis of choroidal malignant melanoma. Surv Ophthalmol. 1977;22(1):29-40.
28 Fuller DG, Snyder WB, Hutton WL, et al. Ultrasonographic features of choroidal malignant melanomas. Arch Ophthalmol. 1979;97(8):1465-1472.
29 Cham MC, Pavlin CJ. Ultrasound detection of posterior scleral bowing in young patients with choroidal melanoma. Can J Ophthalmol. 2000;35(5):263-266.
30 Gragoudas ES, Lane AM, Munzenrider J, et al. Long-term risk of local failure after proton therapy for choroidal/ciliary body melanoma. Trans Am Ophthalmol Soc. 2002;100:43-48.
31 Novak-Andrejcic K, Jancar B, Hawlina M. Echographic follow-up of malignant melanoma of the choroid after brachytherapy with 106Ru. Klin Monatsbl Augenheilkd. 2003;220(12):853-860.
32 Aylward GW, Chang TS, Pautler SE, et al. A long-term follow-up of choroidal osteoma. Arch Ophthalmol. 1998;116(10):1337-1341.
33 Goldstein BG, Miller J. Metastatic calcification of the choroid in a patient with primary hyperparathyroidism. Retina. 1982;2(2):76-79.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 11 Intraocular Tumors
Retinoblastoma
Retinoblastoma is the most common intraocular malignancy of childhood and occurs with a frequency of approximately one in 14,000 to 20,000 live births.1 Ninety percent of cases are diagnosed in children under the age of 3 years. Ultrasonography along with other forms of imaging is invaluable in establishing the diagnosis of retinoblastoma.
Clinical features, symptoms, and signs
While leukocoria is the most common presenting symptom of retinoblastoma, strabismus, decreased vision, ocular inflammation, and other rarer symptoms have also been observed.1 In general, the presentation varies with the stage of the disease at the time of diagnosis. In its earliest clinical stage, retinoblastoma appears as a flat transparent to slightly whitish colored lesion in the sensory retina. Dilated and tortuous feeding retinal vessels may be evident. As the tumor enlarges, it loses its transparency and takes on a creamy yellow to whitish coloration with foci of chalk-like calcification. As it grows beyond the boundary of the sensory retina, retinoblastoma will typically follow either an endophytic or exophytic growth pattern (Figure 11.1). Other growth patterns including mixed and diffuse infiltrative forms (Figure 11.2) are less commonly observed. Necrosis may be a significant component of the tumor. Endophytic retinoblastomas grow from the retina inward towards the vitreous cavity. Vitreous seeding from these friable tumors as well as anterior chamber involvement can simulate endophthalmitis and other inflammatory conditions. In contrast, exophytic retinoblastomas grow from the retina outward into the subretinal space and can cause exudative retinal detachment, sometimes displacing the retina anteriorly behind the lens. Advanced retinoblastoma can present with neovascular glaucoma, corneal edema, spontaneous hyphema, vitreous hemorrhage, pseudohypopyon, and vitreitis.
Diagnostic evaluation
Ultrasonography is helpful in confirming the diagnosis of retinoblastoma and in differentiating the disease from other causes of leukocoria. This is particularly valuable when funduscopic examination is limited in advanced cases. On A-scan, the internal reflectivity of these lesions varies in accordance to the degree of calcification within the tumor. Non-calcified tumors exhibit low to medium internal reflectivity, whereas calcified lesions demonstrate high internal reflectivity. When a significant degree of calcification is present, shadowing of the adjacent sclera and orbit occurs. B-scan ultrasonography typically displays a rounded or irregular intraocular mass. It should be noted that mildly elevated and diffuse lesions have also been reported.2,3 Other associated ultrasonographic findings may include retinal detachment and vitreous opacities. When extraocular extension is present in cases of retinoblastoma, invasion of the optic nerve is the most common route. In cases where extensive calcification is present, tumor involvement of the optic nerve and extraocular extension can be difficult to detect secondary to the shadowing effect. CT and MRI imaging of the orbits should be used in combination with ultrasonography when optic nerve or extraocular invasion is suspected (Figure 11.2). MRI of the optic nerve, orbits, and brain is preferred as this modality offers superior soft tissue resolution and avoids potentially harmful exposure to radiation.
Salient diagnostic findings
The diagnosis of retinoblastoma can generally be suspected based upon the clinical findings observed in a complete ophthalmic examination in the office or an examination performed under anesthesia. The most commonly observed finding is an elevated intraocular mass with characteristic calcification demonstrating either an endophytic or exophytic growth pattern. Other causes of intraocular calcification are listed in Box 11.1.
Differential diagnosis
There are several pediatric ocular conditions that can cause leukocoria and should be considered in the differential diagnosis of retinoblastoma. The conditions that most commonly present a diagnostic challenge include retinopathy of prematurity (ROP), persistent fetal vasculature (PFV), Coats’ disease, toxocariasis, and medulloepithelioma (Table 11.1).
Retinopathy of prematurity
ROP occurs in the setting of known risk factors including: prematurity, low birth weight, and exposure to supplemental oxygenation in the neonatal period. While both ROP and retinoblastoma can present with leukocoria, in ROP the absence of the red reflex is caused by retinal dragging toward fibrovascular tissue in the retinal periphery. Eyes that develop retinoblastoma are usually of normal axial length. In contrast, in ROP it is more common for eyes to have some degree of the axial length shortening. Additionally, ROP is typically a bilateral condition whereas retinoblastoma can be either unilateral or bilateral. In the most advanced cases of ROP, the retina is detached in a funnel-like configuration, resulting in a hyper-reflective retrolental membrane on B-scan. The peripheral retina frequently exhibits a loop or trough-like appearance as a result of traction by the retrolental membrane (Figure 11.3).
Persistent fetal vasculature
The lens is often thin with irregularities in the posterior capsule (Figure 11.4). Eyes usually have some degree of axial length shortening. Calcification may be present, however in contrast to retinoblastoma, there is no discrete mass visualized clinically or with ultrasonography.
Coats’ disease
Coats’ disease is a retinal vascular disorder characterized by telangiectasia, intraretinal exudation, and exudative retinal detachment. Although Coats’ disease can present at any age, it usually is diagnosed in young males between 4 and 10 years of age.4 It is most commonly a unilateral disease process. In the early stages of Coats’ disease, localized, shallow retinal detachments may occur. In more advanced cases, total exudative detachments secondary to leakage from aneurysmal blood vessels are observed. This exudative process results in yellow cholesterol crystal deposition in the subretinal space that can be observed clinically as refractile bodies. These particles are much less reflective than the calcium particles in retinoblastoma. Ultrasonography is helpful in differentiating the two entities, in that in retinoblastoma a distinct tumor can be detected beneath the retinal detachment, whereas no distinct mass is seen in Coats’ disease (Figure 11.5).
Medulloepithelioma
Medulloepithelioma is a congenital neuroepithelial tumor that typically manifests during the first decade of life. It most commonly arises from the ciliary body, however involvement of the iris and optic nerve has also been reported.5–10 On ophthalmic examination, the tumor appears as a lightly pigmented or amelanotic cystic mass. Large cysts may break off from the main tumor and float freely in the anterior chamber or vitreous cavity. Because of their appearance and because medulloepithelioma may present with leukocoria, these tumors are an important consideration in the differential diagnosis of retinoblastoma. A-scan of medulloepithelioma shows mainly high internal reflectivity with a medium spike corresponding to cystic regions of the tumor. On B-scan, medulloepitheliomas typically appear as a dome-shaped, highly reflective mass with irregular internal structures. Cystic spaces can be demonstrated in some lesions (Figure 11.6).
Benign uveal tumors
Circumscribed and diffuse choroidal hemangioma
Clinical features, symptoms, and signs
On ophthalmoscopic examination, circumscribed choroidal hemangiomas appear as an orange choroidal mass with indistinct margins that blend with the surrounding choroid. They are frequently located in the macular region of the posterior pole, and are not usually thicker than 6 mm.11 Although these tumors are highly vascular, dilated and tortuous feeder vessels are not typically observed. Surrounding subretinal fluid leading to exudative retinal detachment with macular involvement is common in symptomatic cases. Retinal hard exudates are minimal or absent. Diffuse choroidal hemangiomas appear as orange, diffuse choroidal thickening that has been likened to a “tomato-catsup fundus.” Focal regions of excessively thickened choroid within the diffuse hemangioma may simulate circumscribed choroidal hemangioma. As with circumscribed hemangiomas, there may be associated exudative retinal detachment that often does not become manifest until adolescence.
Diagnostic evaluation
On A-scan, circumscribed choroidal hemangiomas demonstrate high internal reflectivity with negligible attenuation. This differs from other tumors in the differential diagnosis including malignant melanoma which classically demonstrates low to medium reflectivity on A-scan. On B-scan, circumscribed choroidal hemangioma appears as a dome-shaped choroidal mass with smooth contours. They are hyperechoic with regular internal structure and little internal blood flow. Serous retinal detachment at the tumor margins and calcification on the tumor surface may be present.11 Angiographic studies such as fluorescein and indocyanine green (ICG) can also be diagnostic. Fluorescein angiography demonstrates a hyperfluorescent mass with a fine lacy vascular network of intrinsic vessels in the early phases followed by increasing hyperfluorescence throughout the angiogram with variable leakage in late views.12 With ICG angiography, a rapid increase in hyperfluorescence is seen early on followed by a “washout” effect in the late phase.13 OCT can also be helpful in evaluating secondary changes in the overlying retina such as shallow subretinal fluid or cystoid macular edema (Figure 11.7).
[/not-level-membership-for-opthalmology-category]
