CHAPTER 89 Intradural Tumors
Intradural Tumors
The era of spinal tumor surgery began in London in 1887 when the influential surgeon Dr. Victor Horsley removed a thoracic intradural extramedullary tumor from a 42-year-old British Army General at the urging of the neurologist Dr. William Gowers.1 The patient presented with a 4-year history of progressive symptoms that had culminated in urinary incontinence, spasticity, and paraplegia. Dr. Gowers diagnosed the lesion as a thoracic compressive lesion on the basis of a history and clinical examination and proposed surgery. After a thoracic laminectomy and removal of a compressive fibromyxoma, the patient made a remarkable recovery, regained the ability to walk, returned to work, and lived 20 additional years without recurrence. As a result, Dr. Horsley became a proponent of surgery as a viable treatment option for spinal tumors.
The next advancement in spinal tumor surgery was the first successful intramedullary spinal cord tumor removal in 1907 by Dr. Anton von Eiselsberg in Austria; however, the first published report of a successful intramedullary tumor removal is credited to Dr. Charles Elsberg in 1911 at the New York Neurological Institute.2,3 Soon thereafter in 1916, Elsberg published his influential book Diagnosis and Treatment of Surgical Diseases of the Spinal Cord and Its Membranes. He later published the first large series of intradural spinal cord tumor resections in 1925.4 In his early writings, Elsberg advocated a two-staged surgery for intramedullary tumors: a laminectomy and myelotomy followed 1 week later by removal of the tumor that had delivered through the myelotomy. However, as his experience grew, he transitioned into performing single-stage operations for intramedullary tumor removal. For these significant early contributions he is remembered as an important figure in spinal cord surgery.
Over the ensuing decades, the widespread practice of intraspinal tumor surgery was hindered by difficulty with tumor localization, infections, neurologic morbidity, and poor outcomes. Throughout the mid 20th century, intradural and especially intramedullary tumors were treated with decompressive laminectomy, biopsy, and palliative radiation.5 In the 1960s, technologic advances such as loupe magnification, widespread use of myelography, and bipolar cautery began making good outcomes after surgery feasible and there was a renewed interest in aggressive surgical management.6 Even in the past 30 years, the evolution of technologies such as the operating microscope, microsurgical instruments, magnetic resonance imaging (MRI), ultrasonic cavitation, and electrophysiological monitoring has made aggressive surgical resection a low morbidity mainstay of spinal tumor treatment.
Epidemiology
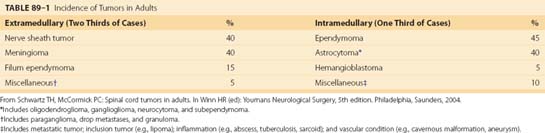
The incidence of intradural spinal tumors is reported to be between 1 and 2 per 100,000 population, and they account for 5% to 10% of central nervous system (CNS) tumors.7–9 In adults, extramedullary tumors predominate, accounting for 60% to 75% of intradural masses with intramedullary tumors accounting for the remainder. In children, however, intramedullary tumors are more common, accounting for greater than 35% of intradural masses.9,10 Intradural tumors occur with equal frequency in both sexes, with the exception of a higher incidence of meningiomas in women. The most frequent intradural-extramedullary tumors are nerve sheath tumors and meningiomas with filum terminale ependymomas being a distant third.9–11 Ependymomas and astrocytomas are the most common intramedullary lesions followed by hemangioblastomas and cavernous malformations.9,10,12 One institution’s large experience is summarized in Table 89–1.
Clinical Presentation
Intradural tumors are typically slow growing, benign lesions that may be asymptomatic or minimally symptomatic for years before presentation. The most common presenting symptoms for intramedullary tumors are sensory deficits, motor weakness, gait ataxia, and pain.10 Pain is often worse at night and awakens the patient from sleep, but pain from a tumor may be difficult to differentiate from common degenerative back pain.13 In patients harboring slow-growing tumors, there is an indolent progression of symptoms from nonspecific pain and sensory disturbance to loss of balance, motor weakness, and other signs of myelopathy.14,15 Patients may present with symptoms at any point along this continuum; however, since the advent of the MRI, tumors are being discovered earlier, resulting in less severe deficits at presentation. Intramedullary tumors can be associated with a suspended dissociated sensory loss.7,16 This pattern of sensory disturbance is attributable to dilatation of the central cord by the tumor or an associated spinal cord syrinx causing an interruption of the pain and temperature fibers crossing in the anterior commissure. In fact, spinal axis imaging with contrast is an important step in the workup of an unexplained spinal cord syrinx in order to rule out an underlying spinal cord tumor.
The most common presenting symptoms for extramedullary tumors are as follows: pain, sensory disturbances, motor deficits, and gait ataxia. Schwannomas are derived from the nerve root sleeve and are more likely to present as unilateral radiculopathy, whereas meningiomas, derived from the dura, are more likely to present with diffuse pain or symptoms of cord compression.17,18 Yet these symptoms are not mutually exclusive, and patients often present with a mixture of nonspecific signs and symptoms.
The presenting signs and symptoms of intradural tumors are also influenced by the tumor’s spinal level.19 Both cervical and thoracic tumors can enlarge or compress the cord, resulting in cord impairment with prominent motor weakness and myelopathy. Cervical tumors are more likely to present with occipitocervical pain, arm pain, hand clumsiness, and sensory disturbances. Thoracic tumors often present with myelopathy and sensory disturbances in the lower body but can also produce pain that is mistakenly attributed to the heart and visceral organs. Tumors of the cauda equina typically cause back and leg pain that is classically worse while lying down and relieved by standing. This is thought to be due to positional pressure fluctuations in the epidural venous plexus.20
Radiologic Diagnosis
In the treatment of intradural tumors, the advent of MRI has allowed earlier tumor discovery, superior lesion characterization, precise localization, and improved surgical planning. MRI has supplanted computed tomography (CT) and contrast myelography as the imaging modality of choice in the diagnosis of intradural tumors. In most cases MRI is now a stand-alone imaging modality with CT for certain tumors, and CT myelogram is reserved for use in patients with contraindications to undergoing an MRI (e.g., those with implanted ferromagnetic objects).21 Plain radiographs are rarely important in the setting of intradural tumors but are useful when looking for abnormal alignment or structural instability in patients who have undergone a previous tumor resection.
In the evaluation of spinal tumors, MRI with gadolinium greatly increases the ability to identify a mass, show its relationship to the spinal cord, identify surrounding vascular structures, and give clues as to the pathologic diagnosis. Additionally, it can help differentiate tumors from less ominous lesions such as arachnoid cysts, lipomas, and neurenteric cysts. On T1-weighted images, intradural tumors are typically isointense or slightly hypointense to the spinal cord. In T2-weighted sequences, cerebrospinal fluid (CSF) has high signal and intramedullary tumors are often hyperintense. Spinal cord edema and syrinx associated with tumors are best identified with T2 images.22 After the intravenous administration of gadolinium (paramagnetic ions), T1-weighted MRI detects contrast that leaks through the compromised blood–spinal cord barrier at the tumor interface. The subsequent delay in T1 relaxation time by the gadolinium within the tumor is interpreted as enhancement. Most intradural tumors display some degree of contrast enhancement.
Extramedullary Tumors
Meningiomas
Intraspinal meningiomas are the most common type of intradural extramedullary tumors and occur predominantly in women (75% to 85%). They typically present in the fifth to seventh decades of life with pain, paresthesias, weakness, and long tract signs.23,24 Meningiomas are homogeneously enhancing, smooth-bordered masses on MRI and can contain calcifications that help differentiate them from nerve sheath tumors (Fig. 89–1). With suspected meningiomas, preoperative CT scans are helpful to determine the presence and extent of tumor calcification, which can significantly affect surgical strategy. Seventy-five percent of intraspinal meningiomas arise in the thoracic spine and are typically dorsolateral to the cord; however, cervical tumors (20%) do occur and are frequently located in the upper cervical region and ventral to the cord.20,25 Lumbar meningiomas are the least common. Spinal meningiomas are thought to arise from arachnoid cap cells in the dura near the nerve root sleeve laterally but may also grow from dural fibroblasts or pial cells, explaining their occasional ventral or dorsal location.26 Meningiomas invariably arise and derive their blood supply from a broad dural base that is often visible as a dural tail on MRI. These tumors are typically benign, encapsulated, slow-growing masses, but other growth patterns are seen including en plaque, atypical, and invasive malignant meningiomas.10,27 Treatment for symptomatic tumors is surgical with the goal of gross total resection; however, total removal may not be possible with en plaque or ventral calcified tumors.27,28 Overall, spinal meningiomas are amenable to surgical removal and have low recurrence rates after complete resection.
Nerve Sheath Tumors
Tumors of the nerve sheath account for up to 30% of spinal neoplasms and are categorized as either schwannomas or neurofibromas.28 They typically arise from the posterior sensory nerve root near its entrance into the neural foramen. Although both tumors are thought to be of Schwann cell origin, their significant histologic, epidemiologic, and biologic differences warrant discussing them separately.
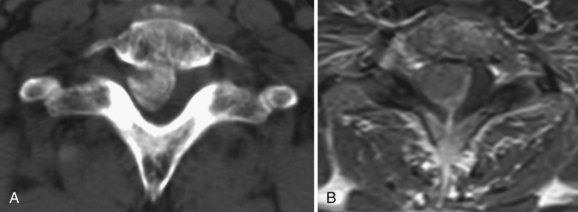
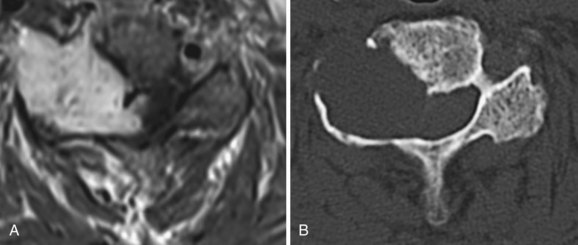
Schwannomas are common intradural extramedullary neoplasms that make up 85% of nerve sheath tumors.18 They occur equally in men and women with a peak age of presentation between the fourth and sixth decades. Most appear as solitary tumors and occur equally throughout the spinal canal. Although rare, there are patients with multiple schwannomas but nearly always in the setting of neurofibromatosis 2 (NF2) and schwannomatosis.29 Radiographically, schwannomas are well-demarcated masses associated with a nerve root, intensely enhance, can include cystic areas, and may grow along the root into the extraspinal space (Fig. 89–2A). Up to 30% of schwannomas grow through the neural foramen, giving the classic “dumbbell” shape on imaging (Fig. 89–2B).18 At surgery, schwannomas are smooth, well-demarcated tumors that are discretely attached to an intact nerve root and can usually be separated from the root without nerve injury. The goal of surgery should be complete tumor removal and can usually be achieved. However, if complete resection is not possible, aggressive debulking with preservation of the root can relieve symptoms and the patient can be monitored radiographically for recurrence.
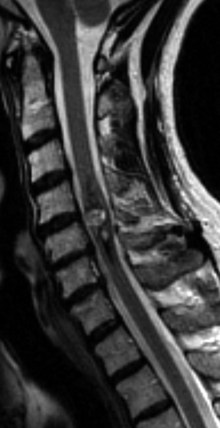
Neurofibromas are less common than schwannomas, accounting for approximately 15% of nerve sheath tumors, and more than half of them are associated with neurofibromatosis 1 (NF1).18,30 Patients with NF1 can harbor single or multiple neurofibromas and are more likely to have malignant tumors.31 Neurofibromas typically, but not exclusively, arise from the sensory root. Consequently, patients most commonly present with pain. Radiographically, neurofibromas are similar to schwannomas but are solid, not cystic. Yet they, too, can extend through the dural root sleeve as a dumbbell tumor (Fig. 89–3). Tumor cells characteristically arise from central nerve root fibers and expand the root, making dissection of the tumor from the root unrealistic. Optimal treatment for symptomatic neurofibromas includes total surgical removal of tumor, often necessitating the sacrifice of the nerve root.28
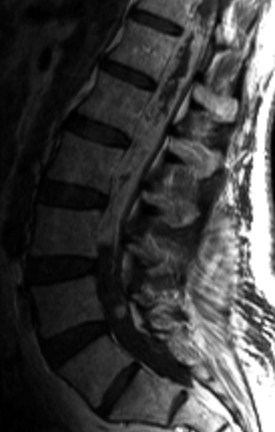
Ependymomas of the Filum Terminale
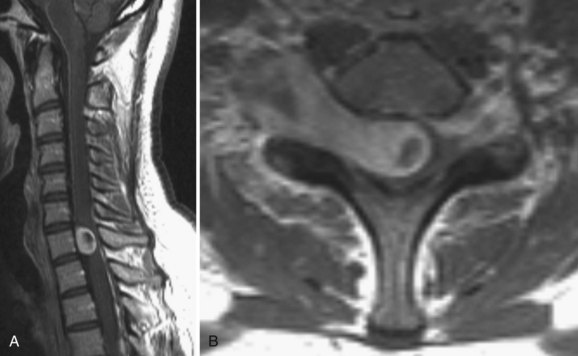
Ependymomas of this region are slow-growing tumors that arise from the filum terminale near the junction with the conus. At the terminal filum, ependymomas are thought to arise from ependymal rests ectopically deposited during development. They typically present in the third to fourth decades of life with slowly progressive low back pain, sensory deficits, and even sphincter and sexual disturbances.10 Histologically, filum terminale ependymomas are overwhelmingly of the benign myxopapillary pathological subtype, but cellular and papillary histiotypes do occur.32 On MRI, these tumors exhibit intense contrast uptake and can be anywhere on a spectrum from small, well-circumscribed tumors at the inferior tip of the conus to massive septated tumors filling the spinal canal (Fig. 89–4). An MRI of the neural axis should be obtained because cauda region ependymomas can disseminate along the neural axis via CSF or be the result of intracranial cranial tumor seeding to the spine. Therapy is surgical with a goal of gross total resection; however, this is often not achievable with massive tumors that are adherent to neural structures. Even after gross total resection of larger tumors, the recurrence rate is up to 20% and patients may need multiple surgeries for recurrences.33,34
Less Common ExtramedullaryTumors
Arachnoid Cysts
Although not tumors, these structures can appear as a cystic mass compressing the spinal cord on MRI. They are composed of arachnoid membranes that have become loculated with CSF under pressure. The most common place for symptomatic arachnoid cysts is in the thoracic region. They typically present with pain and signs of myelopathy. Surgical excision relieves spinal cord compression and is the mainstay of treatment for symptomatic lesions.35
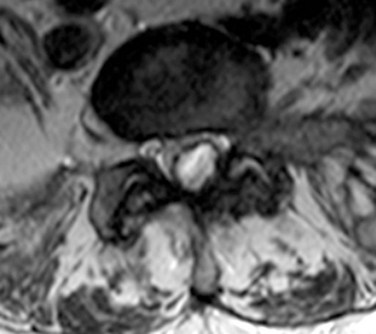
Synovial Cysts
These extradural structures are cystic enlargements of the facet capsule that can enlarge to displace the thecal sac, compress the cord, or impinge nerve roots causing radiculopathy. They are most common in the lumbar region, especially at L4-L5, but have been reported in the cervical spine.36 These cysts are thought to result from excess motion at a vertebral level leading to facet capsule hypertrophy and cystic enlargement. These lesions are filled with synovial fluid and can mimic intradural lesions on plain MRI (Fig. 89–5). Large or persistently symptomatic lesions can be managed with image-guided percutaneous drainage or more definitively with surgical removal.37,38
Intramedullary Tumors
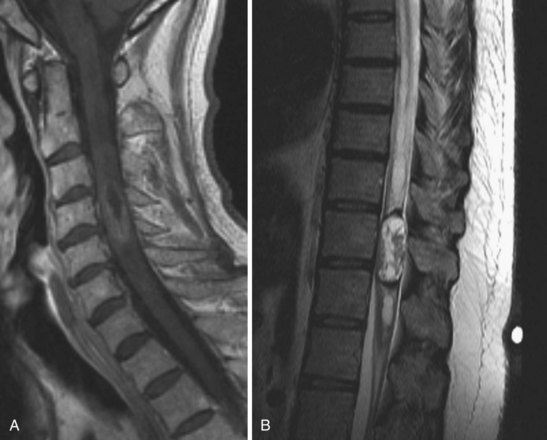
Ependymomas
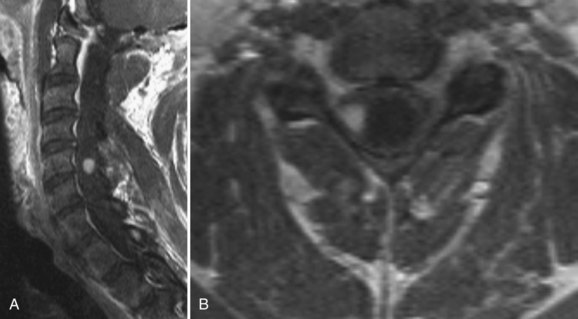
Intramedullary tumors of the spinal cord are rare, accounting for 15% to 30% of intradural spinal lesions. Ependymomas are the most common type of intramedullary spinal cord tumor and usually present in the middle age years with pain, mild sensory changes, motor weakness, and signs of myelopathy.39 There is a curious association of intramedullary ependymomas and NF2. A significant portion of NF2 patients harbor intramedullary ependymomas, and genetic studies on tumor cells have shown NF2 gene mutations in spontaneous intramedullary ependymomas.40–42 Ependymomas of the cord are typically solitary tumors that arise from ependymal lining of the central canal, cause a diffuse enlargement of the cord over several levels, and have an associated syrinx in up to 50% of cases.43,44 Among the four ependymoma histologic subtypes, all are considered benign.12,45,46 On MRI, intramedullary ependymomas are sharply demarcated lesions that are isointense on plain T1 sequences and enhance homogeneously with gadolinium (Fig. 89–6A). Classic radiographic features of spinal cord ependymomas include distinct tumor–spinal cord border, an associated syrinx, cysts within or adjacent to the mass, and hemosiderin deposits or “caps” near the poles of the tumor on T1 and T2 (Fig. 89–6B).47 The treatment of choice is gross total surgical resection. In contrast to intracranial ependymomas, intramedullary spinal cord ependymomas have a good prognosis and a low rate of local recurrence after complete resection, but cases of malignant transformation to malignant pathology after resection have been reported.12,46,48–50
Astrocytoma
Astrocytomas are the second most common intramedullary spinal cord tumor in adults, representing 6% to 8% of intradural tumors. In children, they are the most common histologic type of intramedullary tumors, accounting for 60% to 90% of these lesions.9,51 Patients harboring astrocytomas present most commonly with pain, motor weakness, and gait ataxia that can either be of long duration or of relatively brief onset before presentation.10,52 The average symptom duration before presentation is between 12 and 29 months. Astrocytomas of the spinal cord are infiltrative tumors that are often eccentrically located dorsal or lateral to the central canal. Large tumors can expand and rotate the cord, making identification of landmarks difficult during surgery.12 On MRI, astrocytomas have intermediate T1 signal intensity, can demonstrate heterogenous or homogeneous T1 contrast enhancement, often contain cystic areas, and can rarely grow exophytically into the extramedullary space. The tumor boundaries may be better identified on T2 sequences (Fig. 89–7). In contrast to ependymomas, spinal astrocytomas infiltrate functional cord, have ill-defined borders with normal cord, and are consequently more difficult to remove successfully. Fortunately, intramedullary astrocytomas in adults are largely low grade (Kernohan grade I and II) with approximately 25% considered high grade (Kernohan grade III and IV). In children, where pilocytic astrocytomas predominate, high-grade lesions are even less common, occurring at a rate of 10% to 15%.
Hemangioblastoma
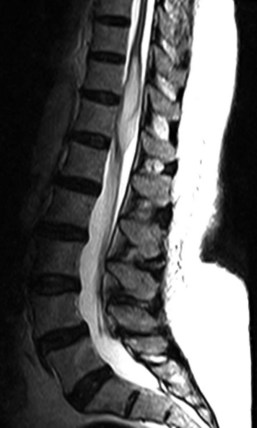
Hemangioblastomas represent 2% to 6% of intramedullary spinal cord tumors and approximately one third are associated with von Hippel-Lindau disease.10,53,54 Spontaneous hemangioblastomas are usually solitary, but patients with von Hippel-Lindau disease (VHL) can harbor multiple hemangioblastomas, especially in the posterior-fossa.54 Hemangioblastomas have a 2 : 1 male-to-female predominance and usually become symptomatic in the third to fourth decades of life. Because of their predilection for the dorsal cord, numerous patients present with posterior column dysfunction or experience it after surgery. Histologically, these tumors consist of a vascular tuft surrounded by pale stromal cells, and there is often an associated cyst that accounts for much of the tumor volume. Radiographically, the tumors can be predominantly cystic masses with a small enhancing nodule of tumor cells located on the cyst wall or a solid homogeneously enhancing mass (Fig. 89–8). They are predominantly dorsal or dorsolaterally located and often grow to and expand the pial surface of the cord. Up to 89% have an associated syrinx that always resolves after tumor removal.43,54 Treatment of spinal hemangioblastomas is surgical resection. Removal of these tumors carries the lowest surgical morbidity and best neurologic outcomes of all the intramedullary tumors.
Less Common Intramedullary Lesions
Lipomas
True intramedullary lipomas are rare lesions composed of metabolically normal fat cells ectopically deposited within the cord during embryologic development. Patients typically present in their 20s to 40s complaining of indolent neurologic symptoms that can rapidly progress over several months.55 MRI is diagnostic, and the tumor signal follows that of normal fat. Treatment of symptomatic lesions is internal debulking to relieve symptoms.56 Complete excision is difficult due to poor dissection planes, and delayed recurrence is common.
Dermoid and Epidermoid Cysts
These tumors arise from an anomalous deposition of ectodermal cells during embryonic neural tube closure, but some cases have been linked to epidural cells implanted into the lumbar canal during lumbar puncture.57 Patients present in the second to fourth decades with typical neurologic complaints of intramedullary masses; however, acute presentations secondary to aseptic meningitis or abscess formation have been reported.58,59 Optimal treatment is complete surgical excision, but the cyst wall is often adherent to the spinal cord substance and incomplete resections are often performed with low recurrence rates.60 Again, expansile duraplasty can be helpful after resection to delay symptoms in the event of a recurrence.
Cavernous Angiomas
These lesions are not tumors but angiographically occult vascular malformations composed of tightly packed venous-like channels without intervening neural tissue. They represent 1% to 3% of intramedullary spinal cord lesions. Spinal cord cavernomas can be asymptomatic, present with slow stepwise decline of neurologic function due to small hemorrhages, or present as an acute neurologic deterioration from a larger bleed.61 Most cavernomas are well-defined single lesions, some have ectopic portions nearly invisible under the pial surface, and rarely they are multiple. Cavernomas are best seen on T2 MRI sequence, often have a mulberry-like appearance, and do not enhance (Fig. 89–9). Patients with symptomatic lesions should undergo surgical excision to prevent neurologic morbidity from future bleeds.62
Metastasis
The spinal cord parenchyma is a rare site of metastasis for systemic cancer and may occur in patients with extensive disseminated disease. The most common tumors that metastasize to the spinal cord are lung cancer (54%), breast cancer (14%), and melanoma (9%).63,64 These patients generally have a grim prognosis related to their primary disease; however, surgical excision of cord lesions should be considered in those with radioresistant tumors and in those with a low systemic burden of disease who have symptomatic, surgically accessible solitary lesions.65
Surgical Treatment
Intradural Extramedullary Tumors
Extramedullary tumors are almost exclusively approached through a posterior midline approach. Anterior tumors may require a facetectomy for adequate tumor exposure, and dumbbell tumors with significant extraspinal extension may necessitate a lateral extracavitary approach.66 Purely anterior surgical approaches are fraught with high complication rates and are necessary only in exceptional cases. To perform a posterior midline approach, the patient is positioned prone and a laminectomy is performed. The laminectomy usually does not need to be wide, and the facets are spared to preserve the structural stability of that bony segment. When the bony exposure is complete, intraoperative ultrasound can be useful to localize the tumor before dural opening.67 Once the dura is opened and the tumor is visualized, the tumor is dissected from the surrounding tissues and care must be taken not to damage the cord parenchyma by excessive manipulation or damage vascular structures compromising cord blood flow. After exposure, removal may be performed en bloc, piecemeal, or with ultrasonic cavitation for internal debulking and, finally, capsule removal. After tumor resection, the spinal cord should be carefully dissected from arachnoid adhesions to the dura that often develop with chronic cord compression and local inflammation. In general, the goal of surgery is maximal tumor removal without neurologic injury. However, some extramedullary tumors force a decision between sacrificing the parent nerve root for complete tumor removal versus subtotal resection with root preservation. This decision is usually based on function of the involved root, tumor pathology, and tumor characteristics.
Nerve sheath tumor removal is dependent on tumor location and characteristics. Most schwannomas are dorsolateral and easily accessed, but more anteriorly located or dumbbell tumors with paraspinal extension may require extended posterolateral exposures.66 Most nerve sheath tumor capsules are adherent to the arachnoid of the nerve root, and this layer must be incised to reach the lesion. The capsule is generally cauterized to shrink and decrease vascular input to the tumor, and then the tumor–nerve root attachment can be visualized. Piecemeal tumor removal or internal debulking with an ultrasonic aspirator should be used with large tumors. Nerve root sacrifice may be required due to tumor inside the root of large tumors encasing it. However, the incidence of permanent neurologic deficit from transection of a tumor-enveloped root is lower than might be expected, usually due to poor preoperative function of the root secondary to the tumor.68,69 Care must be taken to carefully assess root function and weigh the benefit of complete tumor removal against loss of function before and during surgery.
Intramedullary Tumors
The role of surgery in the treatment of intramedullary spinal cord tumors expanded considerably in recent decades. With the advent of the operative microscope, MRI, ultrasonic aspiration, and electrophysiologic monitoring, experienced surgeons can now safely remove or greatly debulk most intramedullary lesions. Neuroprotection is an important consideration during spinal cord tumor surgery, and perioperative steroids are commonly employed. Recently, intraoperative hypothermia using intravenous cooling catheter systems has emerged as a potential neuroprotective adjunct during spinal tumor surgery. Our 4-year experience with intraoperative hypothermia has found it to be safe (unpublished data), but randomized controlled trials will ultimately be necessary to judge its efficacy. Surgery is nearly always performed through a posterior midline approach with laminectomy at the tumor level, as well as one level above and one below. Ultrasound can be useful to confirm the tumor location and confirm it is within the limits of the bony exposure.67,70 Then a midline dural incision is made. With the aid of an operative microscope, the midline is identified and a myelotomy is made in the posterior median sulcus. Of note, the midline is often difficult to identify with larger tumors due to an expanded and often rotated cord, but the midpoint between dorsal root entry zones can be used as a rough estimate of midline in such cases. The myelotomy is deepened until the tumor is identified, and then the resection is guided by the presence or absence of a dissection plane between the tumor and cord parenchyma. Intraoperative frozen pathology can be helpful if suspicion is high for an unusual diagnosis such as demyelinating disease or metastasis; however, frozen sections can be unreliable in differentiating between astrocytomas and ependymomas. Tumor resection is performed to the maximal extent possible while avoiding neurologic injury.
Ependymomas are centrally located, smooth, red-gray, well-demarcated lesions that often have cystic areas at their superior and inferior limits. When the cord is exposed, small blood vessels are commonly seen running across the tumor surface. There is usually a good cleavage plane between tumor and cord, and they can be removed completely in 70% to 95% of cases.49,71,72 Smaller tumors can often be removed in one piece by gentle capsule dissection and light bipolar coagulation to shrink the mass. Larger tumors must be internally decompressed and then the capsule carefully dissected from the cord. The final removal is often achieved by gentle retraction of the superior tumor pole and cauterization of the last arterial feeders. At this point, a common surgical error is to aggressively resect a layer of slightly discolored but ultimately functional tissue that can surround the tumor at the parenchyma-tumor interface. Resection should not continue past obvious tumor into the glial tissue to avoid unnecessary neurologic injury.
Astrocytomas are generally low-grade infiltrating tumors that tend to be asymmetrically located in the spinal cord. Intraoperatively, the tumor appears grossly different than cord parenchyma, but most often tumor cells infiltrate functional cord tissue preventing the development of a safe dissection plane. This makes complete tumor resection hazardous. Some authors have argued for aggressive resection low-grade astrocytomas, but there is an unclear relationship between extent of resection and survival. Therefore resection of obvious tumor without risking neurologic deterioration is recommended.10,49,73,74 Patients harboring high-grade astrocytomas have poor prognosis regardless of the degree of tumor removal, and resection of high-grade lesions is typically conservative. Several long-term survivals have been reported in grade IV astrocytoma patients with tumors below T1 including two patients in the senior author’s personal series.52,73,75
Hemangioblastomas are highly vascular, well-demarcated, often cystic lesions that can rise to the dorsal surface of the spinal cord. These tumors are extremely vascular, so the superficial and deep arterial supply should always be interrupted and the capsule shrunken away from the cord with bipolar coagulation before the tumor is entered or removed. Entering the capsule before interrupting the tumor arterial feeders risks a large hemorrhage. Hemangioblastomas should be completely resected because residual tumors will lead to a recurrence. Preoperative embolization has been advocated by some authors but is not commonly performed because most feeders are surgically accessible.53
Radiation and Chemotherapy
Intradural Extramedullary Tumors
Surgical excision is first-line therapy for extramedullary intradural tumors, with radiation and chemotherapy being rarely indicated for benign pathologies. Adjunctive radiation therapy and chemotherapy is typically reserved for World Health Organization grade III and IV extramedullary tumors. Fractionated radiation is not routinely used for extramedullary low-grade lesions, but some authors advocate radiation for residual or recurrent large filum terminale ependymomas.32,33,76 Radiosurgery, however, has recently been proposed as an alternative to surgery for poor surgical candidates harboring low-grade lesions.77 The studies are small, and follow-up is brief. Delayed radiation-induced spinal cord toxicity is a feared complication of spinal cord radiotherapy, and more follow-up data are necessary to determine its safety, which is of vital importance when treating benign pathologies.77,78
Intramedullary Tumors
Ependymomas are generally amenable to safe, complete removal with low recurrence rates. MRI is a noninvasive method for monitoring for tumor regrowth or seeding of the subarachnoid space and can obviate the need for early radiation after surgery. Radiation may be indicated in the setting of a malignant ependymoma or when a significant portion of a tumor cannot be resected.50,79–81
Intramedullary astrocytomas are typically low-grade tumors treated by surgical resection and monitored for recurrence by clinical and radiographic follow-up.73 In the setting of subtotal tumor removal, some authors have advocated radiation to the residual tumor, but there is no compelling evidence that radiotherapy alters the natural history.73,82 Patients with grade III astrocytomas may be candidates for adjunctive radiation and chemotherapy.83 Malignant astrocytomas of the spinal cord, however, have a poor prognosis and radical resection results in high morbidity; therefore conservative tumor removal for tissue diagnosis followed by radiation therapy is common practice.12,73,84 Alternatively, one group has reported extended survival times for malignant astrocytoma patients treated with radiocordectomy, and long-term survival for spinal cord grade IV astrocytomas has been reported after surgical cordectomy including two long-term survivals in the senior author’s personal series (unpublished).75,85
Stereotactic radiosurgery for intramedullary tumors has been described in small studies with brief follow-up.86,87 In the future, this treatment strategy may prove useful in select cases in which complete surgical excision is not possible or after multiple recurrences. However, more studies are necessary to determine safety and long-term efficacy, especially in light of the proven success of surgery and the low-grade pathologies of most intramedullary tumors.
Outcomes
Intradural Extramedullary Tumors
The surgical morbidity and mortality in the removal of extramedullary tumors is low while the neurologic prognosis is generally good. Postoperative neurologic function is generally predicted by the preoperative neurologic status, patient age, and duration of symptoms.20,23,25 In one large series of intradural extramedullary tumors, there was a 12.3% rate of postoperative neurologic worsening, but most deficits were transient and only 2.3% of the patients had a permanent postoperative loss of function.10 Overall, intradural extramedullary tumors have a recurrence rate of 8.9% to 32% at 5 years with the strongest predictors of recurrence being histologic grade and previous surgical resection.10,88 Meningiomas have a 4% to 8% long-term recurrence rate after gross total resection with higher-grade tumors recurring more frequently. After surgery, recovery of neurologic deficits is common with 80% to 90% of patients showing improvement of preoperative neurologic deficits.17,25 In the absence of NF2, the recurrence rate for schwannomas is 6% to 12%, but in patients with neurofibromatosis it is increased fourfold.18,88,89 Completely resected neurofibromas recur in approximately 12% of patients.90 Large filum terminale ependymomas have a high recurrence rate. Even after gross total resection, the recurrence rate is up to 20% and patients may need multiple operations for revision debulking.33,34
Intramedullary Tumors
The neurologic morbidity for intramedullary tumor removal is significantly higher than for extramedullary tumors. Preoperative neurologic status is the number one predictor of postoperative functional status, but other factors such as aggressive tumor pathology and thoracic location also portend worse functional outcomes.91,92 Up to 57% of postsurgical patients experience a transient neurologic worsening, but only 10% to 20% are left with a new permanent deficit.10,49,71 Dorsal column dysfunction is common after midline myelotomy.39 Despite the new deficits, however, most patients recover to baseline functional status within 1 year.10
After complete resection, ependymomas have good neurologic outcomes, low recurrence rates, and long survival times.12,46,81 Tumors that are not completely resected tend to have less improvement in function and higher recurrence rates.10,71,88,93 Survival rates at 5 years are reported to be between 83% and 95% for postoperative intramedullary ependymoma patients.10,49,94
Astrocytomas are more difficult to completely remove surgically and have a higher recurrence rate. Astrocytoma patients have shorter survival times than those harboring ependymomas. Extent of tumor resection seems to modestly affect local recurrence rates, but this has not been conclusively demonstrated in the literature.10,12,43,74,91 The 5-year survival rate has been reported to be 63% to 77% for grade I and II tumors and 27% for grade III and IV tumors.73 Median survival for high-grade lesions is around 1 year with grade IV tumors having a particularly poor prognosis.73,82,95–97
Hemangioblastomas are amenable to complete resection, have low surgical morbidity, and have low recurrence rates.98–100 The presence or absence of VHL seems to have little impact on outcomes.98 Intramedullary hemangioblastomas patients experience the lowest surgical morbidity and the best neurologic prognosis of all the common intramedullary spinal cord tumors.
Conclusion
Pearls
Pitfalls
Key Points
1 Horsley V, Gowers WR. A case of tumors of the spinal cord: Removal-recovery. Med Chir Trans London. 1888;71:377-428.
The first description of a successful intradural tumor removal.
2 Elsberg CA, Beer E. The operability of intramedullary tumors of the spinal cord: A report of two operations with remarks upon the extrusion of intraspinal tumors. American Journal of Medical Science. 1911;142:636-647.
3 Elsberg CA. Tumors of the spinal cord and the symptoms of irritation and compression of the spinal cord and nerve roots. In: Elsberg, editor. Pathology, Symptomatology, Diagnosis and Treatment. New York: Paul B. Hoeber; 1925:206-239.
A landmark book describing Elsberg’s large early spinal tumor removal experience.
4 Greenwood JJr. Intramedullary Tumors of Spinal Cord. A Follow-up Study after Total Surgical Removal. J Neurosurg. 1963;20:665-668.
5 Klekamp JSM. Surgery of Spinal Tumors. New York: Springer Berlin Heidelberg; 2007.
A detailed description and analysis of a large modern spinal tumor surgical experience.
1 Horsley V, Gowers WR. A case of tumors of the spinal cord: Removal-recovery. Med Chir Trans London. 1888;71:377-428.
2 Eiselsberg AV, Ranzi E. Über die chirurgische Behandlung der Hirnund Rückenmarkstumoren. Arch Klin Chir. 1913;102:309-468.
3 Elsberg CA, Beer E. The operability of intramedullary tumors of the spinal cord: A report of two operations with remarks upon the extrusion of intraspinal tumors. American Journal of Medical Science. 1911;142:636-647.
4 Elsberg CA. Tumors of the spinal cord and the symptoms of irritation and compression of the spinal cord and nerve roots. In: Elsberg, editor. Pathology, Symptomatology, Diagnosis and Treatment. New York: Paul B. Hoeber; 1925:206-239.
5 Wood EH, Berne AS, Taveras JM. The value of radiation therapy in the management of intrinsic tumors of the spinal cord. Radiology. 1954;63:11-24.
6 Greenwood JJr. Intramedullary Tumors of Spinal Cord. A Follow-up Study after Total Surgical Removal. J Neurosurg. 1963;20:665-668.
7 Cooper PR, Epstein F. Radical resection of intramedullary spinal cord tumors in adults. Recent experience in 29 patients. J Neurosurg. 1985;63:492-499.
8 Sasanelli FBE, Kurland LT. Primary intraspinal neoplasms in Rochester, Minnesota, 1935-1981. Neuroepidemiology. 1983;2:156-163.
9 Helseth A, Mork SJ. Primary intraspinal neoplasms in Norway, 1955 to 1986. A population-based survey of 467 patients. J Neurosurg. 1989;71:842-845.
10 Klekamp JSM. Surgery of Spinal Tumors. New York: Springer Berlin Heidelberg; 2007.
11 Miller DC. Surgical pathology of intramedullary spinal cord neoplasms. J Neurooncol. 2000;47:189-194.
12 Cooper PR. Outcome after operative treatment of intramedullary spinal cord tumors in adults: intermediate and long-term results in 51 patients. Neurosurgery. 1989;25:855-859.
13 McGuire RA, Brown MD, Green BA. Intradural spinal tumors and spinal stenosis. Report of two cases. Spine (Phila Pa 1976). 1987;12:1062-1066.
14 Kane PJ, el-Mahdy W, Singh A, Powell MP, Crockard HA. Spinal intradural tumours: Part II–Intramedullary. Br J Neurosurg. 1999;13:558-563.
15 el-Mahdy W, Kane PJ, Powell MP, Crockard HA. Spinal intradural tumours: Part I—Extramedullary. Br J Neurosurg. 1999;13(6):550-557.
16 Quintero-Wolfe SMG, Green BA, Wang MY. Surgical Treatment of Non-Traumatic Myelopathies. In: Lin VW, editor. Spinal Cord Medicine. Demos Medical Publishing, 2010.
17 Gezen F, Kahraman S, Canakci Z, Beduk A. Review of 36 cases of spinal cord meningioma. Spine (Phila Pa 1976). 2000;25(6):727-731.
18 Seppala MT, Haltia MJ, Sankila RJ, Jaaskelainen JE, Heiskanen O. Long-term outcome after removal of spinal schwannoma: a clinicopathological study of 187 cases. J Neurosurg. 1995;83:621-626.
19 Green BA, Eismont F, Klose KJ. Management of Cervical Cord Lesions Including Advances in Rehabilitative Engineering. In: Camins MB, O’Leary PF, editors. Disorders of the Cervical Spine. Baltimore, MD: Williams and Wilkins, 1992.
20 McCormick PC, Post KD, Stein BM. Intradural extramedullary tumors in adults. Neurosurg Clin N Am. 1990;1:591-608.
21 Green BA, Diaz RD, Post MJD. The diagnosis of spinal column and spinal cord tumors with emphasis on the value of computed tomography. In: Post J, editor. Computed Tomography of the Spine. Baltimore, MD: Williams and Wilkins, 1984.
22 Jea AVS, Green BA. Syringomyelia. In Herkowitz R-S, et al, editors: The Spine, 5th Edition, Philadelphia, PA: Elsevier, Inc. WB Saunders Company, 2007.
23 Roux FX, Nataf F, Pinaudeau M, et al. Intraspinal meningiomas: review of 54 cases with discussion of poor prognosis factors and modern therapeutic management. Surg Neurol. 1996;46(5):458-463. discussion 463-464
24 Solero CL, Fornari M, Giombini S, et al. Spinal meningiomas: review of 174 operated cases. Neurosurgery. 1989;25:153-160.
25 Levy WJJr. Bay J, Dohn D: Spinal cord meningioma. J Neurosurg. 1982;57:804-812.
26 Nittner K. Spinal meningiomas, neurinomas, and neurofibromas and hourglass tumors. In: Vinken P, Bruyn B, editors. Handbook of Clinical Neurology. New York: American Elsevier; 1976:177-322.
27 Caroli E, Acqui M, Roperto R, et al. Spinal en plaque meningiomas: a contemporary experience. Neurosurgery. 2004;55(6):1275-1279. discussion 1279
28 Levy WJ, Latchaw J, Hahn JF, et al. Spinal neurofibromas: a report of 66 cases and a comparison with meningiomas. Neurosurgery. 1986;18:331-334.
29 McClatchey AI. Neurofibromatosis. Annu Rev Pathol. 2007;2:191-216.
30 McCormick PC. Surgical management of dumbbell tumors of the cervical spine. Neurosurgery. 1996;38:294-300.
31 Seppala MT, Haltia MJ. Spinal malignant nerve-sheath tumor or cellular schwannoma? A striking difference in prognosis. J Neurosurg. 1993;79:528-532.
32 Celli P, Cervoni L, Cantore G. Ependymoma of the filum terminale: treatment and prognostic factors in a series of 28 cases. Acta Neurochir (Wien). 1993;124:99-103.
33 Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56:883-893.
34 Schweitzer JS, Batzdorf U. Ependymoma of the cauda equina region: diagnosis, treatment, and outcome in 15 patients. Neurosurgery. 1992;30(2):202-207.
35 Kumar K, Malik S, Schulte PA. Symptomatic spinal arachnoid cysts: report of two cases with review of the literature. Spine (Phila Pa 1976). 2003;28:E25-E29.
36 Song JK, Musleh W, Christie SD, Fessler RG. Cervical juxtafacet cysts: case report and literature review. Spine J. 2006;6(3):279-281.
37 Lyons MK, Atkinson JL, Wharen RE, et al. Surgical evaluation and management of lumbar synovial cysts: the Mayo Clinic experience. J Neurosurg. 2000;93(1 Suppl):53-57.
38 Boviatsis EJ, Staurinou LC, Kouyialis AT, et al. Spinal synovial cysts: pathogenesis, diagnosis and surgical treatment in a series of seven cases and literature review. Eur Spine J. 2008;17(6):831-837.
39 Manzano G, Green BA, Vanni S, Levi AD. Contemporary management of adult intramedullary spinal tumors-pathology and neurological outcomes related to surgical resection. Spinal Cord. 2008;46(8):540-546.
40 Birch BD, Johnson JP, Parsa A, et al. Frequent type 2 neurofibromatosis gene transcript mutations in sporadic intramedullary spinal cord ependymomas. Neurosurgery. 1996;39(1):135-140.
41 Ebert C, von Haken M, Meyer-Puttlitz B, et al. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol. 1999;155:627-632.
42 Lee M, Rezai AR, Freed D, Epstein FJ. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996;38:32-37.
43 Samii M, Klekamp J. Surgical results of 100 intramedullary tumors in relation to accompanying syringomyelia. Neurosurgery. 1994;35:865-873. discussion 873
44 Madsen PW, Green B, Bowen BC. Syringomyelia. 3rd ed. Herkowitz H, Garfin S, Balderston R, Eismont FJ, Bell G, Wiesel S, editors. The Spine, vol. II. Philadelphia, PA: WB Saunders. 1992.
45 Epstein FJ, Farmer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg. 1993;79:204-209.
46 McCormick PC, Torres R, Post KD, Stein BM. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
47 Lowe GM. Magnetic resonance imaging of intramedullary spinal cord tumors. J Neurooncol. 2000;47:195-210.
48 Mork SJ, Loken AC. Ependymoma: a follow-up study of 101 cases. Cancer. 1977;40:907-915.
49 Raco A, Esposito V, Lenzi J, et al. Long-term follow-up of intramedullary spinal cord tumors: a series of 202 cases. Neurosurgery. 2005;56:972-981. discussion 972-981
50 Nadeem SQ, Feun LG, Bruce-Gregorios JH, Green B. Post radiation sarcoma (malignant fibrous histiocytoma) of the cervical spine following ependymoma (a case report). J Neurooncol. 1991;11:263-268.
51 Reimer R, Onofrio BM. Astrocytomas of the spinal cord in children and adolescents. J Neurosurg. 1985;63:669-675.
52 Kim MS, Chung CK, Choe G, et al. Intramedullary spinal cord astrocytoma in adults: postoperative outcome. J Neurooncol. 2001;52:85-94.
53 Lee DK, Choe WJ, Chung CK, Kim HJ. Spinal cord hemangioblastoma: surgical strategy and clinical outcome. J Neurooncol. 2003;61:27-34.
54 Lonser RR, Weil RJ, Wanebo JE, et al. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:106-116.
55 Lee M, Rezai AR, Abbott R, et al. Intramedullary spinal cord lipomas. J Neurosurg. 1995;82:394-400.
56 Dyck P. Intramedullary lipoma. Diagnosis and treatment. Spine (Phila Pa 1976). 1992;17:979-981.
57 Halcrow SJ, Crawford PJ, Craft AW. Epidermoid spinal cord tumour after lumbar puncture. Arch Dis Child. 1985;60:978-979.
58 de Baecque C, Snyder DH, Suzuki K. Congenital intramedullary spinal dermoid cyst associated with an Arnold-Chiari malformation. Acta Neuropathol. 1977;38:239-242.
59 Cokca F, Meco O, Arasil E, Unlu A. An intramedullary dermoid cyst abscess due to Brucella abortus biotype 3 at T11-L2 spinal levels. Infection. 1994;22:359-360.
60 Lunardi P, Missori P, Gagliardi FM, Fortuna A. Long-term results of the surgical treatment of spinal dermoid and epidermoid tumors. Neurosurgery. 1989;25:860-864.
61 Deutsch H, Jallo GI, Faktorovich A, Epstein F. Spinal intramedullary cavernoma: clinical presentation and surgical outcome. J Neurosurg. 2000;93(1 Suppl):65-70.
62 Ojemann RG, Crowell RM, Ogilvy CS. Management of cranial and spinal cavernous angiomas (honored guest lecture). Clin Neurosurg. 1993;40:98-123.
63 Mut M, Schiff D, Shaffrey ME. Metastasis to nervous system: spinal epidural and intramedullary metastases. J Neurooncol. 2005;75:43-56.
64 Post MJ, Quencer RM, Green BA, et al. Intramedullary spinal cord metastases, mainly of nonneurogenic origin. AJR Am J Roentgenol. 1987;148:1015-1022.
65 Connolly ESJr, Winfree CJ, McCormick PC, et al. Intramedullary spinal cord metastasis: report of three cases and review of the literature. Surg Neurol. 1996;46:329-337. discussion 337-338
66 Steck JC, Dietze DD, Fessler RG. Posterolateral approach to intradural extramedullary thoracic tumors. J Neurosurg. 1994;81:202-205.
67 Quencer RM, Montalvo MB, Green BA, Eismont FJ. Intraoperative spinal sonography of soft tissue masses of the spinal cord and spinal canal. Yearbook of Critical Care Medicine. 1986.
68 Lot G, George B. Cervical neuromas with extradural components: surgical management in a series of 57 patients. Neurosurgery. 1997;41(4):813-820. discussion 820-822
69 McCormick PC. Anatomic principles of intradural spinal surgery. Clin Neurosurg. 1994;41:204-223.
70 Epstein FJ, Farmer JP, Schneider SJ. Intraoperative ultrasonography: an important surgical adjunct for intramedullary tumors. J Neurosurg. 1991;74:729-733.
71 Chang UK, Choe WJ, Chung SK, et al. Surgical outcome and prognostic factors of spinal intramedullary ependymomas in adults. J Neurooncol. 2002;57:133-139.
72 Ferrante L, Mastronardi L, Celli P, et al. Intramedullary spinal cord ependymomas—a study of 45 cases with long-term follow-up. Acta Neurochir (Wien). 1992;119(1-4):74-79.
73 Epstein FJ, Farmer JP, Freed D. Adult intramedullary astrocytomas of the spinal cord. J Neurosurg. 1992;77:355-359.
74 Minehan KJ, Shaw EG, Scheithauer BW, et al. Spinal cord astrocytoma: pathological and treatment considerations. J Neurosurg. 1995;83:590-595.
75 Marchan EM, Sekula RFJr, Jannetta PJ, Quigley MR. Long-term survival enhanced by cordectomy in a patient with a spinal glioblastoma multiforme and paraplegia. Case report. J Neurosurg Spine. 2007;7:656-659.
76 Whitaker SJ, Bessell EM, Ashley SE, et al. Postoperative radiotherapy in the management of spinal cord ependymoma. J Neurosurg. 1991;74:720-728.
77 Gerszten PC, Burton SA, Ozhasoglu C, et al. Radiosurgery for benign intradural spinal tumors. Neurosurgery. 2008;62:887-895. discussion 895-896
78 Dodd RL, Ryu MR, Kamnerdsupaphon P, et al. CyberKnife radiosurgery for benign intradural extramedullary spinal tumors. Neurosurgery. 2006;58:674-685. discussion 674-685
79 McCormick PC, Stein BM. Intramedullary tumors in adults. Neurosurg Clin N Am. 1990;1:609-630.
80 Lin YH, Huang CI, Wong TT, et al. Treatment of spinal cord ependymomas by surgery with or without postoperative radiotherapy. J Neurooncol. 2005;71:205-210.
81 Lee TT, Gromelski EB, Green BA. Surgical treatment of spinal ependymoma and post-operative radiotherapy. Acta Neurochir (Wien). 1998;140:309-313.
82 Jyothirmayi R, Madhavan J, Nair MK, Rajan B. Conservative surgery and radiotherapy in the treatment of spinal cord astrocytoma. J Neurooncol. 1997;33:205-211.
83 Vaillant B, Loghin M. Treatment of spinal cord tumors. Curr Treat Options Neurol. 2009;11:315-324.
84 Cristante L, Herrmann HD. Surgical management of intramedullary spinal cord tumors: functional outcome and sources of morbidity. Neurosurgery. 1994;35:69-74. discussion 74-76
85 Shirato H, Kamada T, Hida K, et al. The role of radiotherapy in the management of spinal cord glioma. Int J Radiat Oncol Biol Phys. 1995;33:323-328.
86 Moss JM, Choi CY, Adler JRJr, et al. Stereotactic radiosurgical treatment of cranial and spinal hemangioblastomas. Neurosurgery. 2009;65:79-85. discussion 85
87 Ryu SI, Kim DH, Chang SD. Stereotactic radiosurgery for hemangiomas and ependymomas of the spinal cord. Neurosurg Focus. 2003;15:E10.
88 Schick U, Marquardt G, Lorenz R. Recurrence of benign spinal neoplasms. Neurosurg Rev. 2001;24:20-25.
89 Klekamp J, Samii M. Surgery of spinal nerve sheath tumors with special reference to neurofibromatosis. Neurosurgery. 1998;42:279-289. discussion 289-290
90 Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal neurofibroma. J Neurosurg. 1995;82:572-577.
91 Innocenzi G, Salvati M, Cervoni L, et al. Prognostic factors in intramedullary astrocytomas. Clin Neurol Neurosurg. 1997;99:1-5.
92 Nakamura M, Ishii K, Watanabe K, et al. Surgical treatment of intramedullary spinal cord tumors: prognosis and complications. Spinal Cord. 2008;46:282-286.
93 Rawlings CE3rd, Giangaspero F, Burger PC, Bullard DE. Ependymomas: a clinicopathologic study. Surg Neurol. 1988;29:271-281.
94 Lonjon M, Goh KY, Epstein FJ. Intramedullary spinal cord ependymomas in children: treatment, results and follow-up. Pediatr Neurosurg. 1998;29:178-183.
95 Santi M, Mena H, Wong K, et al. Spinal cord malignant astrocytomas. Clinicopathologic features in 36 cases. Cancer. 2003;98:554-561.
96 Ciappetta P, Salvati M, Capoccia G, et al. Spinal glioblastomas: report of seven cases and review of the literature. Neurosurgery. 1991;28:302-306.
97 Cohen AR, Wisoff JH, Allen JC, Epstein F. Malignant astrocytomas of the spinal cord. J Neurosurg. 1989;70:50-54.
98 Van Velthoven V, Reinacher PC, Klisch J, et al. Treatment of intramedullary hemangioblastomas, with special attention to von Hippel-Lindau disease. Neurosurgery. 2003;53:1306-1313. discussion 1313-1314
99 Murota T, Symon L. Surgical management of hemangioblastoma of the spinal cord: a report of 18 cases. Neurosurgery. 1989;25:699-707. discussion 708
100 Pietila TA, Stendel R, Schilling A, et al. Surgical treatment of spinal hemangioblastomas. Acta Neurochir (Wien). 2000;142:879-886.