CHAPTER 99 Intestinal Electrolyte Absorption and Secretion
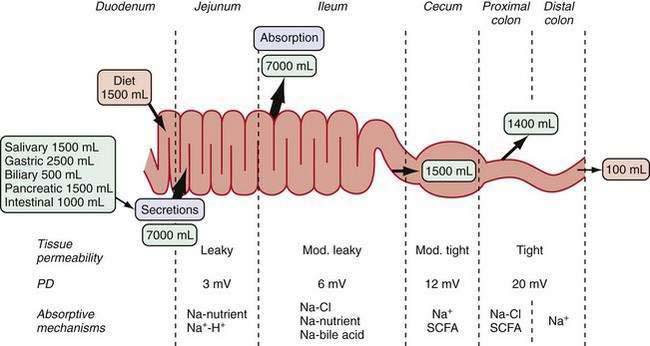
The gastrointestinal (GI) tract processes 8 to 9 L of fluid daily that is derived from oral intake and endogenous exocrine secretions. Intestinal fluid absorption is a process that functions with 98% efficiency, allowing only 100 to 200 mL to be excreted each day. The intestine also extracts nutrients, vitamins, and minerals; excludes destructive antigens and microbes; and excretes waste (Fig. 99-1). This multitasking is achieved by the unique tissue, cellular, and molecular architecture of the small and large intestine in combination with a complex array of intricate regulatory mechanisms (Fig. 99-2). Regulation is accomplished by cross-talk between endocrine and paracrine hormones, neurotransmitters, immunomodulators, and luminal factors. Remarkably, this orchestration proceeds smoothly on a daily basis; however, when the balance is perturbed, as occurs with an enteric infection, diarrhea ensues.
INTESTINAL ARCHITECTURE AND TRANSPORT
The structural and functional design of the intestine is optimally geared to its functions of absorbing nutrients and transporting fluids. In the small intestine, a 600-fold amplification of the absorptive surface is achieved by structural features, such as the circular folds of Kerckring (plicae circulares), villus-crypt architecture, and microvilli. Using a cylinder as the model, it has been estimated that the surface area of the small intestine is about 3300 cm2; the plicae circulare, villi, and microvilli amplify the surface area by factors of 3, 10, and 20, respectively, ultimately giving a surface area of about 2,000,000 cm2. In the large intestine, the spatial separation of crypts and surface cells allows efficient reabsorption of fluid. The overall architecture of the intestinal musculature can influence bulk fluid flow and transit time via changes in motility patterns (see Chapters 97 and 98), but the work of fluid transport occurs in the epithelia.
BASIC EPITHELIAL CELL MODEL
This basic cell model is modified by insertion of transporters into either the apical or basolateral membrane or by the characteristics of tight junctions that determine the unique qualities of a specific epithelial segment. A complex interaction of protein-sorting signals, cytoskeletal elements, and intracellular trafficking processes determines whether a newly synthesized protein is targeted to either the apical or basolateral membrane. For example, proteins with a glycosyl phosphatidyl inositol (GPI) anchor (e.g., alkaline phosphatase or carcinoembryonic antigen) are often associated with lipid rafts, and the GPI anchor serves to direct them toward the apical membrane.1 Membrane proteins destined to be delivered to the basolateral membrane carry specific membrane-sorting signals (amino acid sequences) in their cytoplasmic tails. In contrast, other proteins can insert randomly into either apical or basolateral domain, but they may be retained in the basolateral pole by specific components such as ankyrin.2
Regulation of intracellular trafficking ensures delivery of the right protein to the right membrane and is critical for establishing epithelial polarization and vectorial transport. When tight junctions are disrupted in vitro, diffusion and intermingling of apical and basolateral proteins in the fluid phase of the membrane result in a loss of epithelial cell polarity. There is some evidence that the distribution of Na+ pumps is altered during postischemic injury.3
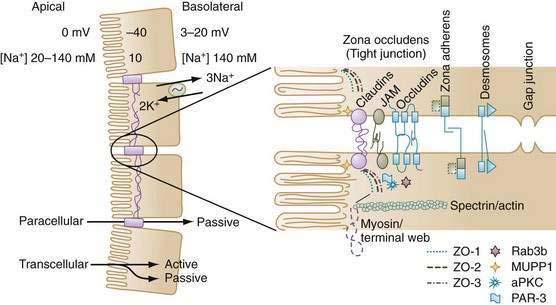
The most prominent feature of epithelial cell polarity is targeting of the Na+,K+-ATPase pump to the basolateral membrane, for which expression of the beta subunit of Na+,K+-ATPase is critical. The Na+ pump is electrogenic, extruding three Na+ ions in exchange for two K+ ions, and thereby maintaining relatively low intracellular Na+ and high intracellular K+ concentrations compared with concentrations of these electrolytes in the extracellular environment (see Fig. 99-2). There also is greater membrane permeability for K+ over Na+, which favors diffusional exit of K+ from the cell over diffusional cellular entry of Na+. These features, in combination with the large number of intracellular proteins with fixed negative charges, lead to the characteristic negative intracellular potential difference compared with either the mucosal or serosal compartments.* Low [Na+] and electronegativity establish a favorable electrochemical gradient for passive Na+ entry into the cell. Functionally, the epithelial cell uses the energy of the favorable Na+ gradient to transport not only Na+ ions but also a variety of nutrients, vitamins, and electrolytes.
These properties provide the basic mechanisms of ion and water transport that apply to all epithelia. In the intestine, differences in transport can be seen along its cephalocaudal length as well as along the surface-crypt axis within a particular segment of intestine. Tissue- and segment-specific nuances arise from structural-functional and regulatory differences of both intracellular and intercellular proteins.4
SEGMENTAL HETEROGENEITY OF TRANSPORT
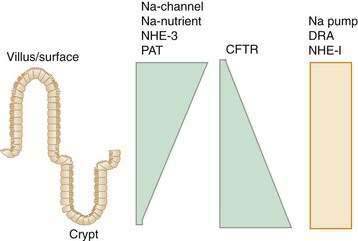
All intestinal segments from the duodenum to the distal colon have mechanisms for absorbing and secreting water and electrolytes. The diverse physiologic functions along the length of the GI tract are supported by the varied array of transporters encountered in its different segments. For example, the glucose- and amino acid-coupled transporters in the jejunum are well suited for absorption of large volumes of nutrients and water. The cecum, proximal colon, and distal colon, however, exhibit distinctly different transporters, with electrogenic Na+ absorption in the distal colon accomplishing the necessary final fluid extraction in preparation of feces.5–8 Different transporter molecules have been identified in specific segments of the GI tract. What is not clear, however, is why an individual transporter is located only in a specific segment of the intestine. For example, the DRA (down-regulated in adenoma) protein is an anion exchanger, and although anion exchange function is recognized in different segments of the intestine, DRA is predominantly expressed in the colon9–10 (see the later discussion of bicarbonate transport).
There also is segmental heterogeneity along the crypt-villus axis. Stem cells near the base of the crypt differentiate and migrate upward to form villus enterocytes in the small intestine or surface colonocytes in the large intestine while undergoing important changes in their transport and barrier properties (Fig. 99-3).11,12 As epithelial cells migrate away from the proliferative zone, the complexity of their tight junctions increases, the microvillus architecture of their apical membrane becomes more pronounced, and underlying cytoskeleton and signaling molecules undergo change; there also is increased expression of Na+ nutrient-coupled transporters, apical Na+-H+ exchangers, and brush border membrane hydrolases. In contrast, the levels of the Na+ pumps remain relatively constant and others, such as the signaling molecule adenylate cyclase and the cyclic adenosine monophosphate (cAMP)-associated Cl− channel CFTR (cystic fibrosis transmembrane conductance regulator), decrease in more mature villus cells.
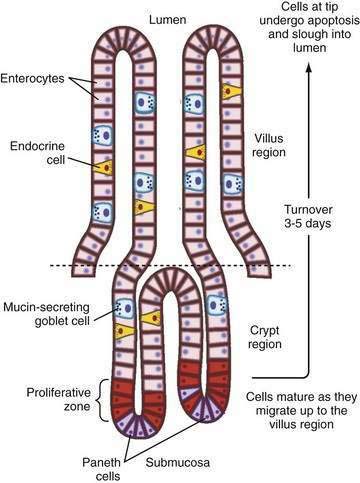
This spatial distribution of transporters (Fig. 99-4) is consistent with a model in which secretory function resides primarily in the crypts and absorption occurs in villus or surface cells. This dichotomy between absorptive surface cells and secretory crypt cells, however, is far from absolute; for example, in the colon, crypts absorb Na+ and fluid, and surface cells secrete Cl−.13,14 Thus, depending on their relative position along the crypt-villus axis, the crosstalk between transporters and their signaling molecules can vary and fine tune intestinal function. This segregation of absorptive and secretory functions might explain why, in diseases that selectively damage villi or surface epithelia—such as enteric infection, inflammatory bowel disease (IBD), and celiac disease—secretion predominates.
TIGHT AND LEAKY EPITHELIA
The paracellular space and junctional complexes between cells define the barrier function of epithelia. Epithelia with a low transepithelial voltage and low resistance are considered leaky, and those that exhibit a high transepithelial voltage and high resistance are considered tight. The tight junctions in villi have higher resistance than do those in crypts. Transepithelial resistance increases in a cephalocaudal direction (see Fig. 99-1).13
Since the 1990s, the model of paracellular transport and tight junctions has rapidly evolved from a static rigid barrier to a dynamic complex structure that is finely regulated (see Fig. 99-2). Movement through the space is exclusively passive, but it is influenced by electrical conductivity, charge selectivity, and its ability to be regulated. Cell-to-cell communications along the paracellular pathway occur in several discrete structures: zona occludens (ZO; tight junction), zona adherens (ZA), desmosomes, and gap junctions. The ZO is composed of several families of proteins that determine its physical and biological properties. For example, claudins belong to a family of 24 membrane-spanning proteins (24-27 kd) that form pores by interactions of the extracellular domains of claudins of adjoining cells; homotypic adhesion claudins are important in determining the charge selectivity of the tight junction.15,16
Additional proteins in the tight junction include occludins, junctional adhesion molecules (JAMs), and scaffolding proteins such as the zona occludens proteins (ZO-1, ZO-2) and multi-PDZ domain protein 1 (MUPP-1). The scaffolding proteins serve to link membrane proteins to an array of protein kinases, phosphatases and, via filamentous actin, to myosin in the terminal web, thereby influencing paracellular permeability.17,18 For example, disruption of tight junctions by enteropathogenic Escherichia coli is specifically associated with protein kinase Cζ activation.19 Another junctional complex that allows cell-to-cell interaction is the zona adherens. In epithelia, the zona adherens primarily is made up of E-cadherins, 120-kd transmembrane glycoproteins, with extracellular motifs that engage in calcium-dependent homotypic interaction with cadherins of adjoining cells. Intracellularly, cadherins bind to a family of adhesion molecules, the catenins, which in turn anchor to a dense actin-filament network. Alterations in cadherin-catenin distribution or function have been implicated in carcinogenesis.20
Desmosomes are junctional complexes that are structurally similar to zona adherens junctions, although instead of actin, they link to intermediate filaments through a dense plaque of intracellular anchor proteins. Gap junctions have a unique function: They bridge gaps between cells, thus allowing neighboring cells to exchange small molecules. They are made up of an assembly of connexins, a four-pass membrane-spanning protein, six of which join to form a hemichannel. When these hemichannels in two adjoining cells are aligned, they form a continuous pore that connects the interior of the two cells.1
TRANSEPITHELIAL TRANSPORT
Our current understanding of the movement of ions, solutes, and fluid across epithelia is gleaned from a combination of in vitro studies using reductionist models of cell lines or isolated epithelial sheets, and from complex in vivo methodologies such as the triple-lumen perfusion technique. All these models underscore that transepithelial ion (largely Na+) movement from the mucosa to the serosa drives fluid absorption, whereas net ion (largely Cl−) movement in the reverse direction drives fluid secretion. Although different approaches help elucidate a complex mechanism, at times they give confounding results. For example, some in vitro studies report decreased Cl− secretion and increased Na+ absorption in the jejunum of cystic fibrosis patients, implying that the intestinal manifestations of the disease are due to hyperabsorption of water. In contrast, in vivo studies show decreases in both Cl− secretion and passive Cl− absorption, suggesting that rather than a hyperabsorption of fluid, the severity of the disease is reflected by decreased fluid absorption.21
WATER MOVEMENT
Although water movement is a major property of the intestine, the mechanism(s) of intestinal water transport have not been clearly delineated. The movement of water is inextricably linked to the movement of solutes, in response to osmotic gradients. The standing-gradient hypothesis of water absorption suggests that even a small increase (2-3 mOsm) in the osmolarity of the intercellular and subepithelial spaces can cause movement of water across the epithelium, both through and around the cells.22 In the early 1950s, “water pores” were postulated to explain transepithelial water movement, but it was not until the remarkable discovery of the aquaporin (AQP) family of water transporters that a role of specific membrane proteins was implicated in erythrocyte and renal water transport.23,24 Although AQP2, AQP3, and AQP7 have been localized to the small intestine and AQP1, AQP3, AQP4, and AQP8 have been localized to the large intestine, the specific intestinal apical water channel, if any, has eluded discovery.25 Wright and associates proposed that the apical Na+-glucose transporter (SGLT) also may be able to transport water, perhaps as much as 5 L/day,26,27 but whether this can compensate for the puzzling lack of functional apical AQPs remains to be determined.
CHANNELS, CARRIERS, AND PUMPS
Small hydrophobic and uncharged molecules move across the lipid bilayer of the cell by diffusion, the rate of transport determined by the concentration gradients and diffusion coefficients (Fig. 99-5). Oxygen, carbon dioxide, fat-soluble vitamins, and unconjugated bile acids are examples of substances transported by diffusion. Because the majority of ions and solutes cannot cross the phospholipid membrane by diffusion, the cell employs an array of distinct integral membrane proteins, including channels, carriers, and pumps to cross cell membranes (Fig. 99-5).1
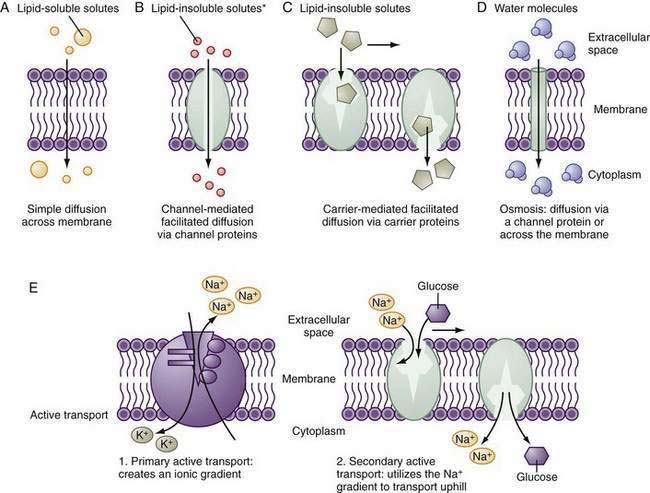
Channels are pores that allow the swift (>106 ions/sec) and controlled (by rapid opening and closing) transit of ions across the membrane, driven by the electrochemical gradient. The advent of molecular cloning techniques, patch clamp methodology (which allows the measurement of function of single channels), and membrane protein crystallography has greatly advanced our knowledge of how these proteins function. Channels tend to be ion selective. For example, Na+ channels exclude K+ despite its same charge and smaller size. Selectivity is determined by the hydration radius of the ion and the physiochemical nature of the pore. The overall transport of a particular ion is determined by the electrochemical gradient, the density of channels, and the gating (open-close time) of the channel; gating may be modulated by voltage, ion, concentration, or intracellular regulation. Mutations of critical residues in the channel protein can have dire functional consequences; for example, in cystic fibrosis, specific mutations of the CFTR affect the ability to transport chloride and bicarbonate.28,29
ION TRANSPORTERS
APICAL SODIUM CHANNEL
In the GI tract, the surface epithelial cells of the distal colon and rectum exhibit electrogenic Na+ absorption against a fairly steep concentration gradient. The downhill electrochemical gradient created by the Na+ pump drives Na+ entry via an apical membrane Na+-specific ion channel (Fig. 99-6), which belongs to the family of epithelial Na+ channels (ENaCs). Members of the ENaC family are found in many epithelia.30–32 They are multimeric proteins composed of α, β, and γ subunits; they exhibit a high sensitivity to the diuretic amiloride; and they are stimulated by mineralocorticoids and cAMP. Colonic ENaCs are inhibited by increases in intracellular Ca2+. Unlike many other channels that regulate transport by gating, ENaCs modulate transport by varying the channel density in the cell membrane; this variation may be accomplished through changes (increases or decreases) in synthesis (e.g., aldosterone) or exocytosis (e.g., cAMP, vasopressin) of the channels. Additionally, both aldosterone and cAMP block the association of ENaC with Nedd4-2, a ubiquitin protein ligase, which normally flags the protein for degradation; this block of degradation increases ENaC. Mutations in this pathway can result in the increased Na+ absorption and hypertension characteristic of Liddle’s syndrome, an autosomal dominant disorder with features of hyperaldosteronism that is a cause of infantile hypertension.
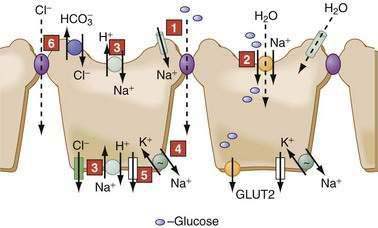
NUTRIENT-COUPLED SODIUM TRANSPORT
Glucose transport processes, elegantly elucidated by Wright and coworkers, provide a good example of nutrient transport.33 Transport across SGLT1 is electrogenic (2 Na+ to 1 glucose), is stereospecific (d-isomer), and transports galactose but not fructose.33 Glucose exit across the basolateral membrane occurs via a separate family of facilitated diffusion carriers, the glucose transporters (GLUT-2) (see Fig. 99-6). Fructose enters the cell via another member of this family, GLUT-5, and exits via GLUT-2.
Although it is clear that Na+ and glucose absorption stimulates water absorption, the mechanism is not fully delineated. The classic explanation is that basolateral exit of glucose creates a hypertonic compartment in the paracellular space, thereby generating an osmotic gradient for the entry of fluid from the lumen. Some enticing evidence suggests that secondary to transcellular transport through SGLT, passive processes triggers the contraction of the actomyosin ring in the terminal web, resulting in increased paracellular permeability to glucose and to water. Additionally, SGLT activation results in a protein kinase-dependent recruitment of GLUT2 to the apical membrane, which then serves as a high-capacity, low-affinity route for sugar entry during feeding.34,35 Evidence suggests that SGLT can serve as a water channel (210 water to 2 Na+ to 1 glucose) and might account for up to 5 L of fluid reabsorption in the fed state.36 How molecular regulation of transporters is translated into net nutrient absorption during feeding in vivo is a critical area of inquiry. For a description of similar advances made in our understanding of amino acid and vitamin transport, see Chapters 100 and 101.
SODIUM-HYDROGEN EXCHANGERS
Ten mammalian isoforms of NHE have been cloned. NHE1-4 and 6-9 exhibit species- and segment-specific distribution in the GI tract.37–39 NHE1 is a plasma membrane protein found in epithelial and nonepithelial cells. It is expressed on epithelial basolateral membranes and functions as the housekeeper regulator of intracellular pH, cell volume, and growth. NHE2 and NHE3 are apical membrane proteins restricted to epithelia and are the major conduits for electroneutral Na+ absorption in the intestine (see Fig. 99-6). NHE2 is expressed throughout the GI tract, but maximal expression is in the proximal colon. NHE3 is considered a marker for the absorptive cells of the small intestine and colon; it is expressed only in the villus or surface cells, and not in the crypts. NHE4 is located in the basolateral membrane and is primarily expressed in gastric parietal and chief cells, where it might have a role in acid secretion. NHE5 and NHE10 are not expressed in the GI tract, and the roles of NHE6-9 remain to be determined.
NHE activity is differentially modulated by neural, paracrine, or endocrine stimuli through intricate scaffolding complexes that include the exchanger itself, a family of NHE regulatory factors (NHERFs) that act as a bridge between the exchanger, and a variety of kinases and phosphatases.40,41 Different stimuli use differing scaffolding complexes to exert their effect. For example, glucocorticoids stimulate Na+ absorption and up-regulate NHE3 but not NHE1, 2, or 4, consistent with their respective roles in vectorial transport and housekeeping. Glucocorticoids act via a serum and glucocorticoid inducible kinase, SGK1; SGK1 stimulates the activity of NHE3 by interacting directly with NHERF2.
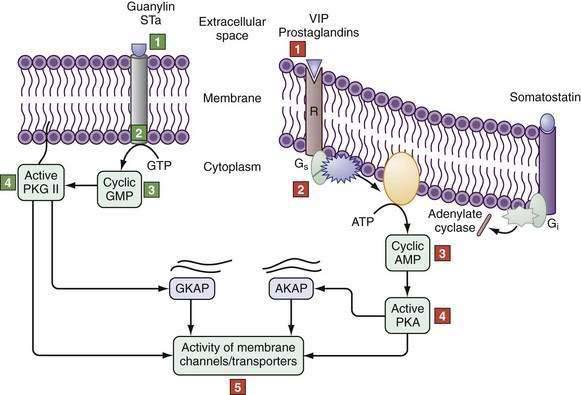
Alternatively, cAMP inhibits NHE3 by activating protein kinase A, which is recruited to the C-terminus of NHE3 by NHERF1, NHERF2, and an additional cytoskeletal protein, ezrin. In this location, PKA induces its inhibitory effect by phosphorylating NHE3. Cyclic guanosine monophosphate (cGMP) can inhibit NHE3 by triggering the formation of a complex between cGMP-dependent protein kinase II (cGKII) and NHERF2, which anchors cGMP kinase protein (GKAP). Activation of guanylate cyclase C by guanylin or E. coli–stable toxin A increases cGMP content near the brush border to locally activate cGKII (Fig. 99-7), which then inhibits NHE3 activity.40
CHLORIDE (ANION) ABSORPTION
The transepithelial, lumen negative potential difference contributes to the passive movement of Cl− and other anions via the paracellular pathway in the jejunum.7 While coupled Cl−-HCO3− and Na-H exchangers govern electroneutral transport in the ileum and proximal colon, sodium-independent Cl−-HCO3− exchange occurs in the distal colon (see Fig. 99-6) (see the discussion of HCO3− transport, later).
CHLORIDE SECRETION
The principal driving force for the secretion of fluid is the transcellular movement of Cl− from the serosal to the luminal compartment. Na+ and water follow passively in response to the ensuing electrical and osmotic gradients (Fig. 99-8). The small and large intestine exhibit a basal rate of Cl− secretion that is maintained by the interplay of cell volume, [Cl]i, and paracrine, autocrine, neuronal, endocrine, luminal, and immune modulators. Disruptions in the balance of these regulatory processes can lead to secretory diarrhea.
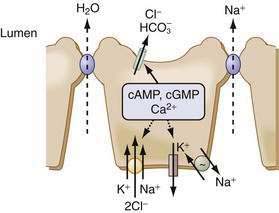
Several epithelia in the GI tract exhibit electrogenic Cl− secretion. Although there are some tissue-specific regulatory differences, the mechanisms underlying this secretion are remarkably similar. The Na+ pump provides the driving force, Cl− enters the cell across the basolateral membrane via an electroneutral cotransporter (NKCC1) that couples the movement of 1Na+:1K+:2Cl−, and Cl− leaves the cell via specific channels on the apical membrane. The Na+ entering the cell via NKCC1 exits via the Na+ pump, and the K+ leaves via K+ channels either on the apical or the basolateral membrane. This complex interplay of transporters is an elegant demonstration of cellular economy. The NKCC1 cotransporter effectively moves 2 Cl− and 1 K+ uphill for the expenditure of a single Na+ ion. The pump-to-leak relation between K+ channels and the Na+ pump helps to maintain the interior of the cell as electronegative, thereby providing the driving force for Cl− exit. Basolateral K+ exit electrically balances the large Cl− flux across the apical membrane. NKCC cotransporters belong to a superfamily of cation transporters and are characterized by their inhibition by the loop diuretics bumetanide and furosemide.42–44 NKCC1 is regulated by many kinases, including a unique PASK (proline-alanine–rich STE20-related kinase), phosphatases, actin-myosin interactions, cell volume, and intracellular Cl−.
CFTR has been localized to the apical membrane of various segments of the small and large intestine, with greater expression in the crypts than the villus or surface cells.45 CFTR is a 250-kd membrane protein belonging to the superfamily of ATP binding cassette proteins. CFTR is a small-conductance (8-10 pS) linear channel, with an ion selectivity of Br− > Cl− > I− > F− that also can transport HCO3−.46,47 It has two membrane-spanning domains, two ATP-binding domains, and a regulatory (R) domain that has many consensus sequences for phosphorylation by kinases, specifically for PKA. Gating of the channel is regulated by sequential binding of ATP to the two domains and phosphorylation and dephosphorylation of the R domain. In secretory diarrhea such as cholera, PKA activates the R domain to increase channel activity while simultaneously stimulating the recruitment of CFTR-bearing endosomes to the apical membrane, increasing the number of channels (Fig. 99-9). Although more than 1000 mutations of CFTR have been identified, approximately 70% of cystic fibrosis patients carry the ΔF508 mutation, a single amino acid deletion that results in improper folding and diversion of the protein to the endoplasmic reticulum-associated degradative pathway rather than to the apical membrane. The protein is pleiotropic and interacts with a variety of other proteins, influencing their expression and regulation, and being modulated, in turn, by mechanisms that are not fully deciphered. Some of this interaction can occur through crosstalk of CFTR with scaffolding proteins it shares with other transporters; for example, the C-terminal of CFTR interacts with NHERF, a protein that also binds to NHE3.
Diminished intestinal fluid secretion in CF−/− mice is associated with goblet cell hyperplasia, increased crypt cell proliferation, Paneth cell abnormalities, and increased inflammation.48 Whether all these changes are secondary to decreased fluid secretion or result from more direct effects of CFTR on other protein function remains to be determined (see later). By combinatorial chemistry, a series of compounds have been designed to specifically inhibit or stimulate CFTR, thereby serving as therapeutic strategies for secretory diarrhea or cystic fibrosis.49
CHLORIDE CHANNELS
CIC Family Channels
Chloride channels of the distinct ClC family,50,51 in particular the widely distributed plasma membrane protein ClC2, are of clinical interest as a potential salvage pathway for Cl− transport in cystic fibrosis patients and as the specific target of lubiprostone, a novel laxative.52 ClC2 is involved in regulation of epithelial transport, intracellular pH, intracellular chloride and cell volume. Because of significant differences in its localization in a diverse set of human and animal models, however, its exact role in chloride secretion is uncertain. Thus, although lubiprostone activates ClC2 in the apical membrane of the human colon carcinoma cell line T84, it cannot rescue Cl− secretion in the CFTR-deficient mouse. This dichotomy suggests that the channel is located on the basolateral membrane of the CF mouse. Further research will be needed to clarify how these channels and their pharmacologic agonist work in regulating chloride secretion in humans.
CALCIUM-ACTIVATED CHLORIDE CHANNELS
A third class of Cl− channels, the calcium-activated Cl− (CLCA) channels, are involved in the diarrhea of rotavirus infection. The rotavirus toxin NSP4 activates calcium-dependent Cl− secretion, especially in the colons of young mammals. Despite an understanding of the regulation of calcium-dependent Cl− secretion in many epithelia, however, the molecular nature of these channels has eluded precise identification; recent evidence points to a group of proteins, the bestrophins, as potential candidates. It remains to be determined if bestrophins are regulated channels per se or regulators of channels or whether they use auxiliary proteins to function as channels.53,54 The CLCA proteins also might play a role in goblet cell function: Mouse CLCA3 is severely reduced in the CF−/− mouse, and its restoration ameliorates the severity of intestinal cystic fibrosis disease.55
POTASSIUM TRANSPORT
A plethora of K+ transport processes help the intestine cope with its need to balance fluid and electrolyte movement.56,57 Potassium secretion and absorption occur along the length of the intestinal tract, although the specific pathways are segment specific. In the small intestine, the luminal negative potential difference drives the passive absorption of K+. In contrast, K+ absorption in the distal colon occurs by primary active transport via H+,K+-ATPase pumps located on the luminal membrane. These H+,K+-ATPase pumps are P-type ATPases, related to the gastric H+,K+-ATPase, and least two colonic isoforms have been identified: a ouabain-sensitive isoform in the crypt cells and a ouabain-insensitive isoform in the surface cells. Depletion of aldosterone and K+ upregulates the ouabain-insensitive H+,K+-ATPase and stimulates K+ absorption.
BICARBONATE TRANSPORT
Bicarbonate is a metabolic product and a critical anion in fluid homeostasis in the intestine. In clinically significant diarrhea, bicarbonate is the major anion in the stool. It is secreted by electrogenic and electroneutral processes in the duodenum, ileum, and colon. Being a metabolic product, intracellular HCO3− can arise from intracellular metabolism, diffusion of CO2 or the action of transporters such as the basolateral Na+-HCO3− cotransporter. Electrogenic HCO3− secretion can occur via apical anion channels, including CFTR; however, the major mechanism for HCO3− secretion in the small and large intestine is inexorably linked to the inward movement of Cl− and occurs through apical Cl−-HCO3− exchangers.58 It is postulated that electrogenic Cl− secretion via CFTR provides luminal Cl−, which is then recycled across the apical membrane in exchange for intracellular HCO3−. SCFA-dependent bicarbonate secretion also has been observed in surface cells of the colon.59
Although the prototype of anion exchangers, the red cell Cl−-HCO3− exchanger (AE1), has been extensively studied, identification of intestinal exchangers is relatively recent. In a reorganization of nomenclature, the more than 360 identified solute carriers (SLC) are now classified in one of 46 families; of these, the SLC4 bicarbonate transporter family encompasses the red cell anion exchanger (AE1) and its epithelial isoforms (e.g., AE2, AE3, and AE4).60–62 The precise role of the AEs in overall intestinal Cl−-HCO3− transport remains to be clarified, and apical and basolateral localizations have been suggested.
Structurally distinct from the SLC4 bicarbonate transporter family is the SLC26 multifunctional anion exchange family. SLC26 exchangers can transport Cl−, HCO3−, sulfate, formate, oxalate, hydroxyl ions, and other anions with differing affinities. Their varied distribution along the GI tract provides them with the flexibility to handle a variety of luminal anions. Two members of this family, SLC26A3 and SLC26A6, are of special interest.10,63–65 The former, first identified as DRA (down-regulated adenoma), is expressed abundantly on the apical membranes of colonocytes, but not enterocytes, and transports more than 2Cl−:1HCO3− ion. Mutations in DRA cause congenital chloride diarrhea, which manifests with severe diarrhea, volume depletion, and metabolic alkalosis. In contrast, SLC26A6, also known as the putative anion transporter 1 (PAT1), is expressed abundantly in the apical membrane of villus enterocytes and less so in the colon. PAT1 transports more than 2HCO3−:Cl− ion; a separate HCO3− conductive pathway might mediate bicarbonate secretion into the duodenum.
SHORT-CHAIN FATTY ACID TRANSPORT
SCFAs are rapidly absorbed in the colon and also greatly enhance Na+ and fluid reabsorption through linked transport mechanisms and by upregulating the expression of NHE3 on the apical membrane of colonocytes.66 SCFAs are weak electrolytes and can be ionized or protonated. Ionized SCFAs need specific carriers, whereas nonionized protonated species can diffuse across the colonocyte membrane; at luminal colonic pH, SCFAs are 95% to 99% ionized.
A picture of the molecular basis of SCFA transport is beginning to emerge.32,67 First, apical NHEs can create an acidic pH microclimate and enhance the diffusion of protonated SCFAs into the cell. Monocarboxylate transporters (MCTs), members of two different SLC families, are involved in electroneutral carrier-mediated SCFA transport. Members of the SLC16 family, specifically SLC16A1 (MCT1), transport 1H:1SCFA and require an ancillary protein for their function.68 High-affinity (SLC5A8) and low-affinity (SLC5A12) Na+-dependent MCTs have been identified in the colon and intestine. Although their molecular identity is unclear, Cl−-butyrate exchangers and SCFA-HCO3− exchangers, functionally coupled to Na+-H+ exchange, might account for SCFA promotion of electroneutral Na+ and Cl− absorption.59,69
PARACRINE IMMUNONEUROENDOCRINE SYSTEM
Extracellular factors from classic paracrine, immunologic, neural, and endocrine systems (PINES) as well as autocrine factors and luminal content regulate intestinal ion transport. The borders separating members of PINES are arbitrary at best, because there is considerable overlap and crosstalk of the underlying factors, and patterns can be altered in disease states (Fig. 99-10). Luminal mechanical (stroking and stretch) or chemical (toxins) stimuli can activate mechanoreceptors and chemoreceptors, respectively, to activate one or more arms of PINES. All these interactions are compounded by many of the factors acting through cell-specific multiple receptors and signaling pathways.
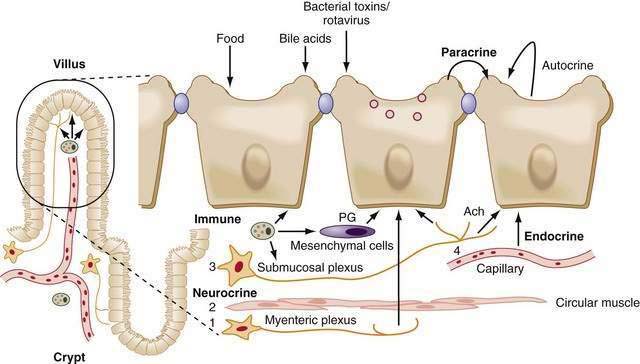
Within the subepithelium, structural elements of PINES, including blood vessels, are in close proximity (see Fig. 99-10), so release of mast cell mediators can easily target neurons and vice versa; this proximity and interplay contributes to the minute-by-minute local regulation necessary in the intestine. Although it is possible to separate the specific effects of an individual component in vitro, clinically they are inextricably intertwined. For example, Verner-Morrison syndrome (pancreatic cholera) is classified as an endocrine-mediated diarrhea, because pancreatic islet cell tumors produce large amounts of vasoactive intestinal peptide (VIP). In the healthy adult, however, VIP is not found in the pancreas but is a peptidergic neurotransmitter in the enteric nervous system that stimulates epithelial cell secretion and smooth muscle relaxation. Alternatively, a single agonist such as cholera toxin directly acts on epithelial cells while simultaneously stimulating neural, paracrine, and immune responses. In a third example, serotonin (5-hydroxytryptamine [5-HT]) released by mucosal enterochromaffin cells acts via distinct receptors on epithelial cells to directly stimulate secretion; it acts on myenteric neurons to release acetylcholine (Ach) and elicit migratory contractions; or it acts on submucosal neurons to release Ach and calcitonin gene-related peptide to stimulate peristalsis and secretory reflexes.
Fluid secretion is the major component in the production of diarrhea, and the organism’s defensive response to intestinal challenge; motility, mucus secretion, and blood flow, all regulated by PINES, are important adjuncts to the process. The involvement of PINES in motility helps to explain diarrhea associated with rapid intestinal transit (e.g., following gastrectomy), altered anorectal motility (e.g., small-volume diarrhea), or decreased motility (e.g., bacterial overgrowth). Decreased motility leads to a increase in the bacteria in the small intestine, which causes diarrhea by a variety of mechanisms (see Chapters 97 and 102). Alternatively, inflammatory mediators such as prostaglandins or bacterial enterotoxins target both the epithelial and muscle layers to elicit a coordinated secretory response, while promoters of absorption such as opiates and enkephalins suppress motility and promote electrolyte absorption. Thus, PINES allow for a coordinated and integrated response to multiple extracellular signals.
EXTRACELLULAR REGULATION
Tables 99-1, 99-2 and 99-3 list the major neurohumoral substances and toxins that modulate intestinal fluid transport. Agents that promote net fluid secretion generally inhibit Na+ absorption and stimulate Cl− secretion, whereas agents that promote net fluid absorption increase Na+ uptake and attenuate Cl− secretion. In a healthy person, net absorption prevails, and when this balance is disrupted, diarrhea can ensue. It is unclear if there is a corollary for a predominant absorptive pattern in a subset of patients with constipation.
Table 99-1 Agents That Stimulate Intestinal Absorption of Fluid and Electrolytes
| Endogenous Absorbagogues |
| α-Adrenergic agonists |
| Aldosterone |
| Angiotensin |
| Enkephalins |
| Glucocorticoids |
| Growth hormone |
| Neuropeptide Y |
| Peptide YY |
| Prolactin |
| Short-chain fatty acids |
| Somatostatin |
| Pharmacologic Agents |
| Berberine |
| Clonidine (α2-agonist) |
| Cyclooxygenase inhibitors |
| Glucocorticoids |
| Lithium |
| Mineralocorticoids |
| Octreotide |
| Opiates |
| Propranolol |
Table 99-2 Endogenous Agonists of Intestinal Secretion
| AGONIST | INTRACELLULAR MEDIATOR | SOURCE |
|---|---|---|
| Acetylcholine | Ca2+ | ENS |
| Adenosine | cAMP | Immune cells |
| Arachidonic acid | cAMP | Immune cells, cell membranes |
| Atrial natriuretic peptide | cGMP | Heart |
| Bombesin | Ca2+ | ?? |
| Bradykinin | cAMP | Immune cells |
| Calcitonin, calcitonin gene-related peptide | ? | ENS |
| Galanin | Ca2+ | |
| Gastric inhibitory polypeptide | ? | ?? |
| Gastrin | Ca2+(PKC/MaPK?) | Endocrine cells |
| Guanylin | cGMP | Goblet, epithelial cells |
| Histamine | Ca2+ | Immune cells |
| Leukotrienes | ? | Immune cells |
| Motilin | Ca2+ | Endocrine M cells |
| Neurotensin | Ca2+ | ENS |
| Nitric oxide | cGMP | Immune cells, mesenchymal cells |
| Peptide histidine isoleucine | cAMP | ENS |
| Platelet activating factor | cAMP | Immune cells |
| Prostaglandins | cAMP | Immune cells, mesenchymal cells |
| Reactive oxygen metabolites | cAMP | Immune cells |
| Secretin | cAMP | Endocrine cells |
| Serotonin | Ca2+ | ENS |
| Substance P | Ca2+ | ENS |
| Vasoactive intestinal polypeptide | cAMP | ENS |
cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; ENS, enteric nervous system.
Table 99-3 Luminal Agents That Stimulate Intestinal Secretion
| AGENT | INTRACELLULAR MEDIATOR |
|---|---|
| Bacterial Enterotoxins | |
| Aeromonas sp. | cAMP |
| Campylobacter jejuni | cAMP |
| Clostridium difficile (toxin A) | Ca2+ |
| Clostridium perfringens | ?? |
| Escherichia coli (heat labile toxin) | cAMP |
| E. coli (heat stable toxin) | cGMP |
| Rotavirus NSP4 | Ca2+ |
| Salmonella sp. | cAMP |
| Vibrio cholerae | |
| accessory cholera enterotoxin | ?? |
| enterotoxin | cAMP |
| zona occludens toxin | ?? |
| Vibrio parahaemolyticus | Ca2+ |
| Yersinia enterocolitica | cGMP |
| Miscellaneous Agents | |
| Bile salts | cAMP/Ca2+ |
| Laxatives | ?? |
| Long-chain fatty acids | cAMP/Ca2+ |
cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; NSP, nonstructural protein.
ENDOCRINE, PARACRINE, JUXTACRINE, AND AUTOCRINE REGULATION
Intestinal transport is modulated by classic endocrine, paracrine, juxtacrine, and autocrine processes. Intestinal endocrine cells are interspersed between epithelial cells and function as sensors that rapidly respond to changes in the luminal environment by releasing secretory granules containing biogenic amines and hormones; these mediators cross the basolateral membrane. These hormones can act either in a classic endocrine manner on distant target cells (via the circulation) or in a local (paracrine) manner by affecting neighboring cells in the intestinal wall. Juxtacrine mediators are those released from nonendocrine cells, such as neural and inflammatory cells, and they affect neighboring cells. Intestinal mesenchymal cells, in particular myofibroblasts, are a rich source of cytokines, chemokines, eicosanoids, and growth factors that can alter intestinal transport. Epithelial cells can self-regulate (autocrine) by secreting factors such as eicosanoids, which act on epithelial cell receptors to alter function.70
NEURAL
Neural input is critical in the regulation of fluid and electrolyte transport (see Fig. 99-10) and involves interactions of the parasympathetic and sympathetic divisions of the autonomic nervous system with the labyrinthine enteric nervous system (ENS) (see Chapters 1, 97, 98 and 120). Cholinergic stimulation of secretion, predominantly through parasympathetic vagal input, and adrenergic stimulation of absorption through prevertebral and sympathetic ganglia have long been recognized as fundamental neural pathways affecting the intestinal epithelium. The ENS is the end controller of neural activity in the intestinal wall, however, independently integrating the regulation of the epithelia, muscles, and blood vessels, with input and modification from the central nervous system.
Like other neural networks, the ENS has reflexes that can have important clinical implications. Target cells for neurons include components of PINES, blood vessels, and, of course, epithelial cells. Sensory input into the ENS comes from changes in the luminal content (e.g., acidity, dietary content, pathogens) or volume (e.g., stretch). Thus, acid or distension can activate TRVP1 vanilloid receptor on capsaicin-sensitive afferent nerves, which in turn evokes secretion by stimulating submucosal neurons and causes vasodilation by directly activating submucosal arterioles.71,72 Endocrine, dendritic, and/or paracrine cells releasing serotonin, adenosine (see later), and other signals are implicated as auxiliary sensors. Primarily cholinergic, interneurons are believed to underlie the ENS-mediated regulation of colonic epithelial responses to distant small intestinal challenges. Motor neurons that innervate epithelial and submucosal cells can be cholinergic or VIPergic, each releasing additional neuroactive substances.
A basal cholinergic secretory drive is tempered by the sympathetic tone; loss of adrenergic sympathetic innervation in diabetic neuropathy is associated with the development of “diabetic diarrhea” and may be corrected by α2-adrenergic agonists.73 As in the brain, the number and variety of compounds that act as neuroactive agents has ballooned, and this has added to the complexity of our elucidating the underlying mechanisms. Thus, neurons can release specific combinations of mediators, such as VIP, cholecystokinin, gastrin-releasing peptide, and ATP, rather than a single substance. Individual neurotransmitters can have biphasic effects varying with concentrations. Furthermore, agents can act as classic neurotransmitters; alternatively, they can act as neuromodulators, fine-tuning the neuronal circuits of presynaptic sites of neurons, or, as in the case of serotonin, they can function as paracrine substances.
IMMUNOLOGIC
The clinical correlation between intestinal inflammation and diarrhea is obvious (see Chapter 2), with ulceration, exudation of protein, changes in motility, and loss of absorptive surface area implicated as causing the fluid losses of (inflammatory) diarrheas. Immunocompetent cells of the intestine reside chiefly in the lamina propria, form the gut-associated lymphoid tissue (GALT), and often are distinct from their systemic counterparts; in noninflamed intestine, T lymphocytes account for 60%, with smaller numbers of B lymphocytes and plasma cells (25% to 30%), macrophages (8% to 10%), mast cells, and polymorphonuclear cells (2% to 5%) (usually eosinophils).74 These cells secrete a vast array of soluble products (chemokines, cytokines, eicosanoids, nucleotides, biogenic amines) whose actions are intimately intertwined with other aspects of PINES. Intestinal inflammation increases the number of immunocytes, the cause of the inflammation determining the type of inflammatory cells; for example, acute bacterial infections increase polymorphonuclear leukocytes, whereas parasitic infections dramatically enlarge the mast cell population and celiac disease is characterized by intraepithelial lymphocytes. In IBD, there is activation of all components of GALT with an increase in immunoglobulin (Ig)G-secreting cells.75,76 Thus, the cause of the inflammatory reaction can determine the type of immunocytes recruited, the range of cytokines released, and the specific effects on transport and motility.
Many inflammatory mediators are potent secretagogues, including peptides (e.g., cytokines, platelet-activating factor, substance P, interferon gamma, kallikreins, and bradykinin), eicosanoids (e.g., arachidonic acid, leukotrienes, and prostaglandins), and oxidants (e.g., superoxides). These mediators either interact directly with epithelia to alter ion transport and barrier function or elicit these effects indirectly by activating other PINES elements (see Fig. 99-10). Prostaglandins are central to the secretory response associated with inflammation, affecting various PINES components, such as enteric neurons, and with mediators such as bradykinin, which liberates arachidonic acid to stimulate prostaglandin production. In interpreting the effects of the inflammatory mediators in normal model systems, it is important to recognize that in vivo, cells damaged by the inflammatory process might not be able to function normally. With this caveat, a few examples are provided.
The mechanisms of secretion resulting from polymorphonuclear infiltration of the mucosa recently have become better understood. Polymorphonuclear leukocytes, responding to chemoattractants (e.g., fMLP [formyl-Met-Leu-Phe]), uniquely present in the inflamed colon, leave the vasculature and interact with epithelial cells.77 The white cells burrow through the intercellular space of the colonic cells in a complex integrin-dependent process. The migrating leukocytes release 5-AMP, which is converted to adenosine by apical membrane enzymes. Adenosine is a potent secretagogue, and this adenosine-stimulated secretion might serve as a mechanism to cleanse the crypt lumen.78 Thus, complex specific immunocyte-epithelial cell interactions are important in alterations of electrolyte secretion associated with mucosal inflammation. Anti-integrin targeted therapy may be effective in treating Crohn’s disease (see Chapter 111).
During inflammation, oxidants such as superoxides, hydrogen peroxide, and hydroxyl radicals released from neutrophils stimulate Cl− secretion; cytokines such as interleukin-1 and interleukin-3 also stimulate secretion.79 In contrast, interferon-γ and TNF-α can cause diarrhea more through an antiabsorptive effect, by down-regulating particular transporters 80–82 or by altering permeability of tight junctions. Similarly, studies on colonic specimens from patients with IBD or ulcerative colitis indicate that diarrhea in these entities results not from stimulated secretion but from increased tissue permeability combined with decreased apical Na+ channel, basolateral K+ channel, and Na+,K+-ATPase expression, which decreases NaCl reabsorptive capacity.76 The multiplicity of transport malfunctions seen in IBD might reflect a sick cell syndrome rather than specific alterations modulated by one or two unique cytokines.
SYSTEMIC EFFECTS
Acid-base balance modulates intestinal electrolyte transport in vivo and in vitro. Changes consistent with metabolic acidosis are potent stimulators of electroneutral NaCl absorption, whereas metabolic alkalosis inhibits this process.83–84 Intracellular bicarbonate concentrations can modulate basal Cl− secretion, and intracellular pH and Pco2 can alter Na+:H+ exchange. Volume status and intestinal blood flow also alter ion transport. Any decrease in intravascular volume, such as with hemorrhage, elicits a series of responses that increase fluid absorption. Cardiopulmonary mechanoreceptors and carotid baroreceptors increase sympathetic input into the ENS, resulting in decreased secretion. Angiotensin II, antidiuretic hormone, and atrial natriuretic peptide also can contribute to regulation of intestinal fluid transport in these conditions.85–86 The metabolic status of the intestine has an impact on its transport capability; a well-fed intestine transports more effectively.87 There also are segmental preferences for metabolic fuels. Although the entire intestinal tract uses glucose, the small bowel effectively uses glutamine, and the colon preferentially uses SCFAs, particularly butyrate.88,89
OSMOTIC EFFECTS
Unlike the kidney, the intestinal epithelium cannot maintain an osmotic gradient. Under normal physiologic conditions, the duodenum and upper jejunum are subject to major fluid shifts as they adjust to dietary intake of hypertonic foods and liquids. Rapid equilibration usually is accomplished by movement of water into the intestinal lumen, and absorptive processes along the remainder of the intestine steadily decrease the luminal volume. The continued presence of a nonabsorbable solute within the intestinal lumen, however, can negate functioning absorptive pathways in the distal intestine. This is the basis for osmotic diarrhea (see Chapter 15).
Carbohydrates, usually disaccharides, are a common source of nonabsorbable solute. Disaccharides must be hydrolyzed to monosaccharides before they can cross the apical membrane of the small intestine, (see Chapters 100 and 101). The most common clinical example of maldigestion resulting from a deficiency of a specific disaccharidase is lactose intolerance, in which the glucose-galactose disaccharide cannot be broken down because of lactase deficiency. Because the human intestine does not naturally possess a lactulase, the disaccharide lactulose reliably increases small intestinal fluid because of luminal hyperosmolarity and bacterial fermentation of the disaccharide. The limited intestinal absorptive capacity for several sugars found in processed foods and drinks, such fructose and sorbitol, can play important, but often overlooked, roles in osmotic diarrhea, bloating, abdominal pain, and irritable bowel syndrome.
The physiology of carbohydrate-induced osmotic diarrhea is complicated by the fact that nonabsorbable solutes in the small bowel can be converted into absorbable solutes by colonic bacteria. Almost all classes of carbohydrates not absorbed by the small intestine are rapidly converted to SCFAs once they cross the ileocecal valve and encounter the colonic bacterial flora; these SCFAs can be absorbed and serve as metabolic fuel for the colon. Thus, depending on the rate of carbohydrate conversion to SCFAs and the colonic capacity for SCFA absorption, small intestinal fluid loss may be compensated by colonic fluid absorption. However, if the capacity of the intestinal flora to convert carbohydrates to SCFAs is maximized, or if the SCFA absorptive capacity of the colon is exceeded, additional unmetabolized carbohydrates could pass through the colon and exacerbate the osmotic effects of nonabsorbable solute.90
In clinical situations in which there is malabsorption or a generalized destruction of the epithelium, solutes normally absorbed readily can remain in the intestinal lumen and thereby contribute an osmotic component to an inflammatory diarrhea or a malabsorptive state. Osmolality is an important factor in patients receiving enteral nutrition (see Chapters 4 and 5). For example, complex carbohydrates provide a significant amount of calories with minimal osmolality compared with simple sugars. Absorption of dipeptides and tripeptides instead of amino acids reduces intestinal osmolality. This balance between calories and osmolality becomes clinically relevant in effectively designing appropriate tube-feeding regimens. Osmolality also is an important factor in designing second-generation oral rehydration therapy (ORT) formulations; by replacing glucose with complex carbohydrates such as rice, intestinal absorption is stimulated further by creating a hypotonic luminal environment, thereby enhancing water absorption. In addition to providing numerous sugar molecules per milliosmole, complex carbohydrates such as rice and amylase-resistant starches have another advantage in ORT: They are metabolized by commensal bacteria in the colon to release SCFAs, which in turn promote fluid absorption (see the earlier discussion of SCFAs).91
SPECIFIC REGULATORY FACTORS
ABSORPTIVE
Intestinal agents that stimulate absorption are listed in Table 99-1.
Mineralocorticoids (e.g., aldosterone) primarily affect electrogenic Na+ absorption in the distal colon and have little effect on the small intestine, which exhibits electroneutral Na+ absorption. Aldosterone increases the activity and numbers of the apical membrane ENaC (see the earlier discussion of Na channels) and stimulates activity of the Na+/K+ pump and SGK1, resulting in an increase in Na+ absorption. Aldosterone increases K+ absorption and K+ secretion.92 Neonates exhibit a correlation between high circulating levels of aldosterone and enhanced colonic Na+ absorption. Clinically, the physiologic role of aldosterone can be seen in the increased colonic absorption after Na+ depletion (aldosterone stimulation) or in the diarrhea associated with Addison’s disease (aldosterone deficiency).
Glucocorticoids are also potent stimulators of Na+ absorption in the small intestine and the colon, in addition to having well-documented anti-inflammatory effects. At low concentrations, glucocorticoids stimulate electroneutral Na+ absorption and suppress electrogenic Na+ absorption, whereas at high concentrations they stimulate both processes. The actions of glucocorticoids are complex and are species and segment specific, and they may be directed at the level of apical Na+ transporters and at the Na+ pump. For example, in the rabbit jejunum, ileum, and colon, glucocorticoid-stimulated increases in Na+ absorption are associated with selective increases in NHE3 but not NHE2 or NHE1 mRNA and protein. Both glucocorticoids and aldosterone evoke rapid cellular responses involving the SGK1 pathway as well as genomic transcriptional effects. These effects might account, in part, for the potent antidiarrheal action of glucocorticoids in a wide variety of clinical settings.38
Catecholamines (e.g., dopamine and epinephrine) acting as α-adrenergic agonists, all have similar absorptive properties. The theoretical basis for the use of clonidine as an antidiarrheal agent, particularly in diabetic diarrhea, is rooted in this adrenergic absorptive pathway.93
The use of plant opiates as antidiarrheal agents dates back two millennia to the early Egyptians and underscores their effectiveness. Elucidating their therapeutic effect led to the characterization of the mammalian opioid peptides—enkephalins, endorphins, and dynorphins—a classic example of molecular mimicry.94 Acting via one of three main opioid receptor subtypes—mu, delta, and kappa—the opiates and opioid peptides decrease secretion and promote nonpropulsive motility patterns, thereby increasing transit time. They can act directly on the epithelial and smooth muscle cells or can modify the electrical and synaptic behavior of ENS neurons. The constipation associated with morphine intake can be due to hyperpolarization of secretomotor neurons and suppression of secretion or to a centrally mediated stimulation of sympathetic noradrenergic discharge, or both. Direct activation of K+ channels and inhibition of Ca2+ channels via a G protein-mediated mechanism underlie these effects. Chronic treatment with opiates leads to tolerance, and diarrhea ensues upon abrupt withdrawal. Clinically, management of constipation in patients receiving opiates as analgesics can be a clinical challenge.
The development of long-acting analogs of somatostatin has transformed this ubiquitously distributed hormone from a physiologically fascinating regulator to a clinically relevant pharmacologic agent (see Chapter 1). In the intestine, enterochromaffin D cells produce somatostatin, which stimulates salt and water absorption in the ileum and colon and blocks the effects of several secretagogues.95,96 Somatostatin analogs such as octreotide are effective in treating several types of diarrheal diseases, particularly endocrine-related secretory diarrhea. Their therapeutic effect is due to a combination of actions, including inhibition of hormone release by tumors, slowing of intestinal transit, and a direct effect on epithelial cells. Paradoxically, elevated somatostatin levels, as encountered in somatostatinomas or with large pharmacologic doses of octreotide, can precipitate diarrhea secondary to steatorrhea.93 Other peptide hormones, including peptide YY, angiotensin II, and insulin, have been implicated as proabsorptive agents, but their physiologic significance is as yet unknown.
SECRETORY
Endogenous agents that stimulate secretion are listed in Table 99-2.
Increased intestinal production of eicosanoids contribute to the diarrhea of IBD (see Chapters 111 and 112). Glucocorticoids can decrease prostaglandin synthesis. Although the 5-ASA (acetylsalicylic acid) class of medications, which are a mainstay of the treatment of IBD, target cyclooxygenase and decrease prostaglandin production, their clinical efficacy probably depends on additional mechanisms of action, including effects on other inflammatory pathways, including nuclear factor κB (NFκB) and reactive oxygen species (ROS).97,98 Leukotrienes are considered also to have a role; the mechanism of their action is less well understood but might involve activation of secretomotor neurons in the subepithelium.
Serotonin
Serotonin plays a critical role in modulating intestinal motility, sensation, and secretion and is responsible for the diarrhea associated with carcinoid tumors. About 95% of the body’s serotonin is produced by enterochromaffin cells, and the remainder is produced by serotoninergic neurons of the myenteric plexus. Sensory receptors on enterochromaffin cells are activated by mechanical stimuli, acidity, invading pathogens, and dietary contents; for example, SGLT-like protein activates enterochromaffin cells, which serve as a glucose sensor to secrete serotonin (see Fig. 99-9).99 Although not completely elucidated, this signaling involves a complex sequence of autocrine and paracrine actions: ATP is released and converted extracellularly to ADP, which in turn activates a purinergic (P2Y) receptor-mediated calcium signaling cascade in the enterochromaffin cell to release 5-HT.99 5-HT then acts in a paracrine manner to stimulate epithelial cells, intrinsic primary afferent neurons (IPANs), and extrinsic primary afferent neurons (EPANs). Specific 5-HT receptor subtypes on different IPANs modulate the secretory reflex. Thus, 5-HT1PR on submucosal IPANs and amplifying presynaptic 5-HT4 receptors cause the release of acetylcholine and calcitonin gene-related peptide, which stimulate peristaltic and secretory reflexes. In contrast, 5-HT3Rs on myentric IPANs trigger the release of acetylcholine to stimulate giant migrating contractions.99–101 Serotonin stimulation of EPANs results in CNS-mediated responses of nausea and discomfort.
The major mechanism of serotonin inactivation is by a serotonin reuptake transporter (SERT) on enterocytes and neurons. Interestingly, SERT may be decreased in patients with diarrhea-predominant irritable bowel syndrome (IBS-D) and ulcerative colitis and might account for their increased colonic motility and diarrhea. There has been a concerted effort to clinically alter intestinal function by pharmacologic manipulation of specific serotonin receptors. Thus, 5-HT3 receptor antagonists, such as alosetron, are used to treat IBS-D, and tegaserod, a partial 5-HT4 agonist, can alleviate constipation associated with IBS. Unfortunately, significant side effects have limited their clinical use (see Chapter 118).93,101,102
Adenosine and related purine nucleotides play unique and complex roles in modulating secretion in vivo. As described earlier, they can stimulate secretion directly as it occurs in response to polymorphonuclear leukocyte infiltration of the mucosa or indirectly via release of 5-HT.99 Adenosine acting via P1 purinoreceptors, however, has been shown to attenuate secretion evoked by mechanical stimuli. Activation or inhibition of different populations of channels can underlie these seemingly opposite effects.
Guanylin, Nitric Oxide, and Reactive Oxygen Metabolites
The search for an endogenous activator of the E. coli heat-stable enterotoxin receptor led to the discovery of guanylin and uroguanylin, another example of molecular mimicry. These small peptides, synthesized in goblet and columnar cells activate membrane guanylate cyclase to increase intracellular cGMP and elicit fluid secretion. The guanylin family of peptides, in conjunction with the atrial natriuretic peptides, calibrate the intestinal-renal acid-base response axis. In contrast, nitric oxide, a neuroimmune regulator, stimulates soluble guanylate cyclase to increase cGMP.103 This enzyme is far more prevalent in the subepithelium of the small intestine, but it is expressed in colonic epithelia. Reactive oxygen metabolites, including oxygen free radicals and hydrogen peroxide, generally are produced by immune cells, but they can be released by epithelial cells under certain conditions. They stimulate fluid secretion (see the discussion of immunologic regulation). Additional agonists stimulate secretion (see Table 99-2). Interestingly, most of these agents also affect intestinal motility.
Microbial Pathogens
Microbial pathogens including bacteria, viruses, and fungi can alter electrolyte transport, increase intestinal permeability, and trigger inflammation to elicit diarrhea. They do this by a variety of mechanisms, including attaching to epithelial cells to insert their own products and alter host cell machinery, and by elaborating enterotoxins, which may be cytotoxic or can capture cell-signaling mechanisms to elicit secretion or disrupt tight junctions (see Table 99-3; see also Chapter 107).104–108 A few examples are provided here.
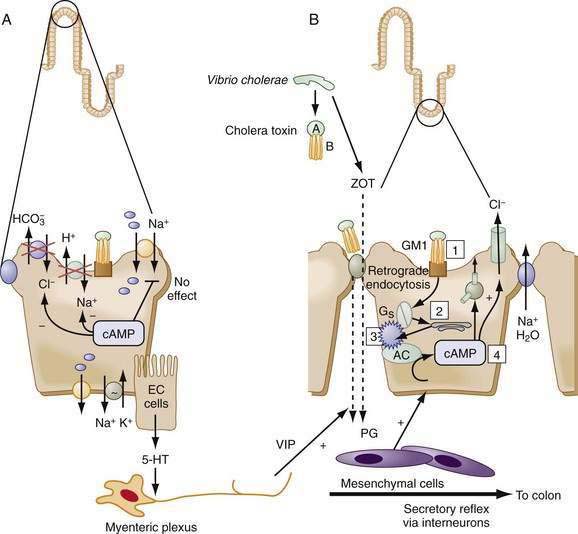
Shigella causes dysentery by release of Shiga cytotoxins, which enter the epithelial cell, inhibit protein synthesis, impair absorption, and damage the mucosa. In contrast, secretory diarrheas, such as those associated with cholera and traveler’s diarrhea, result from noninvasive pathogens, which elaborate enterotoxins that capture and turn on the secretory machinery of the epithelium. The archetypal enterotoxin-mediated diarrhea is cholera. Vibrio cholerae carry a virulence cassette that produces at least three different molecules: an enterotoxin; a zonula occludens toxin (ZOT), which disrupts tight junction permeability; and a channel-like protein.109 The enterotoxin causes an unregulated increase in cAMP, activates CFTR, and inhibits NHE3, thus resulting in copious fluid secretion (see Fig. 99-9 legend for details). Mice lacking CFTR do not respond to cholera toxin. Despite voluminous secretion, specific intestinal Na+-coupled nutrient absorptive (Na+-glucose, Na+-amino acids) pathways are unaltered by the toxin, forming the physiologic basis for ORT. Bacteria such as Salmonella species, Campylobacter jejuni, and E. coli elaborate enterotoxins similar to cholera toxin, thereby employing the cAMP machinery to elicit fluid secretion.
Strains of E. coli and Yersinia enterocolitica associated with traveler’s diarrhea elaborate small-molecular-weight, heat-stable enterotoxins, which increase cGMP to stimulate fluid secretion; signaling cascades distinct from cAMP and cGMP also activate CFTR and inhibit NHE3 (see the discussion of intracellular mediators). Vibrio parahaemolyticus elaborates a thermostable direct hemolysin (TDH) and is a major cause of gastroenteritis; its associated intestinal fluid secretion is attributed to an increase in intracellular calcium and activation of the Ca2+-calmodulin and protein kinase C signaling pathways.93,108
Rotavirus, the major cause of infantile gastroenteritis, induces watery diarrhea. The virus predominantly infects the mature enterocyte of the villus and elaborates an enterotoxin NSP4.110,111 NSP4 can inhibit brush border membrane disaccharidases and SGLT1 activity, thereby limiting Na+-glucose and fluid absorption, with resultant diarrhea. NSP4 in vitro elicits Cl− secretion via a calcium-phospholipase C pathway that stimulates Ca2+-activated Cl− channels; this effects a transient secretion similar to the actions of carbachol. Unlike other secretagogues, NSP4 has no effect on crypt cell secretion, but under favorable electrochemical conditions it stimulates secretion from villus cells. Paradoxically, NSP4 also can stimulate Cl− absorption from villus cells. The role of NSP4 in vivo has yet to be defined.
Many bacterial pathogens use different signaling molecules (e.g., kinases and phosphatases) to perturb the delicate balance of tight junctional proteins and cytoskeletal elements, thus disrupting intestinal permeability. Enteropathogenic (EPEC) and enterohemorrhagic (EHEC) E. coli decrease transepithelial resistance using different signaling cascades. The EPEC strains use a fascinating arsenal to alter host cell responses: They adhere to intestinal cells and in the process recruit a complex network of host cytoskeletal elements. Infectious bacteria use a variety of mechanisms, classified as types I to IV, to infect host cells. EPEC strains use a type III secretion apparatus to insert effector molecules into the host cell and use these molecules to co-opt the cell machinery to cause changes in the actin-myosin network, alter tight-junction proteins, and modulate ion transport processes to decrease absorption and increase fluid accumulation.105,107 The anaerobic bacterium Clostridium difficile, which causes antibiotic-associated pseudomembranous colitis, and Clostridium perfringens, which is associated with food-borne illnesses, alter intestinal permeability by using two distinct processes: The C. difficile toxins A and B interact with the Rho family of cellular proteins to disrupt the perijunctional actin-myosin ring, whereas the tight-junction claudin proteins serve as receptors for C. perfringens enterotoxin (CPE), and binding results in a disruption of tight-junctional fibrils. Other bacteria, including Bacteroides fragilis and V. cholerae, elaborate proteases that attack junctional proteins, including occludins, claudins, and cadherin, to disrupt the integrity of tight junctions.93,108
Many bacterial strains, including EPEC, EHEC, ETEC, Salmonella, and Shigella, trigger a highly specialized cascade to stimulate ion secretion. These pathogens induce the expression of receptors for the peptide neurohormone galanin, whereas uninfected cells do not possess galanin receptors. Galanin in turn activates Cl− secretion via Ca2+-dependent signaling processes.112–114117 The ever-expanding spectrum of toxin-induced mechanisms underscores the importance of delineating the intrinsic regulatory processes and the molecular pathophysiology of infectious diarrhea.
Bile Acids
An increase in colonic bile acids secondary to ileal malabsorption or oral supplementation can cause diarrhea (see Chapters 15 and 64).13,14 Only 7α dihydroxy bile acids such as chenodeoxycholic acid (3α; 7α), but not 7β, are associated with diarrhea. At high concentrations, bile acids act as a detergent and increase intestinal permeability. At more physiologic concentrations, bile salts indirectly increase cAMP and activate mast cells and, more importantly, stimulate epithelial cell Cl− secretion via the Ca2+ and PKCδ cascade.115,116 The ability of bile salts to stimulate Cl− secretion occurs only in the adult and is absent in the neonatal animal.
Long-chain fatty acids are seen in increased concentration in the colonic lumen in conditions such as sprue, when long-chain triglycerides are digested by lipase but the fatty acids are malabsorbed within the small intestine (see Chapters 101 and 104). Hydroxylated fatty acids are more potent secretagogues than the corresponding long-chain fatty acids and arise from colonic bacterial metabolism; ricinoleic acid is the long-chain fatty acid that is derived from oral castor oil, which is a nontoxic oil before it is hydroxylated. Specific fatty acid transporters have been identified in the intestine, and their mechanisms of action in electrolyte secretion are similar to those of bile acids.118,119
INTRACELLULAR MEDIATORS
The barrage of extracellular stimuli need to be translated into an intracellular language so the cell can regulate its transport machinery. The second messenger cascades of the cell include the cyclic nucleotides cAMP and cGMP, intracellular Ca2+, and the inositol phosphate-diacyl glycerol and tyrosine kinases. These messenger systems are common to several organ systems, and many cell-specific and tissue-specific structural and functional nuances contribute to the net biological response. More detailed descriptions of second messenger systems can be found elsewhere, but an overview is provided here.120
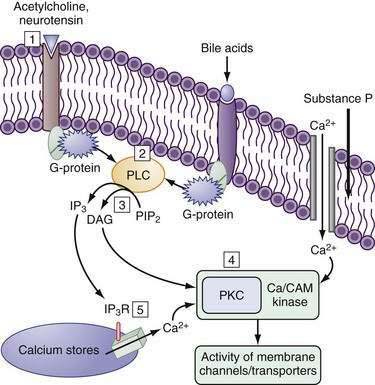
The molecules at almost every step in the signal transduction cascade, from the activating hormone to the receptors, cyclases, kinases, and phosphatases, and, finally, the transporters themselves, exist as multiple isoforms and variants (see Fig. 99-7). These isoforms exhibit differences in species, tissue, cell type, and subcellular distribution and are subject to regulation during their development as well as in response to routine physiologic demands.
Compartmentalization of components of the signaling cascade via cytoskeletal runners, anchoring domains, or sequestration in vesicles as a means of regulation is especially germane to the polarized enterocyte. Localizing transporters and their signaling systems into specific subcellular domains is the norm rather than the exception. This is a dynamic and highly regulatable process. Scaffolding proteins can promote docking of various proteins, kinases, and phosphatases by protein-protein interactions—to each other, to the cytoskeleton, and/or to the membrane (see Fig. 99-7); for example, guanylate cyclase C and the intestinal protein kinase GII have cytoskeletal and membrane interacting domains that bring them in close proximity to CFTR in the brush border membrane (Fig. 99-7). Cholesterol-rich membrane domains such as lipid rafts influence membrane fluidity and anchor specific transporters and their regulators. It should come as no surprise that many anchoring domains serve as multienzyme signaling complexes. Finally, trafficking of transporters into and out of the membrane via endosomal vesicles is an effective way of rapidly altering the Vmax of the transporter. For example, cAMP increases CFTR translocation to the membrane and NHE3 retrieval from the membrane, resulting in an increase in Cl− secretion and decrease in Na+ absorption (see Fig. 99-9). Thus, the cAMP-stimulated increases in Cl− secretion and decreases in Na+ absorption (see Fig. 99-9), respectively, result from an increase in CFTR translocation to, and NHE-3 retrieval from, the membrane.
Generally, agents that elevate intracellular cAMP, cGMP, or Ca2+ increase fluid secretion (see Tables 99-2 and 99-3). They can activate one or more transporters associated with electrogenic Cl− secretion: apical Cl− and K+ channels, basolateral K+ conductances, and NKCC1; they also inhibit the apical NHEs, NHE2 and NHE3. Conversely, fluid absorption is associated with a decrease in these messengers or with an activation of some tyrosine kinase pathways. cAMP plays an additional role in promoting trafficking of transporter-bearing vesicles (CFTR in crypts and distal colonic Na+ channels) to the apical membrane.91,111
The cAMP cascade is triggered by a hormone, such as VIP, binding to a specific member of the superfamily of heptahelical membrane-spanning receptors (7TM–VPAC1 and VPAC2) (see Fig. 99-7).121 Cyclic GMP is generated by the activation of membrane or soluble guanylate cyclases (GCs) by the natriuretic peptides guanylin and uroguanylin (see Fig. 99-7). Guanylins share their receptor GC-C with the heat-stable enterotoxins (see the earlier discussion of guanylin). Hormones and neurotransmitters such as substance P and acetylcholine activate secretion by increasing intracellular Ca2+ (Fig. 99-11; see figure legend for details).
Alberts B, Johnson S, Lewis J, et al. Molecular Biology of the Cell. New York: Garland Science, Taylor & Francis Group; 2007. (Ref 1.)
Alper SL. Molecular physiology of SLC4 anion exchangers. Exp Physiol. 2006;91:153-61. (Ref 61.)
Barrett KE, Seely SJ. Integrative Physiology and Pathophysiology of Intestinal Electrolyte Transport. San Diego: Academic Press; 2006. pp 1931-51. (Ref 4.)
Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931-43. (Ref 13.)
Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397-414. (Ref 101.)
Jentsch TJ, Neagoe I, Scheel O. ClC chloride channels and transporters. Curr Opin Neurobiol. 2005;15:319-25. (Ref 51.)
Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. 2002;82:245-89. (Ref 32.)
Kunzelmann K, McMorran B. First encounter: how pathogens compromise epithelial transport. Physiology (Bethesda). 2004;19:240-4. (Ref 108.)
Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol. 2004;286:C1213-28. (Ref 16.)
Thiagarajah JR, Verkman AS. New drug targets for cholera therapy. Trends Pharmacol Sci. 2005;26:172-5. (Ref 109.)
Rao M. Absorption and secretion of water and electrolytes. In: Ratnaike R, editor. Small Bowel Disorders. London: Hodder Headline Group; 2000:116-34. (Ref 120.)
Rao MC. Oral rehydration therapy: new explanations for an old remedy. Ann Rev Physiol. 2004;66:385-417. (Ref 91.)
Weber CR, Turner JR. Inflammatory bowel disease: is it really just another break in the wall? Gut. 2007;56:6-8. (Ref 82.)
Wright EM, Loo DD, Hirayama BA, Turk E. Surprising versatility of Na+-glucose cotransporters: SLC5. Physiology (Bethesda). 2004;19:370-76. (Ref 33.)
Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411-43. (Ref 38.)
1. Alberts B, Johnson S, Lewis J, et al. Molecular Biology of the Cell. New York: Garland Science, Taylor & Francis Group; 2007.
2. Nelson WJ. Regulation of cell surface polarity from bacteria to mammals. Science. 1992;258:948-55.
3. Kwon O, Corrigan G, Myers BD, et al. Sodium reabsorption and distribution of Na+/K+-ATPase during postischemic injury to the renal allograft. Kidney Int. 1999;55:963-75.
4. Barrett KE, Seely SJ. Integrative physiology and pathophysiology of intestinal electrolyte transport. San Diego: Academic Press; 2006. p 1931-51
5. Sellin JH, De Soignie R. Regulation of NaCl absorption in rabbit proximal colon in vitro. Am J Physiol. 1987;252:G45-51.
6. Sellin JH, DeSoignie R. Rabbit proximal colon: a distinct transport epithelium. Am J Physiol. 1984;246:G603-10.
7. Sellin J, Duffey M. Mechanisms of intestinal chloride absorption. New York: Raven Press; 1990. pp 81-94
8. Hatch M, Freel RW. Electrolyte transport across the rabbit caecum in vitro. Pflugers Arch. 1988;411:333-38.
9. Hoglund P, Haila S, Socha J, et al. Mutations of the down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nature Genet. 1996;14:316-19.
10. Jacob P, Rossmann H, Lamprecht G, et al. Down-regulated in adenoma mediates apical Cl−/HCO3− exchange in rabbit, rat, and human duodenum. Gastroenterology. 2002;122:709-24.
11. Pinto D, Clevers H. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp Cell Res. 2005;306:357-63.
12. Bjerknes M, Cheng H. Gastrointestinal stem cells. II. Intestinal stem cells. Am J Physiol. 2005;289:G381-87.
13. Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931-43.
14. Singh SK, Binder HJ, Boron WF, Geibel JP. Fluid absorption in isolated perfused colonic crypts. J Clin Invest. 1995;96:2373-9.
15. Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol. 1995;269:G467-75.
16. Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol. 2004;286:C1213-28.
17. Cereijido M, Contreras RG, Flores-Benitez D, et al. New diseases derived or associated with the tight junction. Arch Med Res. 2007;38:465-78.
18. Cereijido M, Contreras RG, Shoshani L, et al. Tight junction and polarity interaction in the transporting epithelial phenotype. Biochim Biophys Acta. 2008;1778:770-93.
19. Tomson FL, Koutsouris A, Viswanathan VK, et al. Differing roles of protein kinase C-zeta in disruption of tight junction barrier by enteropathogenic and enterohemorrhagic Escherichia coli. Gastroenterology. 2004;127:859-69.
20. Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233-45.
21. Russo MA, Hogenauer C, Coates SWJr, et al. Abnormal passive chloride absorption in cystic fibrosis jejunum functionally opposes the classic chloride secretory defect. J Clin Invest. 2003;112:118-25.
22. Diamond JM, Bossert WH. Standing-gradient osmotic flow. A mechanism for coupling of water and solute transport in epithelia. J Gen Physiol. 1967;50:2061-83.
23. Verkman AS. Mammalian aquaporins: diverse physiological roles and potential clinical significance. Expert Rev Mol Med. 2008;10:e13.
24. Agre P, Sasaki S, Chrispeels MJ. Aquaporins: a family of water channel proteins. Am J Physiol. 1993;265:F461.
25. Wang KS, Ma T, Filiz F, et al. Colon water transport in transgenic mice lacking aquaporin-4 water channels. Am J Physiol. 2000;279:G463-70.
26. Loo DD, Zeuthen T, Chandy G, Wright EM. Cotransport of water by the Na+/glucose cotransporter. Proc Natl Acad Sci U S A. 1996;93:13367-70.
27. Wright EM, Hirsch JR, Loo DD, Zampighi GA. Regulation of Na+/glucose cotransporters. J Exp Biol. 1997;200:287-93.
28. Quinton PM. The neglected ion: HCO3−. Nature Med. 2001;7:292-3.
29. Quinton PM, Reddy MM. CFTR, a rectifying, non-rectifying anion channel? J Korean Med Sci. 2000;15(Suppl):S17-20.
30. Bonny O, Hummler E. Dysfunction of epithelial sodium transport: from human to mouse. Kidney Int. 2000;57:1313-18.
31. Kunzelmann K. ENaC is inhibited by an increase in the intracellular Cl− concentration mediated through activation of Cl− channels. Pflugers Arch. 2003;445:504-12.
32. Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiology. 2002;82:245-89.
33. Wright EM, Loo DD, Hirayama BA, Turk E. Surprising versatility of Na+-glucose cotransporters: SLC5. Physiology (Bethesda). 2004;19:370-6.
34. Helliwell PA, Richardson M, Affleck J, Kellett GL. Regulation of GLUT5, GLUT2 and intestinal brush-border fructose absorption by the extracellular signal-regulated kinase, p38 mitogen-activated kinase and phosphatidylinositol 3–kinase intracellular signalling pathways: implications for adaptation to diabetes. Biochem J. 2000;350(Pt 1):163-9.
35. Kellett GL, Helliwell PA. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem J. 2000;350(Pt 1):155-62.
36. Naftalin RJ. Osmotic water transport with glucose in GLUT2 and SGLT. Biophys J. 2008;94:3912-23.
37. Keila P, Ghishan F. Na+-H+ Exchange in Mammalian Digestive Tract. San Diego: Academic Press; 2006. p 1847-79
38. Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411-43.
39. Lee SH, Kim T, Park ES, et al. NHE10, an osteoclast-specific member of the Na+/H+ exchanger family, regulates osteoclast differentiation and survival [corrected]. Biochem Biophys Res Commun. 2008;369:320-6.
40. Donowitz M, Cha B, Zachos NC, et al. NHERF family and NHE3 regulation. J Physiol. 2005;567:3-11.
41. Donowitz M, Li X. Regulatory binding partners and complexes of NHE3. Physiol Rev. 2007;87:825-72.
42. Haas M, Forbush B3rd. The Na-K-Cl cotransporter of secretory epithelia. Annu Rev Physiol. 2000;62:515-34.
43. Gimenez I. Molecular mechanisms and regulation of furosemide-sensitive Na-K-Cl cotransporters. Curr Opin Nephrol Hypertens. 2006;15:517-23.
44. Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211-76.
45. Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503-68.
46. Tabcharani JA, Linsdell P, Hanrahan JW. Halide permeation in wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels. J Gen Physiol. 1997;110:341-54.
47. Zhang Z, Chen D, Wheatly MG. Cloning and characterization of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) from crayfish axial muscle. J Exp Biol. 2000;203:3411-23.
48. Clarke LL, Argenzio RA. NaCl transport across equine proximal colon and the effect of endogenous prostanoids. Am J Physiol. 1990;259:G62-9.
49. Verkman AS. Drug discovery and epithelial physiology. Curr Opin Nephrol Hypertens. 2004;13:563-8.
50. Jentsch TJ, Friedrich T, Schriever A, Yamada H. The ClC chloride channel family. Pflugers Arch. 1999;437:783-95.
51. Jentsch TJ, Neagoe I, Scheel O. ClC chloride channels and transporters. Curr Opin Neurobiol. 2005;15:319-25.
52. Cuppoletti J, Malinowska DH, Tewari KP, et al. SPI-0211 activates T84 cell chloride transport and recombinant human ClC-2 chloride currents. Am J Physiol Cell Physiol. 2004;287:C1173-83.
53. Kunzelmann K, Milenkovic VM, Spitzner M, et al. Calcium-dependent chloride conductance in epithelia: is there a contribution by bestrophin? Pflugers Arch. 2007;454:879-89.
54. Fuller CM, Ji HL, Tousson A, et al. Ca2+-activated Cl− channels: a newly emerging anion transport family. Pflugers Arch. 2001;443(Suppl 1):S107-10.
55. Young FD, Newbigging S, Choi C, et al. Amelioration of cystic fibrosis intestinal mucous disease in mice by restoration of mCLCA3. Gastroenterology. 2007;133:1928-37.
56. Schultz SG, Dubinsky WP. Sodium absorption, volume control and potassium channels: in tribute to a great biologist. J Membr Biol. 2001;184:255-61.
57. Heitzmann D, Warth R. Physiology and pathophysiology of potassium channels in gastrointestinal epithelia. Physiol Rev. 2008;88:1119-82.
58. Vidyasagar S, Rajendran VM, Binder HJ. Three distinct mechanisms of HCO3− secretion in rat distal colon. Am J Physiol Cell Physiol. 2004;287:C612-21.
59. Binder HJ, Rajendran V, Sadasivan V, Geibel JP. Bicarbonate secretion: a neglected aspect of colonic ion transport. J Clin Gastroenterol. 2005;39:S53-8.
60. Alrefai WA, Tyagi S, Nazir TM, et al. Human intestinal anion exchanger isoforms: expression, distribution, and membrane localization. Biochim Biophys Acta. 2001;1511:17-27.
61. Alper SL. Molecular physiology of SLC4 anion exchangers. Exp Physiol. 2006;91:153-61.
62. Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 2004;447:710-21.
63. Simpson JE, Schweinfest CW, Shull GE, et al. PAT-1 (Slc26a6) is the predominant apical membrane Cl−/HCO3− exchanger in the upper villous epithelium of the murine duodenum. Am J Physiol. 2007;292:G1079-88.
64. Xu J, Barone S, Petrovic S, et al. Identification of an apical Cl−/HCO3− exchanger in gastric surface mucous and duodenal villus cells. Am J Physiol. 2003;285:G1225-34.
65. Wang Z, Petrovic S, Mann E, Soleimani M. Identification of an apical Cl−/HCO3− exchanger in the small intestine. Am J Physiol. 2002;282:G573-9.
66. Musch MW, Bookstein C, Xie Y, et al. SCFA increase intestinal Na absorption by induction of NHE3 in rat colon and human intestinal C2/bbe cells. Am J Physiol. 2001;280:G687-93.
67. Sellin JH. SCFAs: the enigma of weak electrolyte transport in the colon. News in Physiol Sci. 1999;14:58-64.
68. Halestrap AP, Meredith D. The SLC16 gene family—from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619-28.
69. Ganapathy V, Thangaraju M, Gopal E, et al. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008;10:193-9.
70. Powell DW, Adegboyega PA, Di Mari JF, Mifflin RC. Epithelial cells and their neighbors I. Role of intestinal myofibroblasts in development, repair, and cancer. Am J Physiol. 2005;289:G2-7.
71. Vanner S, Macnaughton WK. Submucosal secretomotor and vasodilator reflexes. Neurogastroenterol Motil. 2004;16(Suppl 1):39-43.
72. Hansen MB, Witte AB. The role of serotonin in intestinal luminal sensing and secretion. Acta Physiol (Oxford). 2008;193:311-23.
73. Chang EB, Bergenstal RM, Field M. Diarrhea in streptozocin-treated rats. Loss of adrenergic regulation of intestinal fluid and electrolyte transport. J Clin Invest. 1985;75:1666-70.
74. Sarto R, Powell D. Mechanisms of diarrhea in intestinal inflammation and hypersensitivity. In: Field M, editor. Diarrheal Diseases. New York: Elsevier; 1991:75-114.
75. MacDermott RP. Alterations in the mucosal immune system in ulcerative colitis and Crohn’s disease. Med Clin North Am. 1994;78:1207-31.
76. Sandle GI. Pathogenesis of diarrhea in ulcerative colitis: new views on an old problem. J Clin Gastroenterol. 2005;39:S49-52.
77. Charrier L, Merlin D. The oligopeptide transporter hPepT1: gateway to the innate immune response. Lab Invest. 2006;86:538-46.
78. Madara JL, Patapoff TW, Gillece-Castro B, et al. 5′-Adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest. 1993;91:2320-5.
79. Chang EB, Musch MW, Mayer L. Interleukins 1 and 3 stimulate anion secretion in chicken intestine. Gastroenterology. 1990;98:1518-24.
80. Rocha F, Musch MW, Lishanskiy L, et al. IFN-γ downregulates expression of Na+/H+ exchangers NHE2 and NHE3 in rat intestine and human Caco-2/bbe cells. Am J Physiol. 2001;280:C1224-32.
81. Clayburgh DR, Barrett TA, Tang Y, et al. Epithelial myosin light chain kinase–dependent barrier dysfunction mediates T cell activation–induced diarrhea in vivo. J Clin Invest. 2005;115:2702-15.
82. Weber CR, Turner JR. Inflammatory bowel disease: is it really just another break in the wall? Gut. 2007;56:6-8.
83. Charney AN, Egnor RW. NaCl absorption in the rabbit ileum. Effect of acid-base variables. Gastroenterology. 1991;100:403-9.
84. Charney AN, Feldman GM. Systemic acid-base disorders and intestinal electrolyte transport. Am J Physiol. 1984;247:G1-12.
85. Levens NR. Control of intestinal absorption by the renin-angiotensin system. Am J Physiol. 1985;249:G3-15.
86. Moriarty KJ, Higgs NB, Lees M, et al. Influence of atrial natriuretic peptide on mammalian large intestine. Gastroenterology. 1990;98:647-53.
87. Penn D, Lebenthal E. Intestinal mucosal energy metabolism—a new approach to therapy of gastrointestinal disease. J Pediatr Gastroenterol Nutr. 1990;10:1-4.
88. Bugaut M, Bentejac M. Biological effects of short-chain fatty acids in nonruminant mammals. Annu Rev Nutr. 1993;13:217-41.
89. Rhoads JM, Keku EO, Quinn J, et al. l-Glutamine stimulates jejunal sodium and chloride absorption in pig rotavirus enteritis. Gastroenterology. 1991;100:683-91.
90. Eherer AJ, Fordtran JS. Fecal osmotic gap and pH in experimental diarrhea of various causes. Gastroenterology. 1992;103:545-51.
91. Rao MC. Oral rehydration therapy: new explanations for an old remedy. Ann Rev Physiol. 2004;66:385-17.
92. Harvey BJ, Alzamora R, Stubbs AK, et al. Rapid responses to aldosterone in the kidney and colon. J Steroid Biochem Mol Biol. 2008;108:310-17.
93. Farthing MJ. Antisecretory drugs for diarrheal disease. Dig Dis. 2006;24:47-58.
94. Wood JD, Galligan JJ. Function of opioids in the enteric nervous system. Neurogastroenterol Motil. 2004;16(Suppl 2):17-28.
95. Dharmsathaphorn K, Racusen L, Dobbins JW. Effect of somatostatin on ion transport in the rat colon. J Clin Invest. 1980;66:813-20.
96. Guandalini S, Kachur JF, Smith PL, et al. In vitro effects of somatostatin on ion transport in rabbit intestine. Am J Physiol. 1980;238:G67-74.
97. Monteleone G, Fina D, Caruso R, Pallone F. New mediators of immunity and inflammation in inflammatory bowel disease. Curr Opin Gastroenterol. 2006;22:361-4.
98. Fantini MC, Monteleone G, Macdonald TT. New players in the cytokine orchestra of inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1419-23.
99. Xue J, Askwith C, Javed NH, Cooke HJ. Autonomic nervous system and secretion across the intestinal mucosal surface. Auton Neurosci. 2007;133:55-63.
100. Galligan JJ, Vanner S. Basic and clinical pharmacology of new motility promoting agents. Neurogastroenterol Motil. 2005;17:643-53.
101. Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397-414.
102. Gershon MD, Liu MT. Serotonin and neuroprotection in functional bowel disorders. Neurogastroenterol Motil. 2007;19(Suppl 2):19-24.
103. Camici M. Guanylin peptides and colorectal cancer (CRC). Biomed Pharmacother. 2008;62:70-6.
104. Petri WAJr, Miller M, Binder HJ, et al. Enteric infections, diarrhea, and their impact on function and development. J Clin Invest. 2008;118:1277-90.
105. Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439-51.
106. Sasaki M, Sitaraman SV, Babbin BA, et al. Invasive Escherichia coli are a feature of Crohn’s disease. Lab Invest. 2007;87:1042-54.
107. Hecht G. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VII. Enteropathogenic Escherichia coli: physiological alterations from an extracellular position. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1-7.
108. Kunzelmann K, McMorran B. First encounter: how pathogens compromise epithelial transport. Physiology (Bethesda). 2004;19:240-4.
109. Thiagarajah JR, Verkman AS. New drug targets for cholera therapy. Trends Pharmacol Sci. 2005;26:172-5.
110. Lorrot M, Vasseur M. How do the rotavirus NSP4 and bacterial enterotoxins lead differently to diarrhea? Virol J. 2007;4:31.
111. Barrett KE. New ways of thinking about (and teaching about) intestinal epithelial function. Adv Physiol Educ. 2008;32:25-34.
112. Mitsukawa K, Lu X, Bartfai T. Galanin, galanin receptors and drug targets. Cell Mol Life Sci. 2008;65:1796-805.
113. Anselmi L, Lakhter A, Hirano AA, et al. Expression of galanin receptor messenger RNAs in different regions of the rat gastrointestinal tract. Peptides. 2005;26:815-19.
114. Anselmi L, Stella SLJr, Lakhter A, et al. Galanin receptors in the rat gastrointestinal tract. Neuropeptides. 2005;39:349-52.
115. Venkatasubramanian J, Nataraja S, Carlos M, et al. Differences in Ca2+ signaling underlie age-specific effects of secretagogues on colonic Cl− transport. Am J Physiol Cell Physiol. 2001;280(3):C646-58.
116. Kanchanapoo J, Ao M, Prasad R, et al. Role of protein kinase C-delta in the age-dependent secretagogue action of bile acids in mammalian colon. Am J Physiol Cell Physiol. 2007;293(6):C1851-61.
117. Gross KJ, Pothoulakis C. Role of neuropeptides in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:918-32.
118. Hajri T, Abumrad NA. Fatty acid transport across membranes: relevance to nutrition and metabolic pathology. Ann Rev Nutr. 2002;22:383-415.
119. Abumrad NA. CD36 may determine our desire for dietary fats. J Clin Invest. 2005;115:2965-7.
120. Rao M. Absorption and secretion of water and electrolytes. In: Ratnaike R, editor. Small Bowel Disorders. London: Hodder Headline Group; 2000:116-34.
121. Muller JM, Debaigt C, Goursaud S, et al. Unconventional binding sites and receptors for VIP and related peptides PACAP and PHI/PHM: an update. Peptides. 2007;28:1655-66.
* There are several potential differences across the epithelium: across the apical membrane into the cell, from the cell interior across the basolateral membrane, across the epithelium, across the mucosa, and across the entire GI tract. By convention, the potential differences across the epithelium, the mucosa, and the entire GI tract generally are considered the same.