41 Intensive Care After Neurosurgery
 Overview
Overview
The benefit of concentration of care in units with sufficient case volume has been well established in different fields of intensive care medicine including trauma,1 neonatology, and specifically neurointensive care.2,3
Treatment of patients with spontaneous intracerebral hemorrhage in a neurointensive care unit is associated with reduced mortality when compared with patients admitted to a general intensive care unit (ICU).4,5 Mortality following aneurysmal subarachnoid hemorrhage (SAH) is lower in centers with a higher case volume.6 Patel and colleagues7 unequivocally showed a 2.15 times increase in the odds of death (adjusted for case mix) for patients with severe traumatic brain injury (TBI) treated in non-neurosurgical centers versus neurosurgical centers. Their report makes a strong case for transferring and treating all patients with severe head injury in a setting with 24-hour neurosurgical facilities. Protocol-driven approaches also improve results.8–10
 Priorities and Goals of Postoperative Neurosurgical Care
Priorities and Goals of Postoperative Neurosurgical Care
Postoperative complications may be systemic or neurosurgical (Table 41-1).
| Systemic Complications | Neurosurgical Complications |
|---|---|
| Coagulation disorders: blood loss, disseminated intravascular coagulation, drug induced Thromboembolic: DVT, pulmonary embolism, myocardial infarction Pulmonary: atelectasis, pneumothorax Hypovolemia: insufficient pre- and perioperative hydration, blood loss Infection: pneumonia, urinary tract infection, catheter sepsis Metabolic: hyperglycemia [steroid induced], diabetes insipidus, hyponatremia Air embolism: sitting position, opening of large cerebral veins during surgery Pressure sores and decubitus ulcers: intraoperative positioning, cervical traction, paraplegia |
Postoperative hematoma: subgaleal, epidural, subdural, intraparenchymal Cerebral ischemia: subarachnoid hemorrhage, vasospasm, vessel occlusion Brain swelling: edema, vasodilation Infection: meningitis, subdural empyema, cerebral abscess Seizures: infection, depressed compound skull fracture, cortical lesions Hydrocephalus: obstruction/resorption Tension pneumocephalus CSF fistula Inverse cerebellar herniation Cranial nerve lesions |
CSF, cerebrospinal fluid; DVT, deep venous thrombosis.
 Prevention and Management of Systemic Complications After Neurosurgery
Prevention and Management of Systemic Complications After Neurosurgery
General Principles and Second Insults
The prevention and management of systemic complications after neurosurgical procedures follows general principles of “intensive care” medicine. It is, however, important to realize that systemic complications and second insults may initiate or aggravate cerebral damage. Aggressive treatment aimed at preventing and limiting second insults is of paramount importance. The main second insults, their causes, and adverse effects on brain homeostasis and function are summarized in Table 41-2, further illustrating the complex interactions between systemic events and CNS function.
| Event | Main Causes | Adverse Effects |
|---|---|---|
| Hypoxemia | Hypoventilation Aspiration atelectasis Pneumothorax Pneumonia Anemia |
Decrease in oxygen delivery and increased risk of ischemic damage |
| Hypotension | Hypovolemia | Decreased CPP, decrease in CBF, increased risk of ischemia |
| Cardiac failure | ||
| Sepsis, spinal cord injury | ||
| Anemia | Blood loss | Decrease in oxygen transport and delivery and increased risk of ischemic damage |
| Hypercapnia | Respiratory depression | Increased CBV, raised ICP |
| Hypocapnia | Hyperventilation, spontaneous or induced | Cerebral vasoconstriction with increased risk of ischemic damage |
| Hyperthermia | Hypermetabolism, stress response, infection | Metabolic requirements may exceed substrate delivery, resulting in energy depletion |
| Central dysregulation | ||
| Hypothermia | Exposure, central dysregulation | May be neuroprotective but can cause significant coagulopathy and electrolyte disturbances |
| Hyperglycemia | IV infusion of dextrose, steroids, stress response | Acidosis, electrolyte disturbances |
| Hypoglycemia | Inadequate nutrition, insulin overdose, pituitary insufficiency | Energy depletion in the brain, seizures |
| Hyponatremia | Inadequate salt intake (hypotonic fluids) Excessive sodium loss (cerebral salt wasting/CSF drainage) Syndrome of inappropriate ADH |
Increased edema, seizures |
| Hypernatremia | Diabetes insipidus | |
| Osmotic agents (mannitol, hypertonic saline) | Lethargy, coma |
ADH, antidiuretic hormone; CBF, cerebral blood flow; CBV, cerebral blood volume; CPP, cerebral perfusion pressure; CSF, cerebrospinal fluid; ICP, intracranial pressure; IV intravenous.
Cardiac Dysfunction
Electrocardiographic (ECG) abnormalities, usually diffuse ST-segment changes mimicking cardiac ischemia and cardiac arrhythmias, may be caused by SAH, TBI, or raised ICP. The devastating effects of a sudden catecholamine release following acute intracranial bleeding have recently received further attention. The left ventricle suffers a typical bulging (indicating ischemic changes and functional impairment) which has been awarded the term Takotsubo syndrome, with reference to the shape of a pot used by ancient Japanese fishermen for catching octopus. The extent of left ventricular dysfunction is variable, but it may lead to extreme cardiac failure and pulmonary edema.11,12
Neurogenic Pulmonary Edema
The development of neurogenic pulmonary edema has been described early in the postoperative period after a variety of neurosurgical procedures, including brain tumors (particularly those resected in the posterior fossa), cysts, hydrocephalus, intracranial hemorrhages, and brainstem lesions.13–16 Although an infrequent event, this is potentially life threatening and requires rapid evaluation and emergent therapy in the ICU. A 9% mortality rate directly attributable to neurogenic pulmonary edema has been reported in a recent review of this condition. Generally this complication appears in the initial 4 hours after the neurologic event and is more common in women than in men, possibly related to the preponderance of cases in patients with SAH.16 The mechanisms underlying this condition are unclear; a sudden central sympathetic discharge may trigger pulmonary venoconstriction, systemic arterial hypertension, increased left ventricle afterload, increased capillary permeability in the pulmonary vascular bed, and simultaneously cause cardiac ischemia and ventricular failure.12,17 Because of these multiple mechanisms, neurogenic pulmonary edema can be interpreted as noncardiogenic or, at least in part, as cardiogenic.18 Both low and high protein content have been reported in the edema fluid.16,19 It is commonly associated with raised ICP, so in addition to therapies directed at intracranial hypertension, therapeutic measures are mostly supportive. To attenuate the massive sympathetic discharge, opioids and sedatives are used. Supplemental oxygen is uniformly required, and tracheal intubation with mechanical ventilation and application of positive end-expiratory pressure (PEEP) has been reported in about 75% of patients.16 Diuretics have been used, provided volume status is adequate, but diuresis causes less effect than in cardiac edema. Most patients require vasoactive drugs.19
Hypercoagulopathy and Thrombosis Prophylaxis
Release of factors from damaged brain tissue may induce local and systemic hypercoagulopathy.20–22 Various studies have confirmed a transient hypercoagulopathy syndrome both in the immediate postoperative phase after brain surgery and in patients with TBI.20,23–25 In patients with a subdural hematoma, consumption of clotting factors may lead to coagulopathy in up to 22% of the patients.26
Deep venous thrombosis (DVT) has been reported to occur in 18% to 50% of neurosurgical cases27 and pulmonary embolism (PE) in 0% to 25%. DVT and PE incidence is particularly high in brain tumor patients. Nevertheless, neurosurgeons tend to underestimate the risk of DVT and PE28 and are sometimes reluctant to routinely prescribe anticoagulant prophylaxis for fear of increasing the risk of postoperative bleeding.29 Options for prevention of thrombosis prophylaxis in neurosurgical patients include both mechanical (graduated compression stockings, intermittent pneumatic compression stockings) and pharmacologic (low dose of classic heparin and low-molecular-weight heparin) therapies. Intuitively, mechanical therapies carry less associated risk, but pharmacologic approaches are more effective in preventing thrombotic complications. Various studies have indeed shown a higher incidence of postoperative hemorrhagic complications,30 but not all are clinically relevant.
Overall, existing evidence, however, shows that the beneficial effects in reducing DVT and in particular PE31,32 outweigh a slightly increased risk of clinically significant hemorrhagic complications with anticoagulant prophylaxis.
These data support the administration of antithrombotic prophylaxis to patients undergoing neurosurgical procedures,33 including those with intracranial hemorrhagic lesions,34 closed TBI,35,36 or high-risk trauma patients.37,38 It has been recommended to remove catheters or drainage tubes in the postoperative phase when anticoagulant effects are low (e.g., just prior to administration of next dose).39
 Prevention and Management of Neurosurgical Postoperative Complications
Prevention and Management of Neurosurgical Postoperative Complications
Supratentorial Procedures
Postoperative Subgaleal Hematoma
Postoperative subgaleal hematoma may occur in up to 11% of procedures. These hematomas generally result from inadvertent damage of the superficial temporal artery, inadequate hemostasis, or hemorrhage from the temporal muscle. If the superficial temporal artery is damaged during the operation, ligation is preferred over coagulation. The occurrence of subgaleal hematomas can be minimized by routine use of postoperative wound drainage for 24 hours. Reoperation for subgaleal hematomas is seldom necessary unless there is a communication with the intracranial compartment, with secondary compression of the brain.40
Intracranial Hemorrhage
Parenchymal hemorrhages are the most frequent cause of postoperative hematomas following supratentorial procedures and generally occur at the site of operation, particularly following partial tumor resection. An increase in systemic BP at the end of surgery may increase the risk of parenchymal hemorrhage. In rare cases, the hematoma may be located distant from the primary site of operation, and cerebellar hematomas have even been described after supratentorial surgery.41,42 The possibility of a postoperative hematoma should be considered in all patients who are not fully alert post anesthesia, as well as in those who exhibit secondary deterioration.
Postoperative Brain Swelling
Modern neuroanesthesiology techniques have diminished the incidence of peri- and postoperative brain swelling. Nevertheless, significant swelling may sometimes occur, causing surgical difficulties and possibly critical problems in the ICU. Predisposing factors are hypercapnia, arterial hypertension, hyponatremia, obstruction of venous drainage, and silent or overt seizures during surgery or in the immediate postoperative phase. Further significant brain swelling after uneventful surgery has been attributed to intracranial hypotension caused by subgaleal suction.42,43 In any patient with brain swelling during the surgical procedure, the possibility of a deep hematoma should be considered, and an urgent computed tomography (CT) scan should be performed. Brain swelling due to vasodilation can be corrected by hyperventilation and barbiturate administration; brain swelling due to cerebral edema should preferentially be treated by mild hyperventilation and osmotic agents.
Tension Pneumocephalus
Some air collection is generally observed on postoperative CT scans.44 In rare circumstances, the rewarming of air in the intracranial compartment postoperatively or continuous air leakage due to a cerebrospinal fluid (CSF) fistula of the skull base may lead to a tension pneumocephalus, with clinical symptomatology including decreasing level of consciousness, signs of raised ICP, and occasionally seizures. Generally, postoperative air accumulations are self-limiting and do not require specific treatment.
Seizures
An epileptic seizure in the direct postoperative phase should be considered a serious complication that may cause significant deterioration secondary to vasodilation, increased cerebral oxygen consumption, and increased brain edema. Occult seizure activity can occur in 15% to 18% of patients with moderate and severe TBI.45 The benefits of prophylactic antiseizure medication should be balanced against risks. In some centers, routine prophylaxis is prescribed in all patients undergoing supratentorial brain surgery. In others, the indications are restricted to patients with a higher risk:
Infratentorial Procedures
The care for patients in the direct postoperative phase following infratentorial procedures poses specific problems. Postoperative complications in the posterior fossa can lead to rapid deterioration because of the relatively small infratentorial volume reserve and the immediate compression of the brainstem, resulting in respiratory insufficiency and acute herniation. Irritation of the brainstem may induce large swings in arterial BP, enhancing the risk of postoperative hemorrhage during hypertensive episodes. Cranial nerves are more susceptible to damage due to surgical manipulation than peripheral nerves.46 Lesions of the lower cranial nerves may lead to a diminished gag reflex, with increased risk of aspiration and pneumonia. After surgery in the cerebellopontine angle, specific attention should be paid to the function of the trigeminal and facial nerves and prophylactic measures to prevent damage of the cornea taken.
After posterior fossa surgery, some patients may develop a syndrome of aseptic meningitis.47 This is characterized by meningeal symptoms, headaches, and an inflammatory response of the CSF in the absence of evidence for infection. The origin of this syndrome has not been fully clarified, but symptoms may resolve sooner with intermittent CSF drainage.
An infrequent transitory complication observed after resection of large midline posterior fossa tumors is cerebellar mutism.47 The exact cause is poorly understood, but a vascular phenomenon has been hypothesized.48
Cerebrovascular Procedures
Postoperative care for patients undergoing cerebrovascular surgery poses specific challenges in neurointensive care. In patients operated for arteriovenous malformations, the risk of seizures is particularly high, and focal deficits may occur secondary to changes in cerebral hemodynamics. Following treatment for a cerebral aneurysm, medical and cerebral complications can occur either related to the disease or to treatment (surgical clipping or endovascular coiling). Medical complications specifically linked to SAH are neurogenic pulmonary edema, cardiac arrhythmias, and ventricular failure.11 Electrolyte disturbances, in particular hyponatremia, are also frequently observed.49
The main cerebral complications are:
Rebleeding occurs mainly in the first weeks after the aneurysmal rupture, and current approaches are to prevent rebleeding by early surgical clipping or endovascular obliteration of the aneurysmal sack. Delayed cerebral ischemia, often due to vasospasm is—besides rebleeding—the most common cause of death and disability due to SAH. The reported incidence of this complication varies widely, but angiographic vasospasm is seen with angiography in over 67% of untreated patients at the time of maximum spasm around the end of the first week.50
Delayed cerebral ischemia (DCI) is considered to be caused by vasospasm. However, not all patients with DCI have vasospasm. Inversely, not all patients with vasospasm develop clinical symptoms and signs of DCI. Recent studies show that DCI cannot always be attributed to vasospasm but more to the occurrence of microthrombosis.51,52 DCI is associated with an activation of the coagulation cascade within a few days after SAH, preceding the time window during which vasospasm occurs. Furthermore, both impaired fibrinolytic activity and inflammatory and endothelium-related processes may lead to the formation of microthrombi, further promoting the development of DCI.
Clinically evident delayed ischemic deficits (DID) affect approximately one third of patients. Various studies have shown a beneficial effect of the administration of oral calcium antagonists in preventing DID.53,54 Beneficial effects of intravenous administration of nimodipine remain unproven.
Following evidence that patients with SAH had reduced blood volume, plasma volume, and erythrocyte mass, triple-H therapy (hypervolemia, hypertension, and hemodilution) was proposed for both prophylaxis and treatment of DID after SAH. Various studies have shown a reduction of DID with triple-H prophylaxis,55 but some debate remains.56,57
The usefulness of triple-H treatment is generally accepted, but it has never been unequivocally demonstrated by a randomized controlled trial to be superior to simple moderate fluid loading. The relative importance of the three components of triple-H therapy is uncertain.58,59 Adequate fluid loading should be considered the most important aspect of early treatment and prophylaxis of DID, but it may be considered reasonable to reserve the more vigorous loading and induced hypertension for situations in which there is clinical evidence of delayed ischemia.59–61
Progressive signs of DID may require more aggressive approaches including angioplasty.62 Transluminal balloon angioplasty is generally recommended, but this requires special equipment and a highly skilled and experienced interventional neuroradiology team. Alternatively, “chemical angioplasty” in which the angiography catheter is used to instill papaverine or nimodipine may be considered.63
Chemical angioplasty often has to be repeated within hours or days and carries complications including pupillary changes, seizures, or respiratory arrest with vertebral artery injection. Alternatively, possibilities of cisternal therapy should be considered, injecting recombinant tissue plasminogen activator (tPA) or urokinase in the basal cisterns to break down the accumulated blood,64 or even nitric oxide donors to improve vascular tone.
Various studies have shown clinical benefit of this approach, with the added benefit of reducing the incidence of hydrocephalus. Acute hydrocephalus after SAH is not uncommon. The reported frequency depends on the criteria used for the diagnosis and ranges from 9%65 up to 67%.66 Spontaneous improvement of hydrocephalus has been reported in approximately half of patients with acute hydrocephalus and impaired consciousness on admission, but it may be difficult to predict spontaneous improvement, because treatment is generally instituted. Evidence exists that in the absence of a hematoma with mass effect or an obstructive element, serial lumbar punctures may be the initial optimal method of treatment, reserving continuous CSF drainage procedures for patients in whom the hydrocephalus does not resolve over time.
 Admission Examination and Monitoring in the Intensive Care Unit
Admission Examination and Monitoring in the Intensive Care Unit
Specific care and monitoring of the postoperative neurosurgical patient requires accurate knowledge of the preoperative situation and the intraoperative procedure, including the surgery, anesthesiology, and any surgical complications or difficulties. Pertinent aspects are summarized in Table 41-3.
TABLE 41-3 Postoperative Intake After Neurosurgical Operations
| Preoperative situation | Neurologic deficit (level of consciousness, focal paresis, cranial nerve lesions, hormonal deficits) Preexisting disease (especially pulmonary and cardiac) Preoperative medication History of seizures Allergy |
| Intraoperative details (anesthesia) | Narcotic agents and antagonists Blood loss and substitution Intraoperative laboratory values Intraoperative second insults, diabetes insipidus, etc. |
| Intraoperative course (surgical) | Indication, approach, and duration of surgery Surgical position Surgical difficulties and complications (brain swelling, difficult hemostasis, temporary or definite vascular occlusion, opening of air sinus) Immobilization/positioning of patient |
| Postoperative instructions (surgeon and anesthetist) | Postoperative medication (e.g., anticonvulsants, antibiotics, steroids, mannitol, antithrombosis prophylaxis) Instructions for postoperative care and monitoring Instructions for removal of drainage, tubes, and stitches Preferred duration of postoperative artificial ventilation Instructions for follow-up CT or MRI examination (if indicated) |
On admission, a full examination of the patient is required; wherever possible, this includes assessment of level of consciousness and neurologic functioning. Medical care for the patient should be provided in joint collaboration between the intensivist and neurosurgeon. Intensive care monitoring includes clinical surveillance, technical monitoring, and follow-up CT or magnetic resonance imaging (MRI). Various approaches to monitoring are summarized in Table 41-4.
TABLE 41-4 Postoperative Monitoring After Intracranial Procedures
| Clinical surveillance | Level of consciousness (Glasgow Coma Scale), pupillary reactivity, focal deficits, cranial nerve lesions |
| Systemic monitoring | Electrocardiogram and heart rate, respiration, pulse oximetry, end-tidal CO2, blood pressure (invasive, noninvasive), temperature, central venous pressure, Swan Ganz catheter |
| Brain-specific monitoring | Intracranial pressure and cerebral perfusion pressure, jugular oximetry, brain oxygen tension monitoring, microdialysis, transcranial Doppler, electroencephalogram, evoked potentials |
| Accesses | Central or peripheral venous catheter, arterial catheter, urinary catheter, gastric tube |
| Laboratory examinations | Blood gases, hematology, electrolytes, glucose and on indication coagulation status |
| Imaging examinations | Chest radiograph (ventilated patients and after lung procedures) Computed tomography or magnetic resonance imaging follow-up (as required) |
Early Evaluation
The level of consciousness should be assessed by the Glasgow Coma Scale (GCS).67 In this scale, standardized assessment of three aspects of responsiveness is performed: the eye, motor and verbal reaction (Table 41-5).
| Eyes | Motor | Verbal |
|---|---|---|
| 1. None | 1. None | 1. None |
| 2. To pain | 2. Abnormal extension | 2. Incomprehensible (groaning) |
| 3. To speech | 3. Abnormal flexion | 3. Inappropriate |
| 4. Spontaneous | 4. Flexion (withdrawal) | 4. Disoriented, confused |
| 5. Localizing | 5. Oriented | |
| 6. Obeying commands |
NOTES: The best score for each response should be documented and communicated in the format described above. Assessment of the best motor score is based on the best response of the arms. For use in individual patients, separate description of the three components of the Glasgow Coma Scale (GCS) is strongly recommended. For purposes of classification, the total GCS can be calculated by adding the best score obtained in each category. The GCS should be annotated to indicate confounding factors: T signifies an intubated patient; S, sedation; P, neuromuscular blockade.
When administration of painful stimuli is necessary to assess the level of responsiveness, standardized administration is required: pressure on the nail bed and supraorbital pressure to test the localizing response of the motor scale (Figure 41-1).
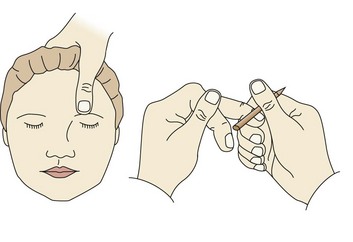
Accurate determination of the full GCS is not always possible because of sedation and paralysis, but when possible, at least the best motor score should be recorded. Approaches to daily interruption of sedation that allow intermittent wake up in ventilated patients not only help care providers to monitor neurologic status but also have been shown to result in better outcome.68 Some authors advocate imputing the eye and verbal scores from the motor score in sedated and/or ventilated patients.69
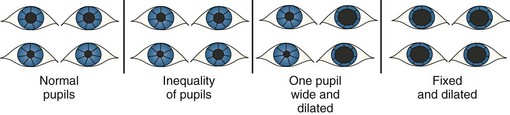
Pressure on the oculomotor nerve leads to a loss of function of the parasympathetic fibers, causing a diminished pupillary response or absent pupillary reactivity, generally initially on the side of a lesion (Figure 41-2). With progressive increase in pressure, both pupils become dilated and unresponsive to light. In patients with a lesion of the optic nerve, the consensual light reflex—contraction of the pupil when a light is shone into the opposite eye—remains intact.
 Systemic Monitoring: Cardiopulmonary, Respiratory Status, and Temperature
Systemic Monitoring: Cardiopulmonary, Respiratory Status, and Temperature
Hypovolemic shock is most common in the setting of multisystem injury or intraoperative blood loss with inadequate replacement. It is important to recognize that tachycardia and signs of peripheral vasoconstriction such as skin pallor and poor capillary refill may precede a drop in BP. Treatment is rapid fluid resuscitation employing isotonic crystalloid fluids, volume expanders, small-volume resuscitation (hypertonic saline), and blood transfusions. Central venous pressure monitoring, or preferably pulmonary artery catheterization, can guide the use of intravenous fluids and vasopressor therapy, aiming for a pulmonary artery wedge pressure of 12 to 14 mm Hg to optimize organ perfusion. After initial volume resuscitation, we suggest a hematocrit of approximately 30% to 33% as optimal in the acute postoperative period in patients in the neurosurgical ICU. Although debate still exists, available evidence suggests that restrictive blood transfusion strategies may be less appropriate in neurointensive care.70–74
After intracranial or spinal cord procedures, we would advocate a more liberal use of blood transfusions than generally recommended in intensive care medicine, aiming at a hemoglobin of at least 5.5 to 6.0 mmol/L (9-10 mg/dL) in order to promote adequate oxygenation of the CNS. This corresponds to the recommendations proposed by Goodnough et al.75 in case of ischemia.
Temperature monitoring is also important, since hypothermia can depress neurologic function to the point of obtundation or coma. Conversely, fever, by increasing metabolic requirements, may exacerbate secondary injury. Mean energy expenditure may be increased up to 200% in patients following brain injury,76 and it would therefore appear appropriate not to risk increasing metabolic requirements even further. Consequently, we recommend that core temperature should be kept lower than 38.0°C, using medications (e.g., acetaminophen, paracetamol, diclofenac) and surface or intravascular cooling.
Hypothermia may be due to adrenal or pituitary insufficiency, hypothalamic disorders, hypoglycemia, or intraoperative exposure. Deliberate hypothermia is sometimes used in complicated cerebrovascular procedures and as second-tier therapy in patients with TBI to reduce ICP. For the indication TBI, hypothermia has been shown to effectively reduce ICP, but uncertainty still exists whether this may translate into an improvement of functional outcome.77,78
Various approaches to cooling have been adopted, but the most frequently used employ surface cooling or gastric lavage with cold fluids. Marion79 reported favorable results with the use of devices for intravascular cooling, and this technique can be expected to become standard for induction of hypothermia in the near future.
Hypothermia has been associated with several complications including cardiovascular instability (mainly arrhythmias), coagulopathy, electrolyte shifts, fluid overload, and increased risk of infection and shivering.80,81 The management of a patient treated with hypothermia over longer periods of time for control of raised ICP can be much more complex than the use of short-term hypothermia post cardiac arrest. Ideally, normothermia could represent the best tradeoff between the dangers of hyperthermia and the complexities and side effects of hypothermia. In practice, a recent trial in neurointensive care comparing conventional treatments with prophylactic normothermia has failed to show benefit.82
Electrolytes and Osmolarity
A direct link exists between plasma osmolarity and water flux into and out of brain cells83,84; if the blood-brain barrier is intact, any decrease in plasma osmolarity will cause an increase of intracellular water in the brain, with potential increase in intracranial pressure, alteration of the transmembrane potential, and so on.85 It is important to prevent the development of hyponatremia, because it may exacerbate the development of brain edema in the postoperative setting. Particularly in pediatric patients undergoing external CSF drainage, replacement of drained CSF by physiologic saline should be considered.
Various factors may contribute to the high risk of electrolyte disorders in neurointensive care:
Glucose
Glucose is an essential substrate for brain metabolism, and every effort should be made to ensure adequate delivery to the nervous tissue. In general intensive care, tight glycemic control has been advocated based on the knowledge that outcome is poorer in the presence of hyperglycemia and following the results of the study by van den Berghe et al.,88 showing reduced mortality in surgical intensive care by keeping glycemia within narrow limits (80-110 mg/dL). These promising findings have, however, been challenged by a more recent trial.89
Although in neurointensive care as well, various studies have demonstrated an association between elevated glucose levels and poorer outcome,90–93 the question whether this association may be causal or simply a marker has remained unanswered. In neurointensive care, the concern is that the injured brain cannot tolerate hypoglycemia, which might result as an adverse event from overenthusiastic glycemic control. There is a strict relationship between the increased use of insulin (for tight glycemic control) and the occurrence of hypoglycemia.94,95
Moreover, lowering blood glucose to “normal” levels may result in unacceptably low levels of glucose in the brain, depriving the most complex organ in the human body of its most essential metabolic substrate. That this concern is real has been demonstrated in microdialysis studies.96–98
 Brain Monitoring and Specific Therapeutic Approaches
Brain Monitoring and Specific Therapeutic Approaches
In comparison to the setting in cardiac intensive care, the possibilities for brain monitoring are still relatively limited.99 In cardiac care, routinely measured parameters include a multitude of pressure indices and a number of different serum markers (e.g., creatine kinase fractions, troponin) to determine if the heart is at risk for further injury. Physiologically, the heart is monitored by electrocardiography and intermittently with echocardiography. In contrast, routine monitoring of the brain is restricted in most centers to ICP and CPP monitoring, but the field is rapidly evolving. Monitoring of cerebral oxygenation is now being increasingly implemented in clinical practice100–102 and continuous EEG performed in some centers.103–105 Magnetic resonance spectroscopy now offers opportunities to noninvasively assess brain metabolism.106,107 Advances in the field of biomarkers are encouraging and offer hope that detection and tracking of pathophysiologic processes in the brain may now be within reach.108,109
As noted, current approaches to brain-specific monitoring include measurements of ICP, cerebral oxygenation, cerebral blood flow (CBF), electrical monitoring, and metabolic monitoring. These specific modalities are discussed in detail in Chapter 31. Here we focus on essential aspects regarding interpretation of monitoring results and therapeutic implications.
Intracranial Pressure and Cerebral Perfusion Pressure
Routine ICP monitoring is not generally indicated in patients with mild or moderate TBI but may be considered when other severe extracranial injuries are present, necessitating anesthesia for surgery, or when the initial CT shows traumatic lesions with space-occupying effects.110 ICP monitoring is further indicated in poor-grade patients with aneurysmal SAH.111–113 Further, it may be considered in patients with other intracranial disorders who are sedated and ventilated and in whom the risk of raised ICP is considered present (postoperative swelling, stroke, Reye syndrome).
ICP monitoring carries a 0.5% risk of hemorrhage and a 2% risk of infection.114 Intracranial hemorrhages are a rare complication of ICP monitoring and are usually caused by multiple punctures in the presence of coagulopathies. The risk of infection is higher in the case of ventricular monitoring, and the rate of infection is proportional to the duration of monitoring.115 Intraventricular catheters are preferable because they are accurate, can be recalibrated, and allow drainage of CSF. Intraparenchymal probes are user friendly and accurate. Less accurate data are provided by subdural catheters,116 and epidural probes are unreliable.117,118 The accuracy of ICP monitoring can be enhanced by use of computer-supported systems.119 Attempts to monitor ICP noninvasively have been unsatisfactory.120,121
Relatively few data exist on routine ICP monitoring in the postoperative situation. In a series of 30 patients after severe TBI and elective craniectomy, 156 instances of raised ICP and/or reduced CPP were recorded.122 These instances were only accompanied by clinical deterioration in 15 cases. Telemetric ICP control has been proposed after posterior fossa surgery.123 In a series of 514 patients after supra- and infratentorial surgery, Constantini et al.124 described raised ICP in 13% and 18% of cases, respectively. Neurologic deterioration occurred in approximately half of the patients suffering ICP rise and was always preceded by the ICP increase. In a large series of 780 patients submitted to routine ICP monitoring after intracranial surgery, 47% required ICP-directed therapy.125 In a report concerning 850 cases, Bullock and associates126 concluded that ICP monitoring allows earlier identification of recurrent hematomas. These data would support a more routine application of ICP monitoring after intracranial surgery, particularly in more complex cases. In some institutions, ICP is routinely measured as part of postoperative surveillance after major neurosurgical procedures, especially when risk of postoperative bleeding exists. Figure 41-3 illustrates a case in which a substantial ICP rise was detected in the first postoperative hours. An enlarging hemorrhage was responsible and required reintervention.
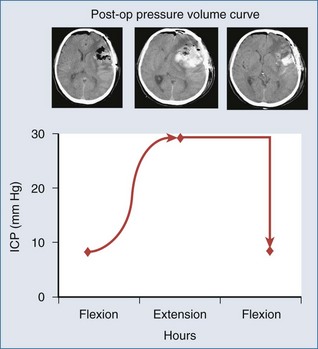
Figure 41-3 Raised intracranial pressure (ICP) as the first indication of a developing postoperative hematoma.
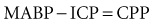
It is important to recognize that physiologic and nonphysiologic wave forms may occur. Technical artifacts and systemic causes should be excluded before specific diagnostic procedures are instituted or ICP-directed therapy initiated or intensified (Table 41-6).
TABLE 41-6 Remediable Extracranial Causes of Intracranial Hypertension
In some patients, the normal pressure autoregulatory mechanisms are disturbed, and the risk exists that increased CPP may worsen cerebral edema. Careful observation of the change in ICP with respect to arterial BP changes is required to determine whether autoregulation is disturbed or intact. For continuous evaluation of the autoregulatory status, it has been proposed to calculate the pressure-reactivity index (PRx) as the moving correlation coefficient between MABP and ICP.127–129 The added value of this approach, however, still requires confirmation.
Treatment of Cerebral Herniation and Elevated Icp
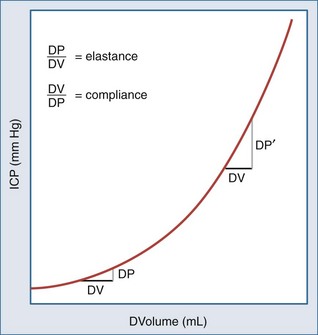
The development of cerebral herniation (tentorial herniation/cerebellar tonsillar herniation) constitutes a neurosurgical emergency. Rapid intervention is required prior to further investigations to determine the cause. According to the concept of the pressure volume curve (Figure 41-4), a small reduction in intracranial volume will already significantly decrease raised ICP and reverse herniation. The emergency measures to be taken include:
The main intracranial causes of raised ICP are:
In the absence of an acute cerebral herniation, elevated ICP is addressed first by ruling out treatable intracranial mass lesions and remediable extracranial causes or monitor malfunction (see Table 41-6).
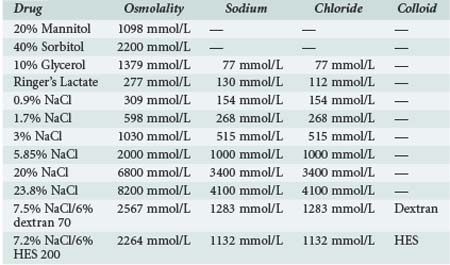
TABLE 41-7 Composition of Different Commercially Available Hypertonic Solutions Used for Treatment of Raised Intracranial Pressure
If these methods fail, second-tier therapies for raised ICP include:
Cerebral Blood Flow
Recent years have seen great advances in approaches to monitoring CBF and CBF-related variables, particularly in the field of neuroimaging. Both CT and MRI techniques have been developed for perfusion imaging and angiography, and possibilities for determining areas of the brain at risk for ischemia are now routinely available to the clinician. These approaches have replaced measurements of CBF with stable xenon CT scanning. Positron emission tomography (PET) studies for CBF and metabolic studies of the brain have largely remained in the domain of research. Thermal diffusion flowmetry has been introduced as a bedside technique for continuously monitoring CBF, but experience is as yet limited.116,130,131 A major drawback of this sensor is that it is not MRI compatible. Transcranial Doppler (TCD) provides a noninvasive assessment of blood flow velocity through the basal cerebral arteries. TCD is widely used for the detection and tracking of cerebral vasospasm,132 but various studies have shown a disappointing correlation when measured flow velocities are compared with direct measurements of CBF.133,134 In patients with stroke, detection of emboli is possible with most current TCD devices.135
Vasopressor therapy may be needed in the postoperative care of patients in the neuro-ICU. Vasopressors are often required in the treatment of SAH and severe TBI (see Chapters 35 and 38). It is important to realize that the pathophysiologic mechanism in these disorders is different, and that commonly employed approaches for treatment of delayed ischemic deficits following aneurysmal SAH cannot be directly translated to the situation of TBI.
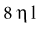
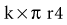
where k = a constant, r = radius of the blood vessel, l = the length of the blood vessel (practically constant), and η = dynamic blood viscosity. The most powerful factor in this equation is the vessel radius.
The vasopressors most frequently used in the care of the postoperative neurosurgical patients are listed in Table 41-8. Dose ranges are provided, but in general it is recommended to titrate the required dose versus the desired BP or CPP.
TABLE 41-8 Vasopressors Commonly Used in the Neurocritical Care Unit
| Agent | Adrenergic Effect | Doses (µg/kg/min) in Adults |
|---|---|---|
| Norepinephrine | Mixed α and β (α >>> β) |
0.02-1.5 |
| Phenylephrine | Pure α | 0.1-9.0 |
| Adrenaline | Mixed α and β (α > β) |
0.1-1 |
NOTE: The use of dopamine, a precursor of norepinephrine, has mainly been abandoned because of its interference with hormone secretion. α, alpha-adrenergic effect; β, beta-adrenergic effect.
Cerebral Oxygenation and Metabolism
Global cerebral oxygenation may be assessed using jugular oximetry, which is discussed in Chapter 31. When hemoglobin concentration and arterial hemoglobin saturation remain constant, AJDO2 may be estimated by simply recording SjvO2. A decrease in SjvO2 indicates that the brain is extracting more oxygen, suggesting that the oxygen supply is inadequate for metabolic demands. Values below 55% indicate an increased oxygen extraction relative to perfusion and suggest the presence of ischemia.136,137
Interpretation of results of jugular oximetry requires that both systemic information (e.g., hemoglobin concentration and arterial saturation) and intracranial data (e.g., CPP) be combined. The technique has limitations: first, continuous monitoring of SjvO2 with fiberoptic devices is prone to artifact; and second, under conditions of anemia or arterio venous shunting, hypoxia may be present at the tissue level despite normal values of jugular saturation.138 Moreover, SjvO2 is a measure of global cerebral oxygenation and does not reflect disturbances due to focal lesions, thus potentially failing to detect ischemia in relevant portions of brain tissue.139
NIRS is a noninvasive technique that permits estimation of oxyhemoglobin (HbO2), deoxyhemoglobin (Hb), and oxidized cytochrome oxidase (CytOx) over the combined arterial, capillary, and venous compartments.140 Various assumptions are made in the calculation algorithm of cerebral oxygen saturation with NIRS that may not always be valid, and uncertainty exists whether NIRS, as claimed, mainly measures the intracranial compartment or that recorded values are “contaminated” by the extracranial compartment.141 The main clinical applications are in neonatology and in coronary or carotid artery surgery.142,143 Recent intracranial surgery and subcutaneous swelling or wounds to the scalp, common in patients with TBI, preclude application of this technique. We do not consider it suitable for routine use in monitoring oxygenation in patients undergoing neurosurgical operations; yet, a noninvasive technique to assess cerebral oxygenation is attractive, and further clinical research should be encouraged.
Brain tissue oxygen tension indicates the balance between oxygen delivered to the tissue and its consumption in a specific area and can indicate regional hypoxia if it falls below 15 to 20 mm Hg.144,145 The diameter of microvascular vessels and diffusion barriers might also influence recorded values.146,147 In TBI, low values of PbrO2 occur in over 50% of patients during the first 24 hours, and depth and duration are related to outcome. Increased hyperventilation has further been shown to reduce PbrO2.139,146 Experimental and clinical evidence suggests that CPP therapy may be targeted towards appropriate levels, based on results of tissue PbrO2 monitoring.148 Non-randomized studies have indicated benefit of an oxygen-targeted treatment protocol.149–151
Microdialysis
The technique of microdialysis allows for measurement of substrate and metabolites (glucose, lactate, pyruvate), amino acids (glutamate), as well as indicators of cerebral damage (glycerol or other proteins as tau and beta amyloid) in the extracellular fluid of the brain.152,153 Dialysate fluid obtained after infusing saline through a semipermeable membrane reflects the composition of the extracellular fluid around the probe. Microdialysis is employed in various specialized neurointensive care units, mainly for research purposes. Technical and logistic considerations, as well as delays in obtaining real-time values, have inhibited the routine application of results toward individualized targeted treatment. The availability of microdialysis catheters with a high cutoff membrane now permit detection of larger molecules and may offer opportunities for tracking the inflammatory response.154–158
Electrical Monitoring
Continuous EEG (cEEG) monitoring has the potential for detecting nonconvulsive status epilepticus in ICU patients. As a primary monitor of brain function, cEEG can be used to titrate continuous infusion of sedative agents, and the technique can further alert the physician to development of focal or global ischemia.159,160 The sensitivity for detecting ischemia and hypoxia is high, but the specificity is low owing to effects of sedative medications. Continuous EEG may permit detection and treatment of such adverse events at an early stage, with a potential positive effect on outcome.161 Electroencephalographic bispectral analysis (BIS) may be useful in assessing the level of sedation in neurocritical care patients.162
In the research setting, interest exists in monitoring cortical spreading depression. Traumatically damaged neurons decrease their firing rates substantially in the early postinjury period. Waves of depolarization result in ionic flux and loss of resting membrane potential, which worsens neurochemical dysregulation and places extra metabolic demands on damaged tissue.163–166 Measurement of evoked potentials,167 assessing the integrity of sensory and motor pathways, may provide diagnostic and prognostic information, but because of the complexity of the technique, it is not recommended for general use.
 Neuroprotection
Neuroprotection
Over the past decades, the concept of neuroprotection has been extended to include treatment started after the onset of an insult, reflecting our increased understanding of progressive pathophysiologic mechanisms causing and/or enhancing secondary brain damage. In neuroprotection, four main approaches can be discerned (Table 41-9).
Strategies Aimed at Improving Metabolism and Microenvironment
Methods for improving metabolism and microenvironment include hypothermia to minimize the effects of energy failure and hyperosmolar therapy to reduce ICP and improve CBF. Hypothermia decreases cerebral blood flow by approximately 5.2% per degree of reduction in body temperature. The cerebral metabolic rate for oxygen (CMRO2) and the arterial jugular venous oxygen difference (AVDO2) fall after the institution of moderate hypothermia. This reflects a reduction in energy requirement and hence less energy loss in the injured brain. Many other effects of hypothermia, such as stabilization of the cell membrane168 and reduction of neurotransmitter turnover, may also contribute to the benefit seen in models of ischemia.169 Consequently, hypothermia is currently seen more as a neuroprotective approach than as a metabolic depressant. The use of hypothermia is therefore not without risks and requires high-level neurointensive care.
Agents Acting on Specific Mechanisms
Increased understanding of the existence of progressive pathophysiologic mechanisms causing or enhancing secondary brain damage has led to the development of a large range of specifically targeted neuroprotective agents aimed at ameliorating such mechanisms, often showing marked beneficial effect in experimental studies.170 Unfortunately, in various fields of neurointensive care, promising experimental results have not translated into clinical efficacy. In addition to the heterogeneity of patient populations, the lack of clinical parameters for effectively identifying mechanistic targets has contributed to these failures. The emerging field of biomarkers and advanced neuroimaging offer hope for the future.
Pluripotent Agents and Combinational Therapies
The realization that various pathophysiologic mechanisms are often concurrently or sequentially active has increased interest in the use of agents with multiple mechanisms; for such agents, the term “dirty drugs” has been coined.171
Corticosteroids, barbiturates, and magnesium are examples of pluripotent neuroprotective agents. Despite their efficacy in treating vasogenic edema, as encountered in brain tumors, corticosteroids are not efficacious in improving cytotoxic edema, as seen after TBI or SAH. Various studies support a neuroprotective effect of magnesium in patients with SAH.172,173 A recent randomized controlled trial, however, could not confirm benefit.174 In TBI, greater mortality and poorer outcome was found in a randomized clinical trial investigating the efficacy of magnesium.175
Erythropoietin (EPO), cyclosporine, and progesterone are agents with neuroprotective potential currently undergoing further clinical evaluation. Rather than seeking a single “silver bullet” agent targeting multiple mechanisms, it may be better to consider combining agents with complementary targets and effects.176 Fundamental to this approach, and in fact to any neuroprotective strategy, would be the accurate detection and tracking of pathophysiologic processes occurring in individual patients, which would provide better evidence for combining or sequential administration of neuroprotective agents.170
 Strategies Promoting Cell Survival and Regeneration
Strategies Promoting Cell Survival and Regeneration
Strategies to promote cell survival and regeneration include cellular replacement, gene therapy, and administration of trophic factors. These approaches are aimed at promoting regeneration and neuroplasticity and may ultimately lead to improved functional recovery.177,178 The potential of these novel approaches is strengthened by promising experimental and clinical results obtained in neurodegenerative diseases including Parkinson’s disease, Huntington’s disease, and stroke.178–181 Promoting cell survival and regeneration is currently the focus of large research efforts that may provide possibilities for further improving outcome in the subacute and chronic phases.
Key Points
Heros RC. Case volume and mortality. J Neurosurg. 2003;99(5):805-806.
Dubey A, Sung WS, Shaya M, et al. Complications of posterior cranial fossa surgery–an institutional experience of 500 patients. Surg Neurol. 2009 Oct;72(4):369-375.
Dankbaar JW, Slooter AJ, Rinkel GJ, Schaaf IC. Effect of different components of triple-H therapy on cerebral perfusion in patients with aneurysmal subarachnoid haemorrhage: a systematic review. Crit Care. 2010;14(1):R23.
Sen J, Belli A, Alborn H, Morgan L, Petzold A, Kitchen N. Triple-H therapy in the management of aneurysmal subarachnoid hemorrhage. Lancet Neurol. 2003;2(10):614-621.
Leal-Noval SR, Munoz-Gomez M, Murillo-Cabezas F. Optimal hemoglobin concentration in patients with subarachnoid hemorrhage, acute ischemic stroke and traumatic brain injury. Curr Opin Crit Care. 2008;14(2):156-162.
Oddo M, Schmidt JM, Carrera E, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. 2008;36(12):3233-3238.
Bhatia A, Gupta AK. Neuromonitoring in the intensive care unit. Intracranial pressure and cerebral blood flow monitoring. Intensive Care Med. 2007;33(7):1263-1271.
Broessner G, Beer R, Lackner P, et al. Prophylactic, endovascularly based, long-term normothermia in ICU patients with severe cerebrovascular disease: bicenter prospective, randomized trial. Stroke. 2009;40(12):657-665.
Margulies S, Hicks R. The Combination Therapies for Traumatic Brain Injury Workshop Leaders. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26(6):925-939.
1 MacKenzie EJ, Rivara FP, Jurkovich GJ, et al. A national evaluation of the effect of trauma center care on mortality. N Engl J Med. 2006;354:366-378.
2 Heros RC. Case volume and mortality. J Neurosurg. 2003;99:805-806.
3 Suarez JI, Zaidat OO, Suri MF, et al. Length of stay and mortality in neurocritically ill patients: impact of a specialized neurocritical care team. Crit Care Med. 2004;32:2311-2317.
4 Diringer MN, Edwards DF. Admission to a neurologic/neurosurgical intensive care unit is associated with reduced mortality rate after intracerebral hemorrhage. Crit Care Med. 2001;29:635-640.
5 Mirski MA, Chang CW, Cowan R. Impact of a neuroscience intensive care unit on neurosurgical patient outcomes and cost of care: evidence-based support for an intensivist-directed specialty ICU model of care. J Neurosurg Anesthesiol. 2001;13:83-92.
6 Cross DT3rd, Tirschwell DL, Clark MA, et al. Mortality rates after subarachnoid hemorrhage: variations according to hospital case volume in 18 states. J Neurosurg. 2003;99:810-817.
7 Patel HC, Bouamra O, Woodford M, King AT, Yates DW, Lecky FE, Trauma Audit and Research Network. Trends in head injury outcome from 1989 to 2003 and the effect of neurosurgical care: an observational study. [published erratum in Lancet 2006; 367: 816]. Lancet. 2005;366:1538-1544.
8 Patel HC, Menon DK, Tebbs S, et al. Specialist neurocritical care and outcome from head injury. Intensive Care Med. 2002;28:547-553.
9 Bulger EM, Nathens AB, Rivara FP, et al. Management of severe head injury: institutional variations in care and effect on outcome. Crit Care Med. 2002;30:1870-1876.
10 Fakhry SM, Trask AL, Waller MA, et al. IRTC Neurotrauma Task Force: management of brain-injured patients by an evidence-based medicine protocol improves outcomes and decreases hospital charges. J Trauma. 2004;56:492-493.
11 Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg. 2006;105:15-20.
12 Macmillan CS, Grant IS, Andrews PJ. Pulmonary and cardiac sequelae of subarachnoid haemorrhage: time for active management? Intensive Care Med. 2002;28:1012-1023.
13 Simon RP, Gean-Marton AD, Sander JE. Medullary lesion inducing pulmonary edema: a magnetic resonance imaging study. Ann Neurol. 1991;30:727-730.
14 Inobe J, Mori T, Ueyama H, et al. Neurogenic pulmonary edema induced by primary medullary hemorrhage: A case report. J Neurol Sci. 2000;172:73-76.
15 Keegan MT, Lanier WL. Pulmonary edema after resection of a fourth ventricle tumor: possible evidence for a medulla-mediated mechanism. Mayo Clin Proc. 1999;74:264-268.
16 Fontes RB, Aguiar PH, Zanetti MV, et al. Acute neurogenic pulmonary edema: case reports and literature review. J Neurosurg Anesthesiol. 2003;15:144-150.
17 Stocchetti N. Wet lungs, broken hearts and difficult therapies after subarachnoid hemorrhage. Crit Care. 2010;14:140.
18 Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med. 2005;353:2788-2796.
19 Smith WS, Matthay MA. Evidence for a hydrostatic mechanism in human neurogenic pulmonary edema. Chest. 1997;111:1326-1333.
20 Harhangi BS, Kompanje EJ, Leebeek FW, Maas AI. Coagulation disorders after traumatic brain injury. Acta Neurochir (Wien). 2008;150:165-175.
21 Halpern CH, Reilly PM, Turtz AR, Stein SC. Traumatic coagulopathy: the effect of brain injury. J Neurotrauma. 2008;25:997-1001.
22 Zehtabchi S, Soghoian S, Liu Y, et al. The association of coagulopathy and traumatic brain injury in patients with isolated head injury. Resuscitation. 2008;76:52-56.
23 Murshid WR, Gader AG. The coagulopathy in acute head injury: comparison of cerebral versus peripheral measurements of haemostatic activation markers. Br J Neurosurg. 2002;16:362-369.
24 Stein SC, Chen XH, Sinson GP, Smith DH. Intravascular coagulation: a major secondary insult in nonfatal traumatic brain injury. J Neurosurg. 2002;97:1373-1377.
25 Saggar V, Mittal RS, Vyas MC. Hemostatic abnormalities in patients with closed head injuries and their role in predicting early mortality. J Neurotrauma. 2009;26:1665-1668.
26 Bershad M, Farhadi S, Suri MF, et al. Coagulopathy and in-hospital deaths in patients with acute subdural hematoma. J Neurosurg. 2008;109:664-669.
27 Hamilton MG, Hull RD, Pineo GF. Venous thromboembolism in neurosurgery and neurology patients: a review. Neurosurgery. 1994;34:280-296.
28 Carman TL, Kanner AA, Barnett GH, Deitcher SR. Prevention of thromboembolism after neurosurgery for brain and spinal tumors. South Med J. 2003;96:17-22.
29 Gnanalingham KK, Holland JP. Attitudes to the use of prophylaxis for thrombo-embolism in neurosurgical patients. J Clin Neurosci. 2003;10:467-469.
30 Agnelli G, Piovella F, Buoncristiani P, et al. Enoxaparin plus compression stockings compared with compression stockings alone in the prevention of venous thromboembolism after elective neurosurgery. N Engl J Med. 1998;339:80-85.
31 Constantini S, Kanner A, Friedman A, et al. Safety of perioperative minidose heparin in patients undergoing brain tumor surgery: a prospective, randomized, double-blind study. J Neurosurg. 2001;94:918-921.
32 Gerlach R, Scheuer T, Beck J, et al. Risk of postoperative hemorrhage after intracranial surgery after early nadroparin administration: result of a prospective study. Neurosurgery. 2003;53:1028-1034.
33 Gerlach R, Krause M, Seifert V, Goerlinger K. Hemostatic and hemorrhagic problems in neurosurgical patients. Acta Neurochir (Wein). 2009;151:873-900.
34 Norwood SH, McAuley CE, Berne JD, et al. Prospective evaluation of the safety of enoxaparin prophylaxis for venous thromboembolism in patients with intracranial hemorrhagic injuries. Arch Surg. 2002;137:696-701. discussion 701-2
35 Norwood SH, Berne JD, Rowe SA, Villareal DH, Ledlie JT. Early venous thromboembolism prophylaxis with enoxaparin in patient with blunt traumatic brain injury. J Trauma. 2008;65:1021-1026.
36 Brain Trauma Foundation, American Association of Neurological Surgeons (AANS), Congress of Neurological Surgeons (CNS), AANS/CNS Joint Section on Neurotrauma and Critical Care. Guidelines for the management of severe traumatic brain injury, 3rd edition. V. Deep vein thrombosis Prophylaxis. J Neurotrauma. 2007;24(Suppl. 1):S32-S36.
37 Piotrowski JJ, Alexander JJ, Brandt CP, et al. Is deep vein thrombosis surveillance warranted in high-risk trauma patients? Am J Surg. 1996;172:210-213.
38 Latronico N, Berardino M. Thromboembolic prophylaxis in head trauma and multiple-trauma patients. Minerva Anestesiol. 2008;74:543-548.
39 Hotta K, Seo N, Kouno Y. Spinal hematoma associated with heparin therapy for venous thromboembolism prophylaxis. Masui. 2007;56:794-800.
40 Palmer JD, Sparrow OC, Iannotti F. Postoperative hematoma: a 5-year survey and identification of avoidable risk factors. Neurosurgery. 1994;35:1061-1064. discussion 1064-5
41 Marquardt G, Setzer M, Schick U, Seifert V. Cerebellar hemorrhage after supratentorial craniotomy. Surg Neurol. 2002;57:241-251. discussion 251-2
42 Honegger J, Zentner J, Spreer J, Carmona H, Schulze-Bonhage A. Cerebellar hemorrhage arising postoperatively as a complication of supratentorial surgery: a retrospective study. J Neurosurg. 2002;96:248-254.
43 Van Roost D, Thees C, Brenke C, et al. Pseudohypoxic brain swelling: a newly defined complication after uneventful brain surgery, probably related to suction drainage. Neurosurgery. 2003;53:1315-1326. discussion 1326-7
44 Schirmer Cm, Heilman CB, Bhardway A. Pneumocephalus: case illustrations and review. Neurocrit Care. 2010. DOI 10.1007/S 12028-010-9363-0
45 Vespa PM, Nuwer MR, Nenov V, et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg. 1999;91:750-760.
46 Menovsky T, van Overbeeke JJ. On the mechanism of transient postoperative deficit of cranial nerves. Surg Neurol. 1999;51:223-226.
47 Dubey A, Sung WS, Shaya M, et al. Complications of posterior cranial fossa surgery–an institutional experience of 500 patients. Surg Neurol. 2009;72:369-375.
48 McMillan HJ, Keene DL, Matzinger MA, et al. Brainstem compression: a predictor of postoperative cerebellar mutism. Childs Nerv Syst. 2009 Jun;25:677-681.
49 Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med. 2006;34:617-624.
50 Lazaridis C, Naval N. Risk factors and medical management of vasospasm after subarachnoid hemorrhage. Neurosurg Clin N Am. 2010;21:353-364.
51 Stein S, Levine J, Nagpal S, Leroux PD. Vasospasm as the sole cause of cerebral ischemia: how strong is the evidence? Neurosurg Focus. 2006;15:E2.
52 Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1761-1770.
53 Rinkel GJE, Feigin Vl, Algra A, et al. Calcium antagonists for aneurysmal subarachnoid hemorrhage. Cochrane Database Syst Rev 2005;1:CD000277.
54 Amenta F, Lanari A, Mignini F, et al. Nicardipine use in cerebrovascular disease: a review of controlled clinical studies. J Neurol Sci. 2009;283:219-223.
55 Dankbaar JW, Slooter AJ, Rinkel GJ, Schaaf IC. Effect of different components of triple-H therapy on cerebral perfusion in patients with aneurysmal subarachnoid hemorrhage: a systematic review. Crit Care. 2010;14:R23.
56 Naval NS, Stevens RD, Mirski MA, Bhardwaj A. Controversies in the management of aneurysmal subarachnoid hemorrhage. Crit Care Med. 2006;34:511-524.
57 Al-Shahi R, White PM, Davenport RJ, Lindsay KW. Subarachnoid haemorrhage. BMJ. 2006;333:235-240.
58 Rinkel GJ, Feigin VL, Algra A, van Gijn J. Circulatory volume expansion therapy for aneurysmal subarachnoid hemorrhage. Cochrane Database Syst Rev 2004;4:CD000483.
59 Raabe A, Beck J, Keller M, et al. Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 2005;103:974-981.
60 Sen J, Belli A, Alborn H, Morgan L, Petzold A, Kitchen N. Triple-H therapy in the management of aneurysmal subarachnoid hemorrhage. Lancet Neurol. 2003;2:614-621.
61 Treggiari MM, Deem S. Which H is the most important in triple-H therapy for cerebral vasospasm? Curr Opin Crit Care. 2009;15:83-86.
62 Keyrouz SG, Diringer MN. Clinical review: prevention and therapy of vasospasm in subarachnoid hemorrhage. Crit Care. 2007;11:220-230.
63 Hänggi D, Turowski B, Beseoglu K, Yong M, Steiger HJ. Intra-arterial nimodipine for severe cerebral vasospasm after aneurysmal subarachnoid hemorrhage: influence on clinical course and cerebral perfusion. AJNR Am J Neuroradiol. 2008;29:1053-1060.
64 Hamada J, Kai Y, Morioka M, et al. Effect on cerebral vasospasm of coil embolization followed by microcatheter intrathecal urokinase infusion into the cisterna magna: a prospective randomized study. Stroke. 2003;34:2549-2554.
65 Germanwala AV, Huang J, Tamargo RJ. Hydrocephalus after aneurysmal subarachnoid hemorrhage. Neurosurg Clin N Am. 2010;21:263-270.
66 Black PM. Hydrocephalus and vasospasm after subarachnoid hemorrhage from ruptured intracranial aneurysms. Neurosurgery. 1986;18:12-16.
67 Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81-84.
68 Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomized controlled trial. Lancet. 2008;12:126-134.
69 Livingston BM, Mackenzie SJ, MacKirdy FN, Howie JC. Should the pre-sedation Glasgow Coma Scale value be used when calculating Acute Physiology and Chronic Health Evaluation scores for sedated patients? Scottish Intensive Care Society Audit Group. Crit Care Med. 2000;28:389-394.
70 Naidech AM, Drescher J, Ault ML, et al. Higher hemoglobin is associated with less cerebral infarction, poor outcome, and death after subarachnoid hemorrhage. Neurosurgery. 2006;59:775-780.
71 Pendem S, Rana S, Manno EM, Gajic O. A review of red cell transfusion in the neurological intensive care unit. Neurocritical Care. 2006;04:63-67.
72 Leal-Noval SR, Munoz-Gomez M, Murillo-Cabezas F. Optimal hemoglobin concentration in patients with subarachnoid hemorrhage, acute ischemic stroke an traumatic brain injury. Curr Opin Crit Care. 2008;14:156-162.
73 Zygun DA, Nortje J, Hutchinson PJ, et al. The effect of red blood cell transfusion on cerebral oxygenation and metabolism after severe traumatic brain injury. Crit Care Med. 2009;37:1074-1078.
74 Sharma D, Vavilala MS. Transfusion improves cerebral oxygenation … but not always. Crit Care Med. 2009;37:1166-1167.
75 Goodnough LT, Bach RG. Anemia, transfusion, and mortality. N Engl J Med. 2001;345:1272-1274.
76 Foley N, Marshall S, Pikul J, Salter K, Teasell R. Hypermetabolism following moderate to severe traumatic acute brain injury: a systematic review. J Neurotrauma. 2008;25:1415-1431.
77 McIntyre L, Fergusson DA, Hebert PC, Moher D, Hutchison JS. Prolonged therapeutic hypothermia after traumatic brain injury in adults. JAMA. 2003;289:2992-2999.
78 Polderman KH, Wesley Ely E, Badr AE, Girbes ARJ. Induced hypothermia in traumatic brain injury: considering the conflicting results of meta-analyses and moving forward. Intensive Care Med. 2004;30:1860-1864.
79 Marion DW. Therapeutic moderate hypothermia and fever. Curr Pharm Des. 2001;7:1533-1536.
80 Zydlewski AWHJ. Hypothermia-induced hypokalemia. Mil Med. 1998;163:719-721.
81 Polderman KH, Peerdeman SM, Girbes AR. Hypophosphatemia and hypomagnesemia induced by cooling in patients with severe head injury. J Neurosurg. 2001;94:697-705.
82 Broessner G, Beer R, Lackner P, et al. Prophylactic, endovascularly based, long-term normothermia in ICU patients with severe cerebrovascular disease: bicenter prospective, randomized trial. Stroke. 2009;40:e657-e665.
83 Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991-1001.
84 Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519-531.
85 Kimelberg HK. Water homeostasis in the brain: basic concepts. Neuroscience. 2004;129:851-860.
86 Schneider HJ, Kreitschmann-Andermahr I, Ghigo E, Stalla GK, Agha A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage. JAMA. 2007;298:1429-1438.
87 Singh S, Bohn D, Carlotti AP, et al. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med. 2002;30:2575-2579.
88 van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359-1367.
89 NICE-SUGAR Study InvestigatorsFinfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283-1297.
90 Van Beek JG, Mushkudiani NA, Steyerberg EW, et al. Prognostic value of admission laboratory parameters in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:315-328.
91 Lannoo E, Van Rietvelde F, Colardyn F, et al. Early predictors of mortality and morbidity after severe closed head injury. J Neurotrauma. 2000;17:403-414.
92 Frontera JA, Fernandez A, Claassen J, et al. Hyperglycemia after SAH: predictors, associated complications, and impact on outcome. Stroke. 2006;37:199-203.
93 Diedler J, Sykora M, Hahn P, et al. Low hemoglobin is associated with poor functional outcome after non-traumatic, supratentorial intracerebral hemorrhage. Crit Care. 14, 2010. DOI10.1186/cc8961
94 Bilotta F, Caramia R, Paoloni FP, Delfini R, Rosa G. Safety and efficacy of intensive insulin therapy in critical neurosurgical patients. Anesthesiology. 2009;110:611-619.
95 De La Rosa GD, Donado JH, Restrepo AH, et al. GICI-HPTU. Strict glycaemic control in patients hospitalised in a mixed medical and surgical intensive care unit: a randomised clinical trial. Grupo de Investigacion en Cuidado intensivo. Crit Care. 2008;12:R120.
96 Vespa P, Boonyaputthikul R, McArthur DL, et al. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med. 2006;34:850-856.
97 Oddo M, Schmidt JM, Carrera E, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. 2008;36:3233-3238.
98 Meierhans R, Bechir M, Ludwig S, et al. Brain metabolism is significantly impaired at blood glucose below 6 mM and brain glucose beneath 1 mM in patients with severe traumatic brain injury. Crit Care. 2010.
99 Adelson DP. Cerebral oximetry in the head-injured patient: is it time for widespread application? Clin Neurosurg. 2007;54:58-63.
100 Stiefel MF, Udoetuk JD, Spiotta AM, et al. Conventional neurocritical care and cerebral oxygenation after traumatic brain injury. J Neurosurg. 2006;105:568-575.
101 Longhi l, Pagan F, Valeriani V, et al. Monitoring brain tissue oxygen tension in brain-injured patients reveals hypoxic episodes in normal-appearing and in peri-focal tissue. Intensive Care Med. 2007;33:2136-2142.
102 Andrews PJ, Citerio G, Longhi L, et al. Neuro-Intensive Care and Emergency Medicine (NICEM) Section of the European Society of Intensive Care Medicine: NICEM consensus on neurological monitoring in acute neurological disease. Intensive Care Med. 2008;34:1362-1370.
103 Scheuer ML. Continuous EEG monitoring in the intensive care unit. Epilepsia. 2002;43(Suppl. 3):114-127.
104 Nuwer MR. ICU EEG monitoring: nonconvulsive seizures, nomenclature, and pathophysiology. Clin Neurophysiol. 2007;118:1653-1654.
105 Oddo M, Carrera E, Claassen J, Mayer SA, Hirsch LJ. Continuous electroencephalography in the medical intensive care unit. Crit Care Med. 2009;37:2051-2056.
106 Garnett MR, Corkill RG, Blamire AM, et al. Altered cellular metabolism following traumatic brain injury: a magnetic resonance spectroscopy study. J Neurotrauma. 2001;18:231-240.
107 Signoretti S, Marmarou A, Aygok GA, et al. Assessment of mitochondrial impairment in traumatic brain injury using high-resolution proton magnetic resonance spectroscopy. J Neurosurg. 2008;108:42-52.
108 Kochanek PM, Berger RP, Bayir H, et al. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr Opin Crit Care. 2008;14:135-141.
109 Stocchetti N, Longhi L. The race for biomarkers in traumatic brain injury: what science promises and the clinicians still expect. Crit Care Med. 2010;38:318-319.
110 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care, AANS/CNS. Guidelines for the management of severe traumatic brain injury. VI. Indications for intracranial pressure monitoring. J Neurotrauma. 2007;24(Suppl. 1):S37-S44.
111 Mack WJ, King RG, Ducruet AF, et al. Intracranial pressure following aneurysmal subarachnoid hemorrhage: monitoring practices and outcome data. Neurosurg Focus. 2003;14:e3.
112 Stocchetti N, Longhi L, Magnoni S, et al. Head injury, subarachnoid hemorrhage and intracranial pressure monitoring in Italy. Acta Neurochir (Wien). 2003;145:761-765.
113 Heuer GG, Smith MJ, Elliott JP, Winn HR, LeRoux PD. Relationship between intracranial pressure and other clinical variables in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 2004;101:408-416.
114 Stocchetti N, Penny KI, Dearden M, et al. European Brain Injury Consortium. Intensive care management of head-injured patients in Europe. A survey from the European Brain Injury Consortium. Intensive Care Med. 2001;27:400-406.
115 Park P, Garton HJ, Kocan MJ, Thompson BG. Risk of infection with prolonged ventricular catheterization. Neurosurgery. 2004;55:594-601.
116 Bhatia A, Gupta AK. Neuromonitoring in the intensive care unit. Intracranial pressure and cerebral blood flow monitoring. Intensive Care Med. 2007;33:1263-1271.
117 Citerio G, Andrews PJ. Intracranial pressure. Part two: clinical applications and technology. Intensive Care Med. 2004;30:1882-1885.
118 Poca MA, Sahuquillo J, Topczewski T, Penarrubia MJ, Muns A. Is intracranial pressure monitoring in the epidural space reliable? Fact and fiction. J Neurosurg. 2007;106:548-556.
119 Zanier ER, Ortolano F, Ghisoni L, et al. Intracranial pressure monitoring in intensive care: clinical advantages of a computerized system over manual recording. Crit Care. 2007;11:R7.
120 Czarnik T, Gawda R, Latka D, et al. Noninvasive measurement of intracranial pressure: is it possible? J Trauma. 2007;62:207-211.
121 Stocchetti N. Could intracranial pressure in traumatic brain injury be measured or predicted noninvasively? Almost. Intensive Care Med. 2007;33:1682-1683.
122 Cotev S, Eimerl D, Feinsod M, Wald U, Perel A. The value of intracranial pressure monitoring after craniotomy and head injury. Crit Care Med. 1981;9:152.
123 Chapman PH, Cosman E, Arnold M. Telemetric ICP monitoring after surgery for posterior fossa and third ventricular tumors. Technical note. J Neurosurg. 1984;60:649-651.
124 Constantini S, Cotev S, Rappaport ZH, Pomeranz S, Shalit MN. Intracranial pressure monitoring after elective intracranial surgery. A retrospective study of 514 consecutive patients. J Neurosurg. 1988;69:540-544.
125 Takagi H, Morii S, Ohwadat T, Yada K. Clinical experience of 780 cases of postoperative ICP monitoring. In: Miller JD, Teasdale G, Rowan JO, Galbraith S, Mendelow AD, editors. ICP VI. Springer Verlag Berlin Heidelberg; 1986:695-700.
126 Bullock R, Hanemann CO, Murray L, Teasdale GM. Recurrent hematomas following craniotomy for traumatic intracranial mass. J Neurosurg. 1990;72:9-14.
127 Zweifel C, Lavinio A, Steiner LA, et al. Continuous monitoring of cerebrovascular pressure reactivity in patients with head injury. Neurosurg Focus. 2008;25:E2.
128 Steiner LA, Coles JP, Johnston AJ, et al. Assessment of cerebrovascular autoregulation in head-injured patients: a validation study. Stroke. 2003;34:2404-2409.
129 Schmidt B, Klingelhöfer J, Perkes I, Czosnyka M. Cerebral autoregulatory response depends on the direction of change in perfusion pressure. J Neurotrauma. 2009;26:651-656.
130 Vajkoczy P, Horn P, Thome C. Regional cerebral blood flow monitoring in the diagnosis of delayed ischaemia following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2003;98:1227-1234.
131 Vajkoczy P, Schomacher M, Czabanka M. Monitoring cerebral blood flow in neurosurgical intensive care. Eur Neurol Dis. 2007;II:2-6.
132 Kincaid MS. Transcranial Doppler ultrasonography: a diagnostic tool of increasing utility. Curr Opin Anaesthesiol. 2008;21:552-559.
133 Minhas PS, Menon DK, Smielewski P, et al. Positron emission tomographic cerebral perfusion disturbances and transcranial Doppler findings among patients with neurological deterioration after subarachnoid hemorrhage. Neurosurgery. 2003;52:1017-1024.
134 Chieregato A, Sabia G, Tanfani A, et al. Xenon-CT and transcranial Doppler in poor-grade or complicated aneurysmatic subarachnoid hemorrhage patients undergoing aggressive management of intracranial hypertension. Intensive Care Med. 2006;32:1143-1150.
135 Chung EML. Transcranial Doppler embolus detection: a primer. Ultrasound. 2006;14:202-210.
136 Robertson CS, Narayan RK, Gokaslan ZL, et al. Cerebral arteriovenous oxygen difference as an estimate of cerebral blood flow in comatose patients. J Neurosurg. 1989;70:222-230.
137 Chieregato A, Calzolari F, Trasforini G, Targa L, Latronico N. Normal jugular bulb oxygen saturation. J Neurol Neurosurg Psychiatry. 2003;74:784-786.
138 Dunn IF, Ellegala DB, Kim DH, et al. Neuromonitoring in neurological critical care. Neurocrit Care. 2006;4:83-92.
139 Coles JP, Fryer TD, Coleman MR, et al. Hyperventilation following head injury: Effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med. 2007;35:568-578.
140 Owen-Reece H, Smith M, Elwell CE, Goldstone JC. Near infrared spectroscopy. Br J Anaesth. 1999;82:418-426.
141 Maas A, Citerio G. Noninvasive monitoring of cerebral oxygenation in traumatic brain injury: a mix of doubts and hope. Intensive Care Med. 2010. accepted for publication
142 Murkin JM, Adams SJ, Novick RJ, et al. Monitoring brain oxygen saturation during coronary bypass surgery: a randomized, prospective study. Anesth Analg. 2007;104:51-58.
143 Pennekamp CW, Bots ML, Kappelle LJ, Moll FL, de Borst GJ. The value of near-infrared spectroscopy measured cerebral oximetry during carotid endarterectomy in perioperative stroke prevention. A review. Eur J Vasc Endovasc Surg. 2009;38:539-545.
144 Van den Brink WA, van Santbrink H, Steyerberg EW, et al. Brain oxygen tension in severe head injury. Neurosurgery. 2000;46:868-878.
145 Zauner A, Daugherty WP, Bullock MR, Warner DS. Brain oxygenation and energy metabolism: part I biological function and pathophysiology. Neurosurgery. 2002;51:289-302.
146 Carmona Suazo JA, Maas AIR, van den Brink WA, et al. CO2 reactivity and brain oxygen pressure monitoring in severe head injury. Crit Care Med. 2000;28:3268-3274.
147 van Santbrink H, Van den Brink WA, Steyerberg EW, et al. Brain tissue oxygen response in severe traumatic brain injury. Acta Neurochir (Wien). 2003;145:429-438.
148 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care, AANS/CNS. Guidelines for the management of severe traumatic brain injury. X. Brain oxygen monitoring and thresholds. J Neurotrauma. 2007;24(Suppl. 1):S65-S70.
149 Meixensberger J, Jaeger M, Vath A, et al. Brain tissue oxygen guided treatment supplementing ICP/CPP therapy after traumatic brain injury. J Neurol Neurosurg Psychiatry. 2003;74:760-764.
150 Stiefel MF, Spiotta A, Gracias VH, et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J Neurosurg. 2005;103:805-811.
151 Adamides AA, Cooper DJ, Rosenfeldt FL, et al. Focal cerebral oxygenation and neurological outcome with or without brain tissue oxygen-guided therapy in patients with traumatic brain injury. Acta Neurochir. 2009;151:1399-1409.
152 Hillered L, Vespa PM, Hovda DA. Translational neurochemical research in acute human brain injury: the current status and potential future for cerebral microdialysis. J Neurotrauma. 2005;22:3-41.
153 Marklund N, Blennow K, Zetterberg H, et al. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110:1227-1237.
154 Hutchinson PJ, O’Connell MT, Nortje J, et al. Cerebral microdialysis methodology—evaluation of 20 kDa and 100 kDa catheters. Physiol Meas. 2005;26:423-428.
155 Helmy A, Carpeneter KL, Skepper JN, et al. Microdialysis of cytokines: methodological considerations, scanning electron microscopy, and determination of relative recovery. J Neurotrauma. 2009;26:549-561.
156 Goodman JC, Robertson CS. Microdialysis: is it ready for prime time? Curr Opin Crit Care. 2009;15:110-117.
157 Hillman J, Åneman O, Persson M, et al. Variations in the response of interleukins in neurosurgical intensive care patients monitored using intracerebral microdialysis. J Neurosurg. 2007;106:820-825.
158 Mellergård P, Åneman O, Sjögren F, Pettersson P, Hillman J. Changes in extracellular concentrations of some cytokines, chemokines, and neurotrophic factors after insertion of intracerebral microdialysis catheters in neurosurgical patients. Neurosurgery. 2008;62:151-158.
159 Claassen J, Mayer SA. Continuous electroencephalographic monitoring in neurocritical care. Curr Neurol Neurosci Rep. 2002;2:534-540.
160 Scheuer ML. Continuous EEG monitoring in the intensive care unit. Epilepsia. 2002;43(Suppl. 3):114-127.
161 Visser MC, Vriens EM, Vandertop WP, et al. Changes in EEG synchronization level during epileptic seizures in ICU patients. J Neurosurg Anesthesiol. 2002;14:265.
162 Citerio G, Cormio M. Sedation in neurointensive care: advances in understanding and practice. Cur Opin Crit Care. 2003;9:120-126.
163 Strong AJ, Fabricius M, Boutelle MG, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738-2743.
164 Strong AJ, Hartings JA, Dreier JP. Cortical spreading depression: an adverse but treatable factor in intensive care? Curr Opin Crit Care. 2007;13:126-133.
165 Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224-3237.
166 Fabricius M, Fuhr S, Bhatia R, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778-790.
167 Guerit JM. Medical technology assessment EEG and evoked potentials in the intensive care unit. Neurophysiol Clin. 1999;29:301-317.
168 Ginsberg MD, Sternau LL, Globus MY, Dietrich WD, Busto R. Therapeutic modulation of brain temperature: relevance to ischemic brain injury. Cerebrovasc Brain Metab Rev. 1992;4:189-225.
169 Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20:904-910.
170 Royo NC, Shimizu S, Schouten JW, Stover JS, McINtosh TK. Pharmacology of traumatic brain injury. Curr Opin Pharmacol. 2003;3:27-32.
171 Barriere H, Belfodil R, Rubera I, et al. CFTR null mutation altered cAMP-sensitive and swelling-activated Cl- currents in primary cultures of mouse nephron. Am J Physiol Renal Physiol. 2003;284:F796-F811.
172 Ma L, Liu WG, Zhang JM, et al. Magnesium sulphate in the management of patients with aneurysmal subarachnoid hemorrhage: a meta analysis of prospective controlled trials. Brain Injury. 2010;24:730-735.
173 Westermaier T, Stetter C, Giles V, et al. Prophylactic intravenous magnesium sulfate for treatment of aneurysmal hemorrhage: a randomized, placebo-controlled, clinical study. Crit Care Med. 2010;38:1284-1290.
174 Wong GK, Poon WS, Chan MT, for the IMASH Investigators. Intravenous magnesium sulphate for aneurysmal subarachnoid hemorrhage (IMASH). A randomized, double-blinded, placebo-controlled, multicenter phase III trial. Stroke. 2010;41:921-926.
175 Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomized controlled study. Lancet Neurol. 2007;6:29-38.
176 Margulies S, Hicks R, The Combination Therapies for Traumatic Brain Injury Workshop Leaders. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26:925-939.
177 Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612-613.
178 Dunnett SB, Bjorklund A, Lindvall O. Cell therapy in Parkinson’s disease—stop or go? Nat Rev Neurosci. 2001;2:365-369.
179 Vink R, Nimmo AJ, Cernak I. An overview of new and novel pharmacotherapies for use in traumatic brain injury. Clin Exp Pharmacol Physiol. 2001;28:919-921.
180 Borlongan CV, Sanberg PR. Neural transplantation for treatment of Parkinson’s disease. Drug Discov Today. 2002;7:674-682.
181 Kondziolka DWL, Geldstein S. Transplantation of cultured of human neuronal cells for patients with stroke. Neurology. 2000;55:565-569.
