[level-membership-for-surgery-category]Chapter 6
Intimal Hyperplasia
Zhihua Jiang, C. Keith Ozaki
Based on a chapter in the seventh edition by Mark G. Davies
Intimal hyperplasia occurs frequently after vascular interventions such as vein bypass grafts, endarterectomies, arteriovenous fistulas, prosthetic bypass grafts, balloon angioplasty, endovascular stents and stent grafts, and solid organ transplantation. Data now suggest that restenosis develops in 5.8% of patients undergoing carotid endarterectomy or receiving carotid stents, though these cases rarely require re-intervention.1 About 30% to 60% of lower extremity arterialized vein grafts fail or are failing within a year,2,3 and intervention is frequently necessary to maintain conduit patency. Finally, 60% of hemodialysis access fistulas fail to mature primarily,4 and obliterative processes near the anastomosis are associated with half of these failures.5,6 Similarly, the durability of prosthetic dialysis grafts remains poor owing to intimal hyperplasia at the outflow vein.6,7 Although molecularly this process begins immediately after the vascular injury that accompanies the procedure, detectable intimal lesions classically form within a few weeks to 2 years.8
Definitions
Intimal Hyperplasia
The blood vessel intima is normally composed of just the thin endothelial cell lining of the vasculature. Intimal hyperplasia reflects additional biomass (cellular and matrix) within this layer. Mammals appear to have evolved intimal hyperplasic processes so that they become survival benefits. For example, hyperplastic intimal growth underlies closure of the ductus arteriosus. Vascular healing and premenopausal occlusion of the uterine arteries are also, in essence, the process of intimal hyperplasia.
Historically, the development of intimal hyperplasia was initially considered a “process of connective tissue hyperplasia.” Later, the concept was revised to “fibroproliferative” intimal thickening.9 Studies in the 1980s identified medial smooth muscle cells as central effectors of this process. Following biochemical insult and/or mechanical trauma, medial smooth muscle cells are activated and undergo a phenotypic switch from a contractile/quiescent to a synthetic/proliferative state. Proteases, particularly matrix metalloproteinases (MMPs), are released by inflammatory infiltrating leukocytes to break the matrix network. Smooth muscle cells are liberated from matrix restriction, and they then migrate from the tunica media to the intimal region, where they proliferate and deposit matrix proteins. Although sharing the same lineage identity with those in the media, smooth muscle cells in the intimal tissue are functionally incapable of organizing the cellular and matrix components. The de novo intimal layer is thus often qualitatively a relatively disordered histologic structure. This grossly fibrous, white material accumulates over time, protruding into the lumen and leading to luminal narrowing and even complete occlusion (Fig. 6-1).
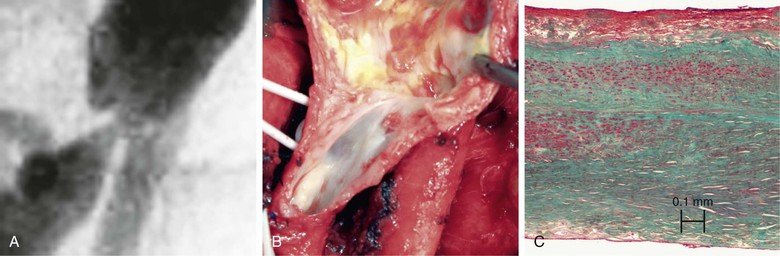
Figure 6-1 A, Digital subtraction angiogram of the distal right limb of an aortofemoral graft showing a filling defect at the junction of the prosthetic graft material and the outflow of the profunda femoris artery. B, Intraoperative photograph of the same field. Note the grossly fibrous, white intimal hyperplasia that corresponds to the outflow lesion. C, Masson trichrome stain preparation of endarterectomized intimal hyperplasia. Green staining corresponds to collagen, whereas red localizes primarily to other matrix components and the cytoplasm of largely smooth muscle cells.
It is currently believed that intimal hyperplasia starts as an acute inflammatory response. Because of the incomplete functional recovery of the repopulated endothelial cells and the phenotypic switch of smooth muscle cells, the acute inflammation often does not completely resolve, leaving behind a chronic inflammation in the intimal tissue. Inflammatory cells, particularly macrophages and T cells, are found in established intimal lesions. In contrast, atherosclerosis is a vascular disease driven by inflammatory mechanisms.10 The inflamed intimal tissue thus can serve as the “soil” for subsequent atherosclerosis to develop.11 Although the early failure of vascular reconstructions is largely caused by intimal hyperplasia, atherosclerosis is the dominant process underlying late failure (>3 years). After coronary artery stenting, atherosclerotic lesion(s) account for more than 70% of cases of recurrence of ischemic symptoms.12
Other Pathologies Underlying Lumen Loss after Vascular Procedures
Wall Remodeling
In addition to the intimal thickening, blood vessels can permanently change the dimensions of the media and adventitia. This change is termed wall remodeling.13 An early description of vascular remodeling comes from Glagov et al14 in 1987. They observed that coronary arteries actively grow to enlarge their cross-sectional area to accommodate the encroachment of atherosclerotic lesions. As the result of this compensatory expansion, appropriate luminal dimensions may be effectively maintained for the diseased vessels. This observation changed the concept of the vessels from inanimate pipes to an active system. It is now well established that the remodeling process involves reorganization/construction of both the medial and adventitial layers. Although leading to an increase in the overall dimension that favors preserving luminal area, remodeling may also proceed in the opposite direction, promoting wall shrinkage and loss of the luminal area.
Several terms have been used to describe remodeling processes. Morphologic terms, such as inward/constrictive remodeling and outward/expansive remodeling, are used to describe the impact on the geometric vascular dimension. Functional terms, such as positive remodeling and negative remodeling, were introduced to emphasize the effect of the remodeling process on the performance of the vessel as a conduit. A challenge in clinical studies is the inability to evaluate the full geometric dimension of the vessel over time because of the inadequate discrimination of the outer vessel wall from the perivascular tissue with imaging approaches such as angiography and computed tomography (CT) scans. This issue has been partially overcome by the availability of the intravascular ultrasound. Wall detail may be described in vivo as long as it is accessible by intravenous ultrasound probes.
Hemodynamic forces, particularly shear stress and wall tension, appear to be primary modulators of the vascular remodeling process. An elevated shear, for example, generally propels an expansion or outward growth, whereas a reduction in shear promotes “shrinkage” or inward growth of the vessel.15 Compared with that of shear forces, the impact of tensile forces on vascular remodeling is less well understood. Generally, wall tension is a factor that favors a negative remodeling process.16 When coupled with intimal hyperplasia, wall remodeling can ameliorate or exacerbate luminal narrowing through its effect on vessel dimension.
Vascular Tone and Recoil
Muscular vessels (arteries and veins) receive autonomic innervation from the central nervous system. Under physiologic conditions, a basal level of activity is transmitted via the sympathetic fibers to maintain a partial state of contraction in blood vessels. This basal level of contractile tension is referred to as vascular tone. Local hemodynamic forces and activation of vascular innervation may finely tune the vascular tone by way of promoting smooth muscle cell contraction or relaxation. Although transient fluctuation in vascular tone is reversible, a persistent increase in vascular tone may lead to structural changes such as the pathology occurring in patients suffering from long-standing arterial hypertension.
In addition to the active adjustment of the vascular caliber by smooth muscle cells, passive contraction of the vascular wall matrix network, consisting of predominantly collagen and elastic fibers, is also a critical determinant of the vessel diameter. Under physiologic conditions, the elastic resilience works to smooth out the pulsatility generated by cardiac ejection. Following the systolic distention, vessels return to their original dimensions as the result of elastic contraction. This phenomenon is termed vascular recoil. In the vessel wall, the intact and well-organized structural components as well as the blood counterbalance the wall shrinkage caused by recoil. In cases in which the wall integrity is destroyed, such as balloon angioplasty, recoil may cause 50% of the loss in acute gain during angioplasty.17,18 To oppose this recoil effect, metal stents were first used in peripheral arteries in 1985,19 and coronary arteries in 1986.20 In comparison with balloon angioplasty alone, the additional placement of stents can offer significant geometric benefits, as reflected by more preservation of the acute gain in lumen diameter and, in selected patients, less frequent requirement for secondary interventions and better quality of life.21
Restenosis
Several months after interventions such as endarterectomy, angioplasty, and bypass, the vascular constructions may become significantly narrowed or may occlude. This recurrence of stenosis and reduction in luminal area is termed restenosis. The causes for restenosis are complex.22 Usually, the primary pathology detected in a vessel with restenosis is intimal hyperplasia, and the loss of luminal area is proportional to the encroachment of the intimal tissue. Geometric remodeling likely also contributes.23 For example, the lumen area loss may be partially or completely compensated if a simultaneous outward remodeling had led to an increase in the geometric dimension. Conversely, significant restenosis may occur because of additional lumen loss caused by inward remodeling even if the amount of intimal hyperplasia is relatively small. Metal stents offer a strategy to limit inward remodeling, although intimal hyperplasia (either through the stent structure or at the ends of a stent graft) often offset these luminal area gains. Under specific circumstances, an improved long-term patency of the stented vessels has been observed.21
Inciting/Modulating Factors for Intimal Hyperplasia
Intimal hyperplasia is a vascular response that heals the injured vessel wall. It can occur in nearly every type of vascular reconstruction, and the initiation and progression of the lesion may be modulated by several factors.
Hemodynamic Stress
Hemodynamic forces, specifically shear stress and wall tensile stress, are well-established initiators and modulators of intimal hyperplasia.24 Vascular cells are equipped to sense and respond to the luminal hemodynamic environment. Several surface receptors (e.g., integrins and vascular endothelial growth factor receptor [VEGFR]), ion channels (e.g., K+ and Ca++), and the cell cytoskeleton may be specialized in mechanosensing and mechanotransduction.25
Under physiologic conditions, the steady laminar blood flow generates on average approximately 15 dyne/cm2 of shear stress in arteries. Endothelial cells sense this physiologic shear force, releasing mediators such as nitric oxide (NO) and Kruppel-like factor-2 to maintain a quiescent state for smooth muscle cells and homeostasis of the whole vessel wall.26 Vascular reconstructions such as vein bypass grafts, stented diseased arteries, and arteriovenous fistulas not only alter the rate of the local blood flow but also frequently induce a disordered flow pattern. Both clinical and experimental observations have demonstrated that disturbed flow and/or low wall shear stress accelerate development of intimal hyperplasia.15,27,28 Endothelial cells respond to these particular hemodynamic conditions by elaborating adhesion molecules, proinflammatory cytokines, and other bioactive substances that in turn enhance cell proliferation and matrix accumulation, leading to robust intimal growth.29,30 On the other hand, laminar, high blood flow (uniform high shear stress) generally exhibits an opposing effect on intimal growth. For example, vein grafts exposed to high flow conditions tend to develop less intima than those with low blood flow.15 Furthermore, augmentation of blood flow in vessels with established intimal hyperplasia induces intimal regression.31 Although the exact mechanisms remain to be fully elucidated, experimental studies suggest that high laminar shear shifts the local vascular milieus from a proinflammatory to an antiinflammatory state, imposing a differential regulation of intimal growth.26 This knowledge has been translated to clinical application, so that creation of a distal fistula to boost the blood flow has led to improvement in the patency rate of lower extremity grafts in selected circumstances.32 Although generally protective, shear may be deleterious when it reaches an extremely high level or becomes disordered.33 Finally, compared with the well-established effect of shear stress, the impact of tensile force on intimal hyperplasia is much less defined. In the cell culture setting, mechanical stretch activates several pathways capable of regulating smooth muscle cell phenotype.34,35 Evidence in vivo suggests that tensile force correlates positively with intimal thickening.16,36
Immune Insults
Solid organ transplantation usually introduces a mismatch in the HLA complex between the transplant and the host immune system, triggering immunologic responses that accelerate formation of diffuse, concentric intimal hyperplastic lesions in the arteries of the allograft that may eventually result in allograft failure. Reflective of this complex immune scenario, allograft vasculopathy is more vulnerable to in-stent restenosis after intervention than the stenotic lesions in native coronary arteries.37 Multiple cell groups, particularly T-cell subsets (CD4+ and CD8+) and B cells, appear to be critical to the pathogenesis of these intimal hyperplastic lesions in the transplant setting. Activated by antigen recognition through either direct (major histocompatibility complex I [MHC I]–dependent) or indirect (MHC II–dependent) pathways, these cells may cause endothelial cell dysfunction via cytolysis, cytokine stimulation (interferon-γ [IFN-γ], interleukin-12 [IL-12]), and specific antibody production.38 Beyond the transplant setting, similar immunologic insult may occur in stented vessels, where immune cells are activated via a response to foreign bodies.39 Finally, risk factors for accelerated intimal hyperplasia identified in clinical studies (hyperlipidemia, diabetes, and smoking) may act by way of augmenting chronic inflammation in the vessel wall.
Genetic Susceptibility
In addition to environmental factors, genetic status stands as a risk factor for intimal hyperplasia. Some of this knowledge was acquired via genome-wide association studies (GWASs). For example, single-nucleotide polymorphisms (SNPs) in the IL-10 gene are associated with higher rate of restenosis in coronary arteries treated with drug-eluting stents.40 Susceptible loci in chromosome 12 have also been documented.41 These genetic discoveries have highlighted the horizon for genetic manipulations as a therapy for inhibiting intimal growth.
Biologic Mechanisms
Intimal hyperplasia is a highly complex process that involves several tissues (perivascular,42 vessel wall, and blood), numerous cell lineages, and multiple molecular signaling networks. Although much has been learned in the past few decades, many mechanistic details remain to be fully elucidated. The current paradigm is founded on the postulate that intimal hyperplasia is a vascular response to injury, with the cellular and molecular events generally mirroring those occurring in the course of wound healing.8,43
Cellular Effectors
Compared with the medial layer, the neointimal lesion contains relatively more extracellular matrix, with a disordered cell-matrix organization that mimics tissue fibrosis immediately beneath the intact endothelial cell lining. However, unlike reactive fibrotic tissue, which is primarily populated with fibroblasts, vascular intimal lesions are dominated by cells that express smooth muscle cell markers, such as α-actin and smooth muscle myosin heavy chain (SM-MHC). Work in the 1980s suggested that these cells are largely medially derived.44 However, in addition to derivations from the tunica media, later studies have identified cells from nonmedial sites, such as the adventitia and perivascular tissue,45 stem cell/progenitor cell niches,46,47 and the circulating blood as suppliers of the intimal cell population.
Once recruited to the intimal lesion, these cells gain a phenotype similar to those derived from the medial smooth muscle cells, making them indistinguishable from each other.48,49 Although vascular wall cells are normally relatively quiescent, the repopulated intimal cells often display a proliferative and synthetic state. Substances that are not produced basally in the vasculature (e.g., cytokines, growth factors, and adhesion molecules) are induced in these neointimal cells.50 Acting through autocrine and paracrine mechanisms, these substances maintain an inflamed state for the intimal tissue, collectively propelling progressive growth of the intimal lesion.
Endothelial Cells
The luminal surface of blood vessels is covered by a monolayer of endothelial cells. Under physiologic conditions, these cells serve as a “physical barrier” that segregates the circulating blood cells and molecules from tissue matrix components, and also function as a “secretory organ,” releasing an array of mediators to maintain homeostasis in the wall and beyond. Lining the luminal surface, endothelial cells produce transmembrane adhesion molecules such as vascular endothelial cadherin (VE-cadherin) and neural cadherin (N-cadherin) for intercellular connections. Interacting with the intracellular cytoskeleton, these molecules form adhesive junctions (e.g., tight junctions, gap junctions, and adherens junctions) with neighboring cells, allowing selective exchange of molecules between blood and tissue.51 Anticoagulants (e.g., heparin, thrombomodulin, prostaglandin I2 [PGI2], and Kruppel-like factor-2) are synthesized and released by these cells to maintain a relatively anticoagulant and thus thrombus-free luminal surface.
In addition to these “direct” biologic effects, endothelial cells govern the homeostasis of the vessel wall via communication with the underlying smooth muscle cells. A well-defined tool for endothelial cells to carry out this function is NO, which has been linked with intimal hyperplasia.52 NO is very lipophilic. Once released, it quickly diffuses into the medial layer and acts directly on smooth muscle cells. The half-life of the endogenous NO is so short (0.1-5 seconds) that the effect on smooth muscle cells can be rapidly turned on or off depending on demand. NO was initially identified as endothelium-derived relaxing factor because of its function to induce vasodilatation. Subsequent studies demonstrated that NO is also a key messenger for endothelial cells to keep the medial smooth muscle cells at a quiescent and contractile phenotype.53
Upon activation by forces such as altered hemodynamics or physical/chemical trauma, endothelial cells convert their transcriptome to a profile that is significantly different from that of their quiescent state.54 Leukocyte and platelet adhesion molecules are upregulated or translocate to the cell membrane. Genes encoding proinflammatory mediators (e.g., chemokines, cytokines, adhesion molecules) are induced or upregulated, whereas those encoding antiinflammatory mediators (e.g., NO, anticoagulants, Kruppel-like factor-2) are downregulated.55 These bioactive substances can trigger a series of events, such as smooth muscle cell activation, inflammatory cell infiltration, and the recruitment of vascular progenitor cells—all contributing to intimal hyperplasia. Finally, studies have now suggested a new mechanism whereby endothelial cells may contribute to intimal hyperplasia through endothelial-mesenchymal transition (EMT).56,57
Smooth Muscle Cells
The intimal hyperplasia tissue, just like the tunica media (though morphologically distinct), is dominated primarily by smooth muscle cells. During the development of intimal hyperplasia, intimal smooth muscle cells display a spectrum of phenotypes with preferential losses and gains of their differentiation markers at various stages. The initial dedifferentiation process leads to a loss of expression of the majority of the differentiation markers, such as α-actin, desmin, and smooth muscle isoforms SM1 and SM2. As the lesion becomes more established, early differentiation markers such as α-actin and desmin are gained relatively quickly. The reproduction of the more mature markers, such as smooth muscle isoforms SM1 and SM2, on the other hand, may be very much delayed.58,59
Although they normally function in the media primarily as the drivers for vascular relaxation and constriction, smooth muscle cells in the intimal lesions serve as effectors that produce cellular and extracellular components to expand the intimal mass. Having switched to the proliferative/synthetic state, smooth muscle cells respond to local cytokines and growth factors by vigorous cell division and matrix production. Experimental studies have demonstrated that the proliferation may start a few hours after an inciting event, reach a peak in a few days, and last for weeks to months.15,60 Genes encoding matrix components (particularly interstitial matrix proteins such as hyaluronic acid, chondroitin, collagens, proteoglycans, fibronectin, and elastin) are greatly upregulated. On the other hand, the production and the activity of the matrix degradation system, which is consisted of proteases (e.g., MMPs, tissue plasminogen [t-PA], urinary plasminogen activator [u-PA], and plasminogen) and their counterpart inhibitors (e.g., tissue inhibitors of metalloproteinases [TIMPs] and plasminogen activator inhibitor-1 [PAI-1]), is modulated to favor a selective deposition of matrix proteins,8,61 leading to an accumulation of de novo interstitial matrix with an altered composition.
In addition to their role of directly making intimal mass, smooth muscle cells synthesize and release a wide spectrum of mediators to the extracellular space.50 Cytokines (e.g., IL-1β and tumor necrosis factor-α [TNF-α]), chemokines (e.g., IL-8 and monocyte chemoattractant proteins [MCPs]), and growth factors (e.g., platelet-derived growth factor [PDGF] and transforming growth factor-β [TGF-β]) have been documented as intimal smooth muscle cell products. The exact profile of these mediators varies according to the stage of intimal hyperplasia development. These mediators may act on smooth muscle cells themselves and other cell lineages through autocrine and/or paracrine function. Although the early mediators serve to amplify the inflammatory response and favor smooth muscle cell migration and proliferation, driving a hyperplastic response with high cellularity,62,63 those released by smooth muscle cells in the established neointima facilitate a fibrotic process.60
Finally, vascular smooth muscle cells may lose their production of contractile proteins through dedifferentiation and acquire a phenotype that overlaps with another vascular cell group, the myofibroblasts. Studies of both systemic hypertension and pulmonary hypertension have demonstrated a critical role for the adventitia and adventitial fibroblasts in the structural remodeling and the resultant pathology of the vessel wall,64,65 and these cells may participate in intimal hyperplasia.66–68 Like medial and intimal smooth muscle cells, myofibroblasts likely stand as cellular effectors that produce and react to various bioactive substances, modulating structural remodeling and intimal growth in the vessel wall.69
Platelets and Leukocytes
Following vascular injury, platelets aggregate on activated endothelial cells and denuded areas to cover the collagens and other matrix components that are exposed to the blood. Fibrin is deposited, together with the activated platelets, forming an adhesive surface to capture circulating leukocytes from the blood stream.70–72 Inflammatory cells such as monocytes, eosinophils, and T cells home to the injured site. Although representing only a relatively small portion in the eventual intimal cell population, inflammatory cells have been identified as central modulators and potential key orchestrators of hyperplastic intimal thickening. For example, clinical studies have demonstrated a positive correlation between leukocyte count and activation with the severity of stenotic and restenotic coronary lesions.73,74 The level of inflammatory markers typically associated with leukocyte activity, particularly C-reactive protein, holds some correlation with clinical outcomes.75,76 Strategies blocking leukocyte adhesion or creating leukopenia significantly inhibit intimal thickening in various experimental settings.72,77 Through their robust ability to produce various inflammatory mediators, including chemokines, cytokines, and growth factors, leukocytes appear to amplify the acute inflammation, contributing to intimal thickening.78
Leukocytes consist of several subpopulations. The quantity of each subset in intimal lesions seems to be determined by the involvement of specific initiating factors and the stage of the pathology. For example, lesions that developed in the setting of hypercholesterolemia or after severe physical injury tend to hold more macrophages than those triggered solely by endothelial denudation.72 Otherwise, neutrophils dominate the inflammatory infiltrates after moderate injury such as endothelial denudation.79 There is also evidence that T cells stimulate intimal hyperplasia via promoting a helper T cell type 1 (Th1) response,80 but this mechanism may be more significant in the pathogenesis of allograft vasculopathy.81 Advances in immunology have led to appreciation of distinct roles for subsets of monocytes and macrophages in the vascular inflammatory response. “Patrolling” and “resident” monocytes have been identified in the peripheral blood.82,83 Upon extravasation into tissue, these cells follow varying pathways to differentiate into M1 and M2 macrophages, performing distinct roles in pathogenesis and tissue repair.84,85 For example, M2 polarization of the resident monocytes has been demonstrated to be indispensable to arterial remodeling triggered by hemodynamic stress.86 Recent research efforts have attempted to identify the key regulators for these processes so that interventional approaches may be developed to guide the kinetics of the inflammatory cell trafficking and intimal lesion progression.87
Progenitor Cells
In addition to smooth muscle cells, several other cell groups are recruited to neointimal lesions. Vascular progenitor cell lines released from bone marrow and other stem cell niches may home to intimal hyperplasia.46,49 Initial evidence suggests that smooth muscle progenitors are released from stem cell niches to the circulation.88 Not only can these cells gain smooth muscle cell markers but they also seemed at first to respond to exogenous stimulation much like smooth muscle cells from tunica media do.89 However, subsequent studies have demonstrated that cells from circulation and bone marrow lack the ability to produce a full repertoire of smooth muscle cell contractile proteins in the intimal lesion90 and that they tend to maintain a sustained inflammatory phenotype.91
A groundbreaking discovery has been the participation of endothelial progenitor cells in reendothelialization of the injured luminal surface.92 Experimental studies revealed that approaches facilitating the homing of these progenitor cells to the lesion site significantly accelerated the recovery of the endothelial cell monolayer and inhibited neointimal hyperplasia.93 Clinical observations support an association of the impaired endothelial progenitor cell function with higher rate of stenosis and restenosis.94 Because the endothelial progenitor cell is a relatively rare element in circulating blood, strategies such as capturing antibodies and adhesion molecules or growth factors to enrich endothelial progenitor cells at lesion sites are being evaluated.95,96
Biochemical Mediators
Studies of vascular wall gene expression have described induction and suppression of numerous genes over the course of the development and progression of intimal hyperplasia. A large number of molecular mediators have been implicated in experimental studies as promising therapeutic targets, and only a selected few are discussed here. Efforts to translate the genomic knowledge to patients have rarely improved the clinical outcome.2 Compelling evidence obtained from both experimental and clinical studies has demonstrated that intimal hyperplasia is a highly complex process.54 It is driven by many networks of molecules functioning with temporal and spatial variations over the course of lesion development, and most of these mediators have significant functional redundancy. Targeting a single molecule with therapeutic interventions has been demonstrated to be unlikely to alter the biologic consequence of such a complex system,2,3,30 although risk factors such as diabetes, smoking, and genetic variations are capable of modulating this system and accelerating intimal hyperplasia development. Further complexity is manifested through the specifics among intimal hyperplasia occurring in response to various vascular events (in-stent restenosis, vein bypass failure, postendarterectomy stenosis, etc.). Each likely holds unique considerations for preventive and therapeutic strategies.
Inflammatory Mediators
Injury to the vessel wall results in aggregation of platelets and activation of vascular cells (e.g., endothelium, smooth muscle cells, and fibroblasts). Once activated, these cells are robust manufactures of bioactive substances, triggering a complex mediator cascade that amplifies the vascular damage and the inflammatory response. P-selectin, which is normally stored in the a-granules of platelets and Weibel-Palade bodies of endothelial cells, is released.97 E-selectin is transcribed, translated, and transported to the surfaces of endothelial cells to facilitate leukocyte rolling as part of the homing process.98,99 Chemokines—monocyte chemoattractant protein-1, RANTES (regulated on activation, normal T cell–expressed and secreted), IL-8, interferon-γ–induced protein [IP-10], and stromal cell–derived factor-1 [SDF-1]—are mobilized to create a concentration gradient to guide leukocyte migration. Other adhesion molecules, such as intracellular adhesion molecules (ICAMs), vascular cell adhesion molecule (VCAM), and integrins, are released, mediating firm leukocyte adhesion. Inflammatory cytokines (e.g., tumor necrosis factor-α and IL-1β) that are normally absent in the vessel wall are produced by activated leukocytes, predominantly monocytes. These cytokines serve as a driving factor for intimal hyperplasia via their mitogenic functions on smooth muscle cells100 and proapoptotic properties for endothelial cells.100–102 In amplification of this traditional dogma, studies have now identified danger-signaling networks as a mechanism for cell activation and the initiation of the inflammatory response. Toll-like receptors (e.g., TLR- 2 and TLR-4) recognize the degraded tissue components, initiating the innate immune response.42 Strategies manipulating the activity of the danger signaling have succeeded in altering the eventual vascular morphology.103–105
In addition to the proinflammatory cytokines, antiinflammatory cytokines such as IL-10 and IL-19 are produced in lesion sites, and these cytokines appear to be protective against lesion progression. In vein grafts, for example, enhanced IL-10 expression is associated with less lesion formation.30,106 Pharmaceutical treatment with recombinant IL-10 has been found to inhibit intimal hyperplasia formed in injured arteries.107 Similar protective effects were also observed for IL-19.108 As part of the regulatory network in the inflammatory response, these counterbalancing entities are primarily produced by the same cell population as the proinflammatory cytokines, specifically macrophages. Through targeting STAT (signal transducer and activator of transcription) and nuclear factor-kappaB (NF-κB) signaling pathways, these molecules inhibit smooth muscle cell proliferation and further induction of other inflammatory mediators, suppressing the hyperplastic vascular response and, thus, intimal growth.107–109
Although this acute inflammation frequently comes to complete resolution in other tissues, it can transition to a chronic state in the vessel wall, perhaps driven by continued hemodynamic or foreign body insult. Studies of inflammatory events occurring in nonvascular tissues support that the stromal microenvironment defined by cytokines (e.g., IL-1β and IL-6) and chemokines (e.g., stromal cell–derived factor-1 and monocyte chemoattractant protein-1) dictate the inflammatory response of complete resolution or chronic inflammation.110 Lipid mediators such as the resolvins and maresins can also downregulate inflammatory systems.111–113 Conversely, in the vascular wall an inflammatory cascade model supports an IL-1β–based system whereby the inflammation is sustained via a self-stimulatory mechanism.114
Growth and Migration Factors
Several growth factors, such as PDGF, basic fibroblastic growth factor, insulin-like growth factor (IGF), vascular endothelial growth factor (VEGF), and TGF-β, are elaborated by factors inciting development of intimal hyperplasia. These molecules propagate their signal exclusively via binding to receptors with tyrosine kinase activity, with their functional specificity being dictated via coupling with various kinases, including the Ras/MAPK, PI3K, Smads, and NF-κB pathways.8
PDGF, basic fibroblastic growth factor, and IGF are potent stimulators for smooth muscle cell proliferation and migration.43,115 In the injured vessel wall, these growth factors are rapidly released from platelet aggregates, inflammatory infiltrates, and damaged vascular cells. Although functioning as chemoattractants and mitogens to medial smooth muscle cells, PDGF, basic fibroblastic growth factor, and IGF may also act on smooth muscle cells and monocytes to enhance the production of metalloproteases such as MMP-2116 and MMP-9,117 triggering first-wave migration and proliferation.61 Using gain- and loss-of-function approaches, several groups have demonstrated a stimulatory role for these growth factors in intimal hyperplasia.43,118,119
Although exhibiting significant functional redundancy, tyrosine kinase growth factors may hold robust specificity in their biologic effects. For example, IGF is relatively more potent in driving cell cycle progression through S phase and subsequent cell division than other smooth muscle mitogens.115 PDGF appears to be the most potent factor that induces dedifferentiation (e.g., phenotypic switch) of the medial smooth muscle cells. Upon activation, the PDGF signaling pathway inhibits expression and production of contractile proteins in smooth muscle cells by interfering with the binding of the transcription factor myocardin to the specific promoter region termed CArG box.120,121
VEGF is an endothelial cell–specific growth factor. Binding to flt-1 or flk-1 receptors, VEGF promotes maturation of endothelial progenitor cells and stimulates proliferation and migration of mature endothelial cells. In case of endothelial cell denudation, it facilitates the repopulation and repair of the endothelial monolayer. Because of these biologic effects, the use of VEGF was evaluated as a strategy to limit the risk of restenosis following angioplasty.122 A series of proof-of-concept studies were undertaken using various experimental models,123–125 although the potential of this approach has not been fully delineated to date.126
Unlike the growth factors described previously, TGF-β appears to be a growth inhibitor for smooth muscle cells. Following stimulation with TGF-β1, cell cycle regulators p21 and p27 are induced, inhibiting cell cycle progression.127,128 In addition, TGF-β may counterbalance PDGF, shifting the phenotypic spectrum of the intimal smooth muscle cells toward the contractile state by promoting expression of contractile proteins such as α-actin, calponin, and smooth muscle myosin heavy chain (SM-MHC).129 Although these functions point toward TGF-β as an important regulator of intimal hyperplasia, its role in accelerating intimal thickening is achieved likely via its regulation of matrix metabolism. For example, it enhances the synthesis of several matrix proteins, such as collagens and elastin. Additionally, proteases that degrade matrix proteins (e.g., plasminogen and MMPs) are inhibited by TGF-β via induction of PAI-1 and TIMPs (see later for details).130,131 Direct evidence supporting this concept is derived from in vivo experiments demonstrating that blocking TGF-β signaling pathways significantly reduced matrix accumulation and thus intimal thickening in both injured arteries132 and vein bypass grafts.133 Although attempts to inhibit the broad TGF-β activities did not bring significant clinical benefit,134 blocking TGF-β signaling in a more selective fashion holds some promise. A chemical compound that specifically inhibits the type I TGF-β receptor Alk5, SM16, has been evaluated. SM16 was found to suppress the fibrotic intimal thickening by 70% in injured common carotid arteries,135 although as with many reports of such findings, no clinical trials have come to fruition.
Matrix Metabolism
A key step in the onset of intimal hyperplasia is the proliferation and migration of smooth muscle cells from the tunica media to the intimal layer. In the intact vessel wall, however, smooth muscle cells are embedded in matrix networks. Liberation of these cells free from the “matrix brakes” is thus indispensable for the subsequent hyperplastic intimal thickening. Supporting this concept, several studies have documented the early upregulation of various proteases that are capable of breaking up the matrix networks.136–140 Of them, matrix metalloproteinases seem to play a central role in this process.141
MMPs are a family of enzymes with significant overlap in their spectrum of substrates. The 23 MMPs are grossly divided into four groups: (1) collagenases (MMP-1, MMP-8, MMP-13, MMP-14); (2) gelatinases (MMP-2, MMP-9); (3) stromelysins (MMP-3, MMP-7, MMP-10, MMP-11); and (4) membranetype MMPs (MT-MMP-1 to MT-MMP-4). MMP-12 is excluded from these groups because it is genetically only distantly related to the other MMPs. Under physiologic conditions, the activity of these MMPs is finely regulated by natural inhibitors, the tissue inhibitor metalloproteinases. TIMP-1 to TIMP-4 are produced in the vessel wall.142 Although both MMPs and TIMPs act redundantly with their family members, specificity may exist in intimal hyperplasia. In experimental studies, overexpression of TIMP-3143 or genetic deletion of MMP-2144 or MMP-9145 attenuated intimal growth. Translation of this experimental success to patient use is being examined in clinical trials.141
In addition to MMPs, t-PA and u-PA are released from the injured vascular cells and the inflammatory infiltrates. These proteases convert plasminogen to plasmin, which in turn degrades fibrin deposited as the result of coagulation. Fibrin is a potent mitogen and chemoattractant for smooth muscle cells.146 This process is normally tightly controlled by PAI-1. In the injured vessel, however, fibrin accumulates locally because of activation of the coagulation cascade and upregulation of PAI-1. In line with these observations, in vivo experiments have demonstrated that genetic deletion of PAI-1 attenuates intimal formation147 and overexpression of PAI-1 exacerbates intimal hyperlasia148 after arterial injury. Lines of evidence point toward inhibition of PAI-1 as a promising therapeutic strategy, but manipulation of PAI-1 activity in vivo is very complex, leading to conflicting results depending on the vascular setting.149
In advanced lesions, up to 80% of the intimal tissue is made of matrix proteins.150 The accumulation of the matrix in intimal lesions is a dynamic process that involves both synthesis and degradation. Although de novo matrix proteins are synthesized, the matrix network formed initially is broken up through protease-mediated matrix degradation. This dynamic matrix metabolism is termed matrix turnover. Matrix turnover can be a physiologic event in normal vessels at a relatively low rate to maintain the integrity of vasculature. During intimal hyperplasia, genes encoding various matrix proteins are upregulated (e.g., TGF-β, angiotensin II [ATII]) and massive amounts of matrix are deposited in the extracellular space. Simultaneously, genes encoding proteases (e.g., MMPs, t-PA, u-PA) that degrade various matrix proteins are also upregulated. As a result, matrix metabolism is shifted toward synthesis, leading to not only an increase in matrix accumulation but also an alteration in matrix composition.151–154
Matrix is the primary structure that provides physical support for the vessel wall. Although they share functional redundancy, the individual matrix components may be specialized and perform specific functions. For example, collagens consist of three functionally distinct groups: fibrillar collagen, nonfibrillar collagen, and network-forming collagen. Fibrillar collagens are primarily responsible for tensile strength and the elastic resilience of the vessel wall, and the other two groups participate in matrix assembly, network formation, and cell-matrix interactions.155 Matrix proteins may also function as signaling molecules for vascular cells. For instance, polymerized type I collagen inhibits the phenotypic transition of smooth muscle cells from the contractile to the synthetic phenotype. Conversely, fragmented or monomeric type I collagen stimulates smooth muscle cell proliferation156 and migration.157 The abnormal composition of matrix components may thus contribute to uncontrolled intimal thickening.
Coagulation Factors
As a mechanism protecting against bleeding, the coagulation system can be rapidly activated when necessary. Initiators for these processes such as tissue factor and von Willebrand factor are stored in the subendothelial space and therefore are physically separated from the circulating blood. Endothelial cells maintain a nonadhesive and anticoagulant luminal surface via regulators suppressing thrombosis and coagulation (e.g., heparin, PGI2, NO, and thrombomodulin). In case of endothelial cell damage and/or denudation, tissue factor, von Willebrand factor, and subendothelial collagen are exposed to blood. Both the coagulation and thrombosis cascades are activated. Through these processes, prothrombin is converted to thrombin. Platelets are activated to aggregate via adhesion molecules (primarily glycoproteins GPIa/IIa and GPIIb/IIIa, von Willebrand factor, and fibronectins), releasing selectins, PDGF, and thromboxane A2. These bioactive substances then trigger cellular events (e.g., platelet activation,158 smooth muscle cell proliferation,159,160 and inflammatory cell recruitment) that are known to stimulate intimal hyperplasia development. Strategies aimed to interrupt these processes, such as the application of thrombomodulin, PGI2, and anticoagulants, successfully inhibited intimal hyperplasia in experimental studies.161–163
Renin-Angiotensin System
ATII holds a key role in maintaining vascular homeostasis. It is produced in the vessel wall by the actions of renin and angiotensin-converting enzyme (ACE), and it binds to receptor AT1 or AT2 to regulate several aspects of cell biology. Signaling through AT1 on smooth muscle cells, ATII stimulates proliferation, promotes the expression of enzymes that produce mediators of inflammation, activates the JAK/STAT pathway, induces expression of protooncogenes (e.g., c-fos), and enhances matrix synthesis via upregulation of the expression and activation of TGF-β.164,165 ATII is therefore a potent factor for vascular inflammation and fibrosis131 and has been evaluated in experimental studies as a promising target for inhibiting development of intimal hyperplasia.166 AT2 receptor is less abundantly expressed than AT1 in the vessel wall. Although recognizing the same ligand as AT1, AT2 generally functions as an antagonist to AT1 and may serve as a protective mechanism for vascular stenosis.167 Indeed, loss of this protective mechanism resulted in more robust intimal hyperplasia in a murine model of arterial injury.168
Preventive and Therapeutic Approaches for Intimal Hyperplasia
Although some of the mechanisms of intimal hyperplasia after vascular intervention have been delineated, few preventative strategies and therapies have been integrated into contemporary clinical practice. Furthermore, the evidence underlying these few approaches is frequently limited. Obstacles to scientific knowledge generation and translation of treatments include (1) limitations in current computer, cell culture, and animal models; (2) lack of quality, noninvasive three-dimensional longitudinal imaging in vivo; and (3) the complexity of the process in humans, in which multiple confound factors cloud investigation. However, advances in highly specific targeted therapies (e.g., small interfering RNA [siRNA], antisense oligodeoxynucleotides, molecular decoys) now provide tools to theoretically affect intimal hyperplasia signaling networks with limited side effects.
Owing to the rapidly dividing cells in the developing neointima, radiation therapy stands as an attractive approach to prevent intimal hyperplasia, especially when delivered locally via catheter as brachytherapy.169–172 Beyond the coronaries, external-beam irradiation has been described for the lower extremity and dialysis access.173–175 Challenges in the approach have been lumen loss at the edges of treatment areas176 and late acute thrombosis177; thus, the techniques are infrequently used in current practice.
The combination of cold therapy with angioplasty (cryoplasty) has been employed in lower extremity arterial interventions on the basis of several reported small series and trials.178,179 However, evidence that such therapy enhances efficacy and durability of angioplasty remains limited.
In selected vascular beds, such as the coronary arteries, drug-eluting stent devices have shown evidence of clinical efficacy in reducing restenosis. These scaffolds provide platforms for local, controlled release of drugs that inhibit neointima formation, and they may offer advantages in restenosis lesions. The Limus family of immunosuppressive agents and the mitotic inhibitor paclitaxel have been loaded onto a variety of devices (polymer based and polymer free). To date the results have been mixed, and it is not clear how performance in one vascular bed relates to that in others. The coronary RAVEL (RAndomized study with the sirolimus-eluting VElocity balloon-expandable stent in the treatment of patients with de novo native coronary artery Lesions) study showed clinical superiority of the sirolimus-coated stents over bare metal stents to 5 years after placement.180 When 47 coronary studies were reviewed (more than 14,500 patients), however, there were no statistically significant differences in rates of death or myocardial infarction between bare metal stents and drug-eluting stents despite the efficacy of the latter in reducing rates of restenosis.181 A self-expanding nitinol paclitaxel drug-eluting stent has been approved by the U.S. Food and Drug Administration (FDA) for use in femoropopliteal arteries on the basis of 1-year outcomes.182 Later efforts have focused on biodegradable stents, drug-coated balloons, and more potent drug delivery systems.183,184
Other anti–intimal hyperplasia strategies have attractive features, but the following have not reached clinical application. The transcription factor E2F holds antiproliferative properties, and this pathway was blocked in the conduit used for vein grafting via a decoy strategy in clinical trials involving both coronary and lower extremity vein grafts. Both studies failed to support clinical efficacy for this compound.2,3 CV-18C3, an IL-1α antagonist, is in clinical phase 2 studies to determine whether treatment with it will reduce restenosis or the time to restenosis in patients undergoing repeat peripheral artery revascularization.
Antithrombotic strategies also hold appeal, and several agents (aspirin, abciximab, cilostazol, clopidogrel) targeting different pathways that activate platelet adhesion, aggregation, and activation had been or are being testing in humans. Despite promise in animal models and in vitro experiments, clopidogrel offered no benefit over aspirin alone in reducing coronary vein graft intimal hyperplasia in a double-blind, placebo–controlled phase 2 clinical trial.185
Finally, of the more complex strategies to abrogate the intimal hyperplastic response, tissue engineering approaches have seen a resurgence after the early work in endothelial cell seeding of prosthetic grafts in the 1980s and 1990s.186–188 More sophisticated cell culture systems, combined with tools to allow for genetic manipulation of the embedded cells (including progenitor cells), now permit construction of artificial blood vessels that theoretically will not succumb to intimal hyperplasia.189 Other emerging strategies are the employment of scaffolds that populate with host cells as they degrade190,191 and the use of deceased donor vein recellularized with autologous stem cells.192
Selected Key References
Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91(1):327–387.
Illustration rich review discussing relationships between disturbed flow and early endothelial cell–based vascular wall adaptations.
Lehoux S, Castier Y, Tedgui A. Molecular mechanisms of the vascular responses to haemodynamic forces. J Intern Med. 2006;259(4):381–392.
Review that summarizes knowledge linking hemodynamic forces with vascular wall biology.
Muto A, Model L, Ziegler K, Eghbalieh SD, Dardik A. Mechanisms of vein graft adaptation to the arterial circulation: insights into the neointimal algorithm and management strategies. Circ J. 2010;74(8):1501–1512.
Summary of venous biology and adaptations to vein graft construction, and potential relationships with vein graft neointimal hyperplasia.
Quint C, Kondo Y, Manson RJ, Lawson JH, Dardik A, Niklason LE. Decellularized tissue-engineered blood vessel as an arterial conduit. Proc Natl Acad Sci U S A. 2011;108(22):9214–9219.
Example contemporary approach to construction of an articifial blood vessel that is resistant to intimal hyperplasia.
Tanaka K, Sata M, Hirata Y, Nagai R. Diverse contribution of bone marrow cells to neointimal hyperplasia after mechanical vascular injuries. Circ Res. 2003;93(8):783–790.
Report in three distinct arterial intimal hyerplasia animal model systems that describes variable contributions of bone marrow–derived cells to neointimal hyerplasia.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Lal BK, et al. Restenosis after carotid artery stenting and endarterectomy: a secondary analysis of CREST, a randomised controlled trial. Lancet Neurol. 2012;11(9):755–763.
2. Conte MS, et al. Results of PREVENT III: a multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J Vasc Surg. 2006;43(4):742–751.
3. Alexander JH, et al. Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery: PREVENT IV: a randomized controlled trial. JAMA. 2005;294(19):2446–2454.
4. Dember LM, et al. Effect of clopidogrel on early failure of arteriovenous fistulas for hemodialysis: a randomized controlled trial. JAMA. 2008;299(18):2164–2171.
5. Beathard GA, et al. Aggressive treatment of early fistula failure. Kidney Int. 2003;64(4):1487–1494.
6. Lee T, et al. Advances and new frontiers in the pathophysiology of venous neointimal hyperplasia and dialysis access stenosis. Adv Chronic Kidney Dis. 2009;16(5):329–338.
7. Hodges TC, et al. Longitudinal comparison of dialysis access methods: risk factors for failure. J Vasc Surg. 1997;26(6):1009–1019.
8. Newby AC, et al. Molecular mechanisms in intimal hyperplasia. J Pathol. 2000;190(3):300–309.
9. Grondin CM, et al. Progressive and late obstruction of an aorto-coronary venous bypass graft. Circulation. 1971;43(5):698–702.
10. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340(2):115–126.
11. Schwartz SM, et al. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77(3):445–465.
12. Motwani JG, et al. Aortocoronary saphenous vein graft disease: pathogenesis, predisposition, and prevention. Circulation. 1998;97(9):916–931.
13. Dzau VJ, et al. Vascular remodeling: mechanisms and implications. J Cardiovasc Pharmacol. 1993;21(Suppl 1):S1–S5.
14. Glagov S, et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371–1375.
15. Jiang Z, et al. A novel vein graft model: adaptation to differential flow environments. Am J Physiol Heart Circ Physiol. 2004;286(1):H240–H245.
16. Schwartz LB, et al. Myointimal thickening in experimental vein grafts is dependent on wall tension. J Vasc Surg. 1992;15(1):176–186.
17. Rozenman Y, et al. Clinical and angiographic predictors of immediate recoil after successful coronary angioplasty and relation to late restenosis. Am J Cardiol. 1993;72(14):1020–1025.
18. Rodriguez AE, et al. Time course and mechanism of early luminal diameter loss after percutaneous transluminal coronary angioplasty. Am J Cardiol. 1995;76(16):1131–1134.
19. Palmaz JC, et al. Expandable intraluminal graft: a preliminary study. Work in progress. Radiology. 1985;156(1):73–77.
20. Serruys PW, et al. Coronary-artery stents. N Engl J Med. 2006;354(5):483–495.
21. Schillinger M, et al. Balloon angioplasty versus implantation of nitinol stents in the superficial femoral artery. N Engl J Med. 2006;354(18):1879–1888.
22. Lee PC, et al. Cellular and molecular mechanisms of coronary artery restenosis. Coron Artery Dis. 1993;4(3):254–259.
23. Lau GT, et al. Lumen loss in the first year in saphenous vein grafts is predominantly a result of negative remodeling of the whole vessel rather than a result of changes in wall thickness. Circulation. 2006;114(1 Suppl):I435–I440.
24. Chiu JJ, et al. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91(1):327–387.
25. Lehoux S, et al. Molecular mechanisms of the vascular responses to haemodynamic forces. J Intern Med. 2006;259(4):381–392.
26. Berk BC. Atheroprotective signaling mechanisms activated by steady laminar flow in endothelial cells. Circulation. 2008;117(8):1082–1089.
27. Wentzel JJ, et al. Relationship between neointimal thickness and shear stress after Wallstent implantation in human coronary arteries. Circulation. 2001;103(13):1740–1745.
28. Richter Y, et al. Dynamic flow alterations dictate leukocyte adhesion and response to endovascular interventions. J Clin Invest. 2004;113(11):1607–1614.
29. Cunningham KS, et al. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest. 2005;85(1):9–23.
30. Jiang Z, et al. Tumor necrosis factor-alpha and the early vein graft. J Vasc Surg. 2007;45(1):169–176.
31. Mattsson EJ, et al. Increased blood flow induces regression of intimal hyperplasia. Arterioscler Thromb Vasc Biol. 1997;17(10):2245–2249.
32. Dardik H, et al. Improved method to create the common ostium variant of the distal arteriovenous fistula for enhancing crural prosthetic graft patency. J Vasc Surg. 1996;24(2):240–248.
33. Paszkowiak JJ, et al. Arterial wall shear stress: observations from the bench to the bedside. Vasc Endovascular Surg. 2003;37(1):47–57.
34. Hellstrand P, et al. Stretch-dependent growth and differentiation in vascular smooth muscle: role of the actin cytoskeleton. Can J Physiol Pharmacol. 2005;83(10):869–875.
35. Cornelissen J, et al. Mechanical stretch induces phosphorylation of p38-MAPK and apoptosis in human saphenous vein. Arterioscler Thromb Vasc Biol. 2004;24(3):451–456.
36. Huynh TT, et al. Alterations in wall tension and shear stress modulate tyrosine kinase signaling and wall remodeling in experimental vein grafts. J Vasc Surg. 1999;29(2):334–344.
37. Lee MS, et al. Cardiac allograft vasculopathy. Rev Cardiovasc Med. 2011;12(3):143–152.
38. Mitchell RN. Graft vascular disease: immune response meets the vessel wall. Annu Rev Pathol. 2009;4:19–47.
39. Costa MA, et al. Molecular basis of restenosis and drug-eluting stents. Circulation. 2005;111(17):2257–2273.
40. Monraats PS, et al. Interleukin 10: a new risk marker for the development of restenosis after percutaneous coronary intervention. Genes Immun. 2007;8(1):44–50.
41. Sampietro ML, et al. A genome-wide association study identifies a region at chromosome 12 as a potential susceptibility locus for restenosis after percutaneous coronary intervention. Hum Mol Genet. 2011;20(23):4748–4757.
42. Nguyen BT, et al. Perivascular innate immune events modulate early murine vein graft adaptations. J Vasc Surg. 2012;57:486–492.
43. Muto A, et al. Mechanisms of vein graft adaptation to the arterial circulation: insights into the neointimal algorithm and management strategies. Circ J. 2010;74(8):1501–1512.
44. Hoofnagle MH, et al. Origin of neointimal smooth muscle: we’ve come full circle. Arterioscler Thromb Vasc Biol. 2006;26(12):2579–2581.
45. Havelka GE, et al. The vascular adventitia: its role in the arterial injury response. Vasc Endovascular Surg. 2011;45(5):381–390.
46. Tanaka K, et al. Diverse contribution of bone marrow cells to neointimal hyperplasia after mechanical vascular injuries. Circ Res. 2003;93(8):783–790.
47. Diao Y, et al. Long-term engraftment of bone marrow-derived cells in the intimal hyperplasia lesion of autologous vein grafts. Am J Pathol. 2008;172(3):839–848.
48. Zalewski A, et al. Diverse origin of intimal cells: smooth muscle cells, myofibroblasts, fibroblasts, and beyond? Circ Res. 2002;91(8):652–655.
49. Sata M, et al. Origin of neointimal cells in autologous vein graft. Arterioscler Thromb Vasc Biol. 2004;24(7):1147–1149.
50. Gerthoffer WT, et al. Secretory functions of smooth muscle: cytokines and growth factors. Mol Interv. 2002;2(7):447–456.
51. Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196(3):430–443.
52. Vavra AK, et al. Insights into the effect of nitric oxide and its metabolites nitrite and nitrate at inhibiting neointimal hyperplasia. Nitric Oxide. 2011;25(1):22–30.
53. Tai SC, et al. Endothelial nitric oxide synthase: a new paradigm for gene regulation in the injured blood vessel. Arterioscler Thromb Vasc Biol. 2004;24(3):405–412.
55. Sana TR, et al. Microarray analysis of primary endothelial cells challenged with different inflammatory and immune cytokines. Cytokine. 2005;29(6):256–269.
56. Frid MG, et al. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. 2002;90(11):1189–1196.
57. Arciniegas E, et al. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293(1):L1–L8.
58. Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75(3):487–517.
59. Ishida M, et al. Immunohistochemical phenotypic alterations of rabbit autologous vein grafts implanted under arterial circulation with or without poor distal runoff-implications of vein graft remodeling. Atherosclerosis. 2001;154(2):345–354.
60. Jiang Z, et al. Established neointimal hyperplasia in vein grafts expands via TGF-β mediated progressive fibrosis. Am J Physiol Heart Circ Physiol. 2009;297(4):200–207.
61. Lemson MS, et al. Intimal hyperplasia in vascular grafts. Eur J Vasc Endovasc Surg. 2000;19(4):336–350.
62. Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100(5):607–621.
63. Bhasin M, et al. Temporal network based analysis of cell specific vein graft transcriptome defines key pathways and hub genes in implantation injury. PLoS One. 2012;7(6):e39123.
64. Stenmark KR, et al. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda). 2006;21:134–145.
65. Strauss BH, et al. Adventitial fibroblasts: defining a role in vessel wall remodeling. Am J Respir Cell Mol Biol. 2000;22(1):1–3.
66. Shi Y, et al. Remodeling of autologous saphenous vein grafts. The role of perivascular myofibroblasts. Circulation. 1997;95(12):2684–2693.
67. Li G, et al. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101(12):1362–1365.
68. Faggin E, et al. Smooth muscle-specific SM22 protein is expressed in the adventitial cells of balloon-injured rabbit carotid artery. Arterioscler Thromb Vasc Biol. 1999;19(6):1393–1404.
69. Sartore S, et al. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. 2001;89(12):1111–1121.
70. Gotoh R, et al. E-selectin blockade decreases adventitial inflammation and attenuates intimal hyperplasia in rat carotid arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2004;24(11):2063–2068.
71. Roque M, et al. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20(2):335–342.
72. Rogers C, et al. A mAb to the beta2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc Natl Acad Sci U S A. 1998;95(17):10134–10139.
73. De SS, et al. Granulocyte activation after coronary angioplasty in humans. Circulation. 1990;82(1):140–146.
74. Neumann FJ, et al. Neutrophil and platelet activation at balloon-injured coronary artery plaque in patients undergoing angioplasty. J Am Coll Cardiol. 1996;27(4):819–824.
75. Walter DH, et al. Preprocedural C-reactive protein levels and cardiovascular events after coronary stent implantation. J Am Coll Cardiol. 2001;37(3):839–846.
76. Wasser K, et al. Inflammation and in-stent restenosis: the role of serum markers and stent characteristics in carotid artery stenting. PLoS One. 2011;6(7):e22683.
77. Miller AM, et al. Inhibition by leukocyte depletion of neointima formation after balloon angioplasty in a rabbit model of restenosis. Cardiovasc Res. 2001;49(4):838–850.
78. Wainwright CL, et al. Inflammation as a key event in the development of neointima following vascular balloon injury. Clin Exp Pharmacol Physiol. 2001;28(11):891–895.
79. Welt FG, et al. Neutrophil, not macrophage, infiltration precedes neointimal thickening in balloon-injured arteries. Arterioscler Thromb Vasc Biol. 2000;20(12):2553–2558.
80. Schwarz JB, et al. Novel role of the CXC chemokine receptor 3 in inflammatory response to arterial injury: involvement of mTORC1. Circ Res. 2009;104(2):189–200.
81. Isobe M, et al. T cell costimulation in the development of cardiac allograft vasculopathy: potential targets for therapeutic interventions. Arterioscler Thromb Vasc Biol. 2006;26(7):1447–1456.
82. Auffray C, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317(5838):666–670.
83. Cros J, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33(3):375–386.
84. Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661.
85. Sica A, et al. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795.
86. Zhou J, et al. CXCR3-dependent accumulation and activation of perivascular macrophages is necessary for homeostatic arterial remodeling to hemodynamic stresses. J Exp Med. 2010;207(9):1951–1966.
87. Moore KJ, et al. Macrophages, atherosclerosis and the potential of netrin-1 as a novel target for future therapeutic intervention. Future Cardiol. 2012;8(3):349–352.
88. Hirschi KK, et al. Smooth muscle stem cells. Anat Rec A Discov Mol Cell Evol Biol. 2004;276(1):22–33.
89. Ross JJ, et al. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J Clin Invest. 2006;116(12):3139–3149.
90. Bentzon JF, et al. Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein E knockout mice. Circulation. 2007;116(18):2053–2061.
91. Iwata H, et al. Bone marrow-derived cells contribute to vascular inflammation but do not differentiate into smooth muscle cell lineages. Circulation. 2010;122(20):2048–2057.
92. Alobaid N, et al. Endothelial progenitor cells and their potential clinical applications in peripheral arterial disease. Endothelium. 2005;12(5–6):243–250.
93. Kong D, et al. Cytokine-induced mobilization of circulating endothelial progenitor cells enhances repair of injured arteries. Circulation. 2004;110(14):2039–2046.
94. Werner N, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353(10):999–1007.
95. Blindt R, et al. A novel drug-eluting stent coated with an integrin-binding cyclic Arg-Gly-Asp peptide inhibits neointimal hyperplasia by recruiting endothelial progenitor cells. J Am Coll Cardiol. 2006;47(9):1786–1795.
96. Aoki J, et al. Endothelial progenitor cell capture by stents coated with antibody against CD34: the HEALING-FIM (Healthy Endothelial Accelerated Lining Inhibits Neointimal Growth-First In Man) Registry. J Am Coll Cardiol. 2005;45(10):1574–1579.
97. Wagner DD. The Weibel-Palade body: the storage granule for von Willebrand factor and P-selectin. Thromb Haemost. 1993;70(1):105–110.
98. Gimbrone MA Jr, et al. Endothelial-dependent mechanisms of leukocyte adhesion in inflammation and atherosclerosis. Ann N Y Acad Sci. 1990;598:77–85.
99. Tedder TF, et al. The selectins: vascular adhesion molecules. FASEB J. 1995;9(10):866–873.
100. Rastogi S, et al. TNF-alpha response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012;19(2):274–283.
101. Schuger L, et al. Cytotoxicity of tumor necrosis factor-alpha for human umbilical vein endothelial cells. Lab Invest. 1989;61(1):62–68.
103. Karper JC, et al. Toll-like receptor 4 is involved in human and mouse vein graft remodeling, and local gene silencing reduces vein graft disease in hypercholesterolemic APOE*3Leiden mice. Arterioscler Thromb Vasc Biol. 2011;31(5):1033–1040.
104. Schoneveld AH, et al. Toll-like receptor 2 stimulation induces intimal hyperplasia and atherosclerotic lesion development. Cardiovasc Res. 2005;66(1):162–169.
105. Zhang LL, et al. PPARgamma attenuates intimal hyperplasia by inhibiting TLR4-mediated inflammation in vascular smooth muscle cells. Cardiovasc Res. 2011;92(3):484–493.
106. Ozaki CK. Cytokines and the early vein graft: strategies to enhance durability. J Vasc Surg. 2007;45(Suppl A):A92–A98.
107. Zimmerman MA, et al. Interleukin-10 attenuates the response to vascular injury. J Surg Res. 2004;121(2):206–213.
108. Tian Y, et al. Expression and suppressive effects of interleukin-19 on vascular smooth muscle cell pathophysiology and development of intimal hyperplasia. Am J Pathol. 2008;173(3):901–909.
109. Feldman LJ, et al. Interleukin-10 inhibits intimal hyperplasia after angioplasty or stent implantation in hypercholesterolemic rabbits. Circulation. 2000;101(8):908–916.
110. Douglas MR, et al. Why does inflammation persist: a dominant role for the stromal microenvironment? Expert Rev Mol Med. 2002;4(25):1–18.
111. Serhan CN, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196(8):1025–1037.
112. Schwab JM, et al. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447(7146):869–874.
113. Serhan CN, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206(1):15–23.
114. Libby P, et al. A cascade model for restenosis. A special case of atherosclerosis progression. Circulation. 1992;86(6 Suppl):III47–III52.
115. Myit S, et al. Different growth properties of neointimal and medial smooth muscle cells in response to growth factors. J Vasc Res. 2003;40(2):97–104.
116. Risinger GM Jr, et al. Matrix metalloproteinase-2 expression by vascular smooth muscle cells is mediated by both stimulatory and inhibitory signals in response to growth factors. J Biol Chem. 2006;281(36):25915–25925.
117. Wagsater D, et al. Effects of PDGF-C and PDGF-D on monocyte migration and MMP-2 and MMP-9 expression. Atherosclerosis. 2009;202(2):415–423.
118. Chen J, et al. PDGF-D contributes to neointimal hyperplasia in rat model of vessel injury. Biochem Biophys Res Commun. 2005;329(3):976–983.
119. Kotani M, et al. Chimeric DNA-RNA hammerhead ribozyme targeting PDGF A-chain mRNA specifically inhibits neointima formation in rat carotid artery after balloon injury. Cardiovasc Res. 2003;57(1):265–276.
120. Kawai-Kowase K, et al. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007;292(1):C59–C69.
121. McDonald OG, et al. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116(1):36–48.
122. Isner JM, et al. Arterial gene therapy for restenosis. Hum Gene Ther. 1996;7(8):989–1011.
123. Hutter R, et al. Vascular endothelial growth factor regulates reendothelialization and neointima formation in a mouse model of arterial injury. Circulation. 2004;110(16):2430–2435.
124. Khurana R, et al. Angiogenesis-dependent and independent phases of intimal hyperplasia. Circulation. 2004;110(16):2436–2443.
125. Ohtani K, et al. Blockade of vascular endothelial growth factor suppresses experimental restenosis after intraluminal injury by inhibiting recruitment of monocyte lineage cells. Circulation. 2004;110(16):2444–2452.
126. Shiojima I, et al. The role of vascular endothelial growth factor in restenosis: the controversy continues. Circulation. 2004;110(16):2283–2286.
127. Hneino M, et al. Density-dependent shift of transforming growth factor-beta-1 from inhibition to stimulation of vascular smooth muscle cell growth is based on unconventional regulation of proliferation, apoptosis and contact inhibition. J Vasc Res. 2009;46(2):85–97.
128. Rensen SS, et al. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15(3):100–108.
129. Hautmann MB, et al. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem. 1997;272(16):10948–10956.
130. Leask A, et al. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18(7):816–827.
131. Ruiz-Ortega M, et al. TGF-beta signaling in vascular fibrosis. Cardiovasc Res. 2007;74(2):196–206.
132. Smith JD, et al. Soluble transforming growth factor-beta type II receptor inhibits negative remodeling, fibroblast transdifferentiation, and intimal lesion formation but not endothelial growth. Circ Res. 1999;84(10):1212–1222.
133. Wolff RA, et al. Antisense to transforming growth factor-beta1 messenger RNA reduces vein graft intimal hyperplasia and monocyte chemotactic protein 1. J Vasc Surg. 2005;41(3):498–508.
134. Holmes DR Jr, et al. Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) trial. Circulation. 2002;106(10):1243–1250.
135. Fu K, et al. SM16, an orally active TGF-beta type I receptor inhibitor prevents myofibroblast induction and vascular fibrosis in the rat carotid injury model. Arterioscler Thromb Vasc Biol. 2008;28(4):665–671.
136. Berceli SA, et al. Differential expression and activity of matrix metalloproteinases during flow-modulated vein graft remodeling. J Vasc Surg. 2004;39(5):1084–1090.
137. Berceli SA, et al. Early differential MMP-2 and -9 dynamics during flow-induced arterial and vein graft adaptations. J Surg Res. 2006;134(2):327–334.
138. Bassiouny HS, et al. Flow regulation of 72-kD collagenase IV (MMP-2) after experimental arterial injury. Circulation. 1998;98(2):157–163.
139. Godin D, et al. Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation. 2000;102(23):2861–2866.
140. Feldman LJ, et al. Differential expression of matrix metalloproteinases after stent implantation and balloon angioplasty in the hypercholesterolemic rabbit. Circulation. 2001;103(25):3117–3122.
141. Newby AC. Matrix metalloproteinase inhibition therapy for vascular diseases. Vascul Pharmacol. 2012;56(5–6):232–244.
142. Chase AJ, et al. Regulation of matrix metalloproteinase (matrixin) genes in blood vessels: a multi-step recruitment model for pathological remodelling. J Vasc Res. 2003;40(4):329–343.
143. George SJ, et al. Inhibition of late vein graft neointima formation in human and porcine models by adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-3. Circulation. 2000;101(3):296–304.
144. Kuzuya M, et al. Deficiency of gelatinase a suppresses smooth muscle cell invasion and development of experimental intimal hyperplasia. Circulation. 2003;108(11):1375–1381.
145. Cho A, et al. Matrix metalloproteinase-9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ Res. 2002;91(9):845–851.
146. Fay WP. Plasminogen activator inhibitor 1, fibrin, and the vascular response to injury. Trends Cardiovasc Med. 2004;14(5):196–202.
147. Otsuka G, et al. Mechanisms of TGF-beta1-induced intimal growth: plasminogen-independent activities of plasminogen activator inhibitor-1 and heterogeneous origin of intimal cells. Circ Res. 2007;100(9):1300–1307.
148. DeYoung MB, et al. Plasminogen activator inhibitor type 1 increases neointima formation in balloon-injured rat carotid arteries. Circulation. 2001;104(16):1972–1981.
150. Ryer EJ, et al. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2006;26(4):780–786.
151. Raines EW. The extracellular matrix can regulate vascular cell migration, proliferation, and survival: relationships to vascular disease. Int J Exp Pathol. 2000;81(3):173–182.
152. Franco CD, et al. Collagens, integrins, and the discoidin domain receptors in arterial occlusive disease. Trends Cardiovasc Med. 2002;12(4):143–148.
153. Zhang WD, et al. Association of smooth muscle cell phenotypic modulation with extracellular matrix alterations during neointima formation in rabbit vein grafts. J Vasc Surg. 1999;30(1):169–183.
154. Bou-Gharios G, et al. Extra-cellular matrix in vascular networks. Cell Prolif. 2004;37(3):207–220.
155. Plenz GA, et al. Vascular collagens: spotlight on the role of type VIII collagen in atherogenesis. Atherosclerosis. 2003;166(1):1–11.
156. Koyama H, et al. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87(6):1069–1078.
157. Carragher NO, et al. Degraded collagen fragments promote rapid disassembly of smooth muscle focal adhesions that correlates with cleavage of pp125(FAK), paxillin, and talin. J Cell Biol. 1999;147(3):619–630.
158. Molino M, et al. Thrombin receptors on human platelets. Initial localization and subsequent redistribution during platelet activation. J Biol Chem. 1997;272(9):6011–6017.
159. Taubman MB, et al. The role of smooth muscle derived tissue factor in mediating thrombosis and arterial injury. Thromb Res. 2008;122(Suppl 1):S78–S81.
160. Walters TK, et al. Thrombin generation following arterial injury is a critical initiating event in the pathogenesis of the proliferative stages of the atherosclerotic process. J Vasc Res. 1994;31(3):173–177.
161. Zoldhelyi P, et al. Local gene transfer of tissue factor pathway inhibitor regulates intimal hyperplasia in atherosclerotic arteries. Proc Natl Acad Sci U S A. 2001;98(7):4078–4083.
162. O’Brien PJ, et al. Direct thrombin inhibitors. J Cardiovasc Pharmacol Ther. 2012;17(1):5–11.
163. Waugh JM, et al. Thrombomodulin overexpression to limit neointima formation. Circulation. 2000;102(3):332–337.
164. Berk BC, et al. Angiotensin II signal transduction in vascular smooth muscle: role of tyrosine kinases. Circ Res. 1997;80(5):607–616.
165. Rautureau Y, et al. Cross-talk between aldosterone and angiotensin signaling in vascular smooth muscle cells. Steroids. 2011;76(9):834–839.
166. Osgood MJ, et al. Role of the renin-angiotensin system in the pathogenesis of intimal hyperplasia: therapeutic potential for prevention of vein graft failure? Ann Vasc Surg. 2012;26(8):1130–1144.
167. Stegbauer J, et al. New insights into angiotensin receptor actions: from blood pressure to aging. Curr Opin Nephrol Hypertens. 2011;20(1):84–88.
168. Akishita M, et al. Inflammation influences vascular remodeling through AT2 receptor expression and signaling. Physiol Genomics. 2000;2(1):13–20.
169. Bottcher HD. Endovascular radioprevention of intimal hyperplasia after percutaneous transluminal angioplasty of peripheral blood vessels. Radiologe. 1994;34(9):519–523.
170. Waksman R. Local catheter-based intracoronary radiation therapy for restenosis. Am J Cardiol. 1996;78(3A):23–28.
171. Drachman DE, et al. Restenosis: Intracoronary brachytherapy. Curr Treat Options Cardiovasc Med. 2002;4(2):109–118.
172. Sun S, et al. Inhibitory effect of brachytherapy on intimal hyperplasia in arteriovenous fistula. J Surg Res. 2003;115(2):200–208.
173. Parikh S, et al. External beam radiation therapy to prevent postangioplasty dialysis access restenosis: a feasibility study. Cardiovasc Radiat Med. 1999;1(1):36–41.
174. Therasse E, et al. External beam radiation to prevent restenosis after superficial femoral artery balloon angioplasty. Circulation. 2005;111(24):3310–3315.
175. Rectenwald JE, et al. External-beam radiation therapy for improved dialysis access patency: feasibility and early safety. Radiat Res. 2001;156(1):53–60.
176. Sabaté M, et al. Geometric vascular remodeling after balloon angioplasty and beta-radiation therapy: a three-dimensional intravascular ultrasound study. Circulation. 1999;100(11):1182–1188.
177. Costa MA, et al. Late coronary occlusion after intracoronary brachytherapy [In Process Citation]. Circulation. 1999;100(8):789–792.
178. Fava M, et al. Cryoplasty for femoropopliteal arterial disease: late angiographic results of initial human experience. J Vasc Interv Radiol. 2004;15(11):1239–1243.
179. Banerjee S, et al. Pilot trial of cryoplasty or conventional balloon post-dilation of nitinol stents for revascularization of peripheral arterial segments: the COBRA trial. J Am Coll Cardiol. 2012;60(15):1352–1359.
180. Morice MC, et al. Long-term clinical outcomes with sirolimus-eluting coronary stents: five-year results of the RAVEL trial. J Am Coll Cardiol. 2007;50(14):1299–1304.
181. Greenhalgh J, et al. Drug-eluting stents versus bare metal stents for angina or acute coronary syndromes. Cochrane Database Syst Rev. 2010;(5):CD004587.
182. Dake MD, et al. Nitinol stents with polymer-free paclitaxel coating for lesions in the superficial femoral and popliteal arteries above the knee: twelve-month safety and effectiveness results from the Zilver PTX single-arm clinical study. J Endovasc Ther. 2011;18(5):613–623.
183. Bosiers M, et al. Refining stent technologies for femoral interventions. J Cardiovasc Surg (Torino). 2012;53(4):465–473.
184. Minar E, et al. Innovative technologies for SFA occlusions: drug coated balloons in SFA lesions. J Cardiovasc Surg (Torino). 2012;53(4):481–486.
185. Kulik A, et al. Aspirin plus clopidogrel versus aspirin alone after coronary artery bypass grafting: the clopidogrel after surgery for coronary artery disease (CASCADE) trial. Circulation. 2010;122(25):2680–2687.
186. Graham LM, et al. Cultured autogenous endothelial cell seeding of prosthetic vascular grafts. Surg Forum. 1979;30:204–206.
187. Herring MB, et al. Endothelial seeding of polytetrafluoroethylene popliteal bypasses. A preliminary report. J Vasc Surg. 1987;6(2):114–118.
188. Nugent HM, et al. Tissue engineering therapy for cardiovascular disease. Circ Res. 2003;92(10):1068–1078.
189. Nishibe T, et al. Optimal prosthetic graft design for small diameter vascular grafts. Vascular. 2007;15(6):356–360.
190. Quint C, et al. Decellularized tissue-engineered blood vessel as an arterial conduit. Proc Natl Acad Sci U S A. 2011;108(22):9214–9219.
191. Wu W, et al. Fast-degrading elastomer enables rapid remodeling of a cell-free synthetic graft into a neoartery. Nat Med. 2012;18(7):1148–1153.
192. Olausson M. Bioengineered vascular graft with autologous stem cells: first use in the clinic. Regen Med. 2012;7(6 Suppl):12–15.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]Chapter 6
Intimal Hyperplasia
Zhihua Jiang, C. Keith Ozaki
Based on a chapter in the seventh edition by Mark G. Davies
Intimal hyperplasia occurs frequently after vascular interventions such as vein bypass grafts, endarterectomies, arteriovenous fistulas, prosthetic bypass grafts, balloon angioplasty, endovascular stents and stent grafts, and solid organ transplantation. Data now suggest that restenosis develops in 5.8% of patients undergoing carotid endarterectomy or receiving carotid stents, though these cases rarely require re-intervention.1 About 30% to 60% of lower extremity arterialized vein grafts fail or are failing within a year,2,3 and intervention is frequently necessary to maintain conduit patency. Finally, 60% of hemodialysis access fistulas fail to mature primarily,4 and obliterative processes near the anastomosis are associated with half of these failures.5,6 Similarly, the durability of prosthetic dialysis grafts remains poor owing to intimal hyperplasia at the outflow vein.6,7 Although molecularly this process begins immediately after the vascular injury that accompanies the procedure, detectable intimal lesions classically form within a few weeks to 2 years.8
Definitions
Intimal Hyperplasia
The blood vessel intima is normally composed of just the thin endothelial cell lining of the vasculature. Intimal hyperplasia reflects additional biomass (cellular and matrix) within this layer. Mammals appear to have evolved intimal hyperplasic processes so that they become survival benefits. For example, hyperplastic intimal growth underlies closure of the ductus arteriosus. Vascular healing and premenopausal occlusion of the uterine arteries are also, in essence, the process of intimal hyperplasia.
Historically, the development of intimal hyperplasia was initially considered a “process of connective tissue hyperplasia.” Later, the concept was revised to “fibroproliferative” intimal thickening.9 Studies in the 1980s identified medial smooth muscle cells as central effectors of this process. Following biochemical insult and/or mechanical trauma, medial smooth muscle cells are activated and undergo a phenotypic switch from a contractile/quiescent to a synthetic/proliferative state. Proteases, particularly matrix metalloproteinases (MMPs), are released by inflammatory infiltrating leukocytes to break the matrix network. Smooth muscle cells are liberated from matrix restriction, and they then migrate from the tunica media to the intimal region, where they proliferate and deposit matrix proteins. Although sharing the same lineage identity with those in the media, smooth muscle cells in the intimal tissue are functionally incapable of organizing the cellular and matrix components. The de novo intimal layer is thus often qualitatively a relatively disordered histologic structure. This grossly fibrous, white material accumulates over time, protruding into the lumen and leading to luminal narrowing and even complete occlusion (Fig. 6-1).
Figure 6-1 A, Digital subtraction angiogram of the distal right limb of an aortofemoral graft showing a filling defect at the junction of the prosthetic graft material and the outflow of the profunda femoris artery. B, Intraoperative photograph of the same field. Note the grossly fibrous, white intimal hyperplasia that corresponds to the outflow lesion. C, Masson trichrome stain preparation of endarterectomized intimal hyperplasia. Green staining corresponds to collagen, whereas red localizes primarily to other matrix components and the cytoplasm of largely smooth muscle cells.
It is currently believed that intimal hyperplasia starts as an acute inflammatory response. Because of the incomplete functional recovery of the repopulated endothelial cells and the phenotypic switch of smooth muscle cells, the acute inflammation often does not completely resolve, leaving behind a chronic inflammation in the intimal tissue. Inflammatory cells, particularly macrophages and T cells, are found in established intimal lesions. In contrast, atherosclerosis is a vascular disease driven by inflammatory mechanisms.10 The inflamed intimal tissue thus can serve as the “soil” for subsequent atherosclerosis to develop.11 Although the early failure of vascular reconstructions is largely caused by intimal hyperplasia, atherosclerosis is the dominant process underlying late failure (>3 years). After coronary artery stenting, atherosclerotic lesion(s) account for more than 70% of cases of recurrence of ischemic symptoms.12
Other Pathologies Underlying Lumen Loss after Vascular Procedures
Wall Remodeling
In addition to the intimal thickening, blood vessels can permanently change the dimensions of the media and adventitia. This change is termed wall remodeling.13 An early description of vascular remodeling comes from Glagov et al14 in 1987. They observed that coronary arteries actively grow to enlarge their cross-sectional area to accommodate the encroachment of atherosclerotic lesions. As the result of this compensatory expansion, appropriate luminal dimensions may be effectively maintained for the diseased vessels. This observation changed the concept of the vessels from inanimate pipes to an active system. It is now well established that the remodeling process involves reorganization/construction of both the medial and adventitial layers. Although leading to an increase in the overall dimension that favors preserving luminal area, remodeling may also proceed in the opposite direction, promoting wall shrinkage and loss of the luminal area.
Several terms have been used to describe remodeling processes. Morphologic terms, such as inward/constrictive remodeling and outward/expansive remodeling, are used to describe the impact on the geometric vascular dimension. Functional terms, such as positive remodeling and negative remodeling, were introduced to emphasize the effect of the remodeling process on the performance of the vessel as a conduit. A challenge in clinical studies is the inability to evaluate the full geometric dimension of the vessel over time because of the inadequate discrimination of the outer vessel wall from the perivascular tissue with imaging approaches such as angiography and computed tomography (CT) scans. This issue has been partially overcome by the availability of the intravascular ultrasound. Wall detail may be described in vivo as long as it is accessible by intravenous ultrasound probes.
Hemodynamic forces, particularly shear stress and wall tension, appear to be primary modulators of the vascular remodeling process. An elevated shear, for example, generally propels an expansion or outward growth, whereas a reduction in shear promotes “shrinkage” or inward growth of the vessel.15 Compared with that of shear forces, the impact of tensile forces on vascular remodeling is less well understood. Generally, wall tension is a factor that favors a negative remodeling process.16 When coupled with intimal hyperplasia, wall remodeling can ameliorate or exacerbate luminal narrowing through its effect on vessel dimension.
Vascular Tone and Recoil
Muscular vessels (arteries and veins) receive autonomic innervation from the central nervous system. Under physiologic conditions, a basal level of activity is transmitted via the sympathetic fibers to maintain a partial state of contraction in blood vessels. This basal level of contractile tension is referred to as vascular tone. Local hemodynamic forces and activation of vascular innervation may finely tune the vascular tone by way of promoting smooth muscle cell contraction or relaxation. Although transient fluctuation in vascular tone is reversible, a persistent increase in vascular tone may lead to structural changes such as the pathology occurring in patients suffering from long-standing arterial hypertension.
In addition to the active adjustment of the vascular caliber by smooth muscle cells, passive contraction of the vascular wall matrix network, consisting of predominantly collagen and elastic fibers, is also a critical determinant of the vessel diameter. Under physiologic conditions, the elastic resilience works to smooth out the pulsatility generated by cardiac ejection. Following the systolic distention, vessels return to their original dimensions as the result of elastic contraction. This phenomenon is termed vascular recoil. In the vessel wall, the intact and well-organized structural components as well as the blood counterbalance the wall shrinkage caused by recoil. In cases in which the wall integrity is destroyed, such as balloon angioplasty, recoil may cause 50% of the loss in acute gain during angioplasty.17,18 To oppose this recoil effect, metal stents were first used in peripheral arteries in 1985,19 and coronary arteries in 1986.20 In comparison with balloon angioplasty alone, the additional placement of stents can offer significant geometric benefits, as reflected by more preservation of the acute gain in lumen diameter and, in selected patients, less frequent requirement for secondary interventions and better quality of life.21
Restenosis
Several months after interventions such as endarterectomy, angioplasty, and bypass, the vascular constructions may become significantly narrowed or may occlude. This recurrence of stenosis and reduction in luminal area is termed restenosis. The causes for restenosis are complex.22 Usually, the primary pathology detected in a vessel with restenosis is intimal hyperplasia, and the loss of luminal area is proportional to the encroachment of the intimal tissue. Geometric remodeling likely also contributes.23 For example, the lumen area loss may be partially or completely compensated if a simultaneous outward remodeling had led to an increase in the geometric dimension. Conversely, significant restenosis may occur because of additional lumen loss caused by inward remodeling even if the amount of intimal hyperplasia is relatively small. Metal stents offer a strategy to limit inward remodeling, although intimal hyperplasia (either through the stent structure or at the ends of a stent graft) often offset these luminal area gains. Under specific circumstances, an improved long-term patency of the stented vessels has been observed.21
Inciting/Modulating Factors for Intimal Hyperplasia
Intimal hyperplasia is a vascular response that heals the injured vessel wall. It can occur in nearly every type of vascular reconstruction, and the initiation and progression of the lesion may be modulated by several factors.
Hemodynamic Stress
Hemodynamic forces, specifically shear stress and wall tensile stress, are well-established initiators and modulators of intimal hyperplasia.24 Vascular cells are equipped to sense and respond to the luminal hemodynamic environment. Several surface receptors (e.g., integrins and vascular endothelial growth factor receptor [VEGFR]), ion channels (e.g., K+ and Ca++), and the cell cytoskeleton may be specialized in mechanosensing and mechanotransduction.25
Under physiologic conditions, the steady laminar blood flow generates on average approximately 15 dyne/cm2 of shear stress in arteries. Endothelial cells sense this physiologic shear force, releasing mediators such as nitric oxide (NO) and Kruppel-like factor-2 to maintain a quiescent state for smooth muscle cells and homeostasis of the whole vessel wall.26 Vascular reconstructions such as vein bypass grafts, stented diseased arteries, and arteriovenous fistulas not only alter the rate of the local blood flow but also frequently induce a disordered flow pattern. Both clinical and experimental observations have demonstrated that disturbed flow and/or low wall shear stress accelerate development of intimal hyperplasia.15,27,28 Endothelial cells respond to these particular hemodynamic conditions by elaborating adhesion molecules, proinflammatory cytokines, and other bioactive substances that in turn enhance cell proliferation and matrix accumulation, leading to robust intimal growth.29,30 On the other hand, laminar, high blood flow (uniform high shear stress) generally exhibits an opposing effect on intimal growth. For example, vein grafts exposed to high flow conditions tend to develop less intima than those with low blood flow.15 Furthermore, augmentation of blood flow in vessels with established intimal hyperplasia induces intimal regression.31 Although the exact mechanisms remain to be fully elucidated, experimental studies suggest that high laminar shear shifts the local vascular milieus from a proinflammatory to an antiinflammatory state, imposing a differential regulation of intimal growth.26 This knowledge has been translated to clinical application, so that creation of a distal fistula to boost the blood flow has led to improvement in the patency rate of lower extremity grafts in selected circumstances.32 Although generally protective, shear may be deleterious when it reaches an extremely high level or becomes disordered.33 Finally, compared with the well-established effect of shear stress, the impact of tensile force on intimal hyperplasia is much less defined. In the cell culture setting, mechanical stretch activates several pathways capable of regulating smooth muscle cell phenotype.34,35 Evidence in vivo suggests that tensile force correlates positively with intimal thickening.16,36
Immune Insults
Solid organ transplantation usually introduces a mismatch in the HLA complex between the transplant and the host immune system, triggering immunologic responses that accelerate formation of diffuse, concentric intimal hyperplastic lesions in the arteries of the allograft that may eventually result in allograft failure. Reflective of this complex immune scenario, allograft vasculopathy is more vulnerable to in-stent restenosis after intervention than the stenotic lesions in native coronary arteries.37 Multiple cell groups, particularly T-cell subsets (CD4+ and CD8+) and B cells, appear to be critical to the pathogenesis of these intimal hyperplastic lesions in the transplant setting. Activated by antigen recognition through either direct (major histocompatibility complex I [MHC I]–dependent) or indirect (MHC II–dependent) pathways, these cells may cause endothelial cell dysfunction via cytolysis, cytokine stimulation (interferon-γ [IFN-γ], interleukin-12 [IL-12]), and specific antibody production.38 Beyond the transplant setting, similar immunologic insult may occur in stented vessels, where immune cells are activated via a response to foreign bodies.39 Finally, risk factors for accelerated intimal hyperplasia identified in clinical studies (hyperlipidemia, diabetes, and smoking) may act by way of augmenting chronic inflammation in the vessel wall.
Genetic Susceptibility
In addition to environmental factors, genetic status stands as a risk factor for intimal hyperplasia. Some of this knowledge was acquired via genome-wide association studies (GWASs). For example, single-nucleotide polymorphisms (SNPs) in the IL-10 gene are associated with higher rate of restenosis in coronary arteries treated with drug-eluting stents.40 Susceptible loci in chromosome 12 have also been documented.41 These genetic discoveries have highlighted the horizon for genetic manipulations as a therapy for inhibiting intimal growth.
Biologic Mechanisms
Intimal hyperplasia is a highly complex process that involves several tissues (perivascular,42 vessel wall, and blood), numerous cell lineages, and multiple molecular signaling networks. Although much has been learned in the past few decades, many mechanistic details remain to be fully elucidated. The current paradigm is founded on the postulate that intimal hyperplasia is a vascular response to injury, with the cellular and molecular events generally mirroring those occurring in the course of wound healing.8,43
Cellular Effectors
Compared with the medial layer, the neointimal lesion contains relatively more extracellular matrix, with a disordered cell-matrix organization that mimics tissue fibrosis immediately beneath the intact endothelial cell lining. However, unlike reactive fibrotic tissue, which is primarily populated with fibroblasts, vascular intimal lesions are dominated by cells that express smooth muscle cell markers, such as α-actin and smooth muscle myosin heavy chain (SM-MHC). Work in the 1980s suggested that these cells are largely medially derived.44 However, in addition to derivations from the tunica media, later studies have identified cells from nonmedial sites, such as the adventitia and perivascular tissue,45 stem cell/progenitor cell niches,46,47 and the circulating blood as suppliers of the intimal cell population.
Once recruited to the intimal lesion, these cells gain a phenotype similar to those derived from the medial smooth muscle cells, making them indistinguishable from each other.48,49 Although vascular wall cells are normally relatively quiescent, the repopulated intimal cells often display a proliferative and synthetic state. Substances that are not produced basally in the vasculature (e.g., cytokines, growth factors, and adhesion molecules) are induced in these neointimal cells.50 Acting through autocrine and paracrine mechanisms, these substances maintain an inflamed state for the intimal tissue, collectively propelling progressive growth of the intimal lesion.
Endothelial Cells
The luminal surface of blood vessels is covered by a monolayer of endothelial cells. Under physiologic conditions, these cells serve as a “physical barrier” that segregates the circulating blood cells and molecules from tissue matrix components, and also function as a “secretory organ,” releasing an array of mediators to maintain homeostasis in the wall and beyond. Lining the luminal surface, endothelial cells produce transmembrane adhesion molecules such as vascular endothelial cadherin (VE-cadherin) and neural cadherin (N-cadherin) for intercellular connections. Interacting with the intracellular cytoskeleton, these molecules form adhesive junctions (e.g., tight junctions, gap junctions, and adherens junctions) with neighboring cells, allowing selective exchange of molecules between blood and tissue.51 Anticoagulants (e.g., heparin, thrombomodulin, prostaglandin I2 [PGI2], and Kruppel-like factor-2) are synthesized and released by these cells to maintain a relatively anticoagulant and thus thrombus-free luminal surface.
In addition to these “direct” biologic effects, endothelial cells govern the homeostasis of the vessel wall via communication with the underlying smooth muscle cells. A well-defined tool for endothelial cells to carry out this function is NO, which has been linked with intimal hyperplasia.52 NO is very lipophilic. Once released, it quickly diffuses into the medial layer and acts directly on smooth muscle cells. The half-life of the endogenous NO is so short (0.1-5 seconds) that the effect on smooth muscle cells can be rapidly turned on or off depending on demand. NO was initially identified as endothelium-derived relaxing factor because of its function to induce vasodilatation. Subsequent studies demonstrated that NO is also a key messenger for endothelial cells to keep the medial smooth muscle cells at a quiescent and contractile phenotype.53
Upon activation by forces such as altered hemodynamics or physical/chemical trauma, endothelial cells convert their transcriptome to a profile that is significantly different from that of their quiescent state.54 Leukocyte and platelet adhesion molecules are upregulated or translocate to the cell membrane. Genes encoding proinflammatory mediators (e.g., chemokines, cytokines, adhesion molecules) are induced or upregulated, whereas those encoding antiinflammatory mediators (e.g., NO, anticoagulants, Kruppel-like factor-2) are downregulated.55 These bioactive substances can trigger a series of events, such as smooth muscle cell activation, inflammatory cell infiltration, and the recruitment of vascular progenitor cells—all contributing to intimal hyperplasia. Finally, studies have now suggested a new mechanism whereby endothelial cells may contribute to intimal hyperplasia through endothelial-mesenchymal transition (EMT).56,57
Smooth Muscle Cells
The intimal hyperplasia tissue, just like the tunica media (though morphologically distinct), is dominated primarily by smooth muscle cells. During the development of intimal hyperplasia, intimal smooth muscle cells display a spectrum of phenotypes with preferential losses and gains of their differentiation markers at various stages. The initial dedifferentiation process leads to a loss of expression of the majority of the differentiation markers, such as α-actin, desmin, and smooth muscle isoforms SM1 and SM2. As the lesion becomes more established, early differentiation markers such as α-actin and desmin are gained relatively quickly. The reproduction of the more mature markers, such as smooth muscle isoforms SM1 and SM2, on the other hand, may be very much delayed.58,59
Although they normally function in the media primarily as the drivers for vascular relaxation and constriction, smooth muscle cells in the intimal lesions serve as effectors that produce cellular and extracellular components to expand the intimal mass. Having switched to the proliferative/synthetic state, smooth muscle cells respond to local cytokines and growth factors by vigorous cell division and matrix production. Experimental studies have demonstrated that the proliferation may start a few hours after an inciting event, reach a peak in a few days, and last for weeks to months.15,60 Genes encoding matrix components (particularly interstitial matrix proteins such as hyaluronic acid, chondroitin, collagens, proteoglycans, fibronectin, and elastin) are greatly upregulated. On the other hand, the production and the activity of the matrix degradation system, which is consisted of proteases (e.g., MMPs, tissue plasminogen [t-PA], urinary plasminogen activator [u-PA], and plasminogen) and their counterpart inhibitors (e.g., tissue inhibitors of metalloproteinases [TIMPs] and plasminogen activator inhibitor-1 [PAI-1]), is modulated to favor a selective deposition of matrix proteins,8,61 leading to an accumulation of de novo interstitial matrix with an altered composition.
In addition to their role of directly making intimal mass, smooth muscle cells synthesize and release a wide spectrum of mediators to the extracellular space.50 Cytokines (e.g., IL-1β and tumor necrosis factor-α [TNF-α]), chemokines (e.g., IL-8 and monocyte chemoattractant proteins [MCPs]), and growth factors (e.g., platelet-derived growth factor [PDGF] and transforming growth factor-β [TGF-β]) have been documented as intimal smooth muscle cell products. The exact profile of these mediators varies according to the stage of intimal hyperplasia development. These mediators may act on smooth muscle cells themselves and other cell lineages through autocrine and/or paracrine function. Although the early mediators serve to amplify the inflammatory response and favor smooth muscle cell migration and proliferation, driving a hyperplastic response with high cellularity,62,63 those released by smooth muscle cells in the established neointima facilitate a fibrotic process.60
Finally, vascular smooth muscle cells may lose their production of contractile proteins through dedifferentiation and acquire a phenotype that overlaps with another vascular cell group, the myofibroblasts. Studies of both systemic hypertension and pulmonary hypertension have demonstrated a critical role for the adventitia and adventitial fibroblasts in the structural remodeling and the resultant pathology of the vessel wall,64,65 and these cells may participate in intimal hyperplasia.66–68 Like medial and intimal smooth muscle cells, myofibroblasts likely stand as cellular effectors that produce and react to various bioactive substances, modulating structural remodeling and intimal growth in the vessel wall.69
Platelets and Leukocytes
Following vascular injury, platelets aggregate on activated endothelial cells and denuded areas to cover the collagens and other matrix components that are exposed to the blood. Fibrin is deposited, together with the activated platelets, forming an adhesive surface to capture circulating leukocytes from the blood stream.70–72 Inflammatory cells such as monocytes, eosinophils, and T cells home to the injured site. Although representing only a relatively small portion in the eventual intimal cell population, inflammatory cells have been identified as central modulators and potential key orchestrators of hyperplastic intimal thickening. For example, clinical studies have demonstrated a positive correlation between leukocyte count and activation with the severity of stenotic and restenotic coronary lesions.73,74 The level of inflammatory markers typically associated with leukocyte activity, particularly C-reactive protein, holds some correlation with clinical outcomes.75,76 Strategies blocking leukocyte adhesion or creating leukopenia significantly inhibit intimal thickening in various experimental settings.72,77 Through their robust ability to produce various inflammatory mediators, including chemokines, cytokines, and growth factors, leukocytes appear to amplify the acute inflammation, contributing to intimal thickening.78
Leukocytes consist of several subpopulations. The quantity of each subset in intimal lesions seems to be determined by the involvement of specific initiating factors and the stage of the pathology. For example, lesions that developed in the setting of hypercholesterolemia or after severe physical injury tend to hold more macrophages than those triggered solely by endothelial denudation.72 Otherwise, neutrophils dominate the inflammatory infiltrates after moderate injury such as endothelial denudation.79 There is also evidence that T cells stimulate intimal hyperplasia via promoting a helper T cell type 1 (Th1) response,80 but this mechanism may be more significant in the pathogenesis of allograft vasculopathy.81 Advances in immunology have led to appreciation of distinct roles for subsets of monocytes and macrophages in the vascular inflammatory response. “Patrolling” and “resident” monocytes have been identified in the peripheral blood.82,83 Upon extravasation into tissue, these cells follow varying pathways to differentiate into M1 and M2 macrophages, performing distinct roles in pathogenesis and tissue repair.84,85 For example, M2 polarization of the resident monocytes has been demonstrated to be indispensable to arterial remodeling triggered by hemodynamic stress.86 Recent research efforts have attempted to identify the key regulators for these processes so that interventional approaches may be developed to guide the kinetics of the inflammatory cell trafficking and intimal lesion progression.87
Progenitor Cells
In addition to smooth muscle cells, several other cell groups are recruited to neointimal lesions. Vascular progenitor cell lines released from bone marrow and other stem cell niches may home to intimal hyperplasia.46,49 Initial evidence suggests that smooth muscle progenitors are released from stem cell niches to the circulation.88 Not only can these cells gain smooth muscle cell markers but they also seemed at first to respond to exogenous stimulation much like smooth muscle cells from tunica media do.89 However, subsequent studies have demonstrated that cells from circulation and bone marrow lack the ability to produce a full repertoire of smooth muscle cell contractile proteins in the intimal lesion90 and that they tend to maintain a sustained inflammatory phenotype.91
A groundbreaking discovery has been the participation of endothelial progenitor cells in reendothelialization of the injured luminal surface.92 Experimental studies revealed that approaches facilitating the homing of these progenitor cells to the lesion site significantly accelerated the recovery of the endothelial cell monolayer and inhibited neointimal hyperplasia.93 Clinical observations support an association of the impaired endothelial progenitor cell function with higher rate of stenosis and restenosis.94 Because the endothelial progenitor cell is a relatively rare element in circulating blood, strategies such as capturing antibodies and adhesion molecules or growth factors to enrich endothelial progenitor cells at lesion sites are being evaluated.95,96
Biochemical Mediators
[/not-level-membership-for-surgery-category]
