[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 109 Intestinal Protozoa
ENTAMOEBA HISTOLYTICA
EPIDEMIOLOGY
Entamoeba histolytica was first linked causally to amebic colitis and liver abscess by Lösch in 1875, and it was named by Schaudinn in 1903 for its ability to destroy host tissues. In 1925, Emil Brumpt proposed the existence of a second, morphologically identical but nonpathogenic Entamoeba species, Entamoeba dispar, to explain why only a minority of people infected with what was then termed E. histolytica develop invasive disease. Although Brumpt’s hypothesis was not accepted during his lifetime, it is now clear that he was correct and E. histolytica (Schaudinn, 1903) was recently reclassified to include two morphologically indistinguishable species: E. histolytica, the cause of invasive amebiasis, and E. dispar, a nonpathogenic intestinal commensal parasite (see later section).1
Entamoeba histolytica is a parasite of global distribution, but most of the morbidity and mortality from amebiasis occurs in Central and South America, Africa, and the Indian subcontinent.2 Fortunately, the majority of the 500 million persons worldwide previously believed to be asymptomatic E. histolytica cyst passers are actually infected with E. dispar. The best current estimate is that E. histolytica causes 34 to 50 million symptomatic infections annually worldwide, resulting in 40,000 to 100,000 deaths each year.3,4 In Dhaka, Bangladesh, where diarrheal diseases are the leading cause of childhood death, 80% of children studied prospectively were infected with E. histolytica at least once during four years of follow-up.5 Furthermore, E. histolytica–associated diarrhea in these children was associated with significantly low weight and height for age.6
E. histolytica has a simple, two-stage life cycle consisting of an infectious cyst and a motile trophozoite (Fig. 109-1). The cyst form measures 5 to 20 µm in diameter and contains four or fewer nuclei. The ameboid trophozoite, which is responsible for tissue invasion, measures 10 to 60 µm (Fig. 109-2) and contains a single nucleus with a central karyosome (Fig. 109-3). The cysts are relatively resistant to chlorination and desiccation, and they can survive in a moist environment for several weeks.
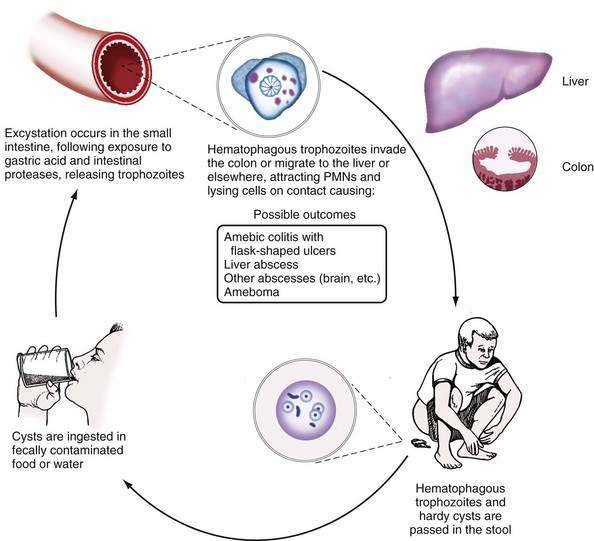
Figure 109-1. Life cycle of Entamoeba histolytica. PMNs, polymorphonuclear neutrophils.
(From Petri WA, Sing U, Ravdin JI. Enteric amebiasis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia: WB Saunders; 1999.)
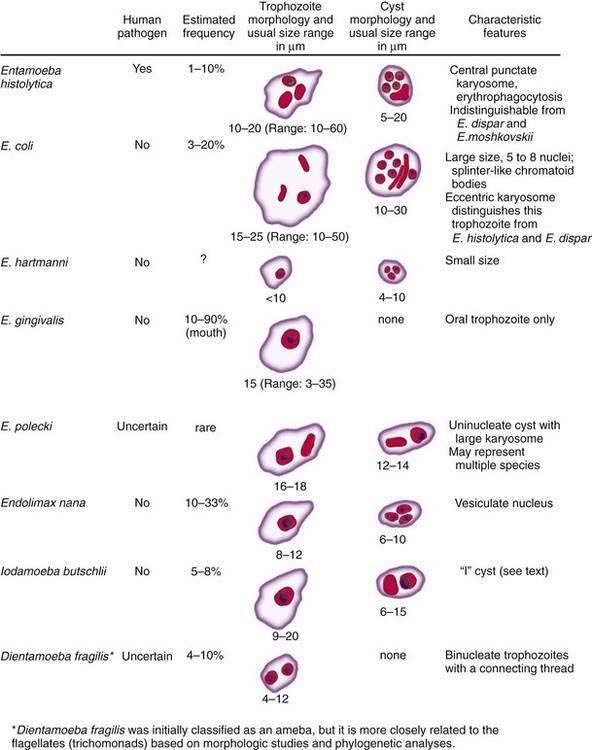
Figure 109-2. Amebae that infect the human gastrointestinal tract. E., entamoeba.
(From Ravdin Jl, Guerrant RL. Current problems in the diagnosis and treatment of amebic infections. Curr Clin Trop Infect Dis 1986; 7:82.)
Infection occurs following ingestion of cysts in fecally contaminated food or water. Within the lumen of the small intestine, the quadrinucleate cyst undergoes nuclear then cytoplasmic division, giving rise to eight trophozoites.7 Only about 10% of infected persons develop invasive disease characterized by invasion of the colonic epithelium by trophozoites.1 Trophozoites that gain access to the bloodstream can spread hematogenously to establish infection at distant sites (most commonly liver abscess, as discussed in Chapter 82). Why some persons develop invasive disease and others remain asymptomatic remains a mystery; parasite and host differences are likely to be important in this regard.
A molecular epidemiologic study that used the polymerase chain reaction (PCR) to amplify a polymorphic region of the E. histolytica genome and assign a genotype to different clinical isolates has demonstrated a correlation between different E. histolytica strains and the outcome of infection.8 The specific underlying genetic differences among ameba strains that are responsible for altered virulence, however, remains unknown. Furthermore, amebic liver abscess is primarily a disease of men, and studies suggest that susceptibility to both intestinal and hepatic amebiasis is linked to human leukocyte antigen (HLA) class II alleles.9,10 For example, the HLA DQB1*0601 allele may be associated with protection from intestinal amebiasis.9 As is the case for genetic differences among E. histolytica strains, however, there is no evidence of a direct causal role for different HLA types; rather, these HLA types are likely to be in linkage disequilibrium with genes in the nearby vicinity that encode the causal factors.
PATHOGENESIS, PATHOLOGY, AND IMMUNOLOGY
Both amebic factors and the host’s inflammatory response contribute to tissue destruction during invasive amebiasis. Microscopy studies have defined a stepwise progression of disease.11–13 After excystation within the lumen of the small intestine, trophozoites adhere to colonic mucins and epithelial cells, largely via an amebic galactose/N-acetyl-d-galactosamine inhibitable surface lectin.14–16 Secreted cysteine proteinases then facilitate tissue invasion by degrading human colonic mucus and extracellular matrix proteins.17–20 Further disruption of the colonic epithelium results directly from contact-dependent cytolysis of epithelial and immune cells and from an acute epithelial cell inflammatory response with recruitment of neutrophils and immune-mediated tissue damage.14,21–25
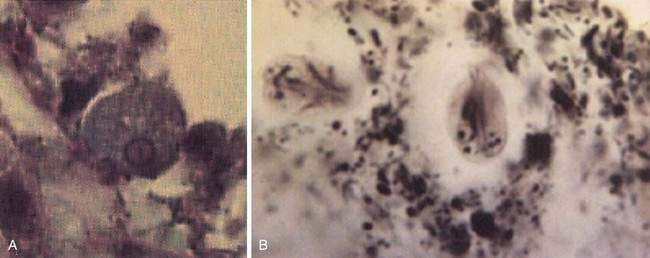
The cecum and ascending colon are affected most commonly, although in severe disease the entire colon may be involved. On gross examination, pathology can range from mucosal thickening to multiple punctate ulcers with normal intervening tissue (Fig. 109-4) to frank necrosis. For unknown reasons, the downward invasion of amebic trophozoites often is halted at the level of the muscularis mucosa. Subsequent lateral spread of amebae undermines the overlying epithelium, resulting in the clean-based, flask-shaped ulcers that characterize amebic colitis.26,27 Early in infection, an influx of neutrophils is typical, but in well-established ulcers, few inflammatory cells are seen.13,26–28 Organisms may be seen ingesting red blood cells (erythrophagocytosis) (Fig. 109-5). At distant sites of infection (e.g., liver abscess), similar pathologic characteristics include central liquefaction of tissue surrounded by a minimal mononuclear cell infiltrate.27–29
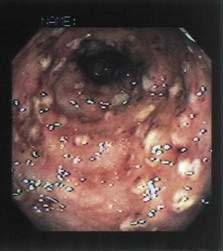
Figure 109-4. Colonoscopic findings in a patient with amebic colitis. Multiple punctate ulcers are visible.
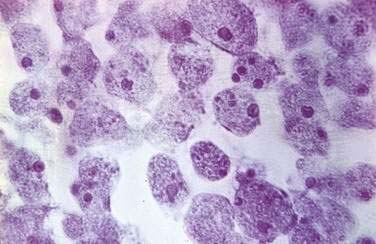
Because more than 90% of persons colonized with E. histolytica spontaneously clear the infection within a year, an effective immune response to amebiasis seems to develop.30 Children with fecal anti-amebic lectin immunoglobulin (Ig)A have short-lived protection from subsequent intestinal infection.5,31,32 The protective role of secretory IgA is not certain, however, and the contributions of humoral and cellular immunity to protection from amebiasis remain unknown. Nearly everyone with invasive amebiasis develops a systemic and a mucosal humoral immune response.33–38 Antibodies alone are unable to clear established infection, however, because asymptomatic cyst passers remain infected for months after anti-amebic antibodies develop.30,33 Passive immunization experiments in a severe combined immunodeficient (SCID) mouse model of liver abscess do suggest an important role for preexisting humoral immunity in protection from infection.39 Reports that patients receiving glucocorticoids may be at increased risk for severe amebic colitis suggest that cellular immunity also plays an important role in control of E. histolytica infection40,41; despite this concern, no increase in disease severity in patients with AIDS has been observed. In fact, in a mouse model of amebic colitis, disease was exacerbated by CD4+ T cells.42
CLINICAL FEATURES
Infection with E. histolytica results in one of three outcomes. Approximately 90% of infected persons remain asymptomatic. The other 10% of infections result in invasive amebiasis characterized by dysentery (amebic colitis) or, in a minority of cases, extraintestinal disease (most commonly amebic liver abscess; see Chapter 82).1,30
In the United States, immigrants from or travelers to endemic regions, male homosexuals, and institutionalized persons are at greatest risk for amebiasis. In addition, malnourished patients, infants, the elderly, pregnant women, and patients receiving glucocorticoids may be at increased risk for fulminant disease.2,40,41 When one or more of these epidemiologic risk factors are present, amebic dysentery should be considered in the differential diagnosis of occult or grossly bloody diarrhea.
The major diagnostic challenge for the clinician seeing a patient with amebic colitis is to distinguish the illness from other causes of bloody diarrhea. The differential diagnosis includes the causes of bacterial dysentery, such as Shigella, Salmonella, and Campylobacter species and enteroinvasive or enterohemorrhagic Escherichia coli, and noninfectious diseases, including inflammatory bowel disease, and ischemic colitis.2,43 In contrast to bacterial dysentery, which typically begins abruptly, amebic colitis begins gradually over one to several weeks (Table 109-1). Although more than 90% of patients with amebic colitis present with diarrhea, abdominal pain can occur without diarrhea; abdominal pain, tenesmus, and fever are highly variable. Weight loss is common because of the chronicity of the illness. Microscopic blood is present in the stool of most patients with amebic dysentery.2,43,44
Table 109-1 Comparison of Amebic Colitis and Invasive Bacterial Dysentery
| FEATURE | AMEBIC COLITIS | BACTERIAL DYSENTERY* |
|---|---|---|
| Travel to or from an endemic area | Yes | Sometimes |
| Usual duration of symptoms | >7 days | 2-7 days |
| Diarrhea | 94-100% | 100% |
| Fecal occult blood | 100% | 40% |
| Abdominal pain | 12-80% | ~50% |
| Weight loss | Common | Unusual |
| Fever >38°C | Minority | Majority |
Adapted from Huston CD, Petri WA. Amebiasis. In: Rakel RE, Bope ET, editors. Conn’s Current Therapy, 2001. Philadelphia: WB Saunders; 2001. pp 50-4.
The most feared complication of amebic dysentery, acute necrotizing colitis with toxic megacolon, occurs in 0.5% of cases. This complication manifests as an acute dilatation of the colon, and 40% of patients die from sepsis unless it is promptly recognized and treated surgically.45,46 Unusual complications include the formation of enterocutaneous, rectovaginal, and enterovesicular fistulas and ameboma. Ameboma, due to intraluminal granulation tissue, can cause bowel obstruction and mimic carcinoma of the colon.2,43
Although a history of dysentery early in the illness is common, dysentery has resolved in most patients by the time of presentation.47–49 Extraintestinal sites of infection are involved and typically result either from direct extension of liver abscesses (e.g., amebic pericarditis or lung abscess) or from hematogenous spread of disease (e.g., brain abscess).2,50
DIAGNOSIS
Because amebiasis patients erroneously treated for inflammatory bowel disease with glucocorticoids can develop fulminant colitis, accurate initial diagnosis is critical.40,41 The gold standard for diagnosis of amebic colitis remains colonoscopy with biopsy, and colonoscopy should be performed whenever infectious causes of bloody diarrhea are strong considerations in the differential diagnosis of ulcerative colitis. Because the cecum and ascending colon are affected most often, colonoscopy is preferred to sigmoidoscopy. Classically, multiple punctate ulcers measuring 2 to 10 mm are seen with essentially normal intervening tissue (see Fig. 109-4); however, the colonic epithelium might simply appear indurated with no visible ulcerations; appear like ulcerative colitis with a myriad of ulcerations and granular, friable mucosa; or, in severe cases where the ulcers have coalesced, the epithelium may appear necrotic. Histologic examination of a biopsy specimen taken from the edge of an ulcer reveals amebic trophozoites and a variable inflammatory infiltrate (see Fig. 109-5).27 Identification of amebae can be aided by periodic acid–Schiff staining of biopsy tissue, which stains trophozoites magenta.
Stool examination for ova and parasites, the traditional method for diagnosing amebiasis, should not be relied upon. Although the presence of amebic trophozoites with ingested erythrocytes strongly correlates with E. histolytica infection, these rarely are present,51 and in the absence of hematophagous trophozoites, microscopy cannot distinguish E. histolytica from E. dispar. Difficulty in distinguishing other nonpathogenic amebae (see later) and white blood cells from E. histolytica also limits the specificity of stool microscopy.52 The sensitivity of microscopy for identification of amebae is at best 60% and it may be reduced by delays in processing of stool samples.52,53 The primary utility of stool microscopy for ova and parasites in a patient with diarrhea, therefore, is to evaluate the stool for other parasitic causes of diarrhea.
Noninvasive methods to accurately differentiate E. histolytica from E. dispar include stool culture with isoenzyme analysis, serum amebic-antibody titers, PCR, and an enzyme-linked immunosorbent assay (ELISA) that detects the amebic lectin antigen in stool samples.54–64 Of these, only serum amebic-antibody titers and the stool ELISA are widely available for clinical use.
Because serum anti-amebic antibodies do not develop in patients infected with E. dispar, serologic tests for amebiasis accurately distinguish E. histolytica and E. dispar infection. From 75% to 85% of patients with acute amebic colitis have detectable anti-amebic antibodies on presentation, and convalescent titers develop in more than 90% of patients.34,35,65 For amebic liver abscess, 70% to 80% of patients have detectable antibody titers on presentation, and convalescent titers develop in more than 90% of patients. Because antiamebic antibodies can persist for years, however, a positive result must be interpreted with caution.34 For persons with known epidemiologic risks (e.g., emigration from or prior travel to an endemic region), a positive result might simply represent infection in the distant past. In the setting of recent travel to an endemic region and a positive antibody titer, diagnosis is confirmed by an appropriate symptomatic response to anti-amebic treatment.
The most specific clinically available test for diagnosis of amebiasis is a stool ELISA to detect the E. histolytica adherence lectin. Only one of the many ELISA tests developed thus far (the E. histolytica II test, TechLab, Blacksburg, Va.) accurately distinguishes E. histolytica from E. dispar.53,60,61 This test’s specificity, when compared with the gold standard of stool culture followed by isoenzyme analysis, was greater than 90%, and it was greater than 85% sensitive for diagnosis of intestinal amebiasis when fresh fecal samples were analyzed without delay.61 In other studies, the sensitivity of this method has been less impressive, emphasizing the need for rapid processing of stool samples.66,67 It also may be possible to use this antigen detection test to diagnose amebic liver abscess, because before treatment is initiated, amebic lectin antigen can be detected in the serum of greater than 90% of patients who have amebic liver abscess.68
TREATMENT
Drugs for treatment of amebiasis are categorized as luminal or tissue amebicides on the basis of the location of their anti-amebic activity (Table 109-2).
Table 109-2 Amebicidal Agents Currently Available in the United States
| AMEBICIDAL AGENT | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| For Luminal Amebiasis | ||
| Paromomycin (Humatin) | 7-day treatment course; may be useful during pregnancy | Frequent gastrointestinal side effects; rare ototoxicity and nephrotoxicity |
| Iodoquinol (Yodoxin) | Inexpensive and effective | 20-day treatment course; contains iodine; rare optic neuritis and atrophy with prolonged use |
| Diloxanide furoate (Furamide) | Available in the United States only from the CDC; frequent gastrointestinal side effects; rare diplopia | |
| For Invasive Intestinal Disease Only | ||
| Tetracyclines, erythromycin | Not effective for liver abscess; frequent gastrointestinal side effects; tetracyclines should not be administered to children or pregnant women | |
| For Both Invasive Intestinal and Extraintestinal Amebiasis | ||
| Metronidazole (Flagyl) | Drug of choice for amebic colitis and liver abscess | Anorexia, nausea, vomiting, and metallic taste in nearly one third of patients; disulfiram-like reaction with alcohol; rare seizures |
| Tinidazole (Tindamax) | Alternative to metronidazole; once daily dosing; now approved for distribution in the United States | Side effects are similar to those with metronidazole |
| Nitazoxanide (Alinia) | Useful alternative if the patient is intolerant of metronidazole or tinidazole | Limited clinical data for amebiasis; rare and reversible conjunctival icterus |
| For Extraintestinal Amebiasis Only | ||
| Chloroquine (Aralen) | Useful only for amebic liver abscess | Occasional headache, pruritus, nausea, alopecia, and myalgias; rare heart block and irreversible retinal injury |
CDC, Centers for Disease Control and Prevention.
Adapted from Huston CD, Petri WA. Amebiasis. In: Rakel RE, Bope ET, editors. Conn’s Current Therapy, 2001. Philadelphia: WB Saunders; 2001. pp 50-4.
The luminal amebicides include iodoquinol, diloxanide furoate, and paromomycin.69,70 Of these, paromomycin, a nonabsorbable aminoglycoside, is preferred because of its safety, short duration of required treatment, and superior efficacy. Its major side effect is diarrhea. Approximately 85% of asymptomatic patients are cured with one course of paromomycin, and, because it is nonabsorbable and has moderate activity against trophozoites that have invaded the colonic mucosa, it might also be useful for single-drug treatment of mild invasive disease during pregnancy.71,72
The tissue amebicides include metronidazole, tinidazole, nitazoxanide, erythromycin, and chloroquine.70,73 Of these, metronidazole and tinidazole are the drugs of choice, with cure rates greater than 90%.74 Nitazoxanide, a new antiparasitic agent, appears to be efficacious, with similar cure rates in several randomized, placebo-controlled trials.73,75–77 Erythromycin has no activity against amebic liver disease, and chloroquine has no activity against intestinal disease.78
Because approximately 10% of asymptomatic cyst passers develop invasive amebiasis, E. histolytica carriers should be treated.1,4 For noninvasive disease, treatment with a luminal agent alone is adequate (e.g., paromomycin 25-35 mg/kg/day in three divided doses for seven days).70 Patients with amebic colitis should first be treated with an oral nitroimidazole (either metronidazole [500-750 mg three times daily for 10 days] or tinidazole [2 grams once daily for three to five days]) to eliminate invasive trophozoites. Metronidazole and tinidazole are believed to be less effective against organisms in the colonic lumen, and subsequent treatment with a luminal agent such as paromomycin is recommended to prevent recurrent disease.70,74
It is also for this reason that the familiar tissue amebicides (e.g., metronidazole) are not recommended as first-line agents for treatment of asymptomatic infection. At the recommended doses of metronidazole and tinidazole, gastrointestinal side effects including nausea and vomiting develop in approximately 30% of patients.74 Because of severe gastrointestinal side effects, simultaneous treatment with a nitroimidazole and a luminal agent generally is not recommended.
Most patients with colitis respond promptly with resolution of diarrhea in two to five days.2
Despite conflicting reports on the safety of the nitroimidazoles for the developing fetus during pregnancy, women with severe disease during pregnancy should probably be treated without delay. As discussed in Chapter 82, metronidazole (750 mg three times a day for 10 days) followed by a luminal agent is also the treatment of choice for amebic liver abscess.70,78
CONTROL AND PREVENTION
Prevention and control of E. histolytica infection depends on interruption of fecal-oral transmission. Water can be made safe for drinking and food preparation by boiling it for one minute, by halogenation (with chlorine or iodine), or by filtration.7 In the United States and Europe, modern water treatment facilities effectively remove E. histolytica. The importance of safe drinking water is highlighted by an outbreak of amebiasis in Tblisi, Republic of Georgia, where there was a water-borne epidemic due to decay of the water treatment facilities following the demise of the Soviet Union.79 In the vast majority of the developing world, however, no modern water treatment facilities exist and none are likely to be constructed in the foreseeable future. Naturally acquired immunity to intestinal amebiasis provides short-lived protection against reinfection, giving hope that a vaccine may be feasible.5,31,32 Because humans and some higher nonhuman primates are the only known hosts for E. histolytica, a vaccine that successfully prevents colonization might enable eradication of the disease.80
OTHER INTESTINAL AMEBAE
Eight species of commensal amebae commonly infect the human gastrointestinal tract (see Fig. 109-2). These include Entamoeba dispar, Entamoeba moshkovskii, Entamoeba coli, Entamoeba hartmanni, Entamoeba gingivalis, Entamoeba polecki, Endolimax nana, and Iodamoeba butschlii. Dientamoeba fragilis (discussed in the following section), previously thought to be an ameba, is more closely related to the flagellated protozoan Trichomonas vaginalis than to the true amebae.7 With the exception of E. gingivalis, which has no known cyst stage, all of these true amebae have simple two-stage life cycles, consisting of an infectious cyst form and a motile trophozoite form.7 All but E. dispar and E. moshkovskii can be differentiated from E. histolytica using light microscopy based on characteristic features of the cyst and trophozoite forms. E. dispar must be differentiated from E. histolytica based on biochemical, antigenic, or genetic differences.1
E. dispar is a nonpathogenic protozoan parasite that is morphologically indistinguishable from Entamoeba histolytica by light microscopy.1 An estimated 450 million people worldwide are infected with E. dispar, and infection with E. dispar is approximately 10 times more prevalent than E. histolytica infection.1,3,4 Although E. dispar has been demonstrated to cause mucosal ulcerations in animal models, it has not been demonstrated to cause human disease and does not require treatment.1 Entamoeba moshkovskii, which is primarily thought to be a free-living ameba, also has cysts and trophozoites indistinguishable from E. dispar and E. histolytica except that trophozoites of E. histolytica might show erythrophagocytosis. Although in some studies a high prevalence of E. moshkovskii infection in humans has been demonstrated, reports are conflicting regarding its pathologic potential.81–83 The primary clinical significance of E. dispar or E. moshkovskii is that they must be distinguished from E. histolytica to enable accurate diagnosis of invasive amebiasis. PCR to amplify small ribosomal RNA (not clinically available), and ELISAs using monoclonal anti-amebic antibodies to detect specific E. histolytica antigens, make accurate diagnosis possible (see the section on E. histolytica diagnosis).55–64
Besides E. dispar, Entamoeba coli is the intestinal commensal most commonly mistaken for E. histolytica. Entamoeba coli trophozoites contain a single nucleus with a prominent karyosome that usually is eccentric in location, distinguishing them from E. histolytica and E. dispar trophozoites, which have a centrally located karyosome. In addition, the cyst form of Entamoeba coli typically contains five to eight nuclei (see Fig. 109-2). Entamoeba coli is nonpathogenic and requires no specific treatment; however, it is a valuable marker of fecal-oral exposure, and it can be found concurrently with E. histolytica in 10% to 30% of patients in endemic regions.7
Entamoeba hartmanni was classified as “small race” E. histolytica for many years. The trophozoites resemble those of E. histolytica except for their small size (less than 10 µm).7 Entamoeba hartmanni now is recognized as a nonpathogen that requires no treatment.
Entamoeba gingivalis is the only ameba found in the oral cavity, where it lives in the anaerobic environment of the gingival crease. The trophozoite is identical in size to that of E. histolytica and contains a single nucleus with a prominent central karyosome (see Fig. 109-2). No cyst form of E. gingivalis has been identified, and oral-oral contact is believed to be its mode of transmission.7,84 Entamoeba gingivalis is associated with poor dental hygiene and periodontal disease, but no causal relationship to periodontitis has been proven.84 The increased frequency of colonization in this setting might simply reflect a more hospitable host environment. E. gingivalis is often associated with periodontal disease in AIDS patients, however, in whom treatment with metronidazole has been reported to be effective.85
Entamoeba polecki, a parasite characterized by a uninucleated cyst, is primarily a parasite of pigs and monkeys that sometimes infects humans. It has been suggested that several distinct uninucleated cyst-producing Entamoeba species can infect humans and it has been proposed that these organisms collectively be termed “E. polecki-like.”86 Infection with E. polecki is rare except in Papua New Guinea where as many as 30% of children were found to be colonized in one study.87 At present, specific treatment of E. polecki–like infections is not routinely recommended, but persons with heavy burdens of this parasite can develop nonspecific gastrointestinal symptoms and might benefit from treatment. Good clinical responses to metronidazole and diloxanide furoate have been reported.88
Endolimax nana is a nonpathogenic intestinal ameba that often infects humans.7 The distribution of E. nana is worldwide, but it is most common in the tropics, where 5% to 33% of persons are infected.89,90 Infection requires no specific treatment, but it serves as a useful marker for fecal-oral exposure. E. nana trophozoites can be distinguished from E. histolytica by their vesiculate nucleus, large irregular karyosome, and relatively small size (8 to 12 µm).7
Iodamoeba butschlii is a nonpathogenic intestinal ameba passed by the fecal-oral route. Trophozoites of I. butschlii contain a single nucleus with a large karyosome (which is distinct from the punctate karyosome of E. histolytica); its cysts contain a single nucleus, and a large, eccentric glycogen mass that stains with iodine (hence the name Iodamoeba). I. butschlii infection requires no treatment.7
GIARDIA INTESTINALIS
EPIDEMIOLOGY
Giardia intestinalis (also called G. lamblia and G. duodenalis) is a ubiquitous flagellated intestinal protozoan. Van Leeuwenhoek accurately described its motile trophozoite form in his own stools in 1681, but it was not until 1915 that Stiles named the species.7
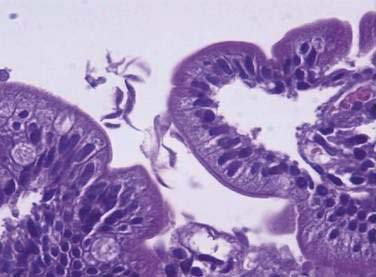
The life cycle of Giardia consists of an infectious cyst form and a motile trophozoite (Fig. 109-6). The cyst is oval (8 to 12 µm long by 7 to 10 µm wide), contains four nuclei, and has a rigid outer wall that protects it from dehydration, extremes of temperature, and chlorination (Fig. 109-7). Giardia cysts can survive in cold water for several weeks.7,91 Ingestion of as few as 10 to 25 cysts can result in infection.91 After ingestion, excystation occurs following exposure to stomach acid and intestinal proteases, each cyst giving rise to two trophozoites. Giardia trophozoites (see Fig. 109-3B) are pear-shaped (10 to 20 µm long by 7 to 10 µm wide), contain two nuclei, have eight flagellae for locomotion, and replicate by binary fission. The trophozoites live in the duodenum, where they adhere to enterocytes. Eventually they encyst, following exposure to alkaline conditions or bile salts, and are excreted in the stool to complete their life cycle.91
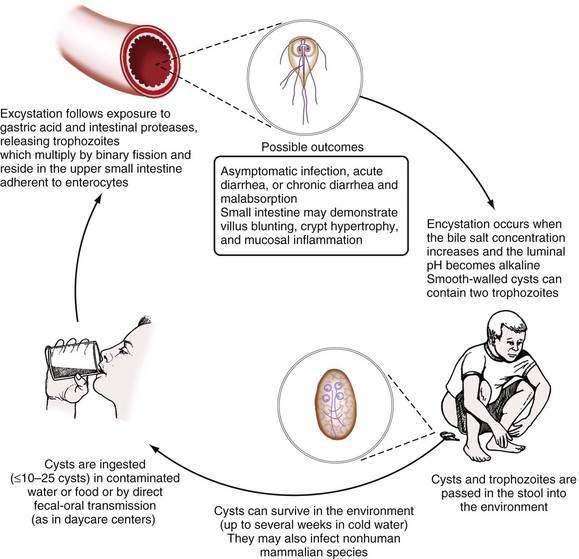
Figure 109-6. Life cycle of Giardia intestinalis.
(From Hill DR, Nash TE. Intestinal flagellate and ciliate infections. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia: WB Saunders; 1999.)
G. intestinalis, which was defined originally as a species by morphology, is more accurately defined as a species complex with at least seven major genotypes (assemblages A through G).92 Of these, only assemblages A and B are known to infect humans. Both of these genotypes also commonly infect cats and dogs, highlighting the importance of these pets as reservoirs for human disease.92 New data suggest that assemblage A isolates may be more virulent than assemblage B isolates.93,94
G. intestinalis is the most commonly identified intestinal parasite in the United States and was identified in 7.2% of stool samples examined by state health departments in 1987.95 Giardiasis occurs in both endemic and epidemic forms via water-borne, food-borne, and person-to-person transmission.96–102 Worldwide, Giardia infects infants more commonly than adults, and in highly endemic regions, essentially all children are infected by two to three years of age.103,104 In the developing world, it is likely that recurrent infantile diarrhea from giardiasis contributes significantly to malnutrition.103 In the United States, children in daycare and sexually active homosexual men have the greatest risk of infection.95,105 During a year-long longitudinal study at a U.S. daycare center, Giardia cysts were identified at some time in the stool of more than 30% of children.102 Additional risk factors for infection include drinking untreated surface water, a shallow well as a residential water source, swimming in any natural body of fresh water, and contact with a person who has giardiasis or contact with a child in daycare.97
PATHOGENESIS, PATHOLOGY, AND IMMUNOLOGY
Giardia causes malabsorptive diarrhea by an unknown mechanism. Trophozoites adhere (perhaps by suction) to the epithelium of the upper small intestine using a disk structure located on their anterior ventral surface.7,91 There is no evidence that trophozoites invade the mucosa,106 but electron microscopy has shown they damage the mucosal brush border.91,107 On biopsy, pathologic changes range from an entirely normal-appearing duodenal mucosa (except for adherent trophozoites), as was found in more than 96% of biopsy specimens in one large study, to severe villus atrophy with a mononuclear cell infiltrate that resembles celiac sprue.106,108,109 The severity of diarrhea appears to correlate with the severity of the pathologic change.91
The host immune response plays a critical role in limiting the severity of giardiasis. When infected with Giardia, persons with common variable immunodeficiency develop severe, protracted diarrhea and malabsorption with sprue-like pathologic changes that resolve with treatment of the Giardia.109 Both systemic and mucosal humoral immune responses can be measured consistently following Giardia infection. High serum anti-giardia IgM, IgG, and IgA titers can be detected, and anti-giardia secretory IgA (s-IgA) can be detected in saliva and in breast milk of infected mothers.110–112 Animal studies suggest that both early and late immune responses are important for control of Giardia infections. Interleukin-6 (IL-6) is important in the early immune response to Giardia in mice, as are mast cells, which might function as IL-6 producers or via another mechanism.113–115 In a B-cell-deficient transgenic mouse model, infection with Giardia does not resolve, confirming the importance of the humoral immune response for clearance of established infections.116 In culture, Giardia trophozoites vary expression of a group of cysteine-rich surface proteins termed variant surface proteins, and in experimental human infections, G. intestinalis isolates have been shown to undergo antigenic variation after approximately two weeks, roughly the time required to mount an initial antibody response.117 Although the role of the variant surface proteins remains unproved, antigenic variation might enable Giardia to evade the host immune response.118
The importance of a cellular immune response also is clear from animal studies. Athymic nude mice are unable to control Giardia muris infection, but reconstitution with immune spleen cells results in partial control. Upon immune reconstitution, however, severe inflammatory changes and villus atrophy develop in the intestine, suggesting that the immune response to infection might contribute to pathologic findings.119
CLINICAL FEATURES
The clinical manifestations of Giardia are highly variable, ranging from asymptomatic infection to severe, chronic diarrhea with malabsorption. In one large study of biopsy-proven giardiasis, only 32% of patients had diarrhea; most had nonspecific gastrointestinal complaints.106 Reported symptoms, in order of decreasing frequency, include diarrhea, fatigue, abdominal cramps, bloating, malodorous stool, flatulence, weight loss, fever, and vomiting (Table 109-3).100,110 During a food-borne outbreak, the mean duration of diarrhea was 16 days, but symptoms resolved spontaneously in nearly half of infected patients after seven to eight days.100 Many patients with clinically apparent giardiasis suffer from lactose intolerance, malabsorption, or both for months following cure of infection.120
Table 109-3 Frequency of Symptom(s) in Patients with Giardiasis100,106,110
| SYMPTOM(S) | FREQUENCY (%) |
|---|---|
| Diarrhea | 32-100 |
| Fatigue | 22-97 |
| Abdominal pain, cramps | 75-83 |
| Flatulence, bloating | 58-79 |
| Weight loss | 60 |
| Anorexia | 45 |
| Vomiting | 17-26 |
| Fever | 12-21 |
As mentioned earlier, the severity of illness depends upon host and parasite factors. Different Giardia isolates have dramatically different abilities to cause disease during experimental human infections.121 Furthermore, certain populations, including children younger than two years and patients with hypogammaglobulinemia, are more likely to develop serious disease.103,109 Despite the importance of cellular immunity for controlling infection in animal models and the increased risk of Giardia infection among sexually active homosexual men, giardiasis is not more common, or severe, or resistant to treatment in patients with AIDS,122 except perhaps when AIDS is advanced.123,124
DIAGNOSIS
Examination of concentrated, iodine-stained, wet stool preparations and modified-trichrome-stained permanent smears has been the conventional approach to identifying Giardia infections (see Fig. 109-3B). Because cysts and trophozoites are present only intermittently in the stool, however, the sensitivity is only about 50%, even with examination of multiple specimens.108 With direct sampling of duodenal contents, such as duodenal aspiration or the string test, sensitivity can be improved to approximately 80%.108 On small intestinal biopsy specimens, identification of trophozoites requires careful examination of multiple microscope fields to ensure accuracy (see Fig. 109-7).106
Numerous molecular tests based on ELISAs or direct immunofluorescent antibody (DFA) microscopy are now widely commercially available to diagnose giardiasis in stool samples.125–127 These assay kits all work well and have sensitivities greater than 90% and specificities approaching 100%.127 Given the inability to exclude giardiasis even with repeated conventional stool examinations and the difficulties of duodenal sampling, the first diagnostic test performed to evaluate for Giardia infection should be a stool ELISA or DFA. The primary role of endoscopy is evaluation for other pathologic conditions.
TREATMENT
Metronidazole (250 mg orally three times a day for five days) is the preferred treatment for giardiasis.70 At this relatively low dosage, metronidazole is generally well tolerated and is 80% to 95% effective at eradicating Giardia.128 The most common side effects of metronidazole are nausea, a metallic taste, and a disulfiram-like reaction with alcohol.
Nitazoxanide appears to be at least as effective as metronidazole and has the advantage of being available in a liquid formulation for use in pediatric patients. The recommended dosage in children is 100 mg (ages 12 to 47 months) or 200 mg (age older than four years) twice daily, and in adults is 500 mg twice daily for three days.73,76,129
Alternative regimens include tinidazole (2 g orally for one dose), quinacrine (2 mg/kg three times a day for five days; maximum 300 mg per day), furazolidone (100 mg orally four times a day for seven to 10 days), or paromomycin (25-35 mg/kg/day in three divided doses for seven days). Single-dose treatment with tinidazole has been used for years in Europe and the developing world and is approved by the U.S. Food and Drug Administration.70 Because paromomycin is not absorbed and there have been conflicting reports regarding the safety of metronidazole and tinidazole for the developing fetus, paromomycin may be especially useful for treatment of giardiasis during pregnancy.70
As noted earlier, many patients have prolonged lactose intolerance following Giardia infection, which can mimic ongoing infection.120 Therefore, the diagnosis should be reconfirmed before repeating therapy. For people in whom therapy fails, repeat therapy with the same drug (e.g., with higher doses of metronidazole) or combination therapy with metronidazole and quinacrine might work.124,128 Nitazoxanide alone also may be effective.130,131 Patients in whom treatment repeatedly fails should be evaluated for common variable immunodeficiency.109,128
CONTROL AND PREVENTION
Control of giardiasis relies on interruption of fecal-oral transmission. Water can be made safe for drinking and food preparation by boiling (for one minute), halogenation (with chlorine or iodine preparations), or filtration.7,128 Because of the low infectious dose of Giardia cysts and the poor hygiene of infants and children, person-to-person spread in daycare centers is much more difficult to control. Temporarily removing infected ill children from daycare is ineffective, perhaps because many infected children remain asymptomatic and go unrecognized.132 In the developing world, endemic giardiasis is unlikely to be controlled until facilities become available for adequate filtration of water and disposal of sewage.
A Giardia vaccine composed of killed G. intestinalis trophozoites has been licensed for use in cats and dogs, but there have been few studies addressing human vaccination for giardiasis.133 Reduced susceptibility of some people living in endemic areas suggests that vaccination may be possible.134
DIENTAMOEBA FRAGILIS
Dientamoeba fragilis is a binucleate organism with an ameboid trophozoite that measures 4 to 12 µm in diameter (see Fig. 109-2). No cyst form has been identified. The organism initially was classified as an ameba, but it is more closely related to the flagellates (trichomonads) based on morphologic studies and phylogenetic analyses of small-subunit rRNA gene sequences. The mode of its transmission remains unknown. The absence of a cyst form makes direct fecal-oral transmission unlikely, because the trophozoite is killed by stomach acid. Because of an association with Enterobius vermicularis (pinworm), some have hypothesized that it is carried in pinworm eggs.135 D. fragilis infection is common throughout the world. D. fragilis was identified in 0.5% of all stool samples examined in a large U.S. study, and the prevalence is as high as 20% to 50% in selected populations.95,136–139
The role of D. fragilis as a pathogen had been controversial. D. fragilis trophozoites do not invade tissue, and many persons infected with D. fragilis are asymptomatic.135 Furthermore, the organism often is identified in the presence of other intestinal parasites, making its role in disease unclear.136,138,139 However, several studies of patients infected only with D. fragilis have found an association with diarrhea, abdominal pain, nausea, weight loss, anorexia, flatus, and malaise that resolved only after eradication.137,140,141 Based on these studies, it now is believed that D. fragilis is pathogenic and that it should be treated. Treatment with iodoquinol (650 mg orally three times a day for 20 days), metronidazole (500 mg to 750 mg three times a day for 10 days), paromomycin (25 to 35 mg/kg/day orally in three divided doses for seven days), or tetracycline (500 mg orally four times a day for 10 days) has been effective.137,141–143
BLASTOCYSTIS HOMINIS
Blastocystis hominis is an intestinal protozoan that commonly infects the human colon. It is of uncertain taxonomic classification. Diameter ranges from 3 to 30 µm. In culture, B. hominis has ameboid, vacuolated, granular,7,144 and cystic forms.145 The distribution of B. hominis is worldwide, but infection is most common in the tropics.90,95,146–148 In a large study of intestinal parasitism in the United States, B. hominis was identified in 2.6% of stool specimens submitted to state health departments; more than 70% of positive samples were from California.95 Among American travelers and expatriates, the prevalence often exceeds 30%.90,148
The significance of B. hominis as a pathogen remains controversial. Several studies have suggested an association with irritable bowel syndrome, but neither cause nor effect has been established and in most series, B. hominis infection is not more common among patients with gastrointestinal complaints than among asymptomatic control subjects.90,148–151 In addition, the parasite burden does not correlate with symptoms.90,146 Nevertheless, multiple studies have used iodoquinol (650 mg orally three times a day for 20 days) or metronidazole (750 mg orally three times a day for 10 days) for treatment of symptomatic patients, with an overall improvement rate of about 50%.89,152 Clinical improvement actually may be due to treatment of unrecognized infections with other organisms, because many people infected with B. hominis simultaneously harbor known pathogens.136,153,154 In one series of patients with B. hominis infection, 84% of patients were found to have at least one recognized pathogen other than B. hominis (E. histolytica, G. intestinalis, or D. fragilis) when repeated stool examinations were obtained.136
CRYPTOSPORIDIUM SPECIES
EPIDEMIOLOGY
First recognized in 1907 by Tyzzer as a gastric infection in mice, Cryptosporidium species are tiny intracellular protozoan parasites (2 to 5 µm) belonging to the phylum Apicomplexa; other medically important Apicomplexan parasites include Plasmodium species, which cause malaria, and Toxoplasma gondii, which causes toxoplasmosis. Cryptosporidium species infect the gastrointestinal epithelium of a wide range of vertebrates. Based on genetic and biologic differences, Cryptosporidium parvum human genotype (genotype 1 or genotype H) was renamed Cryptosporidium hominis in 2002. The name Cryptosporidium parvum was retained for the bovine genotype (genotype 2). Together, C. hominis and C. parvum cause most human infections, but occasionally other species cause human disease, especially in immunocompromised patients.155
Cryptosporidia were brought to prominent medical attention only in the early 1980s because of the devastating disease they caused in patients with advanced HIV infection. However, Cryptosporidium species increasingly are recognized as a cause of self-limited diarrhea, usually lasting one to four weeks, in immunocompetent persons.156,157 In developing countries, children younger than five years of age are affected most frequently.158 In a cohort of children two to five years of age in Dhaka, Bangladesh, 25.7% had at least one symptomatic Cryptosporidium infection during three years of follow-up.159 In industrialized countries, because Cryptosporidium oocysts are small and highly chlorine-resistant, cryptosporidiosis has been associated with water-borne epidemics including numerous chlorinated swimming pool outbreaks. Cryptosporidia were responsible for the largest waterborne outbreak ever recorded that infected more than 400,000 Milwaukee residents in 1993.160,161 A low infectious dose and ready person-to-person spread also has resulted in epidemics in hospitals and daycare centers.162,163 In rural areas, zoonotic infections from direct contact with farm animals have been reported.164 Asymptomatic infection is common and infection appears to be more common in children who carry the DQB1*0301 HLA class II allele or the B*15 HLA class I allele.159
PATHOGENESIS, PATHOLOGY, AND IMMUNOLOGY
Upon ingestion of an infectious dose that may be as low as 1 to 10 oocysts, excystation and release of sporozoa occur in the presence of bile salts in the small intestine. The sporozoites then attach to the intestinal epithelium, which triggers elongation of epithelial cell microvilli on either side of the point of attachment. Fusion of the elongated microvilli with one another encloses the sporozoite within a vacuole located just underneath the brush border inside the epithelial cell. The sporozoites then develop into merozoites, which replicate asexually. After several rounds of asexual replication, the merozoites exit the host cell and invade uninfected neighboring cells. In immunocompetent persons, second-generation merozoites undergo meiosis to yield the male and female micro- and macrogametocytes, respectively. The microgametocytes then divide and exit the cell, whereupon they fertilize the macrogametocytes, forming oocysts that are shed in the feces. Rarely, multiplication has been seen in biliary, respiratory, or even conjunctival epithelium in immunocompromised patients.7
Animal and human studies suggest that both humoral and cellular immune responses aid in the control of Cryptosporidium infections. Cryptosporidial diarrhea is clearly much more severe, if not intractable, in patients with immunoglobulin deficiency, lymphocytic malignancies, or low CD4 counts associated with HIV infection.157,165,166 For unknown reasons, many more rounds of asexual reproduction typically occur in immunocompromised persons before development of the merozoites into micro- and macrogametocytes, which at least in part explains the increased chronicity of infection that is seen in this context.
CLINICAL FEATURES
Following a one-week incubation period (range, 2 to 14 days), a watery, relatively noninflammatory diarrheal illness typically lasts for 10 to 14 days in immunocompetent hosts. Nausea, vomiting, abdominal pain, and mild fever may also be seen. Rarely, respiratory symptoms, pancreatitis, and biliary tract involvement have been reported, the latter in HIV-infected patients (see Chapter 33). Brief recurrence of diarrhea may be seen after improvement.161,167
In immunocompromised patients, particularly those with very low CD4 lymphocyte counts, the diarrheal illness with cryptosporidial infection can be cholera-like, protracted (often for the duration of severe immune compromise), and fatal.157
DIAGNOSIS
Traditionally, cryptosporidial oocysts have been detected with a modified acid-fast stain of the stool (which can also detect Cyclospora and Isospora).168 As with giardiasis, ELISA or direct fluorescence antibody tests of the stool have replaced microscopy as the diagnostic test of choice. Numerous commercial kits using either of these two methods have been developed that have sensitivities and specificities in excess of 90%. These immunodiagnostic tests may be of limited value in testing environmental samples, however, because there is some cross-reactivity with nonhuman cryptosporidial oocysts.127 Occasionally, cryptosporidiosis is diagnosed with intestinal biopsy.
TREATMENT
Nitazoxanide, an antiparasitic agent with broad-spectrum antiprotozoan and anthelminthic activity, is the only known drug with consistent efficacy for treating cryptosporidiosis in immunocompetent patients.73,75,169–171 Unfortunately, failure is common in immunocompromised patients, such as those with advanced HIV infection, although some studies have shown benefit.169,171 The recommended dosage in children is 100 mg (ages 12 to 47 months) or 200 mg (age greater than 4 years) twice daily and in adults is 500 mg twice daily for three days. Nitazoxanide generally is well tolerated. It is converted to the active metabolite tizoxanide, which undergoes conjugation to tizoxanide glucuronide and is excreted in the urine, bile, and feces. Nitazoxanide and tizoxanide are yellow, resulting in yellow urine and, in some patients on prolonged therapy, in yellow discoloration of the eyes that resolves after the drug is discontinued. Additional treatment options in immunocompromised patients not responsive to nitazoxanide include the nonabsorbable aminoglycoside paromomycin or paromomycin in combination with azithromycin.172
Most important in treating HIV-infected patients with cryptosporidiosis is highly active antiretroviral therapy (HAART), because ultimately, improvement of a cryptosporidial illnesses depends on improvement in the immune compromise and the CD4 lymphocyte counts. Finally, papillotomy may be required for biliary obstruction with papillary stenosis from cryptosporidiosis in patients with AIDS (see Chapter 33).
CONTROL AND PREVENTION
Other means for disinfection that are being studied include ultraviolet light and irradiation. In one study, 2.5% glutaraldehyde was effective at inactivating Cryptosporidium oocysts, but only when a relatively low number of oocysts (15,000) were present and after treatment for 10 hours. Due to the corrosiveness of glutaraldehyde, the need to immerse instruments with their contaminating oocysts for this length of time presents a challenge for sterilization of endoscopes.173
Finally, because of the potential substantial long-term impact of cryptosporidial infection on childhood growth and development, control of cryptosporidiosis is critical in developing areas and must receive appropriate high priority in programs directed at improved water and sanitation worldwide.174,175
CYCLOSPORA CAYETANENSIS
EPIDEMIOLOGY
Cyclospora was first reported as a cause of human disease by Ashford in 1979, who described infection of three patients in Papua New Guinea by what was at that time, an unnamed coccidian parasite.176 This parasite came to wider attention when documented as a cause of protracted diarrhea in AIDS patients and persistent diarrhea in nonimmunocompromised patients in New York City and the Caribbean, among expatriates in Nepal, and in an outbreak among house staff in a Chicago hospital.177–179 Finally, in 1993, Ortega and colleagues at Cayetano Heredia University in Peru demonstrated formation of sporozoites (sporulation) within immature oocysts and excystation in vitro and used electron microscopy to demonstrate oocysts containing sporozoites with organelles characteristic of coccidians of the phylum Apicomplexa. They classified the organism as a member of the genus Cyclospora, and it has now been officially named Cyclospora cayetanensis to acknowledge this work.180 Ribosomal DNA analysis of phylogenetic relationships suggest that Cyclospora is closely related to Eimeria.181
Like Cryptosporidium, Cyclospora is being increasingly recognized in immunocompetent as well as immunocompromised persons. The infection is usually highly seasonal (in summer or wet months) and is probably spread via fecal contamination of water and vegetables.182,183 Cyclospora was brought to prominent attention throughout the United States and Canada with repeated outbreaks of diarrheal illnesses occurring in more than 2000 patients every year from 1996 through 2000 in association with consumption of the late spring shipment of Guatemalan raspberries.184,185
PATHOGENESIS, PATHOLOGY, AND IMMUNOLOGY
The histopathologic changes seen in Cyclospora infections are similar to those seen with cryptosporidiosis, with villus blunting and a mild inflammatory infiltrate in the lamina propria predominantly in the small intestine.186
DIAGNOSIS
As with Cryptosporidium, one must consider the diagnosis of Cyclospora in patients with protracted diarrhea. Diagnosis is best made at present with the acid-fast stain. Cyclospora oocysts measure 7 to 10 µm, nearly twice the size of those of Cryptosporidium, which are 4 to 5 µm.187 Cyclospora exhibits striking blue-green autofluorescence when examined under fluorescence microscopy, a characteristic that might have contributed to its initial confusion with cyanobacteria.188 Improved diagnostic methods using PCR have been developed, but they are not currently available for clinical use.189
TREATMENT
In contrast to Cryptosporidium infections, Cyclospora infections are readily treatable, even in immunocompromised patients. The drug of choice is trimethoprim-sulfamethoxazole at a dosage of 160/800 mg twice daily for one week. Treatment promptly eradicates the organism and relieves symptoms.190,191 This treatment is similarly effective in patients with AIDS, although maintenance therapy with a single dose of trimethoprim-sulfamethoxazole three times per week may be needed to prevent relapse.192 Recent data show that ciprofloxacin provides a reasonable alternative in patients unable to tolerate trimethoprim-sulfamethoxazole.193 Nitazoxanide also appears to be effective.75
CONTROL AND PREVENTION
Although readily treatable, Cyclospora infections are extremely difficult to control or prevent because of our limited ability to detect low infectious doses (for humans) of oocysts, which can contaminate products such as raspberries, from which it is very difficult to eradicate. From limited studies, the organism also appears to be relatively chlorine-resistant and thus poses challenges to effective water treatment, much like Cryptosporidium. Elucidation of the reservoir of Cyclospora undoubtedly will enhance our ability to prevent and control the spread of this highly infectious parasite. For example, it remains unclear why it is only the spring rather than the fall shipment of raspberries from Guatemala that has consistently posed problems with spread of Cyclospora infections. Whether this is related to migration of an avian reservoir has been postulated, but not proved.194 Consistent with this, several studies have reported isolation of Cyclospora oocysts from chickens.182,183
ISOSPORA BELLI
EPIDEMIOLOGY
A relative of Cyclospora and Eimeria, I. belli is much larger, with elliptical oocysts measuring 20 to 30 µm long and containing two visible sporocysts that are acid-fast. Like Cyclospora, Isospora oocysts appear to require sporulation outside of the human host before they become infectious. There are no known nonhuman hosts for I. belli, and its distribution appears to be throughout tropical areas around the world. It appears to be a less common cause of diarrhea in children in developing areas than Cryptosporidium and is seen more often in older children, immunocompromised patients, and in institutionalized children in North America.195,196
CLINICAL FEATURES
Similar to Cryptosporidium and Cyclospora infections, Isospora characteristically produces a self-limiting diarrheal illness in immunocompetent persons and in travelers to tropical areas, with watery diarrhea and abdominal pain lasting two to four weeks. In immunocompromised patients, Isospora can produce a protracted sprue-like illness with malabsorption, weight loss, and prolonged diarrhea.196 As with Cryptosporidium and Cyclospora, acalculous cholecystitis also has been reported in patients with AIDS and Isospora infections.
DIAGNOSIS
The diagnosis of Isospora should be suspected in immunocompetent patients with diarrhea lasting longer than five to seven days, especially following travel to tropical or developing areas, and in immunocompromised patients with persistent diarrhea. Unlike other protozoan infections, Isospora infections may be associated with peripheral eosinophilia and with Charcot-Leyden crystals in the stool. The diagnosis of Isospora relies on identification of the large, oval oocysts (20 to 30 µm by 10 to 19 µm) on microscopic examination of concentrated fecal specimens by acid-fast staining. Oocysts also may be seen in biopsy specimens from the small intestine. In contrast to Cryptosporidium and Cyclospora infections, Isospora organisms have been observed invading beyond the epithelium into the lamina propria.155,197
TREATMENT
As with Cyclospora, Isospora infections are readily treated with trimethoprim-sulfamethoxazole; the dosage is 160/800 mg orally four times a day for 10 days, and then two times a day for three weeks, with both symptomatic and parasitologic responses, even in patients with AIDS.70 As previously described with Cyclospora, maintenance of suppressive therapy may be required in patients with AIDS.191 Alternatives to trimethoprim-sulfamethoxazole may include ciprofloxacin.193
MICROSPORIDIA
Microsporidia infections are discussed in detail in Chapter 33.
EPIDEMIOLOGY
Microsporidia, the nontaxonomic term for Enterocytozoon bieneusi, Encephalitozoon (old Septata) intestinalis, and several other nonintestinal members of the phylum Microspora, are important causes of diarrhea, primarily in patients with impaired cell-mediated immunity due to AIDS or organ transplantation.198,199 E. bieneusi causes approximately 90% of cases.200 Microsporidia are identified in as many as 50% of AIDS patients with chronic diarrhea and are the most commonly identified pathogen in most series.199 The prevalence of infection is strongly correlated with decreasing CD4 T lymphocyte counts, although cases are not uncommon in persons with CD4 cell counts greater than 200 cells/mL.157,200,201 Microsporidia is distinctly less common in immunocompetent persons. The reservoir and modes of transmission are not certain.202–204 Epidemiologic data suggest that waterborne, person-to-person, and possibly sexual transmission occur.
PATHOGENESIS, PATHOLOGY, AND IMMUNOLOGY
E. bieneusi enters only the cytoplasm of enterocytes, but E. intestinalis forms a parasitophorous vacuole in enterocytes, endothelial cells, fibroblasts, and macrophages, and it can disseminate to the kidney, prostate gland, and upper respiratory tract. Typically, intestinal pathology is marked by villus atrophy, crypt hyperplasia, and mild inflammation in the lamina propria.198 The importance of cellular immunity in determining both infection and illness with intestinal microsporidia is indicated by its striking predominance in immunocompromised persons after organ transplantation or in those with AIDS.
CLINICAL FEATURES
Although primarily limited to immunocompromised patients, microsporidia cause chronic watery, relatively noninflammatory diarrhea and weight loss, occasionally with abdominal pain, nausea, vomiting, fever, and acalculous cholecystitis or even sclerosing cholangitis.198 E. intestinalis also can cause colitis and disseminate especially to the kidneys or less often to sinuses, bronchi, conjunctivae, or prostate.205 Rarely, cases have been reported of self-limited diarrhea in travelers or health professionals.202–204
DIAGNOSIS
Most laboratories use a modified trichrome stain to identify microsporidia in stool specimens.206 This method requires considerable skill and has limited sensitivity because of the small size of the spores (E. bieneusi measures 1 by 1.5 µm; E. intestinalis is slightly larger). Sensitivity can be improved by initially screening samples with fluorescent chitin stains such as Fungi-Fluor chitin stain (Polysciences, Warrington, Pa.) or Uvitex 2B (Ciba Geigy, Rueil Malmaison, France), and confirming positive results by modified trichrome staining.207,208 In addition, Gram stain and electron microscopy can identify the organisms in intestinal biopsy specimens. Sensitive PCR methods have been developed that enable species differentiation, but use of these methods is currently limited to research applications.204,209
TREATMENT
The response of E. bieneusi to albendazole is poor, but recent data indicate that oral fumagillin (20 mg three times daily for two weeks) may be effective for treatment of intestinal E. bieneusi infection in immunocompromised patients.210 Side effects including neutropenia and thrombocytopenia are common and, given the limited clinical data available, this treatment should be undertaken with caution. E. intestinalis infections (approximately 10% of cases) respond well to albendazole, 400 mg twice daily for three weeks.70 As with all opportunistic infections in patients with AIDS, effective antiretroviral therapy (HAART) is essential for controlling microsporidial infections.
TRYPANOSOMA CRUZI (AMERICAN TRYPANOSOMIASIS OR CHAGAS’ DISEASE)
EPIDEMIOLOGY
Although symptomatic Chagas’ disease has been confined to South and Central America, at least four autochthonous (indigenous) cases, as well as occasional laboratory-acquired and imported cases of acute Chagas’ disease, have occurred in the United States. Increasing numbers of immigrants are presenting with chronic Chagas’ disease, however, and pose distinct risks for transmission of Chagas’ disease.211 In patients surviving acute infection with T. cruzi in whom the chronic form of illness develops, myocardial disease is the most common manifestation. Megaesophagus and megacolon are the most common intestinal manifestations of American trypanosomiasis. Small intestinal dilatation and aperistalsis also are seen. At postmortem examination, even in patients with asymptomatic T. cruzi involvement of the intestine, the small intestine has a significant reduction in submucosal and myenteric autonomic plexuses.
CLINICAL FEATURES
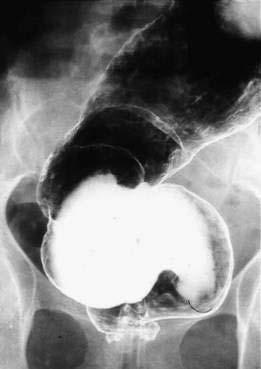
The manifestations of chronic Chagas’ disease depend on the major organ involvement within the body. Most commonly, symptoms are cardiac, manifested primarily as arrhythmias and congestive heart failure. With megaesophagus, the history, barium esophagram, and esophageal motility tracing are indistinguishable from those of achalasia. With megacolon (Fig. 109-8), infrequent bowel movements and chronic constipation are the cardinal symptoms. With dilatation of the small intestine, diarrhea or constipation may be part of the picture. There may be evidence of weight loss and abdominal distention caused by the markedly dilated bowel.
DIAGNOSIS
Diagnosis of chronic Chagas’ disease depends on the presence of a typical clinical and epidemiologic history and on serologic tests, because parasites are only rarely identifiable in the blood or on biopsies of affected organs. Serum antibodies to T. cruzi can be detected either by complement fixation or ELISA, and antibody testing can be requested from the U.S. Centers for Disease Control and Prevention (CDC). Xenodiagnosis has been used but is relatively insensitive, identifying less than 50% of patients infected with chronic Chagas’ disease. In this technique, trypanosome-free laboratory reduviid bugs are allowed to bite suspected victims. The trypanosomes multiply rapidly in the intestinal tract of the insect, and examination of the intestine reveals flagellated trypanosomes in 10 to 30 days. Sensitive and specific PCR-based assays have been developed for diagnosis of acute and chronic Chagas’ disease, but these assays are not available for clinical use.212,213
TREATMENT
Nifurtimox (8 to 10 mg/kg daily in four divided doses for 90 to 120 days) or benznidazole (5 to 7 mg/kg daily in two divided doses for 30 to 90 days) can be used for treatment of Chagas’ disease.70 Both are subject to availability problems, have limited efficacy, and are associated with significant side effects, including gastrointestinal symptoms in 40% to 70% of patients (nausea, vomiting, abdominal pain, and anorexia) and frequent neurologic sequelae.214 Isosorbide dinitrate has been shown to increase esophageal emptying in patients with achalasia resulting from Chagas’ disease and might ameliorate dysphagia.215,216 Most patients with Chagas achalasia are best treated with either balloon dilation of the esophagus or esophagomyotomy. Occasionally, nonperistaltic segments of intestine that are responsible for symptoms need to be resected.
Ali IKM, Mondal U, Roy S, et al. Evidence for a link between parasite genotype and outcome of infection with Entamoeba histolytica. J Clin Microbiol. 2007;45:285-9. (Ref 8.)
Amadi B, Mwiya M, Musuku J, et al. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: A randomised controlled trial. Lancet. 2002;360:1375-80. (Ref 171.)
Duggal P, Haque R, Roy S, et al. Influence of human leukocyte antigen class II alleles on susceptibility to Entamoeba histolytica infection in Bangladeshi children. J Infect Dis. 2004;189:520-6. (Ref 9.)
Haque R, Mollah NU, Ali IKM, et al. Diagnosis of amebic liver abscess and intestinal infection with the TechLab Entamoeba histolytica II antigen detection and antibody tests. J Clin Microbiol. 2000;38:3235-9. (Ref 68.)
Haque R, Mondal D, Duggal P, et al. Entamoeba histolytica infection in children and protection from subsequent amebiasis. Infect Immun. 2006;74:904-9. (Ref 5.)
Haque R, Roy S, Kabir M, et al. Giardia assemblage A infection and diarrhea in Bangladesh. J Infect Dis. 2005;192:2171-3. (Ref 93.)
Johnson EH, Windsor JJ, Clark CG. Emerging from obscurity: Biological, clinical, and diagnostic aspects of Dientamoeba fragilis. Clin Microbiol Rev. 2004;17:553-70. (Ref 135.)
Kirkpatrick BD, Haque R, Duggal P, et al. Association between Cryptosporidium infection and human leukocyte antigen class I and class II alleles. J Infect Dis. 2008;197:474-8. (Ref 159.)
Mackenzie WR, Hoxie NJ, Proctor ME, et al. A massive outbreak in Milwaukee of Cryptosporidium infection transmitted through the public water supply. N Engl J Med. 1994;331:161-7. (Ref 161.)
Monis PT, Andrews RH, Mayrhofer G, Ey PL. Genetic diversity within the morphological species Giardia intestinalis and its relationship to host origin. Infect Gen Evol. 2003;3:29-38. (Ref 92.)
Osterholm MT, Forfang JC, Ristinen TL, et al. An outbreak of foodborne giardiasis. N Engl J Med. 1981;304:24-8. (Ref 100.)
Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Cryptosporidium parvum: A prospective randomized, double-clind, placebo-controlled study of nitazoxanide. J Infect Dis. 2001;184:103-6. (Ref 170.)
Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Giardia intestinalis and Entamoeba histolytica or Entamoeba dispar: A randomized, double-blind placebo-controlled study of nitazoxanide. J Infect Dis. 2002;184:381-4. (Ref 76.)
Verdier RI, Fitzgerald DW, Johnson WD, Pape JW. Trimethoprim-sulfamethoxazole compared with ciprofloxacin for treatment and prophylaxis of Isospora belli and Cyclospora cayetanensis infection in HIV-infected patients. A randomized, controlled trial. Ann Intern Med. 2000;132:885-8. (Ref 193.)
Xiao L, Fayer R, Ryan U, Upton SJ. Cryptosporidium taxonomy: Recent advances and implications for public health. Clin Microbiol Rev. 2004;17:72-97. (Ref 155.)
1. Diamond LS, Clark CG. A redescription of Entamoeba histolytica Schaudinn, 1903 (Emended Walker, 1911) separating it from Entamoeba dispar Brumpt, 1925. J Euk Microbiol. 1993;40:340-4.
2. Haque R, Huston CD, Hughes M, et al. Amebiasis. N Engl J Med. 2003;348:1565-73.
3. Walsh JA. Problems in recognition and diagnosis of amebiasis: Estimation of the global magnitude of morbidity and mortality. Rev Infect Dis. 1986;8:228-38.
4. WHO/PAHO/UNESCO report. A consultation with experts on amoebiasis. Mexico City, Mexico 28-29 January, 1997. WHO Epidemiol Bull. 1997;18:13-14.
5. Haque R, Mondal D, Duggal P, et al. Entamoeba histolytica infection in children and protection from subsequent amebiasis. Infect Immun. 2006;74:904-9.
6. Mondal D, Petri WA, Sack RB, et al. Entamoeba histolytica–associated diarrheal illness is negatively associated with the growth of preschool children: Evidence from a prospective study. Trans R Soc Trop Med Hyg. 2006;100:1032-8.
7. Katz M, Despommier DD, Gwadz R. Parasitic Diseases, 2nd ed. New York: Springer-Verlag; 1989. pp 136-43 and 203-7
8. Ali IKM, Mondal U, Roy S, et al. Evidence for a link between parasite genotype and outcome of infection with Entamoeba histolytica. J Clin Microbiol. 2007;45:285-9.
9. Duggal P, Haque R, Roy S, et al. Influence of human leukocyte antigen class II alleles on susceptibility to Entamoeba histolytica infection in Bangladeshi children. J Infect Dis. 2004;189:520-6.
10. Arellano J, Perez-Rodriguez M, Lopez-Osuna M, et al. Increased frequency of HLA-DR3 and complotype SCO1 in Mexican mestizo children with amoebic abscess of the liver. Parasite Immunol. 1996;18:491-8.
11. Takeuchi A, Phillips BP. Electron microscope studies of experimental Entamoeba histolytica infection in the guinea pig. I. Penetration of the intestinal epithelium by trophozoites. Am J Trop Med Hyg. 1975;24:34-48.
12. Beaver PC, Blanchard JL, Seibold HR. Invasive amebiasis in naturally infected New World and Old World monkeys with and without clinical disease. Am J Trop Med Hyg. 1988;39:343-52.
13. Chadee K, Meerovitch E. Entamoeba histolytica: Early progressive pathology in the cecum of the gerbil (Meriones unguiculatus). Am J Trop Med Hyg. 1985;34:283-91.
14. Ravdin JI, Guerrant RL. Role of adherence in cytopathogenic mechanisms of Entamoeba histolytica. Study with mammalian tissue culture cells and human erythrocytes. J Clin Invest. 1981;68:1305-13.
15. Chadee K, Petri WAJr, Innes DJ, Ravdin JI. Rat and human colonic mucins bind to and inhibit the adherence lectin of Entamoeba histolytica. J Clin Invest. 1987;80:1245-54.
16. Petri WAJr, Smith RD, Schlesinger PH, et al. Isolation of the galactose binding lectin of Entamoeba histolytica. J Clin Invest. 1987;80:1238-44.
17. Moncada D, Keller K, Chadee K. Entamoeba histolytica cysteine proteinases disrupt the polymeric structure of colonic mucin and alter its protective function. Infect Immun. 2003;71:838-44.
18. Keene WE, Petitt MG, Allen S, McKerrow JH. The major neutral proteinase of Entamoeba histolytica. J Exp Med. 1986;163:536-49.
19. Li E, Yang WG, Zhang T, Stanley SLJr. Interaction of laminin with Entamoeba histolytica cysteine proteinases and its effect on amebic pathogenesis. Infect Immun. 1995;63:4150-3.
20. Que X, Reed SL. Cysteine proteinases and the pathogenesis of amebiasis. Clin Microbiol Rev. 2000;13:196-206.
21. Leippe M, Ebel S, Schoenberger OL, et al. Pore-forming peptide of pathogenic Entamoeba histolytica. Proc Nat Acad Sci U S A. 1991;88:7659-63.
22. Huston CD, Houpt ER, Mann BJ, et al. Caspase 3–dependent killing of host cells by the parasite Entamoeba histolytica. Cell Microbiol. 2000;2:617-25.
23. Seydel KB, Li E, Zhang Z, Stanley SL. Epithelial cell–initiated inflammation plays a crucial role in early tissue damage in amebic infection of human intestine. Gastroenterol. 1998;115:1446-53.
24. Zhang Z, Wang L, Seydel KB, et al. Entamoeba histolytica cysteine proteinases with interleukin-1β converting enzyme (ICE) activity cause intestinal inflammation and tissue damage in amoebiasis. Molec Microbiol. 2000;37:542-8.
25. Eckmann L, Reed SL, Smith JR, Kagnoff MF. Entamoeba histolytica trophozoites induce an inflammatory cytokine response by cultured human cells through the paracrine action of cytolytically released interleukin-1α. J Clin Invest. 1995;96:1269-79.
26. Brandt H, Perez-Tamayo R. The pathology of human amebiasis. Hum Pathol. 1970;1:351-85.
27. von Lichtenberg F. Infectious disease: Fungal, protozoal, and helminthic diseases and sarcoidosis: Amebiasis (Entamoeba histolytica). In: Cotran RS, Kumar V, Robbins SL, editors. Robbins Pathologic Basis of Disease. 4th ed. Philadelphia: W.B.: Saunders; 1989:397.
28. Chadee K, Meerovitch E. The pathology of experimentally induced cecal amebiasis in gerbils (Meriones unguiculatus). Liver changes and amebic liver abscess formation. Am J Pathol. 1985;119:485-94.
29. Chadee K, Meerovitch E. The Mongolian gerbil (Meriones unguiculatus) as an experimental host for Entamoeba histolytica. Am J Trop Med Hyg. 1984;33:47-54.
30. Gathiram V, Jackson TF. A longitudinal study of asymptomatic carriers of pathogenic zymodemes of Entamoeba histolytica. South Afr Med J. 1987;72:669-72.
31. Haque R, Ali IM, Sack RB, et al. Amebiasis and mucosal IgA antibody against the Entamoeba histolytica adherence lectin in Bangladeshi children. J Infect Dis. 2001;183:1787-93.
32. Haque R, Duggal P, Ali IM, et al. Innate and acquired resistance to amebiasis in Bangladeshi children. J Infect Dis. 2002;186:547-52.
33. Choudhuri G, Prakash V, Kumar A, et al. Protective immunity to entamoeba histolytica infection in subjects with antiamoebic antibodies residing in a hyperendemic zone. Scand J Infect Dis. 1991;23:771-6.
34. Krupp IM. Antibody response in intestinal and extraintestinal amebiasis. Am J Trop Med. 1970;19:57-62.
35. Ortiz-Ortiz L, Zamacona G, Sepulveda B, Capin NR. Cell-mediated immunity in patients with amebic abscess of the liver. Clin Immunol Immunopathol. 1975;4:127-34.
36. del Muro R, Acosta E, Merino E, et al. Diagnosis of intestinal amebiasis using salivary IgA antibody detection. J Infect Dis. 1990;162:1360-4.
37. Aceti A, Pennica A, Celestino D, et al. Salivary IgA antibody detection in invasive amebiasis and in asymptomatic infection. J Infect Dis. 1991;164:613-15.
38. Abou-el-Magd I, Soong CJ, el-Hawey AM, Ravdin JI. Humoral and mucosal IgA antibody response to a recombinant 52-kDa cysteine-rich portion of the Entamoeba histolytica galactose-inhibitable lectin correlates with detection of native 170-kDa lectin antigen in serum of patients with amebic colitis. J Infect Dis. 1996;174:157-62.
39. Cieslak PR, Virgin HW4th, Stanley SLJr. A severe combined immunodeficient (SCID) mouse model for infection with Entamoeba histolytica. J Exp Med. 1992;176:1605-9.
40. Kanani SR, Knight R. Relapsing amoebic colitis of 12 year’s standing exacerbated by corticosteroids. Brit Med J. 1969;2:613-14.
41. Kanani SR, Knight R. Amoebic dysentery precipitated by corticosteroids. Br Med J. 1969;3:114.
42. Houpt ER, Glembocki DJ, Obrig TG, et al. The mouse model of amebic colitis reveals mouse strain susceptibility to infection and exacerbation of disease by CD4+ T cells. J Immunol. 2002;169:4496-503.
43. Huston CD, Petri WA. Amebiasis. In: Rakel RE, Bope ET, editors. Conn’s Current Therapy, 2001. Philadelphia: WB Saunders; 2001:50-4.
44. Speelman P, McGlaughlin R, Kabir I, Butler T. Differential clinical features and stool findings in shigellosis and amoebic dysentery. Trans R Soc Trop Med Hyg. 1987;81:549-51.
45. Ellyson JH, Bezmalinovic Z, Parks SN, Lewis FRJr. Necrotizing amebic colitis: A frequently fatal complication. Am J Surg. 1986;152:21-6.
46. Aristizabal H, Acevedo J, Botero M. Fulminant amebic colitis. World J Surg. 1991;15:216-21.
47. Kapoor OP, Joshi VR. Multiple amoebic liver abscesses. A study of 56 cases. J Trop Med Hyg. 1972;75:4-6.
48. Katzenstein D, Rickerson V, Braude A. New concepts of amebic liver abscess derived from hepatic imaging, serodiagnosis, and hepatic enzymes in 67 consecutive cases in San Diego. Medicine. 1982;61:237-46.
49. Nordestgaard AG, Stapleford L, Worthen N, et al. Contemporary management of amebic liver abscess. Am Surg. 1992;58:315-20.
50. Kapoor OP, Shah NA. Pericardial amoebiasis following amoebic liver abscess of the left lobe. J Trop Med Hyg. 1972;75:7-10.
51. Gonzalez-Ruiz A, Haque R, Aguirre A, et al. Value of microscopy in the diagnosis of dysentery associated with invasive Entamoeba histolytica. J Clin Pathol. 1994;47:236-9.
52. Krogstad DJ, Spencer HC, Healy GR, et al. Amebiasis: Epidemiologic studies in the United States 1971-1974. Ann Intern Med. 1978;88:89-97.
53. Haque R, Neville LM, Hahn P, Petri WAJr. Rapid diagnosis of Entamoeba infection by using Entamoeba and Entamoeba histolytica stool antigen detection kits. J Clin Microbiol. 1995;33:2558-61.
54. Sargeaunt PG, Williams JE, Greene JD. The differentiation of invasive and noninvasive Entamoeba histolytica by isoenzyme electrophoresis. Trans R Soc Trop Med Hyg. 1978;72:519-21.
55. Strachan WD, Chiodini PL, Spice WM, et al. Immunological differentiation of pathogenic and non-pathogenic isolates of Entamoeba histolytica. Lancet. 1988;1:561-3.
56. Troll H, Marti H, Weiss N. Simple differential detection of Entamoeba histolytica and Entamoeba dispar in fresh stool specimens by sodium acetate–acetic acid–formalin concentration and PCR. J Clin Microbiol. 1997;35:1701-5.
57. Blessman J, Buss H, Ton Nu PA, et al. Real-time PCR for detection and differentiation of Entamoeba histolytica and Entamoeba dispar in fecal samples. J Clin Microbiol. 2002;40:4413-17.
58. Britten D, Wilson SM, McNerney R, et al. An improved colorimetric PCR-based method for detection and differentiation of Entamoeba histolytica and Entamoeba dispar in feces. J Clin Microbiol. 1997;35:1108-11.
59. Petri WAJr, Jackson TF, Gathiram V, et al. Pathogenic and nonpathogenic strains of Entamoeba histolytica can be differentiated by monoclonal antibodies to the galactose-specific adherence lectin. Infect Immun. 1990;58:1802-6.
60. Haque R, Kress K, Wood S, et al. Diagnosis of pathogenic Entamoeba histolytica infection using a stool ELISA based on monoclonal antibodies to the galactose-specific adhesin. J Infect Dis. 1993;167:247-9.
61. Haque R, Ali IK, Akther S, Petri WAJr. Comparison of PCR, isoenzyme analysis, and antigen detection for diagnosis of Entamoeba histolytica infection. J Clin Microbiol. 1998;36:449-52.
62. Ong SJ, Cheng MY, Liu KH, Horng CB. Use of the ProSpecT microplate enzyme immunoassay for the detection of pathogenic and non-pathogenic Entamoeba histolytica in faecal specimens. Trans R Soc Trop Med Hyg. 1996;90:248-9.
63. Jelinek T, Peyerl G, Loscher T, Nothdurft HD. Evaluation of an antigen-capture enzyme immunoassay for detection of Entamoeba histolytica in stool samples. Eur J Clin Microbiol Infect Dis. 1996;15:752-5.
64. Mirelman D, Nuchamowitz Y, Stolarsky T. Comparison of use of enzyme-linked immunosorbent assay–based kits and PCR amplification of rRNA genes for simultaneous detection of Entamoeba histolytica and E. dispar. J Clin Microbiol. 1997;35:2405-7.
65. Ravdin JI, Jackson TFHG, Petri WAJr, et al. Association of serum antibodies to adherence lectin with invasive amebiasis and asymptomatic infection with pathogenic Entamoeba histolytica. J Infect Dis. 1990;162:768-72.
66. Gatti S, Swierczynski G, Robinson F, et al. Amebic infections due to the Entamoeba histolytica–Entamoeba dispar complex: A study of the incidence in a remote rural area of Ecuador. Am J Trop Med Hyg. 2002;67:123-27.
67. Gonin P, Trudel L. Detection and differentiation of Entamoeba histolytica and Entamoeba dispar isolates in clinical samples by PCR and enzyme-linked immunosorbent assay. J Clin Microbiol. 2003;41:237-41.
68. Haque R, Mollah NU, Ali IKM, et al. Diagnosis of amebic liver abscess and intestinal infection with the TechLab Entamoeba histolytica II antigen detection and antibody tests. J Clin Microbiol. 2000;38:3235-9.
69. McAuley JB, Herwaldt BL, Stokes SL, et al. Diloxanide furoate for treating asymptomatic Entamoeba histolytica cyst passers: 14 years’ experience in the United States. Clin Infect Dis. 1992;15:464-8.
70. Drugs for parasitic infections. Med Lett Drugs Ther. 2002;April 1:1-12.
71. Blessmann J, Tannich E. Treatment of asymptomatic intestinal Entamoeba histolytica infection. N Engl J Med. 2002;347:1384.
72. McAuley JB, Juranek DD. Paromomycin in the treatment of mild-to-moderate intestinal amebiasis. Clin Infect Dis. 1992;15:551-3.
73. Nitazoxanide (Alinia)—a new anti-protozoal agent. Med Lett Drugs Ther. 2003;45:29-31.
74. Bassily S, Farid Z, El-Masry NA, Mikhail EM. Treatment of intestinal E. histolytica and G. lamblia with metronidazole, tinidazole, and ornidazole: A comparative study. J Trop Med Hyg. 1987;90:9-12.
75. Diaz E, Mondragon J, Ramirez E, Bernal R. Epidemiology and control of intestinal parasites with nitazoxanide in children in Mexico. Am J Trop Med Hyg. 2003;68:384-5.
76. Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Giardia intestinalis and Entamoeba histolytica or Entamoeba dispar: A randomized, double-blind placebo-controlled study of nitazoxanide. J Infect Dis. 2002;184:381-4.
77. Rossignol JF, Kabil SM, El-Gohary Y, Younis AM. Nitazoxanide in the treatment of amoebiasis. Trans R Soc Trop Med Hyg. 2007;101:1025-31.
78. Powell SJ, Wilmot AJ, Elsdon-Dew R. Further trials of metronidazole in amoebic dysentery and amoebic liver abscess. Ann Trop Med Parasitol. 1967;61:511-4.
79. Barwick RS, Uzicanin A, Lareau S, et al. Outbreak of amebiasis in Tblisi, Republic of Georgia, 1998. Am J Trop Med Hyg. 2002;67:623-31.
80. Huston CD, Petri WAJr. Host-pathogen interaction in amebiasis and progress in vaccine development. Eur J Clin Microbiol Infect Dis. 1998;17:601-14.
81. Ali IKM, Hossain MB, Roy S, et al. Entamoeba moshkovskii infections in children in Bangledesh. Emerg Infect Dis. 2003;9:580-4.
82. Beck DL, Dogan N, Maro V, et al. High prevalence of Entamoeba moshkovskii in a Tanzanian HIV population. Acta Trop. 2008;107:48-9.
83. Fotedar R, Stark D, Marriott D, et al. Entamoeba moshkovskii infections in Sydney, Australia. Eur J Clin Microbiol Infect Dis. 2008;27:133-7.
84. Petri WA, Singh U, Ravdin JI. Enteric amebiasis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. 1st ed. Philadelphia: Churchill Livingstone; 2000:685-702.
85. Lucht E, Evengard B, Skott J, et al. Entamoeba gingivalis in human immunodeficiency virus type 1–infected patients with periodontal disease. Clin Infect Dis. 1998;27:471-3.
86. Verweij JJ, Polderman AM, Clark CG. Genetic variation among human isolates of uninucleated cyst-producing Entamoeba species. J Clin Microbiol. 2001;39:1644-6.
87. Desowitz RS, Barnish G. Entamoeba polecki and other intestinal protozoa in Papua New Guinea highland children. Ann Trop Med Parasitol. 1986;80:399-402.
88. Salaki JS, Shirey JL, Strickland GT. Successful treatment of symptomatic Entamoeba polecki infection. Am J Trop Med Hyg. 1979;28:190-3.
89. Qadri SM, al-Okaili GA, al-Dayel F. Clinical significance of Blastocystis hominis. J Clin Microbiol. 1989;27:2407-9.
90. Herwaldt BL, de Arroyave KR, Wahlquist SP, et al. Infections with intestinal parasites in Peace Corps volunteers in Guatemala. J Clin Microbiol. 1994;32:1376-8.
91. Hill DR. Giardia lamblia. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed. Philadelphia: Churchill Livingstone; 2000:2888-94.
92. Monis PT, Andrews RH, Mayrhofer G, Ey PL. Genetic diversity within the morphological species Giardia intestinalis and its relationship to host origin. Infect Gen Evol. 2003;3:29-38.
93. Haque R, Roy S, Kabir M, et al. Giardia assemblage A infection and diarrhea in Bangladesh. J Infect Dis. 2005;192:2171-3.
94. Sahagun J, Clavel A, Goni P, et al. Correlation between the presence of symptoms and the Giardia duodenalis genotype. Eur J Clin Microbiol Infect Dis. 2008;27:81-3.
95. Kappus KD, Lundgren RGJr, Juranek DD, et al. Intestinal parasitism in the United States: Update on a continuing problem. Am J Trop Med Hyg. 1994;50:705-13.
96. Moore GT, Cross WM, McGuire D, et al. Epidemic giardiasis at a ski resort. N Engl J Med. 1969;281:402-7.
97. Dennis DT, Smith RP, Welch JJ, et al. Endemic giardiasis in New Hampshire: A case-control study of environmental risks. J Infect Dis. 1993;167:1391-5.
98. Isaac-Renton JL, Cordeiro C, Sarafis K, Shahriari H. Characterization of Giardia duodenalis isolates from a waterborne outbreak. J Infect Dis. 1993;167:431-40.
99. Isaac-Renton JL, Lewis LF, Ong CS, Nulsen MF. A second community outbreak of waterborne giardiasis in Canada and serological investigation of patients. Trans R Soc Trop Med Hyg. 1994;88:395-9.
100. Osterholm MT, Forfang JC, Ristinen TL, et al. An outbreak of foodborne giardiasis. N Engl J Med. 1981;304:24-8.
101. White KE, Hedberg CW, Edmonson LM, et al. An outbreak of giardiasis in a nursing home with evidence for multiple modes of transmission. J Infect Dis. 1989;160:298-304.
102. Pickering LK, Woodward WE, DuPont HL, Sullivan P. Occurrence of Giardia lamblia in children in day care centers. J Ped. 1984;104:522-6.
103. Farthing MJ, Mata L, Urrutia JJ, Kronmal RA. Natural history of Giardia infection of infants and children in rural Guatemala and its impact on physical growth. Am J Clin Nutr. 1986;43:395-405.
104. Fraser D, Dagan R, Naggan L, et al. Natural history of Giardia lamblia and Cryptosporidium infections in a cohort of Israeli Bedouin infants: A study of a population in transition. Am J Trop Med Hyg. 1997;57:544-9.
105. Peters CS, Sable R, Janda WM, et al. Prevalence of enteric parasites in homosexual patients attending an outpatient clinic. J Clin Microbiol. 1986;24:684-5.
106. Oberhuber G, Kastner N, Stolte M. Giardiasis: A histologic analysis of 567 cases. Scand J Gastroenterol. 1997;32:48-51.
107. Chavez B, Gonzalez-Mariscal L, Cedillo-Rivera R, Martinez-Palomo A. Giardia lamblia: In vitro cytopathic effect of human isolates. Exp Parasitol. 1995;80:133-8.
108. Kamath KR, Murugasu R. A comparative study of four methods for detecting Giardia lamblia in children with diarrheal disease and malabsorption. Gastroenterol. 1974;66:16-21.
109. Ament ME, Rubin CE. Relation in giardiasis to abnormal intestinal structure and function in gastrointestinal immunodeficiency syndromes. Gastroenterol. 1972;62:216-26.
110. Soliman MM, Taghi-Kilani R, Abou-Shady AF, et al. Comparison of serum antibody responses to Giardia lamblia of symptomatic and asymptomatic patients. Am J Trop Med Hyg. 1998;58:232-9.
111. Nayak N, Ganguly NK, Walia BN, et al. Specific secretory IgA in the milk of Giardia lamblia–infected and uninfected women. J Infect Dis. 1987;155:724-7.
112. Rosales-Borjas DM, Diaz-Rivadeneyra J, Dono-Leyva A, et al. Secretory immune response to membrane antigens during Giardia lamblia infection in humans. Infect Immun. 1998;66:756-9.
113. Zhou P, Li E, Zhu N, et al. Role of interleukin-6 in the control of acute and chronic Giardia lamblia infections in mice. Infect Immun. 2003;71:1566-8.
114. Bienz M, Dai WJ, Welle M, et al. Interleukin-6-deficient mice are highly susceptible to Giardia lamblia infection but exhibit normal intestinal immunoglobulin A responses against the parasite. Infect Immun. 2003;71:1569-73.
115. Li E, Zhou P, Ziva P, Singer SM. Mast cell–dependent control of Giardia lamblia infections in mice. Infect Immun. 2004;72:6642-9.
116. Stager S, Muller N. Giardia lamblia infections in B-cell–deficient transgenic mice. Infect Immun. 1997;65:3944-6.
117. Nash TE, Herrington DA, Levine MM, et al. Antigenic variation of Giardia lamblia in experimental human infections. J Immunol. 1990;144:4362-9.
118. Nash TE. Antigenic variation in Giardia lamblia and the host’s immune response. Phil Trans R Soc London B Biol Sci. 1997;352:1369-75.
119. Hill DR. Giardiasis. Issues in diagnosis and management. Infect Dis Clin N Am. 1993;7:503-25.
120. Pettoello MM, Guandalini S, Ecuba P, et al. Lactose malabsorption in children with symptomatic Giardia lamblia infection: Feasibility of yogurt supplementation. J Ped Gastroenterol Nutr. 1989;9:295-300.
121. Nash TE, Herrington DA, Losonsky GA, Levine MM. Experimental human infections with Giardia lamblia. J Infect Dis. 1987;156:974-84.
122. Smith PD, Lane HC, Gill VJ, et al. Intestinal infections in patients with AIDS. Ann Intern Med. 1988;108:328-33.
123. Aronson NE, Cheney C, Rholl V, et al. Biliary giardiasis in a patient with human immunodeficiency virus. J Clin Gastroenterol. 2001;33:167-70.
124. Nash TE, Ohl CA, Thomas E, et al. Treatment of patients with refractory giardiasis. Clin Infect Dis. 2001;33:22-8.
125. Alles AJ, Waldron MA, Sierra LS, Mattia AR. Prospective comparison of direct immunofluorescence and conventional staining methods for detection of Giardia and Cryptosporidium spp. in human fecal specimens. J Clin Microbiol. 1995;33:1632-4.
126. Zimmerman SK, Needham CA. Comparison of conventional stool concentration and preserved-smear methods with Merifluor Cryptosporidium/Giardia Direct Immunofluorescence Assay and ProSpecT Giardia EZ Microplate Assay for detection of Giardia lamblia. J Clin Microbiol. 1995;33:1942-3.
127. Garcia LS, Shimizu RY. Evaluation of nine immunoassay kits (enzyme immunoassay and direct fluorescence) for detection of Giardia lamblia and Cryptosporidium parvum in human fecal specimens. J Clin Microbiol. 1997;35:1526-9.
128. Hill DR, Nash TE. Intestinal flagellate and ciliate infections. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. 1st ed. Philadelphia: Churchill Livingstone; 2000:703-20.
129. Ortiz JJ, Ayoub A, Gargala G, et al. Randomized clinical study of nitazoxanide compared to metronidazole in the treatment of symptomatic giardiasis in children in Northern Peru. Aliment Pharmacol Ther. 2001;15:1409-15.
130. Adagu IS, Nolder D, Warhurst DC, Rossignol JF. In vitro activity of nitazoxanide and related compounds against isolates of Giardia intestinalis, Entamoeba histolytica, and Trichomonas vaginalis. J Antimicrob Chemotherapy. 2002;2002:103-11.
131. Abboud P, Lemee V, Gargala G, et al. Successful treatment of metronidazole- and albendazole-resistant giardiasis with nitazoxanide in a patient with acquired immunodeficiency syndrome. Clin Infect Dis. 2001;32:1792-4.
132. Bartlett AV, Englender SJ, Jarvis BA, et al. Controlled trial of Giardia lamblia: Control strategies in day care centers. Am J Pub Health. 1991;81:1001-6.
133. Olson ME, Ceri H, Morck DW. Giardia vaccination. Parasitol Today. 2000;16:213-17.
134. Faubert GM. The immune response to Giardia. Parasitol Today. 1996;12:140-5.
135. Johnson EH, Windsor JJ, Clark CG. Emerging from obscurity: Biological, clinical, and diagnostic aspects of Dientamoeba fragilis. Clin Microbiol Rev. 2004;17:553-70.
136. Markell EK, Udkow MP. Blastocystis hominis: Pathogen or fellow traveler? Am J Trop Med Hyg. 1986;35:1023-6.
137. Millet V, Spencer MJ, Chapin M, et al. Dientamoeba fragilis, a protozoan parasite in adult members of a semicommunal group. Digest Dis Sci. 1983;28:335-9.
138. Millet VE, Spencer MJ, Chapin MR, et al. Intestinal protozoan infection in a semicommunal group. Am J Trop Med Hyg. 1983;32:54-60.
139. Spencer MJ, Millet VE, Garcia LS, et al. Parasitic infections in a pediatric population. Ped Infect Dis. 1983;2:110-3.
140. Shein R, Gelb A. Colitis due to Dientamoeba fragilis. Am J Gastroenterol. 1983;78:634-6.
141. Spencer MJ, Garcia LS, Chapin MR. Dientamoeba fragilis. An intestinal pathogen in children? Am J Dis Children. 1979;133:390-3.
142. Dardick KR. Tetracycline treatment of Dientamoeba fragilis. Connect Med. 1983;47:69-70.
143. Vandenberg O, Souayah H, Mouchet F, et al. Treatment of Dientamoeba fragilis infection with paromomycin. Ped Infect Dis J. 2007;26:88-90.
144. Keystone JS, Kozarsky P. Isospora belli, Sarcocystis species, Blastocystis hominis, and Cyclospora. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed. Philadelphia: Churchill Livingstone; 2000:2915-20.
145. Stenzel DJ, Stein B, Lengy J. A cyst-like stage of Blastocystis hominis. Int J Parasitol. 1991;21:613-15.
146. Doyle PW, Helgason MM, Mathias RG, Proctor EM. Epidemiology and pathogenicity of Blastocystis hominis. J Clin Microbiol. 1990;28:116-21.
147. Nimri LF. Evidence of an epidemic of Blastocystis hominis infections in preschool children in northern Jordan. J Clin Microbiol. 1993;31:2706-8.
148. Shlim DR, Hoge CW, Rajah R, et al. Is Blastocystis hominis a cause of diarrhea in travelers? A prospective controlled study in Nepal. Clin Infect Dis. 1995;21:97-101.
149. Giacometti A, Cirioni O, Fiorentini A, et al. Irritable bowel syndrome in patients with Blastocystis hominis infection. Eur J Clin Microbiol Infect Dis. 1999;18:436-9.
150. Yakoob J, Jafri W, Jafri N, et al. Irritable bowel syndrome: In search of an etiology: Role of Blastocystis hominis. Am J Trop Med Hyg. 2004;70:383-5.
151. Udkow MP, Markell EK. Blastocystis hominis: Prevalence in asymptomatic versus symptomatic hosts. J Infect Dis. 1993;168:242-4.
152. Grossman I, Weiss LM, Simon D, et al. Blastocystis hominis in hospital employees. Am J Gastroenterol. 1992;87:729-32.
153. Markell EK, Udkow MP. Association of Blastocystis hominis with human disease? J Clin Microbiol. 1990;28:1085-6.
154. Markell EK. Is there any reason to continue treating Blastocystis infections? Clin Infect Dis. 1995;21:104-5.
155. Xiao L, Fayer R, Ryan U, Upton SJ. Cryptosporidium taxonomy: Recent advances and implications for public health. Clin Microbiol Rev. 2004;17:72-97.
156. Current WL, Reese NC, Ernst JV, et al. Human cryptosporidiosis in immunocompetent and immunodeficient persons. Studies of an outbreak and experimental transmission. N Engl J Med. 1983;308:1252-7.
157. Navin TR, Weber R, Vugia DJ, et al. Declining CD4+ T-lymphocyte counts are associated with increased risk of enteric parasitosis and chronic diarrhea: Results of a 3-year longitudinal study. J Acquir Imm Def Syndr Hum Retrovirol. 1999;20:154-9.
158. Newman RD, Sears CL, Moore SR, et al. Longitudinal study of Cryptosporidium infection in children in northeastern Brazil. J Infect Dis. 1999;180:167-75.
159. Kirkpatrick BD, Haque R, Duggal P, et al. Association between Cryptosporidium infection and human leukocyte antigen class I and class II alleles. J Infect Dis. 2008;197:474-8.
160. LeChevallier MW, Norton WD, Lee RG. Occurrence of Giardia and Cryptosporidium spp. in surface water supplies. Appl Environ Microbiol. 1991;57:2610-16.
161. Mackenzie WR, Hoxie NJ, Proctor ME, et al. A massive outbreak in Milwaukee of cryptosporidium infection transmitted through the public water supply. N Engl J Med. 1994;331:161-7.
162. Koch KL, Phillips DJ, Aber RC, Current WL. Cryptosporidiosis in hospital personnel. Evidence for person-to-person transmission. Ann Int Med. 1985;102:593-6.
163. Cordell RL, Addiss DG. Cryptosporidiosis in child care settings: A review of the literature and recommendations for prevention and control. Ped Infect Dis J. 1994;13:311-17.
164. Miron D, Kenes J, Dagan R. Calves as a source of an outbreak of cryptosporidiosis among young children in an agricultural closed community. Ped Infect Dis J. 1991;10:438-41.
165. Winkelstein JA, Marino MC, Ochs H, et al. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine (Baltimore). 2003;82:373-84.
166. Gentile G, Venditti M, Micozzi A, et al. Cryptosporidiosis in patients with hematologic malignancies. Rev Infect Dis. 1991;13:842-6.
167. Chen XM, Keithly JS, Paya CV, LaRusso NF. Cryptosporidiosis. N Engl J Med. 2002;346:1723-31.
168. Greenberg P, Koch J, Cello J. Diagnosis of Cryptosporidium parvum in patients with severe diarrhea and AIDS. Dig Dis Sci. 1996;41:2286-90.
169. Rossignol JF, Hidalgo H, Feregrino M, et al. A double-blind placebo-controlled study of nitazoxanide in the treatment of cryptosporidial diarrhoea in AIDS patients in Mexico. Trans R Soc Trop Med Hyg. 1998;92:663-6.
170. Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Cryptosporidium parvum: A prospective randomized, double-clind, placebo-controlled study of nitazoxanide. J Infect Dis. 2001;184:103-6.
171. Amadi B, Mwiya M, Musuku J, et al. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: A randomised controlled trial. Lancet. 2002;360:1375-80.
172. White AC, Chappell CL, Hayat CS, et al. Paromomycin for cryptosporidiosis in AIDS: A prospective, double-blind trial. J Infect Dis. 1994;170:419-24.
173. Wilson JA, Margolin AB. The efficacy of three common hospital liquid germicides to inactivate Cryptosporidium parvum oocysts. J Hosp Infect. 1999;42:231-7.
174. Guerrant DI, Moore SR, Lima AAM, et al. Association of early childhood diarrhea and cryptosporidiosis with impaired physical fitness and cognitive function four-seven years later in a poor urban community in northeast Brazil. Am J Trop Med Hyg. 1999;61:707-13.
175. Checkley W, Epstein LD, Gilman RH, et al. Effects of Cryptosporidium parvum infection in Peruvian children: Growth faltering and subsequent catch-up growth. Am J Epidemiol. 1998;148:497-506.
176. Ashford RW. Occurrence of an undescribed coccidian in man in Papua New Guinea. Ann Trop Med Parasitol. 1979;73:497-500.
177. Huang P, Weber JT, Sosin DM, et al. The first reported outbreak of diarrheal illness associated with Cyclospora in the United States. Ann Intern Med. 1995;123:409-14.
178. Long EG, Ebrahimzadeh A, White EH, et al. Alga associated with diarrhea in patients with acquired immunodeficiency syndrome and in travelers. J Clin Microbiol. 1990;28:1101-4.
179. Control CfD. Update: Outbreaks of Cyclospora cayetanensis infection—United States. MMWR. 1996;45:611-2.
180. Ortega YR, Sterling CR, Gilman RH, et al. Cyclospora sp—a new protozoan pathogen of humans. N Engl J Med. 1993;328:1308-12.
181. Relman DA, Schmidt TM, Gajadhar A, et al. Molecular phylogenetic analysis of Cyclospora, the human intestinal pathogen, suggests that it is closely related to Eimeria species. J Infect Dis. 1996;173:440-5.
182. Sherchand JB, Cross JH, Jimba M, et al. Study of Cyclospora cayetanensis in health care facilities, sewage water, and green leafy vegetables in Nepal. Southeast Asian J Trop Med Public Health. 1999;30:58-63.
183. Sherchand JB, Cross JH. Emerging pathogen Cyclospora cayetanensis infection in Nepal. Southeast Asian J Trop Med Public Health. 2001;32:143-50.
184. Herwaldt BL, Ackers ML. An outbreak in 1996 of cyclosporiasis associated with imported raspberries. The Cyclospora Working Group. N Engl J Med. 1997;336:1548-56.
185. Herwaldt BL, Beach MJ. The return of Cyclospora in 1997: Another outbreak of cyclosporiasis in North America associated with imported raspberries. Cyclospora Working Group. Ann Intern Med. 1999;130:210-20.
186. Connor BA, Shlim DR, Scholes JV, et al. Pathologic changes in the small bowel in nine patients with diarrhea associated with a coccidia-like body. Ann Intern Med. 1993;119:377-82.
187. Berlin OG, Novak SM, Porschen RK, et al. Recovery of Cyclospora organisms from patients with prolonged diarrhea. Clin Infect Dis. 1994;18:606-9.
188. Varea M, Clavel A, Doiz O, et al. Fuchsin fluorescence and autofluorescence in Cryptosporidium, Isospora, and Cyclospora oocysts. Int J Parasitol. 1998;28:1881-3.
189. Varma M, Hester JD, Schaeffer FW, et al. Detection of Cyclospora cayetanensis using a quantitative real-time PCR assay. J Microbiol Methods. 2003;53:27-36.
190. Hoge CW, Shlim DR, Ghimire M, et al. Placebo-controlled trial of co-trimoxazole for Cyclospora infections among travellers and foreign residents in Nepal. Lancet. 1995;345:691-3.
191. Pape JW, Verdier RI, Johnson WD. Treatment and prophylaxis of Isospora belli infection in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1989;320:1044-7.
192. Pape JW, Verdier RI, Boncy M, et al. Cyclospora infection in adults infected with HIV. Clinical manifestations, treatment, and prophylaxis. Ann Intern Med. 1994;121:654-7.
193. Verdier RI, Fitzgerald DW, Johnson WD, Pape JW. Trimethoprim-sulfamethoxazole compared with ciprofloxacin for treatment and prophylaxis of Isospora belli and Cyclospora cayetanensis infection in HIV-infected patients. A randomized, controlled trial. Ann Intern Med. 2000;132:885-8.
194. Osterholm MT. Cyclosporiasis and raspberries—lessons for the future. N Engl J Med. 1997;336:1597-8.
195. Godiwala T, Yaeger R. Isospora and traveler’s diarrhea. Ann Intern Med. 1987;106:908-9.
196. DeHovitz JA, Pape JW, Boncy M, Johnson WD. Clinical manifestations and therapy of Isospora belli infection in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1986;315:87-90.
197. Brandborg LL, Goldberg SB, Briedenbach WC. Human coccidiosis—a possible cause of malabsorption. The life cycle in small-bowel mucosal biopsies as a diagnostic feature. N Engl J Med. 1970;283:1306-13.
198. Bryan RT, Weber R, Schwartz DA. Microsporidiosis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. 1st ed. Philadelphia: Churchill Livingstone; 2000:840-51.
199. Didier ES. Microsporidiosis. Clin Infect Dis. 1998;27:1-8.
200. Sobottka I, Schwartz DA, Schottelius J, et al. Prevalence and clinical significance of intestinal microsporidiosis in human immunodeficiency virus–infected patients with and without diarrhea in Germany: A prospective coprodiagnostic study. Clin Infect Dis. 1998;26:475-80.
201. Hutin YJF, Sombardier MN, Liguory O, et al. Risk factors for intestinal microsporidiosis in patients with human immunodeficiency virus infection: A case-control study. J Infect Dis. 1998;178:904-7.
202. Sandfort J, Hannemann A, Gelderblom H, et al. Entercytozoon bieneusi infection in an immunocompetent patient who had acute diarrhea and who was not infected with the human immunodeficiency virus. Clin Infect Dis. 1994;19:514-16.
203. Albrecht H, Sobottka I. Enterocytozoon bieneusi infection in patients who are not infected with human immunodeficiency virus. Clin Infect Dis. 1997;25:344.
204. Muller A, Bialek R, Kamper A, et al. Detection of microsporidia in travelers with diarrhea. J Clin Microbiol. 2001;39:1630-2.
205. Sobottka I, Albrecht H, Schafer H, et al. Disseminated Encephalitozoon (Septata) intestinalis infection in a patient with AIDS: Novel diagnostic approaches and autopsy-confirmed parasitological cure following treatment with albendazole. J Clin Microbiol. 1995;33:2948-52.
206. Weber R, Bryan RT, Owen RL, et al. Improved light-microscopical detection of microsporidia spores in stool and duodenal aspirates. The Enteric Opportunistic Infections Working Group. N Engl J Med. 1992;326:161-6.
207. Berlin OG, Conteas CN, Porschen RK. Rapid epifluorescent technique to detect microsporidia. AIDS. 1996;10:1175-6.
208. van Gool T, Snijders F, Reiss P, et al. Diagnosis of intestinal and disseminated microsporidial infections in patients with HIV by a new rapid fluorescence technique. J Clin Pathol. 1993;46:694-9.
209. Menotti J, Cassinat B, Porcher R, et al. Development of a real-time polymerase-chain-reaction assay for quantitative detection of Enterocytozoon bieneusi DNA in stool specimens from immunocompromised patients with intestinal microsporidiosis. J Infect Dis. 2003;187:1469-74.
210. Molina JM, Tourneur M, Sarfati C, et al. Fumagillin treatment of intestinal microsporidiosis. N Engl J Med. 2002;346:1963-9.
211. Kirchhoff LV. American trypanosomiasis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. 1st ed. Philadelphia: Churchill Livingstone; 2000:785-96.
212. Avila HA, Pereira JB, Theimann O, et al. Detection of Trypanosoma cruzi in blood specimens of chronic chagasic patients by polymerase chain reaction amplification of kinetoplast minicircle DNA: Comparison with serology and xenodiagnosis. J Clin Microbiol. 1993;31:2421-6.
213. Britto C, Cardoso MA, Vanni CM, et al. Polymerase chain reaction detection of Trypanosoma cruzi in human blood samples as a tool for diagnosis and treatment evaluation. Parasitol. 1995;110:241-7.
214. Kirchhoff LV. Changing epidemiology and approaches to therapy for Chagas’ disease. Curr Infect Dis Rep. 2003;5:59-65.
215. Figueiredo MC, Oliveira RB, Iazigi N, Matsuda NM. Short report: Comparison of the effects of sublingual nifedipine and isosorbide dinitrate on oesophageal emptying in patients with Chagasic achalasia. Aliment Pharmacol Ther. 1992;6:507-12.
216. Ferreira-Filho LP, Patto RJ, Troncon LE, Oliveira RB. Use of isosorbide dinitrate for the symptomatic treatment of patients with Chagas’ disease achalasia. A double-blind, crossover trial. Braz J Med Biol Res. 1991;24:1093-8.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 109 Intestinal Protozoa
ENTAMOEBA HISTOLYTICA
EPIDEMIOLOGY
Entamoeba histolytica was first linked causally to amebic colitis and liver abscess by Lösch in 1875, and it was named by Schaudinn in 1903 for its ability to destroy host tissues. In 1925, Emil Brumpt proposed the existence of a second, morphologically identical but nonpathogenic Entamoeba species, Entamoeba dispar, to explain why only a minority of people infected with what was then termed E. histolytica develop invasive disease. Although Brumpt’s hypothesis was not accepted during his lifetime, it is now clear that he was correct and E. histolytica (Schaudinn, 1903) was recently reclassified to include two morphologically indistinguishable species: E. histolytica, the cause of invasive amebiasis, and E. dispar, a nonpathogenic intestinal commensal parasite (see later section).1
Entamoeba histolytica is a parasite of global distribution, but most of the morbidity and mortality from amebiasis occurs in Central and South America, Africa, and the Indian subcontinent.2 Fortunately, the majority of the 500 million persons worldwide previously believed to be asymptomatic E. histolytica cyst passers are actually infected with E. dispar. The best current estimate is that E. histolytica causes 34 to 50 million symptomatic infections annually worldwide, resulting in 40,000 to 100,000 deaths each year.3,4 In Dhaka, Bangladesh, where diarrheal diseases are the leading cause of childhood death, 80% of children studied prospectively were infected with E. histolytica at least once during four years of follow-up.5 Furthermore, E. histolytica–associated diarrhea in these children was associated with significantly low weight and height for age.6
E. histolytica has a simple, two-stage life cycle consisting of an infectious cyst and a motile trophozoite (Fig. 109-1). The cyst form measures 5 to 20 µm in diameter and contains four or fewer nuclei. The ameboid trophozoite, which is responsible for tissue invasion, measures 10 to 60 µm (Fig. 109-2) and contains a single nucleus with a central karyosome (Fig. 109-3). The cysts are relatively resistant to chlorination and desiccation, and they can survive in a moist environment for several weeks.
Figure 109-1. Life cycle of Entamoeba histolytica. PMNs, polymorphonuclear neutrophils.
(From Petri WA, Sing U, Ravdin JI. Enteric amebiasis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia: WB Saunders; 1999.)
Figure 109-2. Amebae that infect the human gastrointestinal tract. E., entamoeba.
(From Ravdin Jl, Guerrant RL. Current problems in the diagnosis and treatment of amebic infections. Curr Clin Trop Infect Dis 1986; 7:82.)
Infection occurs following ingestion of cysts in fecally contaminated food or water. Within the lumen of the small intestine, the quadrinucleate cyst undergoes nuclear then cytoplasmic division, giving rise to eight trophozoites.7 Only about 10% of infected persons develop invasive disease characterized by invasion of the colonic epithelium by trophozoites.1 Trophozoites that gain access to the bloodstream can spread hematogenously to establish infection at distant sites (most commonly liver abscess, as discussed in Chapter 82). Why some persons develop invasive disease and others remain asymptomatic remains a mystery; parasite and host differences are likely to be important in this regard.
A molecular epidemiologic study that used the polymerase chain reaction (PCR) to amplify a polymorphic region of the E. histolytica genome and assign a genotype to different clinical isolates has demonstrated a correlation between different E. histolytica strains and the outcome of infection.8 The specific underlying genetic differences among ameba strains that are responsible for altered virulence, however, remains unknown. Furthermore, amebic liver abscess is primarily a disease of men, and studies suggest that susceptibility to both intestinal and hepatic amebiasis is linked to human leukocyte antigen (HLA) class II alleles.9,10 For example, the HLA DQB1*0601 allele may be associated with protection from intestinal amebiasis.9 As is the case for genetic differences among E. histolytica strains, however, there is no evidence of a direct causal role for different HLA types; rather, these HLA types are likely to be in linkage disequilibrium with genes in the nearby vicinity that encode the causal factors.
PATHOGENESIS, PATHOLOGY, AND IMMUNOLOGY
Both amebic factors and the host’s inflammatory response contribute to tissue destruction during invasive amebiasis. Microscopy studies have defined a stepwise progression of disease.11–13 After excystation within the lumen of the small intestine, trophozoites adhere to colonic mucins and epithelial cells, largely via an amebic galactose/N-acetyl-d-galactosamine inhibitable surface lectin.14–16 Secreted cysteine proteinases then facilitate tissue invasion by degrading human colonic mucus and extracellular matrix proteins.17–20 Further disruption of the colonic epithelium results directly from contact-dependent cytolysis of epithelial and immune cells and from an acute epithelial cell inflammatory response with recruitment of neutrophils and immune-mediated tissue damage.14,21–25
The cecum and ascending colon are affected most commonly, although in severe disease the entire colon may be involved. On gross examination, pathology can range from mucosal thickening to multiple punctate ulcers with normal intervening tissue (Fig. 109-4) to frank necrosis. For unknown reasons, the downward invasion of amebic trophozoites often is halted at the level of the muscularis mucosa. Subsequent lateral spread of amebae undermines the overlying epithelium, resulting in the clean-based, flask-shaped ulcers that characterize amebic colitis.26,27 Early in infection, an influx of neutrophils is typical, but in well-established ulcers, few inflammatory cells are seen.13,26–28 Organisms may be seen ingesting red blood cells (erythrophagocytosis) (Fig. 109-5). At distant sites of infection (e.g., liver abscess), similar pathologic characteristics include central liquefaction of tissue surrounded by a minimal mononuclear cell infiltrate.27–29
Figure 109-4. Colonoscopic findings in a patient with amebic colitis. Multiple punctate ulcers are visible.
Because more than 90% of persons colonized with E. histolytica spontaneously clear the infection within a year, an effective immune response to amebiasis seems to develop.30 Children with fecal anti-amebic lectin immunoglobulin (Ig)A have short-lived protection from subsequent intestinal infection.5,31,32 The protective role of secretory IgA is not certain, however, and the contributions of humoral and cellular immunity to protection from amebiasis remain unknown. Nearly everyone with invasive amebiasis develops a systemic and a mucosal humoral immune response.33–38 Antibodies alone are unable to clear established infection, however, because asymptomatic cyst passers remain infected for months after anti-amebic antibodies develop.30,33 Passive immunization experiments in a severe combined immunodeficient (SCID) mouse model of liver abscess do suggest an important role for preexisting humoral immunity in protection from infection.39 Reports that patients receiving glucocorticoids may be at increased risk for severe amebic colitis suggest that cellular immunity also plays an important role in control of E. histolytica infection40,41; despite this concern, no increase in disease severity in patients with AIDS has been observed. In fact, in a mouse model of amebic colitis, disease was exacerbated by CD4+ T cells.42
CLINICAL FEATURES
Infection with E. histolytica results in one of three outcomes. Approximately 90% of infected persons remain asymptomatic. The other 10% of infections result in invasive amebiasis characterized by dysentery (amebic colitis) or, in a minority of cases, extraintestinal disease (most commonly amebic liver abscess; see Chapter 82).1,30
In the United States, immigrants from or travelers to endemic regions, male homosexuals, and institutionalized persons are at greatest risk for amebiasis. In addition, malnourished patients, infants, the elderly, pregnant women, and patients receiving glucocorticoids may be at increased risk for fulminant disease.2,40,41 When one or more of these epidemiologic risk factors are present, amebic dysentery should be considered in the differential diagnosis of occult or grossly bloody diarrhea.
The major diagnostic challenge for the clinician seeing a patient with amebic colitis is to distinguish the illness from other causes of bloody diarrhea. The differential diagnosis includes the causes of bacterial dysentery, such as Shigella, Salmonella, and Campylobacter species and enteroinvasive or enterohemorrhagic Escherichia coli, and noninfectious diseases, including inflammatory bowel disease, and ischemic colitis.2,43 In contrast to bacterial dysentery, which typically begins abruptly, amebic colitis begins gradually over one to several weeks (Table 109-1). Although more than 90% of patients with amebic colitis present with diarrhea, abdominal pain can occur without diarrhea; abdominal pain, tenesmus, and fever are highly variable. Weight loss is common because of the chronicity of the illness. Microscopic blood is present in the stool of most patients with amebic dysentery.2,43,44
Table 109-1 Comparison of Amebic Colitis and Invasive Bacterial Dysentery
| FEATURE | AMEBIC COLITIS | BACTERIAL DYSENTERY* |
|---|---|---|
| Travel to or from an endemic area | Yes | Sometimes |
| Usual duration of symptoms | >7 days | 2-7 days |
| Diarrhea | 94-100% | 100% |
| Fecal occult blood | 100% | 40% |
| Abdominal pain | 12-80% | ~50% |
| Weight loss | Common | Unusual |
| Fever >38°C | Minority | Majority |
Adapted from Huston CD, Petri WA. Amebiasis. In: Rakel RE, Bope ET, editors. Conn’s Current Therapy, 2001. Philadelphia: WB Saunders; 2001. pp 50-4.
The most feared complication of amebic dysentery, acute necrotizing colitis with toxic megacolon, occurs in 0.5% of cases. This complication manifests as an acute dilatation of the colon, and 40% of patients die from sepsis unless it is promptly recognized and treated surgically.45,46 Unusual complications include the formation of enterocutaneous, rectovaginal, and enterovesicular fistulas and ameboma. Ameboma, due to intraluminal granulation tissue, can cause bowel obstruction and mimic carcinoma of the colon.2,43
Although a history of dysentery early in the illness is common, dysentery has resolved in most patients by the time of presentation.47–49 Extraintestinal sites of infection are involved and typically result either from direct extension of liver abscesses (e.g., amebic pericarditis or lung abscess) or from hematogenous spread of disease (e.g., brain abscess).2,50
DIAGNOSIS
Because amebiasis patients erroneously treated for inflammatory bowel disease with glucocorticoids can develop fulminant colitis, accurate initial diagnosis is critical.40,41 The gold standard for diagnosis of amebic colitis remains colonoscopy with biopsy, and colonoscopy should be performed whenever infectious causes of bloody diarrhea are strong considerations in the differential diagnosis of ulcerative colitis. Because the cecum and ascending colon are affected most often, colonoscopy is preferred to sigmoidoscopy. Classically, multiple punctate ulcers measuring 2 to 10 mm are seen with essentially normal intervening tissue (see Fig. 109-4); however, the colonic epithelium might simply appear indurated with no visible ulcerations; appear like ulcerative colitis with a myriad of ulcerations and granular, friable mucosa; or, in severe cases where the ulcers have coalesced, the epithelium may appear necrotic. Histologic examination of a biopsy specimen taken from the edge of an ulcer reveals amebic trophozoites and a variable inflammatory infiltrate (see Fig. 109-5).27 Identification of amebae can be aided by periodic acid–Schiff staining of biopsy tissue, which stains trophozoites magenta.
Stool examination for ova and parasites, the traditional method for diagnosing amebiasis, should not be relied upon. Although the presence of amebic trophozoites with ingested erythrocytes strongly correlates with E. histolytica infection, these rarely are present,51 and in the absence of hematophagous trophozoites, microscopy cannot distinguish E. histolytica from E. dispar. Difficulty in distinguishing other nonpathogenic amebae (see later) and white blood cells from E. histolytica also limits the specificity of stool microscopy.52 The sensitivity of microscopy for identification of amebae is at best 60% and it may be reduced by delays in processing of stool samples.52,53 The primary utility of stool microscopy for ova and parasites in a patient with diarrhea, therefore, is to evaluate the stool for other parasitic causes of diarrhea.
Noninvasive methods to accurately differentiate E. histolytica from E. dispar include stool culture with isoenzyme analysis, serum amebic-antibody titers, PCR, and an enzyme-linked immunosorbent assay (ELISA) that detects the amebic lectin antigen in stool samples.54–64 Of these, only serum amebic-antibody titers and the stool ELISA are widely available for clinical use.
Because serum anti-amebic antibodies do not develop in patients infected with E. dispar, serologic tests for amebiasis accurately distinguish E. histolytica and E. dispar infection. From 75% to 85% of patients with acute amebic colitis have detectable anti-amebic antibodies on presentation, and convalescent titers develop in more than 90% of patients.34,35,65 For amebic liver abscess, 70% to 80% of patients have detectable antibody titers on presentation, and convalescent titers develop in more than 90% of patients. Because antiamebic antibodies can persist for years, however, a positive result must be interpreted with caution.34 For persons with known epidemiologic risks (e.g., emigration from or prior travel to an endemic region), a positive result might simply represent infection in the distant past. In the setting of recent travel to an endemic region and a positive antibody titer, diagnosis is confirmed by an appropriate symptomatic response to anti-amebic treatment.
The most specific clinically available test for diagnosis of amebiasis is a stool ELISA to detect the E. histolytica adherence lectin. Only one of the many ELISA tests developed thus far (the E. histolytica II test, TechLab, Blacksburg, Va.) accurately distinguishes E. histolytica from E. dispar.53,60,61 This test’s specificity, when compared with the gold standard of stool culture followed by isoenzyme analysis, was greater than 90%, and it was greater than 85% sensitive for diagnosis of intestinal amebiasis when fresh fecal samples were analyzed without delay.61 In other studies, the sensitivity of this method has been less impressive, emphasizing the need for rapid processing of stool samples.66,67 It also may be possible to use this antigen detection test to diagnose amebic liver abscess, because before treatment is initiated, amebic lectin antigen can be detected in the serum of greater than 90% of patients who have amebic liver abscess.68
TREATMENT
Drugs for treatment of amebiasis are categorized as luminal or tissue amebicides on the basis of the location of their anti-amebic activity (Table 109-2).
Table 109-2 Amebicidal Agents Currently Available in the United States
| AMEBICIDAL AGENT | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| For Luminal Amebiasis | ||
| Paromomycin (Humatin) | 7-day treatment course; may be useful during pregnancy | Frequent gastrointestinal side effects; rare ototoxicity and nephrotoxicity |
| Iodoquinol (Yodoxin) | Inexpensive and effective | 20-day treatment course; contains iodine; rare optic neuritis and atrophy with prolonged use |
| Diloxanide furoate (Furamide) | Available in the United States only from the CDC; frequent gastrointestinal side effects; rare diplopia | |
| For Invasive Intestinal Disease Only | ||
| Tetracyclines, erythromycin | Not effective for liver abscess; frequent gastrointestinal side effects; tetracyclines should not be administered to children or pregnant women | |
| For Both Invasive Intestinal and Extraintestinal Amebiasis | ||
| Metronidazole (Flagyl) | Drug of choice for amebic colitis and liver abscess | Anorexia, nausea, vomiting, and metallic taste in nearly one third of patients; disulfiram-like reaction with alcohol; rare seizures |
| Tinidazole (Tindamax) | Alternative to metronidazole; once daily dosing; now approved for distribution in the United States | Side effects are similar to those with metronidazole |
| Nitazoxanide (Alinia) | Useful alternative if the patient is intolerant of metronidazole or tinidazole | Limited clinical data for amebiasis; rare and reversible conjunctival icterus |
| For Extraintestinal Amebiasis Only | ||
| Chloroquine (Aralen) | Useful only for amebic liver abscess | Occasional headache, pruritus, nausea, alopecia, and myalgias; rare heart block and irreversible retinal injury |
CDC, Centers for Disease Control and Prevention.
Adapted from Huston CD, Petri WA. Amebiasis. In: Rakel RE, Bope ET, editors. Conn’s Current Therapy, 2001. Philadelphia: WB Saunders; 2001. pp 50-4.
The luminal amebicides include iodoquinol, diloxanide furoate, and paromomycin.69,70 Of these, paromomycin, a nonabsorbable aminoglycoside, is preferred because of its safety, short duration of required treatment, and superior efficacy. Its major side effect is diarrhea. Approximately 85% of asymptomatic patients are cured with one course of paromomycin, and, because it is nonabsorbable and has moderate activity against trophozoites that have invaded the colonic mucosa, it might also be useful for single-drug treatment of mild invasive disease during pregnancy.71,72
The tissue amebicides include metronidazole, tinidazole, nitazoxanide, erythromycin, and chloroquine.70,73 Of these, metronidazole and tinidazole are the drugs of choice, with cure rates greater than 90%.74 Nitazoxanide, a new antiparasitic agent, appears to be efficacious, with similar cure rates in several randomized, placebo-controlled trials.73,75–77 Erythromycin has no activity against amebic liver disease, and chloroquine has no activity against intestinal disease.78
Because approximately 10% of asymptomatic cyst passers develop invasive amebiasis, E. histolytica carriers should be treated.1,4 For noninvasive disease, treatment with a luminal agent alone is adequate (e.g., paromomycin 25-35 mg/kg/day in three divided doses for seven days).70 Patients with amebic colitis should first be treated with an oral nitroimidazole (either metronidazole [500-750 mg three times daily for 10 days] or tinidazole [2 grams once daily for three to five days]) to eliminate invasive trophozoites. Metronidazole and tinidazole are believed to be less effective against organisms in the colonic lumen, and subsequent treatment with a luminal agent such as paromomycin is recommended to prevent recurrent disease.70,74
It is also for this reason that the familiar tissue amebicides (e.g., metronidazole) are not recommended as first-line agents for treatment of asymptomatic infection. At the recommended doses of metronidazole and tinidazole, gastrointestinal side effects including nausea and vomiting develop in approximately 30% of patients.74 Because of severe gastrointestinal side effects, simultaneous treatment with a nitroimidazole and a luminal agent generally is not recommended.
Most patients with colitis respond promptly with resolution of diarrhea in two to five days.2
Despite conflicting reports on the safety of the nitroimidazoles for the developing fetus during pregnancy, women with severe disease during pregnancy should probably be treated without delay. As discussed in Chapter 82, metronidazole (750 mg three times a day for 10 days) followed by a luminal agent is also the treatment of choice for amebic liver abscess.70,78
CONTROL AND PREVENTION
Prevention and control of E. histolytica infection depends on interruption of fecal-oral transmission. Water can be made safe for drinking and food preparation by boiling it for one minute, by halogenation (with chlorine or iodine), or by filtration.7 In the United States and Europe, modern water treatment facilities effectively remove E. histolytica. The importance of safe drinking water is highlighted by an outbreak of amebiasis in Tblisi, Republic of Georgia, where there was a water-borne epidemic due to decay of the water treatment facilities following the demise of the Soviet Union.79 In the vast majority of the developing world, however, no modern water treatment facilities exist and none are likely to be constructed in the foreseeable future. Naturally acquired immunity to intestinal amebiasis provides short-lived protection against reinfection, giving hope that a vaccine may be feasible.5,31,32 Because humans and some higher nonhuman primates are the only known hosts for E. histolytica, a vaccine that successfully prevents colonization might enable eradication of the disease.80
OTHER INTESTINAL AMEBAE
Eight species of commensal amebae commonly infect the human gastrointestinal tract (see Fig. 109-2). These include Entamoeba dispar, Entamoeba moshkovskii, Entamoeba coli, Entamoeba hartmanni, Entamoeba gingivalis, Entamoeba polecki, Endolimax nana, and Iodamoeba butschlii. Dientamoeba fragilis (discussed in the following section), previously thought to be an ameba, is more closely related to the flagellated protozoan Trichomonas vaginalis than to the true amebae.7 With the exception of E. gingivalis, which has no known cyst stage, all of these true amebae have simple two-stage life cycles, consisting of an infectious cyst form and a motile trophozoite form.7 All but E. dispar and E. moshkovskii can be differentiated from E. histolytica using light microscopy based on characteristic features of the cyst and trophozoite forms. E. dispar must be differentiated from E. histolytica based on biochemical, antigenic, or genetic differences.1
E. dispar is a nonpathogenic protozoan parasite that is morphologically indistinguishable from Entamoeba histolytica by light microscopy.1 An estimated 450 million people worldwide are infected with E. dispar, and infection with E. dispar is approximately 10 times more prevalent than E. histolytica infection.1,3,4 Although E. dispar has been demonstrated to cause mucosal ulcerations in animal models, it has not been demonstrated to cause human disease and does not require treatment.1 Entamoeba moshkovskii, which is primarily thought to be a free-living ameba, also has cysts and trophozoites indistinguishable from E. dispar and E. histolytica except that trophozoites of E. histolytica might show erythrophagocytosis. Although in some studies a high prevalence of E. moshkovskii infection in humans has been demonstrated, reports are conflicting regarding its pathologic potential.81–83 The primary clinical significance of E. dispar or E. moshkovskii is that they must be distinguished from E. histolytica to enable accurate diagnosis of invasive amebiasis. PCR to amplify small ribosomal RNA (not clinically available), and ELISAs using monoclonal anti-amebic antibodies to detect specific E. histolytica antigens, make accurate diagnosis possible (see the section on E. histolytica diagnosis).55–64
Besides E. dispar, Entamoeba coli is the intestinal commensal most commonly mistaken for E. histolytica. Entamoeba coli trophozoites contain a single nucleus with a prominent karyosome that usually is eccentric in location, distinguishing them from E. histolytica and E. dispar trophozoites, which have a centrally located karyosome. In addition, the cyst form of Entamoeba coli typically contains five to eight nuclei (see Fig. 109-2). Entamoeba coli is nonpathogenic and requires no specific treatment; however, it is a valuable marker of fecal-oral exposure, and it can be found concurrently with E. histolytica in 10% to 30% of patients in endemic regions.7
Entamoeba hartmanni was classified as “small race” E. histolytica for many years. The trophozoites resemble those of E. histolytica except for their small size (less than 10 µm).7 Entamoeba hartmanni now is recognized as a nonpathogen that requires no treatment.
Entamoeba gingivalis is the only ameba found in the oral cavity, where it lives in the anaerobic environment of the gingival crease. The trophozoite is identical in size to that of E. histolytica and contains a single nucleus with a prominent central karyosome (see Fig. 109-2). No cyst form of E. gingivalis has been identified, and oral-oral contact is believed to be its mode of transmission.7,84 Entamoeba gingivalis is associated with poor dental hygiene and periodontal disease, but no causal relationship to periodontitis has been proven.84 The increased frequency of colonization in this setting might simply reflect a more hospitable host environment. E. gingivalis is often associated with periodontal disease in AIDS patients, however, in whom treatment with metronidazole has been reported to be effective.85
Entamoeba polecki, a parasite characterized by a uninucleated cyst, is primarily a parasite of pigs and monkeys that sometimes infects humans. It has been suggested that several distinct uninucleated cyst-producing Entamoeba species can infect humans and it has been proposed that these organisms collectively be termed “E. polecki-like.”86 Infection with E. polecki is rare except in Papua New Guinea where as many as 30% of children were found to be colonized in one study.87 At present, specific treatment of E. polecki–like infections is not routinely recommended, but persons with heavy burdens of this parasite can develop nonspecific gastrointestinal symptoms and might benefit from treatment. Good clinical responses to metronidazole and diloxanide furoate have been reported.88
Endolimax nana is a nonpathogenic intestinal ameba that often infects humans.7 The distribution of E. nana is worldwide, but it is most common in the tropics, where 5% to 33% of persons are infected.89,90 Infection requires no specific treatment, but it serves as a useful marker for fecal-oral exposure. E. nana trophozoites can be distinguished from E. histolytica by their vesiculate nucleus, large irregular karyosome, and relatively small size (8 to 12 µm).7
Iodamoeba butschlii is a nonpathogenic intestinal ameba passed by the fecal-oral route. Trophozoites of I. butschlii contain a single nucleus with a large karyosome (which is distinct from the punctate karyosome of E. histolytica); its cysts contain a single nucleus, and a large, eccentric glycogen mass that stains with iodine (hence the name Iodamoeba). I. butschlii infection requires no treatment.7
GIARDIA INTESTINALIS
EPIDEMIOLOGY
Giardia intestinalis (also called G. lamblia and G. duodenalis) is a ubiquitous flagellated intestinal protozoan. Van Leeuwenhoek accurately described its motile trophozoite form in his own stools in 1681, but it was not until 1915 that Stiles named the species.7
The life cycle of Giardia consists of an infectious cyst form and a motile trophozoite (Fig. 109-6). The cyst is oval (8 to 12 µm long by 7 to 10 µm wide), contains four nuclei, and has a rigid outer wall that protects it from dehydration, extremes of temperature, and chlorination (Fig. 109-7). Giardia cysts can survive in cold water for several weeks.7,91 Ingestion of as few as 10 to 25 cysts can result in infection.91 After ingestion, excystation occurs following exposure to stomach acid and intestinal proteases, each cyst giving rise to two trophozoites. Giardia trophozoites (see Fig. 109-3B) are pear-shaped (10 to 20 µm long by 7 to 10 µm wide), contain two nuclei, have eight flagellae for locomotion, and replicate by binary fission. The trophozoites live in the duodenum, where they adhere to enterocytes. Eventually they encyst, following exposure to alkaline conditions or bile salts, and are excreted in the stool to complete their life cycle.91
Figure 109-6. Life cycle of Giardia intestinalis.
(From Hill DR, Nash TE. Intestinal flagellate and ciliate infections. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia: WB Saunders; 1999.)
G. intestinalis, which was defined originally as a species by morphology, is more accurately defined as a species complex with at least seven major genotypes (assemblages A through G).92 Of these, only assemblages A and B are known to infect humans. Both of these genotypes also commonly infect cats and dogs, highlighting the importance of these pets as reservoirs for human disease.92 New data suggest that assemblage A isolates may be more virulent than assemblage B isolates.93,94
G. intestinalis is the most commonly identified intestinal parasite in the United States and was identified in 7.2% of stool samples examined by state health departments in 1987.95 Giardiasis occurs in both endemic and epidemic forms via water-borne, food-borne, and person-to-person transmission.96–102 Worldwide, Giardia infects infants more commonly than adults, and in highly endemic regions, essentially all children are infected by two to three years of age.103,104 In the developing world, it is likely that recurrent infantile diarrhea from giardiasis contributes significantly to malnutrition.103 In the United States, children in daycare and sexually active homosexual men have the greatest risk of infection.95,105 During a year-long longitudinal study at a U.S. daycare center, Giardia cysts were identified at some time in the stool of more than 30% of children.102 Additional risk factors for infection include drinking untreated surface water, a shallow well as a residential water source, swimming in any natural body of fresh water, and contact with a person who has giardiasis or contact with a child in daycare.97
PATHOGENESIS, PATHOLOGY, AND IMMUNOLOGY
Giardia causes malabsorptive diarrhea by an unknown mechanism. Trophozoites adhere (perhaps by suction) to the epithelium of the upper small intestine using a disk structure located on their anterior ventral surface.7,91 There is no evidence that trophozoites invade the mucosa,106 but electron microscopy has shown they damage the mucosal brush border.91,107 On biopsy, pathologic changes range from an entirely normal-appearing duodenal mucosa (except for adherent trophozoites), as was found in more than 96% of biopsy specimens in one large study, to severe villus atrophy with a mononuclear cell infiltrate that resembles celiac sprue.106,108,109 The severity of diarrhea appears to correlate with the severity of the pathologic change.91
The host immune response plays a critical role in limiting the severity of giardiasis. When infected with Giardia, persons with common variable immunodeficiency develop severe, protracted diarrhea and malabsorption with sprue-like pathologic changes that resolve with treatment of the Giardia.109 Both systemic and mucosal humoral immune responses can be measured consistently following Giardia infection. High serum anti-giardia IgM, IgG, and IgA titers can be detected, and anti-giardia secretory IgA (s-IgA) can be detected in saliva and in breast milk of infected mothers.110–112 Animal studies suggest that both early and late immune responses are important for control of Giardia infections. Interleukin-6 (IL-6) is important in the early immune response to Giardia in mice, as are mast cells, which might function as IL-6 producers or via another mechanism.113–115 In a B-cell-deficient transgenic mouse model, infection with Giardia does not resolve, confirming the importance of the humoral immune response for clearance of established infections.116 In culture, Giardia
[/not-level-membership-for-gastroenterology-and-hepatology-category]
